 Haitao Wang
Haitao Wang Jiangping Xu
Jiangping Xu Philip Lazarovici
Philip Lazarovici Remi Quirion
Remi Quirion Wenhua Zheng
Wenhua Zheng- 1Department of Neuropharmacology and Drug Discovery, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou, China
- 2School of Pharmacy Institute for Drug Research, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem, Israel
- 3Douglas Mental Health University Institute, McGill University, Montreal, QC, Canada
- 4Faculty of Health Sciences, University of Macau, Taipa, China
Dopamine is a brain neurotransmitter involved in the pathology of schizophrenia. The dopamine hypothesis states that, in schizophrenia, dopaminergic signal transduction is hyperactive. The cAMP-response element binding protein (CREB) is an intracellular protein that regulates the expression of genes that are important in dopaminergic neurons. Dopamine affects the phosphorylation of CREB via G protein-coupled receptors. Neurotrophins, such as brain derived growth factor (BDNF), are critical regulators during neurodevelopment and synaptic plasticity. The CREB is one of the major regulators of neurotrophin responses since phosphorylated CREB binds to a specific sequence in the promoter of BDNF and regulates its transcription. Moreover, susceptibility genes associated with schizophrenia also target and stimulate the activity of CREB. Abnormalities of CREB expression is observed in the brain of individuals suffering from schizophrenia, and two variants (-933T to C and -413G to A) were found only in schizophrenic patients. The CREB was also involved in the therapy of animal models of schizophrenia. Collectively, these findings suggest a link between CREB and the pathophysiology of schizophrenia. This review provides an overview of CREB structure, expression, and biological functions in the brain and its interaction with dopamine signaling, neurotrophins, and susceptibility genes for schizophrenia. Animal models in which CREB function is modulated, by either overexpression of the protein or knocked down through gene deletion/mutation, implicating CREB in schizophrenia and antipsychotic drugs efficacy are also discussed. Targeting research and drug development on CREB could potentially accelerate the development of novel medications against schizophrenia.
Introduction
The cAMP-response element binding protein (CREB) is localized in the nucleus and acts as a transcription factor, which binds to the cAMP response element (CRE) of the promoters of its target genes, upon phosphorylation at Ser133 by different receptor-activated protein kinases, such as protein kinase A (PKA), calmodulin-dependent protein kinase (CaMK), mitogen-activated protein kinases (MAPK), and other kinases (Alberini, 2009). Once CREB is activated and CREB-binding protein (CBP) is recruited, transcription is initiated (Dyson and Wright, 2016). The activity of CREB in neurons has been correlated with various intracellular processes, including proliferation, differentiation, survival, long-term synaptic potentiation, neurogenesis, and neuronal plasticity (Alberini, 2009; Landeira et al., 2016; Kitagawa et al., 2017). Recent studies propose that CREB is involved in signaling pathways leading to pathogenesis and therapy of certain mental disorders, including schizophrenia, making CREB an important focus of investigation (Ren et al., 2014).
Schizophrenia is a severe mental illness that changes a patient’s way of thinking, feeling, and social behavior (Adan et al., 2017). The onset of typical symptoms usually occurs around puberty or early adulthood (van Rijn et al., 2011). This phenomenon is assumed to be controlled by hormones and a latent immune vulnerability (Walker et al., 2008; Kinney et al., 2010). The current hypothesis of schizophrenia claims that the positive symptoms of the disease are linked to hyperactive dopaminergic activity, mediated by D2 dopamine receptors (D2R) in subcortical brain regions such as the striatum and the nucleus accumbens (Toda and Abi-Dargham, 2007; Lindenmayer et al., 2013), while the deficits in dopamine activity mediated by D1 dopamine receptors (D1R) are responsible for the negative symptoms and cognitive impairment (Toda and Abi-Dargham, 2007; Lindenmayer et al., 2013). Schizophrenia is also considered as a neurodevelopmental disorder, and this is confirmed by epidemiological, developmental, and neuroimaging studies (Owen et al., 2011). Moreover, patient population genetics suggest that schizophrenia may result from a combination of genetic factors and environmental insults, including prenatal infection, perinatal complications, and drug abuse (Stepniak et al., 2014).
The following evidences propose CREB as a convergent dopaminergic signaling protein in schizophrenia: (1) in vitro and animal studies show that dopamine receptor signaling increases the phosphorylation of CREB (Lukasiewicz et al., 2016). Activated CREB promotes the expression of brain-derived neurotrophic factor (BDNF) (Yoo et al., 2017), while on the other hand, BDNF promotes the activation of CREB through tropomyosin receptor kinase (Trk) B receptors (Finkbeiner et al., 1997). Direct relationships were also found between antipsychotic drugs binding to D2R, therapeutic effect, and stimulation of CREB phosphorylation in vitro and in animal models (Konradi et al., 1993; Lee et al., 2010). (2) Data from schizophrenic patients also support the interaction between CREB and BDNF (Palomino et al., 2006; Tejeda and Diaz-Guerra, 2017). As a downstream target of BDNF, protein kinase B (Akt), glycogen synthase kinase 3β (GSK3β), disrupted-in-schizophrenia-protein 1 (DISC1), neuregulin-1 (NRG-1), and dysbindin-1 are common associated susceptibility genes (Zheng et al., 2012). Interestingly, CREB is a substrate for Akt and GSK3β phosphorylation: Akt at Ser133 (Li et al., 2011), while GSK3β at Ser129 (Horike et al., 2008); and (3) besides these studies that implicate CREB in schizophrenia, evidence is provided by patients’ postmortem pathological studies. The protein and gene levels of CREB and the binding activity of CREB to CRE in schizophrenic brains were significantly decreased in the cingulate gyrus (Yuan et al., 2010; Ren et al., 2014), an integral brain limbic system structure, which is involved with emotion, learning, and memory and found to be smaller and with lower neural activity in people with schizophrenia. Therefore, the CREB pathway may represent a promising target for the development of innovative interventions for schizophrenia. In the current review, the role of the CREB signaling pathway in the pathophysiology of schizophrenia will be discussed.
Molecular Structure of CREB
The CREB genes in both mouse and human consist of 11 exons, and 3 isoforms designated α, β, and Δ are produced through alternative splicing (Ichiki, 2006). These isoforms, expressed in most tissues, are identical in function. Primary structure studies show that the full-length sequence of CREB could be divided into four functional domains from the N-terminus to C-terminus, namely (i) Q1 basal transcriptional activity domain; (ii) kinase inducible domain (KID); (iii) a glutamine-rich, Q2 domain for constitutive activation; and iv) a basic region/leucine zipper domain (bZIP) forming a homodimer and responsible for binding to DNA (Xu et al., 2007). The Q1 domain localizes at the N-terminus of CREB, interacts with TATA binding protein, and promotes gene transcription (Felinski and Quinn, 2001). The KID is located in the middle region; central to this region is Ser133 and the phosphorylation of Ser133 by multiple protein kinases is necessary for the activation of CREB (Sun et al., 2016). The upstream protein kinases activating CREB include PKA, Akt, protein kinase C (PKC), calcium/calmodulin-dependent protein kinase II (CaMKII), p90 ribosomal S6 kinase (p90RSK), casein kinase I, and casein kinase II (Wen et al., 2010; Trinh et al., 2013). The Q2 domain is responsible for binding with RNA polymerase II initiation complex. This domain is responsible for the recognition and binding to the canonical CRE, 5′-TGACGTCA-3′ (Altarejos and Montminy, 2011). The carboxy terminal of CREB is a bZIP dimerization domain, which is required for the dimerization of CREB (Schumacher et al., 2000). The Mg2+ ions facilitate the binding activity of bZIP to CRE by more than 25-fold (Schumacher et al., 2000). As mentioned earlier, CREB could be phosphorylated by PKA at Ser133; however, PKA phosphorylation does not alter the secondary structure of CREB, and, therefore, has no effect on the binding of CREB to DNA (Richards et al., 1996). Besides CREB, cAMP response element modulator (CREM) and activating transcription factor-1 (ATF-1) are also members of the CREB family. The structure and biological functions of both CREM and ATF-1 are similar to CREB, which forms heterodimers with ATF-1 or CREM (Wu et al., 1998). However, the interactions of CREB/CREM and CREB/ATF-1 in the pathophysiology of schizophrenia are unknown and will not be further addressed.
Dopamine-Mediated Signaling Affects the Activity of CREB
The CREB is a critical molecule involved in the signal transduction of dopamine receptors. Binding of dopamine to its receptors enhances the phosphorylation of CREB through multiple pathways: (i) binding of dopamine to D1R elevates intracellular cAMP levels and activates PKA followed by the phosphorylation of CREB (Chartoff et al., 2003; Boyd and Mailman, 2012); (ii) binding of dopamine to D2R reduces cAMP production and adenylate cyclase activity followed by reduced phosphorylation of CREB (Boyd and Mailman, 2012). However, repeated treatment with the selective D2R agonist, such as quinpirole, enhances PKA activity and increases phospho-CREB expression in the nucleus accumbens (Culm et al., 2004). Importantly, CREB activation in the nucleus accumbens attenuates prepulse inhibition (PPI) disruption (Culm et al., 2004). Thus, chronic administration of D2R agonist promotes the phosphorylation of CREB, and the mechanism might be related to downregulation of D2R receptors; (iii) D2R is also coupled to phospholipase Cβ (PLCβ). Activation of D2R by its agonist quinpirole causes an elevation of intracellular Ca2+ and activation of PKC. The Ca2+/CaMK and PKC are the upstream protein kinases phosphorylating CREB and, therefore, enhanced Ca2+ and activation of PKC cause the phosphorylation and activation of CREB (Yan et al., 1999). Dopamine- and cAMP-regulated phosphoprotein of molecular weight 32 kDa (DARPP-32) is possibly a molecule that links D2R-mediated signaling and CREB. In DARPP-32 knockout mice, the basal phosphorylation levels of CREB were elevated, and the ability of D2R to induce phosphorylation of CREB was lost (Yan et al., 1999); (iv) Stimulation of D1R and D2R with agonists activates Akt kinase, which translocates to the nucleus. Activated Akt directly phosphorylates CREB at Ser133 in striatal neurons (Brami-Cherrier et al., 2002); (v) CREB stimulates the expression of a number of genes containing CREs (5′-TGACGTCA-3′) in their promoter regions (Montminy, 1997), which may be associated with schizophrenia such as D1R, serotonin transporter, and synapsin 1 (Meyer et al., 1993; Montminy, 1997). It has been also reported that D1R/D2R plays a synergistic role in inducing CREB–DNA binding activities (Kashihara et al., 1999).
Signaling Cascades Regulating the Activity of CREB
Several different protein kinases phosphorylate CREB, making it a convergent target for multiple intracellular signaling cascades. The most important posttranscriptional modification is the phosphorylation of the amino acid residue Ser133 in the KID domain (Guo et al., 2017). This domain contains multiple phosphorylation sites for many canonical signaling pathways, such as Ras/Raf/MAPK/p90RSK, Ca2+/CaMK, PI3K/Akt/GSK3β, and cAMP/PKA pathways (Tang et al., 2014; Cheng et al., 2016; Xi et al., 2016; Zeng et al., 2016; Heckman et al., 2018), which, upon activation, lead to CREB’s antagonistic effects (Figure 1).
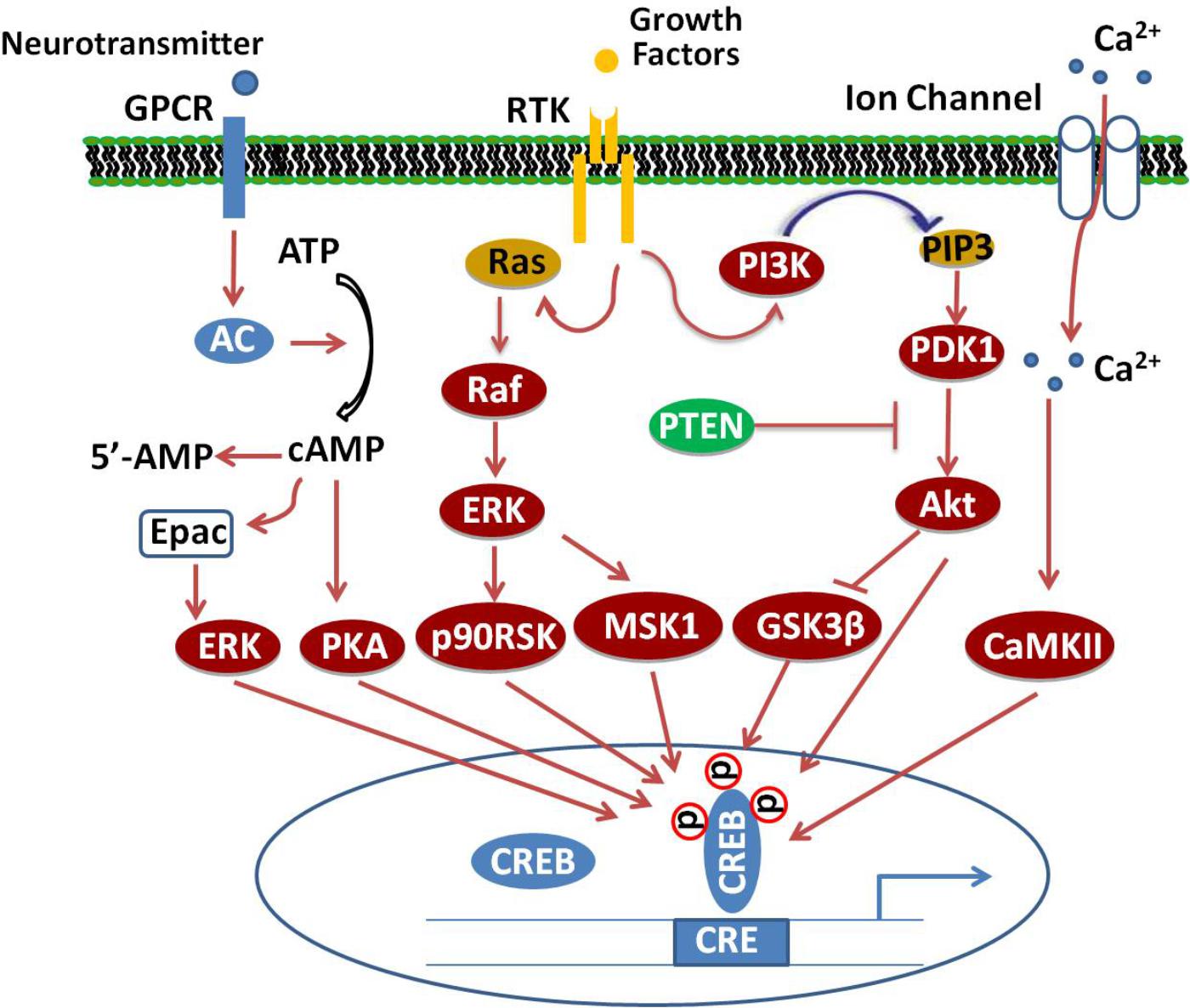
FIGURE 1. Signaling cascade of CREB. Adenylate cyclase (AC) activated upon stimulation of cellular G-protein-coupled receptors (GPCR) by neurotransmitters increases cAMP levels, which, in turn, activate PKA. The catalytic subunits of PKA translocate into the nucleus and phosphorylate CREB at Ser133. Binding of growth factors to receptor tyrosine kinases (RTK) stimulate the activation of PI3K/Akt/GSK3β, Ras/Raf/MEK/ERK/p90/RSK and ERK/MSK1 signaling pathways, which subsequently enhance the phosphorylation of CREB at different sites. Additionally, activation of excitatory NMDA receptors will increase the phosphorylation of CREB through Ca2+/CaMK-dependent pathways.
Both cAMP and cGMP show regulatory functions in mental disorders (Ben Aissa et al., 2016) and both of them could regulate the phosphorylation of CREB. It is well documented that cAMP, through PKA, stimulates the phosphorylation of CREB at Ser133 and causes the activation of CREB (Herold et al., 2011; Heckman et al., 2018), while cGMP activates the downstream protein cGMP-dependent protein kinase G (PKG), which also phosphorylates the transcription factor CREB at Ser133 (Lueptow et al., 2016). This dual phosphorylation by PKA and PKG may amplify the CREB activity (Lu et al., 1999).
In addition to the canonical cAMP–PKA–CREB pathway, the exchange protein directly activated by cAMP (Epac) is another cAMP-binding effector protein. In vitro, binding affinities of cAMP for PKA and Epac are similar (Bos, 2006). cAMP activates the small GTP binding protein Rap1 through Epac and activates extracellular signal-regulated kinase 1/2 (ERK1/2) signaling, which subsequently leads to the phosphorylation of CREB. The cAMP–Epac–ERK–CREB signaling pathway is known to mediate neurotrophic and neuroprotective functions (Grimes et al., 2015).
Cells treated with phosphodiesterase inhibitors will also prevent the degradation and increase of the endogenous level of cAMP (Guo et al., 2017). The activated PKA enters into the nucleus and promotes the binding of phosphorylated CREB to a CRE region (5′-TGACGTCA-3′). The CBP is subsequently recruited and bound to CREB, which co-activates CREB. Activation of CREB consequently stimulates or inhibits the expression of its downstream target genes, including genes involved in metabolism (such as PEPCK, cytochrome c, and aminolevulinate synthase ), transcription (such as ATF-3, STAT3, and c-fos ), cell survival/cell cycle (such as bcl-2, cyclin D1, and cyclin A), and growth factors (such as BDNF and FGF6) (Sakamoto and Frank, 2009). A schematic survey of target genes regulated by CREB is presented in Figure 2. Although CREB could bind to many regulatory gene regions, it may not be critical for the expression of these genes. The most plausible explanation claims that the gene is under regulation by more than one transcription factor and coactivator and, therefore, deletion of one transcription factor is not always effective in the final gene expression (Lemberger et al., 2008).
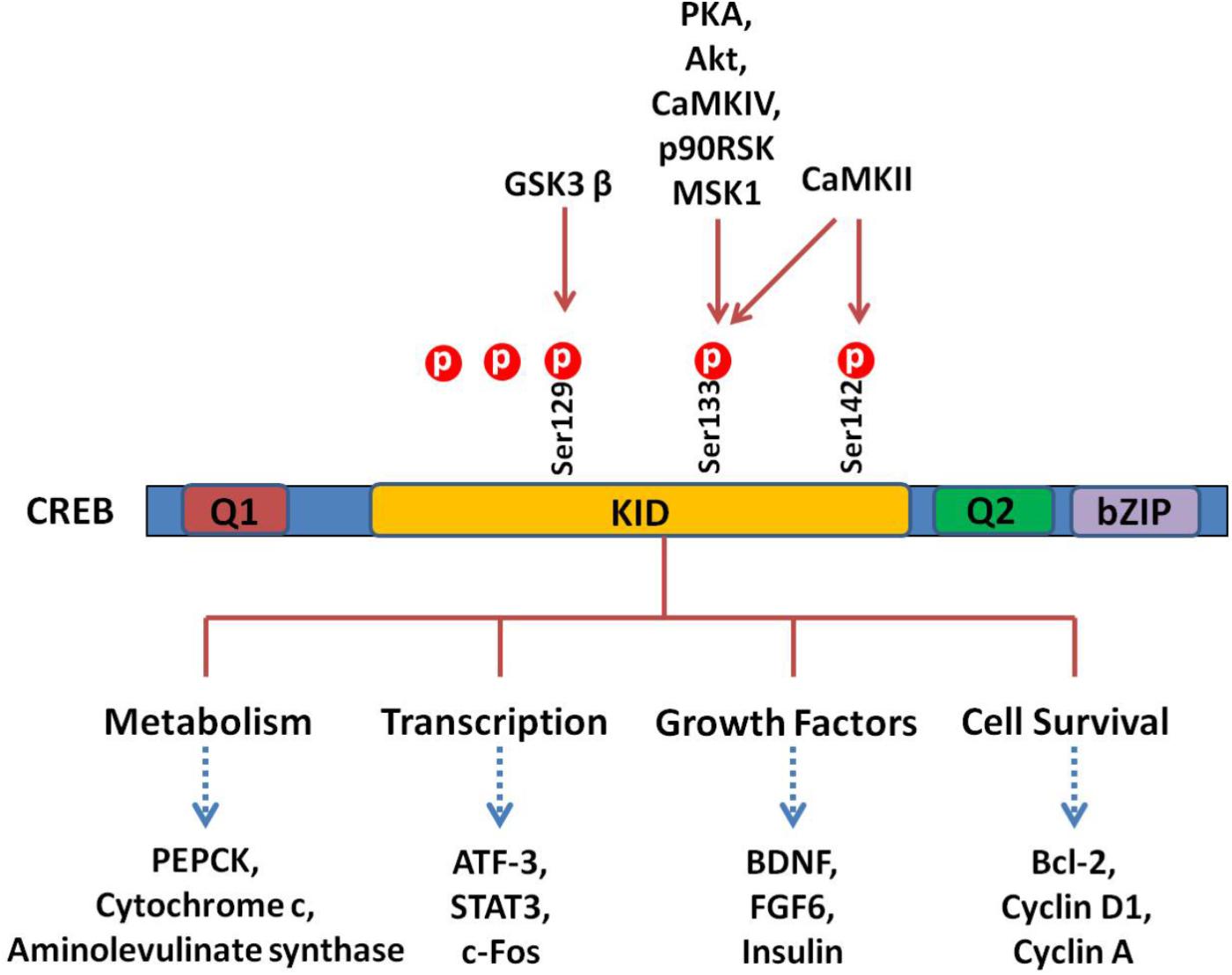
FIGURE 2. CREB and its downstream substrates. The CREB contains Q1, kinase-inducible domain (KID), Q2, and bZIP domains. The crucial event in the activation of CREB is the phosphorylation of Ser133 in KID. This domain could be phoshphorylated by multiple protein kinases such as PKA, Akt, CaMKs, p90RSK, and MSK1. The CREB as a nuclear transcription factor binds to CRE (cAMP response element), regulating transcription activity of its downstream substrates, which regulate neuronal processes, including metabolism and survival and expression of different transcription factors and growth factors.
There are three phosphorylation sites existing in the amino-terminal of CREB, and the phosphorylation of Ser133 is the most studied. The phosphorylation of Ser133 leads to a 10- to 20-fold increase in CREB’s transcriptional activity (Mayr and Montminy, 2001). Functionally, phosphorylation of this site promotes the transcription and expression of CREB target genes (Delghandi et al., 2005). Notably, phosphorylation of Ser133 mainly influences the activity of the trans-activation domain, while it has no effect on the affinity of CRBE to the CRE sequence. Since mutation of Ser133 destroys the transcription activity of this protein (Karin and Smeal, 1992), phosphorylation of Ser133 activates CREB’s trans-activation domain through changing its conformation, resulting in better interaction with other coactivators (Karin and Smeal, 1992; Mayr and Montminy, 2001). It is hypothesized that phosphorylation of CREB makes the trans-activation domain closer to the amino terminal, which interacts with CREB dimers or some other DNA-binding protein(s) (Mayr and Montminy, 2001). The CREB can be phosphorylated at other amino acids than Ser133, including Ser129, Ser142, and Ser143 (Shaywitz and Greenberg, 1999). Different kinases such as CaMK IV phosphorylate CREB at Ser133 only, while CaMK II phosphorylates CREB at both Ser133 and Ser142. Phosphorylation of Ser142 by CaMK II inhibits CREB (Sun et al., 1994), while phosphorylation of Ser133 enhances the CREB trans-activation dramatically. These results indicate that cAMP is a positive stimulator of transcription. Different evidences support the hypothesis that phosphorylation of CREB at Ser142/143 blocks the phosphorylation of CREB at Ser133 and attenuates the binding of CBP to CREB (West et al., 2002).
Phosphatase enzymes, which remove phosphate groups from protein substrates phosphorylated by kinases, are essential to many neuronal functions, because phosphorylation and dephosphorylation reactions serve diverse roles in regulatory signaling networks. In mammals, serine/threonine protein phosphatases include PP1, PP2A, PP2B, and PP2C (Shi, 2009). All of these phosphatases act on CREB and promote the dephosphorylation of CREB. The PP2A in the nucleus is the most effective phosphatase that dephosphorylates CREB (Wadzinski et al., 1993). Specifically, PP2A efficiently dephosphorylates PKA-stimulated CREB phosphorylation and, therefore, attenuates cAMP or PKA-stimulated gene transcription (Wadzinski et al., 1993). Dephosphorylation of Ser133 residue by PP1 also represses CREB activity (Yang et al., 2015) and this effect is involved in long-term depression at glutamatergic synapses (Mauna et al., 2011). The PP2B, a calcium-dependent phosphatase, is also involved in accelerating the decay of phosphorylated CREB at Ser133 (Bito et al., 1996). The PP2B may act on CREB indirectly and the possible target for PP2B is PP1. Synaptic Ca2+ entry triggers the activation of Ca2+/calmodulin signaling and subsequently causes phosphorylation of PP2B, which acts as an inhibitory subunit of PP1 (Bito et al., 1996). The role of PP2C on the dephosphorylation of CREB is not yet clear. Additional proteins such as phosphatase and tensin homolog (PTEN) contribute to the dephosphorylation of CREB. It has been confirmed that CREB is a substrate of PTEN in the nucleus where it is colocalized with phosphorylated CREB and directly dephosphorylates CREB at Ser133 (Gu et al., 2011). The duration and extent of CREB phosphorylation are parallel with the transcriptional regulation of target genes containing the CRE sequence (Hagiwara et al., 1992). The balance between kinases responsible for phosphorylating CREB and phosphatases capable of dephosphorylating CREB determines the degree of CRE-dependent gene transcription (Hagiwara et al., 1992).
CREB-Mediated Transcription in the Nucleus Accumbens (NAc) in Mood Regulation
The CREB is expressed throughout the striatal regions, including the nucleus accumbens (NAc) (Mantamadiotis et al., 2002). The activity of CREB in the NAc is a pivotal modulator of an animal’s behavioral phenotype response to psychiatric stimuli (Barrot et al., 2002). Upon exposure to stress, CREB is stimulated in the NAc, and both D1R- and D2R dopaminergic neurons are involved in the phosphorylation of CREB (Blendy, 2006). Besides dopaminergic neurons, the biological function of CREB in the NAc is also regulated by glutamatergic inputs (Dudman et al., 2003). Current studies indicate that alterations in CREB activity within the NAc represent an important gate between emotional stimuli and animal behavioral response (Barrot et al., 2002). Experimental changes in CREB activity in the NAc are sufficient to change animal behavior. For instance, elevations of CREB activity through viral-mediated gene transfer of CREB within the NAc reduces sensitivity to emotional and stress stimuli (Barrot et al., 2002). Similarly, a sustained activation of CREB in the NAc produces anhedonia-like symptoms, and pro-depression-like symptoms in both acute and chronic stress-induced depression animal models in rats or mice (Pliakas et al., 2001; Newton et al., 2002). Conversely, dominant negative mutant CREB (mCREB) in the rat NAc enhanced cocaine, morphine, and sucrose preference (Barrot et al., 2002; Dinieri et al., 2009). These data demonstrate that CREB activity in the NAc is highly related with drug withdrawal, depression, and other dysphoric states. Additionally, CREB in the NAc also affects anxiety-like behavior in animals. Loss of function of CREB within the NAc produces anxiety-like effects, whereas elevations of CREB function leads to the opposite phenotype (Barrot et al., 2002; Wallace et al., 2009). These studies raise the possibility that modulation of CREB in the NAc contributes to the development of different mood disorders (Carlezon et al., 2005). In animal models of schizophrenia, PKA activity and CREB phosphorylation in the NAc are decreased, and treatment with antipsychotics increases CREB activity in the NAc, and this neuro-adaptive response facilitates the recovery of sensorimotor gating (Culm et al., 2004), which is seriously disrupted in schizophrenic patients. As a transcription factor, activated CREB regulates the transcription of various genes including tyrosine hydroxylase, serotonin 2A receptors, and other genes possibly implicated in schizophrenia (Culm et al., 2004). The CREB phosphorylation in the NAc, after treatment with quinpirole, is proposed to mediate gene transcription and subsequently promoting the recovery of sensorimotor gating (Culm et al., 2004). These studies emphasize on the important role of the target genes of CREB, especially genes specifically expressed in the brain region related to schizophrenia. The CREB and such target genes could be utilized, therefore, in drug discovery efforts.
Interaction Between CREB and Neurotrophins-Mediated Signaling
The “neurodevelopmental hypothesis of schizophrenia” emphasizes that the abnormalities of early brain development increase the risk for the subsequent emergence of clinical symptoms (Schmidt and Mirnics, 2015). In this hypothesis, schizophrenia is associated with a subtle brain lesion that is caused by a combination of genetic and/or early environmental factors and that eventually interacts with normal maturational processes of the synapse and brain, to facilitate symptoms such as psychosis (Schmidt and Mirnics, 2015).
The BDNF is an important neurotrophin that promotes the development of certain populations of neuronal cells and confers neuroprotection under different conditions (Nieto et al., 2013). Dysfunction of BDNF signaling leads to deficits in neuronal growth and synaptic transmission, leading to disorganized brain function, which contributes to the development of schizophrenia (Palomino et al., 2006). Mutual relationships between BDNF and CREB are well documented: BDNF promotes the phosphorylation of CREB, which, in turn, promotes the transcription of BDNF gene. Treatment of neurons with BDNF triggers CREB phosphorylation (Pizzorusso et al., 2000). The BDNF activates CREB, in part, by increasing intracellular Ca2+ that leads to the activation of CaMKIV, which phosphorylates CREB. Neuronal exposure to BDNF also activates the Ras/Erk/Rsk pathway that causes CREB phosphorylation (Finkbeiner et al., 1997). The pathway of BDNF-induced CREB phosphorylation involves BDNF receptor TrkB stimulation, because treatment with the pan Trk antagonist, K252a, completely blocked CREB phosphorylation (Pizzorusso et al., 2000). Neuronal dendritic growth is essential for the neuronal network’s electrophysiological activity, and the dendritic development is impaired in schizophrenia (Glausier and Lewis, 2013). Among different neurotrophins that regulate the development of dendrites, BDNF regulates the dendritic length and complexity of MAPK and CREB signaling (Finsterwald et al., 2010). Moreover, phosphorylation of TrkB induces activation and translocation of MAPKs from the cytoplasm into the nucleus, which subsequently causes the activation of nuclear kinase mitogen- and stress-activated kinase (MSK 1) (Deak et al., 1998). The MSK1 is a protein kinase expressed in the central nervous system (Arthur et al., 2004) and a major CREB (Ser133) kinase that is direct downstream target of BDNF receptors and the MAPK cascade (Daumas et al., 2017). Indeed, MSK1-/- mice displayed deficits in experience-dependent synaptic plasticity (Correa et al., 2012), spatial and recognition memory tasks, and in BDNF-mediated phosphorylation of CREB (Arthur et al., 2004; Karelina et al., 2012). Additionally, BDNF stimulates the binding of CREB to the promoter region of cypin, which is a major PSD-95-binding protein. Enhanced transcription of cypin results in an increase of dendrite branching (Kwon et al., 2011). Maternal inflammation during pregnancy affects the neuronal cell survival in the offspring due to inflammatory factors, which induce axonal loss. The BDNF is a potent pro-survival factor, and the neuroprotective effect of BDNF is associated with the activation of CREB signaling (Fujino et al., 2009; Mohammadi et al., 2018). Cumulatively, these findings show that BDNF plays pivotal roles in both neuronal development and survival, and importantly, CREB is a downstream molecule responsible for some of the neurobiological functions of BDNF.
Interestingly, treatment with BDNF activates CREB through TrkB receptors (Pizzorusso et al., 2000). On the contrary, the expression of BDNF is regulated by CREB (Tao et al., 1998). The PKA enhances the phosphorylation of CREB at Ser133, which leads to the nuclear localization and activation of CREB (Guo et al., 2017). The promoter region of BDNF contains CRE; activated CREB binds to CRE and promotes the transcription of BDNF (Tao et al., 1998). Upregulation of BDNF mRNA expression is parallel to increased phosphorylated CREB expression (Guo et al., 2017), and inhibition of PKA/CREB pathway attenuates the level of BDNF in neurons (Xue et al., 2016).
Biological Functions of CREB in the Central Nervous System
The CREB has been implicated in the regulation of a variety of biological functions (Steven and Seliger, 2016). Biological functions in the brain, such as the contribution of CREB to synaptic plasticity and neurodevelopment, have also been established (Fujino et al., 2009). Immunohistochemical analyses of CREB and ATF-1 indicated that ATF-1 was expressed in trophectoderm and inner cell mass cells at E3.5 of the mice embryo (Bleckmann et al., 2002). The expression of CREB could be detected at E3.5 and its expression increased during later stages in the epiblast and cells derived thereof (Bleckmann et al., 2002). The CREB-null mice, which carry a mutation that knock down all functional isoforms of CREB gene, are smaller than their littermates and face survival problems due to respiratory distress (Rudolph et al., 1998). As CREB knockout is lethal during perinatal development, the function of CREB in the brain in adult mice cannot be investigated in CREB-null mice. To overcome this problem, conditional knockout of CREB in the brain of developing and adult mice was developed using the Cre/loxP system (Mantamadiotis et al., 2002). Absence of CREB in the central nervous system was correlated to upregulation of CREM, but with no significant pathologies. As CREB and CREM play a similar effect in cellular survival, CREM upregulation in the brain is sufficient to maintain neuronal survival. Similarly, loss of only CREB in brain has limited effect on neuronal survival. However, deletion of both CREB and CREM caused non-specific neuronal cell death and progressive neurodegeneration in the brain hippocampus and in the dorsolateral striatum (Dawson and Ginty, 2002; Mantamadiotis et al., 2002). Therefore, it is reasonable to conclude that knockdown of CREB and CREM in neurons of the developing CNS may cause apoptosis, and postnatal ablation of these genes in adulthood may result in neuronal degeneration. Cumulatively, these findings support the concept that CREB expression and transcriptional activity are regulated in both embryonic and mature brain, and it is implicated in neuronal survival as well as in neurogenesis, processes associated with the pathology of schizophrenia.
Additionally, ablation of CREB resulted in neuronal degeneration in hippocampus and striatum (Dawson and Ginty, 2002) contributing to the pathogenesis of neurodegenerative diseases and mood disorders, such as schizophrenia and depression (Wang et al., 2015). It is well established that CREB is necessary for spatial memory (Sekeres et al., 2010). As a binding protein to CREB, conditional knockout of CBP in the mice brain caused significant impairment in spatial, associative, and object-recognition memory (Chen et al., 2010). In Alzheimer’s disease (AD) transgenic mice model, TgCRND8 mice exhibited a profound impairment in the ability in spatial memory. However, increasing CREB function in the CA1 region of dorsal hippocampus rescued the spatial memory deficits (Yiu et al., 2011). In recent experiments, it has been demonstrated that CREB represents an important target for drug development in the therapy of AD (Guo et al., 2017).
Besides its role in neurodegenerative diseases, CREB is also proposed to be involved in the disease process of psychiatric disorders, such as schizophrenia (McGirr et al., 2016), autism (Lyu et al., 2016), drug addiction (Fisher et al., 2016), and depression (Zhou et al., 2016). For example, PPI deficits were observed in rats treated with dopamine D2R agonists and in individuals suffering from schizophrenia, while chronic quinpirole or ropinirole drug treatment produced sustained PPI recovery, requiring CREB activity in the nucleus accumbens of rats (Berger et al., 2011). Recombinant lentivirus LV-CREB133 expressing a dominant negative CREB decreased synapse and spine density, inhibited neurogenesis, and attenuated the expression of synapsin and spinophilin (Zhang et al., 2016). However, LV-VP16-CREB, a constitutively active CREB, increased synapse density and dendrite complexity, enhanced neurogenesis, and increased the expression of synaptic proteins (Zhang et al., 2016). This suggests that CREB is involved in the neuronal plasticity and possibly implicated in modulating schizophrenia-related behaviors. Another example is the link between CREB and autism. The CREB (alpha and delta isoforms)-deficient mice were less active and more inhibited in the behavioral assays of elevated plus maze, open field, and light/dark box (Hebda-Bauer et al., 2004), an effect similar with altered exploratory behavior in autism spectrum disorder (Veenstra-VanderWeele et al., 2012). As an important protein involved in neuronal development and synaptic plasticity, the relationship between CREB and autism is receiving increasing attention (Nuytens et al., 2013; Gao et al., 2016).
It is noteworthy that chronic CREB activation may also cause deleterious consequences. Chronic activation of CREB led to sporadic epileptic seizures and a significant loss of hippocampal neurons (Lopez de Armentia et al., 2007). Chronic enhancement of CREB activity also delayed the retrieval of spatial information (Viosca et al., 2009). Further studies indicate that the pathological consequences resulting from CREB inhibition and CREB activation are mediated through different mechanistic processes (Sakamoto et al., 2011). The CREB inhibition triggers cell death through a pro-apoptotic signal pathway (Zeng et al., 2016), while chronic CREB activation triggers loss of neurons through an excitotoxicity mechanism (Lopez de Armentia et al., 2007). This suggests that the timing of CREB regulation may be a key for the various associative changes that culminate in cellular neuronal responses.
Interactions Between CREB and Susceptibility Genes for Schizophrenia
Schizophrenia is a complicated central nervous disease, and the causes of schizophrenia include genetic factors and gene–environment interactions. In the last decade, a number of chromosomal regions and genes have been studied with molecular biology and genetic analyses. However, there has been no consistent single gene variation confirmed with the development of this illness, and the contribution of genetic factor remains obscure at this time (Tandon et al., 2008). Genome-wide association study (GWAS) provides an unbiased assessment of variation through investigating the entire genome. Many GWAS of schizophrenia have been published in the past 10 years (Bergen and Petryshen, 2012; Visscher et al., 2017), and numerous single nucleotide polymorphisms have been found to be associated with schizophrenia (Giusti-Rodriguez and Sullivan, 2013; Hertzberg et al., 2015). Although it is too premature to link these studies to schizophrenia genetics, current available analyses support that some of the previously implicated pathways such as calcium signaling, CREB signaling, and NMDA receptors are involved in the pathology of schizophrenia. For example, an investigation of the de novo mutations in 623 families with schizophrenia in Bulgaria indicated that synaptic genes, such as genes encoding postsynaptic density proteins, cytoskeleton-associated scaffold proteins, and N-methyl-D-aspartate (NMDA) receptor, were enriched in these mutated genes (Fromer et al., 2014). These results were confirmed by another comprehensive case–control study of schizophrenic patients in Sweden using exome analysis (Purcell et al., 2014). Collectively, GWAS indicates an important role for synaptic genes and genes regulating synaptic plasticity in the risk for schizophrenia. Genetic alterations extracted from GWAS data are shown in Figure 3. Based on these findings, Forero et al. (2016) summarized genes involved in the cAMP/PKA/CREB signaling pathway that could be candidates for schizophrenia. It was suggested that synaptic genes, including CREB1, CREM, PPP3CB, and PRKAR1A, are involved in the etiology of schizophrenia-related psychiatric disorders and regulated by the cAMP/PKA/CREB signaling pathway (Forero et al., 2016). Genetic studies have identified many genes and pathways implicated in schizophrenia, but the genetic liability needs further verification. Using Sanger method and next-generation sequencing, a study at the genome-wide level was performed to test the single nucleotide variants of 10 traditional candidate genes in 727 patients with schizophrenia and 733 controls. Unfortunately, none of the 10 traditional candidate genes had single nucleotide variants showing an association with schizophrenia (Crowley et al., 2013). Consistently, genome-wide array comparative genomic hybridization in five large pedigrees with schizophrenia showed that no linkage exist between any copy number variant and schizophrenia (Timms et al., 2013). In summary, exome studies have indicated that rare de novo and transmitted mutations contribute to the development of schizophrenia. However, it is worthy of our attention that so far there has been no significant association with a gene. The penetrance of de novo mutation to chromatin regulation is yet unknown and deserves further clarification.
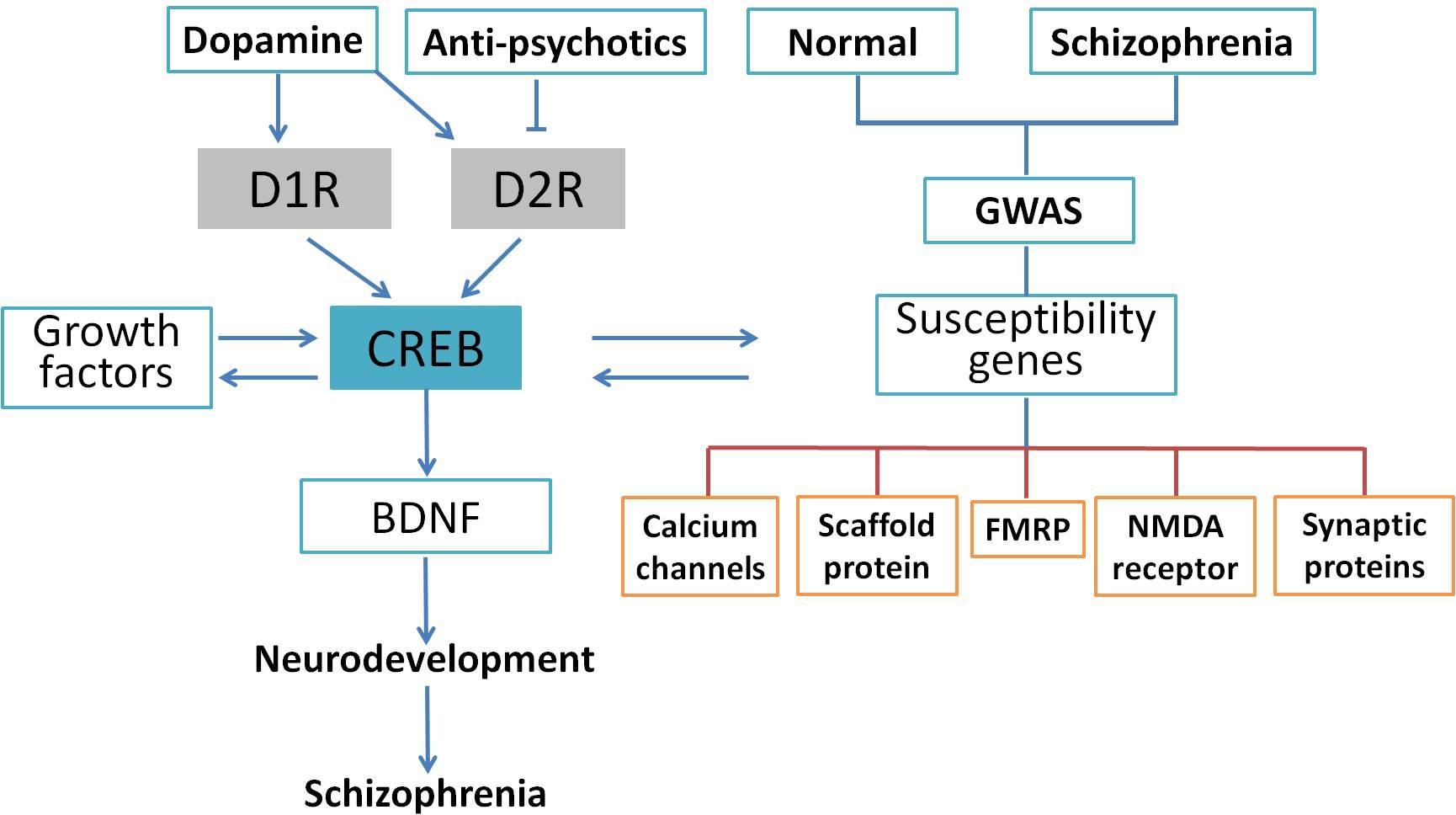
FIGURE 3. CREB as an integrative signaling molecule involved in schizophrenia. Dopamine stimulates the phosphorylation of CREB through D1R-mediated cAMP/PKA pathway or inhibits it through the D2R-mediated cAMP/PKA pathway. The CREB may also affect neurotrophins (BDNF) and other protein expressions and susceptibility genes associated with schizophrenia. The GWAS indicated that gene sets related to calcium channels, activity-regulated cytoskeleton-associated scaffold protein, FMRP, PSD-95, NMDA receptor, and synaptic proteins were potential candidates altered in the schizophrenic patients. The CREB-induced dysregulation in neuronal signaling may lead to neurodevelopmental deficits followed by schizophrenic behavior.
With this background, we would like to address a few studies focusing on NRG-1 and the DISC-1 and dysbindin-1 genes in relation to schizophrenia. These genes play a role in both neural signaling and development, and are associated with schizophrenia (Gong et al., 2014; Mostaid et al., 2017; Prats et al., 2017). It would be interesting to determine the interaction between CREB and these susceptibility genes. The NRG-1 is a member of neuregulin family that acts on the EGFR family of receptors. The NRG-1 is produced in numerous isoforms by alternative splicing, which allows it to perform a wide variety of functions such as regulation of neural development, neurotransmission, and synaptic plasticity through activation of the ErbB receptors. Binding of NRG-1 to ErbB enhanced the phosphorylation of both the ErbB receptors and CREB (Ozaki et al., 2004). The transcription factor early growth response 3 (Egr3) is an immediate-early growth response gene, which is transcriptionally induced by NRG1, and the induction of Egr3 in response to NRG1 is associated with the activation of ERK1/2 and possibly CREB phosphorylation (Herndon et al., 2014).
Collectively, as we previously reviewed (Zheng et al., 2012), all these genes are linked to Akt: (i) DISC1 is a protein involved in the modulation of neuronal proliferation, differentiation, and neuronal outgrowth. Dysregulated expression of DISC1 may predispose individuals to the development of schizophrenia and other psychiatric conditions. DISCI interacts and promotes the activation of Akt (Dahoun et al., 2017); (ii) the PI3K/Akt signaling pathway mediates the neuroprotective effect of NRG1 (Papaleo et al., 2016); (iii) Dysbindin is another protein highly expressed in neural tissues. Dysbindin-1 showed protective effect in neurons against apoptosis through activating the PI3K/Akt signaling in cortical neurons (Zheng et al., 2012). Cumulatively, these findings link the biological effects of these genes with PI3K/Akt signaling pathway. Considering that CREB is one of the downstream key effector modules of the PI3K/Akt signaling pathway (Sun et al., 1994; Li et al., 2011), it is plausible that there is a cross-talk between susceptibility genes and Akt/CREB pathway, although we cannot exclude the possibility that they work independently.
Besides the susceptibility genes listed earlier, there are many other risk-genes that have been identified in schizophrenia (Kato, 2015). It is hypothesized that these susceptibility and/or risk-genes trigger a clinically measurable outcome. The CREB may act as a possible signaling molecule link in the pathophysiology process triggered by the various susceptibility genes. Future human genetic studies will be crucial for shedding light on this concept.
Role of CREB in Therapy of Schizophrenia
Many studies addressed the expression and activity of CREB, a transcription factor regulated by cAMP/PKA, in animal models upon treatment with antipsychotic drugs. The CREB mRNA levels are not regulated by haloperidol in striatum of rats, while haloperidol induces a significant phosphorylation of CREB in striatum, indicating that CREB is transcriptionally active in response to haloperidol (Konradi et al., 1993). In amphetamine-treated rats, haloperidol also induced a distinct immediate early gene (such as c-fos, c-jun) expression and CREB phosphorylation, and these neurochemical changes are associated with behavioral plasticity (Hsieh et al., 2002). In contrast to these findings, the drug olanzapine increased protein levels of CREB and BDNF in the prefrontal cortex, hippocampus, and striatum of adult Wistar rats (Reus et al., 2012). In vitro studies also support this conclusion. In SY5Y neuroblastoma cells, amisulpride, an atypical antipsychotic drug, stimulated neurite outgrowth by regulating D2R-mediated β-arrestin 2 signaling, and increased levels of CREB phosphorylation, with BDNF potentially involved in this process (Park et al., 2011). Similarly, olanzapine treatment increased basal BDNF gene in SY5Y cells. It is supposed that olanzapine activated PKA, PI3K, PKC, and CaMKII signaling pathways and subsequently upregulated BDNF gene transcription via activating CREB (Lee et al., 2010). A 4-week treatment with both olanzapine and lithium in rats led to a 1.4-fold increase of the levels of CREB and BDNF in the dentate gyrus and hippocampal area CA1. These observations support that the activation of CREB and upregulation of BDNF may underlie the neurological actions of olanzapine and lithium (Hammonds and Shim, 2009).
We would like to propose that various antipsychotic drugs may have different roles on CREB phosphorylation in neurons in both culture and in vivo situations. It was reported that haloperidol and eticlopride, selective D2R antagonists, stimulated the phosphorylation of CREB in the dorsal striatum. In contrast, clozapine reduced CREB phosphorylation, indicating that haloperidol and clozapine induce distinct patterns of CREB phosphorylation in the dorsal striatum (Pozzi et al., 2003). On the other hand, neurons at different maturation stages may have distinct phenotypes regarding the phosphorylation of CREB in response to the same antipsychotic drugs. For example, low concentrations (50 nM) of haloperidol and risperidone stimulate the phosphorylation of both ERK and CREB in hippocampal neuron cultures after 25 days in vitro, but not at 10 days (Yang et al., 2004). These different experimental observations may reflect developmental changes in the ratios of expression of the different dopamine receptors in neurons (Yang et al., 2004).
Conclusion and Perspectives
Current studies suggest that CREB is a key integrator of diverse physiological processes in the CNS, including neurotransmission, neurodevelopment, neuronal survival, synaptic plasticity, and memory. Importantly, dysregulation of the CREB signaling has been implicated in a number of disorders in the CNS. Among these disorders, the relevance of CREB to the pathogenesis of schizophrenia has been most intensively investigated. The roles of CREB in the pathology of schizophrenia are depicted schematically in Figure 3. Dopamine, antipsychotic drugs, growth factors, and susceptibility genes could activate CREB and its downstream target BDNF via different pathways. The deficit of CREB/BDNF signaling impairs neurodevelopment and is implicated in the development of schizophrenia.
Given the important role of CREB activation in the CNS, it is reasonable to propose that activation of CREB signaling would have beneficial effects in schizophrenia therapy. However, sustained CREB activation also causes deleterious consequences. Hence, keeping a balance on CREB activation is a reasonable strategy in schizophrenia therapy. Pharmacological interventions, using transcriptional repressors, may serve this purpose. On the other hand, besides Ser133, CREB is also phosphorylated at many other sites. Investigating the relationship between these phosphorylation sites and schizophrenia may lead to novel pharmaceutical approaches. Among these sites, Ser129 and Ser 142 are close to Ser133, and it is tempting to suggest that these two sites will affect the transcription activity or binding of CREB to CRE. Moreover, the activity of CREB is modulated by additional signaling pathways, which due to antagonistic action may be manipulated for schizophrenia therapy. The signaling pathways involved in schizophrenia are complex. Both CREB and BDNF are key molecules implicated in the regulation of mood. Additional basic and clinical research is needed to further identify the specific role of the CREB in the pathology of schizophrenia, as this could shed new light on the effective management of this psychiatric disorder. Furthermore, CREB regulates the transcription of multiple genes, and extensive efforts are required to identify their involvement in the pathogenesis of schizophrenia and their myriad of combinatory effects.
The CREB-mutant mice, especially conditional knockout mice, represent a useful tool for examining its role in mediating neuropsychiatric behaviors. The CREBαΔ mutant mice is homozygous mice with a targeted mutation in CREB; the α and Δ isoforms of CREB are knocked down in this animal model, whereas the CREB β variant is upregulated (Blendy et al., 1996). The functional consequence of this mutation is a reduction of 90% in CRE binding and activity (Walters and Blendy, 2001). Currently, findings with CREBαΔ mutant mice support that enhanced CREB expression and phosphorylation promote neurodevelopment (Malberg and Blendy, 2005), while downregulation of CREB activity in mice would impair neurogenesis (Nakagawa et al., 2002). CREB-mutant mice have been used for the studies on the mechanisms of depression and the therapeutic effects of antidepressants (Conti et al., 2002; Gur et al., 2007). We propose that this animal model is useful for studying the relationship between CREB activation and the etiology of schizophrenia, and it may help to investigate the responses of mice to antipsychotics. However, there are a few disadvantages with this animal model, as CREB is widely expressed in the brain and CREB deficiency in this animal model is not strictly located to a specific neuronal populations (Gass and Riva, 2007). On the other hand, CREB deficiency is occurring throughout the whole body and during the initial fetal development in this animal model, which might cause developmental deficits and/or possible compensatory effects for the loss of CREB (Gass and Riva, 2007).
In summary, targeting CREB proteins might promote synaptic plasticity and neurodevelopment in CNS, which can be beneficial or delay the development of schizophrenia. However, we also would like to point out that the increased risk of suicide should be take into consideration when targeting CREB, as data from human postmortem suggest that the numbers of phosphorylated CREB-positive cells were increased in amygdala in subjects, who had died by suicide, while lithium could significantly decrease the phosphorylation of CREB levels in the same region (Young et al., 2004). Hence, the phosphorylation level of CREB in the brain sections highly related with mood disorders should be maintained at a certain level, namely neither too high, nor too low. Whether or not this strategy can be applied for the treatment of schizophrenia will depend upon further research characterizing the importance of CREB in the pathological processes of schizophrenia.
Author Contributions
HW initiated the research topic and discussed the literature and wrote the draft manuscript. WZ performed the conceptional design and writing of the final manuscript. JX wrote some part of the review. PL and RQ contributed constructive suggestions and extensive language editing.
Funding
This research was supported by National Natural Science Foundation of China (Nos. 81301099, 31771128, and 31371088) and Science and Technology Planning Project of Guangdong Province (No. 2011B050200005). MYRG2016-00052-FHS and MYRG2018-00134-FHS from University of Macau and the Science and Technology Development Fund (FDCT) of Macao (FDCT 021/2015/A1 and 016/2016/A1). PL holds The Jacob Gitlin Chair in Physiology and is affiliated and partially supported by Grass Center for Drug Design and Synthesis of Novel Therapeutics and The Adolph and Klara Brettler Medical Research Center at The Hebrew University of Jerusalem, Israel.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adan, A., Arredondo, A. Y., Capella, M. D., Prat, G., Forero, D. A., and Navarro, J. F. (2017). Neurobiological underpinnings and modulating factors in schizophrenia spectrum disorders with a comorbid substance use disorder: a systematic review. Neurosci. Biobehav. Rev. 75, 361–377. doi: 10.1016/j.neubiorev.2017.01.038
Alberini, C. M. (2009). Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev. 89, 121–145. doi: 10.1152/physrev.00017.2008
Altarejos, J. Y., and Montminy, M. (2011). CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12, 141–151. doi: 10.1038/nrm3072
Arthur, J. S., Fong, A. L., Dwyer, J. M., Davare, M., Reese, E., Obrietan, K., et al. (2004). Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J. Neurosci. 24, 4324–4332. doi: 10.1523/JNEUROSCI.5227-03.2004
Barrot, M., Olivier, J. D., Perrotti, L. I., DiLeone, R. J., Berton, O., Eisch, A. J., et al. (2002). CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc. Natl. Acad. Sci. U.S.A. 99, 11435–11440. doi: 10.1073/pnas.172091899
Ben Aissa, M., Lee, S. H., Bennett, B. M., and Thatcher, G. R. (2016). Targeting NO/cGMP signaling in the CNS for neurodegeneration and Alzheimer’s Disease. Curr. Med. Chem. 23, 2770–2788. doi: 10.2174/0929867323666160812145454
Bergen, S. E., and Petryshen, T. L. (2012). Genome-wide association studies of schizophrenia: does bigger lead to better results? Curr. Opin. Psychiatry 25, 76–82. doi: 10.1097/YCO.0b013e32835035dd
Berger, A. K., Green, T., Siegel, S. J., Nestler, E. J., and Hammer, R. P. Jr. (2011). cAMP response element binding protein phosphorylation in nucleus accumbens underlies sustained recovery of sensorimotor gating following repeated D(2)-like receptor agonist treatment in rats. Biol. Psychiatry 69, 288–294. doi: 10.1016/j.biopsych.2010.08.032
Bito, H., Deisseroth, K., and Tsien, R. W. (1996). CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87, 1203–1214. doi: 10.1016/S0092-8674(00)81816-4
Bleckmann, S. C., Blendy, J. A., Rudolph, D., Monaghan, A. P., Schmid, W., and Schutz, G. (2002). Activating transcription factor 1 and CREB are important for cell survival during early mouse development. Mol. Cell. Biol. 22, 1919–1925. doi: 10.1128/MCB.22.6.1919-1925.2002
Blendy, J. A. (2006). The role of CREB in depression and antidepressant treatment. Biol. Psychiatry 59, 1144–1150. doi: 10.1016/j.biopsych.2005.11.003
Blendy, J. A., Kaestner, K. H., Schmid, W., Gass, P., and Schutz, G. (1996). Targeting of the CREB gene leads to up-regulation of a novel CREB mRNA isoform. EMBO J. 15, 1098–1106.
Bos, J. L. (2006). Epac proteins: multi-purpose cAMP targets. Trends Biochem. Sci. 31, 680–686. doi: 10.1016/j.tibs.2006.10.002
Boyd, K. N., and Mailman, R. B. (2012). Dopamine receptor signaling and current and future antipsychotic drugs. Handb. Exp. Pharmacol. 212, 53–86. doi: 10.1007/978-3-642-25761-2_3
Brami-Cherrier, K., Valjent, E., Garcia, M., Pages, C., Hipskind, R. A., and Caboche, J. (2002). Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: a new route to cAMP response element-binding protein phosphorylation. J. Neurosci. 22, 8911–8921. doi: 10.1523/JNEUROSCI.22-20-08911.2002
Carlezon, W. A. Jr., Duman, R. S., and Nestler, E. J. (2005). The many faces of CREB. Trends Neurosci. 28, 436–445. doi: 10.1016/j.tins.2005.06.005
Chartoff, E. H., Papadopoulou, M., Konradi, C., and Carlezon, W. A. Jr. (2003). Dopamine-dependent increases in phosphorylation of cAMP response element binding protein (CREB) during precipitated morphine withdrawal in primary cultures of rat striatum. J. Neurochem. 87, 107–118. doi: 10.1046/j.1471-4159.2003.01992.x
Chen, G., Zou, X., Watanabe, H., van Deursen, J. M., and Shen, J. (2010). CREB binding protein is required for both short-term and long-term memory formation. J. Neurosci. 30, 13066–13077. doi: 10.1523/JNEUROSCI.2378-10.2010
Cheng, C. Y., Tang, N. Y., Kao, S. T., and Hsieh, C. L. (2016). Ferulic acid administered at various time points protects against cerebral infarction by activating p38 MAPK/p90RSK/CREB/Bcl-2 anti-apoptotic signaling in the subacute phase of cerebral ischemia-reperfusion injury in rats. PLoS One 11:e0155748. doi: 10.1371/journal.pone.0155748
Conti, A. C., Cryan, J. F., Dalvi, A., Lucki, I., and Blendy, J. A. (2002). cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. J. Neurosci. 22, 3262–3268. doi: 10.1523/JNEUROSCI.22-08-03262.2002
Correa, S. A., Hunter, C. J., Palygin, O., Wauters, S. C., Martin, K. J., McKenzie, C., et al. (2012). MSK1 regulates homeostatic and experience-dependent synaptic plasticity. J. Neurosci. 32, 13039–13051. doi: 10.1523/JNEUROSCI.0930-12.2012
Crowley, J. J., Hilliard, C. E., Kim, Y., Morgan, M. B., Lewis, L. R., Muzny, D. M., et al. (2013). Deep resequencing and association analysis of schizophrenia candidate genes. Mol. Psychiatry 18, 138–140. doi: 10.1038/mp.2012.28
Culm, K. E., Lugo-Escobar, N., Hope, B. T., and Hammer, R. P. Jr. (2004). Repeated quinpirole treatment increases cAMP-dependent protein kinase activity and CREB phosphorylation in nucleus accumbens and reverses quinpirole-induced sensorimotor gating deficits in rats. Neuropsychopharmacology. 29, 1823–1830. doi: 10.1038/sj.npp.1300483
Dahoun, T., Trossbach, S. V., Brandon, N. J., Korth, C., and Howes, O. D. (2017). The impact of Disrupted-in-Schizophrenia 1 (DISC1) on the dopaminergic system: a systematic review. Transl. Psychiatry 7:e1015. doi: 10.1038/tp.2016.282
Daumas, S., Hunter, C. J., Mistry, R. B., More, L., Privitera, L., Cooper, D. D., et al. (2017). The kinase function of msk1 regulates BDNF signaling to CREB and basal synaptic transmission, but is not required for hippocampal long-term potentiation or spatial memory. eNeuro 4:ENEURO.0212-16.2017. doi: 10.1523/ENEURO.0212-16.2017
Dawson, T. M., and Ginty, D. D. (2002). CREB family transcription factors inhibit neuronal suicide. Nat. Med. 8, 450–451. doi: 10.1038/nm0502-450
Deak, M., Clifton, A. D., Lucocq, L. M., and Alessi, D. R. (1998). Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 17, 4426–4441. doi: 10.1093/emboj/17.15.4426
Delghandi, M. P., Johannessen, M., and Moens, U. (2005). The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell. Signal. 17, 1343–1351. doi: 10.1016/j.cellsig.2005.02.003
Dinieri, J. A., Nemeth, C. L., Parsegian, A., Carle, T., Gurevich, V. V., Gurevich, E., et al. (2009). Altered sensitivity to rewarding and aversive drugs in mice with inducible disruption of cAMP response element-binding protein function within the nucleus accumbens. J. Neurosci. 29, 1855–1859. doi: 10.1523/JNEUROSCI.5104-08.2009
Dudman, J. T., Eaton, M. E., Rajadhyaksha, A., Macias, W., Taher, M., Barczak, A., et al. (2003). Dopamine D1 receptors mediate CREB phosphorylation via phosphorylation of the NMDA receptor at Ser897-NR1. J. Neurochem. 87, 922–934. doi: 10.1046/j.1471-4159.2003.02067.x
Dyson, H. J., and Wright, P. E. (2016). Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding protein (CBP) and p300. J. Biol. Chem. 291, 6714–6722. doi: 10.1074/jbc.R115.692020
Felinski, E. A., and Quinn, P. G. (2001). The coactivator dTAF(II)110/hTAF(II)135 is sufficient to recruit a polymerase complex and activate basal transcription mediated by CREB. Proc. Natl. Acad. Sci. U.S.A. 98, 13078–13083. doi: 10.1073/pnas.241337698
Finkbeiner, S., Tavazoie, S. F., Maloratsky, A., Jacobs, K. M., Harris, K. M., and Greenberg, M. E. (1997). CREB: a major mediator of neuronal neurotrophin responses. Neuron 19, 1031–1047. doi: 10.1016/S0896-6273(00)80395-5
Finsterwald, C., Fiumelli, H., Cardinaux, J. R., and Martin, J. L. (2010). Regulation of dendritic development by BDNF requires activation of CRTC1 by glutamate. J. Biol. Chem. 285, 28587–28595. doi: 10.1074/jbc.M110.125740
Fisher, M. L., LeMalefant, R. M., Zhou, L., Huang, G., and Turner, J. R. (2016). Distinct roles of CREB within the ventral and dorsal hippocampus in mediating nicotine withdrawal phenotypes. Neuropsychopharmacology 42, 1599–1609. doi: 10.1038/npp.2016.257
Forero, D. A., Herteleer, L., De Zutter, S., Norrback, K. F., Nilsson, L. G., Adolfsson, R., et al. (2016). A network of synaptic genes associated with schizophrenia and bipolar disorder. Schizophr. Res. 172, 68–74. doi: 10.1016/j.schres.2016.02.012
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Fujino, H., Kitaoka, Y., Hayashi, Y., Munemasa, Y., Takeda, H., Kumai, T., et al. (2009). Axonal protection by brain-derived neurotrophic factor associated with CREB phosphorylation in tumor necrosis factor-alpha-induced optic nerve degeneration. Acta Neuropathol. 117, 75–84. doi: 10.1007/s00401-008-0440-9
Gao, J., Wang, X., Sun, H., Cao, Y., Liang, S., Wang, H., et al. (2016). Neuroprotective effects of docosahexaenoic acid on hippocampal cell death and learning and memory impairments in a valproic acid-induced rat autism model. Int. J. Dev. Neurosci. 49, 67–78. doi: 10.1016/j.ijdevneu.2015.11.006
Gass, P., and Riva, M. A. (2007). CREB, neurogenesis and depression. Bioessays 29, 957–961. doi: 10.1002/bies.20658
Giusti-Rodriguez, P., and Sullivan, P. F. (2013). The genomics of schizophrenia: update and implications. J. Clin. Invest. 123, 4557–4563. doi: 10.1172/JCI66031
Glausier, J. R., and Lewis, D. A. (2013). Dendritic spine pathology in schizophrenia. Neuroscience 251, 90–107. doi: 10.1016/j.neuroscience.2012.04.044
Gong, X., Lu, W., Kendrick, K. M., Pu, W., Wang, C., Jin, L., et al. (2014). A brain-wide association study of DISC1 genetic variants reveals a relationship with the structure and functional connectivity of the precuneus in schizophrenia. Hum. Brain Mapp. 35, 5414–5430. doi: 10.1002/hbm.22560
Grimes, M. T., Powell, M., Gutierrez, S. M., Darby-King, A., Harley, C. W., and McLean, J. H. (2015). Epac activation initiates associative odor preference memories in the rat pup. Learn. Mem. 22, 74–82. doi: 10.1101/lm.037101.114
Gu, T., Zhang, Z., Wang, J., Guo, J., Shen, W. H., and Yin, Y. (2011). CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 71, 2821–2825. doi: 10.1158/0008-5472.CAN-10-3399
Guo, H., Cheng, Y., Wang, C., Wu, J., Zou, Z., Niu, B., et al. (2017). FFPM, a PDE4 inhibitor, reverses learning and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB signaling and anti-inflammatory effects. Neuropharmacology. 116, 260–269. doi: 10.1016/j.neuropharm.2017.01.004
Gur, T. L., Conti, A. C., Holden, J., Bechtholt, A. J., Hill, T. E., Lucki, I., et al. (2007). cAMP response element-binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J. Neurosci. 27, 7860–7868. doi: 10.1523/JNEUROSCI.2051-07.2007
Hagiwara, M., Alberts, A., Brindle, P., Meinkoth, J., Feramisco, J., Deng, T., et al. (1992). Transcriptional attenuation following cAMP induction requires PP-1-mediated dephosphorylation of CREB. Cell 70, 105–113. doi: 10.1016/0092-8674(92)90537-M
Hammonds, M. D., and Shim, S. S. (2009). Effects of 4-week treatment with lithium and olanzapine on levels of brain-derived neurotrophic factor, B-cell CLL/lymphoma 2 and phosphorylated cyclic adenosine monophosphate response element-binding protein in the sub-regions of the hippocampus. Basic Clin. Pharmacol. Toxicol. 105, 113–119. doi: 10.1111/j.1742-7843.2009.00416.x
Hebda-Bauer, E. K., Watson, S. J., and Akil, H. (2004). CREB deficient mice show inhibition and low activity in novel environments without changes in stress reactivity. Eur. J. Neurosci. 20, 503–513. doi: 10.1111/j.1460-9568.2004.03487.x
Heckman, P. R. A., Blokland, A., Bollen, E. P. P., and Prickaerts, J. (2018). Phosphodiesterase inhibition and modulation of corticostriatal and hippocampal circuits: clinical overview and translational considerations. Neurosci. Biobehav. Rev. 87, 233–254. doi: 10.1016/j.neubiorev.2018.02.007
Herndon, C. A., Ankenbruck, N., and Fromm, L. (2014). The Erk MAP kinase pathway is activated at muscle spindles and is required for induction of the muscle spindle-specific gene Egr3 by neuregulin1. J. Neurosci. Res. 92, 174–184. doi: 10.1002/jnr.23293
Herold, S., Jagasia, R., Merz, K., Wassmer, K., and Lie, D. C. (2011). CREB signalling regulates early survival, neuronal gene expression and morphological development in adult subventricular zone neurogenesis. Mol. Cell. Neurosci. 46, 79–88. doi: 10.1016/j.mcn.2010.08.008
Hertzberg, L., Katsel, P., Roussos, P., Haroutunian, V., and Domany, E. (2015). Integration of gene expression and GWAS results supports involvement of calcium signaling in Schizophrenia. Schizophr. Res. 164, 92–99. doi: 10.1016/j.schres.2015.02.001
Horike, N., Sakoda, H., Kushiyama, A., Ono, H., Fujishiro, M., Kamata, H., et al. (2008). AMP-activated protein kinase activation increases phosphorylation of glycogen synthase kinase 3beta and thereby reduces cAMP-responsive element transcriptional activity and phosphoenolpyruvate carboxykinase C gene expression in the liver. J. Biol. Chem. 283, 33902–33910. doi: 10.1074/jbc.M802537200
Hsieh, H. C., Li, H. Y., Lin, M. Y., Chiou, Y. F., Lin, S. Y., Wong, C. H., et al. (2002). Spatial and temporal profile of haloperidol-induced immediate-early gene expression and phosphoCREB binding in the dorsal and ventral striatum of amphetamine-sensitized rats. Synapse 45, 230–244. doi: 10.1002/syn.10099
Ichiki, T. (2006). Role of cAMP response element binding protein in cardiovascular remodeling: good, bad, or both? Arterioscler. Thromb. Vasc. Biol. 26, 449–455. doi: 10.1161/01.ATV.0000196747.79349.d1
Karelina, K., Hansen, K. F., Choi, Y. S., DeVries, A. C., Arthur, J. S., and Obrietan, K. (2012). MSK1 regulates environmental enrichment-induced hippocampal plasticity and cognitive enhancement. Learn. Mem. 19, 550–560. doi: 10.1101/lm.025775.112
Karin, M., and Smeal, T. (1992). Control of transcription factors by signal transduction pathways: the beginning of the end. Trends Biochem. Sci. 17, 418–422. doi: 10.1016/0968-0004(92)90012-X
Kashihara, K., Ishihara, T., Akiyama, K., and Abe, K. (1999). D1/D2 receptor synergism on CREB DNA-binding activities in the caudate-putamen of rat. Neurol. Res. 21, 781–784. doi: 10.1080/01616412.1999.11741014
Kato, T. (2015). Whole genome/exome sequencing in mood and psychotic disorders. Psychiatry Clin. Neurosci. 69, 65–76. doi: 10.1111/pcn.12247
Kinney, D. K., Hintz, K., Shearer, E. M., Barch, D. H., Riffin, C., Whitley, K., et al. (2010). A unifying hypothesis of schizophrenia: abnormal immune system development may help explain roles of prenatal hazards, post-pubertal onset, stress, genes, climate, infections, and brain dysfunction. Med. Hypotheses 74, 555–563. doi: 10.1016/j.mehy.2009.09.040
Kitagawa, H., Sugo, N., Morimatsu, M., Arai, Y., Yanagida, T., and Yamamoto, N. (2017). Activity-dependent dynamics of the transcription factor of cAMP-response element binding protein in cortical neurons revealed by single-molecule imaging. J. Neurosci. 37, 1–10. doi: 10.1523/JNEUROSCI.0943-16.2016
Konradi, C., Kobierski, L. A., Nguyen, T. V., Heckers, S., and Hyman, S. E. (1993). The cAMP-response-element-binding protein interacts, but Fos protein does not interact, with the proenkephalin enhancer in rat striatum. Proc. Natl. Acad. Sci. U.S.A. 90, 7005–7009. doi: 10.1073/pnas.90.15.7005
Kwon, M., Fernandez, J. R., Zegarek, G. F., Lo, S. B., and Firestein, B. L. (2011). BDNF-promoted increases in proximal dendrites occur via CREB-dependent transcriptional regulation of cypin. J. Neurosci. 31, 9735–9745. doi: 10.1523/JNEUROSCI.6785-10.2011
Landeira, B. S., Santana, T. T., Araujo, J. A., Tabet, E. I., Tannous, B. A., Schroeder, T., et al. (2016). Activity-independent effects of CREB on neuronal survival and differentiation during mouse cerebral cortex development. Cereb. Cortex 28, 537–548. doi: 10.1093/cercor/bhw387
Lee, J. G., Cho, H. Y., Park, S. W., Seo, M. K., and Kim, Y. H. (2010). Effects of olanzapine on brain-derived neurotrophic factor gene promoter activity in SH-SY5Y neuroblastoma cells. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 1001–1006. doi: 10.1016/j.pnpbp.2010.05.013
Lemberger, T., Parkitna, J. R., Chai, M., Schutz, G., and Engblom, D. (2008). CREB has a context-dependent role in activity-regulated transcription and maintains neuronal cholesterol homeostasis. FASEB J. 22, 2872–2879. doi: 10.1096/fj.08-107888
Li, X. Y., Zhan, X. R., Liu, X. M., and Wang, X. C. (2011). CREB is a regulatory target for the protein kinase Akt/PKB in the differentiation of pancreatic ductal cells into islet beta-cells mediated by hepatocyte growth factor. Biochem. Biophys. Res. Commun. 404, 711–716. doi: 10.1016/j.bbrc.2010.12.048
Lindenmayer, J. P., Nasrallah, H., Pucci, M., James, S., and Citrome, L. (2013). A systematic review of psychostimulant treatment of negative symptoms of schizophrenia: challenges and therapeutic opportunities. Schizophr. Res. 147, 241–252. doi: 10.1016/j.schres.2013.03.019
Lopez de Armentia, M., Jancic, D., Olivares, R., Alarcon, J. M., Kandel, E. R., and Barco, A. (2007). cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J. Neurosci. 27, 13909–13918. doi: 10.1523/JNEUROSCI.3850-07.2007
Lu, Y. F., Kandel, E. R., and Hawkins, R. D. (1999). Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurosci. 19, 10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999
Lueptow, L. M., Zhan, C. G., and O’Donnell, J. M. (2016). Cyclic GMP-mediated memory enhancement in the object recognition test by inhibitors of phosphodiesterase-2 in mice. Psychopharmacology 233, 447–456. doi: 10.1007/s00213-015-4129-1
Lukasiewicz, S., Blasiak, E., Szafran-Pilch, K., and Dziedzicka-Wasylewska, M. (2016). Dopamine D2 and serotonin 5-HT1A receptor interaction in the context of the effects of antipsychotics - in vitro studies. J. Neurochem. 137, 549–560. doi: 10.1111/jnc.13582
Lyu, J. W., Yuan, B., Cheng, T. L., Qiu, Z. L., and Zhou, W. H. (2016). Reciprocal regulation of autism-related genes MeCP2 and PTEN via microRNAs. Sci. Rep. 6:20392. doi: 10.1038/srep20392
Malberg, J. E., and Blendy, J. A. (2005). Antidepressant action: to the nucleus and beyond. Trends Pharmacol. Sci. 26, 631–638. doi: 10.1016/j.tips.2005.10.005
Mantamadiotis, T., Lemberger, T., Bleckmann, S. C., Kern, H., Kretz, O., Martin Villalba, A., et al. (2002). Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 31, 47–54. doi: 10.1038/ng882
Mauna, J. C., Miyamae, T., Pulli, B., and Thiels, E. (2011). Protein phosphatases 1 and 2A are both required for long-term depression and associated dephosphorylation of cAMP response element binding protein in hippocampal area CA1 in vivo. Hippocampus 21, 1093–1104. doi: 10.1002/hipo.20823
Mayr, B., and Montminy, M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609. doi: 10.1038/35085068
McGirr, A., Lipina, T. V., Mun, H. S., Georgiou, J., Al-Amri, A. H., Ng, E., et al. (2016). Specific inhibition of phosphodiesterase-4B results in anxiolysis and facilitates memory acquisition. Neuropsychopharmacology 41, 1080–1092. doi: 10.1038/npp.2015.240
Meyer, T. E., Waeber, G., Lin, J., Beckmann, W., and Habener, J. F. (1993). The promoter of the gene encoding 3’,5’-cyclic adenosine monophosphate (cAMP) response element binding protein contains cAMP response elements: evidence for positive autoregulation of gene transcription. Endocrinology 132, 770–780. doi: 10.1210/endo.132.2.8381074
Mohammadi, A., Amooeian, V. G., and Rashidi, E. (2018). Dysfunction in brain-derived neurotrophic factor signaling pathway and susceptibility to schizophrenia. Curr. Gene Ther. 18, 45–63. doi: 10.2174/1566523218666180302163029
Montminy, M. (1997). Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem. 66, 807–822. doi: 10.1146/annurev.biochem.66.1.807
Mostaid, M. S., Mancuso, S. G., Liu, C., Sundram, S., Pantelis, C., Everall, I. P., et al. (2017). Meta-analysis reveals associations between genetic variation in the 5’ and 3’ regions of Neuregulin-1 and schizophrenia. Transl. Psychiatry 7:e1004. doi: 10.1038/tp.2016.279
Nakagawa, S., Kim, J. E., Lee, R., Malberg, J. E., Chen, J., Steffen, C., et al. (2002). Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J. Neurosci. 22, 3673–3682. doi: 10.1523/JNEUROSCI.22-09-03673.2002
Newton, S. S., Thome, J., Wallace, T. L., Shirayama, Y., Schlesinger, L., Sakai, N., et al. (2002). Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J. Neurosci. 22, 10883–10890. doi: 10.1523/JNEUROSCI.22-24-10883.2002
Nieto, R., Kukuljan, M., and Silva, H. (2013). BDNF and schizophrenia: from neurodevelopment to neuronal plasticity, learning, and memory. Front. Psychiatry 4:45. doi: 10.3389/fpsyt.2013.00045
Nuytens, K., Gantois, I., Stijnen, P., Iscru, E., Laeremans, A., Serneels, L., et al. (2013). Haploinsufficiency of the autism candidate gene Neurobeachin induces autism-like behaviors and affects cellular and molecular processes of synaptic plasticity in mice. Neurobiol. Dis. 51, 144–151. doi: 10.1016/j.nbd.2012.11.004
Owen, M. J., O’Donovan, M. C., Thapar, A., and Craddock, N. (2011). Neurodevelopmental hypothesis of schizophrenia. Br. J. Psychiatry 198, 173–175. doi: 10.1192/bjp.bp.110.084384
Ozaki, M., Itoh, K., Miyakawa, Y., Kishida, H., and Hashikawa, T. (2004). Protein processing and releases of neuregulin-1 are regulated in an activity-dependent manner. J. Neurochem. 91, 176–188. doi: 10.1111/j.1471-4159.2004.02719.x
Palomino, A., Vallejo-Illarramendi, A., Gonzalez-Pinto, A., Aldama, A., Gonzalez-Gomez, C., Mosquera, F., et al. (2006). Decreased levels of plasma BDNF in first-episode schizophrenia and bipolar disorder patients. Schizophr. Res. 86, 321–322. doi: 10.1016/j.schres.2006.05.028
Papaleo, F., Yang, F., Paterson, C., Palumbo, S., Carr, G. V., Wang, Y., et al. (2016). Behavioral, neurophysiological, and synaptic impairment in a transgenic neuregulin1 (NRG1-IV) murine schizophrenia model. J. Neurosci. 36, 4859–4875. doi: 10.1523/JNEUROSCI.4632-15.2016
Park, S. W., Seo, M. K., Cho, H. Y., Lee, J. G., Lee, B. J., Seol, W., et al. (2011). Differential effects of amisulpride and haloperidol on dopamine D2 receptor-mediated signaling in SH-SY5Y cells. Neuropharmacology 61, 761–769. doi: 10.1016/j.neuropharm.2011.05.022
Pizzorusso, T., Ratto, G. M., Putignano, E., and Maffei, L. (2000). Brain-derived neurotrophic factor causes cAMP response element-binding protein phosphorylation in absence of calcium increases in slices and cultured neurons from rat visual cortex. J. Neurosci. 20, 2809–2816. doi: 10.1523/JNEUROSCI.20-08-02809.2000
Pliakas, A. M., Carlson, R. R., Neve, R. L., Konradi, C., Nestler, E. J., Carlezon, W. A., et al. (2001). Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J. Neurosci. 21, 7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001
Pozzi, L., Hakansson, K., Usiello, A., Borgkvist, A., Lindskog, M., Greengard, P., et al. (2003). Opposite regulation by typical and atypical anti-psychotics of ERK1/2, CREB and Elk-1 phosphorylation in mouse dorsal striatum. J. Neurochem. 86, 451–459. doi: 10.1046/j.1471-4159.2003.01851.x
Prats, C., Arias, B., Moya-Higueras, J., Pomarol-Clotet, E., Parellada, M., Gonzalez-Pinto, A., et al. (2017). Evidence of an epistatic effect between Dysbindin-1 and Neuritin-1 genes on the risk for schizophrenia spectrum disorders. Eur. Psychiatry 40, 60–64. doi: 10.1016/j.eurpsy.2016.07.006
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Ren, X., Rizavi, H. S., Khan, M. A., Bhaumik, R., Dwivedi, Y., and Pandey, G. N. (2014). Alteration of cyclic-AMP response element binding protein in the postmortem brain of subjects with bipolar disorder and schizophrenia. J. Affect. Disord. 15, 326–333. doi: 10.1016/j.jad.2013.09.033
Reus, G. Z., Abelaira, H. M., Agostinho, F. R., Ribeiro, K. F., Vitto, M. F., Luciano, T. F., et al. (2012). The administration of olanzapine and fluoxetine has synergistic effects on intracellular survival pathways in the rat brain. J. Psychiatr. Res. 46, 1029–1035. doi: 10.1016/j.jpsychires.2012.04.016
Richards, J. P., Bachinger, H. P., Goodman, R. H., and Brennan, R. G. (1996). Analysis of the structural properties of cAMP-responsive element-binding protein (CREB) and phosphorylated CREB. J. Biol. Chem. 271, 13716–13723. doi: 10.1074/jbc.271.23.13716
Rudolph, D., Tafuri, A., Gass, P., Hammerling, G. J., Arnold, B., and Schutz, G. (1998). Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc. Natl. Acad. Sci. U.S.A. 95, 4481–4486. doi: 10.1073/pnas.95.8.4481
Sakamoto, K., Karelina, K., and Obrietan, K. (2011). CREB: a multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 116, 1–9. doi: 10.1111/j.1471-4159.2010.07080.x
Sakamoto, K. M., and Frank, D. A. (2009). CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin. Cancer Res. 15, 2583–2587. doi: 10.1158/1078-0432.CCR-08-1137
Schmidt, M. J., and Mirnics, K. (2015). Neurodevelopment. Neuropsychopharmacology 40, 190–206. doi: 10.1038/npp.2014.95
Schumacher, M. A., Goodman, R. H., and Brennan, R. G. (2000). The structure of a CREB bZIP.somatostatin CRE complex reveals the basis for selective dimerization and divalent cation-enhanced DNA binding. J. Biol. Chem. 275, 35242–35247. doi: 10.1074/jbc.M007293200
Sekeres, M. J., Neve, R. L., Frankland, P. W., and Josselyn, S. A. (2010). Dorsal hippocampal CREB is both necessary and sufficient for spatial memory. Learn. Mem. 17, 280–283. doi: 10.1101/lm.1785510
Shaywitz, A. J., and Greenberg, M. E. (1999). CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821–861. doi: 10.1146/annurev.biochem.68.1.821
Shi, Y. (2009). Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484. doi: 10.1016/j.cell.2009.10.006
Stepniak, B., Papiol, S., Hammer, C., Ramin, A., Everts, S., Hennig, L., et al. (2014). Accumulated environmental risk determining age at schizophrenia onset: a deep phenotyping-based study. Lancet Psychiatry 1, 444–453. doi: 10.1016/S2215-0366(14)70379-7
Steven, A., and Seliger, B. (2016). Control of CREB expression in tumors: from molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget 7, 35454–35465. doi: 10.18632/oncotarget.7721
Sun, L., Zhao, R., Zhang, L., Zhang, W., He, G., Yang, S., et al. (2016). Prevention of vascular smooth muscle cell proliferation and injury-induced neointimal hyperplasia by CREB-mediated p21 induction: an insight from a plant polyphenol. Biochem. Pharmacol. 103, 40–52. doi: 10.1016/j.bcp.2016.01.015
Sun, P., Enslen, H., Myung, P. S., and Maurer, R. A. (1994). Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 8, 2527–2539. doi: 10.1101/gad.8.21.2527
Tandon, R., Keshavan, M. S., and Nasrallah, H. A. (2008). Schizophrenia, “just the facts” what we know in 2008. 2. Epidemiology and etiology. Schizophr. Res. 102, 1–18. doi: 10.1016/j.schres.2008.04.011
Tang, M., Shi, S., Guo, Y., Xu, W., Wang, L., Chen, Y., et al. (2014). GSK-3/CREB pathway involved in the gx-50’s effect on Alzheimer’s disease. Neuropharmacology 81, 256–266. doi: 10.1016/j.neuropharm.2014.02.008
Tao, X., Finkbeiner, S., Arnold, D. B., Shaywitz, A. J., and Greenberg, M. E. (1998). Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726. doi: 10.1016/S0896-6273(00)81010-7
Tejeda, G. S., and Diaz-Guerra, M. (2017). Integral characterization of defective BDNF/TrkB signalling in neurological and psychiatric disorders leads the way to new therapies. Int. J. Mol. Sci. 18:E268. doi: 10.3390/ijms18020268
Timms, A. E., Dorschner, M. O., Wechsler, J., Choi, K. Y., Kirkwood, R., Girirajan, S., et al. (2013). Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry 70, 582–590. doi: 10.1001/jamapsychiatry.2013.1195
Toda, M., and Abi-Dargham, A. (2007). Dopamine hypothesis of schizophrenia: making sense of it all. Curr. Psychiatry Rep. 9, 329–336. doi: 10.1007/s11920-007-0041-7
Trinh, A. T., Kim, S. H., Chang, H. Y., Mastrocola, A. S., and Tibbetts, R. S. (2013). Cyclin-dependent kinase 1-dependent phosphorylation of cAMP response element-binding protein decreases chromatin occupancy. J. Biol. Chem. 288, 23765–23775. doi: 10.1074/jbc.M113.464057
van Rijn, S., Aleman, A., de Sonneville, L., Sprong, M., Ziermans, T., Schothorst, P., et al. (2011). Neuroendocrine markers of high risk for psychosis: salivary testosterone in adolescent boys with prodromal symptoms. Psychol. Med. 41, 1815–1822. doi: 10.1017/S0033291710002576
Veenstra-VanderWeele, J., Muller, C. L., Iwamoto, H., Sauer, J. E., Owens, W. A., Shah, C. R., et al. (2012). Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc. Natl. Acad. Sci. U.S.A. 109, 5469–5474. doi: 10.1073/pnas.1112345109
Viosca, J., Malleret, G., Bourtchouladze, R., Benito, E., Vronskava, S., Kandel, E. R., et al. (2009). Chronic enhancement of CREB activity in the hippocampus interferes with the retrieval of spatial information. Learn. Mem. 16, 198–209. doi: 10.1101/lm.1220309
Visscher, P. M., Wray, N. R., Zhang, Q., Sklar, P., McCarthy, M. I., Brown, M. A., et al. (2017). 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22. doi: 10.1016/j.ajhg.2017.06.005
Wadzinski, B. E., Wheat, W. H., Jaspers, S., Peruski, L. F. Jr., Lickteig, R. L., Johnson, G. L., et al. (1993). Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol. Cell. Biol. 13, 2822–2834. doi: 10.1128/MCB.13.5.2822
Walker, E., Mittal, V., and Tessner, K. (2008). Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu. Rev. Clin. Psychol. 4, 189–216. doi: 10.1146/annurev.clinpsy.4.022007.141248
Wallace, D. L., Han, M. H., Graham, D. L., Green, T. A., Vialou, V., Iniguez, S. D., et al. (2009). CREB regulation of nucleus accumbens excitability mediates social isolation-induced behavioral deficits. Nat. Neurosci. 12, 200–209. doi: 10.1038/nn.2257
Walters, C. L., and Blendy, J. A. (2001). Different requirements for cAMP response element binding protein in positive and negative reinforcing properties of drugs of abuse. J. Neurosci. 21, 9438–9444. doi: 10.1523/JNEUROSCI.21-23-09438.2001
Wang, H., Quirion, R., Little, P. J., Cheng, Y., Feng, Z. P., Sun, H. S., et al. (2015). Forkhead box O transcription factors as possible mediators in the development of major depression. Neuropharmacology 99, 527–537. doi: 10.1016/j.neuropharm.2015.08.020
Wen, A. Y., Sakamoto, K. M., and Miller, L. S. (2010). The role of the transcription factor CREB in immune function. J. Immunol. 185, 6413–6419. doi: 10.4049/jimmunol.1001829
West, A. E., Griffith, E. C., and Greenberg, M. E. (2002). Regulation of transcription factors by neuronal activity. Nat. Rev. Neurosci. 3, 921–931. doi: 10.1038/nrn987
Wu, X., Spiro, C., Owen, W. G., and McMurray, C. T. (1998). cAMP response element-binding protein monomers cooperatively assemble to form dimers on DNA. J. Biol. Chem. 273, 20820–20827. doi: 10.1074/jbc.273.33.20820
Xi, Y. D., Zhang, D. D., Ding, J., Yu, H. L., Yuan, L. H., Ma, W. W., et al. (2016). Genistein inhibits abeta25-35-Induced synaptic toxicity and regulates CaMKII/CREB pathway in SH-SY5Y Cells. Cell. Mol. Neurobiol. 36, 1151–1159. doi: 10.1007/s10571-015-0311-6
Xu, W., Kasper, L. H., Lerach, S., Jeevan, T., and Brindle, P. K. (2007). Individual CREB-target genes dictate usage of distinct cAMP-responsive coactivation mechanisms. EMBO J. 26, 2890–2903. doi: 10.1038/sj.emboj.7601734
Xue, W., Wang, W., Gong, T., Zhang, H., Tao, W., Xue, L., et al. (2016). PKA-CREB-BDNF signaling regulated long lasting antidepressant activities of Yueju but not ketamine. Sci. Rep. 6:26331. doi: 10.1038/srep26331
Yan, Z., Feng, J., Fienberg, A. A., and Greengard, P. (1999). D(2) dopamine receptors induce mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc. Natl. Acad. Sci. U.S.A. 96, 11607–11612. doi: 10.1073/pnas.96.20.11607
Yang, B. H., Son, H., Kim, S. H., Nam, J. H., Choi, J. H., and Lee, J. S. (2004). Phosphorylation of ERK and CREB in cultured hippocampal neurons after haloperidol and risperidone administration. Psychiatry Clin. Neurosci. 58, 262–267. doi: 10.1111/j.1440-1819.2004.01229.x
Yang, H., Hou, H., Pahng, A., Gu, H., Nairn, A. C., Tang, Y. P., et al. (2015). Protein phosphatase-1 inhibitor-2 Is a novel memory suppressor. J. Neurosci. 35, 15082–15087. doi: 10.1523/JNEUROSCI.1865-15.2015
Yiu, A. P., Rashid, A. J., and Josselyn, S. A. (2011). Increasing CREB function in the CA1 region of dorsal hippocampus rescues the spatial memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 36, 2169–2186. doi: 10.1038/npp.2011.107
Yoo, J. M., Lee, B. D., Sok, D. E., Ma, J. Y., and Kim, M. R. (2017). Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells. Redox Biol. 11, 592–599. doi: 10.1016/j.redox.2016.12.034
Young, L. T., Bezchlibnyk, Y. B., Chen, B., Wang, J. F., and MacQueen, G. M. (2004). Amygdala cyclic adenosine monophosphate response element binding protein phosphorylation in patients with mood disorders: effects of diagnosis, suicide, and drug treatment. Biol. Psychiatry 55, 570–577. doi: 10.1016/j.biopsych.2003.10.023
Yuan, P., Zhou, R., Wang, Y., Li, X., Li, J., Chen, G., et al. (2010). Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J. Affect. Disord. 124, 164–169. doi: 10.1016/j.jad.2009.10.017
Zeng, B., Li, Y., Niu, B., Wang, X., Cheng, Y., Zhou, Z., et al. (2016). Involvement of PI3K/Akt/FoxO3a and PKA/CREB signaling pathways in the protective effect of fluoxetine against corticosterone-induced cytotoxicity in PC12 Cells. J. Mol. Neurosci. 59, 567–578. doi: 10.1007/s12031-016-0779-7
Zhang, J., Cai, C. Y., Wu, H. Y., Zhu, L. J., Luo, C. X., and Zhu, D. Y. (2016). CREB-mediated synaptogenesis and neurogenesis is crucial for the role of 5-HT1a receptors in modulating anxiety behaviors. Sci. Rep. 6:29551. doi: 10.1038/srep29551
Zheng, W., Wang, H., Zeng, Z., Lin, J., Little, P. J., Srivastava, L. K., et al. (2012). The possible role of the Akt signaling pathway in schizophrenia. Brain Res. 1470, 145–158. doi: 10.1016/j.brainres.2012.06.032
Keywords: CREB, schizophrenia, neurotransmitter, neurodevelopment, dopamine
Citation: Wang H, Xu J, Lazarovici P, Quirion R and Zheng W (2018) cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 11:255. doi: 10.3389/fnmol.2018.00255
Received: 03 April 2018; Accepted: 06 July 2018;
Published: 30 August 2018.
Edited by:
Erik Maronde, Goethe-Universität Frankfurt am Main, GermanyReviewed by:
Faramarz Dehghani, Martin Luther University Halle-Wittenberg, GermanyEtsuro Ito, Waseda University, Japan
Copyright © 2018 Wang, Xu, Lazarovici, Quirion and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenhua Zheng, d2VuaHVhemhlbmdAdW1hYy5tbw==