 Oxana V. Galzitskaya
Oxana V. Galzitskaya
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Mol. Neurosci. , 31 January 2020
Sec. Brain Disease Mechanisms
Volume 12 - 2019 | https://doi.org/10.3389/fnmol.2019.00319
This article is part of the Research Topic Strategies and Tools for Modulating Pathologic Protein Self-Assembly in Proteinopathies View all 17 articles
Currently, there is no effective treatment of proteinopathies, as well as their diagnosis in the early stages of the disease until the first clinical symptoms appear. The proposed model of fibrillation of the Aβ peptide and its fragments not only describes molecular rearrangements, but also offers models of processes that occur during the formation of amyloid aggregates. Since this model is also characteristic of other proteins and peptides, a new potential target for drug development in the treatment of Alzheimer’s disease (AD) and other proteinopathies is proposed on the basis of this model. In our opinion, it is oligomers that are promising targets for innovative developments in the treatment of these diseases.
In the process of folding, the protein molecule acquires a unique spatial structure, which is necessary for its biological function. Nevertheless, in cells, there are a number of conditions under which the process of protein folding is disrupted. This leads to the formation of protein oligomers forming insoluble aggregates. A variety of such aggregates are amyloid fibrils. The formation and accumulation of amyloid aggregates in organs and tissues is one of the observed stages of the pathogenesis of diseases, combined into a group of proteinopathies, which includes Alzheimer’s disease (AD), Parkinson’s disease (PD), type 2 diabetes mellitus, and various systemic amyloidoses (Saha et al., 2000; Hardy and Selkoe, 2002; Caughey and Lansbury, 2003; Chiti and Dobson, 2006; Lesné et al., 2006; Shankar et al., 2008).
Currently, there is no effective therapy for proteinopathies, as well as their diagnosis in the early stages of the disease until the first clinical symptoms appear. In addition, a large number of proteins that are not associated with pathological processes are capable of forming amyloid aggregates and fibrils in vitro. This allows us to conclude that the formation of amyloids is a common property of the polypeptide chain (Fändrich and Dobson, 2002). It is also known that amyloid fibrils formed by the same protein can have a high degree of polymorphism (Fändrich et al., 2009). Therefore, the study of the molecular mechanism of the pathogenesis of amyloidosis is one of the urgent and important tasks of modern medicine and molecular biology.
It is extremely alarming that the inefficiency of modern methods of treatment is associated with failures in the development of new drugs for the treatment of AD. The proportion of successful treatment attempts created by drugs during the decade from 2002 to 2012 is 0.4% (Ousset et al., 2014).
Cholinesterase Inhibitors (ChEIs) are a common form of drug treatment of AD, and the three most effective drugs are donepezil, galantamine, and rivastigmine. Side effects when using these drugs are different, but none of them contributes to a significant improvement in cognitive function in patients (Birks, 2006). There is evidence that prolonged exposure to these drugs even accelerates AD (Lu and Tune, 2003). In addition, they effectively increase the level of acetylcholine available for neurotransmission. Memantine is an alternative approved drug that only mildly inhibits the glutamatergic system by binding to N-methyl-D-aspartate receptors (NMDARs; Glasgow et al., 2017), which reduce excess Ca2+ in postsynaptic neurons associated with neurodegenerative diseases (Parsons et al., 2013). Glutamate receptors of the central nervous system play a key role in ensuring the plasticity of neurons and the processes of memory consolidation (under normal conditions). Hyperactivation of the N-methyl-D-aspartate (NMDA) subtype of these receptors leads to the development of neurotoxicity.
Memantine is also effective in combination with ChEIs (Tariot et al., 2004). Non-specific treatments for AD used include antidepressants, such as selective serotonin reuptake inhibitors fluoxetine and paroxetine, which can combine well with ChEI (Aboukhatwa et al., 2010). Other symptoms of AD, such as anxiety and psychosis, may be affected by drugs such as anxiolytics, oxazepam or antipsychotics, risperidone (Ballard and Waite, 2006). Although these drugs are considered effective in the treatment of AD, they nevertheless affect only the symptoms of the disease.
From the point of view of drug targets in the treatment of AD, α-, β- and γ-secretases are studied, which are involved in APP proteolysis to the Aβ peptide. As mentioned above, the disruption of the aggregation of the Aβ peptide can lead to the prevention of plaque formation (Yang et al., 2019). There are several targets associated with the degradation of the Aβ peptide, one of which is neprilysin (Hornung et al., 2019). There are targets that regulate the expression of APP in patients with AD. It is also necessary to include targets related to the phosphorylation and aggregation of tau protein in this incomplete list.
As for β-secretase (BACE1), there are many studies on its inhibition, including docking of a number of flavonoids (Shimmyo et al., 2008), as well as a number of studies on virtual screening (Huang et al., 2005; John et al., 2011); later high-throughput screening (in combination with pharmacophore modeling to clarify), which revealed the reasons for the inhibition of this enzyme (Muthusamy et al., 2013). Studies of mutant forms of BACE1 in mice indicate that there may be serious side effects when inhibiting this particular enzyme. In particular, such effects can be neurodegeneration, which is a serious problem (Yan and Vassar, 2014). As regards γ-secretase, this intramembrane protein is involved not only in the APP proteolysis but also in a number of other processes (Minter et al., 2005). It is clear that inhibition of this enzyme leads to a decrease in the amount of Aβ peptide (He et al., 2010). The situation is complicated by another protein, β-arrestin 2, which apparently regulates γ-secretase, and thus, inhibition of this enzyme can reduce the formation of Aβ peptide plaques (Thathiah et al., 2013). Since the formation of the Aβ peptide is a sequential process from the APP precursor protein that requires the sequential participation of BACE1 and γ-secretase, combination therapy, including both BACE1 inhibitor and γ-secretase modulator, will be more effective than an individual treatment of each individual enzyme during the formation of Aβ peptide (Strömberg et al., 2015).
There are two competing hypotheses about the cause of AD: one of them is the amyloid hypothesis (Tanzi and Bertram, 2005). It is based on the idea that the amyloid Aβ peptide, instead of being synthesized and participating in metabolism, begins to accumulate in the brain and form aggregates in the form of plaques. The accumulation of the peptide leads to pathology, expressed in the death of neuron cells and the appearance of plaques containing this protein. Genetic data are also a source of confirmation of this hypothesis. The Aβ peptide precursor protein (APP) gene is located on chromosome 21, and trisomy of this chromosome in Down syndrome is the reason that AD is often observed in patients with Down syndrome (Masters et al., 1985; Maltsev et al., 2011), which in this case indicates the genetic basis of AD disease. Simple proteolysis is required to convert the Aβ peptide precursor protein (APP) to the Aβ peptide. It should be mentioned that no correlation was found between amyloid plaque formation and neuronal loss (Schmitz et al., 2004). The so-called “channel” hypothesis of AD, first proposed in 1993, states: that oligomers of amyloidogenic proteins make pores into the membrane that causes the influx of Ca2+ ions, an imbalance of ions of other metals, oxidative stress, and finally cell death (Arispe et al., 1993). The second hypothesis is associated with modifications of tau protein. Hyperphosphorylation of tau protein associated with microtubules leads to pathology of neural tangles. Recent studies have shown that there is a connection between these two hypotheses (Small and Duff, 2008; Jin et al., 2011; Maltsev et al., 2014). In addition to this, misfolding of the Aβ peptide and tau protein is observed, which leads to their uncontrolled aggregation. Observation of the pathological process shows that misfolding is distributed from local points by the prion-like mechanism for both tau and Aβ peptide (Bloom, 2014). For tau protein, the formation of polymorphic particles was shown by NMR analysis (Mukrasch et al., 2009).
In the last decade, several researchers have attempted to describe oligomeric particles, which are possibly the precursors of the formation of amyloid fibrils. The direct interest in oligomeric particles is due to the fact that, for example, in the case of AD, oligomers formed by the Aβ peptide are found in the brain tissues of patients suffering from this disease (Roher et al., 1996). A similar role of oligomers (nanomers and dodecamers) was noted in many studies since such particles have the highest toxicity compared to dimers, trimers, and tetramers. More and more facts indicate that in the pathogenesis of neurodegenerative diseases, it is the oligomers, and not the mature fibrils, that pose the greatest danger. Thus, it has recently been demonstrated that oligomers formed by the Aβ(1–42) peptide have a damaging effect on the blood-brain barrier, thereby disrupting brain homeostasis (Brkic et al., 2015).
It is believed that amyloid fibrils are specifically ordered aggregates characterized by the presence of a secondary structure of a certain type—a cross-β structure, in which β-sheets are parallel to the axis of fibril (Makin and Serpell, 2005). In the formation of amyloid aggregates by globular proteins, only part of their amino acid sequence is involved in the formation of the cross-β structure. The introduction of amino acid substitutions into these regions (amyloidogenic region) may affect the ability of the protein to form amyloid fibrils. The determination of amyloidogenic regions is necessary to understand the mechanism of amyloid aggregation and the pathogenesis of neurodegenerative diseases (Selivanova et al., 2016c; Surin et al., 2016).
In addition, it currently remains difficult to establish the spatial structure of fibrillar aggregates due to the limited capabilities of individual physicochemical methods. For example, the structure of amyloid fibrils can be determined using the method of solid-state NMR spectroscopy, however, this method is very time-consuming and ambiguous in the interpretation of the obtained data. Also, despite the fact that amyloidogenic proteins and peptides are the objects of study by a large number of researchers, and the appearance of a large number of publications at the moment there is no general model that describes the molecular mechanism of the formation of amyloid aggregates and fibrils (Fändrich et al., 2011). Protofibrils are not yet available on images from a cryogenic sample, and therefore high-resolution cryo-EM reconstructions from fibrils represent the average values of multiple conformations of protofilaments (Gremer et al., 2017). In this case, averaging as high-resolution information of individual protofilaments, as well as conformational variability in flexible regions, are lost (Seuring et al., 2018).
Using bioinformatics approaches and modern physicochemical methods, we studied the formation of amyloid aggregates of the Aβ(1–40), Aβ(1–42) peptides and their fragments, and a model is proposed that describes the mechanism of the structural organization of amyloid fibrils (Suvorina et al., 2015; Selivanova et al., 2016a,b,c, 2018a,b; Galzitskaya and Selivanova, 2017; Galzitskaya et al., 2018a,b; Galzitskaya, 2019).
Based on the developed kinetic model of amyloid formation, the sizes of primary and secondary nucleus of fibril formation were calculated for two isoforms of the Aβ peptide (Dovidchenko et al., 2014, 2016). Thus, for the Aβ(1–40) peptide, it was found that the size of the primary nucleus is two monomers, and for Aβ(1–42) three monomers. In this case, the size of the secondary nucleus for the Aβ(1–40) peptide is one monomer, and for Aβ(1–42) it is two monomers. Based on the obtained data, a structural model of the primary oligomer underlying the dodecamer was proposed (Dovidchenko et al., 2016).
It was shown by electron microscopy that the key structural element, which is the building block for the formation of amyloid fibrils, is a ring-like oligomer consisting of approximately 12 monomers for Aβ(1–40) and Aβ(1–42) peptides. The diameter of such an oligomer is 8–9 nm, and the height about 3 nm. The inner diameter of the ring oligomer is 3–4 nm. Ring-like oligomers are stacked in a fibril on a ring-to-ring or more frequently ring-on-ring basis with a slight overlap. Oligomers of this type are observed in electron micrographs from the moment when the amyloid formation process begins, and their number gradually decreases. Oligomers interact with each other not only on a ring-to-ring basis but can also attached to the side surface of fibrils due to lateral interactions, thereby increasing its thickness.
We have proposed an oligomer structure for the Aβ peptide and its fragments, which are very different from each other. The Aβ peptide oligomer consists of three primary oligomers that form a ring-like structure in a cross-section. Twelve monomers (56 kDa) form a tubular cylinder with an internal diameter of about 3–4 nm, and the salt bridges stabilize this oligomer: Arg5-Glu22 is formed between the primary oligomers, and Asp23-Lys28 is formed inside the monomeric structure. The regular sizes of the structures observed upon application of these oligomers are present on X-ray diffraction patterns (this is the meridional reflection at 53Å and the equatorial reflection at 55Å). In the case of oligomer for the fragments of Aβ peptide, such an oligomer is formed from 48 peptides that form 12 β-sheets arranged in a tubular cylinder with outer and inner diameters of 6 and 2 nm, respectively (Galzitskaya et al., 2018b).
An important characteristic of amyloid fibrils formed by Aβ(1–40) and Aβ(1–42) peptides is their polymorphism. The model of aggregation of amyloid fibrils that we have proposed is valuable in that it explains this polymorphism. In this case, this is due to a change in the association of oligomers during amyloidogenesis (Figure 1). The structure of the oligomer itself is determined by the amino acid sequence of the monomers. A vivid example of this can be demonstrated by the example of Aβ(1–40) and Aβ(1–42) isoforms and amyloidogenic fragments of the Aβ peptide. The constructions of oligomers differ in the structure of the complex and physicochemical properties (Dovidchenko et al., 2014; Galzitskaya and Selivanova, 2017; Selivanova et al., 2018b; Galzitskaya, 2019; Figure 1).
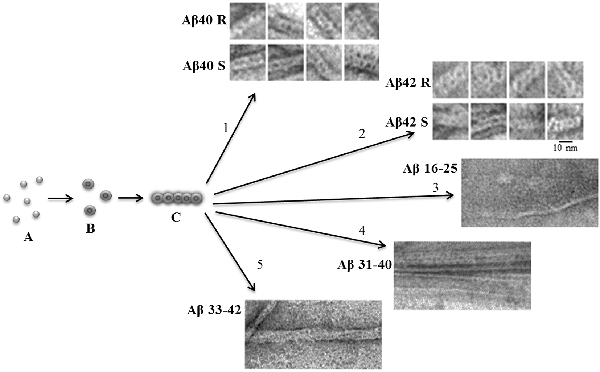
Figure 1. Schematic representation of the possible mechanism of fibril formation by Aβ peptide and its fragments. (A) Monomers; (B) ring-like oligomers; and (C) single fibril. R is a recombinant sample and S is synthetic.
To determine the amyloidogenic regions of the protein polypeptide chain that form intermolecular interactions in amyloid fibrils, an approach was used consisting in the limited proteolysis of mature fibrils and subsequent determination of the amino acid sequence of the obtained peptides using high-resolution mass spectrometry (Selivanova et al., 2016c; Surin et al., 2016; Galzitskaya et al., 2018b).
The data obtained using the approach described above allowed us not only to find out what structural transformations a protein or peptide molecule undergoes in the process of fibril formation, but also to confirm the described model of amyloid formation for Aβ(1–40) and Aβ(1–42) peptides. It should be noted that the experimentally determined amyloidogenic fragments coincided with the predicted sites using the FoldAmyloid program (Garbuzynskiy et al., 2010).
Recent evidence suggests that soluble Aβ peptide oligomers are a major cause of synaptic dysfunction and memory loss in AD. To further address this uncertainty, the neurotoxicity of various isoforms of Aβ peptide was analyzed at the cellular level. The results showed that Aβ(1–42) can form oligomers much faster than Aβ(1–40) oligomers, while Aβ(1–43) and Aβ(1–42) exhibit the highest level of neurotoxicity (Fu et al., 2017).
The EM images clearly show that fibrils are built from oligomeric structures for both the Aβ peptide and its fragments (Figure 1). The structure of Aβ(1–42) fibril, determined using cryo-electron microscopy, does not coincide with the EM images presented in the Supplementary: when increasing the EM images, it is clear that the fibril does not consist of endless beta-sheets obtained using the processing program cryo-EM, and the fibril is constructed of oligomeric structures laid in the same way as in our model (Gremer et al., 2017).
The possible organization of the oligomeric structure may answer the question, why mice do not have AD? In the mouse Aβ peptide, Gly is located instead of Arg5, thereby violating the salt bridge, a bond that stabilizes the layers of primary oligomers. And the presence of Arg13 instead of His13 only prevents the formation of such an oligomer structure. Thus, the replacement of three amino acids at the N-terminus of the murine Aβ peptide results in no signs of AD in mice (Figure 2A). The deletion of Glu22 (Osaka mutant) causes enhanced oligomerization of the Aβ peptide, but not fibrillogenesis (Tomiyama et al., 2008). Again, a salt bridge cannot form, a bond stabilizing the monomeric form of the Aβ peptide. Ala2Thr mutation (Jonsson et al., 2012) can slow down Aβ fibrillogenesis (Lin et al., 2017), but at the same time, Ala2Val mutation (Di Fede et al., 2009) accelerates Aβ fibrillogenesis and is associated with the early onset of AD (Messa et al., 2014), since such mutation leads to stabilization of the N-terminal part of the peptide. English mutation (Janssen et al., 2003) His6Arg promotes fibrillogenesis, enhances cytotoxicity, and increases the average size of Aβ oligomers (Ono et al., 2010). It should be noted that for only a few mutants, the model structure of monomer packing in amyloid fibril was obtained.
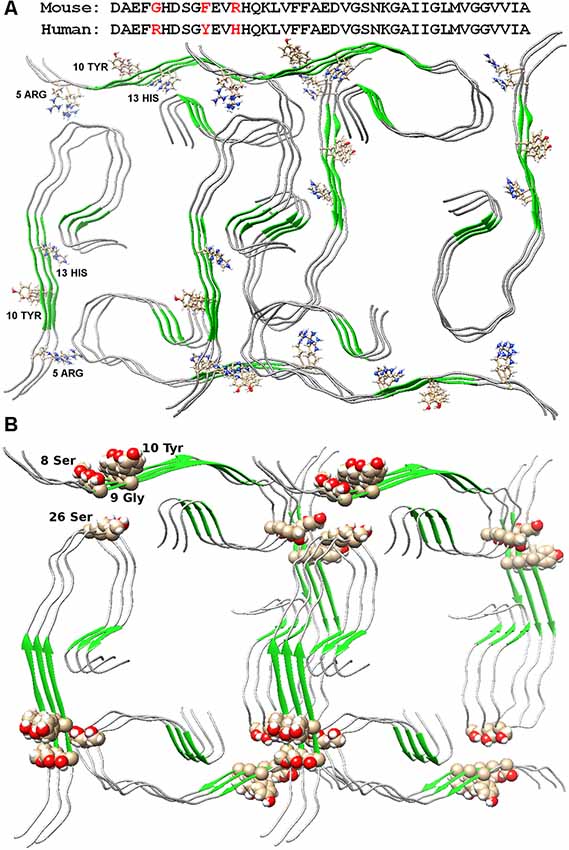
Figure 2. Packing of two dodecamer structures of Aβ(1–42) peptide in fibril. (A) Amino acid sequences of mouse and human Aβ(1–42) peptide are presented. Arg5, Tyr10, and His13 are indicated in the structures. (B) Ser26 and nearby residues are highlighted in the structure.
Five developed antibodies (Gantenerumab, Solanezumab, Aducanumab, Bapineuzumab, Crenezumab) did not reach clinical stage 3. This means that there was a misconception (vision) of the structure of the amyloid against which antibodies were developed. Among the ensemble of oligomers, it is necessary to single out the “correct” oligomer, which is involved in the construction of fibrils. And as we now understand, such an oligomer should be just 56 kDa dodecamer. It is just stable compared with the primary oligomer—tetramer. Murakami (2014) presents in his article a picture with a large pool of oligomers that will participate in the construction of fibrils. But if we take into account that the fibril is built from specific building material, then all the other oligomers should not interest us as a target.
The Ser26Glu mutation was detected in the Aβ peptide (a message from S. Linse at the Amyloid 2019 conference in Lund), which does not lead to cross-seeding, which means that the structures from which the fibrils are built for the wild type and mutant shape are different (Tran et al., 2017). From the point of view of existing structures, such a mutation should not affect the fibril structure in any way, since in both structures of 2016 and 2017, this residue looks at the solvent (Wälti et al., 2016; Gremer et al., 2017). Only in our model, this mutation will prevent the formation of an oligomeric particle, and most likely, the mutant form will have a completely different structure of the building block—the oligomer, so the process of cross-seeding is impossible (Figure 2B).
The study of the reasons and development of neurodegenerative diseases is an important and urgent task of modern medicine. The prevalence of these diseases is from 5 to 15%, depending on the age of the patient. It should also be noted that the spread of this group of diseases is also an acute social problem, since these diseases reduce the quality and life of patients. As a rule, such diseases are diagnosed in the late stages of development, when patients develop the impaired cognitive function. At the moment, the etiology and pathogenesis of various neurodegenerative diseases and proteinopathies are only being clarified, there are almost no diagnostic methods at an early stage of the disease and attempts to develop an effective method of treatment have practically no results. This is a direct consequence of a lack of understanding of key events in the molecular mechanism of the pathogenesis of neurodegenerative diseases and proteinopathies.
Our proposed model of fibrillation of Aβ peptide and its fragments not only describes molecular rearrangements, but also offers models of processes that occur during the formation of amyloid aggregates. In addition, we offer a new potential target for drug development in the treatment of AD. In our opinion, it is “correct” oligomeric complexes that are promising targets for innovative developments in the treatment of this disease.
The datasets generated for this study are available on request to the corresponding author.
The author confirms being the sole contributor of this work and has approved it for publication.
The study was funded by the Russian Science Foundation (grants #18-14-00321 and 14-14-00536).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to A.V. Glyakina for assistance in the preparation of the manuscript, O.M. Selivanova, and A.K. Surin for their help in obtaining EM and mass spectrometry analysis.
Aboukhatwa, M., Dosanjh, L., and Luo, Y. (2010). Antidepressants are a rational complementary therapy for the treatment of Alzheimer’s disease. Mol. Neurodegener. 5:10. doi: 10.1186/1750-1326-5-10
Arispe, N., Rojas, E., and Pollard, H. B. (1993). Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. U S A 90, 567–571. doi: 10.1073/pnas.90.2.567
Ballard, C., and Waite, J. (2006). The effectiveness of atypical antipsychotics for the treatment of aggression and psychosis in Alzheimer’s disease. Cochrane Database Syst. Rev. 4:CD003476. doi: 10.1002/14651858.CD003476.pub2
Birks, J. (2006). Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 1:CD005593. doi: 10.1002/14651858.CD005593
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Brkic, M., Balusu, S., Van Wonterghem, E., Gorlé, N., Benilova, I., Kremer, A., et al. (2015). Amyloid-β oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases. J. Neurosci. 35, 12766–12778. doi: 10.1523/JNEUROSCI.0006-15.2015
Caughey, B., and Lansbury, P. T. (2003). Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298. doi: 10.1146/annurev.neuro.26.010302.081142
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Di Fede, G., Catania, M., Morbin, M., Rossi, G., Suardi, S., Mazzoleni, G., et al. (2009). A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323, 1473–1477. doi: 10.1126/science.1168979
Dovidchenko, N. V., Finkelstein, A. V., and Galzitskaya, O. V. (2014). How to determine the size of folding nuclei of protofibrils from the concentration dependence of the rate and lag-time of aggregation. I. Modeling the amyloid protofibril formation. J. Phys. Chem. B 118, 1189–1197. doi: 10.1021/jp4083294
Dovidchenko, N. V., Glyakina, A. V., Selivanova, O. M., Grigorashvili, E. I., Suvorina, M. Y., Dzhus, U. F., et al. (2016). One of the possible mechanisms of amyloid fibrils formation based on the sizes of primary and secondary folding nuclei of Aβ40 and Aβ42. J. Struct. Biol. 194, 404–414. doi: 10.1016/j.jsb.2016.03.020
Fändrich, M., and Dobson, C. M. (2002). The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 21, 5682–5690. doi: 10.1093/emboj/cdf573
Fändrich, M., Meinhardt, J., and Grigorieff, N. (2009). Structural polymorphism of Alzheimer Aβ and other amyloid fibrils. Prion 3, 89–93. doi: 10.4161/pri.3.2.8859
Fändrich, M., Schmidt, M., and Grigorieff, N. (2011). Recent progress in understanding Alzheimer’s β-amyloid structures. Trends Biochem. Sci. 36, 338–345. doi: 10.1016/j.tibs.2011.02.002
Fu, L., Sun, Y., Guo, Y., Chen, Y., Yu, B., Zhang, H., et al. (2017). Comparison of neurotoxicity of different aggregated forms of Aβ40, Aβ42 and Aβ43 in cell cultures. J. Pept. Sci. 23, 245–251. doi: 10.1002/psc.2975
Galzitskaya, O. (2019). New mechanism of amyloid fibril formation. Curr. Protein Pept. Sci. 20, 630–640. doi: 10.2174/1389203720666190125160937
Galzitskaya, O. V., Galushko, E. I., and Selivanova, O. M. (2018a). Studies of the process of amyloid formation by Aβ peptide. Biochemistry 83, S62–S80. doi: 10.1134/S0006297918140079
Galzitskaya, O. V., Surin, A. K., Glyakina, A. V., Rogachevsky, V. V., and Selivanova, O. M. (2018b). Should the treatment of amyloidosis be personified? Molecular mechanism of amyloid formation by Aβ peptide and its fragments. J. Alzheimers Dis. Rep. 2, 181–199. doi: 10.3233/ADR-180063
Galzitskaya, O. V., and Selivanova, O. M. (2017). Rosetta stone for amyloid fibrils: the key role of ring-like oligomers in amyloidogenesis. J. Alzheimers Dis. 59, 785–795. doi: 10.3233/jad-170230
Garbuzynskiy, S. O., Lobanov, M. Y., and Galzitskaya, O. V. (2010). FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 26, 326–332. doi: 10.1093/bioinformatics/btp691
Glasgow, N. G., Povysheva, N. V., Azofeifa, A. M., and Johnson, J. W. (2017). Memantine and ketamine differentially alter NMDA receptor desensitization. J. Neurosci. 37, 9686–9704. doi: 10.1523/jneurosci.1173-17.2017
Gremer, L., Schölzel, D., Schenk, C., Reinartz, E., Labahn, J., Ravelli, R. B. G., et al. (2017). Fibril structure of amyloid-β(1–42) by cryo-electron microscopy. Science 358, 116–119. doi: 10.1126/science.aao2825
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
He, G., Luo, W., Li, P., Remmers, C., Netzer, W. J., Hendrick, J., et al. (2010). γ-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467, 95–98. doi: 10.1038/nature09325
Hornung, K., Zampar, S., Engel, N., Klafki, H., Liepold, T., Bayer, T. A., et al. (2019). N-terminal truncated Aβ4–42 is a substrate for neprilysin degradation in vitro and in vivo. J. Alzheimers Dis. 67, 849–858. doi: 10.3233/JAD-181134
Huang, D., Lüthi, U., Kolb, P., Edler, K., Cecchini, M., Audetat, S., et al. (2005). Discovery of cell-permeable non-peptide inhibitors of β-secretase by high-throughput docking and continuum electrostatics calculations. J. Med. Chem. 48, 5108–5111. doi: 10.1021/jm050499d
Janssen, J. C., Beck, J. A., Campbell, T. A., Dickinson, A., Fox, N. C., Harvey, R. J., et al. (2003). Early onset familial Alzheimer’s disease: mutation frequency in 31 families. Neurology 60, 235–239. doi: 10.1212/01.wnl.0000042088.22694.e3
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., and Selkoe, D. J. (2011). Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U S A 108, 5819–5824. doi: 10.1073/pnas.1017033108
John, S., Thangapandian, S., Sakkiah, S., and Lee, K. W. (2011). Potent BACE-1 inhibitor design using pharmacophore modeling, in silico screening and molecular docking studies. BMC Bioinformatics 12:S28. doi: 10.1186/1471-2105-12-s1-s28
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., et al. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. doi: 10.1038/nature11283
Lesné, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357. doi: 10.1038/nature04533
Lin, T.-W., Chang, C.-F., Chang, Y.-J., Liao, Y.-H., Yu, H.-M., and Chen, Y.-R. (2017). Alzheimer’s amyloid-β A2T variant and its N-terminal peptides inhibit amyloid-β fibrillization and rescue the induced cytotoxicity. PLoS One 12:e0174561. doi: 10.1371/journal.pone.0174561
Lu, C., and Tune, L. E. (2003). Chronic exposure to anticholinergic medications adversely affects the course of Alzheimer disease. Am. J. Geriatr. Psychiatry 11, 458–461. doi: 10.1176/appi.ajgp.11.4.458
Makin, O. S., and Serpell, L. C. (2005). X-ray diffraction studies of amyloid structure. Methods Mol. Biol. 299, 67–80. doi: 10.1385/1-59259-874-9:067
Maltsev, A. V., Bystryak, S., and Galzitskaya, O. V. (2011). The role of β-amyloid peptide in neurodegenerative diseases. Ageing Res. Rev. 10, 440–452. doi: 10.1016/j.arr.2011.03.002
Maltsev, A. V., Santockyte, R., Bystryak, S., and Galzitskaya, O. V. (2014). Activation of neuronal defense mechanisms in response to pathogenic factors triggering induction of amyloidosis in Alzheimer’s disease. J. Alzheimers Dis. 40, 19–32. doi: 10.3233/jad-131562
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U S A 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
Messa, M., Colombo, L., del Favero, E., Cantù, L., Stoilova, T., Cagnotto, A., et al. (2014). The peculiar role of the A2V mutation in amyloid-β (Aβ) 1–42 molecular assembly. J. Biol. Chem. 289, 24143–24152. doi: 10.1074/jbc.m114.576256
Minter, L. M., Turley, D. M., Das, P., Shin, H. M., Joshi, I., Lawlor, R. G., et al. (2005). Inhibitors of γ-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat. Immunol. 6, 680–688. doi: 10.1038/ni1209x
Mukrasch, M. D., Bibow, S., Korukottu, J., Jeganathan, S., Biernat, J., Griesinger, C., et al. (2009). Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 7:e34. doi: 10.1371/journal.pbio.1000034
Murakami, K. (2014). Conformation-specific antibodies to target amyloid β oligomers and their application to immunotherapy for Alzheimer’s disease. Biosci. Biotechnol. Biochem. 78, 1293–1305. doi: 10.1080/09168451.2014.940275
Muthusamy, K., Singh, K. D., Chinnasamy, S., Nagamani, S., Krishnasamy, G., Thiyagarajan, C., et al. (2013). High throughput virtual screening and E-pharmacophore filtering in the discovery of new BACE-1 inhibitors. Interdiscip. Sci. 5, 119–126. doi: 10.1007/s12539-013-0157-x
Ono, K., Condron, M. M., and Teplow, D. B. (2010). Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 285, 23186–23197. doi: 10.1074/jbc.m109.086496
Ousset, P.-J., Cummings, J., Delrieu, J., Legrand, V., Prins, N., Winblad, B., et al. (2014). Is Alzheimer’s disease drug development broken? What must be improved. J. Prev. Alzheimers Dis. 1, 40–45. doi: 10.14283/jpad.2014.19
Parsons, C. G., Danysz, W., Dekundy, A., and Pulte, I. (2013). Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox. Res. 24, 358–369. doi: 10.1007/s12640-013-9398-z
Roher, A. E., Chaney, M. O., Kuo, Y. M., Webster, S. D., Stine, W. B., Haverkamp, L. J., et al. (1996). Morphology and toxicity of Aβ-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J. Biol. Chem. 271, 20631–20635. doi: 10.1074/jbc.271.34.20631
Saha, A. R., Ninkina, N. N., Hanger, D. P., Anderton, B. H., Davies, A. M., and Buchman, V. L. (2000). Induction of neuronal death by α-synuclein. Eur. J. Neurosci. 12, 3073–3077. doi: 10.1046/j.1460-9568.2000.00210.x
Schmitz, C., Rutten, B. P. F., Pielen, A., Schäfer, S., Wirths, O., Tremp, G., et al. (2004). Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 164, 1495–1502. doi: 10.1016/s0002-9440(10)63235-x
Selivanova, O. M., Gorbunova, E. Y., Mustaeva, L. G., Grigorashvili, E. I., Suvorina, M. Y., Surin, A. K., et al. (2016a). Peptide Aβ(16–25) forms nanofilms in the process of its aggregation. Biochemistry 81, 755–761. doi: 10.1134/S0006297916070129
Selivanova, O. M., Grigorashvili, E. I., Suvorina, M. Y., Dzhus, U. F., Nikulin, A. D., Marchenkov, V. V., et al. (2016b). X-ray diffraction and electron microscopy data for amyloid formation of Aβ40 and Aβ42. Data Brief 8, 108–113. doi: 10.1016/j.dib.2016.05.020
Selivanova, O. M., Surin, A. K., Marchenkov, V. V., Dzhus, U. F., Grigorashvili, E. I., Suvorina, M. Y., et al. (2016c). The mechanism underlying amyloid polymorphism is opened for Alzheimer’s disease amyloid-β peptide. J. Alzheimers Dis. 54, 821–830. doi: 10.3233/jad-160405
Selivanova, O. M., Rogachevsky, V. V., Syrin, A. K., and Galzitskaya, O. V. (2018a). Molecular mechanism of amyloid formation by Aβ peptide: review of own works. Biomed. Khim. 64, 94–109. doi: 10.18097/pbmc20186401094
Selivanova, O. M., Surin, A. K., Ryzhykau, Y. L., Glyakina, A. V., Suvorina, M. Y., Kuklin, A. I., et al. (2018b). To be fibrils or to be nanofilms? oligomers are building blocks for fibril and nanofilm formation of fragments of Aβ peptide. Langmuir 34, 2332–2343. doi: 10.1021/acs.langmuir.7b03393
Seuring, C., Ayyer, K., Filippaki, E., Barthelmess, M., Longchamp, J.-N., Ringler, P., et al. (2018). Femtosecond X-ray coherent diffraction of aligned amyloid fibrils on low background graphene. Nat. Commun. 9:1836. doi: 10.1038/s41467-018-04116-9
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shimmyo, Y., Kihara, T., Akaike, A., Niidome, T., and Sugimoto, H. (2008). Flavonols and flavones as BACE-1 inhibitors: structure-activity relationship in cell-free, cell-based and in silico studies reveal novel pharmacophore features. Biochim. Biophys. Acta 1780, 819–825. doi: 10.1016/j.bbagen.2008.01.017
Small, S. A., and Duff, K. (2008). Linking Aβ and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron 60, 534–542. doi: 10.1016/j.neuron.2008.11.007
Strömberg, K., Eketjäll, S., Georgievska, B., Tunblad, K., Eliason, K., Olsson, F., et al. (2015). Combining an amyloid-β (Aβ) cleaving enzyme inhibitor with a γ-secretase modulator results in an additive reduction of Aβ production. FEBS J. 282, 65–73. doi: 10.1111/febs.13103
Surin, A. K., Grigorashvili, E. I., Suvorina, M. Y., Selivanova, O. M., and Galzitskaya, O. V. (2016). Determination of regions involved in amyloid fibril formation for Aβ(1–40) peptide. Biochemistry 81, 762–769. doi: 10.1134/S0006297916070130
Suvorina, M. Y., Selivanova, O. M., Grigorashvili, E. I., Nikulin, A. D., Marchenkov, V. V., Surin, A. K., et al. (2015). Studies of polymorphism of amyloid-β 42 peptide from different suppliers. J. Alzheimers Dis. 47, 583–593. doi: 10.3233/JAD-150147
Tanzi, R. E., and Bertram, L. (2005). Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. doi: 10.1016/j.cell.2005.02.008
Tariot, P. N., Farlow, M. R., Grossberg, G. T., Graham, S. M., McDonald, S., Gergel, I., et al. (2004). Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 291, 317–324. doi: 10.1001/jama.291.3.317
Thathiah, A., Horré, K., Snellinx, A., Vandewyer, E., Huang, Y., Ciesielska, M., et al. (2013). β-arrestin 2 regulates Aβ generation and γ-secretase activity in Alzheimer’s disease. Nat. Med. 19, 43–49. doi: 10.1038/nm.3023
Tomiyama, T., Nagata, T., Shimada, H., Teraoka, R., Fukushima, A., Kanemitsu, H., et al. (2008). A new amyloid β variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 63, 377–387. doi: 10.1002/ana.21321
Tran, J., Chang, D., Hsu, F., Wang, H., and Guo, Z. (2017). Cross-seeding between Aβ40 and Aβ42 in Alzheimer’s disease. FEBS Lett. 591, 177–185. doi: 10.1002/1873-3468.12526
Wälti, M. A., Ravotti, F., Arai, H., Glabe, C. G., Wall, J. S., Böckmann, A., et al. (2016). Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U S A 113, E4976–E4984. doi: 10.1073/pnas.1600749113
Yan, R., and Vassar, R. (2014). Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 13, 319–329. doi: 10.1016/S1474-4422(13)70276-X
Keywords: amyloid, oligomer, drug, polymorphism, model
Citation: Galzitskaya OV (2020) Oligomers Are Promising Targets for Drug Development in the Treatment of Proteinopathies. Front. Mol. Neurosci. 12:319. doi: 10.3389/fnmol.2019.00319
Received: 24 August 2019; Accepted: 16 December 2019;
Published: 31 January 2020.
Edited by:
Sandra Macedo-Ribeiro, University of Porto, PortugalReviewed by:
Md. Golam Sharoar, University of Connecticut Health Center, United StatesCopyright © 2020 Galzitskaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oxana V. Galzitskaya, b2dhbHppdEB2ZWdhLnByb3RyZXMucnU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.