 Ethan R. Roy
Ethan R. Roy Wei Cao
Wei Cao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Neurosci. , 02 October 2020
Sec. Neurodegeneration
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.577744
This article is part of the Research Topic Infection, Inflammation, Cardiovascular diseases and Neurodegeneration View all 15 articles
Alzheimer’s disease (AD) represents an enormous public health challenge currently and with increasing urgency in the coming decades. Our understanding of the etiology and pathogenesis of AD is rather incomplete, which is manifested in stagnated therapeutic developments. Apart from the well-established Amyloid Hypothesis of AD, gaining traction in recent years is the Pathogen Hypothesis, which postulates a causal role of infectious agents in the development of AD. Particularly, infection by viruses, among a diverse range of microorganisms, has been implicated. Recently, we described a prominent antiviral immune response in human AD brains as well as murine amyloid beta models, which has consequential effects on neuropathology. Such findings expectedly allude to the question about viral infections and AD. In this Perspective, we would like to discuss the molecular mechanism underlying the antiviral immune response, highlight how such pathway directly promotes AD pathogenesis, and depict a multilayered connection between antiviral immune response and other agents and factors relevant to AD. By tying together these threads of evidence, we provide a cohesive perspective on the uprising of antiviral immune response in AD.
Hallmarked by the deposition of β-amyloid plaques and accumulation of neurofibrillary tangles, Alzheimer’s disease (AD) manifests with complex pathophysiology and its etiology remains elusive. Among the many viewpoints for the underlying mechanisms, the Pathogen Hypothesis was proposed initially based on the clinical discoveries of Herpes Simplex Virus-1 (HSV-1) in association with AD, further expanded to include other microbes, and gained experimental support in recent years (Itzhaki et al., 1997, 2016; Itzhaki, 2018). In particular, Aβ has been shown to function as an antimicrobial peptide and, under experimental conditions, protect against microbial infection while seeding Aβ deposition (Kumar et al., 2016; Eimer et al., 2018). Separately, pathogenic involvement of proinflammatory responses in AD is also increasingly being appreciated (Heneka et al., 2015; Ransohoff, 2016). Because of the intimate association between infections and inflammation, the Pathogen Hypothesis putatively links to the neuroinflammation phenomenon in many aspects; however, the precise molecular correlation between these two processes in AD has yet to be established. Here, we intend to make the connections from multiple angles.
Mammalian antiviral innate immune defense mechanism utilize an array of nucleic acid innate immune sensors to detect viral genomes or their replication products, which activate a cascade of signaling events to induce rapid gene expression (Barrat et al., 2016). Among them, the type I IFN (IFN) cytokines, which include multiple IFNα subtypes, IFNβ, IFNε IFNκ, and IFNϖ, instruct the frontline antiviral response (Capobianchi et al., 2015; McNab et al., 2015). All IFN species signal through a common receptor complex and drive the transcription of a large panel of IFN-stimulated genes (ISGs) (Schreiber and Piehler, 2015). ISGs operate in concert to interfere with viral replication through viral genome degradation and blockade of gene expression, protein synthesis and virion assembly, thus conveying protection.
Although normally non-immunogenic, host self-derived nucleic acids can provoke IFN response under pathological conditions such as autoimmune diseases. In systemic lupus erythematosus, immune complexes comprised of a patient’s DNA or RNA and associated autoantibodies activate plasmacytoid dendritic cells (pDCs), a subset of innate immune cells, to stimulate IFN production and flares of systemic inflammation (Gilliet et al., 2008). In psoriasis, antimicrobial peptide cathelicidin LL-37 complexes with nucleic acids and similarly induces aberrant IFN production from pDCs (Lande et al., 2007). In both cases, nucleic acid-containing complexes gain immunogenicity by delivering nucleic acids to the intracellular innate immune sensors in pDCs to activate the signaling cascade for IFN production.
LL-37 and other antimicrobial peptides oligomerize and form pore structures on biological membranes (Arnusch et al., 2007; Xhindoli et al., 2014) (Figure 1). Similarly, oligomerization of Aβ exerts neuronal toxicity as well as antimicrobial function (Kayed et al., 2003; Soscia et al., 2010). By studying oligomers made from various proteins, we not only confirmed this fascinating gain-of-function but also discovered another intrinsic property of protein oligomers – affinity toward negatively charged molecules (Di Domizio et al., 2012b). Complexing soluble oligomers with nucleic acids or glycosaminoglycan, both negatively charged, expedite the formation of amyloid fibrils in vitro. More strikingly, nucleic acid-containing amyloid fibrils are potent inducers of IFN response from pDCs in vitro and in vivo (Di Domizio et al., 2012a). In short, Aβ and LL-37 share several characteristics: oligomerization, cytotoxicity to host and microbe cells, and binding to cofactors, the latter conveys interferongenicity (Figure 1). Not surprisingly then, amyloid-DNA composites present in bacterial biofilm stimulate an IFN response and promote autoimmunity (Gallo et al., 2015). To immune cells, these protein-nucleic acid complexes are indiscriminately sensed as virions to trigger an antiviral immune response.
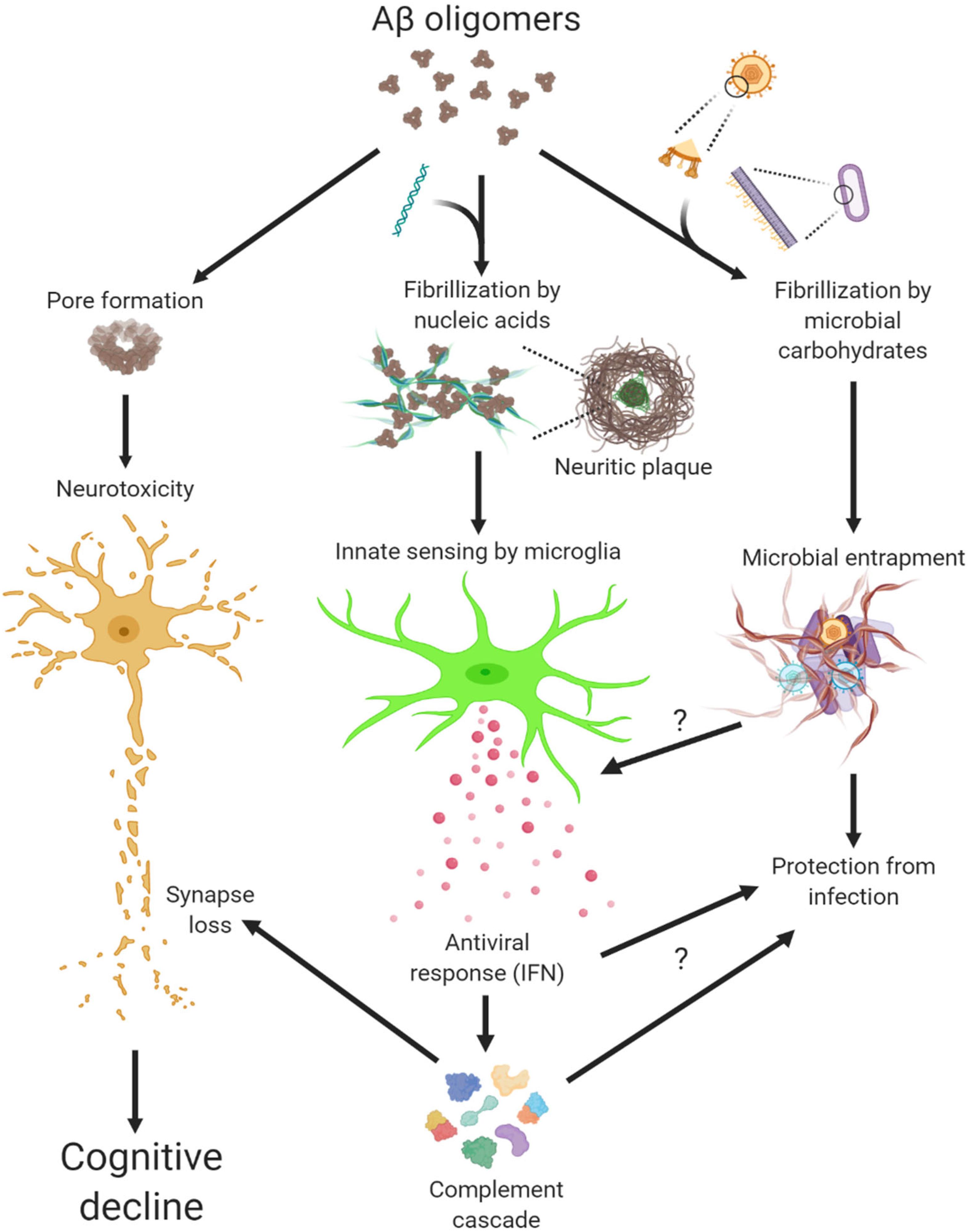
Figure 1. Multiple actions of Aβ oligomers leading to neuropathogenesis in Alzheimer’s disease.
Microglia, the brain resident immune cells, fulfill important functions in trophic support, cell debris removal and tissue surveillance under homeostatic conditions (Li and Barres, 2018; Prinz et al., 2019). However, microglial dysfunction can be a primary cause to neurological disorders, thus disease-associated microglia have received intense attention in recent years. Mouse primary microglia elicited a rapid IFN response to nucleic acid-containing amyloid fibrils in vitro, in a manner similar to that from peripheral immune cells (Roy et al., 2020). Activated microglia expressing Clec7a not only selectively surrounded the amyloid β plaques with sequestered DNA and RNA but also upregulated a large panel of ISGs, indicating profound IFN pathway activation in mouse models of amyloidosis. Thus, IFN constitutes a pivotal element within the neuroinflammatory network of AD. Most recently, the early and persistent IFN and antiviral response was confirmed by an analysis of microglial proteomes from Aβ models (Monasor et al., 2020).
Microglia recruited to amyloid β plaques adopt a disease-associated phenotype, where they acquire a unique molecular state by downregulating homeostatic markers, upregulating immune effector molecules, and express a panel of genes that have been associated with increased risk for AD and other neurodegenerative disorders (Butovsky and Weiner, 2018; Hammond et al., 2018; Wang and Colonna, 2019). IFN was shown to be required for sustaining microglial activation since suppression of IFN signaling significantly reduced the levels of CD68 and Clec7a, markers for microglial activation, and shifted the morphology of microglia toward homeostatic form (Roy et al., 2020). Intriguingly, polymorphisms of ISGs, including OAS1, ITGAM, LAPTM5, and LILRB4, were conjointly identified as a significant risk factor for AD, implicating IFN pathway as a genetic modifier (Salih et al., 2019).
In AD tissue, ISG-expressing microglia preferentially associate with nucleic acid+ neuritic plaques (Roy et al., 2020). Furthermore, the IFN activation signature was correlated with amyloid load and disease severity across a large study cohort represented in the Mt. Sinai Brain Bank. These findings are in line with earlier reports of neuritic plaques sequestering self nucleic acids and microglia expressing IFNα in AD brains (Yamada et al., 1994; Ginsberg et al., 1997). Therefore, a prototypical antiviral immune response is manifested in both pre-clinic models and clinic AD cases. Interestingly, AD patients with rare TREM2 R47H variant, an AD risk factor, have increased presentation of proinflammatory microglia subsets including those enriched with IFN response (Sayed et al., 2020).
Although normal brain lacks detectable IFN expression, transient IFN production in CNS protects against opportunistic viral infections (Nallar and Kalvakolanu, 2014). However, chronic and dysregulated IFN expression is a major driver for type I interferonopathy, a group of hereditary CNS disorders (Hofer and Campbell, 2013; Rodero and Crow, 2016; Schwabenland et al., 2019). Separately, patients receiving systemic IFN treatment or with HIV-induced dementia display increased brain IFN activation, which is associated with cognitive and psychiatric dysfunctions (Gray et al., 1996; Hayley et al., 2013; Dipasquale et al., 2016; Wachholz et al., 2016). These observations collectively suggest a toxic influence of IFN on the brain. When delivered to the brain, IFNβ directly activates microglia, initiates neuroinflammation, and leads to microglia-mediated synapse loss (Roy et al., 2020). Conversely, suppression of IFN signaling in Aβ models rescued the synapse loss in the brain, supporting a direct role of IFN in synapse modification. Interestingly, IFN receptor ablation was previously shown to improve cognitive performance while altering glial phenotypes in APPSWE/PS1ΔE9 mice (Minter et al., 2016).
Complement has been implicated in synaptic pathologies in diverse neurological and neuropsychiatric diseases, in particular neurodegeneration (Hong et al., 2016; Morgan, 2018; Tenner et al., 2018; Wu et al., 2019). Intriguingly, not only do a number of complement genes represent bona fide ISGs, but IFN-stimulated synapse elimination depends on the function of complement C3 (Roy et al., 2020). Not coincidently, synapse loss and memory impairment in mice recovered from West Nile Virus (WNV) infection was shown mediated by persistent microglial activation and the functional involvement of complement C3 (Vasek et al., 2016). Moreover, IFN pathway is robustly correlated with complement cascade in human AD (Roy et al., 2020). Given that synapse loss is clinically associated with cognitive decline (DeKosky and Scheff, 1990; Scheff et al., 1990; Terry et al., 1991), these findings highlight a pathogenic role of antiviral immune response in conjunction with complement cascade in modifying synapses in AD (Figure 1).
The central nervous system (CNS) is protected by a highly complex barrier structure, but is by no means invincible to infections. Many neurotropic viruses can gain access to the brain through blood circulation or peripheral nerves (Swanson and McGavern, 2015). Acute viral infection in the CNS often triggers inflammatory response, where the IFN pathway plays a crucial role in the defense against a wide range of viral pathogens (Paul et al., 2007). However, strong immune reaction also leads to acute meningitis, encephalitis and myelitis, which manifest with behavioral and cognitive disruptions (Swanson and McGavern, 2015). In humans, WNV, HIV-1, Zika virus, and HSV-1 are the most common causes for viral encephalitis. Of note, the elderly population is susceptible to reactivation of latent varicella zoster virus (VZV), which results in shingles (Gilden et al., 2009). VZV is normally controlled by peripheral IFN response, but occasionally it can spread to CNS to cause encephalitis (Gilden et al., 2009; Kim et al., 2017). Fortunately, many people recover from these acute episodes; yet, a significant portion suffer from long term neurological sequelae (Swanson and McGavern, 2015; Klein et al., 2017). Rather than the irreversible damage caused by the pathogens, recent studies suggested a role of chronic inflammation that underlie neurological impairments, including conditions post WNV infection as mentioned earlier (Vasek et al., 2016). Therefore, prior exposure to CNS infections may have long-lasting neurocognitive consequences (Figure 2).
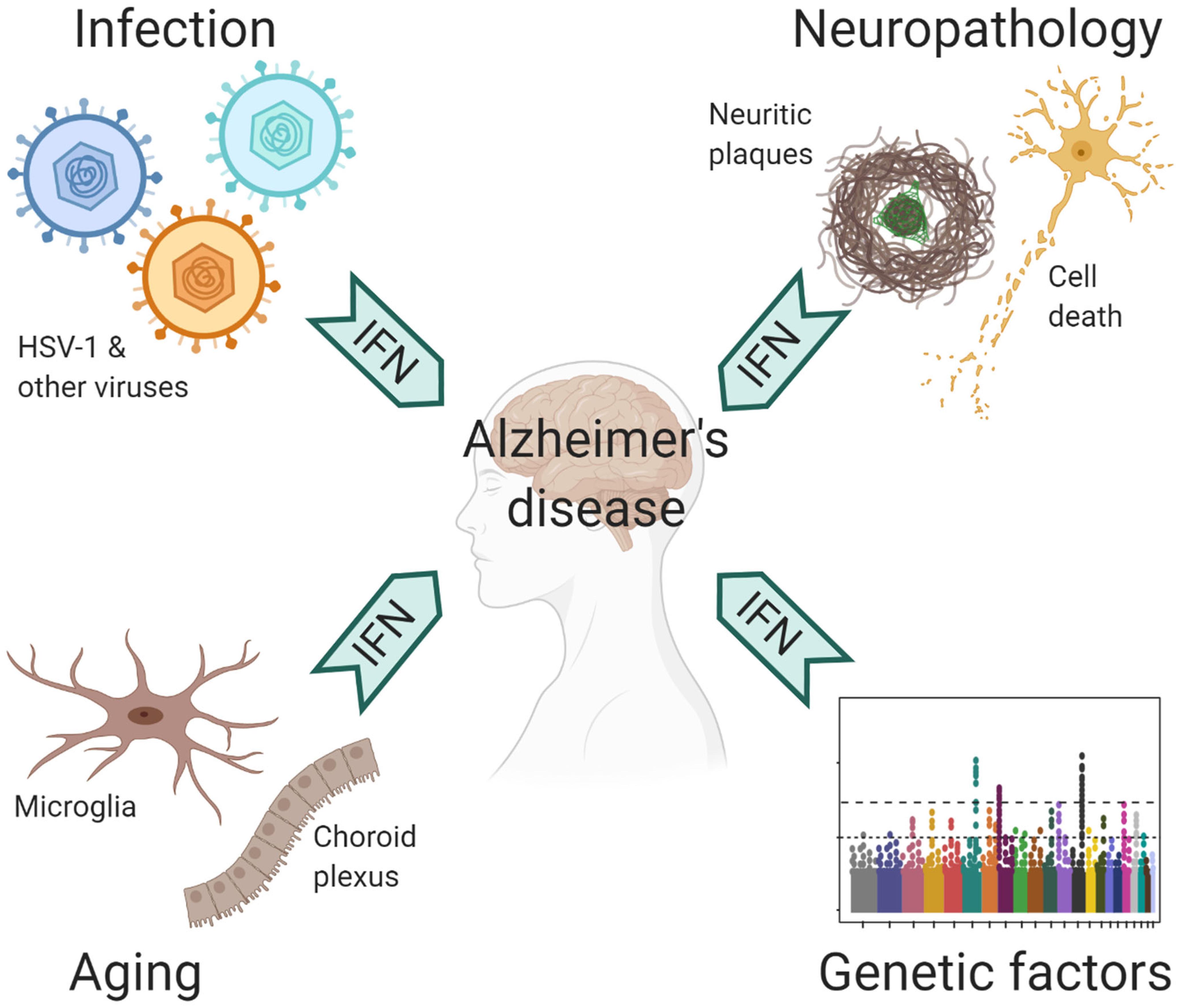
Figure 2. Contributing factors to antiviral immune response in AD.
Although responsible for a common form of acute viral encephalitis, herpes simplex virus mostly carries out latent infection in the general population. Itzhaki et al. (1997) first detected HSV-1 DNA in the brain of 60% cases of apolipoprotein E gene (APOE-ε4) carriers and subsequently postulated the viral concept of AD: HSV-1 likely travels to the brain in middle age, where it remains in a latent state, and accumulation of damage from intermittent reactivation – direct viral action and major inflammatory effects – leads eventually to the development of AD (Itzhaki, 2018). Although the causal relationship between HSV-1 reactivation and AD is extremely difficult to prove in humans, the proposal is consistent with the concept of pathogenic chronic inflammation in CNS, as mentioned above. The IFN signaling, as a result of the cGAS-STING pathway activation in microglia, is indispensable for controlling HSV-1 infection in humans (Dupuis et al., 2003; Reinert et al., 2016). Therefore, IFN response required for resolving periodic HSV-1 activation may conceivably be a contributing factor in AD (Figure 2). Over the years, other viral pathogens have been detected in AD specimens and implicated in AD risk, among which noticeably are members of Herpesviridae. Increased human herpesvirus subtypes (HHV-6 and HHV-7) were independently identified from a multi-scale gene expression network analysis of late-onset AD (Readhead et al., 2018), though the study was later challenged (Jeong and Liu, 2019; Allnutt et al., 2020). On the other hand, while acute HSV-1 infection induced Aβ production in human induced neural stem cells (Cairns et al., 2020), Aβ was shown to play a protective role in brain against experimental infections by Herpesviridae, including HSV-1, and HHV-6 (Eimer et al., 2018). The interaction between Aβ and different types of microbes involves surface carbohydrate recognition, which rapidly seeds amyloid fibrils (Kumar et al., 2016; Eimer et al., 2018; Figure 1). In lieu of immune reaction, it remains to be shown if Aβ-entrapped viruses can be recognized by microglia and elicit effective IFN response.
Under pathological conditions, self-derived molecules can potently trigger inflammatory response in the absence of an infection. As discussed earlier, extracellular amyloid plaques with sequestered nucleic acids are recognized by microglia and elicit an antiviral response analogous to that during viral infection (Figure 1). Of note, sequence analysis of RNA isolated from AD neuritic plaques identified transcripts from cortical neurons, marking a self origin of plaque-associated nucleic acids (Ginsberg et al., 1999). Conceivably, such intrinsic IFN response would deter opportunistic viral infection or reactivation in AD brain, a point that awaits further investigation (Figure 1). On the other hand, dysregulated nucleic acid catabolism also results in aberrant IFN production. For example, mutations of deoxyribonuclease TREX1 or ribonuclease RNASEH2 lead to the accumulation of aberrant cytosolic nucleic acid species, IFN production, and encephalopathy in Aicardi-Goutieres syndrome (Rodero and Crow, 2016). Under neurodegenerative conditions, dead brain cells may release nucleic acids and other alarmin molecules thus stimulating an inflammation response. Although peripheral immune cells discern immunogenic vs. non-immunogenic cell death (Green et al., 2009), how microglia innately respond to different forms of CNS cell death is not known at this time. It is thus important to examine if additional endogenous agents stimulate innate IFN response in AD besides amyloid plaques.
It’s well known that age is the most important risk factor for late-onset AD (LOAD). In normal aging brain, heightened IFN signaling from microglia inside the parenchyma and choroid plexus, an epithelial tissue located within the ventricles, has been shown to be detrimental to neurogenesis and cognitive function (Baruch et al., 2014; Deczkowska et al., 2017). On the other hand, LOAD is a polygenic disease, where a number of risk polymorphism and rare variants exert their functions from microglia and/or involved in immunity (Zhang et al., 2013; Huang et al., 2017; Kunkle et al., 2019). The implication of ISGs as risk factor of AD (Salih et al., 2019) together with IFN upregulation in aging brain suggest that IFN pathway may have a profound influence on AD pathogenesis (Figure 2).
Adult Down syndrome (DS) patients, who mostly carry trisomy 21 in their genome, unanimously develop the neuropathological changes of AD (Lott and Head, 2019). Besides amyloid precursor protein gene, four of the six IFN receptors, IFNAR1, IFNAR2, IFNGR2, and IL10RB, are encoded in the extra chromosome 21, which results in profound peripheral IFN response and autoinflammation in DS patients (Kola and Hertzog, 1997; Sullivan et al., 2016, 2017). Interestingly, mice bearing trisomy 16, which contains many orthologs from human trisomy 21, benefited from receiving antibodies blocking type I IFN (IFNα/β) and type II IFN (IFNγ) at development stage in vivo, whereas blocking IFNγ signaling rescued the premature death of trisomy 16 cortical neurons in vitro (Maroun, 1995; Hallam and Maroun, 1998; Hallam et al., 2000). By contrast, IFNγ blockade did not affect microglial activation nor synapse loss in amyloid β model (Roy et al., 2020). Since both type I and type II IFNs activate an overlapping JAK/STAT pathway to convey antiviral protection (Liu et al., 2012), these findings imply DS as a disease likely more affected by interferon activation.
In summary, we have described the IFN pathway activation, a prototypic antiviral immune response, as a major component of CNS neuroinflammatory network in AD and connected IFN response to various endogenous, pathological, infectious and genetic risk factors that have been implicated in AD pathogenesis. The discussion here is largely focused on the molecular events originated within CNS, but peripheral IFN, manifested as a result of viral infection, autoimmune condition or drug administration, nevertheless affects brain functions (Blank et al., 2016). Therefore, many important questions remain to be investigated to advance our fundamental understanding of AD more in the future.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
WC formulated the concepts and wrote the manuscript. ER contributed to the manuscript writing and figure preparation. All authors contributed to the article and approved the submitted version.
This study was funded by the NIH grant AG057587, BrightFocus ADR, and Brown Foundation 2020 Healthy Aging Initiative to WC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Allnutt, M. A., Johnson, K., Bennett, D. A., Connor, S. M., Troncoso, J. C., Pletnikova, O., et al. (2020). Human herpesvirus 6 detection in Alzheimer’s Disease cases and controls across multiple cohorts. Neuron 105, 1027.e2–1035.e2.
Arnusch, C. J., Branderhorst, H., de Kruijff, B., Liskamp, R. M., Breukink, E., and Pieters, R. J. (2007). Enhanced membrane pore formation by multimeric/oligomeric antimicrobial peptides. Biochemistry 46, 13437–13442. doi: 10.1021/bi7015553
Barrat, F. J., Elkon, K. B., and Fitzgerald, K. A. (2016). Importance of nucleic acid recognition in inflammation and autoimmunity. Annu. Rev. Med. 67, 323–336. doi: 10.1146/annurev-med-052814-023338
Baruch, K., Deczkowska, A., David, E., Castellano, J. M., Miller, O., Kertser, A., et al. (2014). Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93. doi: 10.1126/science.1252945
Blank, T., Detje, C. N., Spiess, A., Hagemeyer, N., Brendecke, S. M., Wolfart, J., et al. (2016). Brain endothelial- and epithelial-specific interferon receptor chain 1 drives virus-induced sickness behavior and cognitive impairment. Immunity 44, 901–912. doi: 10.1016/j.immuni.2016.04.005
Butovsky, O., and Weiner, H. L. (2018). Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 19, 622–635. doi: 10.1038/s41583-018-0057-5
Cairns, D. M., Rouleau, N., Parker, R. N., Walsh, K. G., Gehrke, L., and Kaplan, D. L. (2020). A 3D human brain-like tissue model of herpes-induced Alzheimer’s disease. Sci. Adv. 6:eaay8828. doi: 10.1126/sciadv.aay8828
Capobianchi, M. R., Uleri, E., Caglioti, C., and Dolei, A. (2015). Type I IFN family members: similarity, differences and interaction. Cytokine Growth Factor Rev. 26, 103–111. doi: 10.1016/j.cytogfr.2014.10.011
Deczkowska, A., Matcovitch-Natan, O., Tsitsou-Kampeli, A., Ben-Hamo, S., Dvir-Szternfeld, R., Spinrad, A., et al. (2017). Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat. Commun. 8:717.
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. doi: 10.1002/ana.410270502
Di Domizio, J., Dorta-Estremera, S., Gagea, M., Ganguly, D., Meller, S., Li, P., et al. (2012a). Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 109, 14550–14555. doi: 10.1073/pnas.1206923109
Di Domizio, J., Zhang, R., Stagg, L. J., Gagea, M., Zhuo, M., Ladbury, J. E., et al. (2012b). Binding with nucleic acids or glycosaminoglycans converts soluble protein oligomers to amyloid. J. Biol. Chem. 287, 736–747. doi: 10.1074/jbc.m111.238477
Dipasquale, O., Cooper, E. A., Tibble, J., Voon, V., Baglio, F., Baselli, G., et al. (2016). Interferon-alpha acutely impairs whole-brain functional connectivity network architecture - A preliminary study. Brain Behav. Immun. 58, 31–39. doi: 10.1016/j.bbi.2015.12.011
Dupuis, S., Jouanguy, E., Al-Hajjar, S., Fieschi, C., Al-Mohsen, I. Z., Al-Jumaah, S., et al. (2003). Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33, 388–391. doi: 10.1038/ng1097
Eimer, W. A., Vijaya Kumar, D. K., Navalpur Shanmugam, N. K., Rodriguez, A. S., Mitchell, T., Washicosky, K. J., et al. (2018). Alzheimer’s Disease-associated beta-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99, 56.e3–63.e3.
Gallo, P. M., Rapsinski, G. J., Wilson, R. P., Oppong, G. O., Sriram, U., Goulian, M., et al. (2015). Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity 42, 1171–1184. doi: 10.1016/j.immuni.2015.06.002
Gilden, D., Cohrs, R. J., Mahalingam, R., and Nagel, M. A. (2009). Varicella zoster virus vasculopathies: diverse clinical manifestations, laboratory features, pathogenesis, and treatment. Lancet Neurol. 8, 731–740. doi: 10.1016/s1474-4422(09)70134-6
Gilliet, M., Cao, W., and Liu, Y. J. (2008). Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606. doi: 10.1038/nri2358
Ginsberg, S. D., Crino, P. B., Hemby, S. E., Weingarten, J. A., Lee, V. M., Eberwine, J. H., et al. (1999). Predominance of neuronal mRNAs in individual Alzheimer’s disease senile plaques. Ann. Neurol. 45, 174–181. doi: 10.1002/1531-8249(199902)45:2<174::aid-ana7>3.0.co;2-e
Ginsberg, S. D., Crino, P. B., Lee, V. M., Eberwine, J. H., and Trojanowski, J. Q. (1997). Sequestration of RNA in Alzheimer’s disease neurofibrillary tangles and senile plaques. Ann. Neurol. 41, 200–209.
Gray, F., Scaravilli, F., Everall, I., Chretien, F., An, S., Boche, D., et al. (1996). Neuropathology of early HIV-1 infection. Brain Pathol. 6, 1–15. doi: 10.1007/978-1-4471-1957-9_1
Green, D. R., Ferguson, T., Zitvogel, L., and Kroemer, G. (2009). Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363. doi: 10.1038/nri2545
Hallam, D. M., Capps, N. L., Travelstead, A. L., Brewer, G. J., and Maroun, L. E. (2000). Evidence for an interferon-related inflammatory reaction in the trisomy 16 mouse brain leading to caspase-1-mediated neuronal apoptosis. J. Neuroimmunol. 110, 66–75. doi: 10.1016/s0165-5728(00)00289-7
Hallam, D. M., and Maroun, L. E. (1998). Anti-gamma interferon can prevent the premature death of trisomy 16 mouse cortical neurons in culture. Neurosci. Lett. 252, 17–20. doi: 10.1016/s0304-3940(98)00541-2
Hammond, T. R., Robinton, D., and Stevens, B. (2018). Microglia and the Brain: complementary partners in development and disease. Annu. Rev. Cell. Dev. Biol. 34, 523–544. doi: 10.1146/annurev-cellbio-100616-060509
Hayley, S., Scharf, J., and Anisman, H. (2013). Central administration of murine interferon-alpha induces depressive-like behavioral, brain cytokine and neurochemical alterations in mice: a mini-review and original experiments. Brain Behav. Immun. 31, 115–127. doi: 10.1016/j.bbi.2012.07.023
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405.
Hofer, M. J., and Campbell, I. L. (2013). Type I interferon in neurological disease-the devil from within. Cytokine Growth Factor Rev. 24, 257–267. doi: 10.1016/j.cytogfr.2013.03.006
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Huang, K. L., Marcora, E., Pimenova, A. A., Di Narzo, A. F., Kapoor, M., Jin, S. C., et al. (2017). A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 20, 1052–1061. doi: 10.1038/nn.4587
Itzhaki, R. F. (2018). Corroboration of a major role for herpes simplex virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 10:324. doi: 10.3389/fnagi.2018.00324
Itzhaki, R. F., Lathe, R., Balin, B. J., Ball, M. J., Bearer, E. L., Braak, H., et al. (2016). Microbes and Alzheimer’s Disease. J. Alzheimer’s Dis. 51, 979–984.
Itzhaki, R. F., Lin, W.-R., Shang, D., Wilcock, G. K., Faragher, B., and Jamieson, G. A. (1997). Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349, 241–244. doi: 10.1016/s0140-6736(96)10149-5
Jeong, H. H., and Liu, Z. (2019). Are HHV-6A and HHV-7 really more abundant in Alzheimer’s Disease? Neuron 104, 1034–1035. doi: 10.1016/j.neuron.2019.11.009
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Kim, J.-A., Park, S.-K., Seo, S.-W., Lee, C.-H., and Shin, O. S. (2017). STING Is involved in antiviral immune response against VZV infection via the induction of Type I and III IFN pathways. J. Invest. Dematol. 137, 2101–2109. doi: 10.1016/j.jid.2017.03.041
Klein, R. S., Garber, C., and Howard, N. (2017). Infectious immunity in the central nervous system and brain function. Nat. Immunol. 18, 132–141. doi: 10.1038/ni.3656
Kola, I., and Hertzog, P. J. (1997). Animal models in the study of the biological function of genes on human chromosome 21 and their role in the pathophysiology of Down syndrome. Hum. Mol. Genet. 6, 1713–1727. doi: 10.1093/hmg/6.10.1713
Kumar, D. K., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., Ghofrani, J., et al. (2016). Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 8:340ra372.
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 51, 414–430.
Lande, R., Gregorio, J., Facchinetti, V., Chatterjee, B., Wang, Y. H., Homey, B., et al. (2007). Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449, 564–569.
Li, Q., and Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242. doi: 10.1038/nri.2017.125
Liu, S. Y., Sanchez, D. J., Aliyari, R., Lu, S., and Cheng, G. (2012). Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. U.S.A. 109, 4239–4244. doi: 10.1073/pnas.1114981109
Lott, I. T., and Head, E. (2019). Dementia in down syndrome: unique insights for Alzheimer disease research. Nat. Rev. Neurol. 15, 135–147. doi: 10.1038/s41582-018-0132-6
Maroun, L. E. (1995). Anti-interferon immunoglobulins can improve the trisomy 16 mouse phenotype. Teratology 51, 329–335. doi: 10.1002/tera.1420510509
McNab, F., Mayer-Barber, K., Sher, A., Wack, A., and O’Garra, A. (2015). Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103. doi: 10.1038/nri3787
Minter, M. R., Moore, Z., Zhang, M., Brody, K. M., Jones, N. C., Shultz, S. R., et al. (2016). Deletion of the type-1 interferon receptor in APPSWE/PS1DeltaE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol. Commun. 4:72.
Monasor, L. S., Muller, S. A., Colombo, A. V., Tanrioever, G., Konig, J., Roth, S., et al. (2020). Fibrillar Abeta triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. eLife 9:e54083. doi: 10.7554/eLife.54083
Morgan, B. P. (2018). Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 40, 113–124.
Nallar, S. C., and Kalvakolanu, D. V. (2014). Interferons, signal transduction pathways, and the central nervous system. J. Interferon Cytokine Res. 34, 559–576. doi: 10.1089/jir.2014.0021
Paul, S., Ricour, C., Sommereyns, C., Sorgeloos, F., and Michiels, T. (2007). Type I interferon response in the central nervous system. Biochimie 89, 770–778. doi: 10.1016/j.biochi.2007.02.009
Prinz, M., Jung, S., and Priller, J. (2019). Microglia biology: one century of evolving concepts. Cell 179, 292–311. doi: 10.1016/j.cell.2019.08.053
Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi: 10.1126/science.aag2590
Readhead, B., Haure-Mirande, J. V., Funk, C. C., Richards, M. A., Shannon, P., Haroutunian, V., et al. (2018). Multiscale analysis of independent Alzheimer’s Cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99, 64.e7–82.e7.
Reinert, L. S., Lopusna, K., Winther, H., Sun, C., Thomsen, M. K., Nandakumar, R., et al. (2016). Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 7:13348.
Rodero, M. P., and Crow, Y. J. (2016). Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J. Exp. Med. 213, 2527–2538. doi: 10.1084/jem.20161596
Roy, E. R., Wang, B., Wan, Y. W., Chiu, G., Cole, A., Yin, Z., et al. (2020). Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 130, 1912–1930. doi: 10.1172/jci133737
Salih, D. A., Bayram, S., Guelfi, S., Reynolds, R. H., Shoai, M., Ryten, M., et al. (2019). Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun. 1:fcz022.
Sayed, F. A., Kodama, L., Udeochu, J. C., Fan, L., Carling, G. K., Le, D., et al. (2020). AD-linked R47H-TREM2 mutation induces disease-enhancing proinflammatory microglial states in mice and humans. bioRxiv [Preprint]. doi: 10.1101/2020.07.24.218719
Scheff, S. W., DeKosky, S. T., and Price, D. A. (1990). Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging 11, 29–37. doi: 10.1016/0197-4580(90)90059-9
Schreiber, G., and Piehler, J. (2015). The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol. 36, 139–149. doi: 10.1016/j.it.2015.01.002
Schwabenland, M., Mossad, O., Peres, A. G., Kessler, F., Maron, F. J. M., Harsan, L. A., et al. (2019). Loss of USP18 in microglia induces white matter pathology. Acta Neuropathol. Commun. 7:106.
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505. doi: 10.1371/journal.pone.0009505
Sullivan, K. D., Evans, D., Pandey, A., Hraha, T. H., Smith, K. P., Markham, N., et al. (2017). Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep. 7:14818.
Sullivan, K. D., Lewis, H. C., Hill, A. A., Pandey, A., Jackson, L. P., Cabral, J. M., et al. (2016). Trisomy 21 consistently activates the interferon response. eLife 5:e16220.
Swanson, P. A., and McGavern, D. B. (2015). Viral diseases of the central nervous system. Curr. Opin. Virol. 11, 44–54.
Tenner, A. J., Stevens, B., and Woodruff, T. M. (2018). New tricks for an ancient system: physiological and pathological roles of complement in the CNS. Mol. Immunol. 102, 3–13. doi: 10.1016/j.molimm.2018.06.264
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Vasek, M. J., Garber, C., Dorsey, D., Durrant, D. M., Bollman, B., Soung, A., et al. (2016). A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543. doi: 10.1038/nature18283
Wachholz, S., Esslinger, M., Plumper, J., Manitz, M. P., Juckel, G., and Friebe, A. (2016). Microglia activation is associated with IFN-alpha induced depressive-like behavior. Brain Behav. Immun. 55, 105–113. doi: 10.1016/j.bbi.2015.09.016
Wang, S., and Colonna, M. (2019). Microglia in Alzheimer’s disease: a target for immunotherapy. J. Leukoc Biol. 106, 219–227.
Wu, T., Dejanovic, B., Gandham, V. D., Gogineni, A., Edmonds, R., Schauer, S., et al. (2019). Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 28, 2111.e6–2123.e6.
Xhindoli, D., Pacor, S., Guida, F., Antcheva, N., and Tossi, A. (2014). Native oligomerization determines the mode of action and biological activities of human cathelicidin LL-37. Biochem. J. 457, 263–275. doi: 10.1042/bj20131048
Yamada, T., Horisberger, M. A., Kawaguchi, N., Moroo, I., and Toyoda, T. (1994). Immunohistochemistry using antibodies to α-interferon and its induced protein, MxA, in Alzheimer’s and Parkinson’s disease brain tissues. Neurosci. Lett. 181, 61–64. doi: 10.1016/0304-3940(94)90560-6
Keywords: antiviral, interferon, Alzheimer’s disease, innate immunity, amyloid, pathogen hypothesis, neuroinflammation
Citation: Roy ER and Cao W (2020) Antiviral Immune Response in Alzheimer’s Disease: Connecting the Dots. Front. Neurosci. 14:577744. doi: 10.3389/fnins.2020.577744
Received: 29 June 2020; Accepted: 08 September 2020;
Published: 02 October 2020.
Edited by:
Xian-Le Bu, Third Military Medical University, ChinaReviewed by:
Eric Harold Frost, Université de Sherbrooke, CanadaCopyright © 2020 Roy and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Cao, d2VpLmNhb0BiY20uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.