 Helene H. Jensen
Helene H. Jensen Malene Brohus
Malene Brohus Mette Nyegaard
Mette Nyegaard Michael T. Overgaard
Michael T. Overgaard- 1Section for Biotechnology, Department of Chemistry and Bioscience, Aalborg University, Aalborg, Denmark
- 2Department of Biomedicine, Aarhus University, Aarhus, Denmark
Fluxes of calcium (Ca2+) across cell membranes enable fast cellular responses. Calmodulin (CaM) senses local changes in Ca2+ concentration and relays the information to numerous interaction partners. The critical role of accurate Ca2+ signaling on cellular function is underscored by the fact that there are three independent CaM genes (CALM1-3) in the human genome. All three genes are functional and encode the exact same CaM protein. Moreover, CaM has a completely conserved amino acid sequence across all vertebrates. Given this degree of conservation, it was long thought that mutations in CaM were incompatible with life. It was therefore a big surprise when the first CaM mutations in humans were identified six years ago. Today, more than a dozen human CaM missense mutations have been described, all found in patients with severe cardiac arrhythmias. Biochemical studies have demonstrated differential effects on Ca2+ binding affinities for these CaM variants. Moreover, CaM regulation of central cardiac ion channels is impaired, including the voltage-gated Ca2+ channel, CaV1.2, and the sarcoplasmic reticulum Ca2+ release channel, ryanodine receptor isoform 2, RyR2. Currently, no non-cardiac phenotypes have been described for CaM variant carriers. However, sequencing of large human cohorts reveals a cumulative frequency of additional rare CaM mutations that raise the possibility of CaM variants not exclusively causing severe cardiac arrhythmias. Here, we provide an overview of the identified CaM variants and their known consequences for target regulation and cardiac disease phenotype. We discuss experimental data, patient genotypes and phenotypes as well as which questions remain open to understand this complexity.
Introduction
For generations, a large Swedish family presented with repeated episodes of syncope and cardiac arrest in response to exercise or emotional stress. Several family members were diagnosed with the inherited disorder catecholaminergic polymorphic ventricular tachycardia (CPVT), which is often fatal due to a high risk of ventricular fibrillation and sudden cardiac death (SCD). Indeed, two of the 13 affected individuals died from SCD, both at a young age. In 2012, we linked the disease to a mutation in the gene CALM1, which encodes the calcium (Ca2+) sensor calmodulin (CaM) (Nyegaard et al., 2012). The identification of a human CaM missense mutation came as a dramatic surprise to the CaM research field; CaM is exceptionally conserved across species with all vertebrate CALM genes encoding identical proteins, and human mutations had not previously been reported. The slow evolution of CaM emphasizes the strong selection pressure against even minor changes in the protein sequence (Halling et al., 2016). Further, CaM regulates more than 300 intracellular targets, each interaction with unique facets of binding sites, Ca2+-dependency, target affinity, and functionality (Shen et al., 2005; O’Connell et al., 2010). With this versatility in mind, it was believed that mutations in CaM could not be tolerated.
After our initial finding, a number of CaM mutations have been identified in patients with severe cardiac arrhythmia disorders involving recurrent syncope, ventricular fibrillation, and in some instances SCD under adrenergic stimulation (Table 1). The vast majority of these mutations are de novo and carriers present with disease phenotypes early or very early in childhood, in some cases even before birth. In addition to CPVT, carriers suffer from long QT syndrome (LQTS), and one individual was diagnosed with idiopathic ventricular fibrillation (IVF). The link between CaM mutations and these arrhythmias has primarily been attributed to impaired regulation of the cardiac ryanodine receptor isoform 2 (RyR2), and the cardiac L-type voltage gated Ca2+ channel isoform 1.2 (CaV1.2) (Table 1).
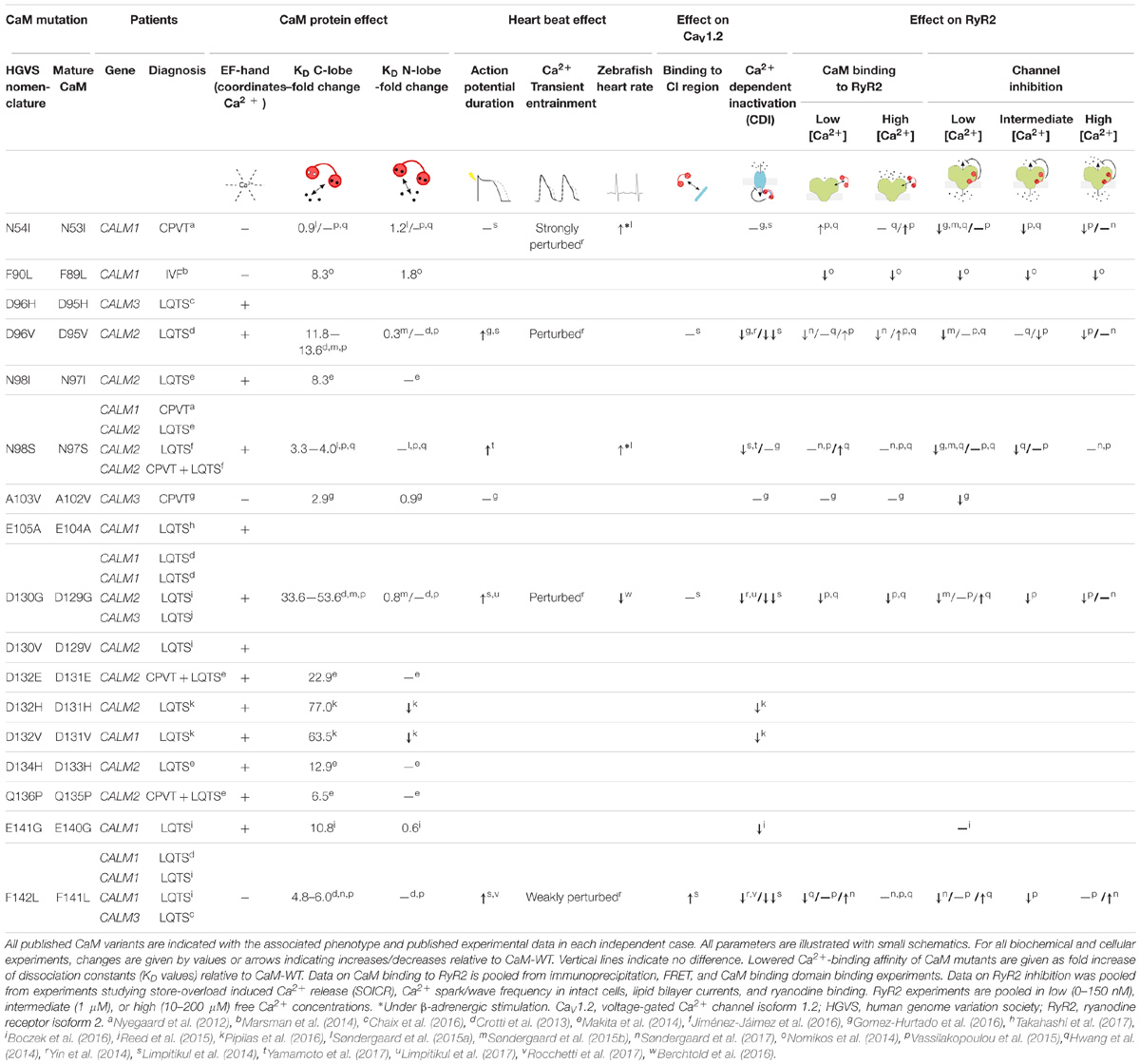
TABLE 1. Pathogenic CaM mutations affect Ca2+ and target binding as well as target regulation.
Since CaM is encoded by three active genes and expressed in all cells, the CaM field is faced with intriguing questions and paradoxes at the genetic and phenotypic level. First, how can a single mutation in one of six CaM-encoding alleles dominantly cause SCD? Second, how can identical missense mutations cause LQTS in one patient and CPVT in another? Third, with an increasing number of new rare CaM missense mutations identified in sequencing databases of large human cohorts, could there be other phenotypes associated with CaM mutations? Improved understanding of the functional impact of CaM mutations may enable predictions of the genotype–phenotype relationship for variants in any of the three CALM genes.
In this review, we summarize and discuss the current knowledge on CaM mutations and their impact on the regulation of CaV1.2 and RyR2, and address the few studies that suggest an involvement of other targets. Finally, we discuss the special genetic context of CaM and the implications for future studies.
CaM, the Cellular Ca2+ Sensing Protein
Fast and compound changes in cytosolic Ca2+ concentration is the foundation for a wide number of cellular responses, including muscle contraction and neuronal firing (Clapham, 2007). Thus, at rest, the cytosolic Ca2+ concentration is maintained at ∼100 nM, but can rapidly increase to more than 100 μM, when Ca2+ channels open in the plasma membrane or in internal stores such as the sarcoplasmic reticulum (SR). Detection of this steep change in Ca2+ concentration depends on Ca2+ binding proteins. CaM is one of the major Ca2+ sensors that relay information on Ca2+ concentration to functionally modulate target proteins (known as calmodulation). CaM is synthesized as a 149 amino acid protein, however, the initiator Met residue is removed upon translation, leaving 148 amino acid residues in the mature protein (Sasagawa et al., 1982). This has led to some confusion in the numbering of CaM variant positions. The Human Genome Variant Sequence (HGVS) nomenclature (den Dunnen et al., 2016) recommends to count the initiator Met as residue number 1, while the CaM protein community tends to leave the residue out as it has no functional role in the mature protein. Throughout this paper we will use the HGVS nomenclature.
The structure of CaM reflects its refined Ca2+ sensing abilities (Kretsinger et al., 1986; Chattopadhyaya et al., 1992). CaM is a 16.7 kDa protein consisting of two lobes connected by a flexible and unstructured or α-helical linker (Figures 1A,B). Each lobe has two EF-hands, which can each coordinate one Ca2+ ion (Figures 1A,B, gray spheres). The C-terminal lobe of CaM binds Ca2+ with six times higher affinity (KD 2.5 μM) than the N-terminal lobe (KD 16 μM), allowing CaM to sense Ca2+ across a wide concentration range (Linse et al., 1991; Søndergaard et al., 2015a). Hydrophobic patches on the inside of each lobe recognize binding motifs on interaction partners, and thereby facilitate CaM binding and target regulation (Tidow and Nissen, 2013). Ca2+ binding to CaM and CaM binding to target proteins allosterically affect the affinity of each other, and targets specifically modulate the conformation of CaM. In this way, the small CaM protein displays a plethora of binding and regulation properties.
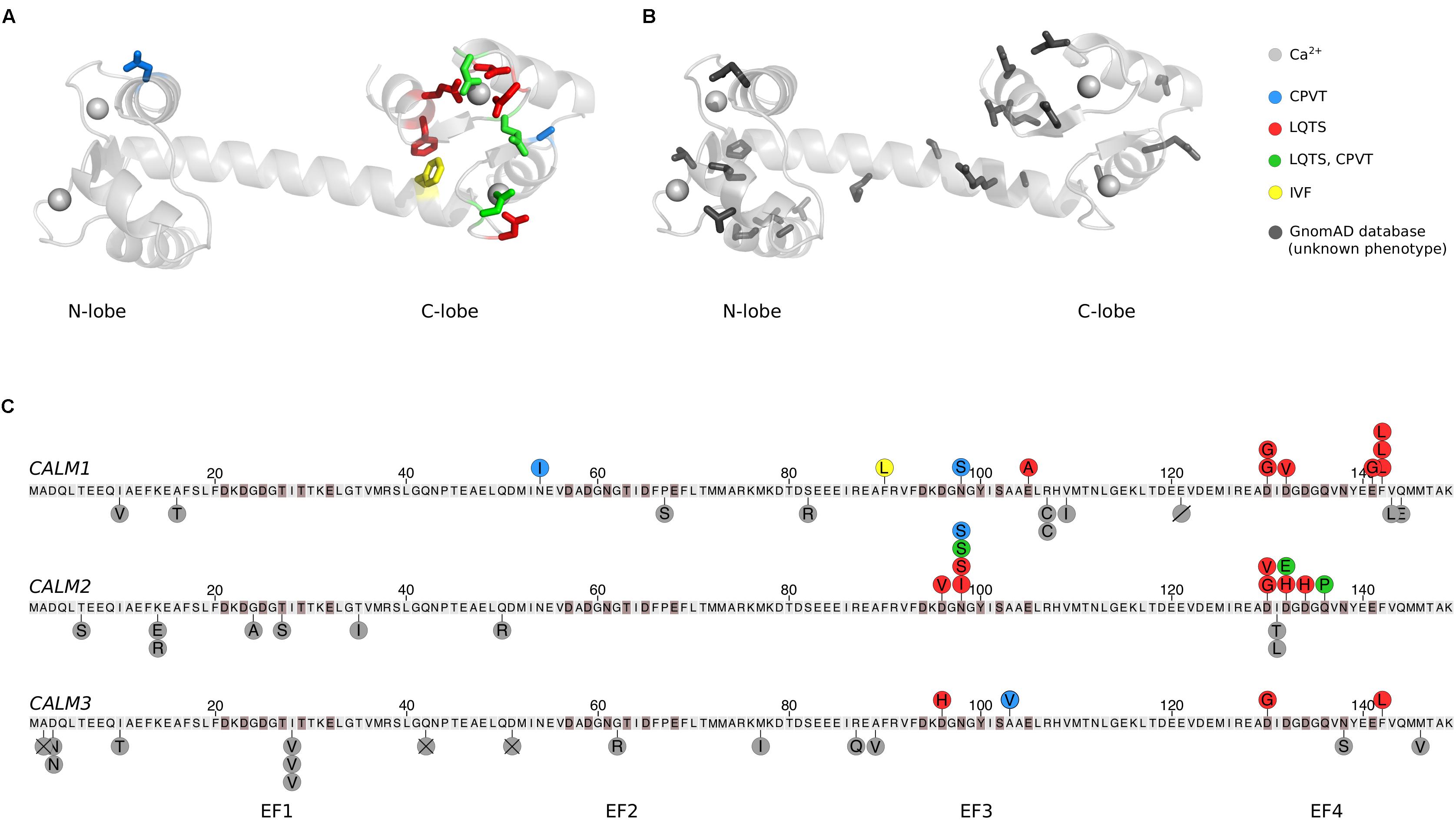
FIGURE 1. Human calmodulin mutations. (A) The calmodulin (CaM) protein has two lobes connected by a flexible α-helical linker. Each lobe has two EF-hands, which each can coordinate a Ca2+ ion (gray spheres), generating four Ca2+ binding sites. Residues highlighted in color and stick representation are mutated in cardiac arrhythmia patients, where they are associated with CPVT, LQTS, IVF, or a combination of CPVT and LQTS as indicated. The structure was visualized in PyMOL (PDB: 1CLL) (Chattopadhyaya et al., 1992). (B) CaM residues shown in dark gray stick representation are found mutated in the GnomAD database. GnomAD includes 277,364 alleles from 138,632 individuals (Lek et al., 2016). (C) Overview of mutations in each of the three human CALM genes, displayed as the translated protein with the initiator-Met as residue 1. Residues highlighted in brown participate in Ca2+ coordination. Published arrhythmogenic human variants are indicated by circles above the sequences, with colors indicating the disease phenotype. Dark gray circles below the sequence are individual allele variations observed in GnomeAD without a known phenotype. Amino acid variants are shown by letters, a slash (deletion), or a cross (frame-shift or premature stop codon). CPVT, catecholaminergic polymorphic ventricular tachycardia; LQTS, long QT syndrome; IVF, idiopathic ventricular fibrillation.
CaM Conservation and Mutations
Despite the remarkable conservation of CaM, 26 cases of arrhythmogenic mutations have now been identified in humans. Their positions in CaM are indicated on a CaM structure in Figure 1A and on the CaM sequence in Figure 1C. Strikingly, the mutations are primarily found in the C-terminal lobe and most affect residues involved in Ca2+ coordination, dramatically reducing Ca2+ affinity (Table 1). One interesting exemption is the mutation initially identified in the large Swedish family, CaM-N54I. This mutation is unique since it (1) resides in the N-terminal lobe and (2) neither coordinates Ca2+ nor is part of the hydrophobic target binding patches. Biochemical and cellular experiments have been employed to model and explain how CaM mutations lead to arrhythmic phenotypes. The results from these studies are discussed in the following.
CaM Mutations Disturb Heart Rhythm
The composite effect of CaM mutations on heart function has been investigated using different experimental model systems. In zebrafish, the CaM-N54I and -N98S mutations caused increased heart rate upon β-adrenergic stimulation, which is in line with the CPVT phenotype observed for patients with these mutations (Søndergaard et al., 2015a). Similarly, the LQTS-mutation D130G increased zebrafish heart rate (Berchtold et al., 2016). In cultures of ventricular cardiomyocytes, expression of LQTS-associated CaM mutations leads to prolonged action potentials (APs), in some cases spilling over to the next stimulation and causing alternans (Limpitikul et al., 2014, 2017; Gomez-Hurtado et al., 2016; Yamamoto et al., 2017). Moreover, CaM-D96V was associated with early after-depolarizations (Gomez-Hurtado et al., 2016). Imaging of Ca2+ fluxes during electrical pacing of cardiomyocytes demonstrated dysregulated Ca2+ concentration in cells expressing LQTS-associated CaM-D96V, -D130G, -F142L, and in particular the CPVT-mutation N54I showed Ca2+ overload, Ca2+ reuptake errors, or alternans (Limpitikul et al., 2014, 2017; Yin et al., 2014).
CaV1.2 and RyR2 are the two major Ca2+ channels involved in Ca2+ handling in the heart. Both channels are essential for stimulating – and importantly, terminating – heart contraction. Briefly, APs stimulate the opening of CaV1.2 in the sarcolemma. This allows Ca2+ to enter the cell. The resulting increase in cytosolic Ca2+ stimulates opening of RyR2, thereby releasing large amounts of Ca2+ from the SR which ultimately leads to heart contraction (Sorensen et al., 2013). CaM binds to both CaV1.2 and RyR2 and is important for the precise and timely gating of the channels in response to changes in Ca2+ concentration. Generally, CPVT is an SR Ca2+ handling disease, most often caused by RyR2 mutations, whereas LQTS involves dysregulation of ion-fluxes across the sarcolemma, e.g., Ca2+ flux disturbances caused by mutations in CaV1.2 (Landstrom et al., 2017).
Impaired Regulation of CaV1.2
CaM is tethered to the intracellular C-terminal tail of CaV1.2 and functions as a Ca2+ sensor to stimulate channel closure when Ca2+ has entered the cell, a process termed calcium-dependent inactivation (CDI) (Brehm and Eckert, 1978; Peterson et al., 1999). Reduction of CDI was observed for several LQTS-causing CaM mutations, including D132H, D132V, and E141G (Boczek et al., 2016; Pipilas et al., 2016), whereas CDI was completely absent for LQTS-associated CaM mutations D96V, D130G, and F142L in HEK293 cells and adult guinea pig cardiomyocytes (Limpitikul et al., 2014). The CaM-N98S mutation is special in the sense that carriers display either CPVT or LQTS arrhythmias or both. Expression of this mutant slightly reduces CDI of CaV1.2, whereas the strictly CPVT-causing mutation CaM-N54I showed no effect (Limpitikul et al., 2014; Yamamoto et al., 2017).
Effects of CaM Mutations on RyR2
RyR2 opening is normally stimulated by the increase in Ca2+ upon CaV1.2 opening. In response to the dramatic increase in cytoplasmic Ca2+ concentration (and the drop in SR luminal Ca2+ concentration), RyR2 closes. CaM acts as a gatekeeper, by modulating the open state probability of RyR2 in response to changes in the Ca2+ concentration (Fabiato, 1985; Xu and Meissner, 2004). Here, we have compiled the diverse experimental approaches used to evaluate whether CaM mutations affect RyR2 regulation under different Ca2+ concentrations (Table 1). The results are mixed and in some cases contradictory. The CPVT-associated CaM mutations, N54I, N98S, and A103V, all showed an increased level of RyR2 opening, that is, decreased inhibition by CaM (Hwang et al., 2014; Søndergaard et al., 2015b; Gomez-Hurtado et al., 2016). These results suggest that the molecular disease mechanism for the CPVT-causing CaM mutations is dysregulation of cardiac SR Ca2+ release, in line with CPVT-causing mutations in RyR2.
Surprisingly, LQTS-associated CaM mutations also caused differences in RyR2 binding and regulation (Hwang et al., 2014; Søndergaard et al., 2015b; Vassilakopoulou et al., 2015). However, in particular at low Ca2+ concentrations, the data are somewhat contradictory (Table 1). Curiously, CaM-F142L showed an increased binding affinity toward RyR2 in the apo-form, as well as an increased inhibitory effect on RyR2 in some assays, an effect opposite of all other CaM mutants analyzed (Søndergaard et al., 2017).
Other Suggested and Potential Targets
Although mutations in CaV1.2 can lead to LQTS, most cases of LQTS can be attributed to mutations in genes encoding the voltage-gated K+ channels KV7.1 (KCNQ1) and KV11.1 (KCNH2), as well as the voltage-gated Na+ channel NaV1.5 (SCN5A) (Modell and Lehmann, 2006). Although regulated by CaM, no clear effects of CaM mutations have been observed on NaV1.5 (Yin et al., 2014; Boczek et al., 2016; Rocchetti et al., 2017). KV7.1 utilizes CaM as a sensor of Ca2+ to stimulate opening (Ghosh et al., 2006). Whereas CaM-F142L did not show any effect on KV7.1 current (IKs) (Rocchetti et al., 2017), CaM-N98S significantly shifted the half-activation of KV7.1 (Sun and MacKinnon, 2017). Further, the small-conductance Ca2+-activated K+ (SK) channel was decreased by several CaM mutations (Yu et al., 2016). Although SK channels play a minor role in ventricular myocytes, they are expressed in atrial myocytes, and interestingly, widely expressed in the nervous system where they play a major role in synaptic transmission (Adelman et al., 2012).
Ca2+/CaM-dependent kinase II (CaMKII) regulates a wide number of pathways and protein targets, but no significant effects were observed on CaMKII with either of the CaM variants N54I, D96V, N98S, D130G, or F142L (Hwang et al., 2014; Berchtold et al., 2016).
Although current literature suggests that Ca2+ channels are the main targets affected by CaM mutations, we hypothesize that other ion channels and potentially other signaling proteins may also be dysregulated if tested in greater detail. But how can one predict which targets are likely affected by CaM mutations? We believe that the Ca2+-dependency of target binding plays an important role. That is, proteins that bind to both the apo- and the Ca2+-form of CaM may be more sensitive to mutations in CaM than targets that only bind the Ca2+-form. Thus, CaM mutations may exert a dominant effect in cases where CaM remains associated with its target when the cell is at rest.
Implications of the Genetic Architecture of CALM Genes
In humans, CaM is encoded by three different and independent loci; on chromosome 2 (CALM2), 14 (CALM1), and 19 (CALM3). Although there are differences in the genomic sequence, the three different transcripts are translated into the exact same protein (Fischer et al., 1988). During the last six years (2012–2018), 26 cases of pathogenic mutations in CaM have been reported, and all three CALM genes are now established major genes for both CPVT and LQTS (Table 1). All identified pathogenic CaM mutations cluster in the C-lobe, except the CPVT-causing variant N54I. Interestingly, this is the mutation with the mildest effect on biophysical parameters of CaM, including Ca2+ binding affinity. It is also the only variant found in a large family. Further, all LQTS-causing CaM mutations strongly reduce the C-lobe Ca2+ affinity, and, except for the F142L mutation, are all located in Ca2+-coordinating residues. This suggests that CaM mutations that strongly affect C-lobe Ca2+ affinity lead to LQTS (Table 1).
To date, there are more published cases of pathogenic mutations in CALM1 (11) and CALM2 (11) compared to CALM3 (4). This could either reflect that the first published mutations were found in CALM1 and CALM2 and thus these two genes were included in genetic screening panels before CALM3. Or, it may be due to some subtle functional differences between the three genes. Quantitative PCR have shown that the three genes are not expressed at equal levels in cardiomyocytes, but the relative levels are not clear: whereas one study found higher levels of CALM3 transcripts in human hearts (Crotti et al., 2013), the CALM1 transcript was the most abundant in human stem cell-derived cardiomyocytes (Rocchetti et al., 2017).
As the number of published pathogenic CaM mutations has increased, several conclusions about genotype–phenotype relationships begin to form. Of particular interest is the CaM-D130G mutation, which has been identified in four unrelated individuals; two carrying the mutation in CALM1, one in CALM2, and one in CALM3, and all four suffering from LQTS. Similarly, the CaM-F142L mutation was found in both CALM1 and CALM3, and all carriers suffered from LQTS. These observations imply that the amino acid position and type of change is important for the phenotype, and not the genetic origin of the transcript (CALM1, −2, or −3). One intriguing observation does, however, challenge this simple genotype–phenotype conclusion. The CaM-N98S mutation was found in CALM1 in one individual, and in three other individuals in CALM2. Interestingly, these four patients present with different phenotypes – either CPVT or LQTS or both – suggesting that we still do not fully understand the underlying mechanisms determining the disease phenotype. These may involve other genomic variants able to shape the phenotype, complex protein regulatory effects, or environmental factors.
Since the protein products from all CALM genes are identical, it is tempting to speculate if a deletion of one allele (equivalent to a loss-of-function mutation) is less pathogenic than missense mutations. This idea immediately poses a therapeutic solution to patients carrying a CaM missense mutation, for example using the CRISPR/Cas9 technology to delete the pathogenic allele. Two studies specifically silenced the mutated CaM allele in patient-derived pluripotent stem cells differentiated into cardiomyocytes. Here, the CaM-D130G and -N98S mutations were silenced in CALM2 with a partial or almost full restoration of CaV1.2 regulation (Limpitikul et al., 2017; Yamamoto et al., 2017). These experiments are proof-of-principle that removal of the diseased allele may be a therapeutic solution. Also, these studies suggest that potential frameshift mutations causing premature stop codons or protein degradation may not be as detrimental as missense mutations. It still needs to be determined, however, if individuals carrying a loss-of-function mutation in a CALM gene are in fact unaffected from disease. Interestingly, a large exome sequencing study [Exome Aggregate Consortium (ExAC)] (Lek et al., 2016), found that CALM1 and CALM2 are intolerant to loss-of-function mutations (pLI = 0.89 and 0.86 respectively).
Digging Deeper May Reveal a Broader Impact
Given the ubiquitous role of CaM, it is striking that all mutations identified are associated with a strong cardiac phenotype. We speculate whether these cases were discovered because of their unusual severity and because cardiologists and geneticists have specifically screened for CALM mutations in populations with cardiac disorders. Looking for CaM mutations in other patient groups may reveal new aspects and consequences of these mutations.
In a database containing variants from a large sequencing effort of almost 140,000 individuals (GnomAD, Lek et al., 2016), additional rare CaM missense mutations are reported (Figures 1B,C, gray residues and circles). The number of coding variants for all three genes is much lower than expected by chance. However, the cumulative frequency of additional rare CaM mutations suggests that CaM variants do not exclusively cause severe cardiac arrhythmias. At present, there is no overlap between variants identified in GnomAD (database variants) and the published pathogenic mutations. Further, the GnomAD missense variants are distributed throughout the entire protein, and more evenly distributed on the three CALM genes (9, 9, and 12 mutations in CALM1, −2, and −3, respectively), compared to the arrhythmogenic CaM variants. Also, all GnomAD variants except two, fall outside Ca2+-coordinating residues. Taken together, we therefore speculate that some of these uncharacterized variants are associated with unknown traits not involving cardiac arrhythmia. Sequencing results from large cohorts with known phenotypes are required to confirm this hypothesis.
We propose that studies of tissues other than cardiac are warranted for future research on the effects of CaM mutations. In particular, CaM expression is high in excitable neuronal cells. Also CaV1.2 is widely expressed in neuronal tissues. Here, CaV1.2 plays a role in cellular firing as well as in gene regulation, and mutations in CaV1.2 have been attributed to psychiatric diseases (Nyegaard et al., 2010; Nanou and Catterall, 2018). Neurons express a number of other CaV channel isoforms, including CaV1.3 and CaV2 variants, which are also regulated by CaM. RyR2 plays a less prominent role in neurons, where the inositol triphosphate receptor (IP3R), which is also regulated by CaM, is the dominating intracellular Ca2+ release channel. Mild neuronal defects have been observed in some patients with CaM mutations, but these effects were suggested to be secondary, resulting from the frequent and severe episodes of syncope or cardiac arrest (Crotti et al., 2013; Boczek et al., 2016; Pipilas et al., 2016).
Conclusion and Outlook
Within the last six years, CaM mutations have emerged as a novel cause of human diseases, the calmodulinopathies. All described pathogenic mutations have been identified in patients suffering from severe arrhythmic disorders, and biochemical as well as cellular studies have demonstrated that particularly the regulation of the Ca2+ channels CaV1.2 and RyR2 are affected by these mutations. Currently, there is a strong correlation between LQTS-causing CaM mutations and Cav1.2 dysregulation, whereas all mutations affect RyR2 function. Given the ubiquitous role of CaM in a vast number of cellular processes, we predict that yet other targets may be affected. Our database search revealed a number of uncharacterized CaM missense mutations with unknown phenotypic consequences present in the population. Future studies will reveal whether other protein targets as well as other disease phenotypes can be assigned to mutations in CaM.
Author Contributions
HJ wrote the first draft of the manuscript. HJ, MB, MN, and MO contributed to the content and writing. HJ and MB prepared the figure and table. All authors have read and approved the manuscript.
Funding
This study was supported by research grants from the Novo Nordic Foundation (NNF15OC0012345 and NNF16OC0023344), the Lundbeck Foundation (R151-2013-14432), and the Danish Council for Independent Research (DFF-4181-00447) to MO, and by a postdoctoral fellowship from the Lundbeck Foundation (R2017-134) to HJ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AP, action potential; CaM, calmodulin; CaV, voltage-gated calcium channel; CDI, calcium-dependent inactivation; CPVT, catecholaminergic polymorphic ventricular tachycardia; IVF, idiopathic ventricular fibrillation; LQTS, long QT syndrome; RyR2, ryanodine receptor isoform 2; SCD, sudden cardiac death; SR, sarcoplasmic reticulum.
References
Adelman, J. P., Maylie, J., and Sah, P. (2012). Small-conductance Ca 2+-activated K+channels: form and function. Annu. Rev. Physiol. 74, 245–269. doi: 10.1146/annurev-physiol-020911-153336
Berchtold, M. W., Zacharias, T., Kulej, K., Wang, K., Torggler, R., Jespersen, T., et al. (2016). The arrhythmogenic calmodulin mutation D129G dysregulates cell growth, calmodulin-dependent kinase II activity, and cardiac function in zebrafish. J. Biol. Chem. 291, 26636–26646. doi: 10.1074/jbc.M116.758680
Boczek, N. J., Gomez-Hurtado, N., Ye, D., Calvert, M. L., Tester, D. J., Kryshtal, D. O., et al. (2016). Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated calmodulin missense variant, E141G. Circ. Cardiovasc. Genet. 9, 136–146. doi: 10.1161/CIRCGENETICS.115.001323
Brehm, P., and Eckert, R. (1978). Calcium entry leads to inactivation of calcium channel in paramecium. Science. 202, 1203–1206. doi: 10.1126/science.103199
Chaix, M. A., Koopmann, T. T., Goyette, P., Alikashani, A., Latour, F., Fatah, M., et al. (2016). Novel CALM3 mutations in pediatric long QT syndrome patients support a CALM3-specific calmodulinopathy. Hear. case reports 2, 250–254. doi: 10.1016/j.hrcr.2016.02.002
Chattopadhyaya, R., Meador, W. E., Means, A. R., and Quiocho, F. A. (1992). Calmodulin structure refined at 1.7 a resolution. J. Mol. Biol. 228, 1177–1192. doi: 10.1016/0022-2836(92)90324-D
Crotti, L., Johnson, C. N., Graf, E., De Ferrari, G. M., Cuneo, B. F., Ovadia, M., et al. (2013). Calmodulin mutations associated with recurrent cardiac arrest in infants. circulation 127, 1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., Mcgowan-Jordan, J., et al. (2016). HGVS Recommendations for the description of sequence variants: 2016 Update. Hum. Mutat. 37, 564–569. doi: 10.1002/humu.22981
Fabiato, A. (1985). Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac purkinje cell. J. Gen. Physiol. 85, 247–289. doi: 10.1085/jgp.85.2.247
Fischer, R., Koller, M., Flura, M., Mathews, S., Strehler-Page, M. A., Krebs, J., et al. (1988). Multiple divergent mRNAs code for a single human calmodulin. J. Biol. Chem. 263, 17055–17062.
Ghosh, S., Nunziato, D. A., and Pitt, G. S. (2006). KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ. Res. 98, 1048–1054. doi: 10.1161/01.RES.0000218863.44140.f2
Gomez-Hurtado, N., Boczek, N. J., Kryshtal, D. O., Johnson, C. N., Sun, J., Nitu, F. R., et al. (2016). Novel CPVT-associated calmodulin mutation in CALM3 (CALM3-A103V) activates arrhythmogenic Ca waves and sparks. Circ. Arrhythmia Electrophysiol. 9:e004161. doi: 10.1161/CIRCEP.116.004161
Halling, D. B., Liebeskind, B. J., Hall, A. W., and Aldrich, R. W. (2016). Conserved properties of individual Ca 2+-binding sites in calmodulin. Proc. Natl. Acad. Sci. U.S.A. 113, E1216–E1225. doi: 10.1073/pnas.1600385113
Hwang, H. S., Nitu, F. R., Yang, Y., Walweel, K., Pereira, L., Johnson, C. N., et al. (2014). Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ. Res. 114, 1114–1124. doi: 10.1161/CIRCRESAHA.114.303391
Jiménez-Jáimez, J., Doza, J. P., Ortega, Á., Macías-Ruiz, R., Perin, F., Rodríguez-Vázquez Del Rey, M. M., et al. (2016). Calmodulin 2 mutation N98S is associated with unexplained cardiac arrest in infants due to low clinical penetrance electrical disorders. PLoS One 11:e0153851. doi: 10.1371/journal.pone.0153851
Kretsinger, R. H., Rudnick, S. E., and Weissman, L. J. (1986). Crystal structure of calmodulin. J. Inorg. Biochem. 28, 289–302. doi: 10.1016/0162-0134(86)80093-9
Landstrom, A. P., Dobrev, D., and Wehrens, X. H. T. (2017). Calcium signaling and cardiac arrhythmias. Circ. Res. 120, 1969–1993. doi: 10.1161/CIRCRESAHA.117.310083
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Limpitikul, W. B., Dick, I. E., Joshi-Mukherjee, R., Overgaard, M. T., George, A. L., and Yue, D. T. (2014). Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca2+ currents and promote proarrhythmic behavior in ventricular myocytes. J. Mol. Cell. Cardiol. 74, 115–124. doi: 10.1016/j.yjmcc.2014.04.022
Limpitikul, W. B., Dick, I. E., Tester, D. J., Boczek, N. J., Limphong, P., Yang, W., et al. (2017). A Precision medicine approach to the rescue of function on malignant calmodulinopathic long-QT syndromenovelty and significance. Circ. Res. 120, 39–48. doi: 10.1161/CIRCRESAHA.116.309283
Linse, S., Helmersson, A., and Forsen, S. (1991). Calcium binding to calmodulin and its globular domains. J. Biol. Chem. 266, 8050–8054.
Makita, N., Yagihara, N., Crotti, L., Johnson, C. N., Beckmann, B. M., Roh, M. S., et al. (2014). Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 7, 466–474. doi: 10.1161/CIRCGENETICS.113.000459
Marsman, R. F., Barc, J., Beekman, L., Alders, M., Dooijes, D., Van Den Wijngaard, A., et al. (2014). A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J. Am. Coll. Cardiol. 63, 259–266. doi: 10.1016/j.jacc.2013.07.091
Modell, S. M., and Lehmann, M. H. (2006). The long QT syndrome family of cardiac ion channelopathies: a huge review. Genet. Med. 8, 143–155. doi: 10.1097/01.gim.0000204468.85308.86
Nanou, E., and Catterall, W. A. (2018). Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron 98, 466–481. doi: 10.1016/j.neuron.2018.03.017
Nomikos, M., Thanassoulas, A., Beck, K., Vassilakopoulou, V., Hu, H., Calver, B. L., et al. (2014). Altered RyR2 regulation by the calmodulin F90L mutation associated with idiopathic ventricular fibrillation and early sudden cardiac death. FEBS Lett. 588, 2898–2902. doi: 10.1016/j.febslet.2014.07.007
Nyegaard, M., Demontis, D., Foldager, L., Hedemand, A., Flint, T. J., Sørensen, K. M., et al. (2010). CACNA1C (rs1006737) is associated with schizophrenia. Mol. Psychiatry 15, 119–121. doi: 10.1038/mp.2009.69
Nyegaard, M., Overgaard, M. T., Søndergaard, M. T., Vranas, M., Behr, E. R., Hildebrandt, L. L., et al. (2012). Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 91, 703–712. doi: 10.1016/j.ajhg.2012.08.015
O’Connell, D. J., Bauer, M. C., O’Brien, J., Johnson, W. M., Divizio, C. A., O’Kane, S. L., et al. (2010). Integrated protein array screening and high throughput validation of 70 novel neural calmodulin-binding proteins. Mol. Cell. Proteomics 9, 1118–1132. doi: 10.1074/mcp.M900324-MCP200
Peterson, B. Z., DeMaria, C. D., and Yue, D. T. (1999). Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22, 549–558. doi: 10.1016/S0896-6273(00)80709-6
Pipilas, D. C., Johnson, C. N., Webster, G., Schlaepfer, J., Fellmann, F., Sekarski, N., et al. (2016). Novel calmodulin mutations associated with congenital long QT syndrome affect calcium current in human cardiomyocytes. Hear. Rhythm 13, 2012–2019. doi: 10.1016/j.hrthm.2016.06.038
Reed, G. J., Boczek, N. J., Etheridge, S. P., and Ackerman, M. J. (2015). CALM3 mutation associated with long QT syndrome. Hear. Rhythm 12, 419-422. doi: 10.1016/j.hrthm.2014.10.035
Rocchetti, M., Sala, L., Dreizehnter, L., Crotti, L., Sinnecker, D., Mura, M., et al. (2017). Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 113, 531–541. doi: 10.1093/cvr/cvx006
Sasagawa, T., Ericsson, L. H., Walsh, K. A., Schreiber, W. E., Fischer, E. H., and Titani, K. (1982). Complete amino acid sequence of human brain calmodulin. Biochemistry 21, 2565–2569. doi: 10.1021/bi00539a041
Shen, X., Valencia, C. A., Szostak, J. W., Szostak, J. W., Dong, B., and Liu, R. (2005). Scanning the human proteome for calmodulin-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 5969–5974. doi: 10.1073/pnas.0407928102
Søndergaard, M. T., Liu, Y., Larsen, K. T., Nani, A., Tian, X., Holt, C., et al. (2017). The arrhythmogenic calmodulin p.Phe142Leu mutation impairs C-domain Ca 2+ binding but not calmodulin-dependent Inhibition of the cardiac ryanodine receptor. J. Biol. Chem. 292, 1385–1395. doi: 10.1074/jbc.M116.766253
Søndergaard, M. T., Sorensen, A. B., Skov, L. L., Kjaer-Sorensen, K., Bauer, M. C., Nyegaard, M., et al. (2015a). Calmodulin mutations causing catecholaminergic polymorphic ventricular tachycardia confer opposing functional and biophysical molecular changes. FEBS J. 282, 803–816. doi: 10.1111/febs.13184
Søndergaard, M. T., Tian, X., Liu, Y., Wang, R., Chazin, W. J., Chen, S. R. W., et al. (2015b). Arrhythmogenic calmodulin mutations affect the activation and termination of cardiac ryanodine receptor-mediated Ca 2+ release. J. Biol. Chem. 290, 26151–26162. doi: 10.1074/jbc.M115.676627
Sorensen, A. B., Søndergaard, M. T., and Overgaard, M. T. (2013). Calmodulin in a heartbeat. FEBS J. 280, 5511–5532. doi: 10.1111/febs.12337
Sun, J., and MacKinnon, R. (2017). Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 169, 1042.e9–1050.e9. doi: 10.1016/j.cell.2017.05.019
Takahashi, K., Ishikawa, T., Makita, N., Takefuta, K., Nabeshima, T., and Nakayashiro, M. (2017). A novel de novo calmodulin mutation in a 6-year-old boy who experienced an aborted cardiac arrest. Hear. Case Rep. 3, 69–72. doi: 10.1016/j.hrcr.2016.09.004
Tidow, H., and Nissen, P. (2013). Structural diversity of calmodulin binding to its target sites. FEBS J. 280, 5551–5565. doi: 10.1111/febs.12296
Vassilakopoulou, V., Calver, B. L., Thanassoulas, A., Beck, K., Hu, H., Buntwal, L., et al. (2015). Distinctive malfunctions of calmodulin mutations associated with heart RyR2-mediated arrhythmic disease. Biochim. Biophys. Acta. Gen. Subj. 1850, 2168–2176. doi: 10.1016/j.bbagen.2015.07.001
Xu, L., and Meissner, G. (2004). Mechanism of calmodulin inhibition of cardiac sarcoplasmic reticulum Ca2+ release channel (ryanodine receptor). Biophys. J. 86, 797–804. doi: 10.1016/S0006-3495(04)74155-7
Yamamoto, Y., Makiyama, T., Harita, T., Sasaki, K., Wuriyanghai, Y., Hayano, M., et al. (2017). Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum. Mol. Genet. 26, 1670–1677. doi: 10.1093/hmg/ddx073
Yin, G., Hassan, F., Haroun, A. R., Murphy, L. L., Crotti, L., Schwartz, P. J., et al. (2014). Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J. Am. Heart Assoc. 3:e000996. doi: 10.1161/JAHA.114.000996
Keywords: calmodulin, cardiac arrhythmia, calmodulinopathy, CPVT, LQTS, CALM1, CALM2, CALM3
Citation: Jensen HH, Brohus M, Nyegaard M and Overgaard MT (2018) Human Calmodulin Mutations. Front. Mol. Neurosci. 11:396. doi: 10.3389/fnmol.2018.00396
Received: 02 September 2018; Accepted: 11 October 2018;
Published: 13 November 2018.
Edited by:
Daniele Dell’Orco, Università degli Studi di Verona, ItalyReviewed by:
David James O’Connell, University College Dublin, IrelandStephan C. F. Neuhauss, Universität Zürich, Switzerland
Copyright © 2018 Jensen, Brohus, Nyegaard and Overgaard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael T. Overgaard, mto@bio.aau.dk