 Michelle E. Watts
Michelle E. Watts Roger Pocock
Roger Pocock Charles Claudianos
Charles Claudianos
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 22 June 2018
Sec. Molecular Signalling and Pathways
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00216
Dynamic metabolic changes occurring in neurons are critically important in directing brain plasticity and cognitive function. In other tissue types, disruptions to metabolism and the resultant changes in cellular oxidative state, such as increased reactive oxygen species (ROS) or induction of hypoxia, are associated with cellular stress. In the brain however, where drastic metabolic shifts occur to support physiological processes, subsequent changes to cellular oxidative state and induction of transcriptional sensors of oxidative stress likely play a significant role in regulating physiological neuronal function. Understanding the role of metabolism and metabolically-regulated genes in neuronal function will be critical in elucidating how cognitive functions are disrupted in pathological conditions where neuronal metabolism is affected. Here, we discuss known mechanisms regulating neuronal metabolism as well as the role of hypoxia and oxidative stress during normal and disrupted neuronal function. We also summarize recent studies implicating a role for metabolism in regulating neuronal plasticity as an emerging neuroscience paradigm.
Regulation of tissue metabolite supply and cellular energy metabolism is essential to maintain healthy cellular and systemic function. This regulation is especially critical to the central nervous system (CNS) where energy consumption is highly dynamic. Within the brain, increased neuronal activity drives increased energy consumption and compensatory metabolic and vasculature changes in turn enhance neuronal function (Roy and Sherrington, 1890). Normal brain function therefore requires metabolism to be tightly regulated both temporally and spatially from a regional level down to the level of a single synapse. Currently our knowledge of the relationship between neuronal activity and oxygen metabolism is poorly understood and it is likely that numerous mechanisms and complex regulatory pathways are yet to be uncovered.
While making up only a small fraction of our total body mass, the brain represents the largest source of energy consumption—accounting for over 20% of total oxygen metabolism. Of this, it is estimated that neurons consume 75%–80% of energy produced in the brain (Hyder et al., 2013). This energy is primarily utilized at the synapse with a large proportion spent in restoration of neuronal membrane potentials following depolarization (Harris et al., 2012). Other neuronal functions such as vesicle recycling, neurotransmitter synthesis and axoplasmic transport also contribute to synaptic energy depletion and the requirement for an elevated metabolic rate in neurons (Attwell and Laughlin, 2001; Rangaraju et al., 2014; Pathak et al., 2015). Energy requirements are therefore not uniform throughout the brain but instead increased in localized regions dependent on neuronal activity. While mechanisms have been identified to modify oxygen supply to brain regions in response to activity there appears to be a role for hypoxia in modulating neuronal function and behavior. Disruption of oxygen metabolism and mitochondrial function are also consistent pathological features of various age-related neurodegenerative diseases associated with cognitive decline (Tabrizi et al., 2000; Silverman et al., 2001; Zhou et al., 2008). Despite this, the underlying molecular mechanisms preceding neurodegeneration remain relatively unknown. In recent years a number of studies have identified links between metabolically regulated genes and behavior, which may provide insight into understanding the role of neuronal oxidative metabolism in both health and disease.
To compensate for varying energy demands throughout the brain and to increase efficiency of metabolite supply, neurovascular and neurometabolic coupling mechanisms have evolved to enhance blood flow and utilization of metabolites in areas of neural activity.
Cerebral blood flow (CBF), blood volume, glucose consumption and oxygen metabolism are all increased within localized regions of activity following neuronal stimulation. Neurovascular coupling, first postulated by Roy and Sherrington (1890) forms the basis of many functional neuroimaging technologies, where areas of neuronal activity are detected by activity-coupled increases in local CBF. While there has been substantial research on neurovascular coupling since this finding, details of the molecular mechanisms are still being uncovered.
Significant evidence suggests neurovascular coupling is mediated through the free radical, nitric oxide (•NO) produced in neurons. Vasodilation is strongly stimulated by •NO through activation of the enzymatic •NO receptor, soluble guanylate cyclase (sGC), producing cGMP and leading to vasodilation by cGMP-dependent kinase signaling (Miki et al., 1977; Archer et al., 1994). Production of •NO by neuronal nitric oxide synthase (nNOS) is tightly coupled to glutamatergic excitation with activation of nNOS being linked to stimulation of ionotrophic glutamate receptors. This principally occurs through NMDA receptors (NMDA-R) due to strong binding between the NMDA-R clustering protein, post-synaptic density protein 95 (PSD-95), and nNOS (Garthwaite et al., 1988; Brenman et al., 1996). Evidence also suggests that •NO is able to spread rapidly beyond the area of directly activated neurons and is likely to be self-regulating as enhanced blood flow inactivates •NO signaling through increased erythrocyte-mediated scavenging of •NO (Steinert et al., 2008; Santos et al., 2011). Astrocytes also play a role in mediating CBF regulation during neuronal activation by triggering Ca2+ release within astrocytic end feet and inducing various downstream Ca2+ signaling pathways known to control vasodilation (Mulligan and MacVicar, 2004; Takano et al., 2006). It recently became clear that astrocytic Ca2+ signaling acts on contractile perictyes surrounding capillaries and not on arterioles (Mishra et al., 2016). The current view on neurovascular coupling, therefore, is that increased CBF is triggered by astrocytic Ca2+ signaling in the capillary bed and by neuronal •NO generated through NMDA-R activation at the arteriolar level (Figures 1a,b; Peppiatt et al., 2006; Mishra et al., 2016).
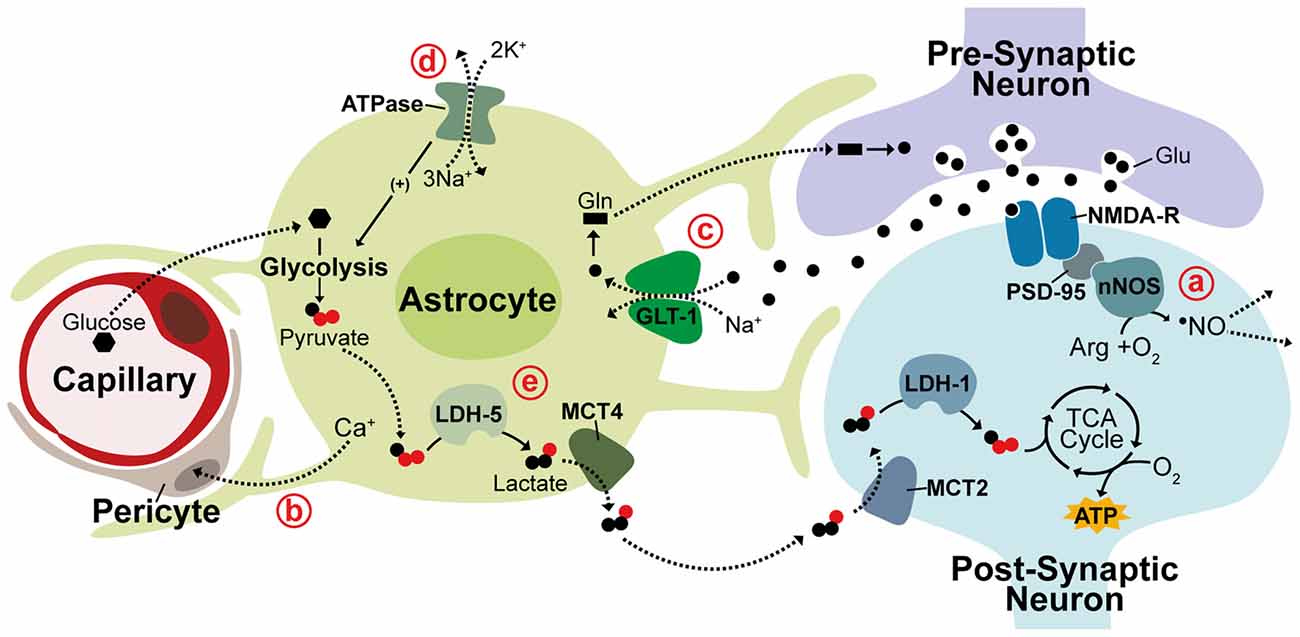
Figure 1. Neurovascular and neurometabolic coupling mechanisms. Schematic illustrating neuronal and astrocytic mechanisms responsible for activity-related blood flow and metabolic changes. (a) NMDA receptors (NMDA-R) are linked to neuronal nitric oxide synthase (nNOS) through post-synaptic density protein 95 (PSD-95) and neurovascular coupling during activity is thought to be triggered through the neuronally-produced vasodilator •NO, which can diffuse rapidly and freely through membranes to act on arterioles. (b) Vasodilation is also thought to be controlled at the capillary level through astrocytic Ca+ signaling acting on contractile perictyes. (c) In the glutamine-glutamate cycle, glutamate (Glu) released into the synaptic cleft is cleared by Na+-dependent astrocytic uptake, primarily through GLT-1. Glutamate is converted to glutamine (Gln) and returned to neurons to replenish neurotransmitter stores. (d,e) The astrocyte-neuron lactate shuttle (ANLS) hypothesis suggests associated increases in astrocytic Na+ concentration triggers activation of Na+/K+ ATPase pumps, promoting glucose uptake and glycolysis. Glycolytically-generated lactate is released and utilized as a substrate for oxidative phosphorylation in neurons during periods of activity. LDH, lactate dehydrogenase; MCT, monocarboxylate transporter. Solid lines indicate enzymatic activity, dashed lines indicate solute movement.
This synergistic function of astrocytes and neurons in CBF regulation is mirrored in their inverse yet complimentary metabolic profiles with astrocytes predominantly metabolizing glucose via glycolysis while neurons rely on oxidative metabolism (Kasischke et al., 2004). Astrocytes closely appose both capillary walls and synaptic clefts and are crucial regulators of neurometabolic coupling during neuronal activity. One of the best-characterized roles of astrocytes in neuronal activation is maintaining neurotransmitter stores through the glutamine-glutamate cycle. Glutamate released into the synaptic cleft during excitation is rapidly cleared by astrocytic uptake, primarily through the Na+-dependent glutamate transporter GLT-1 (EAAT2), causing attenuation of postsynaptic activation (Figure 1c; Danbolt et al., 1992; Bergles and Jahr, 1997). Cleared glutamate is primarily converted by astrocytes into glutamine, which is then released back into extracellular space for neuronal re-uptake and conversion back to glutamate (Hertz et al., 1978; Kvamme, 1998). In the astrocyte-neuron lactate shuttle (ANLS) hypothesis, proposed by Pellerin and Magistretti (1994), a secondary effect of astrocytic glutamate uptake prompts a switch from oxidative metabolism to aerobic glycolysis in astrocytes causing glucose metabolism to be diverted from the tricaboxcylic acid (TCA) cycle to the glycolytic pathway and lactate production. This switch is thought to be triggered by the associated intracellular increase in Na+ concentration, which activates Na+/K+-ATPase pumps stimulating glucose uptake and glycolysis (Figure 1d; Pellerin and Magistretti, 1997). This adaptation seems to support an increased neuronal metabolic load with lactate generated from astrocytic glycolysis being utilized as a substrate for oxidative metabolism in neurons. This hypothesis is supported by numerous studies detecting increased lactate in regions of brain activity as well as evidence that lactate is crucial for synaptic transmission in rat hippocampal slices and sufficient to support synaptic activity in the absence of glucose (Figure 1e; Schurr et al., 1988, 1999; Frahm et al., 1996; Maddock et al., 2009; Suzuki et al., 2011; Schaller et al., 2014; Machler et al., 2016).
This segregated metabolism is supported by distinct gene expression patterns observed in neurons and astrocytes. Differential expression of lactate transporter proteins, monocarboxylate transporters (MCTs), supports shuttling of lactate from astrocytes to neurons. The lactate efflux transporter MCT4 is expressed primarily in astrocytes while MCT2, an isoform that allows for rapid substrate uptake of lactate, is primarily expressed in neurons (Debernardi et al., 2003; Rafiki et al., 2003). Additionally, the lactate dehydrogenase (LDH) isoenzyme, LDH-5, which promotes conversion of pyruvate to lactate is highly expressed in astrocytes but not in neurons while LDH-1, which promotes pyruvate production is found in both neurons and astrocytes (Bittar et al., 1996; Bröer et al., 1997). In support of glycolysis induction in astrocytes, the pyruvate dehydrogenase kinase-4 (PDK4) is expressed at high levels in astrocytes causing its target, pyruvate dehydrogenase (PDH), to remain in an inactive, phosphorylated state thereby decreasing pyruvate entry into the TCA cycle (Halim et al., 2010; Zhang et al., 2014). Correspondingly, astrocytes express higher levels of the glyoxalase enzymes Glo-1 and Glo-2 that detoxify methyglycoxal, a metabolic by-product of glycolysis (Belanger et al., 2011). An enzymatic promoter of glycolysis, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphate 3 (Pfkfb3), is also found to be functional in astrocytes but subject to constant degradation in neurons contributing to the diversion of neuronal glucose from glycolysis to the pentose-phosphate pathway (PPP; Herrero-Mendez et al., 2009; Belanger et al., 2011; Zhang et al., 2014). While there is substantial evidence in support for the ANLS acting as a mechanism for coupling of neuronal activity to neuronal metabolism, contradictory evidence continues the debate of this hypothesis. Glucose uptake and phosphorylation has been shown to preferentially occur in neurons, not astrocytes. Further, neurons metabolize substantial amounts of glucose and increase glucose metabolism in response to activity (Patel et al., 2014; Lundgaard et al., 2015). This contradictory evidence may be due to metabolism being differentially regulated within different neural networks or brain regions. These observations all contribute, however, to mounting evidence suggesting that neurons can sustain and enhance oxidative metabolism to meet energetic requirements during periods of activity.
While there is significant evidence to support enhanced neuronal oxidative metabolism during activity, what remains unclear is what is happens to cellular oxygen concentration following activation. This is partly due to difficulties in recording oxygen concentration as well as from confounds in interpreting oxygen consumption imaging signals. Blood-oxygen-level dependent (BOLD) fMRI which relies on neurovascular coupling to measure regions of brain activity based on measurements of oxyhemeoglobin and deoxyhemeoglobin consistently generates signals with a post-stimulus undershoot (van Zijl et al., 2012). The physiological basis of the BOLD undershoot is heavily debated and is likely stimulus-dependent, one theory however suggests that the BOLD undershoot reflects an uncoupling of CBF and energy metabolism. This is supported by evidence that oxidative metabolism remains elevated post activation after both blood flow and blood volume have returned to baseline (Lu et al., 2004). Consistent with this, numerous studies have reported similar increases in oxidative metabolism indicating that sustained focal activation raises the rate of oxidative metabolism to a new steady state level (Hoge et al., 2005; Mangia et al., 2007; Frahm et al., 2008; Donahue et al., 2009; Lin et al., 2010). With dynamic changes in oxygen metabolism occurring during neuronal activity, dynamic changes are likely to be reflected in levels of oxygen concentration, potentially having secondary effects on protein function and gene expression.
Neurons and neuronal functions are generally viewed as highly sensitive to hypoxia with disruption of oxygen supply to the brain causing detrimental damage within minutes. Although there is not a clearly defined “critical” oxygen tension (PtO2) at which hypoxic damage will occur in neurons, in rat cortex a PtO2 value between 6.8 mm Hg and 8.8 mm Hg has been estimated as a PtO2 where oxidative metabolism will be disrupted (Rolett et al., 2000). Under physiological conditions, PtO2 measurements in rat range from 6 mm Hg to 40 mm Hg within the cortex (6–16 mm Hg in white matter and 19–40 mm Hg in gray matter) and from 1 mm Hg to 60 mm Hg across all brain regions with proximal structures displaying large variations in oxygen tension (Erecińska and Silver, 2001). During embryonic development, oxygen tension is low in the fetal brain (0.076–7.6 mm Hg) and hypoxia is essential for proper embryo morphological development. Within the developing brain, oxygen tension acts as a regulator of neurogenesis with low oxygen promoting progenitor expansion in cortical neurogenic regions and decreasing dopaminergic neurogenesis in the midbrain (Wagenführ et al., 2015, 2016). Additionally, in the adult brain, hypoxic injury caused by ischemic stroke triggers increased neuronal stem cell proliferation and neurogenesis (Arvidsson et al., 2002; Macas et al., 2006; Martí-Fàbregas et al., 2010). This evidence supports a role for hypoxia as a regulatory mechanism in neuronal function and indicates that physiological hypoxia occurring in the adult brain may play a functional role.
Long-term changes in cellular response to hypoxia are mediated through changes in gene expression with hypoxia predicted to regulate around 1%–1.5% of the genome, primarily through the hypoxia-inducible factors (HIFs; Koong et al., 2000; Denko et al., 2003). HIF is a heterodimeric complex consisting of a constitutively expressed β subunit shared by a family of three oxygen-sensitive α subunits. Most widely studied among these is the HIF-1α subunit. HIFα protein is constitutively expressed but is immediately targeted for degradation by HIF prolyl hydroxylases (PHDs) that associate with and hydroxylate two conserved HIFα proline residues in an oxygen dependent manner (Bruick and McKnight, 2001). The Von Hippel-Lindau tumor suppressor ubiquitin ligase complex (pVHL), subsequently recognizes HIFα causing HIFα ubiquitination and protein degradation (Ivan et al., 2001; Jaakkola et al., 2001). During hypoxia, though oxygen-limited inactivation of HIF PHD activity, HIFα is no longer targeted by pVHL and is able to accumulate in the cytoplasm before translocating to the nucleus and acting to promote transcription (Figure 2). Within the nervous system HIF-1α and target genes of HIF-1 are widely expressed under hypoxia, but regulation of HIF-1α can differ among neuronal subtypes (Bergeron et al., 1999; Stroka et al., 2001). Following hypoxia, HIF-1α has been shown both in vitro and in vivo to be significantly upregulated in interneurons but not in pyramidal neurons and in neuronal and non-neuronal cells it has been established that the redox state of a cell contributes to HIF-1α regulation (Welsh et al., 2002; Ramamoorthy and Shi, 2014). Additionally, during in C. elegans development, hypoxia has been shown to cause defects in axonal migration that occur in a neuronal cell-type specific manner and are dependent on stabilization of Hif-1 by either hypoxia or increased reactive oxygen species (ROS; Pocock and Hobert, 2008). Being a primary source of reducing agents, glucose is a major contributor to the redox state of a cell and HIF-1α expression in neurons has been shown to increase in a glucose-dependent manner during hypoxia (Shi and Liu, 2006; Guo et al., 2008). There is also a negative relationship between HIF-1α and ROS levels indicating ROS promotes HIF-1α degradation while a reducing environment stabilizes HIF-1α (Schafer and Buettner, 2001; Niecknig et al., 2012).
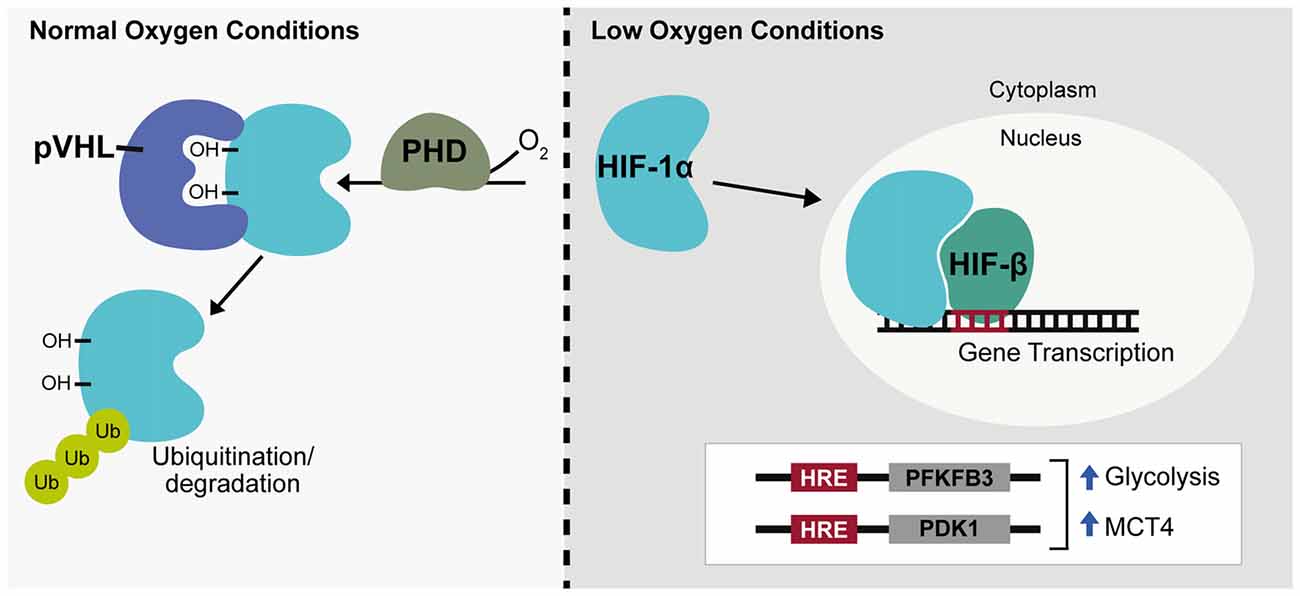
Figure 2. Hypoxia inducible transcription factor regulation. Under normal oxygen conditions hypoxia-inducible factor-1α (HIF-1α) is hydroxylated by prolyl hydroxylase (PHD) enzymes and targeted for ubiquitination by the Von Hippel-Lindau tumor suppresser ubiquitin ligase complex (pVHL). During hypoxia or low oxygen conditions, HIF-1α is stabilized, translocates to the nucleus and associates with HIF-β to promote gene expression, targeting genes containing a hypoxia response element (HRE). HIF-1α acts as a glycolytic enhancer through transcriptional activation of metabolic genes including 6-phosphofructo-2-kinase/fructose-2,6-bisphosphate 3 (PFKFB3) and pyruvate dehydrogenase kinase-1 (PDK1), both positive regulators of glycolysis and monocarboxylate transporter 4 (MCT4), the lactate efflux transporter. Ub, ubiquitin; OH, hydroxyl group.
ROS are highly reactive free radical molecules that can cause cellular damage through oxidation of lipids, proteins and DNA. ROS production primarily occurs through electron leakage at electron transport chain (ETC) complexes I or III during normal oxidative respiration. This causes conversion of 1%–2% of oxygen into the superoxide anion, a precursor to hydrogen peroxide and hydroxyl free radicals. Within the brain, a high neuronal oxidative rate heightens the potential for ROS production and neurons are especially vulnerable to oxidative damage due to low levels of antioxidant enzymes such as glutathione (GSH; Dringen et al., 1999). Neuronal diversion of glucose catabolism from glycolysis to the PPP through Pfkfb3 degradation therefore not only supports oxidative metabolism of lactate but also enhances neuronal antioxidant capacity through production of the reducing agent, NADH. HIF-1α is also involved in this process and acts as a glycolytic enhancer through transcriptional activation of metabolic genes including Pfkfb3 and pyruvate dehydrogenase kinase-1 (PDK1), both positive regulators of glycolysis and the lactate efflux transporter, MCT4 (Figure 2; Minchenko et al., 2002; Kim et al., 2006; Ullah et al., 2006).
As an oxygen-sensitive molecule, which is highly integrated into metabolic processes, HIF-1α is likely to have an important role in brain plasticity, and dysregulation of HIF-1α expression has already been implicated in neuronal activation and learning and memory. In a rat microarray study, seizures induced by injection of Kainate, a potent glutamate-receptor agonist that causes overstimulation of neurons, resulted in a 2.2-fold increase in HIF-1α after 24 h (Hunsberger et al., 2005). In another microarray study HIF-1α was found to be increased 7-fold in mice following environmental enrichment, where mice are exposed to heightened sensory stimulation known to promote neurogenesis and improve performance in memory tasks (Rampon et al., 2000). Elevated HIF-1α levels have also been observed in rats following learning in the Morris water maze and analysis of genes upregulated at early-time points following Morris water maze tests has found an over-representation of HIF-binding sites, hypoxia response elements (HREs), in their promoters (O’Sullivan et al., 2007). These data support a significant role for hypoxia in neuronal activity, potentially though neurovascular uncoupling and enhanced neuronal oxidative metabolism depleting neuronal oxygen levels.
Neurodegenerative disorders encompass a range of conditions characterized by progressive neuronal damage and degeneration as well as neuronal cell death. Although neurodegenerative disorders vary in the neuronal populations and cognitive or motor functions affected, metabolic dysfunction is a unifying pathology underlying many of these disorders. The most prevalent and most extensively studied of these is Alzheimer’s disease (AD) occurring in around 1:10 people aged over 65. AD principally affects short-term working memory and is classified by the presence of two hallmark neuropathologies; extracellular amyloid plaques, formed from aggregation of amyloid (Aβ) peptide, and intraneuronal neurofibrillary tangles formed from aggregation of hyperphosphorylated tau. In AD patients, regional hypometabolism in the brain is a predictor for progressive cognitive decline and reduced cerebral metabolism is associated with carriers of the AD risk allele of the APOE-4 gene (Small et al., 1995; Silverman et al., 2001). At the cellular level, mitochondria (MC) isolated from AD patients display reduced enzymatic activity of the ETC complex IV (cytochrome C oxidase; Parker et al., 1990; Parker and Parks, 1995). Similarly, in mouse models of AD, oxidative respiration is diminished and Aβ is found to localize and progressively accumulate in neuronal MC (Mucke et al., 2000; Manczak et al., 2006; Rhein et al., 2009; Yao et al., 2009). This progressive accumulation of Aβ in MC is associated with reduced oxidative respiration and reduced activity of the rate-limiting TCA cycle enzyme, α-ketoglutarate dehydrogenase complex (KGDHC), and the pyruvate dehydrogenase complex (PDHC), which generates acetyl-CoA for entry into the TCA cycle (Casley et al., 2002). Both metabolic dysfunction and mitochondrial Aβ accumulation appear to occur early in disease progression, preceding the onset of extracellular plaque formation (Wirths et al., 2001; Du et al., 2010). This indicates that early metabolic dysfunction is a key process in AD progression and a potential target for therapeutic intervention.
Also preceding extracellular plaque formation in the AD brain significantly increased ROS production and oxidative stress. Substantially increased ROS activity and oxidative damage is consistently detected in AD patients by various measures (Hensley et al., 1995; Gabbita et al., 1998; Praticò et al., 1998; Calingasan et al., 1999; Greilberger et al., 2008). Increased oxidative stress occurs early in disease progression being observed in patients with mild AD as well as in cases of mild cognitive impairment, at high-risk of developing AD (Baldeiras et al., 2008). The pathological Aβ is also known to be a source of ROS production and a cause of neuronal oxidative damage in AD (Behl et al., 1994; Harris et al., 1995; Bianca et al., 1999).
Related to oxidative stress, and also implicated in AD pathology, is dysregulated homeostasis of redox transition metal ions including zinc, copper and iron (Schrag et al., 2011; Ventriglia et al., 2012; Ayton et al., 2015). Both elevation and deficiency of zinc is associated with AD and evidence suggests that altered compartmentalization of zinc rather than altered zinc levels may be the cause of zinc pathology in AD (Suh et al., 2000; Schrag et al., 2011). This is supported by dysregulation of numerous zinc transporters in AD patient brains (Lovell et al., 2005, 2006; Beyer et al., 2009). Zinc has important roles in normal neuronal function and is co-released along with glutamate at the synapse (Vogt et al., 2000). A major role of zinc is its significant antioxidant capacity, such that zinc deficiency is linked to neuronal oxidative stress (Aimo et al., 2010). Like zinc, copper elevation and copper deficiency have both been associated with AD as well as co-localization of copper with Aβ plaques (Miller et al., 2006; Schrag et al., 2011; Ventriglia et al., 2012). Copper is also modulated by synaptic activation in neurons and both zinc and copper are able to bind Aβ (Schlief et al., 2005; Tõugu et al., 2008). In AD pathology, copper enhances Aβ toxicity and copper:Aβ complexes are a source of ROS production and oxidative damage in neurons (Dikalov et al., 2004; Liu et al., 2008; Ellis et al., 2010).
The redox active iron, although vital for cellular function, is also a pro-oxidant and promotes generation of highly reactive hydroxyl radicals from hydrogen peroxide. Elevated levels of brain iron in the AD brain as well as iron association with Aβ plaques and neurofibrillary tangles have been detected in various studies (Smith et al., 1997; Bartzokis et al., 2000; Raven et al., 2013). Recently, elevated iron has been shown to predict AD progression and elevated iron was linked to the APOE-4 AD risk allele suggesting it may have a pathological role in AD (Ayton et al., 2015).
Another common feature of AD that contributes to AD pathology is vascular dysfunction. Cerebrovascular disease, characterized by disrupted blood flow to the brain, significantly increases AD risk and occurs before Aβ accumulation and cognitive decline (Arvanitakis et al., 2016). In animal models, hypoperfusion also leads to symptoms similar to AD and exacerbates existing AD pathology (Walsh et al., 2002; Wang et al., 2010b). Vascular dysfunction contributes to the pathology of AD due to lower capillary density, meaning narrowed blood vessels and decreased CBF (Hamel et al., 2008). Diminished blood flow reduces metabolite and oxygen supply to the brain and potentially contributes to build-up of Aβ through impaired clearance of neurotoxic molecules (Shibata et al., 2000; Kumar-Singh et al., 2005). Aβ itself is also thought to amplify deficits in CBF and glucose utilization in AD through impairing vasodilation and cerebrovascular autoregulatory mechanisms (Niwa et al., 2002). Cerebrovascular dysfunction can lead to disrupted oxygen metabolism through hypoperfusion-hypoxia and hypoxia in-turn can enhance AD pathology by promoting tau phosphorylation as well as transcriptionally upregulating the HIF-1 target, β-site β-amyloid precursor protein cleavage enzyme 1 (BACE1) that cleaves amyloid precursor protein (APP) to produce Aβ (Figure 3; Sun et al., 2006).
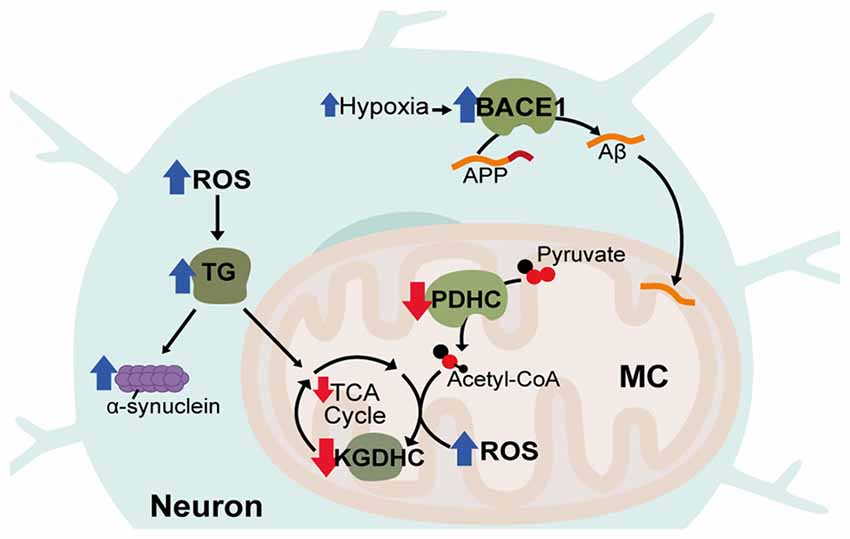
Figure 3. Disrupted metabolic pathways in neurodegenerative diseases. Hypoxia associated with Alzheimer’s Disease (AD) leads to increases in the HIF-1α target, β-site β-amyloid precursor protein cleavage enzyme 1 (BACE1), which cleaves amyloid precursor protein (APP) to produce Aβ. Aβ accumulates in neuronal mitochondria (MC) early in disease progression and disrupts oxidative metabolism. Acetyl-CoA production and tricaboxcylic acid (TCA) cycle entry is decreased in AD through reduced activity of the pyruvate dehydrogenase complex (PDHC). In all three diseases, activity of α-ketoglutarate dehydrogenase complex (KGDHC) is reduced, reactive oxygen species (ROS) is increased and transglutaminase (TG) activity is increased. TG increases α-synuclein aggregation and reduces oxidative respiration.
Aside from rare cases of genetic mutations in familial AD, the major risk factor for developing AD is aging. Correspondingly, AD, shares a number of similarities with other late-onset neurodegenerative disorders including Parkinson’s Disease (PD) and Huntington’s disease (HD). PD is thought to be caused by both genetic and environmental factors and primarily impacts patient motor function. PD involves the formation of protein aggregates consisting mainly of α-synuclein and affects the dopaminergic neurons of the midbrain substantia nigra. HD is an inherited neurodegenerative disorder caused by expanded CAG repeats in the Huntingtin (HTT) gene causing progressive neuronal degeneration and cell death throughout the brain, affecting mood, cognition and motor skills. Inclusions are also found in the HD brain from aggregation of mutant HTT (mHTT) protein. Like AD, both PD and HD are associated with increased oxidative stress as well as decreased activity of the KGDHC enzyme (Tabrizi et al., 2000; Gibson et al., 2003; Klivenyi et al., 2004; Zhou et al., 2008). Also, common to all three disorders is increased activity of transglutaminase (TG; Johnson et al., 1997; Junn et al., 2003; Jeitner et al., 2008). TG catalyzes polyamination post-translational modifications of proteins, is known to be increased by ROS and also attenuates HIF-1 signaling (Campisi et al., 2004; Filiano et al., 2008). TG can decrease oxidative metabolism through modification of glycolytic enzymes and is known to cause oxidative stress in HD and aggregation of α-synuclein in PD (Cooper et al., 1997; Junn et al., 2003; Kim et al., 2005).
Mutations in mitochondrial genes have also been identified in cases of familial PD and exposure to the neurotoxin MPP+, which inhibits ETC Complex I and therefore oxidative respiration, causes permanent Parkinsonism (Langston et al., 1983; Parker and Parks, 2005; Plun-Favreau et al., 2007). Altered metal ion homeostasis may have a role in PD pathology as well with disrupted levels of both zinc and copper observed in PD patients (Brewer et al., 2010; Davies et al., 2014). Similar to Aβ, copper also contributes to α-synuclein aggregation and can contribute to oxidative stress through the formation of reactive copper: α-synuclein complexes (Wang et al., 2010a; Dell’Acqua et al., 2015). α-synuclein is also know to exacerbate mitochondrial dysfunction in the presence of toxic oxidizing agents, with loss of α-synuclein in animal models conferring resistance to mitochondrial toxins (Klivenyi et al., 2006; Norris et al., 2007). Additionally, levels of α-synuclein are increased when oxidative metabolism is inhibited and animal models expressing mutant forms of α-synuclein exhibit neuronal mitochondrial degeneration and cell death (Lee et al., 2002; Martin et al., 2006). In HD, increased oxidative damage to mitochondrial DNA is observed as well as higher frequencies of deletions in the mitochondrial genome and deficits in ETC function with decreased expression of complex II in the striatum and decreased activity of complex IV in striatal and cortical regions (Horton et al., 1995; Polidori et al., 1999). Neuronal mitochondrial permeability is also disrupted by the mHTT protein through increasing sensitivity of the permeability transition pore to Ca2+ concentration, leading to mitochondrial dysfunction and decreased ATP production (Brustovetsky et al., 2003; Milakovic et al., 2006). Vascular deficits and disrupted blood flow is a major pathology of HD as well with altered blood vessel density and size found in cortical gray matter, putamen and striatal brain regions. In HD patients, inclusions of mHTT are also detected in the basal membrane and epithelium of cortical blood vessels and in mouse models of the disease pericytic coverage of cortical and striatal blood vessels is decreased (Drouin-Ouellet et al., 2015; Hsiao et al., 2015).
A number of the metabolic pathologies observed in neurodegenerative disorders are associated with normal aging and may explain the age-related manifestation of neurodegenerative disease phenotypes. While no longer thought to be directly causative of aging, free radicals and oxidative stress accumulate in the aging brain as in neurodegeneration (Smith et al., 1992). Mitochondrial function is also linked to aging due to the association of mitochondrial DNA (mtDNA) haplotypes with longevity and the generation of mtDNA mutator mice that have a premature aging phenotype (Trifunovic et al., 2004; Alexe et al., 2007; Bilal et al., 2008). It has also been shown there is an increased rate of damaging mutations in mtDNA of post-mitotic aging cells as opposed to aging mitotic cells (Greaves et al., 2012). While it has been suggested that the somatic rate of mtDNA mutation is unlikely to have a pathological affect due to redundancy in cell mitochondrial numbers, in post-mitotic neurons mtDNA mutation rates are significantly higher than average and, within the cortex, MC with large mtDNA deletions possess a replicative advantage during mitochondrial expansion (Song et al., 2005; Bender et al., 2006; Kraytsberg et al., 2006; Fukui and Moraes, 2009). Aside from AD and PD, deficiency of zinc is also associated with aging, being decreased in the general elderly population (Pepersack et al., 2001). Diminished CBF occurs in normal aging as well with cortical perfusion found to decrease with age in healthy adults (Chen et al., 2011). An age-dependent reduction in perictyes also occurs in mice and is associated with microvascular changes and neurodegeneration (Bell et al., 2010). Substantial evidence therefore exists supporting disrupted neuronal oxygen supply and oxidative metabolism as a major pathological component of age-related neurodegeneration.
Although it has been well established that metabolic regulation is critical to neuronal function and that metabolic dysfunction is a major pathology in diseases affecting behavior and cognition, there is little known regarding how regulators of metabolism may be involved in neuronal plasticity. A number of studies, however, support a direct role for metabolic regulation and metabolically linked genes in influencing learning and memory. One of the best examples of this is exposure of hypoxia as a modulator of cognitive performance. In C. elegans, hypoxia acts as an enhancer of gustatory sensory perception through Hif-1 dependent induction of the neurotransmitter serotonin within specific sensory neurons (Pocock and Hobert, 2010). In rodent models, exposure to hypobaric hypoxia in adult rats for periods of 7–21 days causes decline in spatial learning similar to aging and is associated with aging-related lipofuscin deposition and ultrastructural changes in MC. Increasing duration of hypobaric hypoxic exposure also positively correlates with increasing expression of aging markers (Biswal et al., 2016). Brief hypoxic exposure (100 s) in rats also causes synaptic arrest of pyramidal CA1 hippocampal neurons and deficits in spatial memory that are both reversed by blockade of receptors for Adenosine, an inhibitory neurotransmitter (Sun et al., 2002). Intermittent hypoxia (90–120 s intervals of 6%–10% O2 for 10 h/day) also produces deficits in acquisition of spatial memory in adult rats that could be prevented by administration of antioxidant (Row et al., 2003; Ward et al., 2009). In contrast, long-term facilitation of motor output in adult rats is enhanced by intermittent hypoxia (3 × 3 min intervals, separated by 5 min hyperoxia) increasing both phrenic amplitude and burst frequency, which was not observed with a continuous hypoxia of the same cumulative duration (Baker and Mitchell, 2000). Differing effects of hypoxia in brain plasticity are likely related to differing exposures as well as measurement of different outputs. Interestingly, mild hypoxia preconditioning confers protection of cognitive abilities during subsequent exposure to severe hypoxia implicating a role for HIFs and transcriptional changes induced by mild hypoxia (Rybnikova et al., 2005). Indeed, neuronal knockout of HIF-1α in mice impairs spatial memory and the stabilization of HIF improves hippocampal memory in fear conditioning (Tomita et al., 2003; Adamcio et al., 2010). Similar learning deficits and age-related changes are also observed in a D-galactose induced model of aging where oxidative injury was the major stimulus for aging (Li et al., 2016).
In learning and memory studies using an inhibitory avoidance paradigm, changes in metabolic gene expression were observed at 24 h, with increased expression of Na+/K+ ATPase, Glut1, Glut3 and, most prominently, lactate transporters MCT1 and MCT4 detected, suggesting transcriptional modulation of neurometabolic coupling occurs following learning (Yao et al., 2009; Tadi et al., 2015). Altered expression of lactate metabolic enzymes and transporters is also related to stress induced improvements in cognitive function. Psychological stress, while harmful under chronic conditions, has evolved to enhance cognitive function and improve reactions to stressful situations through hypothalamic activation of adrenergic receptors and hypothalamic-pituitary-adrenal axis glucocorticoid production (Dong et al., 2017). In a mouse model of stress, induced by activation of the β2 adrenergic receptor (β2AR), cognitive function was improved with short-term (3–5 days) activation while longer activation (>6 days) was harmful. Improved cognitive function following short-term stress induction corresponds with β2AR-dependant increases in LDH A, MCT1 and MCT4 expression, the expression of which was modulated by β-arrestin-1 activation of HIF-1α, downstream of β2AR (Dong et al., 2017).
Altered expression of ETC oxidative phosphorylation genes is also associated with altered behavior in the honeybee. In a study exploring molecular profiles in aggressive honeybee behavior, oxidative phosphorylation was most significantly enriched in association with increased aggression. This was found to be true for aged bees that display increased aggressive behavior as well as following environmentally enhanced aggression by alarm pheromone exposure and genetic-related aggression occurring in the Africanized honeybee population (Alaux et al., 2009). Consistent with this, inhibition of oxidative phosphorylation by treatment with drugs targeting the TCA cycle increased aggression of honeybees measured using an intruder assay (Li-Byarlay et al., 2014). In the same study, cell-type-specific knockdown of ETC complex genes using GAL4 drivers in Drosophila found that neuron-specific, but not glia-specific knockdown of the complex I gene ND20-like, significantly increased aggressive lunging behavior in flies (Li-Byarlay et al., 2014).
Also involved in learning and memory are non-coding miRNA genes which are regulated during neuronal activity by various mechanisms and able to regulate translation of various downstream target genes. A number of miRNAs have been associated with plasticity including the hypoxia-regulated, HIF-1 target, miR-210 that is known to be involved in metabolic regulation. miR-210 is significantly upregulated 24 h after long-term memory formation in the honeybee using an olfactory conditioning paradigm. Upregulation of miR-210 correlated with downregulation of a number of metabolically linked protein-coding genes including Gapdh2, Glucose dehydrogenase, Laccase2 and Aldose reductase-like. Inhibition of miR-210 by treatment of honeybees with miR-210 antogmiR also resulted in reduced memory retention in the olfactory conditioning assay indicating a functional role in learning and memory (Cristino et al., 2014). Considering the sensitivity of neurons and neural structures to hypoxia, Cristino et al. (2014) suggest small changes to oxygen levels in metabolic activity neurons may induce expression of miR-210, which in turn targets key molecules, including plasticity molecules, asparagine synthetase (involved in the biosynthesis of Glutamate) and actin. A follow-up study found that in a human-derived neuronal cell-line, miR-210 targeted neurodegeneration-associated genes as well as other plasticity-related genes within the human transcriptome. This included a number of oxidative metabolism genes, the AD risk-gene APOE as well as the NMDA-R, GRINA, and the human actin homolog, ACTB (Watts et al., 2018). Another hypoxia-regulated miRNA, miR-181c, is also associated with modulating cognitive function in rats. In a model of chronic cerebral hypoperfusion miR-181c was continuously inhibited, correlating with upregulation of its plasticity-related target gene, TRIM2. Hypoperfusion in this model was associated with deficits in spatial learning that were ameliorated by hippocampal overexpression of miR-181c (Fang et al., 2017). These studies all provide support to the hypothesis that metabolically regulated genes are directly involved in the regulation of neuronal plasticity.
While neurovascular coupling mechanisms appear to maintain steady-state oxygen levels in the brain, it is becoming evident that neurovascular uncoupling may in fact have a physiological role in regulating plasticity via oxygen depletion and induction of downstream hypoxia response pathways. Disruptions to hypoxia and oxidative metabolism have also been extensively attributed to neurodegeneration pathology albeit, there is a lack of understanding, as to how these disruptions are triggered and how they may be therapeutically targeted to halt disease progression and improve cognitive and motor functions. Altered behavior, including learning and memory, associated with dysregulation of metabolic genes highlights the importance of understanding the role of oxygen metabolism in neuronal plasticity. Further elucidation of how the hypoxia response pathway and other metabolic genes are involved in neuronal function will be critical in determining the molecular links between cognitive function and oxidative metabolism. This in turn will help elucidate how disrupted metabolism can lead to cognitive deficits and neurodegenerative disease.
MW wrote the manuscript. RP and CC edited the manuscript.
MW was supported by an Australian Government Research Training Program Stipend Scholarship. RP was supported by a National Health and Medical Research Council Senior Research Fellowship (GNT1137645). CC was supported by an Australian Research Council Future Fellowship (FT110100292).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adamcio, B., Sperling, S., Hagemeyer, N., Walkinshaw, G., and Ehrenreich, H. (2010). Hypoxia inducible factor stabilization leads to lasting improvement of hippocampal memory in healthy mice. Behav. Brain Res. 208, 80–84. doi: 10.1016/j.bbr.2009.11.010
Aimo, L., Cherr, G. N., and Oteiza, P. I. (2010). Low extracellular zinc increases neuronal oxidant production through nadph oxidase and nitric oxide synthase activation. Free Radic. Biol. Med. 48, 1577–1587. doi: 10.1016/j.freeradbiomed.2010.02.040
Alaux, C., Sinha, S., Hasadsri, L., Hunt, G. J., Guzmán-Novoa, E., DeGrandi-Hoffman, G., et al. (2009). Honey bee aggression supports a link between gene regulation and behavioral evolution. Proc. Natl. Acad. Sci. U S A 106, 15400–15405. doi: 10.1073/pnas.0907043106
Alexe, G., Fuku, N., Bilal, E., Ueno, H., Nishigaki, Y., Fujita, Y., et al. (2007). Enrichment of longevity phenotype in mtDNA haplogroups D4b2b, D4a, and D5 in the Japanese population. Hum. Genet. 121, 347–356. doi: 10.1007/s00439-007-0330-6
Archer, S. L., Huang, J. M., Hampl, V., Nelson, D. P., Shultz, P. J., and Weir, E. K. (1994). Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. U S A 91, 7583–7587. doi: 10.1073/pnas.91.16.7583
Arvanitakis, Z., Capuano, A. W., Leurgans, S. E., Bennett, D. A., and Schneider, J. A. (2016). Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 15, 934–943. doi: 10.1016/s1474-4422(16)30029-1
Arvidsson, A., Collin, T., Kirik, D., Kokaia, Z., and Lindvall, O. (2002). Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 8, 963–970. doi: 10.1038/nm747
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Ayton, S., Faux, N. G., Bush, A. I., and Alzheimer’s Disease Neuroimaging Initiative (2015). Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 6:6760. doi: 10.1038/ncomms7760
Baker, T. L., and Mitchell, G. S. (2000). Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J. Physiol. 529, 215–219. doi: 10.1111/j.1469-7793.2000.00215.x
Baldeiras, I., Santana, I., Proenca, M. T., Garrucho, M. H., Pascoal, R., Rodrigues, A., et al. (2008). Peripheral oxidative damage in mild cognitive impairment and mild Alzheimer’s disease. J. Alzheimers Dis. 15, 117–128. doi: 10.3233/jad-2008-15110
Bartzokis, G., Sultzer, D., Cummings, J., Holt, L. E., Hance, D. B., Henderson, V. W., et al. (2000). in vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Arch. Gen. Psychiatry 57, 47–53. doi: 10.1001/archpsyc.57.1.47
Behl, C., Davis, J. B., Lesley, R., and Schubert, D. (1994). Hydrogen peroxide mediates amyloid β protein toxicity. Cell 77, 817–827. doi: 10.1016/0092-8674(94)90131-7
Belanger, M., Yang, J., Petit, J. M., Laroche, T., Magistretti, P. J., and Allaman, I. (2011). Role of the glyoxalase system in astrocyte-mediated neuroprotection. J. Neurosci. 31, 18338–18352. doi: 10.1523/JNEUROSCI.1249-11.2011
Bell, R. D., Winkler, E. A., Sagare, A. P., Singh, I., LaRue, B., Deane, R., et al. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. doi: 10.1016/j.neuron.2010.09.043
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Bergeron, M., Yu, A. Y., Solway, K. E., Semenza, G. L., and Sharp, F. R. (1999). Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur. J. Neurosci. 11, 4159–4170. doi: 10.1046/j.1460-9568.1999.00845.x
Bergles, D. E., and Jahr, C. E. (1997). Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19, 1297–1308. doi: 10.1016/s0896-6273(00)80420-1
Beyer, N., Coulson, D. T., Heggarty, S., Ravid, R., Irvine, G. B., Hellemans, J., et al. (2009). ZnT3 mRNA levels are reduced in Alzheimer’s disease post-mortem brain. Mol. Neurodegener. 4:53. doi: 10.1186/1750-1326-4-53
Bianca, V. D., Dusi, S., Bianchini, E., Dal Prà, I., and Rossi, F. (1999). β-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol. Chem. 274, 15493–15499. doi: 10.1074/jbc.274.22.15493
Bilal, E., Rabadan, R., Alexe, G., Fuku, N., Ueno, H., Nishigaki, Y., et al. (2008). Mitochondrial DNA haplogroup D4a is a marker for extreme longevity in Japan. PLoS One 3:e2421. doi: 10.1371/journal.pone.0002421
Biswal, S., Sharma, D., Kumar, K., Nag, T. C., Barhwal, K., Hota, S. K., et al. (2016). Global hypoxia induced impairment in learning and spatial memory is associated with precocious hippocampal aging. Neurob. Learn. Mem. 133, 157–170. doi: 10.1016/j.nlm.2016.05.011
Bittar, P. G., Charnay, Y., Pellerin, L., Bouras, C., and Magistretti, P. J. (1996). Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J. Cereb. Blood Flow Metab. 16, 1079–1089. doi: 10.1097/00004647-199611000-00001
Brenman, J. E., Chao, D. S., Gee, S. H., McGee, A. W., Craven, S. E., Santillano, D. R., et al. (1996). Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell 84, 757–767. doi: 10.1016/s0092-8674(00)81053-3
Brewer, G. J., Kanzer, S. H., Zimmerman, E. A., Molho, E. S., Celmins, D. F., Heckman, S. M., et al. (2010). Subclinical zinc deficiency in Alzheimer’s disease and Parkinson’s disease. Am. J. Alzheimers. Dis. Other Demen. 25, 572–575. doi: 10.1177/1533317510382283
Bröer, S., Rahman, B., Pellegri, G., Pellerin, L., Martin, J. L., Verleysdonk, S., et al. (1997). Comparison of lactate transport in astroglial cells and monocarboxylate transporter 1 (MCT 1) expressing Xenopus laevis oocytes. Expression of two different monocarboxylate transporters in astroglial cells and neurons. J. Biol. Chem. 272, 30096–30102. doi: 10.1074/jbc.272.48.30096
Bruick, R. K., and McKnight, S. L. (2001). A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340. doi: 10.1126/science.1066373
Brustovetsky, N., Brustovetsky, T., Purl, K. J., Capano, M., Crompton, M., and Dubinsky, J. M. (2003). Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J. Neurosci. 23, 4858–4867. doi: 10.1523/JNEUROSCI.23-12-04858.2003
Calingasan, N. Y., Uchida, K., and Gibson, G. E. (1999). Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer’s disease. J. Neurochem. 72, 751–756. doi: 10.1046/j.1471-4159.1999.0720751.x
Campisi, A., Caccamo, D., Li Volti, G., Currò, M., Parisi, G., Avola, R., et al. (2004). Glutamate-evoked redox state alterations are involved in tissue transglutaminase upregulation in primary astrocyte cultures. FEBS Lett. 578, 80–84. doi: 10.1016/j.febslet.2004.10.074
Casley, C. S., Canevari, L., Land, J. M., Clark, J. B., and Sharpe, M. A. (2002). β-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 80, 91–100. doi: 10.1046/j.0022-3042.2001.00681.x
Chen, J. J., Rosas, H. D., and Salat, D. H. (2011). Age-associated reductions in cerebral blood flow are independent from regional atrophy. Neuroimage 55, 468–478. doi: 10.1016/j.neuroimage.2010.12.032
Cooper, A. J., Sheu, K. R., Burke, J. R., Onodera, O., Strittmatter, W. J., Roses, A. D., et al. (1997). Transglutaminase-catalyzed inactivation of glyceraldehyde 3-phosphate dehydrogenase and α-ketoglutarate dehydrogenase complex by polyglutamine domains of pathological length. Proc. Natl. Acad. Sci. U S A 94, 12604–12609. doi: 10.1073/pnas.94.23.12604
Cristino, A. S., Barchuk, A. R., Freitas, F. C., Narayanan, R. K., Biergans, S. D., Zhao, Z., et al. (2014). Neuroligin-associated microRNA-932 targets actin and regulates memory in the honeybee. Nat. Commun. 5:5529. doi: 10.1038/ncomms6529
Danbolt, N. C., Storm-Mathisen, J., and Kanner, B. I. (1992). An [Na+ + K+] coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience 51, 295–310. doi: 10.1016/0306-4522(92)90316-t
Davies, K. M., Bohic, S., Carmona, A., Ortega, R., Cottam, V., Hare, D. J., et al. (2014). Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol. Aging 35, 858–866. doi: 10.1016/j.neurobiolaging.2013.09.034
Debernardi, R., Pierre, K., Lengacher, S., Magistretti, P. J., and Pellerin, L. (2003). Cell-specific expression pattern of monocarboxylate transporters in astrocytes and neurons observed in different mouse brain cortical cell cultures. J. Neurosci. Res. 73, 141–155. doi: 10.1002/jnr.10660
Dell’Acqua, S., Pirota, V., Anzani, C., Rocco, M. M., Nicolis, S., Valensin, D., et al. (2015). Reactivity of copper-α-synuclein peptide complexes relevant to Parkinson’s disease. Metallomics 7, 1091–1102. doi: 10.1039/c4mt00345d
Denko, N. C., Fontana, L. A., Hudson, K. M., Sutphin, P. D., Raychaudhuri, S., Altman, R., et al. (2003). Investigating hypoxic tumor physiology through gene expression patterns. Oncogene 22, 5907–5914. doi: 10.1038/sj.onc.1206703
Dikalov, S. I., Vitek, M. P., and Mason, R. P. (2004). Cupric-amyloid β peptide complex stimulates oxidation of ascorbate and generation of hydroxyl radical. Free Radic. Biol. Med. 36, 340–347. doi: 10.1016/j.freeradbiomed.2003.11.004
Donahue, M. J., Stevens, R. D., de Boorder, M., Pekar, J. J., Hendrikse, J., and van Zijl, P. C. (2009). Hemodynamic changes after visual stimulation and breath holding provide evidence for an uncoupling of cerebral blood flow and volume from oxygen metabolism. J. Cereb. Blood Flow Metab. 29, 176–185. doi: 10.1038/jcbfm.2008.109
Dong, J. H., Wang, Y. J., Cui, M., Wang, X. J., Zheng, W. S., Ma, M. L., et al. (2017). Adaptive activation of a stress response pathway improves learning and memory through Gs and β-arrestin-1-regulated lactate metabolism. Biol. Psychiatry 81, 654–670. doi: 10.1016/j.biopsych.2016.09.025
Dringen, R., Kussmaul, L., Gutterer, J. M., Hirrlinger, J., and Hamprecht, B. (1999). The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 72, 2523–2530. doi: 10.1046/j.1471-4159.1999.0722523.x
Drouin-Ouellet, J., Sawiak, S. J., Cisbani, G., Lagacé, M., Kuan, W. L., Saint-Pierre, M., et al. (2015). Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: potential implications for its pathophysiology. Ann. Neurol. 78, 160–177. doi: 10.1002/ana.24406
Du, H., Guo, L., Yan, S., Sosunov, A. A., McKhann, G. M., and Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U S A 107, 18670–18675. doi: 10.1073/pnas.1006586107
Ellis, G., Fang, E., Maheshwari, M., Roltsch, E., Holcomb, L., Zimmer, D., et al. (2010). Lipid oxidation and modification of amyloid-β (Aβ) in vitro and in vivo. J. Alzheimers Dis. 22, 593–607. doi: 10.3233/JAD-2010-100960
Erecińska, M., and Silver, I. A. (2001). Tissue oxygen tension and brain sensitivity to hypoxia. Respir. Physiol. 128, 263–276. doi: 10.1016/s0034-5687(01)00306-1
Fang, C., Li, Q., Min, G., Liu, M., Cui, J., Sun, J., et al. (2017). MicroRNA-181c ameliorates cognitive impairment induced by chronic cerebral hypoperfusion in rats. Mol. Neurobiol. 54, 8370–8385. doi: 10.1007/s12035-016-0268-6
Filiano, A. J., Bailey, C. D., Tucholski, J., Gundemir, S., and Johnson, G. V. (2008). Transglutaminase 2 protects against ischemic insult, interacts with HIF1β, and attenuates HIF1 signaling. FASEB J. 22, 2662–2675. doi: 10.1096/fj.07-097709
Frahm, J., Baudewig, J., Kallenberg, K., Kastrup, A., Merboldt, K. D., and Dechent, P. (2008). The post-stimulation undershoot in BOLD fMRI of human brain is not caused by elevated cerebral blood volume. Neuroimage 40, 473–481. doi: 10.1016/j.neuroimage.2007.12.005
Frahm, J., Kruger, G., Merboldt, K. D., and Kleinschmidt, A. (1996). Dynamic uncoupling and recoupling of perfusion and oxidative metabolism during focal brain activation in man. Magn. Reson. Med. 35, 143–148. doi: 10.1002/mrm.1910350202
Fukui, H., and Moraes, C. T. (2009). Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 18, 1028–1036. doi: 10.1093/hmg/ddn437
Gabbita, S. P., Lovell, M. A., and Markesbery, W. R. (1998). Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 71, 2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x
Garthwaite, J., Charles, S. L., and Chess-Williams, R. (1988). Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 336, 385–388. doi: 10.1038/336385a0
Gibson, G. E., Kingsbury, A. E., Xu, H., Lindsay, J. G., Daniel, S., Foster, O. J., et al. (2003). Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson’s disease. Neurochem. Int. 43, 129–135. doi: 10.1016/s0197-0186(02)00225-5
Greaves, L. C., Elson, J. L., Nooteboom, M., Grady, J. P., Taylor, G. A., Taylor, R. W., et al. (2012). Comparison of mitochondrial mutation spectra in ageing human colonic epithelium and disease: absence of evidence for purifying selection in somatic mitochondrial DNA point mutations. PLoS Genet. 8:e1003082. doi: 10.1371/journal.pgen.1003082
Greilberger, J., Koidl, C., Greilberger, M., Lamprecht, M., Schroecksnadel, K., Leblhuber, F., et al. (2008). Malondialdehyde, carbonyl proteins and albumin-disulphide as useful oxidative markers in mild cognitive impairment and Alzheimer’s disease. Free Radic. Res. 42, 633–638. doi: 10.1080/10715760802255764
Guo, S., Bragina, O., Xu, Y., Cao, Z., Chen, H., Zhou, B., et al. (2008). Glucose up-regulates HIF-1 α expression in primary cortical neurons in response to hypoxia through maintaining cellular redox status. J. Neurochem. 105, 1849–1860. doi: 10.1111/j.1471-4159.2008.05287.x
Halim, N. D., McFate, T., Mohyeldin, A., Okagaki, P., Korotchkina, L. G., Patel, M. S., et al. (2010). Phosphorylation status of pyruvate dehydrogenase distinguishes metabolic phenotypes of cultured rat brain astrocytes and neurons. Glia 58, 1168–1176. doi: 10.1002/glia.20996
Hamel, E., Nicolakakis, N., Aboulkassim, T., Ongali, B., and Tong, X. K. (2008). Oxidative stress and cerebrovascular dysfunction in mouse models of Alzheimer’s disease. Exp. Physiol. 93, 116–120. doi: 10.1113/expphysiol.2007.038729
Harris, M. E., Hensley, K., Butterfield, D. A., Leedle, R. A., and Carney, J. M. (1995). Direct evidence of oxidative injury produced by the Alzheimer’s β-amyloid peptide (1–40) in cultured hippocampal neurons. Exp. Neurol. 131, 193–202. doi: 10.1016/0014-4886(95)90041-1
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Hensley, K., Hall, N., Subramaniam, R., Cole, P., Harris, M., Aksenov, M., et al. (1995). Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 65, 2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x
Herrero-Mendez, A., Almeida, A., Fernández, E., Maestre, C., Moncada, S., and Bolanos, J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747–752. doi: 10.1038/ncb1881
Hertz, L., Schousboe, A., Boechler, N., Mukerji, S., and Fedoroff, S. (1978). Kinetic characteristics of the glutamate uptake into normal astrocytes in cultures. Neurochem. Res. 3, 1–14. doi: 10.1007/bf00964356
Hoge, R. D., Franceschini, M. A., Covolan, R. J., Huppert, T., Mandeville, J. B., and Boas, D. A. (2005). Simultaneous recording of task-induced changes in blood oxygenation, volume, and flow using diffuse optical imaging and arterial spin-labeling MRI. Neuroimage 25, 701–707. doi: 10.1016/j.neuroimage.2004.12.032
Horton, T. M., Graham, B. H., Corral-Debrinski, M., Shoffner, J. M., Kaufman, A. E., Beal, M. F., et al. (1995). Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology 45, 1879–1883. doi: 10.1212/wnl.45.10.1879
Hsiao, H. Y., Chen, Y. C., Huang, C. H., Chen, C. C., Hsu, Y. H., Chen, H. M., et al. (2015). Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann. Neurol. 78, 178–192. doi: 10.1002/ana.24428
Hunsberger, J. G., Bennett, A. H., Selvanayagam, E., Duman, R. S., and Newton, S. S. (2005). Gene profiling the response to kainic acid induced seizures. Mol. Brain Res. 141, 95–112. doi: 10.1016/j.molbrainres.2005.08.005
Hyder, F., Rothman, D. L., and Bennett, M. R. (2013). Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. U S A 110, 3549–3554. doi: 10.1073/pnas.1214912110
Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., et al. (2001). HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. doi: 10.1126/science.1059817
Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J., et al. (2001). Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. doi: 10.1126/science.1059796
Jeitner, T. M., Matson, W. R., Folk, J. E., Blass, J. P., and Cooper, A. J. (2008). Increased levels of γ-glutamylamines in Huntington disease CSF. J. Neurochem. 106, 37–44. doi: 10.1111/j.1471-4159.2008.05350.x
Johnson, G. V., Cox, T. M., Lockhart, J. P., Zinnerman, M. D., Miller, M. L., and Powers, R. E. (1997). Transglutaminase activity is increased in Alzheimer’s disease brain. Brain Res. 751, 323–329. doi: 10.1016/s0006-8993(96)01431-x
Junn, E., Ronchetti, R. D., Quezado, M. M., Kim, S. Y., and Mouradian, M. M. (2003). Tissue transglutaminase-induced aggregation of α-synuclein: implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U S A 100, 2047–2052. doi: 10.1073/pnas.0438021100
Kasischke, K. A., Vishwasrao, H. D., Fisher, P. J., Zipfel, W. R., and Webb, W. W. (2004). Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305, 99–103. doi: 10.1126/science.1096485
Kim, S. Y., Marekov, L., Bubber, P., Browne, S. E., Stavrovskaya, I., Lee, J., et al. (2005). Mitochondrial aconitase is a transglutaminase 2 substrate: transglutamination is a probable mechanism contributing to high-molecular-weight aggregates of aconitase and loss of aconitase activity in Huntington disease brain. Neurochem. Res. 30, 1245–1255. doi: 10.1007/s11064-005-8796-x
Kim, J. W., Tchernyshyov, I., Semenza, G. L., and Dang, C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. doi: 10.1016/j.cmet.2006.02.002
Klivenyi, P., Siwek, D., Gardian, G., Yang, L., Starkov, A., Cleren, C., et al. (2006). Mice lacking α-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 21, 541–548. doi: 10.1016/j.nbd.2005.08.018
Klivenyi, P., Starkov, A. A., Calingasan, N. Y., Gardian, G., Browne, S. E., Yang, L., et al. (2004). Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity. J. Neurochem. 88, 1352–1360. doi: 10.1046/j.1471-4159.2003.02263.x
Koong, A. C., Denko, N. C., Hudson, K. M., Schindler, C., Swiersz, L., Koch, C., et al. (2000). Candidate genes for the hypoxic tumor phenotype. Cancer Res. 60, 883–887. Available online at: http://cancerres.aacrjournals.org/content/60/4/883.long
Kraytsberg, Y., Kudryavtseva, E., McKee, A. C., Geula, C., Kowall, N. W., and Khrapko, K. (2006). Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38, 518–520. doi: 10.1038/ng1778
Kumar-Singh, S., Pirici, D., McGowan, E., Serneels, S., Ceuterick, C., Hardy, J., et al. (2005). Dense-core plaques in Tg2576 and PSAPP mouse models of Alzheimer’s disease are centered on vessel walls. Am. J. Pathol. 167, 527–543. doi: 10.1016/s0002-9440(10)62995-1
Kvamme, E. (1998). Synthesis of glutamate and its regulation. Prog. Brain Res. 116, 73–85. doi: 10.1016/s0079-6123(08)60431-8
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (1983). Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. doi: 10.1126/science.6823561
Lee, H. J., Shin, S. Y., Choi, C., Lee, Y. H., and Lee, S. J. (2002). Formation and removal of α-synuclein aggregates in cells exposed to mitochondrial inhibitors. J. Biol. Chem. 277, 5411–5417. doi: 10.1074/jbc.M105326200
Li, X., Chen, Y., Shao, S., Tang, Q., Chen, W., Chen, Y., et al. (2016). Oxidative stress induces the decline of brain EPO expression in aging rats. Exp. Gerontol. 83, 89–93. doi: 10.1016/j.exger.2016.07.012
Li-Byarlay, H., Rittschof, C. C., Massey, J. H., Pittendrigh, B. R., and Robinson, G. E. (2014). Socially responsive effects of brain oxidative metabolism on aggression. Proc. Natl. Acad. Sci. U S A 111, 12533–12537. doi: 10.1073/pnas.1412306111
Lin, A. L., Fox, P. T., Hardies, J., Duong, T. Q., and Gao, J. H. (2010). Nonlinear coupling between cerebral blood flow, oxygen consumption and ATP production in human visual cortex. Proc. Natl. Acad. Sci. U S A 107, 8446–8451. doi: 10.1073/pnas.0909711107
Liu, L., Komatsu, H., Murray, I. V., and Axelsen, P. H. (2008). Promotion of amyloid β protein misfolding and fibrillogenesis by a lipid oxidation product. J. Mol. Biol. 377, 1236–1250. doi: 10.1016/j.jmb.2008.01.057
Lovell, M. A., Smith, J. L., and Markesbery, W. R. (2006). Elevated zinc transporter-6 in mild cognitive impairment, Alzheimer disease, and pick disease. J. Neuropathol. Exp. Neurol. 65, 489–498. doi: 10.1097/01.jnen.0000229237.98124.91
Lovell, M. A., Smith, J. L., Xiong, S., and Markesbery, W. R. (2005). Alterations in zinc transporter protein-1 (ZnT-1) in the brain of subjects with mild cognitive impairment, early and late-stage Alzheimer’s disease. Neurotox. Res. 7, 265–271. doi: 10.1007/bf03033884
Lu, H., Golay, X., Pekar, J. J., and Van Zijl, P. C. (2004). Sustained poststimulus elevation in cerebral oxygen utilization after vascular recovery. J. Cereb. Blood Flow Metab. 24, 764–770. doi: 10.1097/01.wcb.0000124322.60992.5c
Lundgaard, I., Li, B., Xie, L., Kang, H., Sanggaard, S., Haswell, J. D., et al. (2015). Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun. 6:6807. doi: 10.1038/ncomms7807
Macas, J., Nern, C., Plate, K. H., and Momma, S. (2006). Increased generation of neuronal progenitors after ischemic injury in the aged adult human forebrain. J. Neurosci. 26, 13114–13119. doi: 10.1523/JNEUROSCI.4667-06.2006
Machler, P., Wyss, M. T., Elsayed, M., Stobart, J., Gutierrez, R., von Faber-Castell, A., et al. (2016). In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 23, 94–102. doi: 10.1016/j.cmet.2015.10.010
Maddock, R. J., Buonocore, M. H., Copeland, L. E., and Richards, A. L. (2009). Elevated brain lactate responses to neural activation in panic disorder: a dynamic 1H-MRS study. Mol. Psychiatry 14, 537–545. doi: 10.1038/sj.mp.4002137
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Mangia, S., Tkác, I., Gruetter, R., Van de Moortele, P. F., Maraviglia, B., and Uğurbil, K. (2007). Sustained neuronal activation raises oxidative metabolism to a new steady-state level: evidence from 1H NMR spectroscopy in the human visual cortex. J. Cereb. Blood Flow Metab. 27, 1055–1063. doi: 10.1038/sj.jcbfm.9600401
Martí-Fàbregas, J., Romaguera-Ros, M., Gómez-Pinedo, U., Martínez-Ramírez, S., Jiménez-Xarrié, E., Marín, R., et al. (2010). Proliferation in the human ipsilateral subventricular zone after ischemic stroke. Neurology 74, 357–365. doi: 10.1212/WNL.0b013e3181cbccec
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., et al. (2006). Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50. doi: 10.1523/JNEUROSCI.4308-05.2006
Miki, N., Kawabe, Y., and Kuriyama, K. (1977). Activation of cerebral guanylate cyclase by nitric oxide. Biochem. Biophys. Res. Commun. 75, 851–856. doi: 10.1016/0006-291x(77)91460-7
Milakovic, T., Quintanilla, R. A., and Johnson, G. V. (2006). Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: functional consequences. J. Biol. Chem. 281, 34785–34795. doi: 10.1074/jbc.M603845200
Miller, L. M., Wang, Q., Telivala, T. P., Smith, R. J., Lanzirotti, A., and Miklossy, J. (2006). Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 155, 30–37. doi: 10.1016/j.jsb.2005.09.004
Minchenko, A., Leshchinsky, I., Opentanova, I., Sang, N., Srinivas, V., Armstead, V., et al. (2002). Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J. Biol. Chem. 277, 6183–6187. doi: 10.1074/jbc.M110978200
Mishra, A., Reynolds, J. P., Chen, Y., Gourine, A. V., Rusakov, D. A., and Attwell, D. (2016). Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 19, 1619–1627. doi: 10.1038/nn.4428
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of aβ 1–b42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000
Mulligan, S. J., and MacVicar, B. A. (2004). Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431, 195–199. doi: 10.1038/nature02827
Niecknig, H., Tug, S., Reyes, B. D., Kirsch, M., Fandrey, J., and Berchner-Pfannschmidt, U. (2012). Role of reactive oxygen species in the regulation of HIF-1 by prolyl hydroxylase 2 under mild hypoxia. Free Radic. Res. 46, 705–717. doi: 10.3109/10715762.2012.669041
Niwa, K., Kazama, K., Younkin, L., Younkin, S. G., Carlson, G. A., and Iadecola, C. (2002). Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am. J. Physiol. Heart Circ. Physiol. 283, H315–H323. doi: 10.1152/ajpheart.00022.2002
Norris, E. H., Uryu, K., Leight, S., Giasson, B. I., Trojanowski, J. Q., and Lee, V. M. (2007). Pesticide exposure exacerbates α-synucleinopathy in an A53T transgenic mouse model. Am. J. Pathol. 170, 658–666. doi: 10.2353/ajpath.2007.060359
O’Sullivan, N. C., McGettigan, P. A., Sheridan, G. K., Pickering, M., Conboy, L., O’Connor, J. J., et al. (2007). Temporal change in gene expression in the rat dentate gyrus following passive avoidance learning. J. Neurochem. 101, 1085–1098. doi: 10.1111/j.1471-4159.2006.04418.x
Parker, W. D. Jr., Filley, C. M., and Parks, J. K. (1990). Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 40, 1302–1303. doi: 10.1212/wnl.40.8.1302
Parker, W. D. Jr., and Parks, J. K. (1995). Cytochrome C oxidase in Alzheimer’s disease brain: purification and characterization. Neurology 45, 482–486. doi: 10.1212/wnl.45.3.482
Parker, W. D. Jr., and Parks, J. K. (2005). Mitochondrial ND5 mutations in idiopathic Parkinson’s disease. Biochem. Biophys. Res. Commun. 326, 667–669. doi: 10.1016/j.bbrc.2004.11.093
Patel, A. B., Lai, J. C., Chowdhury, G. M., Hyder, F., Rothman, D. L., Shulman, R. G., et al. (2014). Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc. Natl. Acad. Sci. U S A 111, 5385–5390. doi: 10.1073/pnas.1403576111
Pathak, D., Shields, L. Y., Mendelsohn, B. A., Haddad, D., Lin, W., Gerencser, A. A., et al. (2015). The role of mitochondrially derived ATP in synaptic vesicle recycling. J. Biol. Chem. 290, 22325–22336. doi: 10.1074/jbc.M115.656405
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U S A 91, 10625–10629. doi: 10.1073/pnas.91.22.10625
Pellerin, L., and Magistretti, P. J. (1997). Glutamate uptake stimulates Na+,K+-ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J. Neurochem. 69, 2132–2137. doi: 10.1046/j.1471-4159.1997.69052132.x
Pepersack, T., Rotsaert, P., Benoit, F., Willems, D., Fuss, M., Bourdoux, P., et al. (2001). Prevalence of zinc deficiency and its clinical relevance among hospitalised elderly. Arch. Gerontol. Geriatr. 33, 243–253. doi: 10.1016/s0167-4943(01)00186-8
Peppiatt, C. M., Howarth, C., Mobbs, P., and Attwell, D. (2006). Bidirectional control of CNS capillary diameter by pericytes. Nature 443, 700–704. doi: 10.1038/nature05193
Plun-Favreau, H., Klupsch, K., Moisoi, N., Gandhi, S., Kjaer, S., Frith, D., et al. (2007). The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell Biol. 9, 1243–1252. doi: 10.1038/ncb1644
Pocock, R., and Hobert, O. (2008). Oxygen levels affect axon guidance and neuronal migration in Caenorhabditis elegans. Nat. Neurosci. 11, 894–900. doi: 10.1038/nn.2152
Pocock, R., and Hobert, O. (2010). Hypoxia activates a latent circuit for processing gustatory information in C. elegans. Nat. Neurosci. 13, 610–614. doi: 10.1038/nn.2537
Polidori, M. C., Mecocci, P., Browne, S. E., Senin, U., and Beal, M. F. (1999). Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci. Lett. 272, 53–56. doi: 10.1016/s0304-3940(99)00578-9
Praticò, D., Lee, V. M.-Y., Trojanowski, J. Q., Rokach, J., and Fitzgerald, G. A. (1998). Increased F2-isoprostanes in Alzheimer’s disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 12, 1777–1783. doi: 10.1096/fasebj.12.15.1777
Rafiki, A., Boulland, J. L., Halestrap, A. P., Ottersen, O. P., and Bergersen, L. (2003). Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience 122, 677–688. doi: 10.1016/j.neuroscience.2003.08.040
Ramamoorthy, P., and Shi, H. (2014). Ischemia induces different levels of hypoxia inducible factor-1α protein expression in interneurons and pyramidal neurons. Acta Neuropathol. Commun. 2:51. doi: 10.1186/2051-5960-2-51
Rampon, C., Jiang, C. H., Dong, H., Tang, Y. P., Lockhart, D. J., Schultz, P. G., et al. (2000). Effects of environmental enrichment on gene expression in the brain. Proc. Natl. Acad. Sci. U S A 97, 12880–12884. doi: 10.1073/pnas.97.23.12880
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Raven, E. P., Lu, P. H., Tishler, T. A., Heydari, P., and Bartzokis, G. (2013). Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J. Alzheimers Dis. 37, 127–136. doi: 10.3233/JAD-130209
Rhein, V., Song, X., Wiesner, A., Ittner, L. M., Baysang, G., Meier, F., et al. (2009). Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. U S A 106, 20057–20062. doi: 10.1073/pnas.0905529106
Rolett, E. L., Azzawi, A., Liu, K. J., Yongbi, M. N., Swartz, H. M., and Dunn, J. F. (2000). Critical oxygen tension in rat brain: a combined 31P-NMR and EPR oximetry study. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R9–R16. doi: 10.1152/ajpregu.2000.279.1.r9
Row, B. W., Liu, R., Xu, W., Kheirandish, L., and Gozal, D. (2003). Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am. J. Respir. Crit. Care Med. 167, 1548–1553. doi: 10.1164/rccm.200209-1050oc
Roy, C. S., and Sherrington, C. S. (1890). On the regulation of the blood-supply of the brain. J. Physiol. 11, 85.17–158.17. doi: 10.1113/jphysiol.1890.sp000321
Rybnikova, E., Vataeva, L., Tyulkova, E., Gluschenko, T., Otellin, V., Pelto-Huikko, M., et al. (2005). Mild hypoxia preconditioning prevents impairment of passive avoidance learning and suppression of brain NGFI-A expression induced by severe hypoxia. Behav. Brain Res. 160, 107–114. doi: 10.1016/j.bbr.2004.11.023
Santos, R. M., Lourenco, C. F., Pomerleau, F., Huettl, P., Gerhardt, G. A., Laranjinha, J., et al. (2011). Brain nitric oxide inactivation is governed by the vasculature. Antioxid. Redox Signal. 14, 1011–1021. doi: 10.1089/ars.2010.3297
Schafer, F. Q., and Buettner, G. R. (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191–1212. doi: 10.1016/s0891-5849(01)00480-4
Schaller, B., Xin, L., O’Brien, K., Magill, A. W., and Gruetter, R. (2014). Are glutamate and lactate increases ubiquitous to physiological activation? A 1H functional MR spectroscopy study during motor activation in human brain at 7Tesla. Neuroimage 93, 138–145. doi: 10.1016/j.neuroimage.2014.02.016
Schlief, M. L., Craig, A. M., and Gitlin, J. D. (2005). NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 25, 239–246. doi: 10.1523/JNEUROSCI.3699-04.2005
Schrag, M., Mueller, C., Oyoyo, U., Smith, M. A., and Kirsch, W. M. (2011). Iron, zinc and copper in the Alzheimer’s disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 94, 296–306. doi: 10.1016/j.pneurobio.2011.05.001
Schurr, A., Miller, J. J., Payne, R. S., and Rigor, B. M. (1999). An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J. Neurosci. 19, 34–39. doi: 10.1523/JNEUROSCI.19-01-00034.1999
Schurr, A., West, C. A., and Rigor, B. M. (1988). Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 240, 1326–1328. doi: 10.1126/science.3375817
Shi, H., and Liu, K. J. (2006). Effects of glucose concentration on redox status in rat primary cortical neurons under hypoxia. Neurosci. Lett. 410, 57–61. doi: 10.1016/j.neulet.2006.09.066
Shibata, M., Yamada, S., Kumar, S. R., Calero, M., Bading, J., Frangione, B., et al. (2000). Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499. doi: 10.1172/jci10498
Silverman, D. H., Small, G. W., Chang, C. Y., Lu, C. S., Kung De Aburto, M. A., Chen, W., et al. (2001). Positron emission tomography in evaluation of dementia: regional brain metabolism and long-term outcome. JAMA 286, 2120–2127. doi: 10.1001/jama.286.17.2120
Small, G. W., Mazziotta, J. C., Collins, M. T., Baxter, L. R., Phelps, M. E., Mandelkern, M. A., et al. (1995). Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273, 942–947. doi: 10.1001/jama.1995.03520360056039
Smith, C. D., Carney, J. M., Tatsumo, T., Stadtman, E. R., Floyd, R. A., and Markesbery, W. R. (1992). Protein oxidation in aging brain. Ann. N Y Acad. Sci. 663, 110–119. doi: 10.1111/j.1749-6632.1992.tb38654.x
Smith, M. A., Harris, P. L., Sayre, L. M., and Perry, G. (1997). Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U S A 94, 9866–9868. doi: 10.1073/pnas.94.18.9866
Song, X., Deng, J. H., Liu, C. J., and Bai, Y. (2005). Specific point mutations may not accumulate with aging in the mouse mitochondrial DNA control region. Gene 350, 193–199. doi: 10.1016/j.gene.2005.02.008
Steinert, J. R., Kopp-Scheinpflug, C., Baker, C., Challiss, R. A., Mistry, R., Haustein, M. D., et al. (2008). Nitric oxide is a volume transmitter regulating postsynaptic excitability at a glutamatergic synapse. Neuron 60, 642–656. doi: 10.1016/j.neuron.2008.08.025
Stroka, D. M., Burkhardt, T., Desbaillets, I., Wenger, R. H., Neil, D. A., Bauer, C., et al. (2001). HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 15, 2445–2453. doi: 10.1096/fj.01-0125com
Suh, S. W., Jensen, K. B., Jensen, M. S., Silva, D. S., Kesslak, P. J., Danscher, G., et al. (2000). Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 852, 274–278. doi: 10.1016/s0006-8993(99)02096-x
Sun, X., He, G., Qing, H., Zhou, W., Dobie, F., Cai, F., et al. (2006). Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. U S A 103, 18727–18732. doi: 10.1073/pnas.0606298103
Sun, M. K., Xu, H., and Alkon, D. L. (2002). Pharmacological protection of synaptic function, spatial learning, and memory from transient hypoxia in rats. J. Pharmacol. Exp. Ther. 300, 408–416. doi: 10.1124/jpet.300.2.408
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. doi: 10.1016/j.cell.2011.02.018
Tabrizi, S. J., Workman, J., Hart, P. E., Mangiarini, L., Mahal, A., Bates, G., et al. (2000). Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann. Neurol. 47, 80–86. doi: 10.1002/1531-8249(200001)47:1<80::aid-ana13>3.3.co;2-b
Tadi, M., Allaman, I., Lengacher, S., Grenningloh, G., and Magistretti, P. J. (2015). Learning-induced gene expression in the hippocampus reveals a role of neuron -astrocyte metabolic coupling in long term memory. PLoS One 10:e0141568. doi: 10.1371/journal.pone.0141568
Takano, T., Tian, G. F., Peng, W., Lou, N., Libionka, W., Han, X., et al. (2006). Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 9, 260–267. doi: 10.1038/nn1623
Tomita, S., Ueno, M., Sakamoto, M., Kitahama, Y., Ueki, M., Maekawa, N., et al. (2003). Defective brain development in mice lacking the Hif-1α gene in neural cells. Mol. Cell. Biol. 23, 6739–6749. doi: 10.1128/mcb.23.19.6739-6749.2003
Tõugu, V., Karafin, A., and Palumaa, P. (2008). Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-β peptide. J. Neurochem. 104, 1249–1259. doi: 10.1111/j.1471-4159.2007.05061.x
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
Ullah, M. S., Davies, A. J., and Halestrap, A. P. (2006). The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1α-dependent mechanism. J. Biol. Chem. 281, 9030–9037. doi: 10.1074/jbc.M511397200
van Zijl, P. C., Hua, J., and Lu, H. (2012). The BOLD post-stimulus undershoot, one of the most debated issues in fMRI. Neuroimage 62, 1092–1102. doi: 10.1016/j.neuroimage.2012.01.029
Ventriglia, M., Bucossi, S., Panetta, V., and Squitti, R. (2012). Copper in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimers Dis. 30, 981–984. doi: 10.3233/JAD-2012-120244
Vogt, K., Mellor, J., Tong, G., and Nicoll, R. (2000). The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 26, 187–196. doi: 10.1016/s0896-6273(00)81149-6
Wagenführ, L., Meyer, A. K., Braunschweig, L., Marrone, L., and Storch, A. (2015). Brain oxygen tension controls the expansion of outer subventricular zone-like basal progenitors in the developing mouse brain. Development 142, 2904–2915. doi: 10.1242/dev.121939
Wagenführ, L., Meyer, A. K., Marrone, L., and Storch, A. (2016). Oxygen tension within the neurogenic niche regulates dopaminergic neurogenesis in the developing midbrain. Stem Cells Dev. 25, 227–238. doi: 10.1089/scd.2015.0214
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wang, X., Moualla, D., Wright, J. A., and Brown, D. R. (2010a). Copper binding regulates intracellular α-synuclein localisation, aggregation and toxicity. J. Neurochem. 113, 704–714. doi: 10.1111/j.1471-4159.2010.06638.x
Wang, X., Xing, A., Xu, C., Cai, Q., Liu, H., and Li, L. (2010b). Cerebrovascular hypoperfusion induces spatial memory impairment, synaptic changes, and amyloid-β oligomerization in rats. J. Alzheimers Dis. 21, 813–822. doi: 10.3233/jad-2010-100216
Ward, C. P., McCoy, J. G., McKenna, J. T., Connolly, N. P., McCarley, R. W., and Strecker, R. E. (2009). Spatial learning and memory deficits following exposure to 24 h of sleep fragmentation or intermittent hypoxia in a rat model of obstructive sleep apnea. Brain Res. 1294, 128–137. doi: 10.1016/j.brainres.2009.07.064
Watts, M. E., Williams, S. M., Nithianantharajah, J., and Claudianos, C. (2018). Hypoxia-induced MicroRNA-210 targets neurodegenerative pathways. Noncoding RNA 4:E10. doi: 10.3390/ncrna4020010
Welsh, S. J., Bellamy, W. T., Briehl, M. M., and Powis, G. (2002). The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1α protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 62, 5089–5095. Available online at: http://cancerres.aacrjournals.org/content/62/17/5089
Wirths, O., Multhaup, G., Czech, C., Blanchard, V., Moussaoui, S., Tremp, G., et al. (2001). Intraneuronal Aβ accumulation precedes plaque formation in β-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 306, 116–120. doi: 10.1016/s0304-3940(01)01876-6
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 106, 14670–14675. doi: 10.1073/pnas.0903563106
Zhang, B., Yin, C. P., Zhao, Q., and Yue, S. W. (2014). Upregulation of HIF-1α by hypoxia protect neuroblastoma cells from apoptosis by promoting survivin expression. Asian Pac. J. Cancer Prev. 15, 8251–8257. doi: 10.7314/apjcp.2014.15.19.8251
Keywords: oxidative metabolism, hypoxia, neurometabolism, plasticity, neurodegeneration
Citation: Watts ME, Pocock R and Claudianos C (2018) Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 11:216. doi: 10.3389/fnmol.2018.00216
Received: 14 May 2018; Accepted: 01 June 2018;
Published: 22 June 2018.
Edited by:
Julie A. Chowen, Hospital Infantil Universitario Niño Jesús, SpainReviewed by:
Maria Angeles Arevalo, Consejo Superior de Investigaciones Científicas (CSIC), SpainCopyright © 2018 Watts, Pocock and Claudianos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles Claudianos, Y2hhcmxlcy5jbGF1ZGlhbm9zQGFudS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.