
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 28 June 2019
Sec. Molecular Signalling and Pathways
Volume 12 - 2019 | https://doi.org/10.3389/fnmol.2019.00163
This article is part of the Research Topic Lithium and Neurodegeneration View all 5 articles
 Ashleigh Duthie1
Ashleigh Duthie1 Lidy van Aalten2Cara MacDonald1
Lidy van Aalten2Cara MacDonald1 Alison McNeilly2Jennifer Gallagher2John Geddes3
Alison McNeilly2Jennifer Gallagher2John Geddes3 Simon Lovestone3†
Simon Lovestone3† Calum Sutherland2*
Calum Sutherland2*Lithium has been used for decades to treat Bipolar Disorder. Some of its therapeutic benefits may be through inhibition of Glycogen Synthase Kinase (GSK)-3. Enhanced GSK3 activity associates with development of Alzheimer’s disease (AD), therefore lithium is a currently used therapeutic with potential to be repurposed for prevention of Dementia. An important step toward a clinical trial for AD prevention using lithium is to establish the dose of lithium that blocks GSK3 in Mild Cognitive Impairment (MCI), a high-risk condition for progression to AD. We investigated volunteer recruitment, retention, and tolerance in this population, and assessed biomarkers of GSK3 in MCI compared to control and after lithium treatment. Recruitment was close to target, with higher than anticipated interest. Drop out was not related to lithium blood concentration. Indeed, 33% of the withdrawals were in the first week of very low dose lithium. Most made it through to the highest dose of lithium with no adverse events. We analyzed 18 potential biomarkers of GSK3 biology in rat PBMCs, but only four of these gave a robust reproducible baseline signal. The only biomarker that was modified by acute lithium injection in the rat was the inhibitory phosphorylation of Ser9 of GSK3beta (enhanced in PBMCs) and this associated with reduced activity of GSK3beta. In contrast to the rat PBMC preparations the protein quality of the human PBMC preparations was extremely variable. There was no difference between GSK3 biomarkers in MCI and control PBMC preparations and no significant effect of chronic lithium on the robust GSK3 biomarkers, indicating that the dose reached may not be sufficient to modify these markers. In summary, the high interest from the MCI population, and the lack of any adverse events, suggest that it would be relatively straightforward and safe to recruit to a larger clinical trial within this dosing regimen. However, it is clear that we will need an improved PBMC isolation process along with more robust, sensitive, and validated biomarkers of GSK3 function, in order to use GSK3 pathway regulation in human PBMC preparations as a biomarker of GSK3 inhibitor efficacy, within a clinical trial setting.
The pharmacological treatment of Dementia is limited to the treatment of symptoms, primarily due to the ever increasing number of failed clinical trials (Hardy, 2009; Wang et al., 2016; Amanatkar et al., 2017), while the paucity of non-amyloid targeting drugs in development is highly concerning (Cummings et al., 2017). In contrast, there is increasing awareness of the importance of identifying high risk groups and developing Dementia prevention strategies.
Mild Cognitive impairment (MCI) can be thought of as a prodromal phase of Dementia with a significant proportion of MCI patients converting to Dementia. Interventions in MCI may provide the best opportunities to delay or prevent the development of irreversible pathology and debilitating symptoms (Petersen et al., 2001). In order to generate novel therapeutics as rapidly as possible one approach is to investigate the “repositioning” of therapeutics currently used for other neurological conditions, with acceptable safety and side effect profiles, that have mechanistic potential to target the molecular pathways in brain associated with early AD development (e.g., amyloid, tangle, inflammation, etc.) (Corbett et al., 2012).
Glycogen Synthase Kinase-3 (GSK3) influences many important cellular processes (Jope and Johnson, 2004; Kockeritz et al., 2006; Sutherland, 2011), and its dysregulation has been linked to the pathophysiology of numerous age-related diseases including Alzheimer’s disease (AD) and inflammation (Cohen and Goedert, 2004; O’Brien and Klein, 2007; Hooper et al., 2008; Sutherland, 2011; Gao et al., 2012; Beurel et al., 2013; Russo et al., 2013). Importantly, partial deletion (pharmacological or genetic) of GSK3 reduces the severity of disease in many preclinical models, including models of familial AD (Jope et al., 2007; Hooper et al., 2008; Sutherland, 2011; Avrahami et al., 2013; Ly et al., 2013). Furthermore, transgenic overexpression of GSK3, specifically in mouse brain, promotes AD-like symptoms and generates pathology (Lucas et al., 2001). Importantly, GSK3 activation is directly linked to the development of cognitive impairment independent of AD pathology (King et al., 2013). This implies that GSK3 inhibition could have clinical benefits from early stage development right through to late stage disease. However, the wide spectrum of important biological actions influenced by GSK3 activity has questioned the clinical safety of a global GSK3 inhibition approach, especially as genetic ablation of GSK3 has serious health implications including disruption of brain development and repair (Jiang et al., 2005; Kim et al., 2009). However, GSK3 in human dementia is related to abnormally high GSK3 activity in adults, and no GSK3 inhibitor toxicology studies have been reported in an adult preclinical model with excessive GSK3 activity.
The safety concerns around GSK3 inhibition are doubly surprising since a GSK3 inhibitor is in everyday clinical use without the side-effects observed in some animal models of GSK3 ablation. Lithium is a well characterized (if non-selective) inhibitor of GSK3 (Klein and Melton, 1996; O’Brien and Klein, 2007, 2009; Can et al., 2014) with a well-known and acceptable side effect profile. Indeed, it has been used effectively for decades in the treatment of mood disorder, and although the precise mechanisms of action remain the subject of some debate there is evidence for at least some of its neuroprotective actions being mediated by inhibition of GSK3 (O’Brien and Klein, 2007, 2009; Malhi et al., 2013; Can et al., 2014). Importantly, patients prescribed long term lithium for Bipolar disorder have lower incidence of all forms of dementia compared to short term use or alternative therapies (Sutherland and Duthie, 2015). This is tantalizing evidence that lithium, or other GSK3 inhibitors, have the potential to prevent conversion to dementia.
Lithium is generally well tolerated as a long-term therapy for Bipolar disease in humans. However, toxicity can lead to ataxia and confusion, and in severe cases it can be life threatening. For this reason, lithium serum levels are routinely monitored in clinical practice and kept within a narrow therapeutic range of 0.4–0.75 mM/l (McKnight et al., 2012). Clinical studies giving lithium to elderly people with AD (at doses relevant to treatment of Bipolar disease) had relatively few side effects (Macdonald et al., 2008; Hampel et al., 2009). A study with lithium in people with MCI reported excellent volunteer retention and tolerability up to 1 year of lithium administration (Forlenza et al., 2011a). This treatment indicated a trend toward cognitive benefit and 91% tolerability but was too short to include effects on conversion to Dementia (Forlenza et al., 2011a). It seems reasonable therefore to suggest that lithium is a clinically viable GSK3 inhibitor that is available for immediate use in trials to evaluate GSK3 inhibition as an anti-dementia therapeutic agent.
The therapeutic range of lithium contains the IC50 (50% inhibition constant) for direct GSK3 inhibition in vitro [approx. 0.5 mM (Davies et al., 2000)], suggesting that pharmacologically relevant GSK3 inhibition would be achieved by the doses used to treat Bipolar disease. However, lithium regulates GSK3 activity by several distinct mechanisms (Sutherland and Duthie, 2015), which may mean that inhibition of GSK3 in patients will occur with even lower levels of lithium than predicted from in vitro obtained IC50 values. Therefore, experimentally evaluating the lithium concentration required to inhibit GSK3 in relevant human populations becomes a valuable exercise prior to balancing safety concerns with therapeutic benefits. However, measuring changes in GSK3 activity in humans is challenging, and almost impossible in intact brain.
The studies where lithium administration to rodent models of dementia has beneficial actions on brain imaging, behavior, or AD pathology [for example (O’Brien et al., 2004; Noble et al., 2005; Zhang et al., 2011; Sudduth et al., 2012; Yu et al., 2012; Chen et al., 2013; de Cristobal et al., 2014)] measure the N-terminal inhibitory phosphorylation status of GSK3 in a tissue lysate to assess GSK3 activity status. However, this modification does not inhibit GSK3 against all of its downstream targets (Frame et al., 2001), and thus is not sufficient to provide a comprehensive assessment of GSK3 activity. In reality, GSK3 inhibition may occur at lithium concentrations lower than those required to regulate this phosphorylation or to competitively inhibit GSK3 activity, and specific targets may be regulated by different mechanisms of GSK3 regulation (Robertson et al., 2018). There is evidence that therapeutic doses of lithium are sufficient to increase GSK3 N-terminal phosphorylation in human blood cells in Bipolar disorder (Li et al., 2007) or MCI (Forlenza et al., 2011b).
Alternative “potential biomarkers” of GSK3 activity status are detailed in Table 1. There are several phosphorylation sites on GSK3 that influence GSK3 activity, including Tyr 279/216 and Thr390 (GSK3β only) (Table 1), and total GSK3 isoform mRNA or protein can also vary in some disease states. One can also monitor the signaling pathways that regulate phosphorylation of these sites, such as the PI3K-Akt pathway (Cross et al., 1995). Activation of these pathways may be extrapolated to an assumption of inhibition of GSK3 (Table 1). Possibly more informative is quantitative assessment of phosphorylation of direct downstream targets of GSK3 (Table 1; Sutherland, 2011). However, this depends on antibody availability and protein detection in the tissues of interest.

Table 1. Potential molecular biomarkers of GSK3 activity, classified by the relationship to GSK3 function.
The current study aimed to assess and validate many of the potential biomarkers for GSK3 activity in Table 1, in rodent Peripheral Blood Mononuclear cells (PBMCs). We then took robust biomarkers from that study to generate pilot information on inhibition of GSK3 by lithium in people with MCI. Importantly we also aimed to extend the evidence that recruiting and retaining this population to a trial which involved taking lithium, a drug not normally prescribed to MCI, was achievable. We performed three studies, one where we gave rats an acute dose of lithium and measured GSK3 biomarkers in PBMCs, hippocampus and cortex, one (Arm 1) where we compared GSK3 biomarkers in PBMCs between volunteers with MCI and age-matched controls, and one (Arm 2) where we performed a step-wise increasing lithium dosing experiment in MCI volunteers over 12 weeks.
Vacutainers for blood collection were purchased from BD (Wokingham, United Kingdom) (367869), Optiprep media (D1556), lithium chloride (203637), and sodium chloride (S7653) were from Sigma (Gillingham, United Kingdom), Mono-Poly resolving media from MP Biomedicals (Santa Ana, United States) (0916980). Protease inhibitor tablets were from Roche (11836170001) and Bradford protein assay reagent was from Biorad (Watford, United Kingdom) (500-0006). The nitrocellulose (Amersham Protran) membrane and Phosphocellulose P81 paper were from GE Healthcare Life Sciences (Little Chalfort, United Kingdom) (10600002), the Enhanced Chemiluminescence (ECL) Kit used was from Amersham (RPN2209) and CL-Xposure film from Thermo Scientific (Waltham, United States). The radioisotope, γ-32P-ATP was purchased from Perkin Elmer (Waltham, United States). All other chemicals were of the highest grade available. All antibodies used in this study are detailed in Table 2.
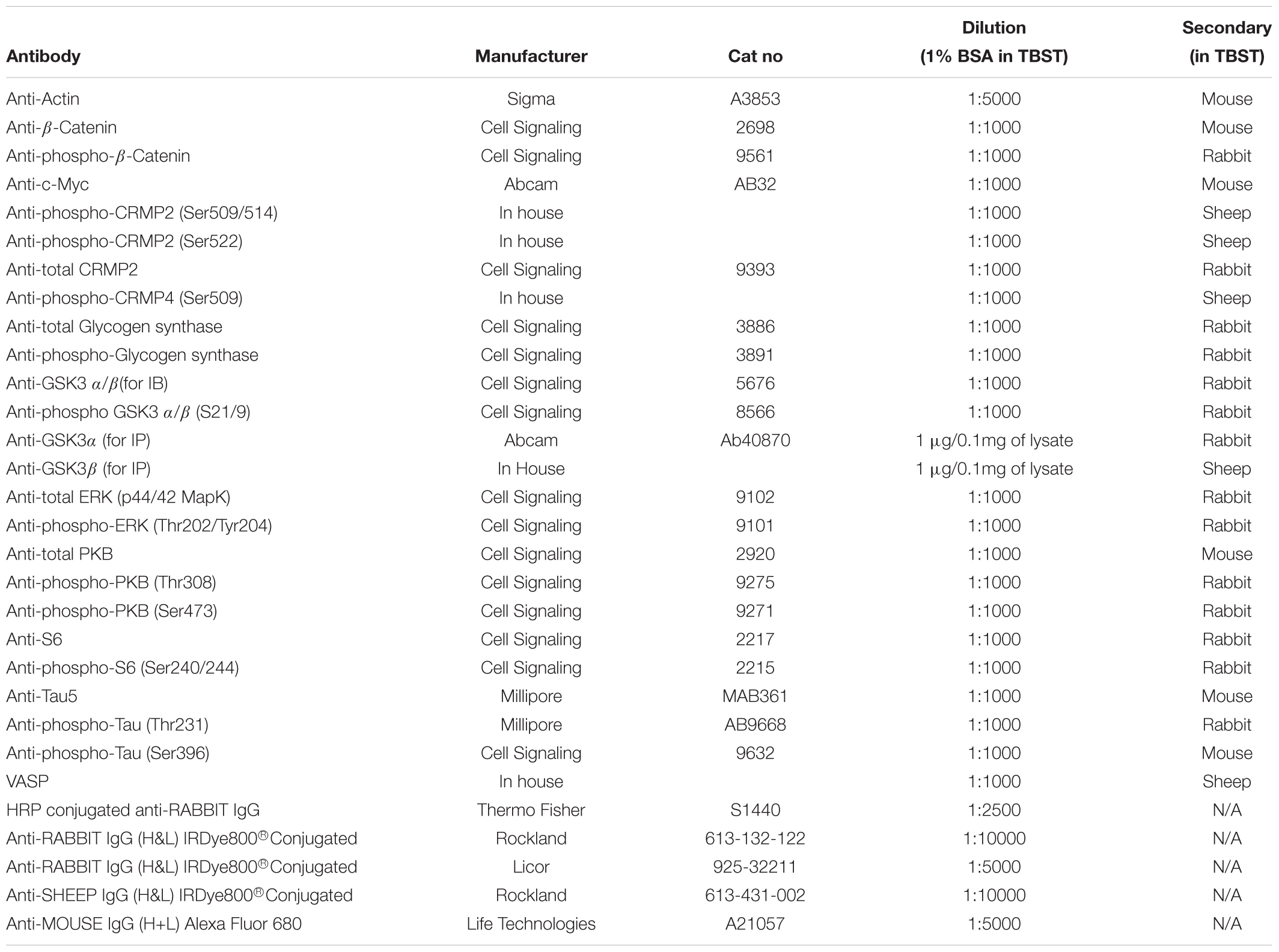
Table 2. Antibodies used in this work.
All experiments were performed using male Wistar rats (Harlan Ltd., United Kingdom), initial body weight 150–175 g. Animals were housed in cages of four (12:12 h light: dark cycle) at an ambient temperature of 22 ± 1°C and 50% humidity, with ad libitum access to food and water. All animal procedures were approved by the University of Dundee Ethical Review Process and were performed in accordance with UK Home Office regulations (under the auspices of Project License PIL60/4280). Animals were assigned at random to one of three treatment groups (i) Control (Saline), (ii) Low dose lithium chloride (2 nmoles/kg i.p.), or (iii) High dose lithium chloride (4 nmoles/kg i.p.). 3 h post injection animals were sacrificed by cervical dislocation and blood samples collected by cardiac puncture for the preparation of PBMCs and plasma lithium analysis. Brain regions (hippocampus, hypothalamus, cortex, and cerebellum) and liver samples were isolated and snap frozen in liquid nitrogen for subsequent biochemical analysis.
One milliliter of whole rat blood was collected from each animal on Sodium Heparin. Samples were spun and serum transferred to 1.5 ml Eppendorf tubes prior to running on an AVL 9180 Electrolyte Analyser, alongside an appropriate lithium standard curve.
Peripheral Blood Mononuclear cells were collected from the rat blood samples immediately after collection using OptiPrep density gradient medium using the flotation technique as per manufacturer’s instructions. Protein was harvested from the cell pellet in lysis buffer [25 mM Tris–HCl (pH 7.4), 50 mM NaF, 0.1 M NaCl, 1 mM EDTA, 5 mM EGTA, 1% Triton-X, 10 mM Na-pyrophsphate, 0.1% beta-mercaptoethanol, 1 mM Na-vanadate, protease inhibitor cocktail tablet (Roche 11836170001) and 0.27M sucrose] and snap frozen in liquid N2 prior to storage at -80°C.
There were two arms to the human study, firstly a comparison of GSK3 biomarkers in PBMCs between volunteers with MCI and age-matched controls. This included a comparison of the biomarkers from the same individual in two samples taken 12 weeks apart. Secondly, an intervention arm, where volunteers with MCI were given lithium tablets for 9 weeks, with a step wise increase from 100 to 200 mg/d then 400 mg/d for 3 weeks each, followed by a 3 week wash out period (Figure 1). Blood was taken at baseline, 1 and 3 weeks after lithium dose change as detailed in Figure 1. The study was approved by the North of Scotland Research Ethics committee, number16/NS/006.
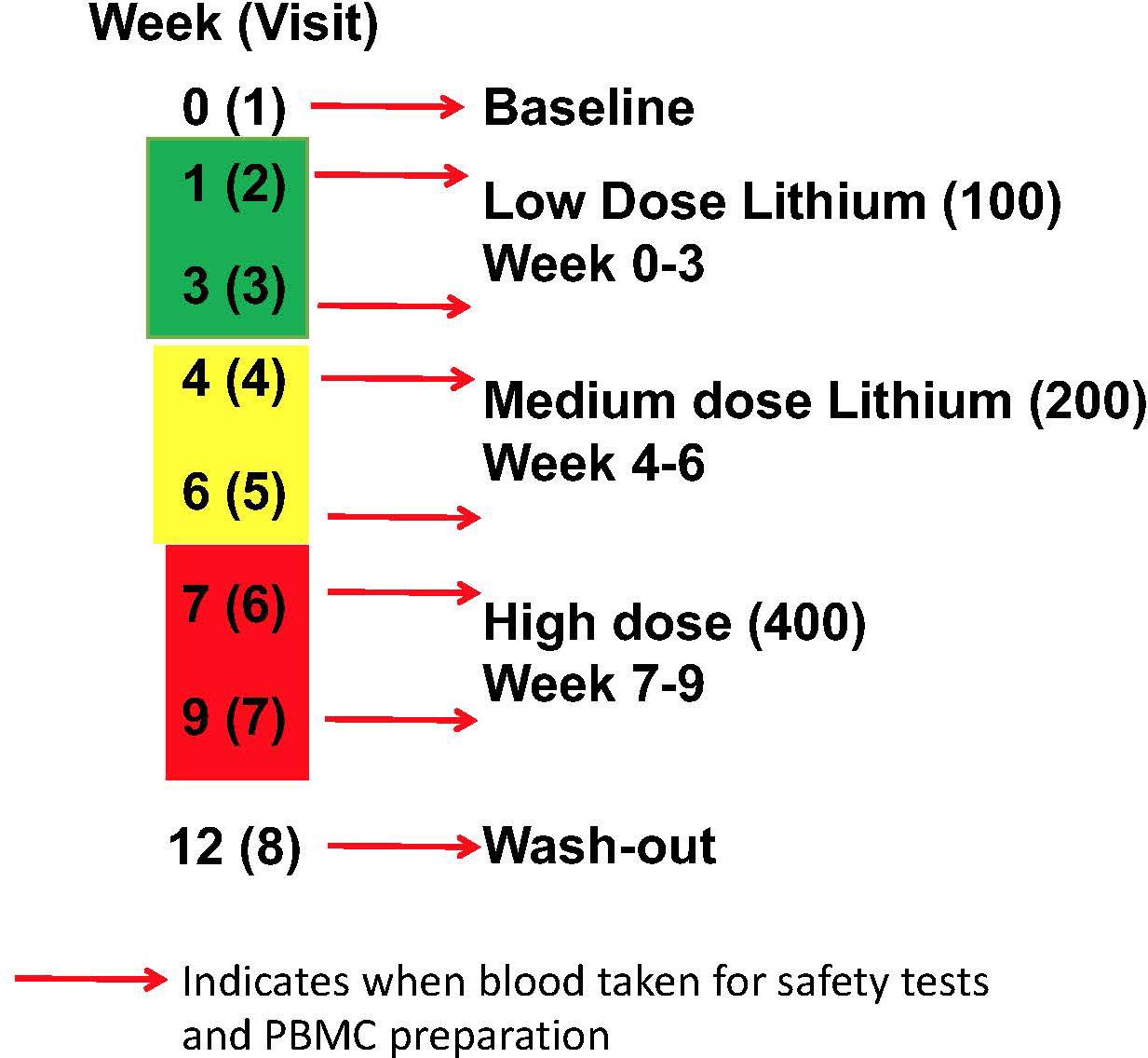
Figure 1. Lithium intervention Study Design (Arm 2). Eleven volunteers were given low dose (100 mg daily) for 3 weeks, then medium dose (200 mg daily) for 3 weeks, high dose (400 mg daily) for 3 weeks, prior to a 3 week washout period. Blood samples were taken for safety measures, PBMC preparation and Lithium measure at baseline (visit 1), 1 and 3 weeks after starting each dose (visit 2–7), and after washout (visit 8).
Participants over age 55 and with a diagnosis of MCI were recruited from Tayside Old Age Psychiatry and Geriatric Medicine outpatient clinics and by using Join Dementia Research (JDR) Database. Of 830 clinic contacts screened via consultant caseloads, 67 MCI patients were identified and invited by letter plus 4 contacts identified through existing local research lists and 3 via JDR. MCI was defined according to Petersen’s criteria (Petersen et al., 2001). Two brief standardized questionnaires [Bristol Activities of Daily Living Schedule and Geriatric Depression Scale Short Form (15 point version)] were administered to check diagnosis and existing cognitive assessments were documented from case file. The control group was recruited via two routes, firstly through Join Dementia Research and secondly by approaching spouses/partners/relatives of similar age to the MCI group but who lack an MCI diagnosis. Exclusions for the Study Arm 1 group were as follows; Dementia, Diabetes, current medication of Lithium or NSAIDS, BMI < 18 or > 30, incapacity to consent, Rheumatoid Arthritis and Autoimmune Inflammatory disorders and for Study Arm 2 the following additional exclusion criteria were applied: contraindication to Lithium Carbonate (Priadel) according to Summary of Product Characteristics, Moderate renal failure (defined by eGFR < 60 mL/min/1.73 m2) or severe Renal failure (defined by eGFR < 29 mL/min/1.73 m2), epilepsy, and suicidality. Screening included baseline blood tests for renal function and thyroid function.
Safety assessments were face to face and occurred at each scheduled visit at weeks 1, 3, 4, 6, 7, 9, and 12. Participants were asked about general health and the presence of any common side effects and a Lithium side effects rating scale (LISERS) completed where any side effects were reported. The blister pack was examined for compliance. Blood lithium concentration and renal function were assessed as standard clinical safety measures in line with guidelines for routine clinical practice. Lithium > 0.7 mmMol/L or significant deterioration in renal function (defined by eGFR falling to < 60 mL/min/1.73 m2) or acute kidney injury [diagnosed by acute rise in creatinine blood result according to NICE Guidelines for Acute Kidney Injury (Levin et al., 2008)] would result in withdrawal from lithium and entry to washout.
Volunteer blood (13–18 ml) was collected in Na-heparin containing vacuettes, kept at room temperature and processed within 6 h of collection. In sterile test tubes 3.5 ml of blood was carefully layered on top of 3 ml Mono-poly resolving media. These were centrifuged for 45 min, 800 ×g in a swinging bucket rotor at room temperature without brake (Beckman Coulter Allegra X-12 centrifuge). The plasma layer at the top of the tube was then removed and discarded using a Pasteur’s pipette and the top layer of cells containing the mononuclear leucocytes was collected and combined in a clean test tube. The cell suspension was washed with three volumes of PBS and centrifuged 10 min, at room temperature, 250 ×g with brake on. The cell pellet was washed in PBS and centrifuged again. An equal volume of PBS was added to the pellet, the cells were resuspended gently and the cell mixture transferred to a cryovial and snap frozen in liquid N2 prior to storage at -80°C.
The PBMC pellets were lysed on ice in lysis buffer. Following centrifugation to remove insoluble material, supernatants were collected and protein concentrations were determined using the Bradford method. Lysates were made up to equal concentrations with lithium dodecyl sulfate sample buffer added and subjected to SDS-polyacrylamide gel electrophoresis on 4–12% NuPAGE polyacrylamide gels (Novex, Life Technologies), then either stained using Coomassie Blue (SimplyBlueTMSafe Stain, Invitrogen, CA, United States), or transferred to nitrocellulose membrane. Membranes were blocked in 5% (w/v) BSA, incubated with primary antibodies overnight at 4°C, and then Alexa-fluor secondary antibodies for 1 h at room temperature. Proteins were visualized using the Licor Odyssey. When probing for phospho Akt(Ser473), HRP secondary antibody was used, detection achieved with ECL and proteins visualized on film using different exposure times, typically between 10 s and 10 min.
Protein bands were quantified using the Image Studio software (Licor). Odyssey images were quantified directly while ECL images on film were scanned using a desktop scanner and imported into the software. The background was determined around each box individually, either left and right, top and bottom, or both with varying width, dependent upon the layout of each blot, but kept consistent per blot. Normalization was done by generating a ratio against a loading control (usually actin), or for Phosphorylation sites a ratio against the total protein signal of the protein of study where antibodies were available. The differences between groups were determined by using a Student t test. Analyses were performed using Excel (Microsoft Corp.).
A total of 200 μl of (50:50) Protein A agarose beads in PBS were conjugated to 20 μg of either anti-GSK3β or anti-GSK3α by incubating for 1 h at room temperature with shaking. Unbound antibody was removed by washing with lysis buffer and 5 μl of the (50:50) anti-GSK3β-beads incubated with 100 μl of 1 μg/μl cell lysate overnight at 4°C with shaking. GSK3β immunocomplexes were isolated by centrifugation at 3000 ×g for 5 min. Supernatant was incubated with 5 μl of (50:50) anti-GSK3α-beads for 4 h at 4°C prior to isolation of the GSK3α immunocomplexes by centrifugation at 3000 ×g for 5 min. All immunocomplexes were washed with 1 ml of lysis buffer, followed by 1 ml of kinase assay buffer (25 mM TRIS, 0.1 mM EDTA, 5% glycerol) containing 0.5 M NaCl and then 1 ml of kinase assay buffer before resuspending to 10 μl in assay buffer.
Each washed immunocomplex was incubated with PGS2 peptide (10 μM final), MgCl2 (10 mM final), γ - 32P-ATP (0.1 mM final, 0.5 × 106 CPM/nmole) in assay buffer in a final volume of 50 μl at 30°C for 20 min. The assay was stopped by spotting the assay mix onto P81 phosphocellulose paper and placing in 10 mM Phosphoric Acid (PA). Papers were washed 4× 5 min in PA then once in acetone and air dried. Phosphate incorporation into the GSK3 substrate was quantified by counting the paper in a scintillation counter. One unit of activity of each protein kinase was calculated as the amount required to transfer 1 nmole of phosphate/min. The differences between groups were determined by using a Student t test. Analyses were performed using Excel (Microsoft Corp.).
The recruitment strategy is provided in Figure 2. Study Arm 1 contained six MCI and eleven controls (all providing two blood samples, 12 weeks apart), and Study Arm 2 recruited eleven MCI volunteers for lithium treatment (eight blood samples across 12 weeks). The number of participants excluded due to BMI above 30 was greater than anticipated from this age group and was the most common reason for exclusion.
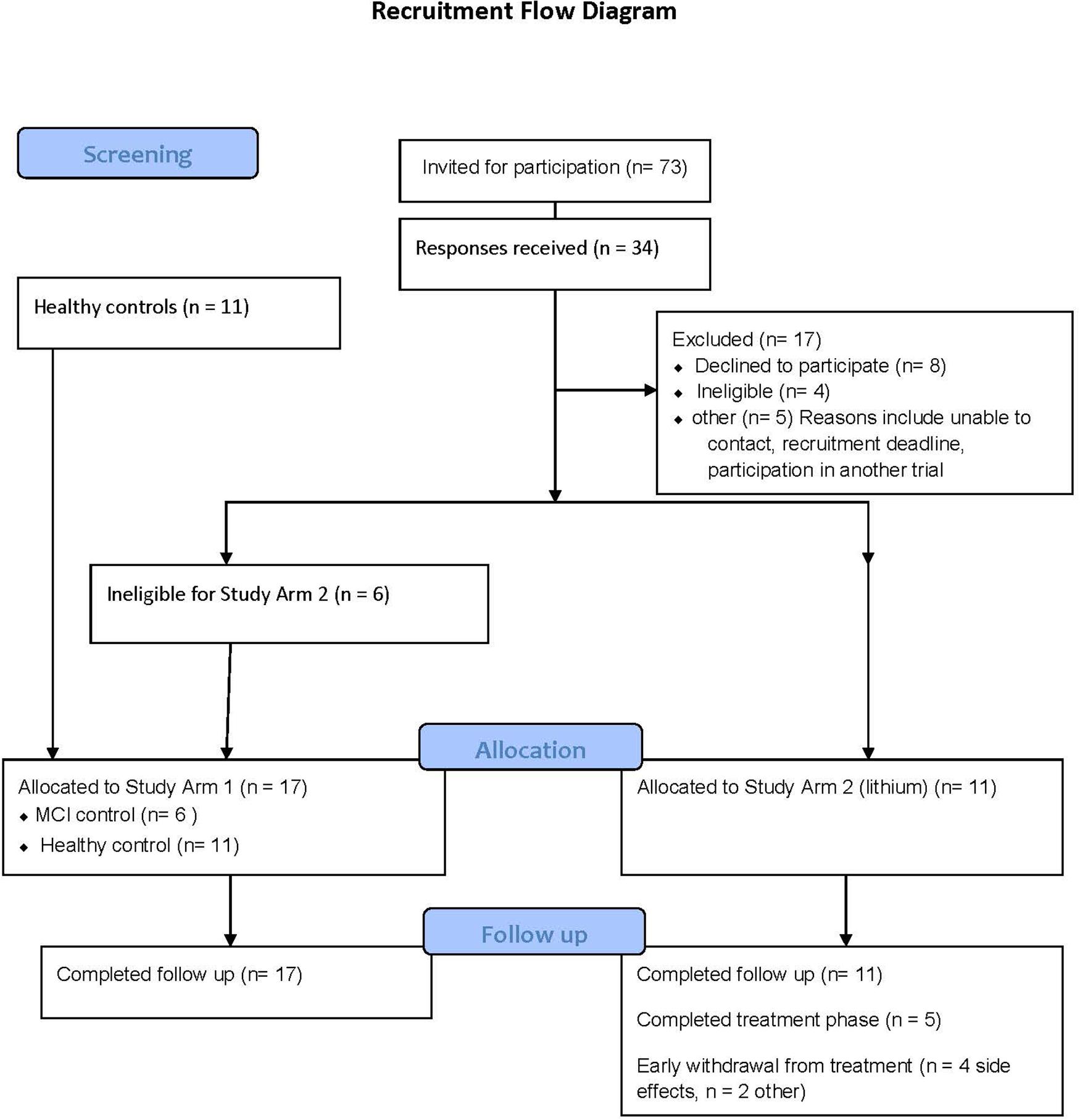
Figure 2. Consort diagram detailing the recruitment process for the clinical studies.
There were no retainment issues in Study Arm 1, with no withdrawals and no adverse events reported. In contrast, of the eleven participants in Arm 2, six withdrew from the lithium titration phase (54%) due to reported side effects (n = 4), non-compliance (n = 1) and altered blood results (n = 1). All participants completed a lithium wash out visit (>3 weeks after last lithium dose). Interestingly, there was no association between lithium blood concentration and withdrawal (withdrawals had plasma lithium in the range 0.2–0.61 mmol/L, mean 0.38 mmol/L, while non-withdrawals plasma lithium ranged from 0.33–0.54 mmol/L, mean 0.42 mmol/L). The mean duration of lithium titration before withdrawal was 5 weeks (note; high dose lithium only started in week 6). Table 3 provides the plasma lithium of all participants through the study period, including those that withdrew. Five of the volunteers provided the full set of seven blood samples for lithium measure (Table 3).
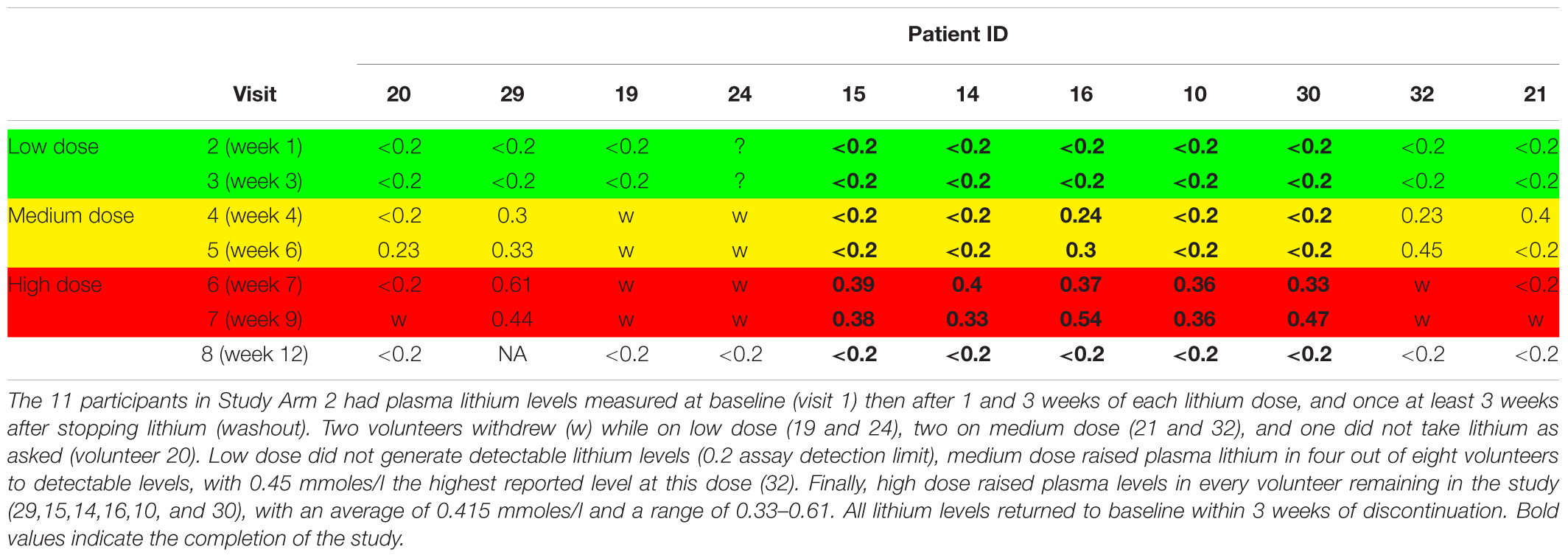
Table 3. Plasma lithium level (mmoles/l) during stepwise increase in lithium dose in volunteers.
There were no serious adverse events reported. Six participants experienced side effects (54%) with two of these continuing with the lithium titration phase to completion. Side effects reported at either moderate or severe intensity included: fatigue, metallic taste, dry mouth, nausea, decreased concentration, insomnia, or confusion. Feeling drowsy or lethargic during the day was the most commonly reported side effect with five of the six participants reporting this in either mild, moderate or severe intensity.
Only one individual of the eleven given lithium exhibited a change in renal function (mild reduction in eGFR to 57 mls/min with accompanying mild increase in creatinine). Examination of the case files revealed that while normal at trial initiation, this individual had previously had similar mild impairment of renal function on blood testing. There were no instances of electrolyte disturbance or other renal problems during the trial.
Our initial study was to investigate whether the GSK3 biomarkers in Table 1 could be detected in a PBMC preparation from rats, and whether detectable markers respond to lithium administration. Additionally, we wanted to try to investigate whether any lithium induced biomarker responses in the PBMCs associated with changes in the brain. Ultimately, we wanted to generate a focussed, validated, panel of GSK3 biomarkers as a readout for GSK3 function in humans to investigate early dementia, and for establishing efficacy of lithium dose.
However, we could detect only eight of the biomarkers from Table 1 in rat PBMCs either using immunoblotting, loading up to 50 μg of total protein on the gels, or by immunoprecipitation of GSK3 isoforms from cell lysates and assay (Figure 3A, for examples of immunoblots, and Table 4 for IP assay data). Importantly, only four of these biomarkers were detected robustly (signal > twice background), and reproducibly from multiple separate PBMC preparations (Table 5). These were Phospho-Ser21/9 of GSK3, total GSK3 (both by immunoblot, Figure 3A), and GSK3α and GSK3β activity (measured by IP-assay, Table 4). We are confident that the blotting reagents were suitable for detection of each protein (positive controls included in Figure 3A and biomarkers were detectable in brain tissues, Figure 3). Therefore, the major GSK3 regulatory pathways and the best characterized GSK3 substrates to date all proved difficult to detect robustly in PBMCs by immunoblotting.
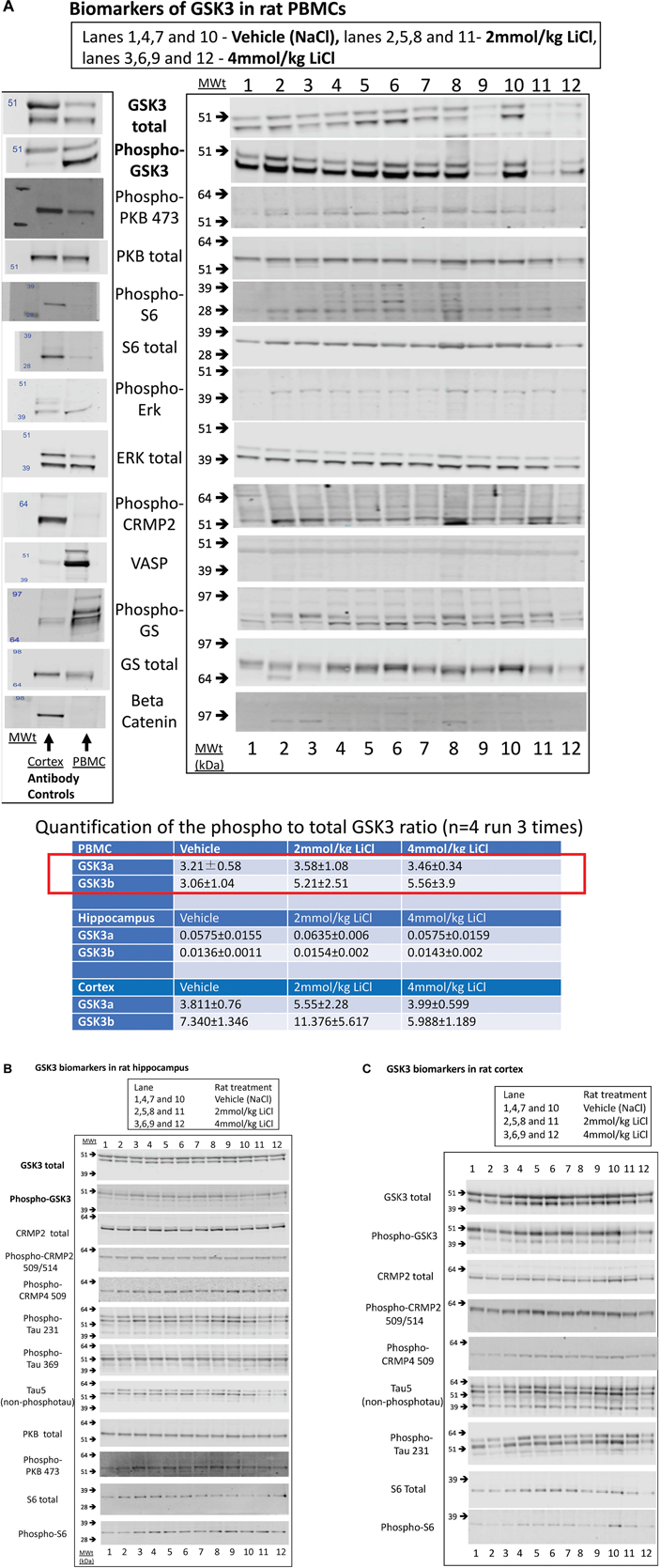
Figure 3. Biomarkers of GSK3 in rat tissues following lithium injection. Rats were given an i.p. injection of vehicle or lithium chloride as detailed in section “Materials and Methods,” and tissues isolated 3 h later. Representative examples of Western blots of rat protein lysates isolated from (A) rat PBMCs, (B) Hippocampus, or (C) Cortex, are provided using the antibodies stated. In (A), we also provide a comparison of vehicle treated rat PBMC lysate with rat brain cortex lysate with every antibody as evidence for the utility of each antibody (antibody control). The ratio of expression of GSK3 phosphorylation at the N-terminal inhibitory site to the overall isoform expression was quantified from the images as detailed in section “Materials and Methods,” and is provided for each tissue (±SD) in the table inserted into A. The red box highlights the major biomarker changes in response to lithium, although only the P-GSK3β-total GSK3β ratio was significantly increased by lithium (p < 0.05, student t test).
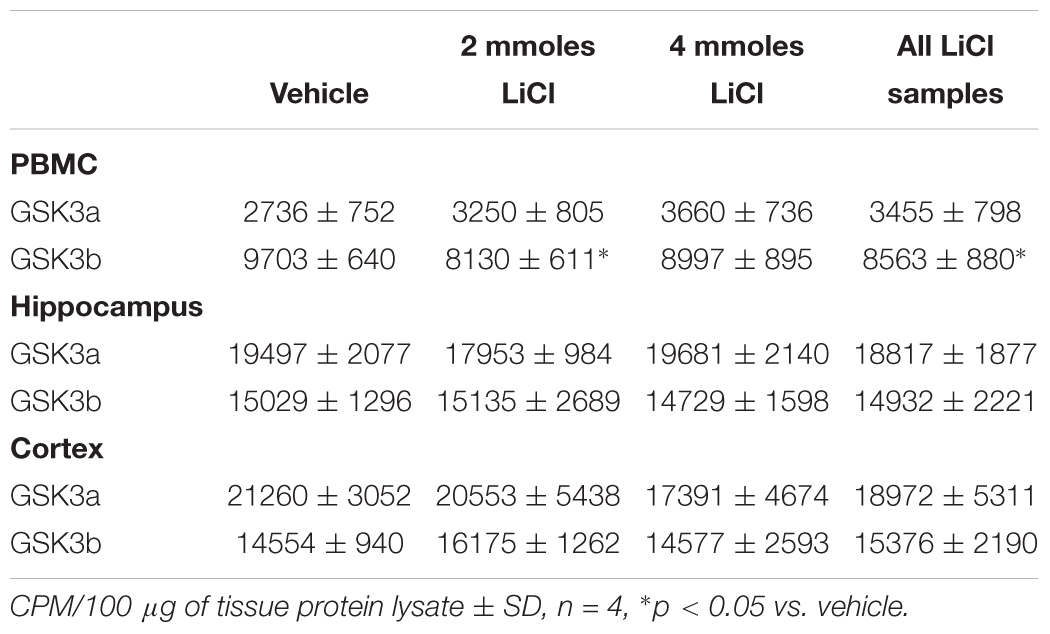
Table 4. GSK3 specific activity (measured by IP assay) in rat tissue lysates.
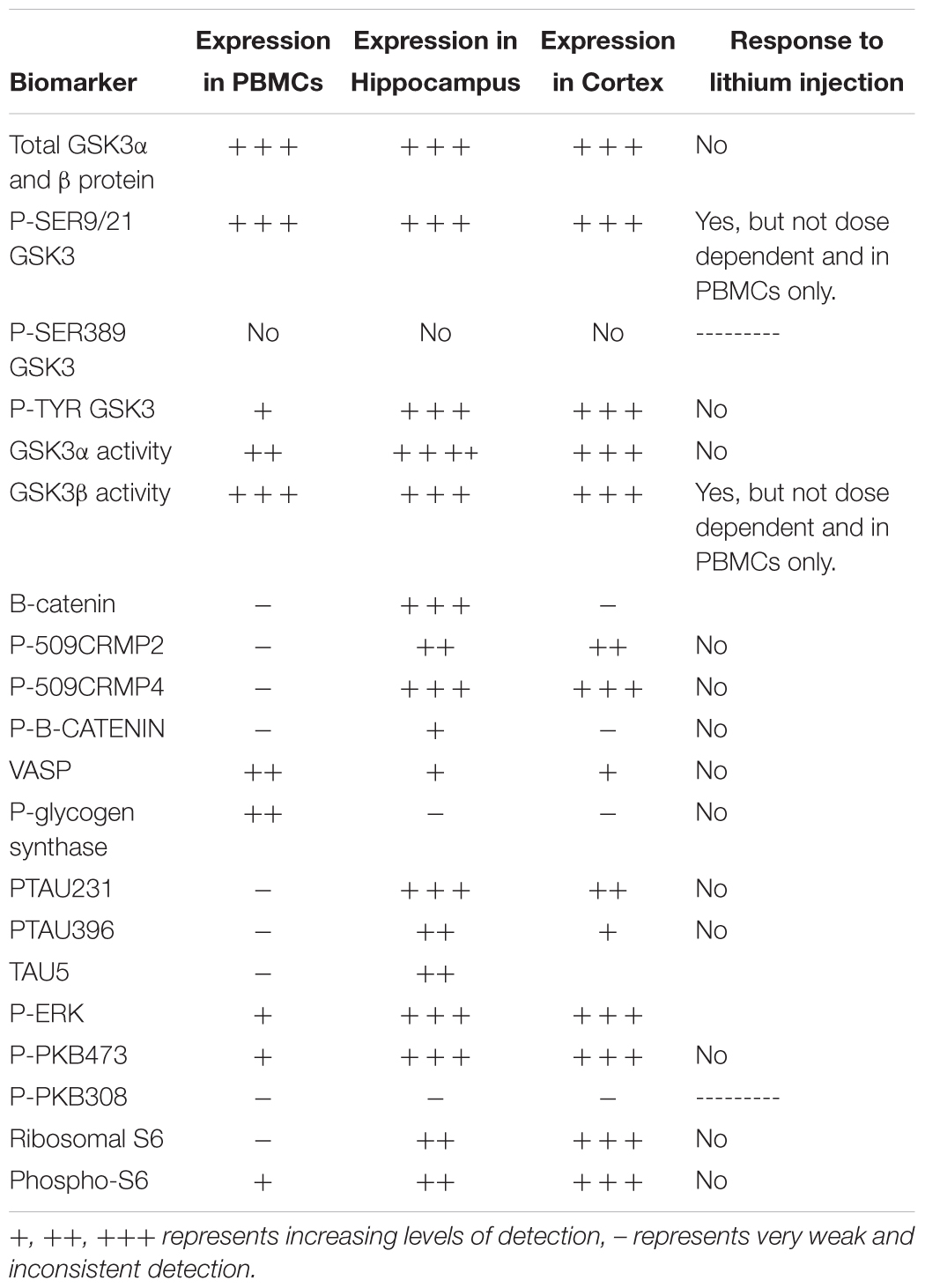
Table 5. Expression of potential biomarkers of GSK3 function in rat tissues.
We next investigated whether any of these markers were influenced by a single i.p. injection of LiCl vs. NaCl (control). 3 h after injection the blood lithium levels in the rats were raised in a dose dependent fashion, with roughly twice the plasma lithium concentration achieved by doubling the lithium dose (Table 6). Cell based studies had previously suggested that 0.5–1 mM Lithium is required to acutely reduce cellular GSK3 activity by around 50%, and we were achieving these levels in the rat blood with a single injection. Therefore, we monitored the potential GSK3 biomarkers in protein lysates isolated from PBMCs (Figure 3A) and two brain regions (Figures 3B,C), in order to assess the response to Lithium injection (summary in Table 5). In addition, we immunoprecipitated, and assayed the specific activity of the two GSK3 isoforms from the same tissues (Table 4), as lithium is reported to enhance covalent inhibitory modification of GSK3. The phosphorylation of Ser9 of GSK3β was enhanced in PBMCs by lithium injection (see insert table in Figure 3A for quantification of immunoblots), and consistent with this, the activity of GSK3β (but not GSK3α) was significantly reduced in PBMCs (Tables 4, 5). This indicates that a plasma lithium level of 0.83 mM was sufficient to enhance Ser9 phosphorylation and at least partly inhibit GSK3β activity, and could be achieved by this single lithium injection. Interestingly, double the plasma level of lithium (1.6 mM) did not further increase the degree of Ser9 phosphorylation nor enhance the inhibition of GSK3β, suggesting the effect had plateaued. In addition, this plasma level of lithium did not significantly alter any of the measurable GSK3 biomarkers studied in the hippocampus, or cortex (Figures 3B,C, quantified in table within Figure 3A). This may mean the brain does not get exposed to the same level of lithium as the blood, or the time point we examined was not optimal for effects on the brain.
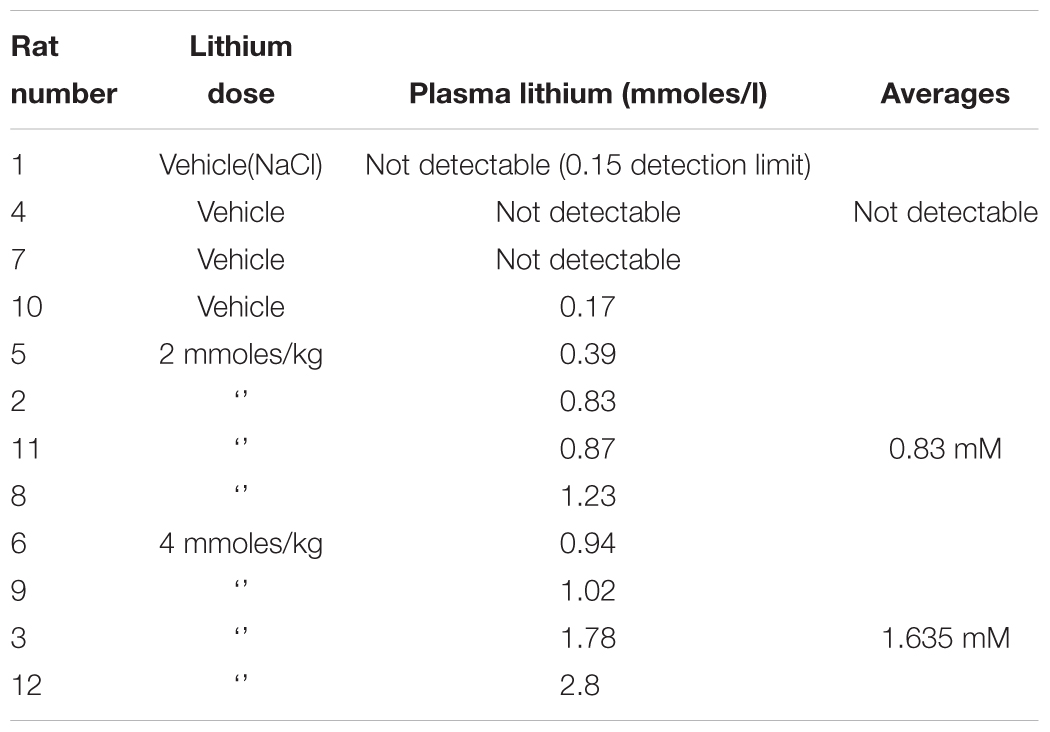
Table 6. Rat plasma lithium levels achieved after a single i.p. dose of lithium chloride.
The next step in our study examined the GSK3 biomarkers, identified in the rat study, in PBMCs isolated from human volunteers. In Study Arm 1 we wanted: (1) to establish that the GSK3 activity biomarkers detectable in rat PBMCs could also be detected in cells isolated from the human population of greatest interest (i.e., MCI), (2) to investigate the reproducibility of marker detection in two PBMC preparations generated from the same individual but taken 12 weeks apart, and finally, (3) to ask whether there were major differences in any of these markers that associated with MCI diagnosis. We recruited six patients with MCI (mean age 78, range 77–81) and eleven non-MCI, age-matched controls (mean age 76, range 65–84) to this arm. In addition, we had baseline samples from the 11 MCI volunteers recruited to Arm 2 below (making n = 17 for the MCI group).
A major finding of this work was that the consistency of protein recovery and quality between the human PBMC preparations, even those generated from samples obtained from the same individual, was varied (Figure 4A for protein stains). In some cases, total protein recovery was relatively low (e.g., visit 1 volunteer X6), in others there was clear evidence of substantial degradation (e.g., visit 2 volunteer 03, visit 1 volunteer 04, visits 1 and 2 volunteer X3, visit 2 volunteer 09). The Coomassie stained gels emphasized that 13 of the 45 PBMC isolates investigated had noticeable overall protein degradation. Nevertheless, we went on to examine several of the biomarkers from Table 1, in particular the four which were robustly detected in rat PBMCs. There was also variability in the biomarkers across the samples, even between samples from the same individuals, mostly associated with protein quality (Figure 4B), however, we were able to confirm which biomarkers could be detected in human PBMCs. In addition to phosphorylation of Ser9/21 of GSK3, total GSK3 and the activity of the GSK3 isoforms, we could also detect the GSK3 substrate c-myc (Figure 4B). Due to the poor quality of protein isolation we quantified the protein staining in every sample (22 healthy and 12 MCI in Study arm 1, plus the baseline PBMC preparations from the 11 MCI volunteers in Study Arm 2 below, so n = 22 and 23, respectively), and discarded any samples with less than 20% of the average protein level by protein staining (Figure 4A, e.g., visit 1 volunteer X6), or samples with < 10% of the average actin staining (Figure 4B, e.g., visit 2 Volunteer 03). We then quantified the immunoblot densities for the remaining samples (control group n = 14, MCI group n = 18), for every detectable biomarker. The phosphorylation of GSK3β at Ser-9 (47 kDa) and the activity of GSK3b were the only biomarkers that were significantly affected by lithium in the rat study (Figure 3A). In the human PBMCs we generated a P-GSK3β to actin ratio for every sample. The healthy control samples had a P-GSK3β:actin ratio of 0.0283 ± 0.018 (n = 14) and the MCI samples had a ratio of 0.0295 ratio ± 0.015 (n = 18). These data sets were not significantly different (p > 0.05), implying that development of MCI does not alter this aspect of GSK3 regulation, at least in PBMCs. Similarly, an IP assay of GSK3 activity indicated a possible trend of increased GSK3β activity in the MCI group, however, this did not reach significance (Figure 4C). The GSK3 substrate, c-myc, was detected in human PBMCs (Figure 4B), however, there was not a significant difference in the c-myc:actin ratio between controls and MCI (0.143 ± 0.1 controls, vs. 0.129 ± 0.1 MCI, p = 0.77). This protein is degraded upon phosphorylation by GSK3, hence the data would suggest no significant increase in GSK3 activity in MCI PBMCs.
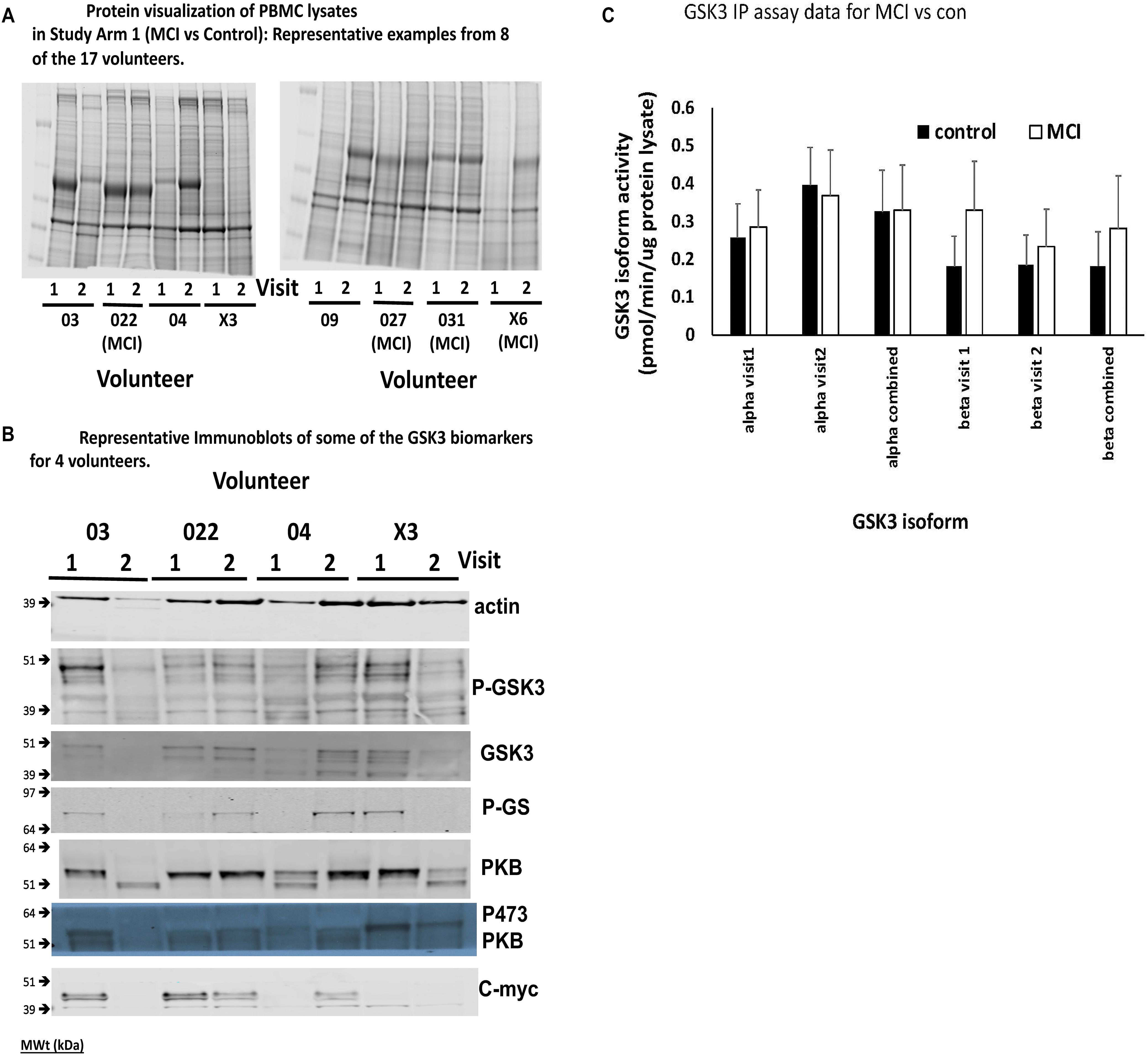
Figure 4. Comparison of Biomarkers of GSK3 in human PBMCs isolated from volunteers with MCI and controls (Study Arm 1). Protein lysates were prepared from PBMCs isolated from two groups of human volunteers, one with a diagnosis of MCI, the other age matched but with no MCI. In each case the volunteer provided two blood samples for PBMC preparation, taken 12 weeks apart (Visit 1 and 2). (A) Representative Coomassie protein staining of PBMC lysates from 8 of the 17 volunteers in Study Arm 1, where equal total protein was loaded on the gel. (B) Representative immunoblots of biomarkers within the PBMCs of four of the volunteers in Study arm 1. (C) Immunoprecipitation and assay of GSK3 isoforms from 50 μg of protein lysate in each of the PBMC samples. The specific activity was quantified for GSK3alpha and GSK3beta from all PBMC preparations and the average activity (±SE) for the MCI and control groups at each visit, as well as the two visits combined, is given.
Although the GSK3 biomarkers were not significantly altered in PBMCs in people with MCI we proposed that they could still be of use to monitor GSK3 activity status in response to interventions which work through modulating GSK3, such as lithium. Therefore, we performed study Arm 2 generating up to eight different PBMC preparations from each of the 11 volunteers, taken at various stages during a step wise increasing lithium dosing schedule (Figure 1). Separate blood samples were taken at every time point to measure plasma lithium and isolate PBMC preparations, respectively, and so biomarker detection in the PBMC preparations could be linked to changes in blood lithium (Table 3). Two volunteers withdrew while on low dose (ID 19 and 24), two on medium dose (ID 21 and 32), one did not take the lithium (ID 20) and one failed to attend on more than one occasion. This left five volunteers who completed the full 8 sample set (Figures 1, 2). Low dose lithium did not generate detectable plasma lithium levels (Table 3), medium dose raised plasma lithium to detectable levels in only four out of the eight volunteers that remained at this stage, with 0.45 mmol/l the highest reported level on medium dose (ID 32). Finally, high dose treatment raised plasma levels in every volunteer (ID 29,15,14,16,10, and 30), with an average of 0.415 mmol/l and a range of 0.33–0.61 in these six volunteers (Table 3). All lithium levels returned to baseline within 3 weeks of discontinuation. Five volunteers (ID 10, 14, 15, 16, and 30) provided all eight PBMC preparations (baseline, six samples during dosing and a final washout sample, Figure 1). The PBMC preparations had significant variation in the quality of protein recovery, even across the samples isolated from the same individual (Figure 5A), however, this was not related to the lithium dose at time of blood collection. We examined the expression of the GSK3 biomarkers detectable in rat and human PBMCs, across these eight samples for each of the five volunteers. Quantitation of these immunoblots was problematic due to the high variability in loading controls (despite matching loading for total protein), which implies protein degradation or dephosphorylation in many of the samples. Indeed, it is worth noting that the variability and quality of the P-GSK3 and P-PKB signals in Figure 5B were very sensitive to even minor variations in protein (Figure 5A) and housekeeping protein (Figure 5B, actin). For example compare protein, b-actin, P-GSK3 and P-PKB in volunteer D15, visit 2, 3, and 4 (protein and b-actin degradation) with volunteer D14, visit 6, 7, and 8. Therefore, although we found no significant effect of lithium on the phosphorylation of GSK3 and PKB (Figure 5B and quantified in Supplementary Table), or the activity of either GSK3 isoform (GSK3 beta activity shown in Figure 5C, p > 0.05), the extensive variation in human PBMCs may have greatly reduced the power to detect effect sizes of those seen in the rat study. It should be noted that the highest blood lithium levels (visits 6 and 7 in Figure 5) never reached the lithium levels in the acute dosing rat study, and this implies that 0.33–0.61 mM plasma lithium is not sufficient to regulate the inhibitory phosphorylation of GSK3β in human PBMCs.
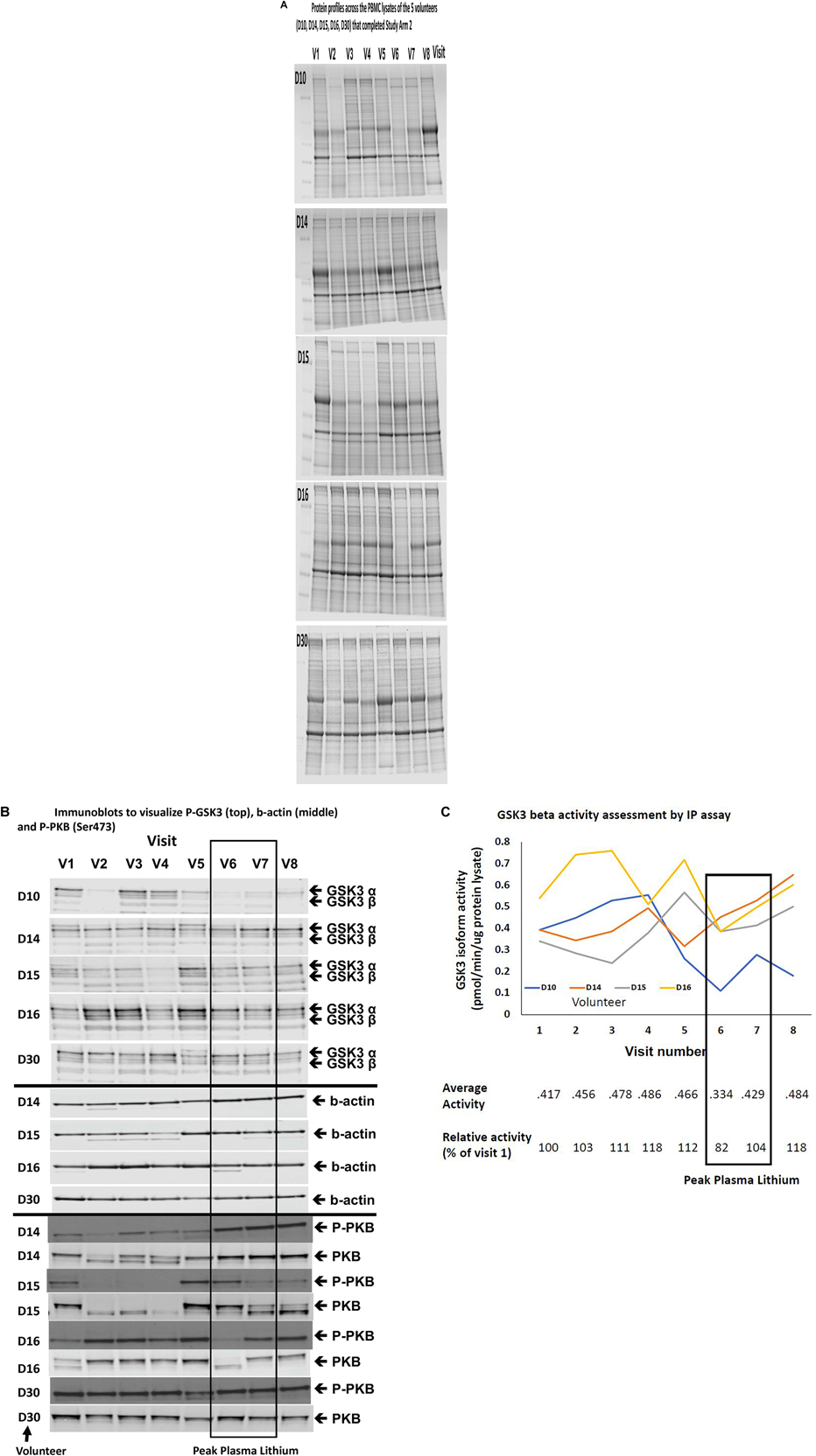
Figure 5. Comparison of Biomarkers of GSK3 in human PBMCs isolated from volunteers with MCI treated with a stepwise increasing dose of Lithium (Study Arm 2). Protein lysates were prepared from PBMCs isolated from volunteers with MCI who underwent lithium treatment (see Figure 1). (A) Protein profiles (Coomassie staining) from the PBMC lysates of the five volunteers that completed eight visits. Equal amounts of protein were loaded in each lane (by Bradford assay). (B) Immunoblot of all of the lysates from the same five volunteers using the phosphoser9/21 GSK3 antibody. The box around visit 6 and 7 highlights the samples with the peak plasma lithium levels (Table 3). (C) GSK3β isoform specific activity in PBMCs isolated across the eight visits for four of the five volunteers that completed the study. The average GSK3β activity for each visit and the average GSK3β activity relative to the baseline activity for each volunteer are given under the graph. The box highlights samples (6 and 7) with the peak plasma lithium levels (Table 3).
These data emphasize the need to improve the standard PBMC isolation process, identify further GSK3 activity biomarkers, and increase volunteer numbers to utilize PBMC preparations for monitoring GSK3 activity.
This was a preliminary study to investigate the feasibility and methodology around performing GSK3 functional biomarker development in a highly relevant human population. The study had several research goals, firstly to validate which of the numerous potential biomarkers of GSK3 biology could be detected in PBMCs, secondly to establish whether expression of these detectable biomarkers was affected by a diagnosis of MCI, thirdly, to investigate whether expression of these biomarkers was modified by treatment with lithium, at a dose up to that used to treat Bipolar disorder, and finally to add to the growing information on tolerance of lithium in the MCI population. The hope was that we would generate information to inform the design of a larger clinical assessment of lithium treatment for the prevention of progression from MCI to dementia (Sutherland and Duthie, 2015). Validated biomarkers of GSK3 function in human PBMCs could have potential use in multiple other conditions, including diabetes, inflammation, and cancer (Wagman et al., 2004; Hernandez et al., 2009; Castillo-Quan et al., 2016; Jope et al., 2017; Robertson et al., 2018). GSK3 inhibition is considered to have potential therapeutic value in all of these conditions, therefore robust, easily attainable markers of GSK3 may have clinical utility in diagnosis, prognosis and response to therapy (such as response to lithium).
Although there are a host of potential biomarkers for GSK3 function in cells and tissues, none are, in themselves, ideal for accurate and complete assessment of GSK3 activity. The main biomarker used in numerous studies is the phosphorylation status of Ser21 of GSK3α, and the equivalent residue Ser9 in GSK3β (Hye et al., 2005; Li et al., 2007; Soutar et al., 2010). However, while this modification is associated with GSK3 activity (Sutherland et al., 1993) it is a semi-quantitative, comparative measure and only assesses GSK3 activity toward primed substrates (Frame et al., 2001), which may not represent all of the clinically relevant GSK3 substrates (e.g., tau). Importantly, it does not reflect GSK3 regulation by other mechanisms, including by small molecule inhibition. Although some small molecule inhibitors such as lithium do enhance this phosphorylation it is unlikely to be as potent an inhibitory mechanism as direct inhibition through the active site of GSK3, and the two mechanisms may function at distinct lithium concentrations. Therefore, monitoring this phosphorylation site provides incomplete information on overall GSK3 regulation and function. Additional approaches include measuring upstream regulatory pathways, such as the PI3K-Akt pathway which regulates cellular GSK3 in response to insulin and some growth factors. However, other pathways including the Ras-MAPK, PKA and mTOR-S6K can complicate the input of the PI3K-PKB regulation (Eldar-Finkelman et al., 1995), and the degree of upstream pathway activation does not necessarily reflect the degree of GSK3 inhibition. Therefore, we believe that the most accurate approach is to combine assessment of the Ser21/9 phosphorylation status, the inherent activity of GSK3 isoforms (by IP assay), while simultaneously measuring the phosphorylation of a range of substrates known to be directly targeted by GSK3. This would provide a more quantitative and complete assessment of GSK3 functional status. Indeed, many of the targets of GSK3 become targets for the β-TrCP ubiquitination system, thereby directing them to the proteasome for destruction (Robertson et al., 2018). Therefore, there may be a panel of GSK3 targets whose protein levels provide a functional measure of GSK3 activity steady state level. What is clear is that in order to develop accurate tools to monitor GSK3 activity in clinical trials we need validated, selective and sensitive biomarkers of GSK3, either in tissues of importance to disease or in surrogate tissues (such as blood cells). This was the starting point for our investigation of the range of potential biomarkers of GSK3 function listed in Table 1.
Unfortunately, our study failed to achieve the desired outcome of establishing a panel of biomarkers for GSK3 that could be used to track early disease or drug response, and determine the appropriate dose of lithium for inhibiting GSK3 in MCI volunteers. This was mainly due to the unexpected poor quality of biomarker detection from PBMCs, which meant our studies were underpowered. However, we established a number of methodological issues and the data does question the clinical utility of most of the biomarkers listed in Table 1. Firstly, very few of them were detectable (by immunoblot) in PBMCs in rats or humans. Secondly, and probably most importantly, the quality and reproducibility of the signaling proteins (especially phosphoproteins) isolated from human PBMCs using standard protocols was disappointingly poor. Finally, our study in the rats indicated that there may be a complex non-linear relationship between plasma lithium level, GSK3 regulation in PBMCs and GSK3 regulation in brain.
Most of the biomarkers from Table 1 are only readily detectable in tissues where their function is important, (e.g., glycogen synthase is most highly expressed in muscle or liver tissues, while tau is neuronally enriched), and as such it is perhaps not surprising that many are not highly expressed in blood cells. The list of potential “GSK3 biomarkers” is actually much greater than that listed in Table 1, with more than 100 substrates of GSK3 reported in the literature (Sutherland, 2011). However, those in Table 1 are the best validated targets, and/or those with the most sensitive and specific antibodies, to facilitate detection by immunoblotting. We propose that it is now time to widen the search for well validated GSK3 targets to aid the development of a “GSK3 biomarker panel” for application to multiple tissues and species (Robertson et al., 2018), and it may be sensible to move to quantitative proteomic technology rather than relying on immunoblotting.
We would also propose that improved protocols to isolate human PBMCs (or other blood cells) are required to permit quantification of metabolic signaling pathways. Molecules such as GSK3 are regulated by a wide variety of extracellular cues. This may include some of the chemicals involved in PBMC isolation, or volunteer specific characteristics such as hormonal or diurnal cycles and dietary or stress influences on the day of blood collection. Indeed, the high degree of variation in our human PBMC preparations compared to the rat preparations would point to these latter influences as worthy of consideration.
Previously, increased GSK3 protein levels were found in white blood cells in AD and MCI patients (Hye et al., 2005), while reduced GSK3 phosphorylation was found in platelets of MCI patients (Forlenza et al., 2011b). We were not able to confirm these important findings possibly due to the poor quality of our PBMC protein preparations. It is also worth noting that the relationship between plasma lithium and GSK3β phosphorylation and activity in rat PBMCs was unusual. There was a similar effect on GSK3 inhibition at 0.83 and 1.6 mM plasma lithium. This may mean that the maximal dose dependent effect on GSK3β phosphorylation was below 0.83 mM, however, the degree of effect on GSK3 was not major (<twofold enhancement of phosphorylation and around 15% inhibition). This may be related to the fact we examined a single time point or the nature of this biomarker (phosphorylation rather than direct measure of GSK3 activity). For example, if GSK3β was 50% phosphorylated in control animals then the maximum possible effect of lithium on phosphorylation would be a twofold increase. As stated earlier, the IP assay is perhaps not the ideal biomarker for effects of small molecules, as lithium would be washed away during the IP process but it should still assess activity changes due to the inhibitory phosphorylation. This is why we believe that the accurate assessment of the pharmacological relationship between plasma lithium and GSK3 regulation would be best achieved with biomarkers that directly monitor GSK3 specific targets rather than simply the phosphorylation of GSK3.
It was concerning that we did not detect any effects of lithium on any GSK3 biomarkers in the brain of the rats despite achieving relatively high plasma levels of 1.6 mM lithium (albeit acutely). In this case we were monitoring several markers that we know are modified by GSK3 inhibition in cell culture (Cole and Sutherland, 2008), and in animal models of GSK3 deficiency (Soutar et al., 2010). This implies that this acute level of plasma lithium is not sufficient to regulate GSK3 in these brain areas. Alternatively, it is possible that either the injection resulted in a very transient increase in plasma lithium that is not sufficient to alter these biomarkers, or that the time point after injection was not optimal for effects on brain biomarkers. In essence a much more extensive study is needed to truly establish the lithium dose required to modify GSK3 activity in these brain areas.
Despite these major technical issues around the assessment of current GSK3 biomarkers in human PBMCs we were encouraged by the interest of the MCI patient population, as well as the relative safety of lithium in this population. This is consistent with previous studies in MCI (Forlenza et al., 2011a,b) and other conditions (Li et al., 2007; Hampel et al., 2009; Diniz et al., 2011; McKnight et al., 2012), where lithium has been found to be more safe and certainly manageable than current perception may have us believe (Macdonald et al., 2008). We should also keep in mind that lithium is a low-cost drug and even continuous monitoring for the known side effects is relatively cheap. Therefore, we provide further evidence that preforming an efficacy trial on the effect of lithium on progression from MCI to dementia is not only feasible but worthwhile. Within that study one could expand the investigation of GSK3 biomarkers from a simple readout for diagnosis, or drug action, to investigate any association with conversion from MCI to dementia. As such we are actively working to address the technical issues listed above.
The study was approved by the North of Scotland Research Ethics Committee, number 16/NS/006. All animal procedures were approved by the University of Dundee Ethical Review Process and were performed in accordance with UK Home Office regulations (under the auspices of Project License PIL60/4280).
CS, AD, JG, and SL raised the funding for the study. CS was the Chief Investigator, contributing to data collection, analysis, and writing of the manuscript. AD was the Principle Clinical Investigator in Dundee and contributed to writing of the manuscript. SL was the Principle Clinical Investigator in Oxford. AD, CM, SL, and JG were responsible for volunteer recruitment, clinical ethics, and volunteer safety monitoring as well as contributing to study design, clinical data analysis, and writing of the manuscript. LvA, AM, and JGall were responsible for all rodent and lab work, data storage, and contributed to the writing of the manuscript.
This work was supported by the Chief Scientist Office, project grant ETM/401.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are extremely grateful to all volunteers who gave up their time to participate in the study, in particular to those volunteers who undertook the lithium dose study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2019.00163/full#supplementary-material
Amanatkar, H. R., Papagiannopoulos, B., and Grossberg, G. T. (2017). Analysis of recent failures of disease modifying therapies in Alzheimer’s disease suggesting a new methodology for future studies. Expert Rev. Neurother. 17, 7–16. doi: 10.1080/14737175.2016.1194203
Avrahami, L., Farfara, D., Shaham-Kol, M., Vassar, R., Frenkel, D., and Eldar-Finkelman, H. (2013). Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J. Biol. Chem. 288, 1295–1306. doi: 10.1074/jbc.M112.409250
Beurel, E., Kaidanovich-Beilin, O., Yeh, W. I., Song, L., Palomo, V., Michalek, S. M., et al. (2013). Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J. Immunol. 190, 5000–5011. doi: 10.4049/jimmunol.1203057
Can, A., Schulze, T. G., and Gould, T. D. (2014). Molecular actions and clinical pharmacogenetics of lithium therapy. Pharmacol. Biochem. Behav. 123, 3–16. doi: 10.1016/j.pbb.2014.02.004
Castillo-Quan, J. I., Li, L., Kinghorn, K. J., Ivanov, D. K., Tain, L. S., Slack, C., et al. (2016). Lithium promotes longevity through GSK3/NRF2-dependent hormesis. Cell Rep. 15, 638–650. doi: 10.1016/j.celrep.2016.03.041
Chen, X., Sun, W., Pan, Y., Yang, Q., Cao, K., Zhang, J., et al. (2013). Lithium ameliorates open-field and elevated plus maze behaviors, and brain phospho-glycogen synthase kinase 3-beta expression in fragile X syndrome model mice. Neurosciences 18, 356–362.
Cohen, P., and Goedert, M. (2004). GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug Discov. 3, 479–487. doi: 10.1038/nrd1415
Cole, A. R., and Sutherland, C. (2008). Measuring GSK3 expression and activity in cells. Methods Mol. Biol. 468, 45–65. doi: 10.1007/978-1-59745-249-6_4
Corbett, A., Pickett, J., Burns, A., Corcoran, J., Dunnett, S. B., Edison, P., et al. (2012). Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 11, 833–846.
Cross, D. A. E., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995). Inhibition of GSK3 by insulin mediated by protein kinase B. Nature 378, 785–789. doi: 10.1038/378785a0
Cummings, J., Lee, G., Mortsdorf, T., Ritter, A., and Zhong, K. (2017). Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement. 3, 367–384. doi: 10.1016/j.trci.2017.05.002
Davies, S. P., Reddy, H., Caviano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105. doi: 10.1042/bj3510095
de Cristobal, J., Garcia-Garcia, L., Delgado, M., Pozo, M. A., and Medina, M. (2014). A Longitudinal FDG-PET Study of Transgenic Mice Overexpressing GSK- 3beta in the Brain. Curr. Alzheimer Res. 11, 175–181. doi: 10.2174/156720501102140313145704
Diniz, B. S., Talib, L. L., Joaquim, H. P. G., de Paula, V. R. J., Gattaz, W. F., and Forlenza, O. V. (2011). Platelet GSK3B activity in patients with late-life depression: marker of depressive episode severity and cognitive impairment? World J. Biol. Psychia. 12, 216–222. doi: 10.3109/15622975.2010.551408
Eldar-Finkelman, H., Seger, R., Vandenheede, J. R., and Krebs, E. G. (1995). Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J. Biol. Chem. 270, 987–990. doi: 10.1074/jbc.270.3.987
Forlenza, O. V., Diniz, B. S., Radanovic, M., Santos, F. S., Talib, L. L., and Gattaz, W. F. (2011a). Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Brit. J. Psychiat. 198, 351–356. doi: 10.1192/bjp.bp.110.080044
Forlenza, O. V., Torres, C. A., Talib, L. L., de Paula, V. J., Joaquim, H. P. G., Diniz, B. S., et al. (2011b). Increased platelet GSK3B activity in patients with mild cognitive impairment and Alzheimer’s disease. J. Psychiatr. Res. 45, 220–224. doi: 10.1016/j.jpsychires.2010.06.002
Frame, S., Cohen, P., and Biondi, R. M. (2001). A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell. 7, 1321–1327. doi: 10.1016/s1097-2765(01)00253-2
Gao, C., Holscher, C., Liu, Y., and Li, L. (2012). GSK3: a key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev. Neurosci. 23, 1–11. doi: 10.1515/rns.2011.061
Hampel, H., Ewers, M., Burger, K., Annas, P., Mortberg, A., Bogstedt, A., et al. (2009). Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931. doi: 10.4088/jcp.08m04606
Hardy, J. (2009). The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J. Neurochem. 110, 1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x
Hernandez, F., Nido, J. D., Avila, J., and Villanueva, N. (2009). GSK3 inhibitors and disease. Mini. Rev. Med. Chem. 9, 1024–1029. doi: 10.2174/138955709788922647
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 104, 1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x
Hye, A., Kerr, F., Archer, N., Foy, C., Poppe, M., Brown, R., et al. (2005). Glycogen synthase kinase-3 is increased in white cells early in Alzheimer’s disease. Neurosci. Lett. 373, 1–4. doi: 10.1016/j.neulet.2004.10.031
Jiang, H., Guo, W., Liang, X., and Rao, Y. (2005). Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120, 123–135. doi: 10.1016/s0092-8674(04)01258-9
Jope, R. S., Cheng, Y., Lowell, J. A., Worthen, R. J., Sitbon, Y. H., and Beurel, E. (2017). Stressed and inflamed, can GSK3 be blamed? Trends Biochem. Sci. 42, 180–192. doi: 10.1016/j.tibs.2016.10.009
Jope, R. S., and Johnson, G. V. (2004). The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29, 95–102. doi: 10.1016/j.tibs.2003.12.004
Jope, R. S., Yuskaitis, C. J., and Beurel, E. (2007). Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 32, 577–595. doi: 10.1007/s11064-006-9128-5
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., et al. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397. doi: 10.1038/nn.2408
King, M. K., Pardo, M., Cheng, Y., Downey, K., Jope, R. S., and Beurel, E. (2013). Glycogen synthase kinase-3 inhibitors: rescuers of cognitive impairments. Pharmacol. Ther. 141, 1–12. doi: 10.1016/j.pharmthera.2013.07.010
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459. doi: 10.1073/pnas.93.16.8455
Kockeritz, L., Doble, B., Patel, S., and Woodgett, J. R. (2006). Glycogen synthase kinase-3–an overview of an over-achieving protein kinase. Curr. Drug Targets 7, 1377–1388. doi: 10.2174/1389450110607011377
Levin, A., Hemmelgarn, B., Culleton, B., Tobe, S., McFarlane, P., and Ruzicka, M. (2008). Guidelines for the management of chronic kidney disease. CMAJ 179, 1154–1162.
Li, X., Friedman, A. B., Zhu, W., Wang, L., Boswell, S., May, R. S., et al. (2007). Lithium regulates glycogen synthase kinase-3beta in human peripheral blood mononuclear cells: implication in the treatment of bipolar disorder. Biol. Psychiatry 61, 216–222. doi: 10.1016/j.biopsych.2006.02.027
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39. doi: 10.1093/emboj/20.1.27
Ly, P. T., Wu, Y., Zou, H., Wang, R., Zhou, W., Kinoshita, A., et al. (2013). Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 123, 224–235. doi: 10.1172/JCI64516
Macdonald, A., Briggs, K., Poppe, M., Higgins, A., Velayudhan, L., and Lovestone, S. (2008). A feasibility and tolerability study of lithium in alzheimer’s disease. Int. J. Geriatr. Psychiatry 23, 704–711. doi: 10.1002/gps.1964
Malhi, G. S., Tanious, M., Das, P., Coulston, C. M., and Berk, M. (2013). Potential mechanisms of action of lithium in bipolar disorder. current understanding. CNS Drugs 27, 135–153. doi: 10.1007/s40263-013-0039-0
McKnight, R. F., Adida, M., Budge, K., Stockton, S., Goodwin, G. M., and Geddes, J. R. (2012). Lithium toxicity profile: a systematic review and meta-analysis. Lancet 379, 721–728. doi: 10.1016/S0140-6736(11)61516-X
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., et al. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995. doi: 10.1073/pnas.0500466102
O’Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S., Piccolo, S., et al. (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798. doi: 10.1523/jneurosci.4753-03.2004
O’Brien, W. T., and Klein, P. S. (2007). Regulation of glycogen synthase kinase-3 in patients with affective disorders. Biol. Psychiatry 61, 139–141. doi: 10.1016/j.biopsych.2006.11.008
O’Brien, W. T., and Klein, P. S. (2009). Validating GSK3 as an in vivo target of lithium action. Biochem. Soc. Trans. 37, 1133–1138. doi: 10.1042/BST0371133
Petersen, R. C., Doody, R., Kurz, A., Mohs, R. C., Morris, J. C., Rabins, P. V., et al. (2001). Current concepts in mild cognitive impairment. Arch. Neurol. 58, 1985–1992.
Robertson, H., Hayes, J. D., and Sutherland, C. (2018). A partnership with the proteasome; the destructive nature of GSK3. Biochem. Pharmacol. 147, 77–92. doi: 10.1016/j.bcp.2017.10.016
Russo, A., Schmid, E., Nurbaeva, M. K., Yang, W., Yan, J., Bhandaru, M., et al. (2013). PKB/SGK-dependent GSK3-phosphorylation in the regulation of LPS-induced Ca(2+) increase in mouse dendritic cells. Biochem. Biophys. Res. Commun. 437, 336–341. doi: 10.1016/j.bbrc.2013.06.075
Soutar, M. P., Kim, W. Y., Williamson, R., Peggie, M., Hastie, C. J., McLauchlan, H., et al. (2010). Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 115, 974–983. doi: 10.1111/j.1471-4159.2010.06988.x
Sudduth, T. L., Wilson, J. G., Everhart, A., Colton, C. A., and Wilcock, D. M. (2012). Lithium treatment of APPSwDI/NOS2-/- mice leads to reduced hyperphosphorylated tau, increased amyloid deposition and altered inflammatory phenotype. PLoS One 7:e31993. doi: 10.1371/journal.pone.0031993
Sutherland, C. (2011). What are the bona fide substrates of GSK3? Int. J. Alz. Dis. 2011:505607. doi: 10.4061/2011/505607
Sutherland, C., and Duthie, A. C. (2015). Invited commentary on. Lithium treatment and risk for dementia in adults with bipolar disorder. Br. J. Psychiatry 207, 52–54. doi: 10.1192/bjp.bp.114.161729
Sutherland, C., Leighton, I. A., and Cohen, P. (1993). Inactivation of glycogen synthase kinase-3b by phosphorylation; new kinase connections in insulin and growth factor signalling. Biochem. J. 296, 15–19. doi: 10.1042/bj2960015
Wagman, A. S., Johnson, K. W., and Bussiere, D. E. (2004). Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes. Curr. Pharm. Des 10, 1105–1137. doi: 10.2174/1381612043452668
Wang, J., Tan, L., and Yu, J. T. (2016). Prevention trials in alzheimer’s disease: current status and future perspectives. J. Alzheimers Dis. 50, 927–945. doi: 10.3233/JAD-150826
Yu, F., Zhang, Y., and Chuang, D. M. (2012). Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J. Neurotrauma 29, 2342–2351. doi: 10.1089/neu.2012.2449
Keywords: lithium, GSK3, clinical trial, biomarker, safety
Citation: Duthie A, van Aalten L, MacDonald C, McNeilly A, Gallagher J, Geddes J, Lovestone S and Sutherland C (2019) Recruitment, Retainment, and Biomarkers of Response; A Pilot Trial of Lithium in Humans With Mild Cognitive Impairment. Front. Mol. Neurosci. 12:163. doi: 10.3389/fnmol.2019.00163
Received: 27 August 2018; Accepted: 12 June 2019;
Published: 28 June 2019.
Edited by:
Gianluca Serafini, Ospedale Policlinico San Martino (IRCCS), ItalyReviewed by:
Andrea B. Maier, The University of Melbourne, AustraliaCopyright © 2019 Duthie, van Aalten, MacDonald, McNeilly, Gallagher, Geddes, Lovestone and Sutherland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Calum Sutherland, Yy5kLnN1dGhlcmxhbmRAZHVuZGVlLmFjLnVr
†Present address: Simon Lovestone, Janssen-Cilag, High Wycombe, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.