 Jenice Linde
Jenice Linde Geraldine Zimmer-Bensch
Geraldine Zimmer-Bensch- 1Division of Functional Epigenetics in the Animal Model, Institute for Biology II, RWTH Aachen University, Aachen, Germany
- 2Research Training Group 2416 MultiSenses – MultiScales, RWTH Aachen University, Aachen, Germany
Neuropsychiatric diseases, such as mood disorders, schizophrenia, and autism, represent multifactorial disorders, differing in causes, disease onset, severity, and symptoms. A common feature of numerous neuropsychiatric conditions are defects in the cortical inhibitory GABAergic system. The balance of excitation and inhibition is fundamental for proper and efficient information processing in the cerebral cortex. Thus, altered inhibition is suggested to account for pathological symptoms like cognitive impairments and dysfunctional multisensory integration. While it became apparent that most of these diseases have a clear genetic component, environmental influences emerged as an impact of disease manifestation, onset, and severity. Epigenetic mechanisms of transcriptional control, such as DNA methylation, are known to be responsive to external stimuli, and are suspected to be implicated in the functional impairments of GABAergic interneurons, and hence, the pathophysiology of neuropsychiatric diseases. Here, we provide an overview about the multifaceted functional implications of DNA methylation and DNA methyltransferases in cortical interneuron development and function in health and disease. Apart from the regulation of gamma-aminobutyric acid-related genes and genes relevant for interneuron development, we discuss the role of DNA methylation-dependent regulation of synaptic transmission by the modulation of endocytosis-related genes as potential pathophysiological mechanisms underlying neuropsychiatric conditions. Deciphering the hierarchy and mechanisms of changes in epigenetic signatures is crucial to develop effective strategies for treatment and prevention.
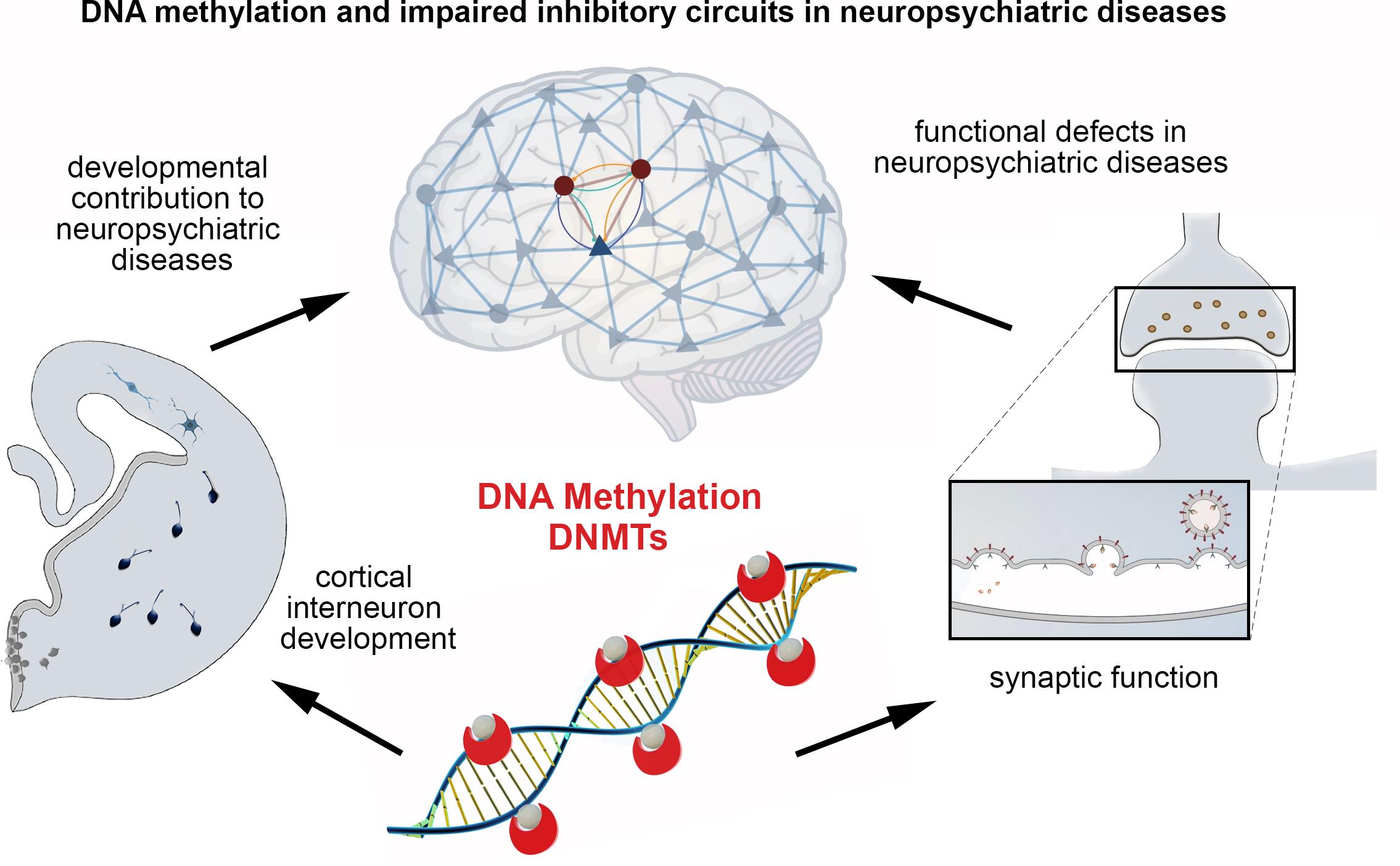
Graphical Abstract. Main conclusions of the minireview.
Introduction
Neuropsychiatric and neurological diseases, like schizophrenia, autism spectrum disorder (ASD), and epilepsy, represent multifactorial disorders, which seem to emerge from a combination of both, genetic predisposition and environmental impacts. For these disorders, evidence was provided that insults during development can likewise contribute to disease manifestation (Tsuang et al., 2001; Brown, 2011; Risch et al., 2014; Bölte et al., 2019). Considering the observation that numerous disease susceptibility genes are linked to neurodevelopmental processes has caused a debate whether schizophrenia, ASD, and epilepsy should be classified as neuropsychiatric or neurodevelopmental diseases (Bolton et al., 1998; Price et al., 2000; Lewis and Levitt, 2002; DiCicco-Bloom et al., 2006; Tuchman and Cuccaro, 2011). Indeed, the age at disease onset can vary tremendously, ranging from patients being few months to several years old. Besides genetic causes (Krebs et al., 2000; Klei et al., 2012; Pourcain et al., 2018), environmental contributions might provide an explanation for the differing onsets as well as for the various degrees of disease severity.
Epigenetic mechanisms of transcriptional control, such as histone modifications, DNA methylation, and non-coding RNAs, provide an attractive hypothesis for the causal relationship of extrinsic incidents and the afflicted persons’ disease development. DNA methylation was reported to be affected distinctively in different neuronal subsets across various neuropsychiatric conditions (Feng and Fan, 2009; Gräff et al., 2011), and emerges to mediate a broad spectrum of biological effects. For this, we focus on DNA methylation here. DNA methylation was traditionally considered a repressive epigenetic mark, impeding transcription factor binding either directly, or indirectly by the action of methyl-CpG-binding domain proteins (Curradi et al., 2002). This view has been challenged by recent studies, suggesting that DNA methylation could also create new motifs for transcription factor binding sites in addition to interaction sites for histone modifying complexes (Smith and Meissner, 2013; Jang et al., 2017). Moreover, DNA methylation is relevant for alternative promoter choice and alternative splicing, giving rise to different protein isoforms (Maunakea et al., 2010, 2013). As DNA methylation was reported to be changed in response to altered neuronal activity (Guo et al., 2011), it could likely mediate or contribute to the integration of external cues into pathological cell features, resulting in impaired neuronal functionality. Moreover, DNA methylation is known to act on diverse aspects of neuronal development, such as differentiation, migration and survival regulation (Hutnick et al., 2009; Chestnut et al., 2011; Pensold et al., 2017; Symmank and Zimmer, 2017), supporting the assumption that it might further be implicated in or mediate the developmental contribution to disease manifestation.
DNA methylation is dynamically regulated by DNA methyltransferases (DNMTs) and ten-eleven translocation (TET) proteins. While DNMTs catalyze cytosine methylation, TET proteins initiate active demethylation by oxidizing 5mC–5hmC and further oxidation forms, which are then actively demethylated by thymine DNA glycosylase (TDG)-mediated base excision repair (Ito et al., 2011; Kaas et al., 2013; Kohli and Zhang, 2013; Wu and Zhang, 2014).
DNMT1 and DNMT3a are the two main DNMTs being expressed in developing and adult neurons. Apart from cell fate specification, neuronal migration, and survival regulation during brain development, DNA methylation and DNMTs were reported to regulate synaptic function and plasticity, being involved in learning and memory regulation (Levenson et al., 2006; Nelson et al., 2008; Meadows et al., 2015, 2016; Sweatt, 2016, 2017). Thereby they seem to exert in part redundant (Feng et al., 2010) but also distinctive functions (Morris et al., 2016). Similarly, TET enzymes were identified to regulate neuronal development (Santiago et al., 2014), synaptic plasticity and memory extinction (Rudenko et al., 2013), as well as gene expression in response to global synaptic activity changes (Yu et al., 2015).
Dysregulated expression of DNMTs and TETs, as well as alterations in both, DNA methylation and demethylation networks, were found in brains of patients suffering from different neuropsychiatric diseases including schizophrenia and ASD, but also epilepsy (Huang and Akbarian, 2007; Zhubi et al., 2009; Guidotti et al., 2011; Dong et al., 2012; Grayson and Guidotti, 2013; Guidotti and Grayson, 2014; Benes, 2015). Based on their implications in healthy brain development and function, it is conceivable that disease-related changes in DNA methylation and demethylation, as well as DNMT and TET function contribute to the symptoms seen in these diseases.
Such symptoms include substantial restrictions in learning and memory formation, disorientation, hallucinations, and problems with communication, which point toward mayor impairments regarding the patients’ sensory cortical information processing. Indeed, one crucial hallmark of neuropsychiatric diseases is defective multisensory integration, which is important for perception, cognitive processing, and complex behaviors, and might thus be the basis for the former mentioned symptoms.
Multisensory processing is known to critically rely on the proper function of inhibitory gamma-aminobutyric acid (GABA) – containing interneurons in the cerebral cortex (Olcese et al., 2013), which synchronize surrounding glutamatergic pyramidal neurons, enabling local neural assemblies (Hensch, 2005; Klausberger and Somogyi, 2008).
The group of cortical inhibitory GABAergic interneurons is enormously diverse in matters of morphological, molecular and functional features (Tremblay et al., 2016). The most abundant subset of cortical interneurons are parvalbumin (PV)-expressing interneurons, which include the fast-spiking chandelier and basket cells (Rudy et al., 2011). Due to the essential role inhibitory interneurons have in cortical information processing (Defelipe et al., 2013), deficits in the development and/or function of GABAergic interneurons are likely implicated in causing or mediating the pathological symptoms in neuropsychiatric diseases. In support of that, alterations in the cortical GABAergic system represent a common denominator of different neuropsychiatric disorders, and human and mouse genetic studies provided evidence for the critical role of altered GABAergic circuit formation in schizophrenia, epilepsy, and autism (Marín, 2012). Especially for schizophrenia and ASD, disturbed interneuron development would explain a variety of the associated pathological symptoms (Lewis and Levitt, 2002; Marín, 2012).
Of note, alterations in DNA methylation signatures of GABA-related genes and/or changed expression of DNMTs in GABAergic interneurons were reported for diverse neuropsychiatric diseases (Veldic et al., 2004; Guidotti et al., 2011). Given that DNMTs and DNA methylation not only regulate cortical interneuron functionality (Pensold et al., 2020) but also their development (Pensold et al., 2017; Symmank et al., 2018, 2020), an implication of altered DNA methylation in inhibitory interneurons for disease manifestation seems plausible. On the other hand, a dysregulated epigenetic machinery could also constitute a secondary effect of disease progression. To explore the potential of epigenetic key regulators as targets for therapeutic interventions, we need to draw a conclusive picture, shedding light on the detailed mechanisms underlying transcriptional dysregulation and impaired physiology of the different neuronal subtypes in neuropsychiatric disorders.
Defects of the Gabaergic System Contribute to Neuropsychiatric Disorders
In this paragraph, the evidence for the implication of GABAergic defects in different neuropsychiatric and neurological diseases will be discussed in more detail.
Altered levels of GABA and GABA-mediated inhibition were found in vivo using magnetic resonance spectroscopy (MRS) in patients suffering from epilepsy or depression (Chang et al., 2003), and in brain tissue of epileptic patients (Treiman, 2001). Moreover, changes in GABAergic function became apparent in genetic and acquired animal models for epilepsy (Treiman, 2001). Concordantly, both GABA antagonists and therapeutics, that downregulate GABA synthesis, can elicit seizures in otherwise unaffected patients. On the other hand, GABA agonists and GABA-mimetic agents act as anticonvulsants (Treiman, 2001) and antidepressants (Sanacora et al., 2000), underlining the implication of GABA in eliciting neurological symptoms.
Altered GABA levels were further detected in post mortem brains of schizophrenia patients (Benes, 2015). In line with this, people suffering from schizophrenia were found to have reduced inhibitory interneuron numbers (Bakhshi and Chance, 2015). On a molecular level, the expression levels of GAD67 (an isoform of the GABA-producing glutamate decarboxylase) and GAT1 (a GABA membrane transporter) mRNAs were found to be noticeably decreased (Lewis et al., 2005). Accordingly, inhibitory cortical interneurons displayed lower neurotransmitter levels and reduced firing capacities in these patients. The following reduction of inhibition of excitatory neurons leads to a systemic disinhibition, resulting in abnormally high activity levels (Bakhshi and Chance, 2015). These aberrant properties were particularly striking in the dorsolateral prefrontal cortex (DLPFC). Additionally, a reduction of REELIN on mRNA and protein levels was evident (Impagnatiello et al., 1998). Under physiological conditions, REELIN mediates neuronal migration and positioning (Franco et al., 2011; Sekine et al., 2014). A mis-positioning of neurons in DLPFC might contribute to personality changes, e.g., expressed as extremely disorganized behavior and paranoid thinking, and impairments on tasks of executive function seen in schizophrenic patients (Bakhshi and Chance, 2015). Furthermore, working memory and attention deficits as well reflect dysfunctional DLPFC activity (Lewis et al., 2004, 2005).
Research on the molecular basis of ASD embraces both investigating ASD and underlying causes in general as well as the role of individual genes, whose aberrant expression or function lead to ASD as a comorbidity. In patients suffering from ASD, several genetic variations were identified as potential causative factors (Marín, 2012). Albeit some of the affected genes are expressed ubiquitously in the brain, restricting the expression of mutated genes to the GABAergic system is already sufficient to induce the expected phenotype, involving stereotypic and compulsive behavior, motor dysfunction and changes in social behavior (Chao et al., 2010). Also, in different mouse models of ASD, a reduced number of inhibitory interneurons throughout the neocortex was found as a common denominator (Gogolla et al., 2009). Based on this it is not surprising that ASD patients have a high comorbidity with epilepsy (Canitano, 2007). Altered levels of GABA and GABA-related synaptic transmission in ASD patients are probably due to the reported changes in mRNA and protein levels of different GABA-receptor-subunits (Fatemi et al., 2010), and a decrease of available GABA synthesizing enzymes GAD65 and GAD67 (Coghlan et al., 2012).
Overall, the described changes of the GABAergic system in the pathological context render neuropsychiatric diseases “interneuropathies”. In addition to working memory, the GABAergic system decisively influences multisensory integration (Olcese et al., 2013), which is virtually inoperable in patients with neuropsychiatric diseases. Multisensory processing is a key feature of higher cognitive power, enabling an individuum to form coherent multimodal objects out of spatio-temporal cohesive inputs. The ability of primary and higher cortical areas to integrate unisensory information and build a meaningful representation of events is the basis of many cognitive capacities such as learning and the expression of behavior (Murray et al., 2016). Hence, impairments in interneuron development and function as seen in neuropsychiatric diseases likely affect multisensory processing. Thus, the described aberrations in the GABAergic system in patients suffering from neuropsychiatric conditions provide a potential explanation for many of the psychiatric symptoms.
How are these aberrations in the GABAergic system caused or mediated? There is increasing evidence that epigenetic mechanisms of gene regulation like DNA methylation in inhibitory GABAergic interneurons are implicated in the pathophysiology of neuropsychiatric diseases like schizophrenia and autism (Huang and Akbarian, 2007; Connor and Akbarian, 2008). To understand the relevance DNMTs and DNA methylation have in the etiology of neuropsychiatric diseases, their implication in interneuron development and adult function in the healthy brain has to be dissected.
DNMT Function in Interneuron Development
DNA methyltransferase-dependent DNA methylation in developing interneurons seems to be implicated in schizophrenia, as prenatal stress in mice elevates Dnmt1 and Dnmt3a expression in GABAergic interneurons and induces behaviors indicative of a schizophrenia-like phenotype in their offspring (Matrisciano et al., 2013). Neuronal development is a multifaceted process involving differentiation from neural progenitor cells, migration of post-mitotic neurons to their target regions, morphological maturation, and synapse formation. While in the dorsal telencephalon DNA methylation has been proposed to control the neurogenic versus glial cell fate in cortical progenitors (Miller and Gauthier, 2007), such investigations have not been executed so far for the basal telencephalon, from where inhibitory interneurons originate.
At post-mitotic level, DNMT1 was described to regulate cortical interneuron migration by promoting the migratory morphology and survival (Pensold et al., 2017) in part through modulating Pak6 and Lhx1 expression as downstream targets (Symmank et al., 2018, 2019, 2020). Interestingly, these genes are DNA methylation-independently regulated by DNMT1 via a crosstalk with histone modifications (Symmank et al., 2019, 2020). In line with this, DNMTs and DNA methylation were further shown to be essential for the maturation of diverse other neuronal subtypes (Fan et al., 2001; Hutnick et al., 2009; Chestnut et al., 2011; Rhee et al., 2012), including cortical excitatory neurons (Hutnick et al., 2009; Feng et al., 2010), and dentate gyrus neurons (Noguchi et al., 2016).
DNA Methylation and DNMTs in Interneuron Functionality in Health and Disease
Alterations in DNA methylation signatures of synapse- and GABA-related genes were reported for ASD (Nardone et al., 2017) and schizophrenia (Costa et al., 2003; Veldic et al., 2004; Ruzicka et al., 2007). The pathophysiology of the Rett’s syndrome, a form of ASD with symptoms like impaired language skills, cognitive deficits, and stereotypic behavior (Chahrour and Zoghbi, 2007), can be traced back to a loss-of-function mutation of methyl-CpG-binding protein 2 (MECP2; Amir et al., 1999). Consequently, a systemic MECP2 knockout reproduces many of the neurological symptoms of Rett’s syndrome in mice (Chen et al., 2001; Guy et al., 2001). Strikingly, most of the symptoms can already be seen when MECP2 is deleted only in GABAergic cells (Chao et al., 2010), resulting in a defect of GABAergic synapses (Medrihan et al., 2008). This underlines the functional relevance of DNA methylation in GABAergic interneurons for neuropsychiatric diseases.
Changed DNA methylation signatures in GABAergic interneurons were further reported for schizophrenia patients, with genes like REELIN and GAD1, relevant for GABAergic neurotransmission as well as interneuron function and development, displaying elevated DNA methylation levels and reduced expression, being suggested to account for impaired interneuron function (Veldic et al., 2004; Ruzicka et al., 2007).
That DNMT1-dependent DNA methylation affects cortical interneuron functionality, was recently shown in mice. Dnmt1 deletion in PV interneurons leads to increased inhibition, which however, seems not to be primarily caused by DNA methylation-dependent regulation of GABA- or synapse-related genes (Pensold et al., 2020). Instead, repressive DNMT1-dependent DNA methylation was identified to act on endocytosis-related gene expression in parvalbuminergic cortical interneurons. Functional validation experiments proposed that DNA methylation catalyzed by DNMT1 restricts clathrin-mediated endocytosis at pre-synapses, and through this synaptic vesicle recycling and GABAergic transmission (Pensold et al., 2020). Data obtained from human hippocampal biopsies of patients with temporal lobe epilepsy revealed a correlation between DNA methylation-dependent transcriptional regulation of endocytosis-related genes with the patients’ seizure rates (Pensold et al., 2020). This corroborates the connection between DNA methylation, endocytosis regulation and synaptic functionality.
DNA methylation-dependent regulation of endocytosis represents a novel mechanism for orchestrating synaptic transmission and might help us understand the pathophysiology of disorders characterized by abnormal synaptic transmission, as seen in different neuropsychiatric disorders (Ramocki and Zoghbi, 2008; Südhof, 2008). In support of this, it was recently reported that in addition to pyramidal neurons, PV cells in the DLPFC of schizophrenia patients display significant transcriptional changes of genes related to clathrin-mediated endocytosis signaling (Enwright et al., 2018). Another study analyzed genome-wide DNA methylation signatures in the frontal cortex of subjects diagnosed with schizophrenia and control subjects (Wockner et al., 2014). In cortical tissue of schizophrenia patients, significant DNA methylation changes were determined for genes related to endocytosis, including CLTC, DNM1, DNM3, RAB7A, WIPF1, ZFYVE9, HGS, SPG20 and RAC1 (Wockner et al., 2014). These genes were identified to be transcriptionally regulated by DNMT1-dependent DNA methylation in cortical PV interneurons in mice (Pensold et al., 2020). This proposes that DNA methylation-mediated alterations in endocytosis-related gene expression seem to be implicated in the pathophysiology of schizophrenia, which could consequently contribute to the impaired synaptic functionality in addition to the reported changes in GABA-related gene expression. However, the latter could alternatively represent a secondary consequence of dysregulated DNMT function to compensate for endocytosis-mediated alterations of synaptic transmission.
The observed changes in DNA methylation signatures in schizophrenia were hypothesized to be mediated by increased expression of DNMT1, as elevated DNMT1 mRNA levels were found in post mortem schizophrenic brains (Veldic et al., 2004; Dong et al., 2015). However, a general increase in DNMT1 expression does not explain site-specific effects. Alternatively, changes in DNMT1 targeting to specific gene loci could account as disease causing mechanisms. Site-specific DNA methylation by DNMT1 was proposed to be mediated by long non-coding RNAs (lncRNAs), preventing or promoting DNMT1 binding (Chalei et al., 2014). Indeed, there is accumulating evidence for the significance of lncRNAs impairment in several neuropsychiatric diseases including ASD, schizophrenia, intellectual disability, major depressive disorder and others (Hosseini et al., 2019).
Moreover, it should be kept in mind that DNMT1 was described to act non-canonically on gene expression by interactions with histone modifiers (Symmank and Zimmer, 2017; Symmank et al., 2018). DNMTs and DNA methylation interfere at different levels with histone modifications, which likewise modulate the specificity of transcriptional changes. Hence, a more global and combinatorial analysis of expression profiles and epigenetic signatures of post mortem brain material, favorably at single cell level, might help to get better insights into the role of DNMTs in mediating the patho-mechanisms of schizophrenia and other neuropsychiatric conditions at subcellular level.
Discussion and Conclusion
For numerous neuropsychiatric diseases, abnormalities of the GABAergic system were identified. In fact, a dysfunction or dysgenesis of inhibitory interneurons in mouse models were observed to elicit pathological phenotypes, such as multimodal integration impairments. This coincides with inhibitory interneurons being pivotal for multisensory processing in the cerebral cortex. Next to genetic predispositions, environmental impact during development and resulting changes in the epigenetic landscape have more recently been considered as contributors to the diseases’ cause and course. Alterations in gene expression, relevant for cortical interneuron functionality, have been related to changes in DNA methylation signatures and DNMT1 expression levels in schizophrenia patients. Also, a mouse model of maternal adversity found that neonatal stress induced DNA methylation changes of genes related to synapse formation and function (Oh et al., 2013; Tordjman et al., 2014). Phenotypically, these mice showed neuropsychiatric symptoms, such as anxiety, elevated stress reactivity, and impairments of vocal communication, which could be affected by malfunctioning multimodal integration. Hence, a connection of epigenetic modulation and multisensory processing is highly likely and a noteworthy matter of future research.
DNMT1-dependent DNA methylation likewise regulates interneuron function in the healthy (mouse) brain, acting on synaptic transmission by modulating endocytosis, which might be affected in disease. Deciphering disease-relevant mechanisms requires more integrative analyses of patients’ brain samples, including the profiling of both, transcriptomic as well as epigenomic signatures. Therefore, studies analyzing changes in DNA methylation patterns need to be complemented by histone modification profiling and lncRNA expression and interaction analysis, to draw a conclusive picture of the hierarchy of epigenetic networks. Moreover, cell type-specific investigation of excitatory and inhibitory neurons of the different cortical layers within the distinct brain regions is elementary, to approach network changes and dynamics.
To relate the transcriptome to electrophysiological properties of cells, Patch-Sequencing techniques reach increasing popularity, which enables to correlate individual firing properties and eventually morphology with molecular features (Cadwell et al., 2016; Fuzik et al., 2016). The ongoing improvement of sequencing-based single cell approaches might render the comprehensive and parallel analysis of the epigenome, transcriptome, and electrophysiological features tangible. This will help to decipher which epigenetic mechanisms act as drivers or passengers in neuropsychiatric diseases.
Author Contributions
Both authors were responsible for the article’s conceptual design, and wrote the manuscript, and agree to be accountable for the content of the work.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 368482240/GRK2416.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Bakhshi, K., and Chance, S. A. (2015). The neuropathology of schizophrenia: a selective review of past studies and emerging themes in brain structure and cytoarchitecture. Neuroscience 303, 82–102. doi: 10.1016/j.neuroscience.2015.06.028
Benes, F. M. (2015). The GABA system in schizophrenia: cells, molecules and microcircuitry. Schizophr. Res. 167, 1–3. doi: 10.1016/j.schres.2015.07.017
Bölte, S., Girdler, S., and Marschik, P. B. (2019). The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell. Mol. Life Sci. 76, 1275–1297. doi: 10.1007/s00018-018-2988-4
Bolton, P. F., Pickles, A., Murphy, M., and Rutter, M. (1998). Autism, affective and other psychiatric disorders: patterns of familial aggregation. Psychol. Med. 28, 385–395. doi: 10.1017/S0033291797006004
Brown, A. S. (2011). The environment and susceptibility to schizophrenia. Progr. Neurobiol. 93, 23–58. doi: 10.1016/j.pneurobio.2010.09.003
Cadwell, C. R., Palasantza, A., Jiang, X., Berens, P., Deng, Q., Yilmaz, M., et al. (2016). Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat. Biotechnol. 34, 199–203. doi: 10.1038/nbt.3445
Canitano, R. (2007). Epilepsy in autism spectrum disorders. Eur. Child Adolesc. Psychiatry 16, 61–66. doi: 10.1007/s00787-006-0563-2
Chahrour, M., and Zoghbi, H. Y. (2007). The story of rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chalei, V., Sansom, S. N., Kong, L., Lee, S., Montiel, J. F., Vance, K. W., et al. (2014). The long non-coding RNA Dali is an epigenetic regulator of neural differentiation. ELife 3, 1–24. doi: 10.7554/eLife.04530
Chang, L., Cloak, C. C., and Ernst, T. (2003). Magnetic resonance spectroscopy studies of GABA in neuropsychiatric disorders. J. Clin. Psychiatry 64, 7–14.
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Chestnut, B. A., Chang, Q., Price, A., Lesuisse, C., Wong, M., and Martin, L. J. (2011). Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 31, 16619–16636. doi: 10.1523/JNEUROSCI.1639-11.2011
Coghlan, S., Horder, J., Inkster, B., Mendez, M. A., Murphy, D. G., and Nutt, D. J. (2012). GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci. Biobehav. Rev. 36, 2044–2055. doi: 10.1016/j.neubiorev.2012.07.005
Connor, C. M., and Akbarian, S. (2008). DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics 3, 55–58. doi: 10.4161/epi.3.2.5938
Costa, E., Grayson, D. R., and Guidotti, A. (2003). Epigenetic downregulation of GABAergic function in schizophrenia: potential for pharmacological intervention? Mol. Intervent. 3, 220–229. doi: 10.1124/mi.3.4.220
Curradi, M., Izzo, A., Badaracco, G., and Landsberger, N. (2002). Molecular mechanisms of gene silencing mediated by DNA methylation. Mol. Cell. Biol. 22, 3157–3173. doi: 10.1128/MCB.22.9.3157-3173.2002
Defelipe, J., López-Cruz, P. L., Benavides-Piccione, R., Bielza, C., Larrañaga, P., Anderson, S., et al. (2013). New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat. Rev. Neurosci. 14, 202–216. doi: 10.1038/nrn3444
DiCicco-Bloom, E., Lord, C., Zwaigenbaum, L., Courchesne, E., Dager, S. R., Schmitz, C., et al. (2006). The developmental neurobiology of autism spectrum disorder. J. Neurosci. 26, 6897–6906. doi: 10.1523/JNEUROSCI.1712-06.2006
Dong, E., Gavin, D. P., Chen, Y., and Davis, J. (2012). Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl. Psychiatry 2, 1–7. doi: 10.1038/tp.2012.86
Dong, E., Ruzicka, W. B., Grayson, D. R., and Guidotti, A. (2015). DNA-methyltransferase1 (DNMT1) binding to CpG rich GABAergic and BDNF promoters is increased in the brain of schizophrenia and bipolar disorder patients. Schizophr. Res. 167, 35–41. doi: 10.1016/j.schres.2014.10.030
Enwright, J. F., Huo, Z., Arion, D., Corradi, J. P., Tseng, G., and Lewis, D. A. (2018). Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia. Mol. Psychiatry 23, 1606–1613. doi: 10.1038/mp.2017.216
Fan, G., Beard, C., Chen, R. Z., Csankovszki, G., Sun, Y., Siniaia, M., et al. (2001). DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J. Neurosci. 21, 788–797. doi: 10.1523/jneurosci.21-03-00788.2001
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., Rooney, R. J., Patel, D. H., and Thuras, P. D. (2010). mRNA and protein levels for GABA A a4, a5, b1 and GABA B R1 receptors are altered in brains from subjects with autism. J. Autism Dev. Dis. 40, 743–750. doi: 10.1007/s10803-009-0924-z
Feng, J., and Fan, G. (2009). The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int. Rev. Neurobiol. 89, 67–84. doi: 10.1016/S0074-7742(09)89004-1
Feng, J., Zhou, Y., Campbell, S. L., Le, T., Li, E., Sweatt, J. D., et al. (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Publish. Group 13, 423–430. doi: 10.1038/nn.2514
Franco, S. J., Martinez-Garay, I., Gil-Sanz, C., Harkins-Perry, S. R., and Müller, U. (2011). Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron 69, 482–497. doi: 10.1016/j.neuron.2011.01.003
Fuzik, J., Zeisel, A., Mate, Z., Calvigioni, D., Yanagawa, Y., Szabo, G., et al. (2016). Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat. Biotechnol. 34, 175–183. doi: 10.1038/nbt.3443
Gogolla, N., LeBlanc, J. J., Quast, K. B., Südhof, T. C., Fagiolini, M., and Hensch, T. K. (2009). Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 1, 172–181. doi: 10.1007/s11689-009-9023-x
Gräff, J., Kim, D., Dobbin, M. M., and Li-Huei, T. (2011). Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 91, 603–649. doi: 10.1152/physrev.00012.2010
Grayson, D. R., and Guidotti, A. (2013). The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology 38, 138–166. doi: 10.1038/npp.2012.125
Guidotti, A., Auta, J., Chen, Y., Davis, J. M., Dong, E., Gavin, D. P., et al. (2011). Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 60, 1007–1016.
Guidotti, A., and Grayson, D. R. (2014). DNA methylation and demethylation as targets for antipsychotic therapy. Dialogues Clin. Neurosci. 16, 419–429.
Guo, J. U., Ma, D. K., Mo, H., Ball, M. P., Jang, M. H., Bonaguidi, M. A., et al. (2011). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 14, 1345–1351. doi: 10.1038/nn.2900
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Hensch, T. K. (2005). Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 6, 877–888. doi: 10.1038/nrn1787
Hosseini, E., Bagheri-Hosseinabadi, Z., De Toma, I., Jafarisani, M., and Sadeghi, I. (2019). The importance of long non-coding RNAs in neuropsychiatric disorders. Mol. Aspects Med. 70, 127–140. doi: 10.1016/j.mam.2019.07.004
Huang, H.-S., and Akbarian, S. (2007). GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PLoS One 2:e809. doi: 10.1371/journal.pone.0000809
Hutnick, L. K., Golshani, P., Namihira, M., Xue, Z., Matynia, A., Yang, X. W., et al. (2009). DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum. Mol. Genet. 18, 2875–2888.
Impagnatiello, F., Guidotti, A. R., Pesold, C., Dwivedi, Y., Caruncho, H., Pisu, M. G., et al. (1998). A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 95, 15718–15723. doi: 10.1073/pnas.95.26.15718
Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. doi: 10.1126/science.1210597
Jang, H. S., Shin, W. J., Lee, J. E., and Do, J. T. (2017). CpG and Non-CpG methylation in epigenetic gene regulation and brain function. Genes 8:148. doi: 10.3390/genes8060148
Kaas, G. A., Zhong, C., Eason, D. E., Ross, D. L., Vachhani, R. V., Ming, G.-L., et al. (2013). TET1 controls CNS 5-Methylcytosine Hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 79, 1086–1093. doi: 10.1016/j.neuron.2013.08.032
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. doi: 10.1126/science.1149381
Klei, L., Sanders, S. J., Murtha, M. T., Hus, V., Lowe, J. K., Willsey, A. J., et al. (2012). Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 3:9. doi: 10.1186/2040-2392-3-9
Kohli, R. M., and Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. doi: 10.1038/nature12750
Krebs, M. O., Guillin, O., Bourdel, M. C., Schwartz, J. C., Olie, J. P., Poirier, M. F., et al. (2000). Brain derived neurotrophic factor (BDNF) gene variants association with age at onset and therapeutic response in schizophrenia. Mol. Psychiatry 5, 558–562. doi: 10.1038/sj.mp.4000749
Levenson, J. M., Roth, T. L., Lubin, F. D., Miller, C. A., Huang, I. C., Desai, P., et al. (2006). Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 281, 15763–15773. doi: 10.1074/jbc.M511767200
Lewis, D. A., Hashimoto, T., and Volk, D. W. (2005). Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324. doi: 10.1038/nrn1648
Lewis, D. A., and Levitt, P. (2002). Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 25, 409–432. doi: 10.1146/annurev.neuro.25.112701.142754
Lewis, D. A., Volk, D. W., and Hashimoto, T. (2004). Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology 174, 143–150. doi: 10.1007/s00213-003-1673-x
Marín, O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13, 107–120. doi: 10.1038/nrn3155
Matrisciano, F., Tueting, P., Dalal, I., Kadriu, B., Grayson, D. R., Davis, J. M., et al. (2013). Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 68, 184–194. doi: 10.1016/j.neuropharm.2012.04.013
Maunakea, A. K., Chepelev, I., Cui, K., and Zhao, K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. doi: 10.1038/cr.2013.110
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., Dsouza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi: 10.1038/nature09165
Meadows, J. P., Guzman-Karlsson, M. C., Phillips, S., Brown, J. A., Strange, S. K., Sweatt, J. D., et al. (2016). Dynamic DNA methylation regulates neuronal intrinsic membrane excitability. Sci. Signal. 9, ra83–ra83. doi: 10.1126/scisignal.aaf5642
Meadows, J. P., Guzman-Karlsson, M. C., Phillips, S., Holleman, C., Posey, J. L., Day, J. J., et al. (2015). DNA methylation regulates neuronal glutamatergic synaptic scaling. Sci. Signal. 8:ra61. doi: 10.1126/scisignal.aab0715
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., et al. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 99, 112–121. doi: 10.1152/jn.00826.2007
Miller, F. D., and Gauthier, A. S. (2007). Timing is everything: making neurons versus glia in the developing cortex. Neuron 54, 357–369. doi: 10.1016/j.neuron.2007.04.019
Morris, M. J., Na, E. S., Autry, A. E., and Monteggia, L. M. (2016). Impact of DNMT1 and DNMT3a forebrain knockout on depressive- and anxiety like behavior in mice. Neurobiol. Learn. Mem. 135, 139–145. doi: 10.1016/j.nlm.2016.08.012
Murray, M. M., Lewkowicz, D. J., Amedi, A., and Wallace, M. T. (2016). Multisensory processes: a balancing act across the lifespan. Trends Neurosci. 39, 567–579. doi: 10.1016/j.tins.2016.05.003
Nardone, S., Sams, D. S., Zito, A., Reuveni, E., and Elliott, E. (2017). Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cereb. Cortex 27, 5739–5754.
Nelson, E. D., Kavalali, E. T., and Monteggia, L. M. (2008). Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J. Neurosci. 28, 395–406. doi: 10.1523/JNEUROSCI.3796-07.2008
Noguchi, H., Murao, N., Kimura, A., Matsuda, T., Namihira, M., and Nakashima, K. (2016). DNA methyltransferase 1 is indispensable for development of the hippocampal dentate gyrus. J. Neurosci. 36, 6050–6068. doi: 10.1523/JNEUROSCI.0512-16.2016
Oh, J. E., Chambwe, N., Klein, S., Gal, J., Andrews, S., Gleason, G., et al. (2013). Differential gene body methylation and reduced expression of cell adhesion and neurotransmitter receptor genes in adverse maternal environment. Transl. Psychiatry 3:e218. doi: 10.1038/tp.2012.130
Olcese, U., Iurilli, G., and Medini, P. (2013). Cellular and synaptic architecture of multisensory integration in the mouse neocortex. Neuron 79, 579–593.
Pensold, D., Reichard, J., Van Loo, K. M. J., Ciganok, N., Hahn, A., Bayer, C., et al. (2020). DNA methylation-mediated modulation of endocytosis as potential mechanism for synaptic function regulation in murine inhibitory cortical interneurons. Cereb. Cortex 30, 3921–3937. doi: 10.1093/cercor/bhaa009
Pensold, D., Symmank, J., Hahn, A., Lingner, T., Salinas-Riester, G., Downie, B., et al. (2017). The DNA methyltransferase 1 (DNMT1) controls the shape and dynamics of migrating poa-derived interneurons fated for the murine cerebral cortex. Cereb. Cortex 27, 5696–5714. doi: 10.1093/cercor/bhw341
Pourcain, B. S., Robinson, E. B., Anttila, V., Sullivan, B. B., Maller, J., Golding, J., et al. (2018). ASD and schizophrenia show distinct developmental profiles in common genetic overlap with population-based social communication difficulties. Mol. Psychiatry 23, 263–270. doi: 10.1038/mp.2016.198
Price, B. H., Adams, R. D., and Coyle, J. T. (2000). Neurology and psychiatry: closing the great divide. Neurology 54, 8–14. doi: 10.1212/wnl.54.1.8
Ramocki, M. B., and Zoghbi, H. Y. (2008). Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455, 912–918. doi: 10.1038/nature07457
Rhee, K. D., Yu, J., Zhao, C. Y., Fan, G., and Yang, X. J. (2012). Dnmt1-dependent DNA methylation is essential for photoreceptor terminal differentiation and retinal neuron survival. Cell Death Dis. 3:e427. doi: 10.1038/cddis.2012.165
Risch, N., Hoffmann, T. J., Anderson, M., Croen, L. A., Grether, J. K., and Windham, G. C. (2014). Familial recurrence of autism spectrum disorder: evaluating genetic and environmental contributions. Am. J. Psychiatry 171, 1206–1213. doi: 10.1176/appi.ajp.2014.13101359
Rudenko, A., Dawlaty, M. M., Seo, J., Cheng, A. W., Meng, J., Le, T., et al. (2013). Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 79, 1109–1122. doi: 10.1016/j.neuron.2013.08.003
Rudy, B., Fishell, G., Lee, S. H., and Hjerling-Leffler, J. (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 71, 45–61. doi: 10.1002/dneu.20853
Ruzicka, W. B., Zhubi, A., Veldic, M., Grayson, D. R., Costa, E., and Guidotti, A. (2007). Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry 12, 385–397. doi: 10.1038/sj.mp.4001954
Sanacora, G., Mason, G. F., and Krystal, J. H. (2000). Impairment of GABAergic transmission in depression: new insights from neuroimaging studies. Crit. Rev. Neurobiol. 14, 23–45. doi: 10.1615/CritRevNeurobiol.v14.i1.20
Santiago, M., Antunes, C., Guedes, M., Sousa, N., and Marques, C. J. (2014). TET enzymes and DNA hydroxymethylation in neural development and function - How critical are they? Genomics 104, 334–340. doi: 10.1016/j.ygeno.2014.08.018
Sekine, K., Kubo, K. I., and Nakajima, K. (2014). How does Reelin control neuronal migration and layer formation in the developing mammalian neocortex? Neurosci. Res. 86, 50–58. doi: 10.1016/j.neures.2014.06.004
Smith, Z. D., and Meissner, A. (2013). DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 14, 204–220. doi: 10.1038/nrg3354
Südhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi: 10.1038/nature07456
Sweatt, J. D. (2016). Dynamic DNA methylation controls glutamate receptor trafficking and synaptic scaling. J. Neurochem. 137, 312–330. doi: 10.1111/jnc.13564
Sweatt, J. D. (2017). Layered-up regulation in the developing brain. Nature 551, 448–449. doi: 10.1038/d41586-017-07269-7
Symmank, J., Bayer, C., Reichard, J., Pensold, D., and Zimmer-Bensch, G. (2020). Neuronal Lhx1 expression is regulated by DNMT1-dependent modulation of histone marks. Epigenetics. doi: 10.1080/15592294.2020.1767372 [Epub ahead of print].
Symmank, J., Bayer, C., Schmidt, C., Hahn, A., Pensold, D., and Zimmer-Bensch, G. (2018). DNMT1 modulates interneuron morphology by regulating Pak6 expression through crosstalk with histone modifications. Epigenetics 13, 536–556. doi: 10.1080/15592294.2018.1475980
Symmank, J., Gölling, V., Gerstmann, K., and Zimmer, G. (2019). The transcription factor LHX1 regulates the survival and directed migration of POA-derived cortical interneurons. Cereb. Cortex 29, 1644–1658. doi: 10.1093/cercor/bhy063
Symmank, J., and Zimmer, G. (2017). Regulation of neuronal survival by DNA methyltransferases. Neural Regen. Res. 12, 1768–1775. doi: 10.4103/1673-5374.219027
Tordjman, S., Somogyi, E., Coulon, N., Kermarrec, S., Cohen, D., Bronsard, G., et al. (2014). Gene × environment interactions in autism spectrum disorders: role of epigenetic mechanisms. Front. Psychiatry 5:53. doi: 10.3389/fpsyt.2014.00053
Treiman, D. M. (2001). GABAergic mechanisms in epilepsy. Epilepsia 42(Suppl. 3), 8–12. doi: 10.1046/j.1528-1157.2001.042Suppl.3008.x
Tremblay, R., Lee, S., and Rudy, B. (2016). GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292. doi: 10.1016/j.neuron.2016.06.033
Tsuang, M. T., Stone, W. S., and Faraone, S. V. (2001). Genes, environment and schizophrenia. Br. J. Psychiatry Suppl. 40, s18–s24. doi: 10.1192/bjp.178.40.s18
Tuchman, R., and Cuccaro, M. (2011). Epilepsy and autism: neurodevelopmental perspective. Curr. Neurol. Neursci. Rep. 11, 428–434. doi: 10.1007/s11910-011-0195-x
Veldic, M., Caruncho, H. J., Liu, W. S., Davis, J., Satta, R., Grayson, D. R., et al. (2004). DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc. Natl. Acad. Sci. U.S.A. 101, 348–353. doi: 10.1073/pnas.2637013100
Wockner, L. F., Noble, E. P., Lawford, B. R., Young, R. M. D., Morris, C. P., Whitehall, V. L. J., et al. (2014). Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 4:e339. doi: 10.1038/tp.2013.111
Wu, H., and Zhang, Y. (2014). Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68. doi: 10.1016/j.cell.2013.12.019
Yu, H., Su, Y., Shin, J., Zhong, C., Guo, J. U., Weng, Y. L., et al. (2015). Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat. Neurosci. 18, 836–843. doi: 10.1038/nn.4008
Zhubi, A., Veldic, M., Puri, N. V., Kadriu, B., Caruncho, H., Loza, I., et al. (2009). An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr. Res. 111, 115–122. doi: 10.1016/j.schres.2009.03.020
Keywords: inhibitory interneurons, GABA, schizophrenia, autism, cerebral cortex, DNA methylation
Citation: Linde J and Zimmer-Bensch G (2020) DNA Methylation-Dependent Dysregulation of GABAergic Interneuron Functionality in Neuropsychiatric Diseases. Front. Neurosci. 14:586133. doi: 10.3389/fnins.2020.586133
Received: 22 July 2020; Accepted: 25 August 2020;
Published: 16 September 2020.
Edited by:
Yan Jiang, Fudan University, ChinaReviewed by:
Nathalie Dehorter, Australian National University, AustraliaDaniel Vogt, Michigan State University, United States
Copyright © 2020 Linde and Zimmer-Bensch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geraldine Zimmer-Bensch, emltbWVyQGJpbzIucnd0aC1hYWNoZW4uZGU=; emltbWVyX2dlcmFsZGluZUB5YWhvby5kZQ==