 Shalini S. Rao
Shalini S. Rao Paul Anthony Adlard
Paul Anthony Adlard- Division of Mental Health, The Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, VIC, Australia
There is an emerging link between the accumulation of iron in the brain and abnormal tau pathology in a number of neurodegenerative disorders, such as Alzheimer’s disease (AD). Studies have demonstrated that iron can regulate tau phosphorylation by inducing the activity of multiple kinases that promote tau hyperphosphorylation and potentially also by impacting protein phosphatase 2A activity. Iron is also reported to induce the aggregation of hyperphosphorylated tau, possibly through a direct interaction via a putative iron binding motif in the tau protein, facilitating the formation of neurofibrillary tangles (NFTs). Furthermore, in human studies high levels of iron have been reported to co-localize with tau in NFT-bearing neurons. These data, together with our own work showing that tau has a role in mediating cellular iron efflux, provide evidence supporting a critical tau:iron interaction that may impact both the symptomatic presentation and the progression of disease. Importantly, this may also have relevance for therapeutic directions, and indeed, the use of iron chelators such as deferiprone and deferoxamine have been reported to alleviate the phenotypes, reduce phosphorylated tau levels and stabilize iron regulation in various animal models. As these compounds are also moving towards clinical translation, then it is imperative that we understand the intersection between iron and tau in neurodegeneration. In this article, we provide an overview of the key pathological and biochemical interactions between tau and iron. We also review the role of iron and tau in disease pathology and the potential of metal-based therapies for tauopathies.
Introduction
Neurofibrillary tangles (NFTs) are a pathological hallmark of a class of disorders known as tauopathies, that includes Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), Parkinson’s disease (PD), Huntington’s disease (HD), Pick’s disease (PiD) and frontotemporal dementia with parkinsonism-17 (FTDP-17; Goedert et al., 2012). The primary constituent of NFTs is paired helical filaments (PHFs) composed of the hyperphosphorylated tau protein, an intrinsically unfolded and highly soluble phosphoprotein that is encoded by the MAPT gene (Weingarten et al., 1975; Spillantini and Goedert, 2013). Over the years, in vitro and in vivo studies have identified novel functions of tau in both normal physiology and disease, with reported roles in axonal transport, protein trafficking, cognitive function (Wang and Mandelkow, 2016) and interactions with proteins such as α-synuclein (the main component of Lewy Bodies (LBs) in PD; Lei et al., 2010) and amyloid-ß (Aß; the primary constituent of plaques found in AD; Bloom, 2014). Furthermore, it is well established that mutations in the MAPT gene are the primary cause of disease in FTDP-17 patients (Hutton et al., 1998). Whilst much is known about the regulation of tau, such as the effects of various kinases and phosphatases on the phosphorylation state of the protein which can then lead to conformational changes in protein structure (Zhu et al., 2015), a loss of tau function and the subsequent formation of PHFs and NFTs (Morris et al., 2011), there remains much to learn about this protein and the mechanisms through which it can influence both healthy and pathological aging. One such example is the emerging interaction between tau and iron. Dysregulation in cerebral iron is reported to be associated with the progression of tau-mediated neurodegeneration, with elevations in iron in affected brain regions that correlate both with the progression of neurodegeneration and the formation of NFTs in tauopathies (Andrasi et al., 1995; Duce et al., 2010). Further to this, reducing the burden of iron has been shown to alleviate behavioral deficits and reduce the formation of NFTs in animal models (Fine et al., 2012; Prasanthi et al., 2012; Guo et al., 2013a). In this review, we will provide a brief overview of the structure and function of tau and also iron metabolism, as these topics have been reviewed extensively. The main scope of this article is to revise the role of iron and tau in disease, to examine the proposed interaction between iron and tau and to review the potential of metal chelation as a therapeutic strategy for tauopathies.
Tau Physiology
In the human brain, tau is encoded by the MAPT gene on chromosome 17q21 and is predominately localized in the axons of neurons and to a lesser extent in dendrites and glial cells (Binder et al., 1985). The alternative splicing of MAPT generates six tau isoforms (as outlined in Figure 1), which differ by one or two 29 amino acid N-terminal inserts and by the number of microtubule binding domains (MBD; either 3 or 4, referred to as either 3R or 4R tau respectively; Goedert et al., 1989a,b; Andreadis et al., 1992). Under physiological conditions, 3R and 4R tau isoforms are expressed equally in most regions of the brain (Goedert and Jakes, 1990). However, alterations in this ratio (normally an increase in 4R tau) are observed in FTD-17 and PSP (Goode et al., 2000). Tau is unusually hydrophilic; as a phosphoprotein the longest tau isoform contains 84 putative phosphorylation sites, which includes 45 serines, 35 threonines and 4 tyrosines (Goedert et al., 1993; Chen et al., 2004; Götz et al., 2010; Mandelkow and Mandelkow, 2012). The structure of tau is subdivided into three domains, all of which are important in facilitating the interaction of tau with microtubules: (i) the projection domain located in the N-terminal region, determines the spacing between microtubules; (ii) the proline rich domain, comprised of up to seven PXXP motifs that are mainly serine-proline (SP) or threonine-proline (TP) motifs (targets of proline-directed kinases); and (iii) the assembly domain, located in the C-terminal half of the protein and consisting of the MBD, which interacts directly with microtubules (Weingarten et al., 1975; Cleveland et al., 1977; Steiner et al., 1990; Chen et al., 1992; Gustke et al., 1994; Lee et al., 1998). The assembly domain is of particular importance in understanding tau pathology as this region can seed and form PHFs (Pérez et al., 1996; Arrasate et al., 1999; Mukrasch et al., 2005). This characteristic of tau is quite counterintuitive to its physiological nature as tau is natively unfolded in solution and has low tendency for aggregation, as evidenced by various biophysical methods such as circular dichroism (CD; Schweers et al., 1994), nuclear magnetic resonance (NMR; Mukrasch et al., 2009) and small angle X-ray scattering (Mylonas et al., 2008). The MBD is also reported to interact with other proteins such as α-synuclein (Jensen et al., 1999), apolipoprotein E (ApoE; Strittmatter et al., 1994; Huang et al., 1995) and presenilin 1 (Takashima et al., 1998b). Though the function of tau is well established in microtubule stabilization and regulation, it is also involved in regulating axonal transport by interacting with p150, the major component of the dynein–activator complex dynactin (Magnani et al., 2007; Dixit et al., 2008). Further studies speculate a role for tau in neurotransmission via the Fyn pathway (Stamer et al., 2002; Ittner et al., 2010; Lei et al., 2012) and iron metabolism by the trafficking of APP (Lei et al., 2012). Tau function is regulated by its degree of phosphorylation, which modulates the binding of tau to microtubules and axonal transport (Sengupta et al., 1998; Abraha et al., 2000; Alonso Adel et al., 2004; Haase et al., 2004; Cuchillo-Ibanez et al., 2008). For example, phosphorylation of tau at Ser262, Thr231 and Ser235 inhibits its binding to microtubules by 35%, 25% and 10%, respectively (Sengupta et al., 1998), whilst phosphorylation at Thr231, Ser396 and Ser422 promotes aggregation of tau into filaments (Alonso Adel et al., 2004). As ~50% of tau phosphorylation sites are followed by proline; kinases of the proline-directed protein kinases (PDPK) family play a significant role in tau phosphorylation. The main PDPKs of tau include glycogen synthase kinase 3ß (GSK3ß), cyclin dependent kinase-5 (CDK5) and mitogen-activated protein kinases (MAPK). Phosphorylation sites not followed by proline are phosphorylated by non-PDPKs, include protein kinase A (PKA) and microtubule affinity regulating kinases (MARK; Dolan and Johnson, 2010). Tau phosphorylation sites are subject to the activity of more than one kinase as a form on inter-regulation between kinases (Martin et al., 2013). Indeed, a combination of PDPKs and non-PDPKS work synergistically to modulate tau phosphorylation under both physiological and pathological conditions (Martin et al., 2013). Candidate phosphatases of tau include serine/threonine protein phosphatase PP1, PP2A and PP2B (Hanger et al., 2009). PP2A is the main regulator of tau phosphorylation, which accounts for ~70% of tau phosphatase activity in the brain (Goedert et al., 1995; Wang et al., 1995, 1996). In a similar manner, PP2A activity is regulated by Pin 1, a phosphorylation dependent cis/trans isomerase which interacts with SP and TP motifs to regulate tau dephosphorylation (Liou et al., 2003). Though tau undergoes several post-translational modifications such as glycation, ubiquitination and truncation (Gong et al., 2005); aggregates of hyperphosphorylated tau are the primary components of NFTs and this pathway has been studied extensively in understanding tau dysregulation.
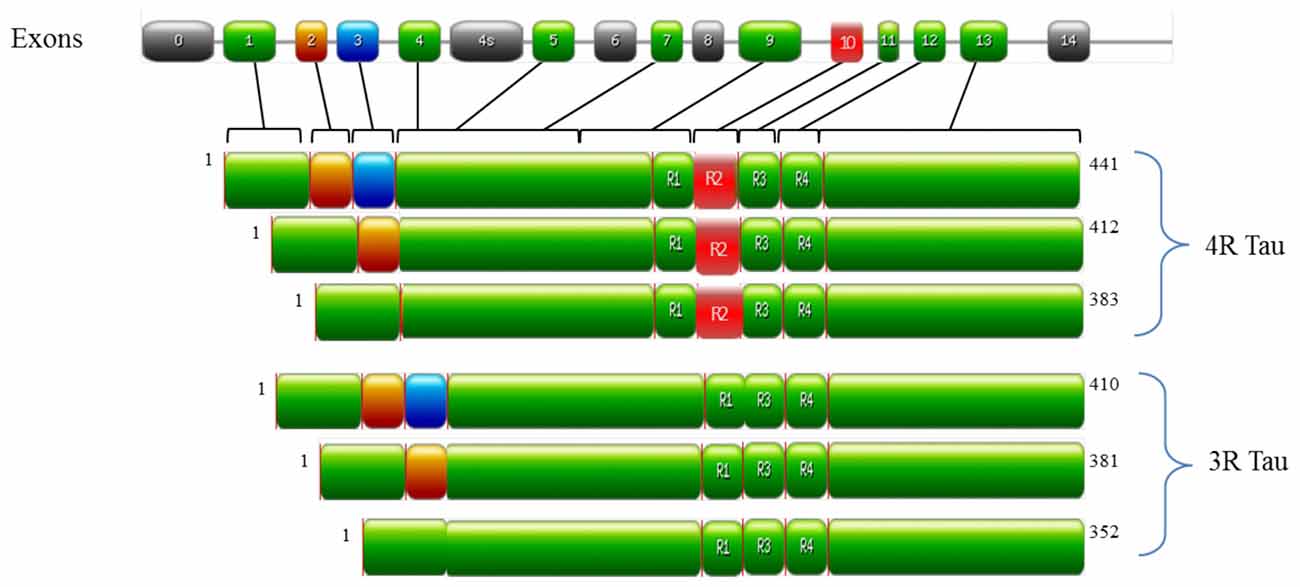
Figure 1. Structure of MAPT and the six tau isoforms expressed in an adult human brain. MAPT consists of 16 exons (E). Alternative mRNA splicing of E2 (orange), E3 (blue) and E10 (red) generates six tau isoforms ranging from 352–441 amino acids, The alternative splicing of additional exons (E1, E4, E5, E7, E9, E11, E12 and E13) are shown in green. E0, is part of the promoter and E14 are non-coding (gray). E6 and E8 (gray) are not transcribed in human brain. E4a (gray) is expressed only in the peripheral nervous system. The repeats motifs of tau (R1–R4) are shown, with three isoforms having four repeats each (4R Tau) and three isoforms having three repeats each (3R tau). Each repeat is 31 or 32 amino acids in length. Image was generated with PROSITE (Hulo et al., 2008).
Tau Pathology
Whilst the etiology of tau-mediated neurodegeneration is still unknown, it is generally accepted that tau hyperphosphorylation is one the key steps in disease progression which may lead to a “loss of tau function” and/or “gain of toxic function.” In familial tauopathies, MAPT mutations appear to be the primary cause of tau mediated neurodegeneration. However, in the case of sporadic tauopathies, the events or processes which trigger degeneration remain elusive.
Tau Mutations
The most direct cause and effect link is established between MAPT mutations and the development of FTDP-17, PSP HD and corticobasal degeneration (CBD; Clark et al., 1998; Coppola et al., 2012; Kouri et al., 2014). To date there are 80 mutations, of which 51 are missense mutations (that alter sequencing or splicing of tau), located within exons 9 to 13 of MAPT. A majority of the missense mutations are clustered around the MBD of tau and effectively reduce the binding affinity of tau to microtubules (such as G272V, P301L/S, V337M, G389R and R406W; Hasegawa et al., 1998; Hong et al., 1998) and potentially retard axonal transport (Zhang et al., 2004; Gilley et al., 2012). Furthermore, these mutations are reported to generate tau mutants more prone to aggregation (Alonso Adel et al., 2004), possibly by increasing the amount of free soluble tau (unbound tau), which is a favorable substrate for hyperphosphorylation (Sengupta et al., 2006). Other mutations cause a change in the 3R:4R tau ratio (N279K, ΔN296, N296N, N296H, S305N and S305S)—and this is typically an increase in 4R-tau (Hong et al., 1998; Delisle et al., 1999; Hasegawa et al., 1999; Spillantini et al., 2000; Stanford et al., 2000; Iseki et al., 2001). Despite the heterogeneity of tauopathies caused by MAPT mutations, they all appear to reflect the burden of tau pathology and cause degeneration in brain regions known to serve specific cognitive, executive or motor functions. The research linking MAPT mutations to the development of tauopathies has led to the generation of animal models over-expressing tau mutations which develop phenotypes and biochemical changes associated with tauopathies. As such, they are valuable tools in understanding the mechanisms of tau-mediated neurodegeneration.
A Balancing Act: Tau Hyperphosphorylation
Tau is highly phosphorylated in the later stages of neurodegeneration, which is speculated to be a result of an imbalance between kinase and phosphatase activity. Normal tau contains 2–3 moles of phosphate per mole of tau (Ksiezak-Reding et al., 1992), which is significantly increased in a range of human tauopathies (Ksiezak-Reding et al., 1992; Wray et al., 2008). As a majority of tau phosphorylation sites are followed by proline residues (Martin et al., 2011), it is not surprising that the activity of PDPK kinases such as GSK3β, CDK5 and MAPK are strongly linked to development of tau-mediated neurodegeneration. Indeed, these kinases are found to be associated with early tau deposits and tangles in brain samples from PSP, Pick’s Disease and AD (Pei et al., 1998; Perry et al., 1999; Ferrer et al., 2001a,b, 2002). Overexpression of GSK3β and the CDK-5 regulatory protein p25 in transgenic mouse models causes spatial learning deficits correlating with increased tau phosphorylation (Patrick et al., 1999; Lucas et al., 2001; Hernández et al., 2002; Noble et al., 2003; Li et al., 2004; de Barreda et al., 2010; Gómez de Barreda et al., 2010). Conversely suppression of tau kinases prevents tau hyperphosphorylation and neurodegeneration (Hong et al., 1997; Engel et al., 2006). These studies provide strong evidence for the aberrant activation of tau kinases in tauopathies. In addition, PP2A may drive tau hyperphosphorylation, as its activity is significantly compromised in AD brains and animal models of FTD (Liu et al., 2005; Tanimukai et al., 2005; Khandelwal et al., 2012). Tau dephosphorylation is achieved through the direct binding of PP2A to the MBD of tau (Sontag et al., 1999; Xu et al., 2008), as its hyperphosphorylation leads to a conformational change in its structure, this may prevent the binding of PP2A to tau. Furthermore, several MAPT mutations such as P301L and R406W are reported to reduce the binding affinity of PP2A to tau (Goedert et al., 2000). As a heterotrimeric holoenzyme, PP2A is comprised of a scaffolding A subunit, a variable regulatory B subunit and a catalytic C subunit. The activity of PP2A can be inhibited by the phosphorylation of the catalytic subunit at tyrosine-307 (Tyr307). Phosphorylated PP2A is found to co-localize with tangles in both AD and PD brains (Liu et al., 2008; Arif et al., 2014). The dysregulation of PP2A is also reported to modulate the activity of tau kinases mentioned above, both in vivo and in vitro (Pei et al., 2003; Wang et al., 2010; Louis et al., 2011). For example, PP2A dysregulation upregulates GSK3β activity, which may participate in inhibitory phosphorylation of PP2A (Yao et al., 2011). Conversely, the inhibition of GSK3β reverses PP2A inhibition and reduces phosphorylated tau levels in vitro (Martin et al., 2009). The phosphatase is also regulated by methylation; removal of the methyl group inactivates PP2A (Xing et al., 2008), which is also evidenced in AD brains (Sontag et al., 2004a,b). Dysregulation of methylated PP2A is also reported to be implicated in APP processing (Sontag et al., 2007); demethylated PP2A is found to drive Aβ induced neurotoxicity in vivo, whereas regulated PP2A methylation protected against neurodegeneration (Nicholls et al., 2016).
Tangled
Neurodegeneration attributed solely to tau dysfunction such as in PSP may be driven by MAPT mutations and dysregulation in tau phosphorylation. However, in other tauopathies such as AD and PD, neurodegeneration may be mediated through interactions with key disease-relevant proteins such as ApoE4, APP or α-synuclein. Senile plaques (comprised of Aβ) and NFTs are the hallmark pathology of AD, though the accumulation of plaques does not correlate with age-dependent cognitive impairment, NFT formation on the other hand strongly correlates with brain degeneration and cognitive function (Wilcock and Esiri, 1982; Arriagada et al., 1992; Itoh et al., 1996; Perez-Nievas et al., 2013). It is suggested, however, that Aβ may drive tau-mediated neurodegeneration. Injection of synthetic Aβ in brains of transgenic mice overexpressing the tau P301L mutation, increases NFT formation 5-fold in regions near the injection sites (Götz et al., 2001). Furthermore, Aβ deposits develop prior to tangle pathology; though NFT formation is more pronounced in mice co-expressing mutant APP and tau (Lewis et al., 2001; Hurtado et al., 2010), a similar profile is observed in mice expressing both mutant APP and wild type (WT) tau (Umeda et al., 2014). The knockdown of tau in APP/PS1 mice also eliminated AD-like pathology and prevented cognitive impairment (Leroy et al., 2012). Notably, Aβ is speculated to activate tau kinases GSK3β, CDK-5 and MAPK, leading to tau hyperphosphorylation (Takashima et al., 1996, 1998a; Ferreira et al., 1997) causing an increase in free soluble tau, which may be a critical component of Aβ induced neurotoxicity. In a Drosophila model of AD, administration of lithium (a GSK3β inhibitor) significantly reduced the effects of Aβ toxicity, decreasing tau phosphorylation and neuronal dysfunction (Folwell et al., 2010). Tau may also be necessary for Aβ-induced neurotoxicity; in transgenic mice expressing human APP, tau suppression resulted in an inhibition of Aβ production, and associated neuronal dysfunction (Roberson et al., 2007). Tau is also implicated in axonal transport deficits induced by Aβ via activation of GSK3β, impairing tau function and facilitating aberrant neuronal activity (Vossel et al., 2015). Further, in primary neurons from transgenic mice expressing familial AD-linked forms of human APP, suppression of tau abolished Aβ-induced GSK3β activation and thereby inhibited transport deficits (Vossel et al., 2015). The ApoE4 protein is a major genetic risk factor for AD and is associated with both senile plaques and NFTs in AD (Strittmatter et al., 1993). A recent study has demonstrated a significant effect of ApoE4 on tau-mediated neurodegeneration; in mice expressing the P301S tau mutation and ApoE4, a more severe phenotype and pathology was observed in an age dependent manner, compared to P301S mice with ApoE4 knocked down (Shi et al., 2017). In this model, the mislocalization of tau was evident as early as 3 months of age, a process which occurs through abnormal tau hyperphosphorylation. In another model expressing human ApoE4, phosphorylated levels of tau are found to correlate with ApoE4 in regions vulnerable to neurodegeneration, such as the hippocampus and cortex (Brecht et al., 2004). The interaction between ApoE4 and tau may arise through the ability of ApoE4 to modulate tau phosphorylation (Tesseur et al., 2000; Huang et al., 2001; Brecht et al., 2004). Transfection of ApoE4 in neural stem cells causes demethylation of the catalytic subunit of PP2A resulting in PP2A inactivation. Furthermore, ApoE4 is reported to modulate PP2A at the transcriptional level by regulating PP2A expression levels. These changes were also observed in post-mortem AD brain samples (Korwek et al., 2009; Theendakara et al., 2017). Thus, ApoE4 may drive tau hyperphosphorylation in familial AD, by modulating the regulatory pathways of tau and exacerbating tau-mediated neurodegeneration. In PD, α-synuclein is a pre-synaptic soluble protein that is strongly linked to pathology and is found to co-localize with tau in LBs (Arima et al., 1999; Ishizawa et al., 2003). Tau hyperphosphorylation is reported to be induced by PKA, which is triggered by the binding of α-synuclein to tau (Jensen et al., 1999). In transgenic mouse models expressing A53T human α-syn (a point mutation linked to autosomal dominant early onset PD) and in double transgenic mouse models expressing WT α-syn and P301L mutant tau, α-syn was reported to induce tau fibrillization (Giasson et al., 2003). In a Drosophila model of PD, α-syn and tau augmented neurotoxicity through the disruption of cytoskeletal organization, axonal transport and aberrant synaptic organization (Roy and Jackson, 2014). These data demonstrate that tau and other key disease proteins may work together in tauopathies, contributing to disease progression.
The Tau:Iron Interaction
The current view of tau-mediated neurodegeneration revolves around there being a loss of tau function. As outlined above, the aberrant activation of tau kinases and MAPT mutations appear the most causative reasons for tau dysregulation in tauopathies. However, there is a new perspective emerging speculating a pivotal role for iron in tau pathology. Elevated iron levels are found in brain regions which accumulate PHFs and NFTs; such as the cortex and hippocampus in AD (Andrasi et al., 1995; Duce et al., 2010) and the basal ganglia and brainstem in PSP (Pérez et al., 1998) and is predominately concentrated within PHFs and NFTs in AD and PSP (Good et al., 1992; Smith et al., 1997; Pérez et al., 1998). Iron exists in two oxidation states: ferrous (Fe2+) (redox active) and (Fe3+) (redox-inert state); Fe3+ is associated with NFTs in AD and PSP (Smith et al., 1997; Pérez et al., 1998; Yamamoto et al., 2002) and can induce the aggregation of hyperphosphorylated tau. Reducing Fe3+ to Fe2+ can reverse the aggregation of tau and solubilized tau species isolated from AD brains (Yamamoto et al., 2002). This interaction may occur via a putative iron binding motif located within the MBD of tau (Smith et al., 1997; Pérez et al., 1998)—Fe3+ binds strongly to His residues (as demonstrated in a series of NMR experiments, Nair et al., 2010) and modification of His residues in tau isolated from AD brain prevents the interaction between iron and tau (Smith et al., 1997). Recent electrochemical studies have reported a direct interaction using X-ray photoelectron spectroscopy and CD (Ahmadi et al., 2017). This study suggested that Fe2+ may mediate reversible conformational changes in tau to promote its aggregation through Thr residues, conversely Fe3+ interactions are less pronounced but irreversible as observed by CD. Furthermore, the hyperphosphorylation of tau may not affect the interaction between tau and Fe3+, but Thr phosphorylation can modulate interactions between Fe2+ and tau (Ahmadi et al., 2017), which may explain why previous studies have speculated no interaction between Fe and tau. As noted earlier, tau hyperphosphorylation is one of the key steps in NFT formation and it can be modulated by iron through the aberrant activation of tau kinases such as GSK3ß, CDK-5 and MAPK (Egaña et al., 2003; Lovell et al., 2004; Muñoz et al., 2006; Bautista et al., 2016). Though there is debate that this process may not be the primary cause of aggregation, as hyperphosphorylated tau is evident during animal hibernation and anesthesia-induced hypothermia (Arendt et al., 2003; Planel et al., 2007), the evidence suggests that iron may act as a co-factor for tau aggregation and the phosphorylated state of tau may induce conformational changes of the protein to mediate tau:iron interactions. Consequently, tau dysregulation has been implicated in iron dyshomeostasis, as intracellular iron accumulation has been linked to a loss of tau function (Duce et al., 2010; Lei et al., 2012). Tau-KO mice accumulate iron in the cortex, hippocampus and substantia nigra causing age-dependent neurodegeneration (Lei et al., 2012), which was the result of impaired trafficking of APP to the membrane, where it interacts with ferroportin to export iron from the cell.
Iron Metabolism
Iron is the most abundant transition metal in the brain and is vital for various neuronal functions such as oxygen transport and mitochondrial respiration (Wrigglesworth and Baum, 1988; Que et al., 2008). Iron metabolism is strictly regulated, with 1–2 mg of iron absorbed and excreted from our bodies daily (Bothwell et al., 1979). Total iron homeostasis is regulated via absorption in the gastrointestinal tract and a series of protein interactions in the crypts of Lieberkühn. These proteins include a product of the hemeochromatosis gene (HFE), transferrin (Tf) one of the major iron transporters in the body, transferrin receptor (TfR) and iron regulatory proteins (IRPs). Iron exists in two oxidation states: Fe2+ or Fe3+. Fe2+ is highly reactive and participates in many cellular functions such as myelin synthesis, neurotransmitter synthesis and oxidative metabolism, whereas Fe3+ is stored in ferritin (Ft, the main iron storage protein) in its redox-inert state (Que et al., 2008). Fe3+ binds to Tf to form holo-Tf, which allows it to be transported through the blood brain barrier and cell membranes via transferrin receptor-mediator endocytosis (Finch and Huebers, 1982; Swaiman and Machen, 1984; Andrews, 2000). The low pH of the endosome releases Tf bound iron and it is reduced to Fe2+ by the six-transmembrane epithelial antigen prostate 1–4 (STEAP 1–4) and released to the labile iron pool (LIP) in the cytosol via the divalent metal transporter 1 (DMT1; Andrews, 1999; Pelizzoni et al., 2012). It is then either transported to the mitochondria to supply iron for heme and iron-sulfur biosynthesis or stored in Ft (Eisenstein, 2000). Export of iron from the cell occurs through the intramembrane protein ferroportin (Ftn). It is stabilized through several molecular interactions that includes tau, ceruloplasmin (Cp) and APP (Figure 2). Iron homeostasis is tightly regulated by IRPs, which act as the cells iron sensor. IRPs bind to iron-response elements (IREs) located in the stem-loop structures on 3′ or 5′ of untranslated mRNA of proteins involved in iron metabolism such as those mentioned above. In cases of low cellular iron, IREs bind to the 3′ end of DMT1 and TfR to prevent degradation of the proteins and increase iron levels and bind to the 5′ end of Ft to prevent Ft synthesis. When iron is abundant in the cell, iron binds to IRPs and thereby causing the degradation of TfR and DMT1 and allowing the expression of Ft.
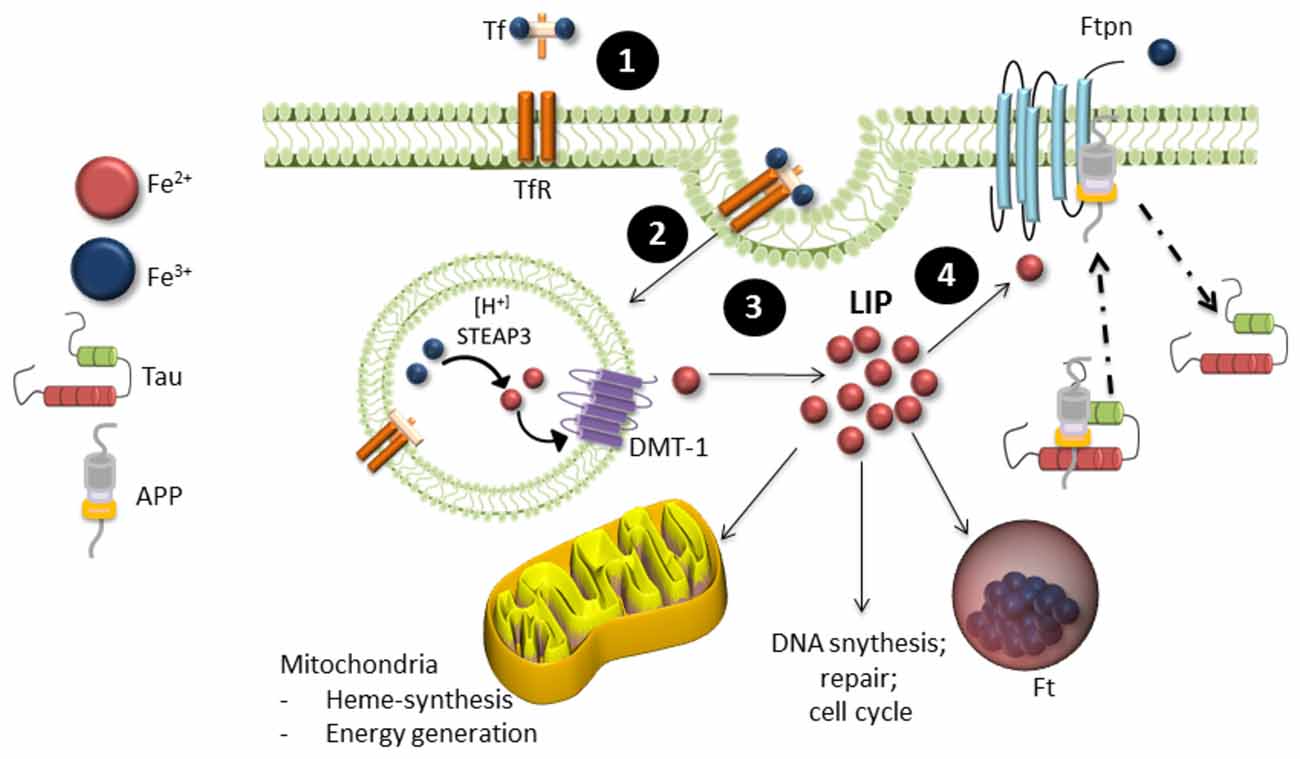
Figure 2. Iron metabolism in neurons. (1) Ferric (Fe3+) iron bound to transferrin (Tf) binds to transferrin receptors (TfR) on the cell surface, which undergoes (2). Endocytosis. (3) Internalization of Tf-TfR complex causes ferric iron to be released and quickly reduced by ferric reductase (STEAP3). Ferrous iron (Fe2+) is then transported across the endosomal membrane into the cytosol by the divalent metal transporter 1 (DMT-1) and is released in the labile iron pool (LIP). The LIP provides a source of iron available for neuronal function, where it is either stored in ferritin (Ft) in its ferric form or proceeds to the mitochondria for biological processing. (4) Iron is exported from neurons by ferroportin (Ftpn), which is stabilized by an unknown mechanism involving tau, ceruloplasmin and APP. Iron is then recirculated by apo-transferrin (not shown).
Iron in Neurodegeneration
Iron distribution in the brain is heterogeneous and certain regions such as the extrapyramidal regions (basal ganglia, substantia nigra and red nucleus) tend to accumulate more iron compared to other regions (Hallgren and Sourander, 1958). In the normal aging process iron accumulates predominantly in glial cells in the cortex, cerebellum, basal ganglia, substantia nigra, hippocampus and amygdala (Connor et al., 1992; Bartzokis et al., 2007). As such, brain iron accumulation is generally accepted to be a function of age, but whether it is the primary cause of neurodegenerative disorders or a result of brain degeneration is unknown. The role of iron in neurodegeneration is well documented in a class of disorders referred to as Neurodegeneration with Brain Iron Accumulation (NBIA), characterized by excess iron deposits in the extrapyramidal regions of the brain (Schneider and Bhatia, 2013). Interestingly, tau pathology is evident in the cortex, substantia nigra and globus pallidus of several NBIA cases (Arber et al., 2016), thereby demonstrating a causal link between iron dysregulation and tau. In tauopathies, several iron-associated proteins are reported to be augmented such a DMT-1, TfR and Ft. In PD, DMT-1 is upregulated in dopaminergic neurons in the substantia nigra (Qian and Wang, 1998) and increased Ft levels correlate with elevated iron levels in PSP and HD in postmortem brains (Dexter et al., 1992). In PSP and AD, tangles reportedly co-localize with Ft (Grundke-Iqbal et al., 1990; Pérez et al., 1998), suggesting that iron stored in Ft may facilitate tau aggregation, as Fe3+ induces tau aggregation (Yamamoto et al., 2002). Indeed, the predominant source of iron in tauopathies is speculated to be in the form of hemosiderin (Foroutan et al., 2013)—an iron storage complex comprised primarily of Ft and denatured Ft (Iancu, 1992). Iron dysregulation is hypothesized to be a source of oxidative stress in tauopathies, as the interaction between NFTs and iron is reported to act as a source for reactive-oxygen species (ROS) in neurons (Smith et al., 1997; Pérez et al., 1998; Sayre et al., 2000; Foroutan et al., 2013). Furthermore, iron is reported to interact with key disease-relevant proteins such as ApoE4, APP and α-synuclein, which as mentioned previously all interact with tau. Levels of CSF ApoE4 were reported to correlate strongly with CSF Ft (which reflects brain iron levels) in AD patients (Ayton et al., 2015), both of which are elevated in AD (Earley et al., 2000; Ayton et al., 2015). Iron is reported to upregulate intracellular ApoE in primary cortical neurons treated with iron (II) sulfate (Xu et al., 2016). In addition, carriers of the ApoE4 accumulate significantly higher levels of cortical iron, compared to patients with no ApoE4 in mild cognitive impairment (van Bergen et al., 2016). Together this data suggests ApoE4 mediated neurodegeneration is associated with brain iron levels, which in turn promotes NFT formation. Iron levels can also modulate APP expression; as a metalloprotein, increased iron levels lead to an increase in the expression of APP (Rogers et al., 2002), which may facilitate Aβ formation (Bandyopadhyay et al., 2006; Cho et al., 2010) and promote tau pathology. However, iron is also involved in the cleavage of APP via IRE within the protein and is found to modulate secretase cleavage of APP (Bodovitz et al., 1995; Bandyopadhyay et al., 2006). In rat primary cortical neurons, iron treatment resulted in the cellular retention of APP, thereby generating APP products susceptible to amyloidogenic processing in tauopathies (Chen et al., 2018). In PD, α-syn, (similar to APP) contains an IRE (Friedlich et al., 2007) and may therefore be similarly regulated by pathological iron overload. The aggregation of α-syn is also induced by Fe3+ through a direct interaction in the C-terminal region of the protein (Castellani et al., 2000; Giasson et al., 2000) and is postulated to be a source of ROS generation observed in PD (Levin et al., 2011).
Evidence For an Iron:Tau Interaction From Preclinical and Clinical Trials/Animal Models and Therapies
Iron chelation therapy has been effective in clinical iron overload disorders such as ß-thalassemia major reducing morbidity and mortality (Ladis et al., 2005). Thus, the use of iron chelators such as deferoxamine and deferiprone for the treatment of tauopathies is speculated to have promising outcomes for neurodegenerative disorders such as AD (Adlard and Bush, 2018). Indeed, animal models have provided valuable insights in assessing the efficacy of iron chelation therapy for tauopathies. In transgenic mice overexpressing the P301L tau mutation, intranasal administration of deferoxamine resulted in a significant decrease in GSK3β levels, though no changes in phosphorylated tau was observed (Fine et al., 2012). However, deferoxamine restored markers of oxidation and inflammation compared to age-matched controls (Fine et al., 2012). In APP/PS1 mice, deferoxamine improved cognitive function and reduced phosphorylated tau levels and the aberrant activity of GSK3β and CDK-5 (Guo et al., 2013b). In white male rabbits, AD pathology was induced through a cholesterol-enriched diet, followed by deferiprone administration. Iron chelation reportedly reduced phosphorylated tau levels and Aβ (Prasanthi et al., 2012). A more recent study in A53T mice (a model for PD), reported improved motor function following deferiprone treatment (Carboni et al., 2017). Clinical trials have gone some way to validate the results from these animal studies in demonstrating the potential benefits of iron chelation therapy for neurodegeneration. A small deferoxamine trial in AD patients was reported to slow down the clinical progression of dementia in AD over a 24-month period (Crapper McLachlan et al., 1991). The study included two sample groups: 23 patients in the non-treatment group and 25 patients treated with deferoxamine. The rate of clinical progression of AD was measured by several behavioral assessments which included the Wechsler Adult Intelligence Scale-revised, the Wechsler Memory Scale form 1, the Western Aphasia Battery and a recorded behavioral assessment conducted in the patient’s home that was used to assess daily activities such as motor skills, left/right orientation and counting money. The primary outcome from this study was that the rate of decline in non-treated patients was twice that of patients treated with deferoxamine, which was recorded as a decrease/increase in daily activities. However, a significant number of participants dropped out from the study—resulting in n = 14 non-treated and n = 21 treated patients—and many failed to complete neuropsychiatric evaluations after the baseline assessment. The principal finding then, was based upon the recorded behavioral assessment conducted in the patient’s home. Due to the small sample population and the limited neuropsychological assessments, then the implications from this trial were limited. Furthermore, the toxicity of deferoxamine was an issue, with 30% of patients developing side effects that included lethargy, anorexia, nausea and abdominal pain, which was reversed by briefly stopping deferoxamine treatment. This may be one reason why this trial was not followed up. Other reported side effects of long term deferoxamine treatment include, audiovisual impairments, hypersensitivity reactions (Bene et al., 1989) and potentially death. The authors concluded that there was a need to develop safe and effective chelators for therapeutic use. While the aim of this study was to reduce aluminum levels in AD patients, deferoxamine has the capacity to bind several metals such as zinc, copper and iron; the affinity for iron is six times higher than that of aluminum (Gotsbacher et al., 2017), therefore it is postulated that the effects seen in the study may have been a result of iron chelation. Indeed, targeting iron has been reported to have therapeutic benefits in clinical trials of PD patients (tau is also disrupted in PD and in animal models of PD). Phase I clinical trials with a delayed start paradigm of deferiprone (30 mg/kg/day) in PD patients showed significant clinical improvements. Assessment of motor indications at 6 and 12 months using the Unified PD Rate Scale (UPDRS) showed improvements in motor function in early start patients (n = 20) compared to delayed start patients (n = 19; Devos et al., 2014). Deferiprone was also found to reduce iron deposits in the substantia nigra and putamen as measured by MRI, correlating with reduced CSF-Ft levels. Importantly, this study demonstrated that deferiprone was able to target iron in a region-specific manner; MRI analysis found no alterations in iron in unaffected brain regions such as the caudate or pallidum and no adverse effects on systemic levels of metals as measured by serum levels of Ft, Tf and ceruloplasmin. Furthermore, this study demonstrated the safety and tolerability of iron chelation therapy over a long-term period, as it was conducted over 18 months. (Devos et al., 2014). A more recent Phase II clinical trial of deferiprone in PD patients reported reduced iron levels in dentate and caudate nucleus compared to placebo following deferiprone treatment (Martin-Bastida et al., 2017). However, no difference was observed in the substantia nigra or putamen at 6 months following 20 mg/kg or 30 mg/kg deferiprone compared to the placebo group. Furthermore, no statistically significant improvements in motor function as assessed by UPDRS were observed in this trial. While both Devos et al. (2014) and Martin-Bastida et al. (2017) used the same dose of deferiprone, the discrepancies between the studies may be due to the small sample size (n = 5–7/group) in the latter study. Evidently more research is required in understanding the utility of iron chelators in neurodegenerative disorders, though the above studies have highlighted the potential benefits of chelating iron in the clinical setting. While deferiprone is a compound that was created in 1982 (Kontoghiorghes, 1982) for the treatment of thalassaemia, FDA approval for therapeutic use of the compound was only obtained in 2011. Deferiprone and deferoxamine are both high affinity iron chelators (Kd ≈ 10−35 M; Kd ≈ 10−31 respectively; Origa et al., 2005). However unlike deferoxamine, deferiprone is relatively small in size, crosses the blood brain barrier (Baksi and Singh, 2017) and can redistribute iron to apotransferrin (Kontoghiorghe et al., 2013)—thereby preventing systemic iron loss. As mentioned previously however, potentially serious side effects can result from the use of high affinity chelators; whilst the toxicity of deferiprone is less than that of deferoxamine, clinical studies have reported joint pains and gastrointestinal symptoms in 21% of patients with thalassemia major (Sajid et al., 2009). Deferoxamine has also been reported to decrease iron absorption and its metabolites are reported to have chelation properties (Kontoghiorghes, 1990; Lee et al., 1993), which can potentially cause iron deficiency. Furthermore, high affinity chelators are required to compete for iron with endogenous proteins such as transferrin, lactoferrin and heme containing proteins (Kontoghiorghe and Kontoghiorghes, 2016). This may have implications for the pathways associated with the formation of these iron-protein complexes such as the transferrin-iron complex, which is one of the efficient antioxidant systems located in the blood stream (Kontoghiorghe et al., 2013); the affinity of deferoxamine for iron is greater than transferrin (Aisen et al., 1978) and can therefore potentially interfere with such physiological iron-protein interactions. These issues highlight the need for “moderate” and selective iron chelators, which target abnormal iron accumulation/localization. One such compound is PBT434, which is an orally bioavailable and moderate affinity iron chelator that was designed to inhibit the aggregation of alpha-synuclein and tau. This compound is currently in phase one human clinical trial for parkinsonian diseases and has been previously tested in mouse models of PD (Finkelstein et al., 2017). In the MPTP mouse model of PD, iron levels are elevated in the substantia nigra and correlate with the accumulation of aggregated α-syn. MPTP mice treated with PBT434 (30 mg/kg/day; oral gavage) had reduced iron levels in substantia nigra compared to untreated MPTP mice, in addition to increased levels of ferroportin, reduced α-syn aggregates and improved motor function (Finkelstein et al., 2017). This study demonstrates that moderate affinity iron chelators, which should confer a more favorable safety profile compared to high affinity compounds, are sufficient to produce significant positive biological effects in mouse models of neurodegeneration. Whether this translates to efficacy in human clinical trials, however, has yet to be determined.
Conclusion
Tau dysfunction in AD, PSP and FTD-17 (and potentially other diseases) coupled with elevated iron levels appear to drive neurodegeneration. Whilst the precise relationship between iron and tau is still unclear, there is accumulating evidence supporting the existence of a tau:iron interaction. Understanding this will help elucidate the extent to which this interaction contributes to neurodegeneration. Furthermore, given the multitude of disease-specific and other critical pathways that iron can modulate, then pharmacologically targeting this abundant metal ion may represent a tractable therapeutic approach for slowing the progression of neurodegeneration in a number of different neurological disorders as well as for promoting successful brain aging.
Author Contributions
SR wrote the manuscript and PA edited the manuscript.
Conflict of Interest Statement
PA is a shareholder and paid consultant for Prana Biotechnology.
The remaining author SR declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/14-3048r1). SR is supported by a Parkinson’s Victoria—Argyrou Family PhD Partner Scholarship.
References
Abraha, A., Ghoshal, N., Gamblin, T. C., Cryns, V., Berry, R. W., Kuret, J., et al. (2000). C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J. Cell Sci. 113, 3737–3745. Available online: http://jcs.biologists.org/content/113/21/3737.article-info
Adlard, P. A., and Bush, A. I. (2018). Metals and Alzheimer’s disease: how far have we come in the clinic? J. Alzheimers Dis. 62, 1369–1379. doi: 10.3233/JAD-170662
Ahmadi, S., Ebralidze, I. I., She, Z., and Kraatz, H. B. (2017). Electrochemical studies of tau protein-iron interactions—potential implications for Alzheimer’s disease. Electrochim. Acta 236, 384–393. doi: 10.1016/j.electacta.2017.03.175
Aisen, P., Leibman, A., and Zweier, J. (1978). Stoichiometric and site characteristics of the binding of iron to human transferrin. J. Biol. Chem. 253, 1930–1937.
Alonso Adel, C., Mederlyova, A., Novak, M., Grundke-Iqbal, I., and Iqbal, K. (2004). Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 279, 34873–34881. doi: 10.1074/jbc.M405131200
Andrasi, E., Farkas, E., Scheibler, H., Reffy, A., and Bezur, L. (1995). Al, Zn, Cu, Mn and Fe levels in brain in Alzheimer’s disease. Arch. Gerontol. Geriatr. 21, 89–97. doi: 10.1016/0167-4943(95)00643-y
Andreadis, A., Brown, W. M., and Kosik, K. S. (1992). Structure and novel exons of the human .tau. gene. Biochemistry 31, 10626–10633. doi: 10.1021/bi00158a027
Andrews, N. C. (1999). The iron transporter DMT1. Int. J. Biochem. Cell Biol. 31, 991–994. doi: 10.1016/S1357-2725(99)00065-5
Andrews, N. C. (2000). Iron homeostasis: insights from genetics and animal models. Nat. Rev. Genet. 1, 208–217. doi: 10.1038/35042073
Arber, C. E., Li, A., Houlden, H., and Wray, S. (2016). Review: insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol. Appl. Neurobiol. 42, 220–241. doi: 10.1111/nan.12242
Arendt, T., Stieler, J., Strijkstra, A. M., Hut, R. A., Rüdiger, J., Van der Zee, E. A., et al. (2003). Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J. Neurosci. 23, 6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003
Arif, M., Kazim, S. F., Grundke-Iqbal, I., Garruto, R. M., and Iqbal, K. (2014). Tau pathology involves protein phosphatase 2A in parkinsonism-dementia of Guam. Proc. Natl. Acad. Sci. U S A 111, 1144–1149. doi: 10.1073/pnas.1322614111
Arima, K., Hirai, S., Sunohara, N., Aoto, K., Izumiyama, Y., Uéda, K., et al. (1999). Cellular co-localization of phosphorylated tau- and NACP/α-synuclein-epitopes in lewy bodies in sporadic Parkinson’s disease and in dementia with Lewy bodies. Brain Res. 843, 53–61. doi: 10.1016/s0006-8993(99)01848-x
Arrasate, M., Pérez, M., Armas-Portela, R., and Avila, J. (1999). Polymerization of tau peptides into fibrillar structures. The effect of FTDP-17 mutations. FEBS Lett. 446, 199–202. doi: 10.1016/s0014-5793(99)00210-0
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639. doi: 10.1212/wnl.42.3.631
Ayton, S., Faux, N. G., Bush, A. I., and Alzheimer’s Disease Neuroimaging Initiative. (2015). Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 6:6760. doi: 10.1038/ncomms7760
Baksi, S., and Singh, N. (2017). α-Synuclein impairs ferritinophagy in the retinal pigment epithelium: Implications for retinal iron dyshomeostasis in Parkinson’s disease. Sci. Rep. 7:12843. doi: 10.1038/s41598-017-12862-x
Bandyopadhyay, S., Huang, X., Cho, H., Greig, N. H., Youdim, M. B., and Rogers, J. T. (2006). Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5′untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J. Neural Transm. Suppl. 71, 237–247. doi: 10.1007/978-3-211-33328-0_25
Bartzokis, G., Tishler, T. A., Lu, P. H., Villablanca, P., Altshuler, L. L., Carter, M., et al. (2007). Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiol. Aging 28, 414–423. doi: 10.1016/j.neurobiolaging.2006.02.005
Bautista, E., Vergara, P., and Segovia, J. (2016). Iron-induced oxidative stress activates AKT and ERK1/2 and decreases Dyrk1B and PRMT1 in neuroblastoma SH-SY5Y cells. J. Trace Elem. Med. Biol. 34, 62–69. doi: 10.1016/j.jtemb.2015.11.005
Bene, C., Manzler, A., Bene, D., and Kranias, G. (1989). Irreversible ocular toxicity from single “challenge” dose of deferoxamine. Clin. Nephrol. 31, 45–48.
Binder, L. I., Frankfurter, A., and Rebhun, L. I. (1985). The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378. doi: 10.1083/jcb.101.4.1371
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Bodovitz, S., Falduto, M. T., Frail, D. E., and Klein, W. L. (1995). Iron levels modulate α-secretase cleavage of amyloid precursor protein. J. Neurochem. 64, 307–315. doi: 10.1046/j.1471-4159.1995.64010307.x
Bothwell, T. H., Charlton, R., Cook, J., and Finch, C. (1979). Iron Metabolism in Man. Oxford: Blackwell.
Brecht, W. J., Harris, F. M., Chang, S., Tesseur, I., Yu, G. Q., Xu, Q., et al. (2004). Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 24, 2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004
Carboni, E., Tatenhorst, L., Tonges, L., Barski, E., Dambeck, V., Bahr, M., et al. (2017). Deferiprone rescues behavioral deficits induced by mild iron exposure in a mouse model of α-synuclein aggregation. Neuromolecular Med. 19, 309–321. doi: 10.1007/s12017-017-8447-9
Castellani, R. J., Siedlak, S. L., Perry, G., and Smith, M. A. (2000). Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 100, 111–114. doi: 10.1007/s004010050001
Chen, Y. T., Chen, W. Y., Huang, X. T., Xu, Y. C., and Zhang, H. Y. (2018). Iron dysregulates APP processing accompanying with sAPPα cellular retention and β-secretase inhibition in rat cortical neurons. Acta Pharmacol. Sin. 39, 177–183. doi: 10.1038/aps.2017.113
Chen, F., David, D., Ferrari, A., and Gotz, J. (2004). Posttranslational modifications of tau–role in human tauopathies and modeling in transgenic animals. Curr. Drug. Targets 5, 503–515. doi: 10.2174/1389450043345236
Chen, J., Kanai, Y., Cowan, N. J., and Hirokawa, N. (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360, 674–677. doi: 10.1038/360674a0
Cho, H. H., Cahill, C. M., Vanderburg, C. R., Scherzer, C. R., Wang, B., Huang, X., et al. (2010). Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 285, 31217–31232. doi: 10.1074/jbc.M110.149161
Clark, L. N., Poorkaj, P., Wszolek, Z., Geschwind, D. H., Nasreddine, Z. S., Miller, B., et al. (1998). Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc. Natl. Acad. Sci. U S A 95, 13103–13107. doi: 10.1073/pnas.95.22.13103
Cleveland, D. W., Hwo, S. Y., and Kirschner, M. W. (1977). Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 116, 207–225. doi: 10.1016/0022-2836(77)90213-3
Connor, J. R., Snyder, B. S., Beard, J. L., Fine, R. E., and Mufson, E. J. (1992). Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J. Neurosci. Res. 31, 327–335. doi: 10.1002/jnr.490310214
Coppola, G., Chinnathambi, S., Lee, J. J., Dombroski, B. A., Baker, M. C., Soto-Ortolaza, A. I., et al. (2012). Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum. Mol. Genet. 21, 3500–3512. doi: 10.1093/hmg/dds161
Crapper McLachlan, D. R., Dalton, A. J., Kruck, T. P., Bell, M. Y., Smith, W. L., Kalow, W., et al. (1991). Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308. doi: 10.1016/0140-6736(91)92978-b
Cuchillo-Ibanez, I., Seereeram, A., Byers, H. L., Leung, K. Y., Ward, M. A., Anderton, B. H., et al. (2008). Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 22, 3186–3195. doi: 10.1096/fj.08-109181
de Barreda, E. G., Pérez, M., Ramos, P. G., de Cristobal, J., Martín-Maestro, P., Morán, A., et al. (2010). Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol. Dis. 37, 622–629. doi: 10.1016/j.nbd.2009.11.017
Delisle, M.-B., Murrell, J. R., Richardson, R., Trofatter, J. A., Rascol, O., Soulages, X., et al. (1999). A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol. 98, 62–77. doi: 10.1007/s004010051052
Devos, D., Moreau, C., Devedjian, J. C., Kluza, J., Petrault, M., Laloux, C., et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 21, 195–210. doi: 10.1089/ars.2013.5593
Dexter, D. T., Jenner, P., Schapira, A. H., and Marsden, C. D. (1992). Alterations in levels of iron, ferritinm, and other trace metals in neurodegenerative diseases affecting the basal ganglia. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann. Neurol. 32, S94–S100. doi: 10.1002/ana.410320716
Dixit, R., Ross, J. L., Goldman, Y. E., and Holzbaur, E. L. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. doi: 10.1126/science.1152993
Dolan, P. J., and Johnson, G. V. (2010). The role of tau kinases in Alzheimer’s disease. Curr. Opin. Drug Discov. Devel. 13, 595–603.
Duce, J. A., Tsatsanis, A., Cater, M. A., James, S. A., Robb, E., Wikhe, K., et al. (2010). Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142, 857–867. doi: 10.1016/j.cell.2010.08.014
Earley, C. J., Connor, J. R., Beard, J. L., Malecki, E. A., Epstein, D. K., and Allen, R. P. (2000). Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology 54, 1698–1700. doi: 10.1212/wnl.54.8.1698
Egaña, J. T., Zambrano, C., Nuñez, M. T., Gonzalez-Billault, C., and Maccioni, R. B. (2003). Iron-induced oxidative stress modify tau phosphorylation patterns in hippocampal cell cultures. Biometals 16, 215–223. doi: 10.1023/A:1020727218493
Eisenstein, R. S. (2000). Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu. Rev. Nutr. 20, 627–662. doi: 10.1146/annurev.nutr.20.1.627
Engel, T., Goni-Oliver, P., Lucas, J. J., Avila, J., and Hernández, F. (2006). Chronic lithium administration to FTDP-17 tau and GSK-3β overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 99, 1445–1455. doi: 10.1111/j.1471-4159.2006.04139.x
Ferreira, A., Lu, Q., Orecchio, L., and Kosik, K. S. (1997). Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A β. Mol. Cell. Neurosci. 9, 220–234. doi: 10.1006/mcne.1997.0615
Ferrer, I., Barrachina, M., and Puig, B. (2002). Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 104, 583–591. doi: 10.1007/s00401-002-0587-8
Ferrer, I., Blanco, R., Carmona, M., Puig, B., Barrachina, M., Gomez, C., et al. (2001a). Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson’s disease and Dementia with Lewy bodies. J. Neural Transm. 108, 1383–1396. doi: 10.1007/s007020100015
Ferrer, I., Blanco, R., Carmona, M., Ribera, R., Goutan, E., Puig, B., et al. (2001b). Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 11, 144–158. doi: 10.1111/j.1750-3639.2001.tb00387.x
Finch, C. A., and Huebers, H. (1982). Perspectives in iron metabolism. N. Engl. J. Med. 306, 1520–1528. doi: 10.1056/NEJM198206243062504
Fine, J., Baillargeon, A., Renner, D., Hoerster, N., Tokarev, J., Colton, S., et al. (2012). Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1α, and phosphorylates GSK3β in P301L tau transgenic mice. Exp. Brain Res. 219, 381–390. doi: 10.1007/s00221-012-3101-0
Finkelstein, D. I., Billings, J. L., Adlard, P. A., Ayton, S., Sedjahtera, A., Masters, C. L., et al. (2017). The novel compound PBT434 prevents iron mediated neurodegeneration and α-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 5:53. doi: 10.1186/s40478-017-0456-2
Folwell, J., Cowan, C. M., Ubhi, K. K., Shiabh, H., Newman, T. A., Shepherd, D., et al. (2010). Aβ exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer’s disease. Exp. Neurol. 223, 401–409. doi: 10.1016/j.expneurol.2009.09.014
Foroutan, P., Murray, M. E., Fujioka, S., Schweitzer, K. J., Dickson, D. W., Wszolek, Z. K., et al. (2013). Progressive supranuclear palsy: high-field-strength MR microscopy in the human substantia nigra and globus pallidus. Radiology 266, 280–288. doi: 10.1148/radiol.12102273
Friedlich, A. L., Tanzi, R. E., and Rogers, J. T. (2007). The 5′-untranslated region of Parkinson’s disease α-synuclein messengerRNA contains a predicted iron responsive element. Mol. Psychiatry 12, 222–223. doi: 10.1038/sj.mp.4001937
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Giasson, B. I., Forman, M. S., Higuchi, M., Golbe, L. I., Graves, C. L., Kotzbauer, P. T., et al. (2003). Initiation and synergistic fibrillization of tau and α-synuclein. Science 300, 636–640. doi: 10.1126/science.1082324
Gilley, J., Seereeram, A., Ando, K., Mosely, S., Andrews, S., Kerschensteiner, M., et al. (2012). Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a “P301L” tau knockin mouse. Neurobiol. Aging 33, 621.e1–621.e15. doi: 10.1016/j.neurobiolaging.2011.02.014
Goedert, M., Ghetti, B., and Spillantini, M. G. (2012). Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006254. doi: 10.1101/cshperspect.a006254
Goedert, M., and Jakes, R. (1990). Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230.
Goedert, M., Jakes, R., Crowther, R. A., Six, J., Lübke, U., Vandermeeren, M., et al. (1993). The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Natl. Acad. Sci. U S A 90, 5066–5070. doi: 10.1073/pnas.90.11.5066
Goedert, M., Jakes, R., Qi, Z., Wang, J. H., and Cohen, P. (1995). Protein phosphatase 2A is the major enzyme in brain that dephosphorylates tau protein phosphorylated by proline-directed protein kinases or cyclic AMP-dependent protein kinase. Nature 65, 2804–2807. doi: 10.1046/j.1471-4159.1995.65062804.x
Goedert, M., Satumtira, S., Jakes, R., Smith, M. J., Kamibayashi, C., White, C. L., et al. (2000). Reduced binding of protein phosphatase 2A to tau protein with frontotemporal dementia and parkinsonism linked to chromosome 17 mutations. J. Neurochem. 75, 2155–2162. doi: 10.1046/j.1471-4159.2000.0752155.x
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989a). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526. doi: 10.1016/0896-6273(89)90210-9
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., and Crowther, R. A. (1989b). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399.
Gómez de Barreda, E., Pérez, M., Gómez Ramos, P., de Cristobal, J., Martín-Maestro, P., Morán, A., et al. (2010). Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol. Dis. 37, 622–629. doi: 10.1016/j.nbd.2009.11.017
Gong, C. X., Liu, F., Grundke-Iqbal, I., and Iqbal, K. (2005). Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural Transm. 112, 813–838. doi: 10.1007/s00702-004-0221-0
Goode, B. L., Chau, M., Denis, P. E., and Feinstein, S. C. (2000). Structural and functional differences between 3-repeat and 4-repeat tau isoforms. implications for normal tau function and the onset of neurodegenetative disease. J. Biol. Chem. 275, 38182–38189. doi: 10.1074/jbc.M007489200
Good, P. F., Perl, D. P., Bierer, L. M., and Schmeidler, J. (1992). Selective accumulation of aluminum and iron in the neurofibrillary tangles of Alzheimer’s disease: a laser microprobe (LAMMA) study. Ann. Neurol. 31, 286–292. doi: 10.1002/ana.410310310
Gotsbacher, M. P., Telfer, T. J., Witting, P. K., Double, K. L., Finkelstein, D. I., and Codd, R. (2017). Analogues of desferrioxamine B designed to attenuate iron-mediated neurodegeneration: synthesis, characterisation and activity in the MPTP-mouse model of Parkinson’s disease. Metallomics 9, 852–864. doi: 10.1039/c7mt00039a
Götz, J., Chen, F., van Dorpe, J., and Nitsch, R. M. (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ 42 fibrils. Science 293, 1491–1495. doi: 10.1126/science.1062097
Götz, J., Gladbach, A., Pennanen, L., van Eersel, J., Schild, A., David, D., et al. (2010). Animal models reveal role for tau phosphorylation in human disease. Biochim. Biophys. Acta 1802, 860–871. doi: 10.1016/j.bbadis.2009.09.008
Grundke-Iqbal, I., Fleming, J., Tung, Y. C., Lassmann, H., Iqbal, K., and Joshi, J. G. (1990). Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta Neuropathol. 81, 105–110. doi: 10.1007/bf00334497
Guo, C., Wang, T., Zheng, W., Shan, Z. Y., Teng, W. P., and Wang, Z. Y. (2013a). Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 562–575. doi: 10.1016/j.neurobiolaging.2012.05.009
Guo, C., Wang, P., Zhong, M. L., Wang, T., Huang, X. S., Li, J. Y., et al. (2013b). Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 62, 165–172. doi: 10.1016/j.neuint.2012.12.005
Gustke, N., Trinczek, B., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (1994). Domains of tau protein and interactions with microtubules. Biochemistry 33, 9511–9522. doi: 10.1021/bi00198a017
Haase, C., Stieler, J. T., Arendt, T., and Holzer, M. (2004). Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 88, 1509–1520. doi: 10.1046/j.1471-4159.2003.02287.x
Hallgren, B., and Sourander, P. (1958). The effect of age on the non-haemin iron in the human brain. J. Neurochem. 3, 41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x
Hanger, D. P., Anderton, B. H., and Noble, W. (2009). Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112–119. doi: 10.1016/j.molmed.2009.01.003
Hasegawa, M., Smith, M. J., and Goedert, M. (1998). Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 437, 207–210. doi: 10.1016/s0014-5793(98)01217-4
Hasegawa, M., Smith, M. J., Iijima, M., Tabira, T., and Goedert, M. (1999). FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett. 443, 93–96. doi: 10.1016/s0014-5793(98)01696-2
Hernández, F., Borrell, J., Guaza, C., Avila, J., and Lucas, J. J. (2002). Spatial learning deficit in transgenic mice that conditionally over-express GSK-3β in the brain but do not form tau filaments. J. Neurochem. 83, 1529–1533. doi: 10.1046/j.1471-4159.2002.01269.x
Hong, M., Chen, D. C., Klein, P. S., and Lee, V. M. (1997). Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 272, 25326–25332. doi: 10.1074/jbc.272.40.25326
Hong, M., Zhukareva, V., Vogelsberg-Ragaglia, V., Wszolek, Z., Reed, L., Miller, B. I., et al. (1998). Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282, 1914–1917. doi: 10.1126/science.282.5395.1914
Huang, Y., Liu, X. Q., Wyss-Coray, T., Brecht, W. J., Sanan, D. A., and Mahley, R. W. (2001). Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. U S A 98, 8838–8843. doi: 10.1073/pnas.151254698
Huang, D. Y., Weisgraber, K. H., Goedert, M., Saunders, A. M., Roses, A. D., and Strittmatter, W. J. (1995). ApoE3 binding to tau tandem repeat I is abolished by tau serine262 phosphorylation. Neurosci. Lett. 192, 209–212. doi: 10.1016/0304-3940(95)11649-h
Hulo, N., Bairoch, A., Bulliard, V., Cerutti, L., Cuche, B. A., de Castro, E., et al. (2008). The 20 years of PROSITE. Nucleic Acids Res. 36, D245–D249. doi: 10.1093/nar/gkm977
Hurtado, D. E., Molina-Porcel, L., Iba, M., Aboagye, A. K., Paul, S. M., Trojanowski, J. Q., et al. (2010). Aβ accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 177, 1977–1988. doi: 10.2353/ajpath.2010.100346
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Iancu, T. C. (1992). Ferritin and hemosiderin in pathological tissues. Electron Microsc. Rev. 5, 209–229. doi: 10.1016/0892-0354(92)90011-e
Iseki, E., Matsumura, T., Marui, W., Hino, H., Odawara, T., Sugiyama, N., et al. (2001). Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol. 102, 285–292.
Ishizawa, T., Mattila, P., Davies, P., Wang, D., and Dickson, D. W. (2003). Colocalization of tau and α-synuclein epitopes in Lewy bodies. J. Neuropathol. Exp. Neurol. 62, 389–397. doi: 10.1093/jnen/62.4.389
Itoh, Y., Yamada, M., Yoshida, R., Suematsu, N., Oka, T., Matsushita, M., et al. (1996). Dementia characterized by abundant neurofibrillary tangles and scarce senile plaques: a quantitative pathological study. Eur. Neurol. 36, 94–97. doi: 10.1159/000117216
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jensen, P. H., Hager, H., Nielsen, M. S., Hojrup, P., Gliemann, J., and Jakes, R. (1999). α-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 274, 25481–25489. doi: 10.1074/jbc.274.36.25481
Khandelwal, P. J., Dumanis, S. B., Herman, A. M., Rebeck, G. W., and Moussa, C. E. (2012). Wild type and P301L mutant Tau promote neuro-inflammation and α-Synuclein accumulation in lentiviral gene delivery models. Mol. Cell. Neurosci. 49, 44–53. doi: 10.1016/j.mcn.2011.09.002
Kontoghiorghe, C. N., and Kontoghiorghes, G. J. (2016). New developments and controversies in iron metabolism and iron chelation therapy. World J. Methodol. 6, 1–19. doi: 10.5662/wjm.v6.i1.1
Kontoghiorghe, C. N., Kolnagou, A., and Kontoghiorghes, G. J. (2013). Potential clinical applications of chelating drugs in diseases targeting transferrin-bound iron and other metals. Expert Opin. Investig. Drugs 22, 591–618. doi: 10.1517/13543784.2013.787408
Kontoghiorghes, G. J. (1982). The Design of Orally Active Iron Chelators for the Treatment of Thalassaemia. Colchester, UK: University of Essex.
Kontoghiorghes, G. J. (1990). Chelators affecting iron absorption in mice. Arzneimittelforschung 40, 1332–1335.
Korwek, K. M., Trotter, J. H., Ladu, M. J., Sullivan, P. M., and Weeber, E. J. (2009). ApoE isoform-dependent changes in hippocampal synaptic function. Mol. Neurodegener. 4:21. doi: 10.1186/1750-1326-4-21
Kouri, N., Carlomagno, Y., Baker, M., Liesinger, A. M., Caselli, R. J., Wszolek, Z. K., et al. (2014). Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol. 127, 271–282. doi: 10.1007/s00401-013-1193-7
Ksiezak-Reding, H., Liu, W. K., and Yen, S. H. (1992). Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res. 597, 209–219. doi: 10.1016/0006-8993(92)91476-u
Ladis, V., Chouliaras, G., Berdousi, H., Kanavakis, E., and Kattamis, C. (2005). Longitudinal study of survival and causes of death in patients with thalassemia major in Greece. Ann. N Y Acad. Sci. 1054, 445–450. doi: 10.1196/annals.1345.067
Lee, G., Newman, S. T., Gard, D. L., Band, H., and Panchamoorthy, G. (1998). Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 111, 3167–3177.
Lee, P., Mohammed, N., Marshall, L., Abeysinghe, R. D., Hider, R. C., Porter, J. B., et al. (1993). Intravenous infusion pharmacokinetics of desferrioxamine in thalassaemic patients. Drug Metab. Dispos. 21, 640–644.
Lei, P., Ayton, S., Finkelstein, D. I., Adlard, P. A., Masters, C. L., and Bush, A. I. (2010). Tau protein: relevance to Parkinson’s disease. Int. J. Biochem. Cell Biol. 42, 1775–1778. doi: 10.1016/j.biocel.2010.07.016
Lei, P., Ayton, S., Finkelstein, D. I., Spoerri, L., Ciccotosto, G. D., Wright, D. K., et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295. doi: 10.1038/nm.2613
Leroy, K., Ando, K., Laporte, V., Dedecker, R., Suain, V., Authelet, M., et al. (2012). Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 181, 1928–1940. doi: 10.1016/j.ajpath.2012.08.012
Levin, J., Högen, T., Hillmer, A. S., Bader, B., Schmidt, F., Kamp, F., et al. (2011). Generation of ferric iron links oxidative stress to α-synuclein oligomer formation. J. Parkinsons Dis. 1, 205–216. doi: 10.3233/JPD-2011-11040
Lewis, J., Dickson, D. W., Lin, W. L., Chisholm, L., Corral, A., Jones, G., et al. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. doi: 10.1126/science.1058189
Li, B., Ryder, J., Su, Y., Moore, S. A. Jr., Liu, F., Solenberg, P., et al. (2004). Overexpression of GSK3βS9A resulted in tau hyperphosphorylation and morphology reminiscent of pretangle-like neurons in the brain of PDGSK3β transgenic mice. Transgenic Res. 13, 385–396. doi: 10.1023/b:trag.0000040039.44899.6f
Liou, Y. C., Sun, A., Ryo, A., Zhou, X. Z., Yu, Z. X., Huang, H. K., et al. (2003). Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424, 556–561. doi: 10.1038/nature01832
Liu, F., Grundke-Iqbal, I., Iqbal, K., and Gong, C. X. (2005). Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 22, 1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x
Liu, R., Zhou, X. W., Tanila, H., Bjorkdahl, C., Wang, J. Z., Guan, Z. Z., et al. (2008). Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology. J. Cell. Mol. Med. 12, 241–257. doi: 10.1111/j.1582-4934.2008.00249.x
Louis, J. V., Martens, E., Borghgraef, P., Lambrecht, C., Sents, W., Longin, S., et al. (2011). Mice lacking phosphatase PP2A subunit PR61/B’delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3β. Proc. Natl. Acad. Sci. U S A 108, 6957–6962. doi: 10.1073/pnas.1018777108
Lovell, M. A., Xiong, S., Xie, C., Davies, P., and Markesbery, W. R. (2004). Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimers Dis. 6, 659–671; discussion 673–681. doi: 10.3233/jad-2004-6610
Lucas, J. J., Hernández, F., Gómez-Ramos, P., Morán, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J. 20, 27–39. doi: 10.1093/emboj/20.1.27
Magnani, E., Fan, J., Gasparini, L., Golding, M., Williams, M., Schiavo, G., et al. (2007). Interaction of tau protein with the dynactin complex. EMBO J. 26, 4546–4554. doi: 10.1038/sj.emboj.7601878
Mandelkow, E. M., and Mandelkow, E. (2012). Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2:a006247. doi: 10.1101/cshperspect.a006247
Martin, L., Latypova, X., and Terro, F. (2011). Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem. Int. 58, 458–471. doi: 10.1016/j.neuint.2010.12.023
Martin, L., Latypova, X., Wilson, C. M., Magnaudeix, A., Perrin, M. L., Yardin, C., et al. (2013). Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res. Rev. 12, 289–309. doi: 10.1016/j.arr.2012.06.003
Martin, L., Magnaudeix, A., Esclaire, F., Yardin, C., and Terro, F. (2009). Inhibition of glycogen synthase kinase-3β downregulates total tau proteins in cultured neurons and its reversal by the blockade of protein phosphatase-2A. Brain Res. 1252, 66–75. doi: 10.1016/j.brainres.2008.11.057
Martin-Bastida, A., Ward, R. J., Newbould, R., Piccini, P., Sharp, D., Kabba, C., et al. (2017). Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 7:1398. doi: 10.1038/s41598-017-01402-2
Morris, M., Maeda, S., Vossel, K., and Mucke, L. (2011). The many faces of tau. Neuron 70, 410–426. doi: 10.1016/j.neuron.2011.04.009
Mukrasch, M. D., Bibow, S., Korukottu, J., Jeganathan, S., Biernat, J., Griesinger, C., et al. (2009). Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 7:e34. doi: 10.1371/journal.pbio.1000034
Mukrasch, M. D., Biernat, J., von Bergen, M., Griesinger, C., Mandelkow, E., and Zweckstetter, M. (2005). Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 280, 24978–24986. doi: 10.1074/jbc.M501565200
Muñoz, P., Zavala, G., Castillo, K., Aguirre, P., Hidalgo, C., and Núñez, M. T. (2006). Effect of iron on the activation of the MAPK/ERK pathway in PC12 neuroblastoma cells. Biol. Res. 39, 189–190. doi: 10.4067/s0716-97602006000100021
Mylonas, E., Hascher, A., Bernadó, P., Blackledge, M., Mandelkow, E., and Svergun, D. I. (2008). Domain conformation of tau protein studied by solution small-angle X-ray scattering. Biochemistry 47, 10345–10353. doi: 10.1021/bi800900d
Nair, N. G., Perry, G., Smith, M. A., and Reddy, V. P. (2010). NMR studies of zinc, copper, and iron binding to histidine, the principal metal ion complexing site of amyloid-β peptide. J. Alzheimers Dis. 20, 57–66. doi: 10.3233/JAD-2010-1346
Nicholls, R. E., Sontag, J. M., Zhang, H., Staniszewski, A., Yan, S., Kim, C. Y., et al. (2016). PP2A methylation controls sensitivity and resistance to β-amyloid-induced cognitive and electrophysiological impairments. Proc. Natl. Acad. Sci. U S A 113, 3347–3352. doi: 10.1073/pnas.1521018113
Noble, W., Olm, V., Takata, K., Casey, E., Mary, O., Meyerson, J., et al. (2003). Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38, 555–565. doi: 10.1016/s0896-6273(03)00259-9
Origa, R., Bina, P., Agus, A., Crobu, G., Defraia, E., Dessì, C., et al. (2005). Combined therapy with deferiprone and desferrioxamine in thalassemia major. Haematologica 90, 1309–1314. Available online: http://www.haematologica.org/content/90/10/1309.article-info
Patrick, G. N., Zukerberg, L., Nikolic, M., de la Monte, S., Dikkes, P., and Tsai, L. H. (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. doi: 10.1038/45159
Pei, J. J., Gong, C. X., An, W. L., Winblad, B., Cowburn, R. F., Grundke-Iqbal, I., et al. (2003). Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2 and p70 S6, similar to that in Alzheimer’s disease. Am. J. Pathol. 163, 845–858. doi: 10.1016/s0002-9440(10)63445-1
Pei, J. J., Grundke-Iqbal, I., Iqbal, K., Bogdanovic, N., Winblad, B., and Cowburn, R. F. (1998). Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res. 797, 267–277. doi: 10.1016/s0006-8993(98)00296-0
Pelizzoni, I., Zacchetti, D., Smith, C. P., Grohovaz, F., and Codazzi, F. (2012). Expression of divalent metal transporter 1 in primary hippocampal neurons: reconsidering its role in non-transferrin-bound iron influx. J. Neurochem. 120, 269–278. doi: 10.1111/j.1471-4159.2011.07578.x
Pérez, M., Valpuesta, J. M., de Garcini, E. M., Quintana, C., Arrasate, M., Carrascosa, J. L., et al. (1998). Ferritin is associated with the aberrant tau filaments present in progressive supranuclear palsy. Am. J. Pathol. 152, 1531–1539.
Pérez, M., Valpuesta, J. M., Medina, M., Montejo de Garcini, E., and Avila, J. (1996). Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J. Neurochem. 67, 1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x
Perez-Nievas, B. G., Stein, T. D., Tai, H. C., Dols-Icardo, O., Scotton, T. C., Barroeta-Espar, I., et al. (2013). Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526. doi: 10.1093/brain/awt171
Perry, G., Roder, H., Nunomura, A., Takeda, A., Friedlich, A. L., Zhu, X., et al. (1999). Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 10, 2411–2415. doi: 10.1097/00001756-199908020-00035
Planel, E., Richter, K. E., Nolan, C. E., Finley, J. E., Liu, L., Wen, Y., et al. (2007). Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J. Neurosci. 27, 3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007
Prasanthi, J. R., Schrag, M., Dasari, B., Marwarha, G., Dickson, A., Kirsch, W. M., et al. (2012). Deferiprone reduces amyloid-β and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol-enriched diet. J. Alzheimers Dis. 30, 167–182. doi: 10.3233/JAD-2012-111346
Qian, Z. M., and Wang, Q. (1998). Expression of iron transport proteins and excessive iron accumulation in the brain in neurodegenerative disorders. Brain Res. Rev. 27, 257–267. doi: 10.1016/s0165-0173(98)00012-5
Que, E. L., Domaille, D. W., and Chang, C. J. (2008). Metals in neurobiology: probing their chemistry and biology with molecular imaging. Chem. Rev. 108, 1517–1549. doi: 10.1021/cr078203u
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rogers, J. T., Randall, J. D., Cahill, C. M., Eder, P. S., Huang, X., Gunshin, H., et al. (2002). An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 277, 45518–45528. doi: 10.1074/jbc.m207435200
Roy, B., and Jackson, G. R. (2014). Interactions between Tau and α-synuclein augment neurotoxicity in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 23, 3008–3023. doi: 10.1093/hmg/ddu011
Sajid, R., Ghani, F., Adil, S., and Khurshid, M. (2009). Oral iron chelation therapy with deferiprone in patients with Thalassemia Major. J. Pak. Med. Assoc. 59, 388–390. Available online: https://ecommons.aku.edu/cgi/viewcontent.cgi?referer=http://scholar.google.com.au/&httpsredir=1&article=1037&context=pakistan_fhs_mc_pathol_microbiol
Sayre, L. M., Perry, G., Harris, P. L., Liu, Y., Schubert, K. A., and Smith, M. A. (2000). in situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J. Neurochem. 74, 270–279. doi: 10.1046/j.1471-4159.2000.0740270.x
Schneider, S. A., and Bhatia, K. P. (2013). Excess iron harms the brain: the syndromes of neurodegeneration with brain iron accumulation (NBIA). J. Neural Transm. 120, 695–703. doi: 10.1007/s00702-012-0922-8
Schweers, O., Schönbrunn-Hanebeck, E., Marx, A., and Mandelkow, E. (1994). Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for β-structure. J. Biol. Chem. 269, 24290–24297.
Sengupta, A., Grundke-Iqbal, I., and Iqbal, K. (2006). Regulation of phosphorylation of tau by protein kinases in rat brain. Neurochem. Res. 31, 1473–1480. doi: 10.1007/s11064-006-9205-9
Sengupta, A., Kabat, J., Novak, M., Wu, Q., Grundke-Iqbal, I., and Iqbal, K. (1998). Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 357, 299–309. doi: 10.1006/abbi.1998.0813
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Smith, M. A., Harris, P. L., Sayre, L. M., and Perry, G. (1997). Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U S A 94, 9866–9868. doi: 10.1073/pnas.94.18.9866
Sontag, E., Hladik, C., Montgomery, L., Luangpirom, A., Mudrak, I., Ogris, E., et al. (2004a). Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 63, 1080–1091. doi: 10.1093/jnen/63.10.1080
Sontag, E., Luangpirom, A., Hladik, C., Mudrak, I., Ogris, E., Speciale, S., et al. (2004b). Altered expression levels of the protein phosphatase 2A ABαC enzyme are associated with Alzheimer disease pathology. J. Neuropathol. Exp. Neurol. 63, 287–301. doi: 10.1093/jnen/63.4.287
Sontag, E., Nunbhakdi-Craig, V., Lee, G., Brandt, R., Kamibayashi, C., Kuret, J., et al. (1999). Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem. 274, 25490–25498. doi: 10.1074/jbc.274.36.25490
Sontag, E., Nunbhakdi-Craig, V., Sontag, J. M., Diaz-Arrastia, R., Ogris, E., Dayal, S., et al. (2007). Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci. 27, 2751–2759. doi: 10.1523/JNEUROSCI.3316-06.2007
Spillantini, M. G., and Goedert, M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. doi: 10.1016/S1474-4422(13)70090-5
Spillantini, M. G., Yoshida, H., Rizzini, C., Lantos, P. L., Khan, N., Rossor, M. N., et al. (2000). A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann. Neurol. 48, 939–943. doi: 10.1002/1531-8249(200012)48:6<939::aid-ana17>3.3.co;2-t
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2002). Tau blocks traffic of organelles, neurofilaments and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063. doi: 10.1083/jcb.200108057
Stanford, P. M., Halliday, G. M., Brooks, W. S., Kwok, J. B., Storey, C. E., Creasey, H., et al. (2000). Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene. Brain 123, 880–893. doi: 10.1093/brain/123.5.880
Steiner, B., Mandelkow, E. M., Biernat, J., Gustke, N., Meyer, H. E., Schmidt, B., et al. (1990). Phosphorylation of microtubule-associated protein tau: identification of the site for Ca2+-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J. 9, 3539–3544.
Strittmatter, W. J., Saunders, A. M., Goedert, M., Weisgraber, K. H., Dong, L. M., Jakes, R., et al. (1994). Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc. Natl. Acad. Sci. U S A 91, 11183–11186. doi: 10.1073/pnas.91.23.11183
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993). Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U S A 90, 1977–1981. doi: 10.1073/pnas.90.5.1977
Swaiman, K. F., and Machen, V. L. (1984). Iron uptake by mammalian cortical neurons. Ann. Neurol. 16, 66–70. doi: 10.1002/ana.410160113
Takashima, A., Honda, T., Yasutake, K., Michel, G., Murayama, O., Murayama, M., et al. (1998a). Activation of tau protein kinase I/glycogen synthase kinase-3β by amyloid β peptide (25–35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res. 31, 317–323. doi: 10.1016/s0168-0102(98)00061-3
Takashima, A., Murayama, M., Murayama, O., Kohno, T., Honda, T., Yasutake, K., et al. (1998b). Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc. Natl. Acad. Sci. U S A 95, 9637–9641. doi: 10.1073/pnas.95.16.9637
Takashima, A., Noguchi, K., Michel, G., Mercken, M., Hoshi, M., Ishiguro, K., et al. (1996). Exposure of rat hippocampal neurons to amyloid β peptide (25–35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 β. Neurosci. Lett. 203, 33–36. doi: 10.1016/0304-3940(95)12257-5
Tanimukai, H., Grundke-Iqbal, I., and Iqbal, K. (2005). Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am. J. Pathol. 166, 1761–1771. doi: 10.1016/s0002-9440(10)62486-8
Tesseur, I., Van Dorpe, J., Spittaels, K., Van den Haute, C., Moechars, D., and Van Leuven, F. (2000). Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am. J. Pathol. 156, 951–964. doi: 10.1016/s0002-9440(10)64963-2
Theendakara, V., Bredesen, D. E., and Rao, R. V. (2017). Downregulation of protein phosphatase 2A by apolipoprotein E: Implications for Alzheimer’s disease. Mol. Cell. Neurosci. 83, 83–91. doi: 10.1016/j.mcn.2017.07.002
Umeda, T., Maekawa, S., Kimura, T., Takashima, A., Tomiyama, T., and Mori, H. (2014). Neurofibrillary tangle formation by introducing wild-type human tau into APP transgenic mice. Acta Neuropathol. 127, 685–698. doi: 10.1007/s00401-014-1259-1
van Bergen, J. M., Li, X., Hua, J., Schreiner, S. J., Steininger, S. C., Quevenco, F. C., et al. (2016). Colocalization of cerebral iron with Amyloid β in Mild Cognitive Impairment. Sci. Rep. 6:35514. doi: 10.1038/srep35514
Vossel, K. A., Xu, J. C., Fomenko, V., Miyamoto, T., Suberbielle, E., Knox, J. A., et al. (2015). Tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. J. Cell Biol. 209, 419–433. doi: 10.1083/jcb.201407065
Wang, X., Blanchard, J., Kohlbrenner, E., Clement, N., Linden, R. M., Radu, A., et al. (2010). The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J. 24, 4420–4432. doi: 10.1096/fj.10-158477
Wang, J. Z., Gong, C. X., Zaidi, T., Grundke-Iqbal, I., and Iqbal, K. (1995). Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J. Biol. Chem. 270, 4854–4860. doi: 10.1074/jbc.270.9.4854
Wang, J. Z., Grundke-Iqbal, I., and Iqbal, K. (1996). Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Mol. Brain Res. 38, 200–208. doi: 10.1016/0169-328x(95)00316-k
Wang, Y., and Mandelkow, E. (2016). Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21. doi: 10.1038/nrn.2015.1
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U S A 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Wilcock, G. K., and Esiri, M. M. (1982). Plaques, tangles and dementia. A quantitative study. J. Neurol. Sci. 56, 343–356. doi: 10.1016/0022-510x(82)90155-1
Wray, S., Saxton, M., Anderton, B. H., and Hanger, D. P. (2008). Direct analysis of tau from PSP brain identifies new phosphorylation sites and a major fragment of N-terminally cleaved tau containing four microtubule-binding repeats. J. Neurochem. 105, 2343–2352. doi: 10.1111/j.1471-4159.2008.05321.x
Wrigglesworth, J. M., and Baum, H. (1988). “Iron-dependent enzymes in the brain,” in Neurochemical and Behavioural Aspects, ed. M. B. H. Youdinl (New York, NY: Taylor & Francis), 25.
Xing, Y., Li, Z., Chen, Y., Stock, J. B., Jeffrey, P. D., and Shi, Y. (2008). Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell 133, 154–163. doi: 10.1016/j.cell.2008.02.041
Xu, Y., Chen, Y., Zhang, P., Jeffrey, P. D., and Shi, Y. (2008). Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol. Cell 31, 873–885. doi: 10.1016/j.molcel.2008.08.006
Xu, H., Perreau, V. M., Dent, K. A., Bush, A. I., Finkelstein, D. I., and Adlard, P. A. (2016). Iron regulates apolipoprotein E expression and secretion in neurons and astrocytes. J. Alzheimers Dis. 51, 471–487. doi: 10.3233/jad-150797
Yamamoto, A., Shin, R. W., Hasegawa, K., Naiki, H., Sato, H., Yoshimasu, F., et al. (2002). Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 82, 1137–1147. doi: 10.1046/j.1471-4159.2002.t01-1-01061.x
Yao, X. Q., Zhang, X. X., Yin, Y. Y., Liu, B., Luo, D. J., Liu, D., et al. (2011). Glycogen synthase kinase-3β regulates Tyr307 phosphorylation of protein phosphatase-2A via protein tyrosine phosphatase 1B but not Src. Biochem. J. 437, 335–344. doi: 10.1042/BJ20110347
Zhang, B., Higuchi, M., Yoshiyama, Y., Ishihara, T., Forman, M. S., Martinez, D., et al. (2004). Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J. Neurosci. 24, 4657–4667. doi: 10.1523/JNEUROSCI.0797-04.2004
Keywords: tau, iron, Alzheimer’s disease, metal, deferiprone
Citation: Rao SS and Adlard PA (2018) Untangling Tau and Iron: Exploring the Interaction Between Iron and Tau in Neurodegeneration. Front. Mol. Neurosci. 11:276. doi: 10.3389/fnmol.2018.00276
Received: 28 May 2018; Accepted: 20 July 2018;
Published: 17 August 2018.
Edited by:
Andreas Martin Grabrucker, University of Limerick, IrelandReviewed by:
Jorge Busciglio, University of California, Irvine, United StatesMarta Bolós, Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), Spain
Copyright © 2018 Rao and Adlard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Anthony Adlard, paul.adlard@florey.edu.au