Corrigendum: Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis
 Luke McAlary
Luke McAlary Steven S. Plotkin
Steven S. Plotkin Justin J. Yerbury
Justin J. Yerbury Neil R. Cashman
Neil R. Cashman- 1Illawarra Health and Medical Research Institute, University of Wollongong, Wollongong, NSW, Australia
- 2Molecular Horizons and School of Chemistry and Molecular Bioscience, Faculty of Science, Medicine and Health, University of Wollongong, Wollongong, NSW, Australia
- 3Department of Physics and Astronomy, University of British Columbia, Vancouver, BC, Canada
- 4Genome Sciences and Technology Program, University of British Columbia, Vancouver, BC, Canada
- 5Djavad Mowafaghian Centre for Brain Health, University of British Columbia, Vancouver, BC, Canada
The discovery that prion protein can misfold into a pathological conformation that encodes structural information capable of both propagation and inducing severe neuropathology has revolutionized our understanding of neurodegenerative disease. Many neurodegenerative diseases with a protein misfolding component are now classified as “prion-like” owing to the propagation of both symptoms and protein aggregation pathology in affected individuals. The neuromuscular disorder amyotrophic lateral sclerosis (ALS) is characterized by protein inclusions formed by either TAR DNA-binding protein of 43 kDa (TDP-43), Cu/Zn superoxide dismutase (SOD1), or fused in sarcoma (FUS), in both upper and lower motor neurons. Evidence from in vitro, cell culture, and in vivo studies has provided strong evidence to support the involvement of a prion-like mechanism in ALS. In this article, we review the evidence suggesting that prion-like propagation of protein aggregation is a primary pathomechanism in ALS, focusing on the key proteins and genes involved in disease (TDP-43, SOD1, FUS, and C9orf72). In each case, we discuss the evidence ranging from biophysical studies to in vivo examinations of prion-like spreading. We suggest that the idiopathic nature of ALS may stem from its prion-like nature and that elucidation of the specific propagating protein assemblies is paramount to developing effective therapies.
Neurodegenerative Disease: A Common Pathophysiology
Neurodegenerative diseases are characterized by the selective and progressive loss of neurons, leading to the development of neurological dysfunction which can include memory, behavioral, and movement deficits (Erkkinen et al., 2018). Neurodegenerative diseases include Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), spinocerebellar ataxia’s, Huntington’s disease, multiple system atrophy, dementia with Lewy bodies, corticobasal degeneration, chronic traumatic encephalopathy, and prion diseases [e.g., Creutzfeldt-Jakob Disease (CJD), fatal familial insomnia, or kuru]. Collectively, these diseases exact a significant and growing toll on human well-being both socially and economically (Prince et al., 2013), making the development of effective therapies essential to societal health and well-being.
Although all of the above listed diseases have in common the progressive degeneration of neurons, they typically manifest clinically with different symptoms due to a characteristic set of neuronal subtypes specifically vulnerable in each condition (Erkkinen et al., 2018). For example, a patient suffering from canonical ALS will exhibit preferential degeneration of motor neurons in the spinal cord/brainstem and motor cortex, however, neurons in other central nervous system (CNS) regions will generally remain unaffected (Ravits and La Spada, 2009). Another example is that in PD dopaminergic neurons within the substantia nigra preferentially degenerate (Brichta and Greengard, 2014). Although the vulnerability of certain neurons to undergo death appears to be disease-specific, the key markers for this death are observed across many diseases (Chi et al., 2018). One such key marker is mitochondrial dysfunction, which can be a consequence of the accumulation of misfolded proteins like SOD1 (Magrané et al., 2012). Maintenance of mitochondrial protein homeostasis is especially important for neurons, owing to their large size and high energy requirements (reviewed in Baker et al., 2011).
A unifying feature of all the above mentioned neurodegenerative diseases is that the cell and neuroanatomical region specificity of degeneration is often accompanied by the aberrant misfolding, aggregation, and deposition of specific proteins (Chiti and Dobson, 2017). Protein misfolding is defined as the adoption of a non-native conformation by a protein; Protein aggregation is the aberrant accumulation of a protein into multimeric soluble or insoluble non-native structures; Protein deposition is the formation of large insoluble deposits of proteins (Chiti and Dobson, 2017). Examples of specific disease-associated aggregating proteins include amyloid-β (Glenner and Wong, 1984) and τ-protein (Grundke-Iqbal et al., 1986) in AD, α-synuclein in PD (Spillantini et al., 1997) and multiple system atrophy (Tu et al., 1998; Wakabayashi et al., 1998), poly-Q expansions in huntingtin protein in Huntington’s disease (DiFiglia et al., 1997), and either Cu/Zn superoxide dismutase (Rosen et al., 1993), TAR-DNA-binding protein of 43 kDa (TDP-43; Neumann et al., 2006), or fused in sarcoma (FUS; Kwiatkowski et al., 2009; Vance et al., 2009) in ALS.
It was previously thought that the aggregation of specific proteins is exclusively associated with specific disorders, however, there is evidence for some overlap between misfolding and aggregating proteins and various syndromes (Jellinger, 2012). Indeed, aberrant localization and aggregation of TDP-43 are being associated with an increasing number of neurodegenerative diseases (reviewed in Gao et al., 2018), and SOD1 positive inclusions have been reported in PD cases (Trist et al., 2017). Furthermore, the fact that patients suffering from FTD can have protein inclusion pathology that is positive for either TDP-43, τ-protein, or FUS (Ling et al., 2013), and that ~15% of ALS patients are estimated to develop cognitive deficits meeting FTD criteria (Ringholz et al., 2005), is striking evidence of both pathological and symptomatic overlap. This has led to the suggestion that both ALS and FTD perhaps exist as extreme syndromes of a single disease spectrum (Ling et al., 2013).
Amyotrophic Lateral Sclerosis—Clinical and Pathological Manifestation
ALS is a neuromuscular disorder in which the upper (motor cortex) and lower (spinal) motor neurons, responsible for motor function, progressively degenerate, ultimately resulting in patient mortality via asphyxiation or inanition (Hardiman et al., 2017). The disease is considered to comprise two main categories on the basis of familial history of disease or lack thereof. Sporadic ALS (sALS) accounts for approximately 90% of cases (Taylor et al., 2016), and the remaining 10% are classed as familial (fALS). Collectively, ALS is estimated to have an annual incidence rate of 2.4 per 100,000 people in Europe, and is suggested to affect a susceptible population rather than being generally related with aging (Logroscino et al., 2010). Clinical diagnosis of ALS is often difficult due to significant variability in patient presentation and prognosis, as well as lack of a definitive biomarker for disease (Al-Chalabi et al., 2016; Grad et al., 2017). Generally, ALS patients will initially show symptoms of either lower motor neuron degeneration (weakness with muscle wasting, fasciculation, cramps) or upper motor neuron degeneration (weakness and spasticity), however, both regions of the CNS are ultimately affected (Tartaglia et al., 2007; Van den Berg-Vos et al., 2009). A characteristic feature of ALS is that often, regardless of the initial site of onset, symptoms spread to nearby contiguous anatomical regions in the CNS in a spatiotemporal manner (Ravits et al., 2007; Ravits and La Spada, 2009). There are cases in which the spread of symptoms has been reported to be discontiguous, however, they remain a minority (Walhout et al., 2018; Zhenfei et al., 2019). To better understand the relationship between symptoms and pathology, it will be important to examine discontiguous and contiguous spread in relation to the patterns of pathology in patients.
A key pathological hallmark of ALS is the deposition of proteins into ubiquitinated, and sometimes hyperphosphorylated, cytoplasmic inclusions in motor neurons and glia in the spinal cord/brainstem and motor cortex. Other hallmarks include the degeneration of motor neurons in the spinal cord and motor cortex, as well as atrophy of the corresponding denervated skeletal muscles. The pathological protein inclusions observed in patients are immunoreactive for either TDP-43 (Neumann et al., 2006), SOD1 (Rosen et al., 1993), or FUS (Kwiatkowski et al., 2009; Vance et al., 2009), of which the vast majority of cases show TDP-43 (97% of cases), rather than SOD1 (2%) or FUS (1%), pathology (Ling et al., 2013). Notably, aggregates of SOD1 or FUS are associated with mutations in cognate genes, whereas TDP-43 aggregates can be made from wild-type protein in sALS as well as associated with mutations in TDP-43 in fALS. TDP-43 pathology can also be observed as a downstream consequence of other ALS-implicated gene mutations (e.g., C9orf72).
Genetics of ALS—Interplay of RNA Metabolism, Cytoskeletal Dynamics, and Proteostasis
Both sALS and fALS are clinically indistinguishable from each other (Hardiman et al., 2011), whereas the genetics of ALS are heterogeneous (Taylor et al., 2016), involving numerous genes that control various biochemical processes and cellular pathways (Taylor et al., 2016). As our understanding of the functions of these genes has increased, it has become apparent that they can be broadly classified into three major groups; genes associated with RNA metabolism, genes associated with cytoskeletal dynamics, and genes associated with protein homeostasis (proteostasis; Taylor et al., 2016). There is also a great deal of overlap between these important cellular processes, making it difficult to designate causal pathomechanism(s).
Disturbances in RNA metabolism in ALS have been keenly studied owing to the discovery that TDP-43 and FUS, which are RNA-binding proteins (RBPs), form neuronal inclusions and harbor mutations that are ALS causative (Neumann et al., 2006; Kwiatkowski et al., 2009; Vance et al., 2009). An increasing number of RNA metabolism-related genes have been identified to carry ALS-associated mutations, including heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) and heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1; Kim et al., 2013), matrin-3 (Johnson et al., 2014), TIA-1 cytotoxic granule-associated RNA binding protein (Mackenzie et al., 2017), and TATA-box binding protein associated factor 15 (TAF-15; Couthouis et al., 2011; Ticozzi et al., 2011). Furthermore, the most common identified cause for fALS are hexanucleotide (GGGGCC) repeat expansions in the chromosome 9 open reading frame 72 (C9orf72) gene (DeJesus-Hernandez et al., 2011; Renton et al., 2011), that potentially lead to toxicity caused by the synthesis of large non-coding RNAs that form sense and anti-sense foci in cells that can sequester RBPs and prevent them from carrying out their biological functions (Donnelly et al., 2013; Gendron et al., 2013; Lagier-Tourenne et al., 2013; Mizielinska et al., 2013; Zu et al., 2013). Many of the above mentioned ALS-associated RBPs take part in the formation of ribonucleoprotein (RNP) granules and biomolecular condensates in cells (Banani et al., 2017), and there is growing evidence that mutations affect the dynamics of formation and dissolution of these structures (Murakami et al., 2015; Patel et al., 2015; Lee et al., 2016; Lin et al., 2016; Martinez et al., 2016; Boeynaems et al., 2017; Mackenzie et al., 2017). It is suggested that this dysregulation of RNP granules can result in significant alterations to alternative splicing, mRNA transcription, and RNA transport, which can result in widespread toxicity in cells (Butti and Patten, 2019). An overlap with proteostasis mechanisms is also possible with ALS-associated mutations in RBPs as they can act to promote a liquid-to-solid transition of biomolecular condensates, leading to the formation of pathological aggregates in cells (Murakami et al., 2015; Patel et al., 2015; Mackenzie et al., 2017; Sun and Chakrabartty, 2017; Gasset-Rosa et al., 2019). Cytoskeletal dynamics can also be affected by ALS-associated RBP mutants, for example, ALS-associated mutations in TDP-43 can alter the transport dynamics and viscosity of transport RNPs (Alami et al., 2014; Gopal et al., 2017), potentially resulting in impaired delivery of mRNA to the axons of motor neurons.
Motor neurons are significantly larger than other cell types, owing to the requirement to extend axons anatomically long distances throughout the motor cortex, spinal cord and periphery. Therefore, disruption of the mechanisms that traffic protein and nucleotides to distal cellular compartments would be expected to impair proper cellular functioning. Indeed, ALS-associated mutations have been identified in cytoskeletal components and regulatory elements, such as heavy neurofilament (Al-Chalabi et al., 1999), peripherin (Gros-Louis et al., 2004), NIMA-related kinase 1 (NEK1; Kenna et al., 2016), profilin-1, and kinesin family member 5A (KIF5A; Brenner et al., 2018), amongst others (Puls et al., 2003; Wu et al., 2012; Smith et al., 2014). The mechanisms by which mutations in cytoskeletal components can cause ALS are not fully understood but are thought to be a combination of loss-of-function and gain-of-toxic-function that can overlap with both RNA metabolism and proteostasis (Hensel and Claus, 2017). In the case of profilin-1, in its normal role it binds to actin and it affects the overall structure of the cytoskeleton. In terms of RNA metabolism, it was found that ALS-associated profilin-1 mutants differentially localized to stress granules and that stress granule dynamics were altered (Figley et al., 2014). In the case of proteostasis, expression of ALS-associated profilin-1 mutants has been shown to lead to the formation and seeding of TDP-43 positive proteinaceous aggregates in cultured cells (Wu et al., 2012; Smith et al., 2015; Tanaka and Hasegawa, 2016).
That proteinaceous aggregates are the primary characteristic marker for ALS suggests that, at the very least, proteostasis is a key mechanism affected in ALS. Indeed, there are several identified ALS-associated mutations in genes associated with autophagy (Maruyama et al., 2010; Fecto et al., 2011), and the ubiquitin proteasome system (UPS; Johnson et al., 2010; Deng et al., 2011; Williams et al., 2016). Furthermore, autophagy and the UPS have been suggested to be altered in sALS on the basis of studies of post-mortem tissue (Sasaki, 2011; Kabashi et al., 2012). Interestingly, TDP-43, SOD1, and FUS, which are the primary components of inclusions in ALS patient tissue (Ling et al., 2013), are shown to be supersaturated (expressed at levels higher than their predicted solubility) in spinal motor neurons (Ciryam et al., 2017). Following this notion, it has been suggested that the average supersaturation of the entire proteome of spinal motor neurons is greater than that of the ALS-resistant ocular motor neurons (Yerbury et al., 2019), providing important physico-chemical insight into the selective vulnerability of spinal motor neurons to proteotoxicity. Again, overlap is observed between proteostasis and both RNA metabolism and cytoskeletal dynamics. For example, the UPS protein ubiquilin-2 has been observed to control the dynamics of both stress granule formation and RNA/FUS complexes (Alexander et al., 2018), and mutant SOD1 can interact with the stress granule forming protein Ras GTPase-activating protein-binding protein 1 (G3BP1) to impair proper stress granule formation (Gal et al., 2016).
Collectively, RNA metabolism, cytoskeletal dynamics, and proteostasis should not be viewed as separate pathologically altered pathways, but as an interconnected network. However, regardless of whether RNA metabolism, cytoskeletal dynamics, or proteostasis are perturbed in ALS, the most prominent pathology is the formation of proteinaceous aggregates in neurons and glial support cells composed of TDP-43, or SOD1, or FUS in a mutually exclusive manner (Farrawell et al., 2015). This is supported by the finding that TDP-43 and FUS, despite both being RNA binding proteins participating in stress granule formation, generally do not appear to co-localize in pathological ALS inclusions (Ling et al., 2013). Furthermore, the neuroanatomical spread of pathology and symptoms indicates that there are propagating agents in ALS (Table 1). Owing to the spatiotemporal spread of pathology in ALS, as well as PD and AD, it has been suggest that a key mechanism in neurodegenerative disease pathobiology is the “prion-like” spread of protein misfolding and aggregation, pathways of which would be different for the three major inclusion forming proteins (Farrawell et al., 2015) to account for the exclusivity noted above.
TABLE 1
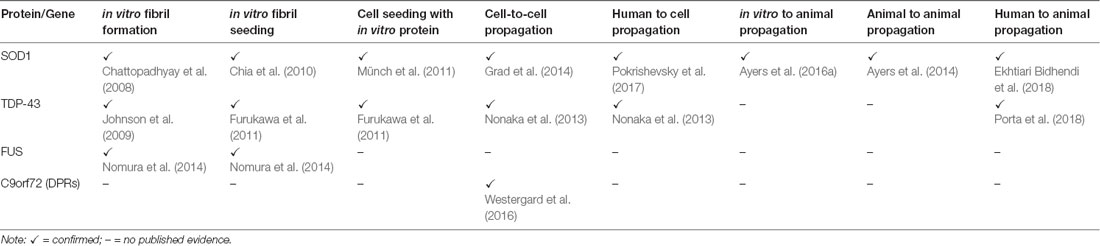
Table 1. Evidence supporting the prion-like characteristics of amyotrophic lateral sclerosis (ALS)-associated proteins.
Prion Disease and Prion Theory
Classical understanding of progressive and infectious diseases mandated that a nucleic acid component be present to facilitate replication of the infectious particles. This idea has since been challenged owing to the discovery that a class of neurological disorders, known as the prion diseases, possess an infectious life cycle and spread in the CNS in the absence of any microbial nucleic acid component. The landmark finding was that the infectious particle responsible for scrapie, a neurodegenerative disease that affects sheep and goats, was proteinaceous in origin, resulting from the misfolding of prion protein (PrPC) into a pathological conformation (PrPSc; Prusiner, 1982). Furthermore, PrPSc was found to be able to catalyze the misfolding of PrPC, thus generating more conformational copies of PrPSc that can then spread throughout cells, tissues, and even between organisms. This notion of the propagation of protein misfolding has been subsequently applied to neurodegenerative diseases, providing a possible explanation for the protein aggregation pathology and progressive spatiotemporal degeneration observed in these diseases.
Prion diseases include CJD, Bovine Spongiform Encephalopathy, Fatal Familial Insomnia, Kuru, and Gerstmann–Sträussler–Scheinker disease, amongst others (Geschwind, 2015). The clinical manifestations and pathological profiles of prion diseases are highly similar to those of other neurodegenerative diseases (Prusiner, 2001). In both cases they are invariably fatal; The clinical symptoms are mainly associated with neurological functioning and can include dementia, ataxia, involuntary movements, weakness, and spasticity, although, specific prion diseases will manifest distinct symptoms from this list (Glatzel et al., 2005). The neuropathological similarities of prion diseases with other neurodegenerative diseases include substantial neuronal loss, accumulation of proteins into aggregates, gliosis, and general cerebral atrophy (Prusiner, 2001).
Biophysical examination of PrPC has suggested that its aggregation is preceded by a conformational change from a structure composed mostly of α-helical elements to a β-sheet rich conformation, where the β-sheet rich conformation is thought to be the disease-associated PrPSc form (Pan et al., 1993). The β-sheet rich form is capable of interacting with and converting the normal α-helix rich form to the β-sheet rich form in a template-directed manner. Furthermore, the β-sheet rich form of PrP is capable of polymerizing into amyloid-like fibrils (McKinley et al., 1991). This is a similar kinetic process that occurs to amyloid-β peptides to generate amyloid fibrils (Arosio et al., 2015). The process is understood as an initial monomeric misfolding event, followed by the formation of small disordered oligomers, where oligomers nucleate the formation of the larger ordered amyloid fibrils. Interestingly, in vitro studies have shown that many neurodegenerative disease-associated proteins are capable of forming amyloid-like fibrils (Eisenberg and Jucker, 2012; Knowles et al., 2014). In ALS this includes TDP-43 (Chen et al., 2010; Guo et al., 2011; Saini and Chauhan, 2011, 2014; Jiang et al., 2013; Wang et al., 2013; Zhang et al., 2013; Mompeán et al., 2014; Sun et al., 2014; Cao et al., 2019), SOD1 (Chattopadhyay et al., 2008, 2015; Münch and Bertolotti, 2010; Lang et al., 2012; Chan et al., 2013; Ivanova et al., 2014; Abdolvahabi et al., 2016; McAlary et al., 2016), FUS (Kato et al., 2012; Murakami et al., 2015; Patel et al., 2015; Murray et al., 2017; Luo et al., 2018), amongst others (Molliex et al., 2015; Mackenzie et al., 2017).
Within the schema of prion theory, there exists a notion that different structural conformations of the prion agent may confer specific modes of spread, different abilities to recruit normal protein, and different levels and mechanisms of cellular toxicity to recipient species (Morales, 2017). Together, these characteristics are used to define different “strains” of prion. An integral part of the prion strain idea is that the ability of a prion strain to transmit disease is dependent on the conformation of the prion particle, the amino acid sequence of the template-competent substrate, post-translational modification of the substrate, and the cell type (substrate expression level and cellular capacity for degradation of misfolded/aggregated protein; Telling et al., 1996; Safar et al., 1998; Legname et al., 2006). Conformation of the prion assembly is especially important, as it has been established that strains that are more likely to fragment into a greater number of seeds are the most capable of spreading (Tanaka et al., 2006). In relation to ALS, it is interesting to note that the main aggregating proteins (SOD1, TDP-43, FUS) are supersaturated in spinal motor neurons (Ciryam et al., 2017), suggesting that motor neurons may provide an efficient environment for ALS-associated prion-like seeds to form and propagate. Indeed, the combination of the spatiotemporal spread of symptoms in ALS with both the evidence of protein aggregation and ability of the aggregating proteins to form amyloid-like structures has led to the suggestion that a prion-like mechanism is active in ALS (Ayers and Cashman, 2018). Below we review the evidence for prion-like characteristics of SOD1, TDP-43, FUS, and C9orf72 associated dipeptide repeats, as they relate to ALS.
Superoxide Dismutase-1
SOD1 Structure and Function
Superoxide dismutase-1 (SOD1) was the first gene linked to familial ALS (Rosen et al., 1993), and since this initial discovery, there have been over 160 fALS-associated mutations identified in the Sod1 gene (Abel et al., 2013). In its natively folded state, SOD1 is a 32 kDa homodimer, where each 153 amino acid subunit coordinates a zinc ion, copper ion, and contains an intramolecular disulfide bond. The maturation pathway (Figure 1A) of SOD1, from nascent polypeptide to native dimeric conformation, is complex for such a small protein and has been extensively studied (Banci et al., 2012, 2013; Wright et al., 2016; Luchinat et al., 2017; Sala et al., 2019). Following synthesis, SOD1 assumes a partially folded state with no metal cofactors and reduced cysteines [E, E(SH)]. This nascent state is further stabilized by the binding of a zinc (Zn) ligand to form E, Zn(SH). The copper chaperone for SOD1 (CCS) then associates with the Zn-bound intermediate monomer, forming a heterodimer that facilitates Cu delivery and intramolecular disulfide formation to form a mature SOD1 monomer [Cu, Zn(SS)]. Interestingly, this interaction can be prohibited or stalled by SOD1-fALS mutations (Wright et al., 2016). Disulfide formation via CCS can also occur through a Cu-independent pathway (Banci et al., 2013) and can be promoted using small molecules (Capper et al., 2018). Recent work has also suggested that CCS performs a chaperone function to stabilize E, E-SOD12SH SOD1 and promote Zn-binding mediated by its own SOD-like domain (Luchinat et al., 2017). The final step is dimerization of mature monomers to form the highly stable enzyme. Once natively folded, SOD1 acts to catalyze the dismutation of superoxide anions to hydrogen peroxide and molecular oxygen.
FIGURE 1
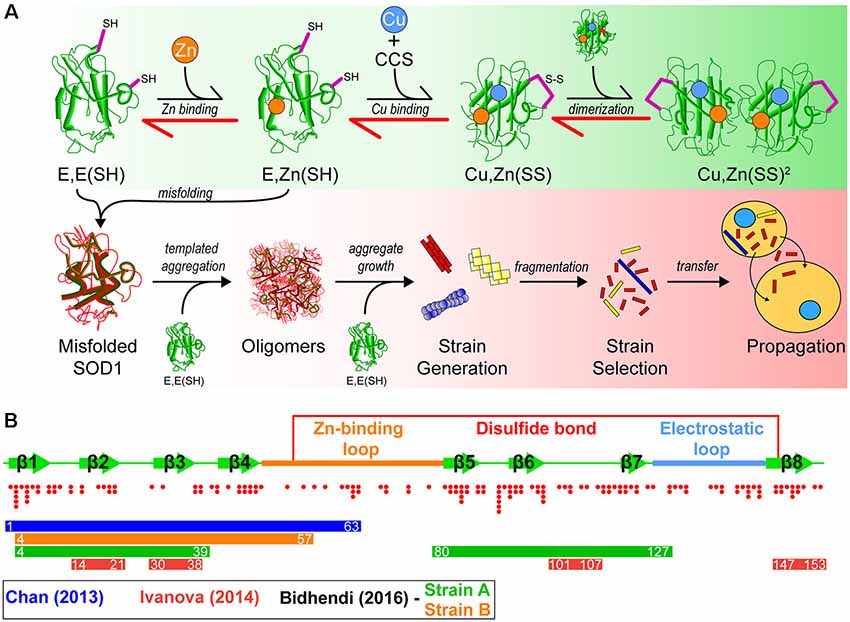
Figure 1. A model of superoxide dismutase-1 (SOD1) aggregation and prion-like propagation. (A) The native folding (green background) and off-folding pathways (red background) of SOD1. When undergoing native folding, SOD1 is initially in a metal-free intermediate state [E, E(SH)] which is primed for Zn binding. Following Zn binding, the copper chaperone for SOD1 (CCS) heterodimerizes with a SOD1 monomer to implant Cu and facilitate the oxidation of the disulfide bond between Cys57 and Cys146 to form a mature SOD1 monomer [Cu, Zn(SS)]. Subsequent dimerization of two mature monomers gives the fully folded SOD1 enzyme. In the off-folding pathway (red background) amyotrophic lateral sclerosis (ALS)-associated mutations have the ability to push the folding back towards the nascent intermediates (red arrows), which are more aggregation prone than mature states. Misfolded SOD1 is capable of recruiting nascent SOD1 states to form oligomers. Oligomers can grow in size to form larger aggregates that may have strain-like properties. Strains that are prone to fragment are the most likely to propagate to adjacent cells and seed naïve SOD1. (B) Graphic representation of the SOD1 primary structure with important features. SOD1 is composed of 8 beta-strands and contains two major loop regions. The Zn-binding loop is responsible for coordination of Zn and, to a lesser extent, Cu. The electrostatic loop is responsible for guidance of superoxide substrate to the active site of the enzyme. A disulfide bond is formed between Cys57 and Cys146 in the mature protein. Red dots below the graphic represent the location and number of ALS-associated mutations that have been identified to date within SOD1. Coloured bars represent potential cores or contributing regions of the protein to the formation of aggregate strains from corresponding colour coded publications.
Loss of Function or Gain of Toxic Function?
Owing to the role of SOD1 as an antioxidant enzyme, it was initially thought that the fALS-associated mutations in SOD1 were inducing a loss of enzymatic function, leading to toxicity mediated by increased concentrations of oxygen radicals in cells. However, continued research revealed that this was not the case as overexpression of human mutant SOD1-G93A in a transgenic mouse line showed increased dismutase activity combined with rapid mortality in comparison to wild-type mice (Gurney et al., 1994). Another study knocked out SOD1 from mice, showing slightly increased neuronal cell death following axonal injury, but normal motor neuron development (Reaume et al., 1996), indicating that SOD1 is not essential to cell survival or development, and its antioxidant activity may be compensated for by other proteins (including SOD2 and SOD3). Furthermore, in vitro studies of several SOD1-fALS mutants found no correlation between relative enzymatic activity and clinical pathology (Borchelt et al., 1995; Ratovitski et al., 1999), and that many SOD1-fALS mutants possess significant enzymatic activity (Borchelt et al., 1994). Ruling out a loss-of-function mechanism for SOD1-fALS led to research into a gain-of-toxic-function mechanism, focused on the misfolding and aggregation of SOD1 (Figure 1A).
SOD1 Pathological Features in Patients
Immunohistochemical studies of post-mortem tissue from both fALS and sALS patients with SOD1 misfolding-specific antibodies have revealed a potential role for the misfolding of WT SOD1 in all ALS (Bosco et al., 2010; Forsberg et al., 2010, 2011, 2019; Pokrishevsky et al., 2012; Paré et al., 2018), however, this is a contentious idea (Liu et al., 2009; Kerman et al., 2010; Da Cruz et al., 2017). Regardless, discrete SOD1 inclusions are a key component of fALS with SOD1 mutations. Currently, there are discrepancies as to the exact structural nature of the SOD1 inclusions found in patient tissue. Electron microscopy imaging of protein inclusions harvested from ALS patients suggests that they are distinct from classic amyloid, having an amorphous or granular structure (Kato et al., 2000) that does not stain with Congo red, and stains relatively weakly with thioflavin-S (Kerman et al., 2010). Whether amyloid-like elements exist in inclusions composed of SOD1 in SOD1-fALS patients remains to be fully elucidated, although it is important to note that SOD1 has the ability to form amyloid-like fibrils in vitro (Chattopadhyay et al., 2008, 2015; Münch and Bertolotti, 2010; Lang et al., 2012; Chan et al., 2013; Ivanova et al., 2014; Abdolvahabi et al., 2016; McAlary et al., 2016). Furthermore, aggregated SOD1 in human mutant SOD1-expressing transgenic mice is observed to have an ordered microscopic and molecular structure (Ayers et al., 2014, 2016b; Bergh et al., 2015; Lang et al., 2015; Bidhendi et al., 2016, 2018).
Amyloid Aggregation of SOD1
The first evidence that SOD1 may act in a prion-like manner came from protein only in vitro assays utilizing recombinant wild-type (WT) SOD1 and fALS-mutants. Initially, SOD1 aggregation assays utilized high temperatures, extreme pH, and organic solvents to induce aggregate assembly (DiDonato et al., 2003; Stathopulos et al., 2003; Banci et al., 2008), conditions with little relevance to the intracellular environment. Modern recombinant SOD1 in vitro aggregation protocols rely upon destabilizing SOD1 via reduction of the disulfide and chelation of the metals under physiological salt and pH (Chattopadhyay et al., 2008; Lang et al., 2012; Chan et al., 2013; Ivanova et al., 2014; Abdolvahabi et al., 2016; McAlary et al., 2016), which are events more likely to occur in the reducing and metal-poor intracellular environment. Utilizing denaturing conditions, Chia et al. (2010) demonstrated that preformed recombinant SOD1 fibrils or spinal cord homogenates from multiple SOD1 transgenic mice were capable of seeding the aggregation of recombinant WT or mutant Cu, Zn-SOD1SS in vitro. This requirement to destabilize SOD1 state to promote fibril formation and seeding suggests that immature SOD1 may be the major molecular species prone to aggregation and a primary template for SOD1 seeded aggregation. Indeed, careful investigations into the in vivo aggregation of SOD1 mutants in transgenic mice have been shown to have a striking similarity to these cell-free recombinant systems, suggesting E, E(SH) SOD1 is the primary aggregation substrate, and that aggregate growth is fragmentation dependent both in vivo and in vitro (Lang et al., 2015).
Interestingly, SOD1 has multiple sequence segments that have characteristics favorable to form amyloid fibrils (Ivanova et al., 2014). These include amino acid sequence segments 14VQGIINFE21, 30KVWGSIKGL38, 101DSVISLS107, and 147GVIGIAQ153, as determined by using the 3D profile method (see Münch and Bertolotti, 2010). The 3D profile method uses a sliding window of every six amino acid peptides from a queried protein sequence to generate structures of these peptides, where these peptide structures are computationally mapped onto ensembles created from the amyloidogenic NNQQNY peptide sequence from Sup35 yeast prion-protein. Peptides with strong binding energies are considered to be potentially amyloidogenic (Thompson et al., 2006). Assays on these synthesized peptides have shown that all have the ability to form fibrils in vitro, and seed full-length SOD1 aggregation (Ivanova et al., 2014). Of particular note is segment 30KVWGSIKGL38 which has been recently shown to be able to form oligomers that are neurotoxic (Sangwan et al., 2017, 2018), and also contains the only tryptophan residue in SOD1 (W32), which is implicated in its aggregation and prion-like propagation (Taylor et al., 2007; Grad et al., 2011, 2014; Pokrishevsky et al., 2018). That SOD1 can form fibrils which may be nucleated by several different regions of the protein or controlled by specific residues, has implications for the existence of SOD1 prion-like strains (Figure 1B).
Intra- and Inter-cellular Prion-Like Spread of SOD1 Aggregation
The ability of cellular SOD1 to misfold and/or aggregate in a prion-like manner in living cells was first demonstrated using the addition of recombinant SOD1 aggregates to cultured cells overexpressing ALS mutant SOD1 variants (Münch et al., 2011). It was suggested that the exogenous SOD1 aggregates were entering the cells via macropinocytosis and escaping the macropinosomes to seed aggregation of cytosolic SOD1 (Münch et al., 2011), a finding that was substantiated in following publications (Holmes et al., 2013; Grad et al., 2014; Zeineddine et al., 2015). Shortly following the first study reporting intracellular seeding of SOD1, another study demonstrated the ability of endogenous wild-type SOD1 to become misfolded when ALS-associated mutant SOD1 was overexpressed in cultured cells (Grad et al., 2011). It was also determined that the prion-like propagation of SOD1 misfolding required the unique tryptophan at position 32 in both seed and substrate molecules (Grad et al., 2011). However, although a potential mechanism of cellular uptake and seeding was suggested, it was not known if or how aggregated or misfolded SOD1 may exit cells and spread. Grad et al. (2014) demonstrated the intercellular propagation of SOD1 misfolding and aggregation, and established two main mechanisms by which spread was occurring: release of aggregates from dying cells, and/or packaging of misfolded protein seed into or on extracellular vesicles called exosomes. More recently, it has been shown that extracellular vesicles purified from the neural tissues of SOD1-G93A mice carry misfolded SOD1 (Silverman et al., 2019), providing further evidence that this mechanism may play an important role in prion-like propagation of protein aggregation in ALS. Additionally, homogenates generated from spinal cord samples of SOD1-fALS patients were recently found to seed the aggregation of ALS mutant SOD1-GFP protein expressed in human embryonic kidney cells (Ayers et al., 2014; Pokrishevsky et al., 2017).
In vivo Seeding and Spread of SOD1 Aggregation
Evidence showing the prion-like propagation of SOD1 in vivo shortly followed the cultured cell experiments, providing information on the existence of different transmissible strains of SOD1. Initially, transgenic mice expressing low levels of the SOD1-G85R ALS mutant C-terminally tagged with yellow fluorescent protein (SOD1-G85R:YFP) were used as a model owing to them not developing SOD1-associated ALS-like pathology or symptoms when the transgene was heterozygous (Wang et al., 2009). Spinal cord homogenates harvested from mice overexpressing SOD1 mutants were injected into the sciatic nerve, cervical spinal cord, or cerebral ventricles of SOD1-G85R:YFP mice in an attempt to induce ALS-like pathology and symptoms (Ayers et al., 2014). Potent seeding was observed following injection into the mice, resulting in the formation of SOD1-G85R:YFP aggregates in neurons in spinal cord and motor cortex. The formation of SOD1-G85R:YFP aggregates was accompanied by the typical ALS phenotypes of motor dysfunction and paralysis. Later, this same model was used to examine the anatomical pathways by which SOD1 aggregates may traverse the CNS, finding that aggregation spread through synaptically connected cells in the spinal cord (Ayers et al., 2016b), providing useful information on possible mechanisms of prion-like transmission in ALS.
Considering that slight differences in the conformation of PrPSc can result in different pathological activities in prion diseases (Prusiner, 2013), it has been suggested that this could also occur in regards to SOD1 aggregation in fALS. Evidence for the existence of SOD1 strains in vivo was first shown by binary epitope mapping the insoluble material from spinal cords of transgenic mice overexpressing WT, G85R, D90A, or G93A SOD1 variants (Bergh et al., 2015), in which two strains (denoted strain A or strain B) were identified on the basis of differential epitope exposure. Mice expressing WT, G85R, and G93A were found to contain strain A aggregates, whereas D90A expressing mice contained either strain A or B aggregates (Bergh et al., 2015). Additionally, strain B aggregates correlated with a more rapid disease progression in mice and were more structurally unstable than strain A aggregates, suggesting aggregate fragmentation as an important mechanism in prion-like spreading (Bergh et al., 2015; Lang et al., 2015). These strains were found to be transmissible to SOD1-G85R transgenic mice through injection of fractions enriched with insoluble SOD1 from strain A or B into the spinal lumbar spinal cord, resulting in strain propagation as measured by binary epitope mapping and ALS-like symptoms (Bidhendi et al., 2016). Similar results were also obtained using the aforementioned SOD1-G85R:YFP mice, where injections of recombinant WT SOD1 protein resulted in aggregates with a thread-like morphology in recipient mice, in comparison to more amorphous features from injections using homogenates from other SOD1-fALS mice (Ayers et al., 2016a). More recently, transmission of strain A was found to occur when enriched insoluble material from the spinal cord of a SOD1-G127X patient was injected into transgenic mice harboring the same mutation (Ekhtiari Bidhendi et al., 2018). Collectively, the combined cell-free, in vitro, and in vivo studies strongly suggest that SOD1 acts in a prion-like manner in SOD1-fALS.
TDP-43 Prion Evidence
TDP-43 Structure and Function
As mentioned previously, aggregated cytoplasmic TDP-43 is observed in the majority of ALS cases (Ling et al., 2013). TDP-43 is a DNA/RNA-binding protein that has various roles in RNA metabolism, including trafficking, splicing, and degradation (Buratti et al., 2001, 2005; Hefferon et al., 2004; Mercado et al., 2005). The protein contains both a nuclear export signal (NES) and a nuclear localization signal (NLS), however, it is predominantly localized to the nucleus. Structurally, the protein is composed of 4 domains; an N-terminal domain (NTD) with a ubiquitin-like fold (Mompeán et al., 2016), tandem RNA-recognition motifs (RRM), and a prion-like (on the basis of sequence properties similar to yeast prion proteins) C-terminal low-complexity domain (LCD) rich in Gln/Asn residues (Figure 2A). The architecture of TDP-43 plays a significant role in its ability to interact with other TDP-43 molecules and other proteins. For example, the NTD can mediate the formation of physiological TDP-43 multimers (Afroz et al., 2017; Mompeán et al., 2017; Tsoi et al., 2017; Wang et al., 2018), and the LCD mediates TDP-43’s ability to form or take part in biomolecular condensates (Conicella et al., 2016; Li et al., 2018), such as stress granules (Bentmann et al., 2012).
FIGURE 2
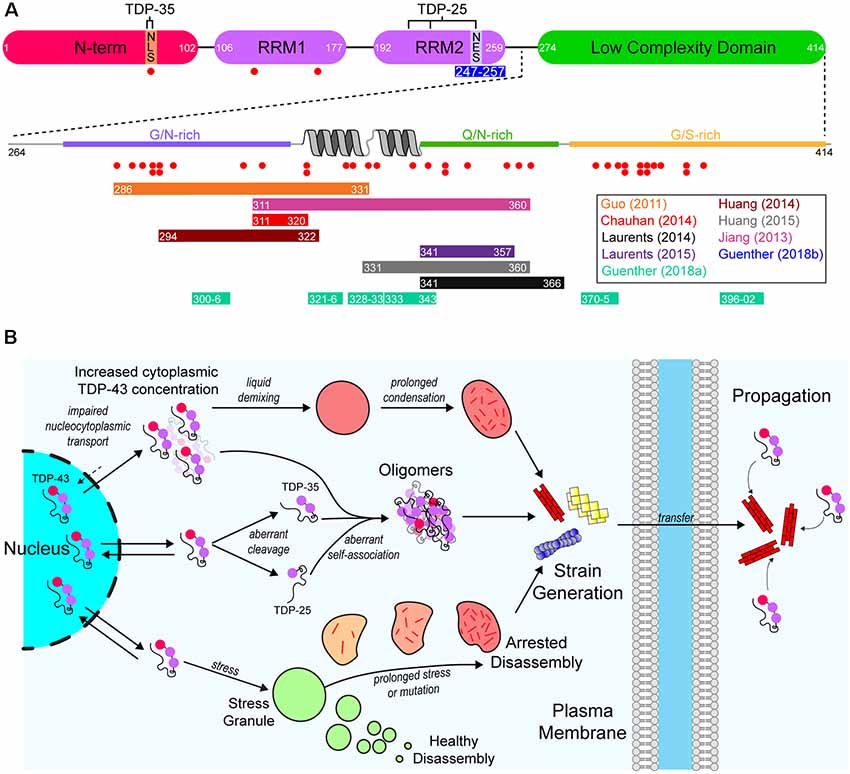
Figure 2. A model of the aggregation and prion-like propagation of TAR DNA-binding protein of 43 kDa (TDP-43) in ALS. (A) TDP-43 is composed of four domains which include the ubiquitin-like N-terminal domain (NTD), tandem RNA-recognition motifs (RRM), and a C-terminal low complexity domain (LCD). The NTD contains the nuclear localization signal (NLS) of TDP-43, whereas the nuclear export signal (NES) is in RRM2. The pathological 35 kDa (TDP-35) and 25 kDa (TDP-25) are formed via aberrant caspase-mediated cleavage at the NLS and in RRM2 respectively. The blue bar below RRM2 represents an amyloidogenic segment. The LCD is a prion-like domain due to it being enriched for G/N/Q/S residues. It also contains a helix-turn-helix motif which is important for association with other TDP-43 LCDs. Red dots below both the main TDP-43 graphic and the LCD expansion represent ALS-associated mutations. Colored bars represent experimentally identified regions of the protein that are capable of forming amyloid-like structures. Bar colors correspond to the publications listed on the right. (B) TDP-43 is a primarily nuclear-localized protein, however, it shuttles between the nucleus and cytoplasm. Impairment of nucleocytoplasmic transport (top) can lead to the cytoplasmic concentration of TDP-43 increasing, leading to liquid demixing to form biomolecular condensates distinct from stress granules. Prolonged residency of TDP-43 in these condensates can lead to the formation of aggregates. Aberrant cleavage (middle) leads to the formation of the highly aggregation-prone TDP-35 and TDP-25 pathological fragments. These fragments, and even full-length TDP-43 can aberrantly self-associate to form oligomers that may grow to become larger aggregates. Also, TDP-43 is known to inhabit stress granules (bottom). Given prolonged stress or genetic mutations, stress granules can persist for longer than necessary and can fail to disassemble, which leads to the formation of aggregates. Each of these possible pathways may also result in strains of TDP-43 aggregates that can propagate from cell-to-cell and seed further aggregation.
TDP-43 Pathological Features in Patients
Pathologically, TDP-43 is observed to be mislocalized from the nucleus to the cytoplasm in degenerating neurons, where it forms large ubiquitinated and hyperphosphorylated cytoplasmic inclusions (Neumann et al., 2006). Although there is some debate, strong evidence indicates that at least some of the TDP-43 inclusions observed in patient tissue contain amyloid structures (Mori et al., 2008; Bigio et al., 2013; Nonaka et al., 2013; Porta et al., 2018; Laferrière et al., 2019). Indeed, immunohistochemical examination of sALS patient tissue suggests that maturation of some TDP-43 inclusions proceeds first through small linear “wisps” in the cytoplasm that then go on to form thicker thread-like aggregates, which then grow to form skein-like inclusions (Mori et al., 2008). Additionally, more exhaustive processing of patient tissue to reduce background fluorescence shows that TDP-43 inclusions in ALS are strongly thioflavin-S fluorescent (Bigio et al., 2013). Furthermore, inclusions in sALS patient tissue that are observed to be non-linear at the micron scale are found to have a linear filamentous structure at the scale of tens of nanometers (Lowe et al., 1988; Lowe, 1994), suggesting potential amyloid-like aggregation is associated with TDP-43-related ALS. Biochemical characterization of the insoluble fractions of ALS patient CNS tissue showed that TDP-43 is aberrantly cleaved into 35 kDa and 25 kDa C-terminal fragments (Neumann et al., 2006) which are aggregation-prone and toxic in cell overexpression models (Igaz et al., 2009; Zhang et al., 2009; Yang et al., 2010; Nonaka et al., 2013). Although the TDP-43 C-terminal fragments are associated with ALS and FTD, what causes their fragmentation and what their pathological role might be is still debated (reviewed in Berning and Walker, 2019). It is interesting to note that genetic screening has identified more than 50 mutations in TDP-43 that are ALS or FTD-associated, with most of these mutations being localized to the LCD (Abel et al., 2013), a domain which comprises the bulk mass of the pathological C-terminal fragments.
Amyloid Aggregation of TDP-43
Regarding the aggregation propensity of TDP-43, the LCD is of particular interest since it is predicted to be a prion-like domain (PrLD) on the basis of similarity to the yeast prion protein Sup35 sequence physicochemical properties, which is enriched for asparagine, glutamine, tyrosine and glycine residues (King et al., 2012). Some PrLDs are capable of forming amyloid owing to their amino acid sequence being enriched for polar and uncharged amino acids such as glycine, glutamine, tyrosine, and asparagine (Alberti et al., 2009; Toombs et al., 2010; King et al., 2012). Indeed, application of the 3D-profile method (Thompson et al., 2006) to the TDP-43 LCD identified several regions that could potentially form amyloid fibrils, with subsequent crystallography experiments showing that 6 of these segments (300GNNQGSN306, 321AMMAAA326, 328AALQSS333, 333SWGMMGMLASQ343, 370GNNSYS375, and 396GFNGGFG402) could form steric zippers (Guenther et al., 2018a), a structure that is characteristic of the spines of amyloid fibrils (Sawaya et al., 2007; Eisenberg and Jucker, 2012). Other studies, using longer peptide segments, have also identified amyloid-forming regions within the TDP-43 LCD (Chen et al., 2010; Guo et al., 2011; Jiang et al., 2013, 2016; Mompeán et al., 2014; Cao et al., 2019; Figure 2A). A recent study has used cryo-electron microscopy to determine the structure of several TDP-43 LCD polymorphic fibrillar assemblies occurring from two segments of the LCD corresponding to amino acid residues 311–360 and 286–331 (Cao et al., 2019). This has implications for the prion-like nature of TDP-43 as different fibrillar polymorphs may have different prion-like characteristics.
Other than the LCD, a segment of RRM2 (247DLIIKGISVHI257) has been shown to form several steric zipper polymorphs (Guenther et al., 2018b), suggesting this domain may also contribute to pathological aggregation at least when RRM2 is partially folded (Tavella et al., 2018). Additionally, the size of the TDP-43 fragments (35 kDa or 25 kDa) influences the aggregation and degradation pathways of TDP-43 (Kasu et al., 2018). On the other hand, the majority of evidence for TDP-43 to form amyloid comes from purified protein studies utilizing C-terminal fragments that are either pathological fragments (35 or 25 kDa) or just the LCD (amino acids 274–414), as well as just the RRMs or small segments thereof (Guenther et al., 2018b; Agrawal et al., 2019; Cao et al., 2019). Less work has been carried out examining the ability of full-length TDP-43 to form amyloid in these assays (Furukawa et al., 2011; Vogler et al., 2018), which may be due to the difficulty in purifying and handling full-length recombinant TDP-43. It appears as though the full-length protein is capable of forming amyloid at low micromolar concentrations (Furukawa et al., 2011), and also capable of being seeded by cell culture-derived TDP-43 positive myo-granules (Vogler et al., 2018). Collectively, the above evidence suggests that, at the least, the pathologic fragments of TDP-43 are capable of forming amyloid and that the LCD makes a significant contribution to the formation of TDP-43 amyloid-like fibrils (Figure 2A).
Contribution of Phase Transitions to TDP-43 Amyloid Formation
Considering the importance of the TDP-43 LCD to the formation of both amyloid (Chen et al., 2010; Guo et al., 2011; Jiang et al., 2013, 2016; Mompeán et al., 2014) and biomolecular condensates (Conicella et al., 2016; Li et al., 2018), it is interesting to consider the relationship between these phenomena. Previous studies investigating ALS-associated mutations in both FUS (Murakami et al., 2015; Patel et al., 2015) and hnRNPA1 (Molliex et al., 2015) have highlighted their capacity to mature/age from a liquid-like state to more solid gels composed of fibrillar structures under conditions that promote phase transition. In the case of TDP-43, recent work has suggested that phase transitions of purified TDP-43 LCD promote the formation of amyloid-like fibrils (Babinchak et al., 2019), however, it is important to note that in some cases fibrils generated from the phase transition of PrLDs can be sensitive to sodium-dodecyl-sulfate (SDS; Lin et al., 2015), whereas typical amyloid is resistant to SDS denaturation (Kryndushkin et al., 2003; Halfmann and Lindquist, 2008). Indeed, recent work has highlighted the contribution of small sequence segments containing aromatic residues within PrLDs, called “low-complexity aromatic-rich kinked segments” (LARKS), to reversible phase transition and fiber formation (Hughes et al., 2018). Importantly, LARKS can form fibers that are calculated to have weaker binding energies than the steric zippers that form amyloid spines, evidenced by the LARKS-formed fibrils being susceptible to dissolution by gentle heat (Hughes et al., 2018). It has been suggested that post-translational modification or mutation of the TDP-43 LCD can result in the strengthening of LARKS interactions to become irreversible (Guenther et al., 2018a), although these experiments were performed using small peptide segments of the TDP-43 LCD. It remains to be established if full-length TDP-43 that has undergone maturation in its liquid-liquid phase-separated state forms typical steric zipper amyloid structures, although it is highly likely.
Cell culture studies of the ability of TDP-43 to form biomolecular condensates, and examination of the consequences of condensate formation, have been difficult for several reasons. First, the induction of stress granules requires stressors such as oxidative stress, heat stress, or osmotic stress (Aulas et al., 2017). These stressors not only affect cell health, making the long term monitoring of condensate maturation into aggregates difficult but also induce condensates with different protein constituents dependent on the stressor or cell type used (Aulas et al., 2017). Methods to overcome this have recently been introduced with the usage of light-sensitive proteins to promote protein-protein interactions of proteins capable of undergoing phase transitions (Shin et al., 2017, 2018; Dine et al., 2018; Bracha et al., 2019; Zhang et al., 2019). Replacement of the dimerization domain on G3BP1 with the light-sensitive Cryptochrome-2 (CRY2) protein allowed for the light-inducible generation of biomolecular condensates with similar biochemical and physical properties to stress granules without the addition of stressors (Zhang et al., 2019). The authors found that repetitive or sustained induction of the light-inducible condensates resulted in their evolution to inclusions with the pathological hallmarks of ALS, such as the deposition of SQSTM1 and the hyperphosphorylation of TDP-43 (Zhang et al., 2019). This work suggests that the initial site of TDP-43 aggregate nucleation may be in stress granules or perhaps another biomolecular condensate (Figure 2B), although, the authors did not examine if the inclusions formed in this assay were capable of transmitting aggregation pathology to naïve cells. A more recent study has suggested that either addition of preformed TDP-43 amyloid-like fibrils or higher than typical TDP-43 levels in the cytoplasm can provoke the formation of cytoplasmic TDP-43 condensates independent of stress granules (Gasset-Rosa et al., 2019), suggesting that nucleation of TDP-43 aggregates may occur in the absence of typical membraneless-organelles. It remains to be established if the aggregates generated by phase transitions can seed the further aggregation of TDP-43.
Intra- and Inter-cellular Prion-Like Spread of TDP-43 Aggregation
Overexpression of untagged or fluorescently-tagged TDP-43 in cultured mammalian cells can result in its mislocalization and aggregation in the cytoplasm (Johnson et al., 2009; Nonaka et al., 2009; Yamashita et al., 2009; Zhang et al., 2009, 2011; Barmada et al., 2010; Wang et al., 2010, 2013; Xu et al., 2010; Yang et al., 2010; Che et al., 2011, 2015; Furukawa et al., 2011; Gregory et al., 2012; Hong et al., 2012; Chang et al., 2013; Walker et al., 2013; Farrawell et al., 2015; Jiang et al., 2016; Tanaka and Hasegawa, 2016; Afroz et al., 2017; Zeineddine et al., 2017). Furthermore, disruption of the TDP-43 NLS leads to its accumulation and aggregation in the cytoplasm (a mutant denoted as TDP-43ΔNLS; Winton et al., 2008). Indeed, the first evidence for the seeded aggregation of TDP-43 in cells came from experiments utilizing an overexpression of TDP-43 with a C-terminal hemagglutinin (HA) tag (Furukawa et al., 2011). Furukawa et al. (2011) generated recombinant full-length TDP-43 and induced its fibrillar aggregation by vigorous agitation and subsequently introduced these fibrils to cells using lipofection. They found that introduction of TDP-43 aggregates to cells induced the aggregation of the HA-tagged TDP-43 in the cell cytoplasm, and that these cellular aggregates were polyubiquitinated and sarkosyl insoluble (Furukawa et al., 2011). The first evidence of the intercellular transmission of TDP-43 aggregation and relevance to patient pathology came when Nonaka et al. (2013) induced the aggregation of overexpressed HA-tagged TDP-43 in a neuronal cell line through lipofection of the insoluble protein fraction of human sALS or TDP-43 positive FTD patient brain samples. They observed that addition of either the FTD or sALS samples to cells resulted in the formation of HA-tagged positive TDP-43 positive inclusions. Additionally, they determined that the inclusions formed by the brain fractions in cultured cells could be used to seed further aggregation in cultures of naïve cells (Nonaka et al., 2013), providing substantial evidence supporting a pathological prion-like propagation of TDP-43 in FTD and ALS. Later studies showed similar results for the cell-to-cell spread of aggregation using stable cultures (Porta et al., 2018; Laferrière et al., 2019) overexpression cultures (Smethurst et al., 2016), co-cultures (Zeineddine et al., 2017), and even showed the axonal uptake and transport of TDP-43 seeds (Feiler et al., 2015).
Currently, cell cultured-based experiments provide the most well-established examinations of pathologic TDP-43 prion-like strains. Sequential deletion of amino acid segments, 20 residues in size, from the C-terminal region of TDP-43 highlighted amino acids 274–313 or 314–353 as being important for the cellular aggregation of TDP-43 (Shimonaka et al., 2016), suggesting that separate regions of the protein could be responsible for aggregation. Indeed, injection of amyloid fibrils composed of synthetic peptides composed of residues 274–313 or 314–353 resulted in the template-directed aggregation of wild-type TDP-43 or TDP-43ΔNLS indicating that more than one region of the C-terminal region of the protein was capable of seeding pathologic aggregation (Shimonaka et al., 2016), however, the authors of this study did not note differences in aggregation characteristics. Although this provides evidence of possible strain-like characteristics of TDP-43, synthetic peptides provide little relevance to the pathological structures that may exist in disease. More recently, strong evidence for the existence of TDP-43 strains in human FTD patients was provided by Laferrière et al. (2019) when they carefully processed human tissue from FTD patients and ALS patients to enrich for pathological TDP-43. Examination of the size of the enriched TDP-43 assemblies from patients using velocity sedimentation showed that disease subtypes had different sized assemblies present, and that material from different disease subtypes as capable of inducing different levels of toxicity in HEK cells stably expressing HA-tagged TDP-43 and primary mouse cortical neurons (Laferrière et al., 2019). Most importantly, the authors showed that the toxicity of the TDP-43 assemblies to cultured cells correlated with the severity of the disease determined in the donor patient. It will be interesting to see if the disease severity will correlate with an in vivo model of TDP-43 prion-like propagation in future work.
In vivo Seeding and Spread of TDP-43 Aggregation
Evidence supporting the prion-like propagation of TDP-43 aggregation in vivo occurred only recently. Porta et al. (2018) screened the prion-like properties of FTD patient brain extracts using cultured cells. Samples that had seeding activity were directly injected into the brains of mice expressing human TDP-43 with a mutated NLS signal under a doxycycline controllable promoter in their forebrain neurons (Igaz et al., 2011; Alfieri et al., 2014). It was found that injection of FTD patient-derived TDP-43 enriched samples led to the formation of pathologically relevant TDP-43 inclusions in the recipient mice (Porta et al., 2018). Additionally, careful immunohistochemical analysis of the injected mice showed that pathology appeared to spread through connected neuroanatomical tracts in a time-dependent manner, and interestingly, that in some cases endogenous mouse TDP-43 was colocalized with human TDP-43 inclusions. The authors then went on to inject seeding-competent, human-derived FTD material into non-transgenic wild-type mice, finding that the endogenous mouse TDP-43 could be seeded also, but to a lesser extent comparative to the doxycycline-regulated human TDP-43 transgenic mice (Porta et al., 2018). It was also determined that mice expressing a C-terminal fragment under a doxycycline regulatable element had a lower efficiency of seeding after injection with the FTD patient-derived samples, suggesting that the N-terminal regions of TDP-43 play an important role in its self-recognition and prion-like aggregation. Although the study by Porta et al. (2018) provides strong evidence for the prion-like role of TDP-43 in FTD, the experimental transmissibility of ALS patient-derived TDP-43 aggregates remains to be established.
Fused in Sarcoma and C9orf72
Fused in Sarcoma
Other than TDP-43 or SOD1, FUS plays an active role in the etiology of ALS owing to its presence in inclusions in a small percentage of ALS patients (Ling et al., 2013), and it containing several ALS-associated mutations (Kwiatkowski et al., 2009; Vance et al., 2009). Pathologically, FUS mostly forms basophilic inclusion bodies and was first found to play a role in a form of FTD called basophilic inclusion body disease (Munoz et al., 2009). Previous immunohistochemical analysis of basophilic inclusions in aytpical ALS patient tissue identified key constituents of stress granules [poly(A)-binding protein, TIA-1, and ribosomal protein S6] but not markers of other biomolecular condensates such as processing bodies or ribonucleoprotein particles (Fujita et al., 2008). Importantly, this work showed for the first time that stress granules play an important role in ALS pathogenesis.
Similar to TDP-43, FUS is an RBP which plays crucial roles in RNA metabolism (Belly et al., 2005; Fujii and Takumi, 2005; Andersson et al., 2008), but is also important for the DNA-damage repair response (Wang et al., 2008, 2013). Structurally, FUS belongs to the ten-eleven translocation (TET) protein family which are characterized by their N-terminal QSGY-rich domains, conserved RRMs, RGG-rich regions, and a C-terminal zinc finger motif (Morohoshi et al., 1998; Iko et al., 2004). Importantly, the N-terminal QSGY-rich domain (residues 1–165) of FUS is predicted to be a prion-like domain (PrLD; King et al., 2012), suggesting that this domain plays a role in the pathological aggregation of FUS observed in patients. However, the majority of ALS-associated mutations identified in FUS are localized to the N-terminal RGG-rich region (residues 165–276) or a short stretch (residues 495–526) of the C-terminal RGG-rich region (residues 449–526) which contains the FUS NLS (Abel et al., 2013); only one ALS-associated mutation (G156E) has been identified in the PrLD.
Examination of purified FUS PrLD has shown that it can undergo a phase transition to form hydrogels composed of amyloid-like fibrils (Han et al., 2012; Kato et al., 2012; Kwon et al., 2013). Additionally, EGFP-tagged full-length FUS can form biomolecular condensates in which the G156E mutation can exacerbate a transition from liquid to fibrous solid (Patel et al., 2015). Interestingly, although the ability of purified FUS to form aggregates is established, evidence suggesting that these assemblies can seed the aggregation of soluble FUS protein has only recently been established (Nomura et al., 2014). Nomura et al. (2014) generated purified GST-tagged FUS wild-type and the G156E mutant, finding that the G156E mutant was more prone to form fibrils and that these fibrils were capable of seeding purified wild-type FUS. They then went on to transfect rat-hippocampal primary neurons with both G156E FUS and wild-type FUS constructs, finding that inclusions would only form for the mutant and that these inclusions would recruit the plasmid derived wild-type FUS (Nomura et al., 2014). Other than the work by Nomura et al. (2014), there has been little research carried out on examining the prion-like characteristics of FUS. Future work examining if FUS has bona-fide prion-like characteristics (seeding from exogenous aggregates, cell-to-cell propagation) will be important in determining if a prion-like mechanism is common across the main aggregation-prone proteins in ALS (SOD1, TDP-43, FUS).
C9orf72
Hexanucleotide repeat (GGGGCC) expansions in C9orf72 were first identified in DeJesus-Hernandez et al. (2011) and Renton et al. (2011) and are now thought to be responsible for approximately 40% of fALS cases (Majounie et al., 2012). Typically, the number of repeats in the healthy population is ≤11, whereas the number of repeats in an individual suffering from C9orf72-associated fALS can number in the thousands (van Blitterswijk et al., 2013; Suh et al., 2015). Although exact functions for the protein encoded by C9orf72 is yet to be fully elucidated, there is evidence to suggest that it has a role as a guanine nucleotide exchange factor for small GTPases (Wu et al., 2011; Levine et al., 2013; Iyer et al., 2018). The protein encoded by C9orf72 is also implicated in other cellular functions such as autophagy (Sellier et al., 2016; Webster et al., 2016) and vesicular transport (Aoki et al., 2017). Although there is little known about the function(s) of the C9orf72 protein, understanding of the mechanisms by which disease arises consequently from repeat expansions has proceeded rapidly.
There are now thought to be several mechanisms by which repeat expansions in C9orf72 cause toxicity, including a loss of function, transcription into long repetitive RNA that forms foci composed of sense or antisense RNA (Donnelly et al., 2013; Gendron et al., 2013; Lagier-Tourenne et al., 2013; Mizielinska et al., 2013; Zu et al., 2013), and the translation of repeat RNA into five different dipeptide repeat (DPR) proteins (poly-GR, poly-GP, poly-GA, poly-PR, and poly-PA; Mori et al., 2013). Currently, there is little evidence to suggest a prion-like propagation of any of the DPRs generated in C9orf72-associated ALS. One study by Westergard et al. (2016) used a battery of cell culture experiments to examine DPR spread between cells. They determined that all DPRs, with the exception of poly-PR, were capable of transfer between cells through anterograde and retrograde mechanisms, and that this transfer can occur through both exosome-dependent and exosome-independent mechanisms Modes of transfer of prion-like material is summarized in Figure 3. Although this work provides strong evidence for the cell-to-cell transfer of certain DPRs, it is still not established if DPRs can assume a conformation with prion-like characteristics or if they can seed further aggregation of other DPRs.
FIGURE 3
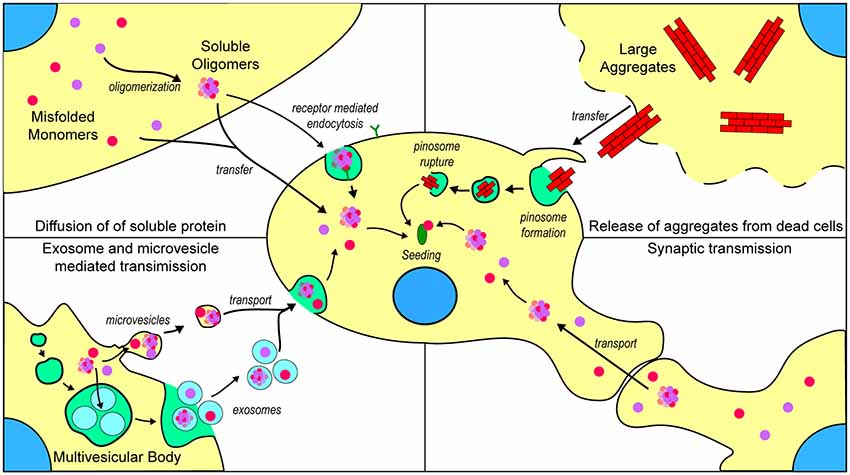
Figure 3. Mechanisms by which prion-like ALS-associated protein aggregates may transfer between cells. (Top left) Once formed, misfolded monomers and soluble oligomers can potentially exit the cell through diffusion and enter nearby cells either through diffusion or receptor-mediated endocytosis. (Top right) Cells that are dead or dying can potentially release larger insoluble aggregates that can be taken up into nearby naïve cells through micropinocytosis. (Bottom left) Misfolded and oligomeric proteins are capable of being loaded into either microvesicles or exosomes for transport to nearby cells. (Bottom right) Misfolded monomers or soluble oligomers are capable of being transported across axon terminals to naïve cells.
Perspective and Conclusion
It is now established that prion-like mechanisms play key roles in the cell-specific nature and cell-to-cell spread of pathology in neurodegenerative disease (Prusiner, 2013). In particular, ALS shows the hallmarks of prion-like propagation owing to the spread of both symptoms and pathology throughout affected tissues (evidence is summarized in Table 1). Different types of research including biophysical, biochemical, and biomedical studies have increased our understanding of the contribution of prion-like mechanisms to ALS. Cell-free in vitro examination of the aggregation and seeding characteristics of prion-like proteins associate with ALS have provided useful information on the kinetics of aggregation and the biophysical traits of the substrate and seed species. Cell culture experiments have helped establish the cellular mechanisms by which proteinaceous aggregation may occur, and how cells may facilitate the transmission of the prion-like particles. Research using animal models has been effective at establishing the biochemical and phenotypic profiles of some prion-like proteins and strains in ALS and has also helped in determining if material from patients suffering ALS or FTD have prion-like (PrP-like) properties (Ayers et al., 2014, 2016a,b; Bergh et al., 2015; Lang et al., 2015; Bidhendi et al., 2016, 2018; Laferrière et al., 2019).
Although great progress has been made in the understanding of the prion-like nature of ALS, what remains to be elucidated are high-resolution structural models of specific ALS-associated prion strains that are related to disease pathology. While recent work has identified several fibrillar polymorphs of the TDP-43 low complexity domain (Cao et al., 2019), it remains to be established if these conformational assemblies have relevance to pathogenicity in vivo, missing a crucial link between conformation of the amyloid-like assemblies and their toxicity. If we are to develop effective therapeutics against such polymorphic protein assemblies, we need to know what to target. Future experiments investigating the roles of prion-like propagation in human disease should combine in vivo models of seeding and neuroanatomical spread, cell culture studies of seeding potency of different strains, and the structural resolving power of cryo-electron microscopy for structure determination.
The existence of prion-like conformational strains in ALS provides not only a challenging hurdle to overcome experimentally and therapeutically, but also provides opportunities to develop effective therapies. With access to disease-specific structural models of prion-like aggregates, researchers have the capability to perform large-scale structure-based small molecule docking screens, which may identify compounds that could alleviate or prevent the occurrence or spread of pathology in prion-like diseases (Jiang et al., 2013). Furthermore, structure-based design of monoclonal antibodies targeted against misfolding specific epitopes is a growing space with great potential. Given a prion strain structure, an antibody could be rationally designed to target a specific conformation of protein assembly to prevent further spread or promote clearance (Peng et al., 2018; Silverman et al., 2018). An advantage of this approach is that the use of monoclonal antibodies would overcome target distraction through their high binding specificity.
Lastly, the role of prion-like propagation of pathologic proteins in ALS is likely tied to the mostly idiopathic nature of the disease. Whilst there are mutations that are ALS-causative, the majority of cases remain sporadic, suggesting that an environmental trigger may exist. From this, a question that must be asked is whether or not all, or a set, of sporadic cases of ALS are the result of a still-unidentified prion-like agent in the environment. Recent research has provided evidence supporting the Braak hypothesis (Braak et al., 2003) that pathologically aggregated α-synuclein can propagate from the gut, through the vagus nerve, to the ventral midbrain (Kim et al., 2019). Furthermore, some patients who received human growth hormone from patients suffering from Creutzfeld-Jakob disease (CJD) developed CJD themselves (Gibbs et al., 1985). Surprisingly, some of the patients who received growth hormone from CJD sufferers were found to have amyloid-β pathology, suggesting that amyloid-β had also propagated in these cases (Jaunmuktane et al., 2015). Considering the above evidence, there is a possibility for similar mechanisms to be occurring in ALS, however, there is currently no strong evidence supporting this hypothesis. Epidemiological studies may provide further insight into links between the populations that develop sporadic ALS beyond just genetic background.
Author Contributions
LM wrote the initial manuscript. SP edited and revised the manuscript. JY edited and revised the manuscript. NC conceived, edited and revised the manuscript.
Funding
LM is supported by funding from The Motor Neurone Disease Research Institute of Australia (Dr. Paul Brock MND NSW Research Grant). SP acknowledges the Canadian Institutes of Health Research Transitional Operating Grant 2682, and the Alberta Prion Research Institute, Research Team Program Grant PTM13007. JY is supported by an NHMRC Career Development Fellowship (1084144) and a Dementia Teams Grant (1095215). NC acknowledges the Canadian Consortium for Neurodegeneration in Aging (CCNA) and Brain Canada.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdolvahabi, A., Shi, Y., Chuprin, A., Rasouli, S., and Shaw, B. F. (2016). Stochastic formation of fibrillar and amorphous superoxide dismutase oligomers linked to amyotrophic lateral sclerosis. ACS Chem. Neurosci. 7, 799–810. doi: 10.1021/acschemneuro.6b00048
Abel, O., Shatunov, A., Jones, A. R., Andersen, P. M., Powell, J. F., and Al-Chalabi, A. (2013). Development of a smartphone app for a genetics website: the amyotrophic lateral sclerosis online genetics database (ALSoD). JMIR Mhealth Uhealth 1:e18. doi: 10.2196/mhealth.2706
Afroz, T., Hock, E. M., Ernst, P., Foglieni, C., Jambeau, M., Gilhespy, L. A. B., et al. (2017). Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 8:45. doi: 10.1038/s41467-017-00062-0
Agrawal, S., Kuo, P.-H., Chu, L.-Y., Golzarroshan, B., Jain, M., and Yuan, H. S. (2019). RNA recognition motifs of disease-linked RNA-binding proteins contribute to amyloid formation. Sci. Rep. 9:6171. doi: 10.1038/s41598-019-42367-8
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S. W., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Alberti, S., Halfmann, R., King, O., Kapila, A., and Lindquist, S. (2009). A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137, 146–158. doi: 10.1016/j.cell.2009.02.044
Al-Chalabi, A., Andersen, P. M., Nilsson, P., Chioza, B., Andersson, J. L., Russ, C., et al. (1999). Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum. Mol. Genet. 8, 157–164. doi: 10.1093/hmg/8.2.157
Al-Chalabi, A., Hardiman, O., Kiernan, M. C., Chiò, A., Rix-Brooks, B., and van den Berg, L. H. (2016). Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 15, 1182–1194. doi: 10.1016/S1474-4422(16)30199-5
Alexander, E. J., Ghanbari Niaki, A., Zhang, T., Sarkar, J., Liu, Y., Nirujogi, R. S., et al. (2018). Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc. Natl. Acad. Sci. U S A 115, E11485–E11494. doi: 10.1073/pnas.1811997115
Alfieri, J. A., Pino, N. S., and Igaz, L. M. (2014). Reversible behavioral phenotypes in a conditional mouse model of TDP-43 proteinopathies. J. Neurosci. 34, 15244–15259. doi: 10.1523/JNEUROSCI.1918-14.2014
Andersson, M. K., Stahlberg, A., Arvidsson, Y., Olofsson, A., Semb, H., Stenman, G., et al. (2008). The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 9:37. doi: 10.1186/1471-2121-9-37
Aoki, Y., Manzano, R., Lee, Y., Dafinca, R., Aoki, M., Douglas, A. G. L., et al. (2017). C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897. doi: 10.1093/brain/awx024
Arosio, P., Knowles, T. P., and Linse, S. (2015). On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 17, 7606–7618. doi: 10.1039/c4cp05563b
Aulas, A., Fay, M. M., Lyons, S. M., Achorn, C. A., Kedersha, N., Anderson, P., et al. (2017). Stress-specific differences in assembly and composition of stress granules and related foci. J. Cell Sci. 130, 927–937. doi: 10.1242/jcs.199240
Ayers, J. I., Diamond, J., Sari, A., Fromholt, S., Galaleldeen, A., Ostrow, L. W., et al. (2016a). Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS. Acta Neuropathol. 132, 827–840. doi: 10.1007/s00401-016-1623-4
Ayers, J. I., Fromholt, S. E., O’Neal, V. M., Diamond, J. H., and Borchelt, D. R. (2016b). Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 131, 103–114. doi: 10.1007/s00401-015-1514-0
Ayers, J. I., and Cashman, N. R. (2018). Chapter 18-Prion-like mechanisms in amyotrophic lateral sclerosis. Handbook of Clin. Neurol. 153, 337–354. doi: 10.1016/B978-0-444-63945-5.00018-0
Ayers, J. I., Fromholt, S., Koch, M., DeBosier, A., McMahon, B., Xu, G., et al. (2014). Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 128, 791–803. doi: 10.1007/s00401-014-1342-7
Babinchak, W. M., Haider, R., Dumm, B. K., Sarkar, P., Surewicz, K., Choi, J. K., et al. (2019). The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 294, 6306–6317. doi: 10.1074/jbc.ra118.007222
Baker, M. J., Tatsuta, T., and Langer, T. (2011). Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 3:a007559. doi: 10.1101/cshperspect.a007559
Banani, S. F., Lee, H. O., Hyman, A. A., and Rosen, M. K. (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298. doi: 10.1038/nrm.2017.7
Banci, L., Barbieri, L., Bertini, I., Luchinat, E., Secci, E., Zhao, Y., et al. (2013). Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat. Chem. Biol. 9, 297–299. doi: 10.1038/nchembio.1202
Banci, L., Bertini, I., Boca, M., Girotto, S., Martinelli, M., Valentine, J. S., et al. (2008). SOD1 and amyotrophic lateral sclerosis: mutations and oligomerization. PLoS One 3:e1677. doi: 10.1371/journal.pone.0001677
Banci, L., Bertini, I., Cantini, F., Kozyreva, T., Massagni, C., Palumaa, P., et al. (2012). Human superoxide dismutase 1 (hSOD1) maturation through interaction with human copper chaperone for SOD1 (hCCS). Proc. Natl. Acad. Sci. U S A 109, 13555–13560. doi: 10.1073/pnas.1207493109
Barmada, S. J., Skibinski, G., Korb, E., Rao, E. J., Wu, J. Y., and Finkbeiner, S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 30, 639–649. doi: 10.1523/JNEUROSCI.4988-09.2010
Belly, A., Moreau-Gachelin, F., Sadoul, R., and Goldberg, Y. (2005). Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci. Lett. 379, 152–157. doi: 10.1016/j.neulet.2004.12.071
Bentmann, E., Neumann, M., Tahirovic, S., Rodde, R., Dormann, D., and Haass, C. (2012). Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094. doi: 10.1074/jbc.m111.328757
Bergh, J., Zetterstrom, P., Andersen, P. M., Brannstrom, T., Graffmo, K. S., Jonsson, P. A., et al. (2015). Structural and kinetic analysis of protein-aggregate strains in vivo using binary epitope mapping. Proc. Natl. Acad. Sci. U S A 112, 4489–4494. doi: 10.1073/pnas.1419228112
Berning, B. A., and Walker, A. K. (2019). The pathobiology of TDP-43 C-terminal fragments in ALS and FTLD. Front. Neurosci. 13:335. doi: 10.3389/fnins.2019.00335
Bidhendi, E. E., Bergh, J., Zetterstrom, P., Andersen, P. M., Marklund, S. L., and Brannstrom, T. (2016). Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis-like disease. J. Clin. Invest. 126, 2249–2253. doi: 10.1172/JCI84360
Bidhendi, E. E., Bergh, J., Zetterström, P., Forsberg, K., Pakkenberg, B., Andersen, P. M., et al. (2018). Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 136, 939–953. doi: 10.1007/s00401-018-1915-y
Bigio, E. H., Wu, J. Y., Deng, H.-X., Bit-Ivan, E. N., Mao, Q., Ganti, R., et al. (2013). Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid. Acta Neuropathol. 125, 463–465. doi: 10.1007/s00401-013-1089-6
Boeynaems, S., Bogaert, E., Kovacs, D., Konijnenberg, A., Timmerman, E., Volkov, A., et al. (2017). Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell 65, 1044.e5–1055.e5. doi: 10.1016/j.molcel.2017.02.013
Borchelt, D. R., Guarnieri, M., Wong, P. C., Lee, M. K., Slunt, H. S., Xu, Z. S., et al. (1995). Superoxide dismutase 1 subunits with mutations linked to familial amyotrophic lateral sclerosis do not affect wild-type subunit function. J. Biol. Chem. 270, 3234–3238. doi: 10.1074/jbc.270.7.3234
Borchelt, D. R., Lee, M. K., Slunt, H. S., Guarnieri, M., Xu, Z. S., Wong, P. C., et al. (1994). Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. U S A 91, 8292–8296. doi: 10.1073/pnas.91.17.8292
Bosco, D. A., Morfini, G., Karabacak, N. M., Song, Y., Gros-Louis, F., Pasinelli, P., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403. doi: 10.1038/nn.2660
Braak, H., Tredici, K. D., Rüb, U., de Vos, R. A. I., Jansen Steur, E. N. H., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Bracha, D., Walls, M. T., Wei, M.-T., Zhu, L., Kurian, M., Avalos, J. L., et al. (2019). Mapping local and global liquid phase behavior in living cells using photo-oligomerizable seeds. Cell 176:407. doi: 10.1016/j.cell.2018.12.026
Brenner, D., Yilmaz, R., Müller, K., Grehl, T., Petri, S., Meyer, T., et al. (2018). Hot-spot KIF5A mutations cause familial ALS. Brain 141, 688–697. doi: 10.1093/brain/awx370
Brichta, L., and Greengard, P. (2014). Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: an update. Front. Neuroanat. 8:152. doi: 10.3389/fnana.2014.00152
Buratti, E., Brindisi, A., Giombi, M., Tisminetzky, S., Ayala, Y. M., and Baralle, F. E. (2005). TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 280, 37572–37584. doi: 10.1074/jbc.M505557200
Buratti, E., Dörk, T., Zuccato, E., Pagani, F., Romano, M., and Baralle, F. E. (2001). Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784. doi: 10.1093/emboj/20.7.1774
Butti, Z., and Patten, S. A. (2019). RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 9:712. doi: 10.3389/fgene.2018.00712
Cao, Q., Boyer, D. R., Sawaya, M. R., Ge, P., and Eisenberg, D. S. (2019). Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 26, 619–627. doi: 10.1038/s41594-019-0248-4
Capper, M. J., Wright, G. S. A., Barbieri, L., Luchinat, E., Mercatelli, E., McAlary, L., et al. (2018). The cysteine-reactive small molecule ebselen facilitates effective SOD1 maturation. Nat. Commun. 9:1693. doi: 10.1038/s41467-018-04114-x
Chan, P. K., Chattopadhyay, M., Sharma, S., Souda, P., Gralla, E. B., Borchelt, D. R., et al. (2013). Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc. Natl. Acad. Sci. U S A 110, 10934–10939. doi: 10.1073/pnas.1309613110
Chang, H. Y., Hou, S. C., Way, T. D., Wong, C. H., and Wang, I. F. (2013). Heat-shock protein dysregulation is associated with functional and pathological TDP-43 aggregation. Nat. Commun. 4:2757. doi: 10.1038/ncomms3757
Chattopadhyay, M., Durazo, A., Sohn, S. H., Strong, C. D., Gralla, E. B., Whitelegge, J. P., et al. (2008). Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. U S A 105, 18663–18668. doi: 10.1073/pnas.0807058105
Chattopadhyay, M., Nwadibia, E., Strong, C. D., Gralla, E. B., Valentine, J. S., and Whitelegge, J. P. (2015). The disulfide bond, but not zinc or dimerization, controls initiation and seeded growth in amyotrophic lateral sclerosis-linked Cu,Zn superoxide dismutase (SOD1) fibrillation. J. Biol. Chem. 290, 30624–30636. doi: 10.1074/jbc.m115.666503
Che, M. X., Jiang, L. L., Li, H. Y., Jiang, Y. J., and Hu, H. Y. (2015). TDP-35 sequesters TDP-43 into cytoplasmic inclusions through binding with RNA. FEBS Lett. 589, 1920–1928. doi: 10.1016/j.febslet.2015.06.009
Che, M. X., Jiang, Y. J., Xie, Y. Y., Jiang, L. L., and Hu, H. Y. (2011). Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J. 25, 2344–2353. doi: 10.1096/fj.10-174482
Chen, A. K., Lin, R. Y., Hsieh, E. Z., Tu, P. H., Chen, R. P., Liao, T. Y., et al. (2010). Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 132, 1186–1187. doi: 10.1021/ja9066207
Chi, H., Chang, H.-Y., and Sang, T.-K. (2018). Neuronal cell death mechanisms in major neurodegenerative diseases. Int. J. Mol. Sci. 19:E3082. doi: 10.3390/ijms19103082
Chia, R., Tattum, M. H., Jones, S., Collinge, J., Fisher, E. M. C., and Jackson, G. S. (2010). Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS One 5:e10627. doi: 10.1371/journal.pone.0010627
Chiti, F., and Dobson, C. M. (2017). Protein misfolding, amyloid formation and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. doi: 10.1146/annurev-biochem-061516-045115
Ciryam, P., Lambert-Smith, I. A., Bean, D. M., Freer, R., Cid, F., Tartaglia, G. G., et al. (2017). Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. U S A 114, E3935–E3943. doi: 10.1073/pnas.1613854114
Conicella, A. E., Zerze, G. H., Mittal, J., and Fawzi, N. L. (2016). ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24, 1537–1549. doi: 10.1016/j.str.2016.07.007
Couthouis, J., Hart, M. P., Shorter, J., DeJesus-Hernandez, M., Erion, R., Oristano, R., et al. (2011). A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. U S A 108, 20881–20890. doi: 10.1073/pnas.1109434108
Da Cruz, S., Bui, A., Saberi, S., Lee, S. K., Stauffer, J., McAlonis-Downes, M., et al. (2017). Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol. 134, 97–111. doi: 10.1007/s00401-017-1688-8
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, H. X., Chen, W., Hong, S. T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset alS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
DiDonato, M., Craig, L., Huff, M. E., Thayer, M. M., Cardoso, R. M., Kassmann, C. J., et al. (2003). ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J. Mol. Biol. 332, 601–615. doi: 10.1016/s0022-2836(03)00889-1
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Dine, E., Gil, A. A., Uribe, G., Brangwynne, C. P., and Toettcher, J. E. (2018). Protein phase separation provides long-term memory of transient spatial stimuli. Cell Syst. 6, 655.e5–663.e5. doi: 10.1016/j.cels.2018.05.002
Donnelly, C. J., Zhang, P. W., Pham, J. T., Haeusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015
Eisenberg, D., and Jucker, M. (2012). The amyloid state of proteins in human diseases. Cell 148, 1188–1203. doi: 10.1016/j.cell.2012.02.022
Ekhtiari Bidhendi, E., Bergh, J., Zetterström, P., Forsberg, K., Pakkenberg, B., Andersen, P. M., et al. (2018). Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 136, 939–953. doi: 10.1007/s00401-018-1915-y
Erkkinen, M. G., Kim, M. O., and Geschwind, M. D. (2018). Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 10:a033118. doi: 10.1101/cshperspect.a033118
Farrawell, N. E., Lambert-Smith, I. A., Warraich, S. T., Blair, I. P., Saunders, D. N., Hatters, D. M., et al. (2015). Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 5:13416. doi: 10.1038/srep13416
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Feiler, M. S., Strobel, B., Freischmidt, A., Helferich, A. M., Kappel, J., Brewer, B. M., et al. (2015). TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 211, 897–911. doi: 10.1083/jcb.201504057
Figley, M. D., Bieri, G., Kolaitis, R. M., Taylor, J. P., and Gitler, A. D. (2014). Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J. Neurosci. 34, 8083–8097. doi: 10.1523/jneurosci.0543-14.2014
Forsberg, K., Andersen, P. M., Marklund, S. L., and Brannstrom, T. (2011). Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 121, 623–634. doi: 10.1007/s00401-011-0805-3
Forsberg, K., Graffmo, K., Pakkenberg, B., Weber, M., Nielsen, M., Marklund, S., et al. (2019). Misfolded SOD1 inclusions in patients with mutations in C9orf72 and other ALS/FTD-associated genes. J. Neurol. Neurosurg. Psychiatry 90, 861–869. doi: 10.1136/jnnp-2018-319386
Forsberg, K., Jonsson, P. A., Andersen, P. M., Bergemalm, D., Graffmo, K. S., Hultdin, M., et al. (2010). Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 5:e11552. doi: 10.1371/journal.pone.0011552
Fujii, R., and Takumi, T. (2005). TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 118, 5755–5765. doi: 10.1242/jcs.02692
Fujita, K., Ito, H., Nakano, S., Kinoshita, Y., Wate, R., and Kusaka, H. (2008). Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol. 116, 439–445. doi: 10.1007/s00401-008-0415-x
Furukawa, Y., Kaneko, K., and Nukina, N. (2011). Molecular properties of TAR DNA binding protein-43 fragments are dependent upon its cleavage site. Biochim. Biophys. Acta 1812, 1577–1583. doi: 10.1016/j.bbadis.2011.09.005
Furukawa, Y., Kaneko, K., Watanabe, S., Yamanaka, K., and Nukina, N. (2011). A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 286, 18664–18672. doi: 10.1074/jbc.M111.231209
Gal, J., Kuang, L., Barnett, K. R., Zhu, B. Z., Shissler, S. C., Korotkov, K. V., et al. (2016). ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 132, 563–576. doi: 10.1007/s00401-016-1601-x
Gao, J., Wang, L., Huntley, M. L., Perry, G., and Wang, X. (2018). Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. doi: 10.1111/jnc.14327 [Epub ahead of print].
Gasset-Rosa, F., Lu, S., Yu, H., Chen, C., Melamed, Z., Guo, L., et al. (2019). Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102, 339.e7–357.e7. doi: 10.1016/j.neuron.2019.02.038
Gendron, T. F., Bieniek, K. F., Zhang, Y. J., Jansen-West, K., Ash, P. E., Caulfield, T., et al. (2013). Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844. doi: 10.1007/s00401-013-1192-8
Gibbs, C. J. Jr., Joy, A., Heffner, R., Franko, M., Miyazaki, M., Asher, D. M., et al. (1985). Clinical and pathological features and laboratory confirmation of Creutzfeldt-Jakob disease in a recipient of pituitary-derived human growth hormone. N. Engl. J. Med. 313, 734–738. doi: 10.1056/NEJM198509193131207
Glatzel, M., Stoeck, K., Seeger, H., Lührs, T., and Aguzzi, A. (2005). Human prion diseases: molecular and clinical aspects. JAMA Neurology 62, 545–552. doi: 10.1001/archneur.62.4.545
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/s0006-291x(84)80190-4
Gopal, P. P., Nirschl, J. J., Klinman, E., and Holzbaur, E. L. F. (2017). Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci. U S A 114, E2466–E2475. doi: 10.1073/pnas.1614462114
Grad, L. I., Guest, W. C., Yanai, A., Pokrishevsky, E., Neill, M. A., Gibbs, E., et al. (2011). Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. U S A 108, 16398–16403. doi: 10.1073/pnas.1102645108
Grad, L. I., Yerbury, J. J., Turner, B. J., Guest, W. C., Pokrishevsky, E., O’Neill, M. A., et al. (2014). Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U S A 111, 3620–3625. doi: 10.1073/pnas.1312245111
Grad, L. I., Rouleau, G. A., Ravits, J., and Cashman, N. R. (2017). Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb. Perspect. Med. 7:a024117. doi: 10.1101/cshperspect.a024117
Gregory, J. M., Barros, T. P., Meehan, S., Dobson, C. M., and Luheshi, L. M. (2012). The aggregation and neurotoxicity of TDP-43 and its ALS-associated 25 kDa fragment are differentially affected by molecular chaperones in Drosophila. PLoS One 7:e31899. doi: 10.1371/journal.pone.0031899
Gros-Louis, F., Larivière, R., Gowing, G., Laurent, S., Camu, W., Bouchard, J. P., et al. (2004). A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J. Biol. Chem. 279, 45951–45956. doi: 10.1074/jbc.M408139200
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U S A 83, 4913–4917. doi: 10.1073/pnas.83.13.4913
Guenther, E. L., Cao, Q., Trinh, H., Lu, J., Sawaya, M. R., Cascio, D., et al. (2018a). Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat. Struct. Mol. Biol. 25, 463–471. doi: 10.1038/s41594-018-0064-2
Guenther, E. L., Ge, P., Trinh, H., Sawaya, M. R., Cascio, D., Boyer, D. R., et al. (2018b). Atomic-level evidence for packing and positional amyloid polymorphism by segment from TDP-43 RRM2. Nat. Struct. Mol. Biol. 25, 311–319. doi: 10.1038/s41594-018-0045-5
Guo, W., Chen, Y., Zhou, X., Kar, A., Ray, P., Chen, X., et al. (2011). An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 18, 822–830. doi: 10.1038/nsmb.2053
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Halfmann, R., and Lindquist, S. (2008). Screening for amyloid aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. J. Vis. Exp. 17:838. doi: 10.3791/838
Han, T. W., Kato, M., Xie, S., Wu Leeju, C., Mirzaei, H., Pei, J., et al. (2012). Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779. doi: 10.1016/j.cell.2012.04.016
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 3:17071. doi: 10.1038/nrdp.2017.71
Hardiman, O., van den Berg, L. H., and Kiernan, M. C. (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 639–649. doi: 10.1038/nrneurol.2011.153
Hefferon, T. W., Groman, J. D., Yurk, C. E., and Cutting, G. R. (2004). A variable dinucleotide repeat in the CFTR gene contributes to phenotype diversity by forming RNA secondary structures that alter splicing. Proc. Natl. Acad. Sci. U S A 101, 3504–3509. doi: 10.1073/pnas.0400182101
Hensel, N., and Claus, P. (2017). The Actin cytoskeleton in SMA and ALS: how does it contribute to motoneuron degeneration? Neuroscientist 24, 54–72. doi: 10.1177/1073858417705059
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U S A 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Hong, K., Li, Y., Duan, W., Guo, Y., Jiang, H., Li, W., et al. (2012). Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 530, 144–149. doi: 10.1016/j.neulet.2012.10.003
Hughes, M. P., Sawaya, M. R., Boyer, D. R., Goldschmidt, L., Rodriguez, J. A., Cascio, D., et al. (2018). Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 359, 698–701. doi: 10.1126/science.aan6398
Igaz, L. M., Kwong, L. K., Chen-Plotkin, A., Winton, M. J., Unger, T. L., Xu, Y., et al. (2009). Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J. Biol. Chem. 284, 8516–8524. doi: 10.1074/jbc.M809462200
Igaz, L. M., Kwong, L. K., Lee, E. B., Chen-Plotkin, A., Swanson, E., Unger, T., et al. (2011). Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest. 121, 726–738. doi: 10.1172/JCI44867
Iko, Y., Kodama, T. S., Kasai, N., Oyama, T., Morita, E. H., Muto, T., et al. (2004). Domain architectures and characterization of an RNA-binding protein, TLS. J. Biol. Chem. 279, 44834–44840. doi: 10.1074/jbc.M408552200
Ivanova, M. I., Sievers, S. A., Guenther, E. L., Johnson, L. M., Winkler, D. D., Galaleldeen, A., et al. (2014). Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc. Natl. Acad. Sci. U S A 111, 197–201. doi: 10.1073/pnas.1320786110
Iyer, S., Subramanian, V., and Acharya, K. R. (2018). C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ 6:e5815. doi: 10.7717/peerj.5815
Jaunmuktane, Z., Mead, S., Ellis, M., Wadsworth, J. D., Nicoll, A. J., Kenny, J., et al. (2015). Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525, 247–250. doi: 10.1038/nature15369
Jellinger, K. A. (2012). Interaction between pathogenic proteins in neurodegenerative disorders. J. Cell. Mol. Med. 16, 1166–1183. doi: 10.1111/j.1582-4934.2011.01507.x
Jiang, L. L., Che, M. X., Zhao, J., Zhou, C. J., Xie, M. Y., Li, H. Y., et al. (2013). Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 288, 19614–19624. doi: 10.1074/jbc.M113.463828
Jiang, L., Liu, C., Leibly, D., Landau, M., Zhao, M., Hughes, M. P., et al. (2013). Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid β. Elife 2:e00857. doi: 10.7554/eLife.00857
Jiang, L. L., Zhao, J., Yin, X. F., He, W. T., Yang, H., Che, M. X., et al. (2016). Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci. Rep. 6:23928. doi: 10.1038/srep23928
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Johnson, J. O., Pioro, E. P., Boehringer, A., Chia, R., Feit, H., Renton, A. E., et al. (2014). Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 17, 664–666. doi: 10.1038/nn.3688
Johnson, B. S., Snead, D., Lee, J. J., McCaffery, J. M., Shorter, J., and Gitler, A. D. (2009). TDP-43 is intrinsically aggregation-prone and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339. doi: 10.1074/jbc.M109.010264
Kabashi, E., Agar, J. N., Strong, M. J., and Durham, H. D. (2012). Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 367–371. doi: 10.3109/17482968.2012.686511
Kasu, Y. A. T., Alemu, S., Lamari, A., Loew, N., and Brower, C. S. (2018). The N termini of TAR DNA-binding protein 43 (TDP43) C-terminal fragments influence degradation, aggregation propensity and morphology. Mol. Cell. Biol. 38:e00243-18. doi: 10.1128/MCB.00243-18
Kato, M., Han, T. W., Xie, S., Shi, K., Du, X., Wu, L. C., et al. (2012). Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767. doi: 10.1016/j.cell.2012.04.017
Kato, S., Takikawa, M., Nakashima, K., Hirano, A., Cleveland, D. W., Kusaka, H., et al. (2000). New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons and astrocytes. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 163–184. doi: 10.1080/14660820050515160
Kenna, K. P., van Doormaal, P. T., Dekker, A. M., Ticozzi, N., Kenna, B. J., Diekstra, F. P., et al. (2016). NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48, 1037–1042. doi: 10.1038/ng.3626
Kerman, A., Liu, H. N., Croul, S., Bilbao, J., Rogaeva, E., Zinman, L., et al. (2010). Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 119, 335–344. doi: 10.1007/s00401-010-0646-5
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Kim, S., Kwon, S.-H., Kam, T.-I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103, 627.e7–641.e7. doi: 10.1016/j.neuron.2019.05.035
King, O. D., Gitler, A. D., and Shorter, J. (2012). The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80. doi: 10.1016/j.brainres.2012.01.016
Knowles, T. P., Vendruscolo, M., and Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. doi: 10.1038/nrm3810
Kryndushkin, D. S., Alexandrov, I. M., Ter-Avanesyan, M. D., and Kushnirov, V. V. (2003). Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278, 49636–49643. doi: 10.1074/jbc.M307996200
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Kwon, I., Kato, M., Xiang, S., Wu, L., Theodoropoulos, P., Mirzaei, H., et al. (2013). Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155, 1049–1060. doi: 10.1016/j.cell.2013.10.033
Laferrière, F., Maniecka, Z., Pérez-Berlanga, M., Hruska-Plochan, M., Gilhespy, L., Hock, E. M., et al. (2019). TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 22, 65–77. doi: 10.1038/s41593-018-0294-y
Lagier-Tourenne, C., Baughn, M., Rigo, F., Sun, S., Liu, P., Li, H. R., et al. (2013). Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U S A 110, E4530–E4539. doi: 10.1073/pnas.1318835110
Lang, L., Kurnik, M., Danielsson, J., and Oliveberg, M. (2012). Fibrillation precursor of superoxide dismutase 1 revealed by gradual tuning of the protein-folding equilibrium. Proc. Natl. Acad. Sci. U S A 109, 17868–17873. doi: 10.1073/pnas.1201795109
Lang, L., Zetterström, P., Brännström, T., Marklund, S. L., Danielsson, J., and Oliveberg, M. (2015). SOD1 aggregation in ALS mice shows simplistic test tube behavior. Proc. Natl. Acad. Sci. U S A 112, 9878–9883. doi: 10.1073/pnas.1503328112
Lee, K. H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics and function of membrane-less organelles. Cell 167, 774.e17–788.e17. doi: 10.1016/j.cell.2016.10.002
Legname, G., Nguyen, H. O., Peretz, D., Cohen, F. E., DeArmond, S. J., and Prusiner, S. B. (2006). Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl. Acad. Sci. U S A 103, 19105–19110. doi: 10.1073/pnas.0608970103
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Li, H. R., Chiang, W. C., Chou, P. C., Wang, W. J., and Huang, J. R. (2018). TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 293, 6090–6098. doi: 10.1074/jbc.AC117.001037
Lin, Y., Mori, E., Kato, M., Xiang, S., Wu, L., Kwon, I., et al. (2016). Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167, 789.e12–802.e12. doi: 10.1016/j.cell.2016.10.003
Lin, Y., Protter, D. S., Rosen, M. K., and Parker, R. (2015). Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219. doi: 10.1016/j.molcel.2015.08.018
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, H. N., Sanelli, T., Horne, P., Pioro, E. P., Strong, M. J., Rogaeva, E., et al. (2009). Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 66, 75–80. doi: 10.1002/ana.21704
Logroscino, G., Traynor, B. J., Hardiman, O., Chiò, A., Mitchell, D., Swingler, R. J., et al. (2010). Incidence of amyotrophic lateral sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 81, 385–390. doi: 10.1136/jnnp.2009.183525
Lowe, J. (1994). New pathological findings in amyotrophic lateral sclerosis. J. Neurol. Sci. 124, 38–51. doi: 10.1016/0022-510x(94)90175-9
Lowe, J., Lennox, G., Jefferson, D., Morrell, K., McQuire, D., Gray, T., et al. (1988). A filamentous inclusion body within anterior horn neurones in motor neurone disease defined by immunocytochemical localisation of ubiquitin. Neurosci. Lett. 94, 203–210. doi: 10.1016/0304-3940(88)90296-0
Luchinat, E., Barbieri, L., and Banci, L. (2017). A molecular chaperone activity of CCS restores the maturation of SOD1 fALS mutants. Sci. Rep. 7:17433. doi: 10.1038/s41598-017-17815-y
Luo, F., Gui, X., Zhou, H., Gu, J., Li, Y., Liu, X., et al. (2018). Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 25, 341–346. doi: 10.1038/s41594-018-0050-8
Münch, C., and Bertolotti, A. (2010). Exposure of hydrophobic surfaces initiates aggregation of diverse ALS-causing superoxide dismutase-1 mutants. J. Mol. Biol. 399, 512–525. doi: 10.1016/j.jmb.2010.04.019
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808.e9–816.e9. doi: 10.1016/j.neuron.2017.07.025
Magrané, J., Sahawneh, M. A., Przedborski, S., Estévez, Á. G., and Manfredi, G. (2012). Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J. Neurosci. 32, 229–242. doi: 10.1523/JNEUROSCI.1233-11.2012
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. doi: 10.1016/S1474-4422(12)70043-1
Martinez, F. J., Pratt, G. A., Van Nostrand, E. L., Batra, R., Huelga, S. C., Kapeli, K., et al. (2016). Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron 92, 780–795. doi: 10.1016/j.neuron.2016.09.050
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
McAlary, L., Aquilina, J. A., and Yerbury, J. J. (2016). Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not in vitro aggregation propensity. Front. Neurosci. 10:499. doi: 10.3389/fnins.2016.00499
McKinley, M. P., Meyer, R. K., Kenaga, L., Rahbar, F., Cotter, R., Serban, A., et al. (1991). Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J. Virol. 65, 1340–1351.
Mercado, P. A., Ayala, Y. M., Romano, M., Buratti, E., and Baralle, F. E. (2005). Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 33, 6000–6010. doi: 10.1093/nar/gki897
Mizielinska, S., Lashley, T., Norona, F. E., Clayton, E. L., Ridler, C. E., Fratta, P., et al. (2013). C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126, 845–857. doi: 10.1007/s00401-013-1200-z
Molliex, A., Temirov, J., Lee, J., Coughlin, M., Kanagaraj, A. P., Kim, H. J., et al. (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. doi: 10.1016/j.cell.2015.09.015
Mompeán, M., Buratti, E., Guarnaccia, C., Brito, R. M., Chakrabartty, A., Baralle, F. E., et al. (2014). Structural characterization of the minimal segment of TDP-43 competent for aggregation. Arch. Biochem. Biophys. 545, 53–62. doi: 10.1016/j.abb.2014.01.007
Mompeán, M., Romano, V., Pantoja-Uceda, D., Stuani, C., Baralle, F. E., Buratti, E., et al. (2016). The TDP-43 N-terminal domain structure at high resolution. FEBS J. 283, 1242–1260. doi: 10.1111/febs.13651
Mompeán, M., Romano, V., Pantoja-Uceda, D., Stuani, C., Baralle, F. E., Buratti, E., et al. (2017). Point mutations in the N-terminal domain of transactive response DNA-binding protein 43 kDa (TDP-43) compromise its stability, dimerization and functions. J. Biol. Chem. 292, 11992–12006. doi: 10.1074/jbc.m117.775965
Morales, R. (2017). Prion strains in mammals: different conformations leading to disease. PLoS Pathog. 13:e1006323. doi: 10.1371/journal.ppat.1006323
Mori, F., Tanji, K., Zhang, H. X., Nishihira, Y., Tan, C. F., Takahashi, H., et al. (2008). Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 116, 193–203. doi: 10.1007/s00401-008-0396-9
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morohoshi, F., Ootsuka, Y., Arai, K., Ichikawa, H., Mitani, S., Munakata, N., et al. (1998). Genomic structure of the human RBP56/hTAFII68 and FUS/TLS genes. Gene 221, 191–198. doi: 10.1016/s0378-1119(98)00463-6
Münch, C., O’Brien, J., and Bertolotti, A. (2011). Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. U S A 108, 3548–3553. doi: 10.1073/pnas.1017275108
Munoz, D. G., Neumann, M., Kusaka, H., Yokota, O., Ishihara, K., Terada, S., et al. (2009). FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 118, 617–627. doi: 10.1007/s00401-009-0598-9
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030
Murray, D. T., Kato, M., Lin, Y., Thurber, K. R., Hung, I., McKnight, S. L., et al. (2017). Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 171, 615.e16–627.e16. doi: 10.1016/j.cell.2017.08.048
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nomura, T., Watanabe, S., Kaneko, K., Yamanaka, K., Nukina, N., and Furukawa, Y. (2014). Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 289, 1192–1202. doi: 10.1074/jbc.M113.516492
Nonaka, T., Kametani, F., Arai, T., Akiyama, H., and Hasegawa, M. (2009). Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 18, 3353–3364. doi: 10.1093/hmg/ddp275
Nonaka, T., Masuda-Suzukake, M., Arai, T., Hasegawa, Y., Akatsu, H., Obi, T., et al. (2013). Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134. doi: 10.1016/j.celrep.2013.06.007
Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., et al. (1993). Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U S A 90, 10962–10966. doi: 10.1073/pnas.90.23.10962
Paré, B., Lehmann, M., Beaudin, M., Nordström, U., Saikali, S., Julien, J.-P., et al. (2018). Misfolded SOD1 pathology in sporadic amyotrophic lateral sclerosis. Sci. Rep. 8:14223. doi: 10.1038/s41598-018-31773-z
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Peng, X., Cashman, N. R., and Plotkin, S. S. (2018). Prediction of misfolding-specific epitopes in SOD1 using collective coordinates. J. Phys. Chem. B 122, 11662–11676. doi: 10.1021/acs.jpcb.8b07680
Pokrishevsky, E., Grad, L. I., Yousefi, M., Wang, J., Mackenzie, I. R., and Cashman, N. R. (2012). Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 7:e35050. doi: 10.1371/journal.pone.0035050
Pokrishevsky, E., Hong, R. H., Mackenzie, I. R., and Cashman, N. R. (2017). Spinal cord homogenates from SOD1 familial amyotrophic lateral sclerosis induce SOD1 aggregation in living cells. PLoS One 12:e0184384. doi: 10.1371/journal.pone.0184384
Pokrishevsky, E., McAlary, L., Farrawell, N. E., Zhao, B., Sher, M., Yerbury, J. J., et al. (2018). Tryptophan 32-mediated SOD1 aggregation is attenuated by pyrimidine-like compounds in living cells. Sci. Rep. 8:15590. doi: 10.1038/s41598-018-32835-y
Porta, S., Xu, Y., Restrepo, C. R., Kwong, L. K., Zhang, B., Brown, H. J., et al. (2018). Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 9:4220. doi: 10.1038/s41467-018-06548-9
Prince, M., Bryce, R., Albanese, E., Wimo, A., Ribeiro, W., and Ferri, C. P. (2013). The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 9, 63.e2–75.e2. doi: 10.1016/j.jalz.2012.11.007
Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. doi: 10.1126/science.6801762
Prusiner, S. B. (2001). Neurodegenerative diseases and prions. N. Engl. J. Med. 344, 1516–1526. doi: 10.1056/NEJM200105173442006
Prusiner, S. B. (2013). Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 47, 601–623. doi: 10.1146/annurev-genet-110711-155524
Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456. doi: 10.1038/ng1123
Ratovitski, T., Corson, L. B., Strain, J., Wong, P., Cleveland, D. W., Culotta, V. C., et al. (1999). Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum. Mol. Genet. 8, 1451–1460. doi: 10.1093/hmg/8.8.1451
Ravits, J. M., and La Spada, A. R. (2009). ALS motor phenotype heterogeneity, focality and spread: deconstructing motor neuron degeneration. Neurology 73, 805–811. doi: 10.1212/WNL.0b013e3181b6bbbd
Ravits, J., Paul, P., and Jorg, C. (2007). Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68, 1571–1575. doi: 10.1212/01.wnl.0000260965.20021.47
Reaume, A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. F., et al. (1996). Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13, 43–47. doi: 10.1038/ng0596-43
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ringholz, G. M., Appel, S. H., Bradshaw, M., Cooke, N. A., Mosnik, D. M., and Schulz, P. E. (2005). Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590. doi: 10.1212/01.wnl.0000172911.39167.b6
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Safar, J., Wille, H., Itri, V., Groth, D., Serban, H., Torchia, M., et al. (1998). Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 4, 1157–1165. doi: 10.1038/2654
Saini, A., and Chauhan, V. S. (2011). Delineation of the core aggregation sequences of TDP-43 C-terminal fragment. Chembiochem 12, 2495–2501. doi: 10.1002/cbic.201100427
Saini, A., and Chauhan, V. S. (2014). Self-assembling properties of peptides derived from TDP-43 C-terminal fragment. Langmuir 30, 3845–3856. doi: 10.1021/la404710w
Sala, F. A., Wright, G. S. A., Antonyuk, S. V., Garratt, R. C., and Hasnain, S. S. (2019). Molecular recognition and maturation of SOD1 by its evolutionarily destabilised cognate chaperone hCCS. PLOS Biol. 17:e3000141. doi: 10.1371/journal.pbio.3000141
Sangwan, S., Sawaya, M. R., Murray, K. A., Hughes, M. P., and Eisenberg, D. S. (2018). Atomic structures of corkscrew-forming segments of SOD1 reveal varied oligomer conformations. Protein Sci. 27, 1231–1242. doi: 10.1002/pro.3391
Sangwan, S., Zhao, A., Adams, K. L., Jayson, C. K., Sawaya, M. R., Guenther, E. L., et al. (2017). Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. U S A 114, 8770–8775. doi: 10.1073/pnas.1705091114
Sasaki, S. (2011). Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 70, 349–359. doi: 10.1097/nen.0b013e3182160690
Sawaya, M. R., Sambashivan, S., Nelson, R., Ivanova, M. I., Sievers, S. A., Apostol, M. I., et al. (2007). Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457. doi: 10.1038/nature05695
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Shimonaka, S., Nonaka, T., Suzuki, G., Hisanaga, S., and Hasegawa, M. (2016). Templated Aggregation of TAR DNA-binding Protein of 43 kDa (TDP-43) by Seeding with TDP-43 Peptide Fibrils. J. Biol. Chem. 291, 8896–8907. doi: 10.1074/jbc.m115.713552
Shin, Y., Berry, J., Pannucci, N., Haataja, M. P., Toettcher, J. E., and Brangwynne, C. P. (2017). Spatiotemporal control of intracellular phase transitions using light-activated optoDroplets. Cell 168, 159.e14–171.e14. doi: 10.1016/j.cell.2016.11.054
Shin, Y., Chang, Y.-C., Lee, D. S. W., Berry, J., Sanders, D. W., Ronceray, P., et al. (2018). Liquid nuclear condensates mechanically sense and restructure the genome. Cell 175, 1481.e13–1491.e13. doi: 10.1016/j.cell.2018.10.057
Silverman, J. M., Christy, D., Shyu, C. C., Moon, K. M., Fernando, S., Gidden, Z., et al. (2019). CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)(G93A) ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 294, 3744–3759. doi: 10.1074/jbc.ra118.004825
Silverman, J. M., Gibbs, E., Peng, X., Martens, K. M., Balducci, C., Wang, J., et al. (2018). A rational structured epitope defines a distinct subclass of toxic amyloid-β oligomers. ACS Chem. Neurosci. 9, 1591–1606. doi: 10.1021/acschemneuro.7b00469
Smethurst, P., Newcombe, J., Troakes, C., Simone, R., Chen, Y. R., Patani, R., et al. (2016). In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 96, 236–247. doi: 10.1016/j.nbd.2016.08.007
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Smith, B. N., Vance, C., Scotter, E. L., Troakes, C., Wong, C. H., Topp, S., et al. (2015). Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol. Aging 36, 1602.e17–1602.e27. doi: 10.1016/j.neurobiolaging.2014.10.032
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stathopulos, P. B., Rumfeldt, J. A. O., Scholz, G. A., Irani, R. A., Frey, H. E., Hallewell, R. A., et al. (2003). Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc. Natl. Acad. Sci. U S A 100, 7021–7026. doi: 10.1073/pnas.1237797100
Suh, E., Lee, E. B., Neal, D., Wood, E. M., Toledo, J. B., Rennert, L., et al. (2015). Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta Neuropathol. 130, 363–372. doi: 10.1007/s00401-015-1445-9
Sun, Y., and Chakrabartty, A. (2017). Phase to phase with TDP-43. Biochemistry 56, 809–823. doi: 10.1021/acs.biochem.6b01088
Sun, C. S., Wang, C. Y., Chen, B. P., He, R. Y., Liu, G. C., Wang, C. H., et al. (2014). The influence of pathological mutations and proline substitutions in TDP-43 glycine-rich peptides on its amyloid properties and cellular toxicity. PLoS One 9:e103644. doi: 10.1371/journal.pone.0103644
Tanaka, M., Collins, S. R., Toyama, B. H., and Weissman, J. S. (2006). The physical basis of how prion conformations determine strain phenotypes. Nature 442, 585–589. doi: 10.1038/nature04922
Tanaka, Y., and Hasegawa, M. (2016). Profilin 1 mutants form aggregates that induce accumulation of prion-like TDP-43. Prion 10, 283–289. doi: 10.1080/19336896.2016.1207033
Tartaglia, M. C., Rowe, A., Findlater, K., Orange, J. B., Grace, G., and Strong, M. J. (2007). Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: examination of symptoms and signs at disease onset and during follow-up. Arch. Neurol. 64, 232–236. doi: 10.1001/archneur.64.2.232
Tavella, D., Zitzewitz, J. A., and Massi, F. (2018). Characterization of TDP-43 RRM2 partially folded states and their significance to ALS pathogenesis. Biophys. J. 115, 1673–1680. doi: 10.1016/j.bpj.2018.09.011
Taylor, J. P., Brown, R. H. Jr., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Taylor, D. M., Gibbs, B. F., Kabashi, E., Minotti, S., Durham, H. D., and Agar, J. N. (2007). Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 282, 16329–16335. doi: 10.1074/jbc.M610119200
Telling, G. C., Parchi, P., DeArmond, S. J., Cortelli, P., Montagna, P., Gabizon, R., et al. (1996). Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274, 2079–2082. doi: 10.1126/science.274.5295.2079
Thompson, M. J., Sievers, S. A., Karanicolas, J., Ivanova, M. I., Baker, D., and Eisenberg, D. (2006). The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. U S A 103, 4074–4078. doi: 10.1073/pnas.0511295103
Ticozzi, N., Vance, C., Leclerc, A. L., Keagle, P., Glass, J. D., McKenna-Yasek, D., et al. (2011). Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 3, 285–290. doi: 10.1002/ajmg.b.31158
Toombs, J. A., McCarty, B. R., and Ross, E. D. (2010). Compositional determinants of prion formation in yeast. Mol. Cell. Biol. 30, 319–332. doi: 10.1128/MCB.01140-09
Trist, B. G., Davies, K. M., Cottam, V., Genoud, S., Ortega, R., Roudeau, S., et al. (2017). Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 134, 113–127. doi: 10.1007/s00401-017-1726-6
Tsoi, P. S., Choi, K.-J., Leonard, P. G., Sizovs, A., Moosa, M. M., MacKenzie, K. R., et al. (2017). The N-terminal domain of ALS-linked TDP-43 assembles without misfolding. Angew. Chem. Int. Ed Engl. 56, 12590–12593. doi: 10.1002/anie.201706769
Tu, P. H., Galvin, J. E., Baba, M., Giasson, B., Tomita, T., Leight, S., et al. (1998). Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann. Neurol. 44, 415–422. doi: 10.1002/ana.410440324
van Blitterswijk, M., DeJesus-Hernandez, M., Niemantsverdriet, E., Murray, M. E., Heckman, M. G., Diehl, N. N., et al. (2013). Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 12, 978–988. doi: 10.1016/s1474-4422(13)70210-2
Van den Berg-Vos, R. M., Visser, J., Kalmijn, S., Fischer, K., de Visser, M., de Jong, V., et al. (2009). A long-term prospective study of the natural course of sporadic adult-onset lower motor neuron syndromes. Arch. Neurol. 66, 751–757. doi: 10.1001/archneurol.2009.91
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vogler, T. O., Wheeler, J. R., Nguyen, E. D., Hughes, M. P., Britson, K. A., Lester, E., et al. (2018). TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 563, 508–513. doi: 10.1038/s41586-018-0665-2
Wakabayashi, K., Yoshimoto, M., Tsuji, S., and Takahashi, H. (1998). α-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 249, 180–182. doi: 10.1016/s0304-3940(98)00407-8
Walhout, R., Verstraete, E., van den Heuvel, M. P., Veldink, J. H., and van den Berg, L. H. (2018). Patterns of symptom development in patients with motor neuron disease. Amyotroph. Lateral Scler. Frontotemporal Degener. 19, 21–28. doi: 10.1080/21678421.2017.1386688
Walker, A. K., Soo, K. Y., Sundaramoorthy, V., Parakh, S., Ma, Y., Farg, M. A., et al. (2013). ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One 8:e81170. doi: 10.1371/journal.pone.0081170
Wang, X., Arai, S., Song, X., Reichart, D., Du, K., Pascual, G., et al. (2008). Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130. doi: 10.1038/nature06992
Wang, A., Conicella, A. E., Schmidt, H. B., Martin, E. W., Rhoads, S. N., Reeb, A. N., et al. (2018). A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation and RNA splicing. EMBO J. 37:e97452. doi: 10.15252/embj.201797452
Wang, X., Fan, H., Ying, Z., Li, B., Wang, H., and Wang, G. (2010). Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci. Lett. 469, 112–116. doi: 10.1016/j.neulet.2009.11.055
Wang, J., Farr, G. W., Zeiss, C. J., Rodriguez-Gil, D. J., Wilson, J. H., Furtak, K., et al. (2009). Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc. Natl. Acad. Sci. U S A 106, 1392–1397. doi: 10.1073/pnas.0813045106
Wang, Y. T., Kuo, P. H., Chiang, C. H., Liang, J. R., Chen, Y. R., Wang, S., et al. (2013). The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem. 288, 9049–9057. doi: 10.1074/jbc.m112.438564
Wang, W. Y., Pan, L., Su, S. C., Quinn, E. J., Sasaki, M., Jimenez, J. C., et al. (2013). Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 16, 1383–1391. doi: 10.1038/nn.3514
Webster, C. P., Smith, E. F., Bauer, C. S., Moller, A., Hautbergue, G. M., Ferraiuolo, L., et al. (2016). The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 35, 1656–1676. doi: 10.15252/embj.201694401
Westergard, T., Jensen, B. K., Wen, X., Cai, J., Kropf, E., Iacovitti, L., et al. (2016). Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 17, 645–652. doi: 10.1016/j.celrep.2016.09.032
Williams, K. L., Topp, S., Yang, S., Smith, B., Fifita, J. A., Warraich, S. T., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 7:11253. doi: 10.1038/ncomms11253
Winton, M. J., Igaz, L. M., Wong, M. M., Kwong, L. K., Trojanowski, J. Q., and Lee, V. M. (2008). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration and aggregate formation. J. Biol. Chem. 283, 13302–13309. doi: 10.1074/jbc.m800342200
Wright, G. S. A., Antonyuk, S. V., and Hasnain, S. S. (2016). A faulty interaction between SOD1 and hCCS in neurodegenerative disease. Sci. Rep. 6:27691. doi: 10.1038/srep27691
Wu, X., Bradley, M. J., Cai, Y., Kummel, D., De La Cruz, E. M., Barr, F. A., et al. (2011). Insights regarding guanine nucleotide exchange from the structure of a DENN-domain protein complexed with its Rab GTPase substrate. Proc. Natl. Acad. Sci. U S A 108, 18672–18677. doi: 10.1073/pnas.1110415108
Wu, C. H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Xu, Y. F., Gendron, T. F., Zhang, Y. J., Lin, W. L., D’Alton, S., Sheng, H., et al. (2010). Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 30, 10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010
Yamashita, M., Nonaka, T., Arai, T., Kametani, F., Buchman, V. L., Ninkina, N., et al. (2009). Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Lett. 583, 2419–2424. doi: 10.1016/j.febslet.2009.06.042
Yang, C., Tan, W., Whittle, C., Qiu, L., Cao, L., Akbarian, S., et al. (2010). The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One 5:e15878. doi: 10.1371/journal.pone.0015878
Yerbury, J. J., Ooi, L., Blair, I. P., Ciryam, P., Dobson, C. M., and Vendruscolo, M. (2019). The metastability of the proteome of spinal motor neurons underlies their selective vulnerability in ALS. Neurosci. Lett. 704, 89–94. doi: 10.1016/j.neulet.2019.04.001
Zeineddine, R., Pundavela, J. F., Corcoran, L., Stewart, E. M., Do-Ha, D., Bax, M., et al. (2015). SOD1 protein aggregates stimulate macropinocytosis in neurons to facilitate their propagation. Mol. Neurodegener. 10:57. doi: 10.1186/s13024-015-0053-4
Zeineddine, R., Whiten, D. R., Farrawell, N. E., McAlary, L., Hanspal, M. A., Kumita, J. R., et al. (2017). Flow cytometric measurement of the cellular propagation of TDP-43 aggregation. Prion 11, 195–204. doi: 10.1080/19336896.2017.1314426
Zhang, Y. J., Caulfield, T., Xu, Y. F., Gendron, T. F., Hubbard, J., Stetler, C., et al. (2013). The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 22, 3112–3122. doi: 10.1093/hmg/ddt166
Zhang, P., Fan, B., Yang, P., Temirov, J., Messing, J., Kim, H. J., et al. (2019). Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. Elife 8:e39578. doi: 10.7554/elife.39578
Zhang, T., Mullane, P. C., Periz, G., and Wang, J. (2011). TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum. Mol. Genet. 20, 1952–1965. doi: 10.1093/hmg/ddr076
Zhang, Y. J., Xu, Y. F., Cook, C., Gendron, T. F., Roettges, P., Link, C. D., et al. (2009). Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U S A 106, 7607–7612. doi: 10.1073/pnas.0900688106
Zhenfei, L., Shiru, D., Xiaomeng, Z., Cuifang, C., and Yaling, L. (2019). Discontiguous or contiguous spread patterns affect the functional staging in patients with sporadic amyotrophic lateral sclerosis. Front. Neurol. 10:523. doi: 10.3389/fneur.2019.00523
Keywords: amyotrophic lateral sclerosis, protein misfolding, protein aggregation, prion, proteostasis
Citation: McAlary L, Plotkin SS, Yerbury JJ and Cashman NR (2019) Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 12:262. doi: 10.3389/fnmol.2019.00262
Received: 22 August 2019; Accepted: 14 October 2019;
Published: 01 November 2019.
Edited by:
Tiago F. Outeiro, University Medical Center Goettingen, GermanyReviewed by:
Hidefumi Ito, Wakayama Medical University, JapanGen Matsumoto, Nagasaki University, Japan
Copyright © 2019 McAlary, Plotkin, Yerbury and Cashman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neil R. Cashman, neil.cashman@vch.ca