 Jiejie Wang
Jiejie Wang Raymond A. Swanson
Raymond A. Swanson- Department of Neurology, University of California, San Francisco, and San Francisco Veterans Affairs Health Care System, San Francisco, CA, United States
Excitotoxicity is classically attributed to Ca2+ influx through NMDA receptors (NMDAr), leading to production of nitric oxide by neuronal nitric oxide synthase and superoxide by mitochondria, which react to form highly cytotoxic peroxynitrite. More recent observations warrant revision of the classic view and help to explain some otherwise puzzling aspects of excitotoxic cell injury. Studies using pharmacological and genetic approaches show that superoxide produced by NMDAr activation originates primarily from NADPH oxidase rather than from mitochondria. As NADPH oxidase is localized to the plasma membrane, this also provides an explanation for the extracellular release of superoxide and cell-to-cell “spread” of excitotoxic injury observed in vitro and in vivo. The signaling pathway linking NMDAr to NADPH oxidase involves Ca2+ influx, phosphoinositol-3-kinase, and protein kinase Cζ, and interventions at any of these steps can prevent superoxide production and excitotoxic injury. Ca2+ influx specifically through NMDAr is normally required to induce excitotoxicity, through a mechanism presumed to involve privileged Ca2+ access to local signaling domains. However, experiments using selective blockade of the NMDAr ion channel and artificial reconstitution of Ca2+ by other routes indicate that the special effects of NMDAr activation are attributable instead to concurrent non-ionotropic NMDAr signaling by agonist binding to NMDAr. The non-ionotropic signaling driving NADPH oxidase activation is mediated in part by phosphoinositol-3-kinase binding to the C-terminal domain of GluN2B receptor subunits. These more recently identified aspects of excitotoxicity expand our appreciation of the complexity of excitotoxic processes and suggest novel approaches for limiting neuronal injury.
Introduction
The term “excitotoxicity” was first used in reference to rapid neuronal death caused by glutamate receptor activation (Olney et al., 1971). The term has subsequently been used in reference to glutamate receptor-mediated cell death of other cell types, to describe more protracted cell death processes, and with activation of several different glutamate receptor subtypes. The present review focuses specifically on aspects of rapid neuronal death induced by pathological stimulation of NMDA-type glutamate receptors. This process is widely attributed to Ca2+ influx, leading to superoxide and nitric oxide production, which together generate the cytotoxic reactive oxygen species, peroxynitrite. However, recent studies have identified several additional complexities that challenge this classical view and identify novel ways to suppress excitotoxic neuronal death. These complexities arise from interactions between superoxide and nitric oxide, the sources of superoxide formation, and the newly appreciated role of non-ionotropic NMDA receptor (NMDAr) signaling.
Superoxide and Nitric Oxide in Excitotoxicity: Can You Tell the Difference?
An intriguing aspect of excitotoxic cell death is that it requires the combined effects of two independently generated “gaseous” reactive oxygen species: nitric oxide and superoxide. Nitric oxide (NO) is non-polar, lipid permeable, and has a relatively long half-life and diffusion distance in brain (Pacher et al., 2007). Superoxide (O2–), by contrast, is polar, largely lipid impermeable, and has much shorter half-life and mean diffusion distance. Nitric oxide is generated in a subset of neurons by neuronal nitric oxide synthase. It is considered a reactive oxygen species (ROS) because it contains an unpaired electron, but it is not intrinsically a highly reactive molecule in biological systems. Nevertheless, studies using both pharmacological and genetic abrogation of nitric oxide production showed near-complete suppression of excitotoxic neuronal death (Dawson et al., 1991, 1996).
Superoxide, despite its name, is not a powerful oxidant and in fact behaves as a mild reductant under physiological conditions (Pacher et al., 2007). At roughly the same time that nitric oxide was identified as excitotoxic intermediate, studies using electron paramagnetic resonance and other methods demonstrated that superoxide was likewise produced by neurons during NMDAr stimulation, and that scavenging superoxide could likewise block excitotoxic death (Lafon-Cazal et al., 1993; Reynolds and Hastings, 1995; Patel et al., 1996). While at first unclear how negating either one or the other of these reactive intermediates could prevent excitotoxic injury, the observations were reconciled by the understanding that nitric oxide and superoxide combine at an extraordinarily fast rate to form the much more toxic reactive species, peroxynitrite (ONOO–) (Beckman et al., 1990; Radi, 2018). Peroxynitrite has subsequently become recognized as the primarily species responsible for the lipid peroxidation, DNA damage, protein nitration, and cell death that occur during excitoxicity (Lipton et al., 1994; Pacher et al., 2007; Campolo et al., 2020). Though highlighted more than 15 years ago, it remains poorly recognized how this interaction between superoxide and nitric oxide complicates the interpretation of experiments evaluating excitotoxicity, and specifically whether it is superoxide or nitric oxide that is increased (or decreased) in any particular setting (Pacher et al., 2007). Increased production of either superoxide alone or nitric oxide alone is sufficient to increase peroxynitrite production, as long as a minimal, basal level of the other species is present. Markers of peroxynitrite reaction products, such as 3-nitrotyrosine, thus increase whether the underlying cause of peroxynitrite formation is elevated superoxide, elevated nitric oxide, or both (Radi, 2018). Only with measures that assess the actual flux of these reactive intermediates can these possibilities be distinguished. As further detailed in the section “Signaling pathways underlying NOX2 activation by NMDA receptors,” the issue gains significance because interventions that are thought to act by blocking nitric oxide formation may in fact function by blocking superoxide formation, and vice versa.
Sources of Excitotoxic Superoxide Production
Although nitric oxide is produced almost exclusively by nitric oxide synthase, superoxide can originate from multiple sources. Mitochondria can generate superoxide as a byproduct of normal respiration, by one electron addition to oxygen at the level of electron transport chain or any of several mitochondrial dehydrogenases (Andreyev et al., 2005). Importantly, mitochondria can also indirectly elevate superoxide levels when their normal superoxide scavenging functions are impaired. These scavenging functions require active regeneration of mitochondrial NADPH from NADP+ by electrons from mitochondrial dehydrogenases or electron transport complexes (Andreyev et al., 2005), such that impaired mitochondrial function can limit this scavenging function.
The first evidence that mitochondria could be a source of NMDA-induced superoxide elevation was based on mitochondrial localization of oxidant-sensitive fluorescent indicators together with a reduction in this mitochondrial signal with mitochondrial inhibitors (Dugan et al., 1995; Reynolds and Hastings, 1995; Bindokas et al., 1996; Duan et al., 2007). However, a reduction in the mitochondrial fluorescent dye signal in response to mitochondrial inhibitors does not establish mitochondria as the oxidant source. This is in part because the inhibitors also cause mitochondrial and plasma membrane depolarization, with resultant dye efflux and reduced signal from the mitochondria, independent of any change in actual superoxide levels (Nicholls, 2006). Mitochondrial inhibitors also reduce the oxidant scavenging capacity of mitochondria, which is extremely difficult to distinguish from a true increase in superoxide production. Moreover, the localization of oxidized dyes or any other oxidant signal to the mitochondria do not provide definitive evidence that mitochondria are the source of production, as demonstrated by oxidation of mitoSOX in neuronal mitochondria by xanthine/xanthine oxidase added to the culture medium (Johnson-Cadwell et al., 2007). Last, the calcium influx induced by NMDAr stimulation causes mitochondrial depolarization, and mitochondrial depolarization acts to reduce rather than increase superoxide production in other settings (Nicholls and Ward, 2000).
Nevertheless, many subsequent studies have provided additional, indirect evidence for mitochondria as a source of excitotoxic superoxide formation (Sengpiel et al., 1998; Starkov et al., 2004; Stanika et al., 2009; Nguyen et al., 2011; Wang et al., 2013b). Perhaps the strongest evidence comes from two studies in which genetic and pharmacological inhibition of the mitochondrial calcium uniporter was found to reduce excitotoxic death (Qiu et al., 2013; Angelova et al., 2019), however, neither of these studies evaluated actual superoxide production, and a biophysical mechanism by which Ca2+ movement into mitochondria drives superoxide production remains to be established.
A second source of superoxide production is the ubiquitous enzyme, nicotinamide adenine dinucleotide phosphate oxidase (abbreviated as NOX). NOX is a membrane-associated enzyme that transfers an electron from NADPH on one side of the membrane to molecular oxygen on the other side, forming O2– (Figure 1). NOX was originally described in oocytes and neutrophils, but has subsequently been identified in most other cell types including neurons (Tejada-Simon et al., 2005; Bedard and Krause, 2007). NOX is composed of catalytic and regulatory subunits which, upon activation, coalesce with an assembly subunit at a cell membrane. The dominant NOX isoform in both neurons and neutrophils is NOX2, which contains the gp91 catalytic subunit and the p47phox assembly subunit (Bedard and Krause, 2007). Electron micrographs show NOX2 to be localized to neuronal cell bodies and processes that also express NMDA receptors (Girouard et al., 2009; Wang et al., 2013a).
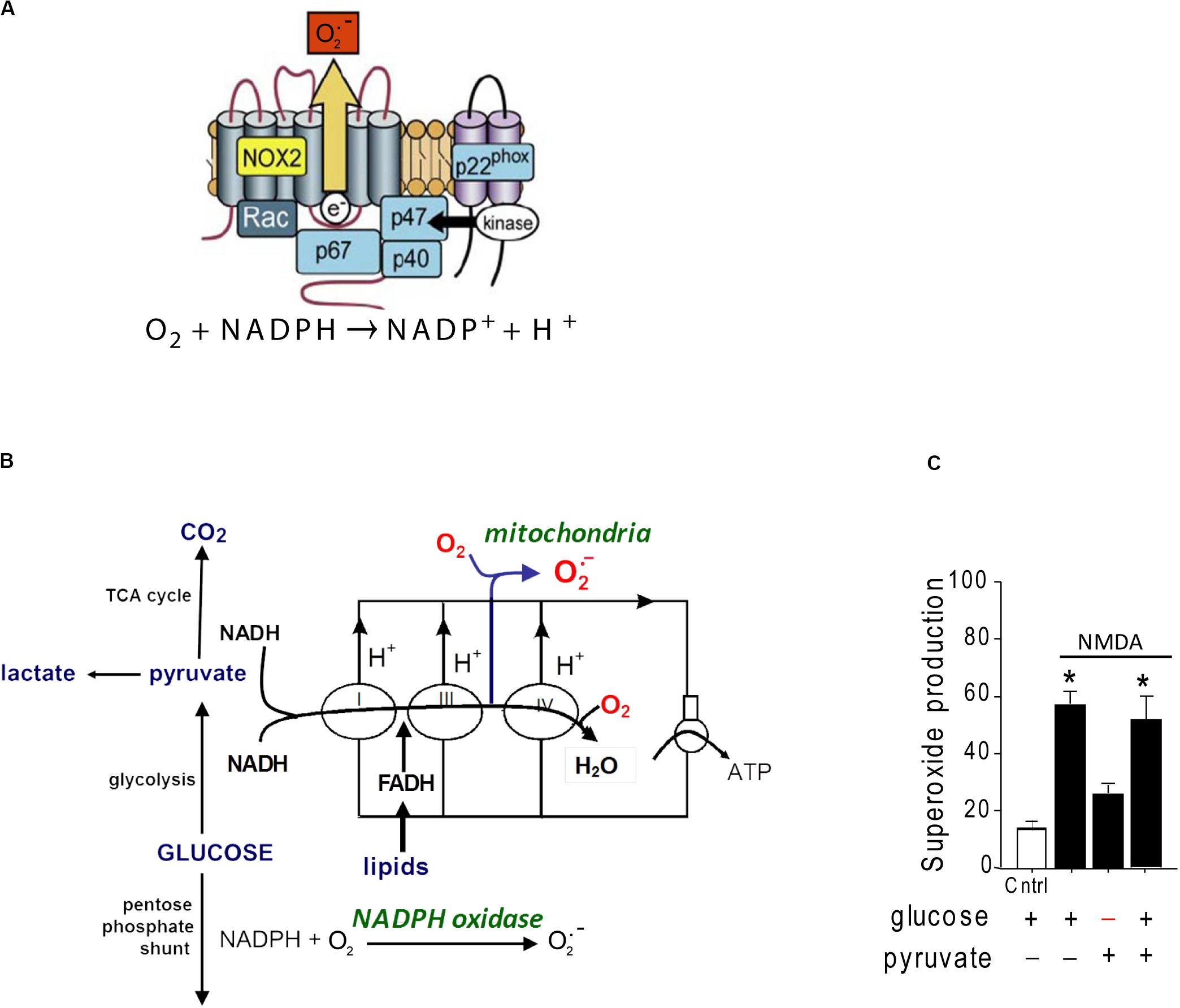
Figure 1. Structure and glucose dependence of NADPH oxidase. (A) NADPH oxidase is a multi-subunit enzyme that uses electrons derived from NADPH on one side of a membrane to generate superoxide on the other side. The NOX2 isoform of NADPH oxidase diagrammed here is the dominant form expressed by neurons. NOX2 requires activation of both the GTPase rac and the p47phox subunit to be catalytically functional. p47phox functions as an organizing subunit that bring the cytosolic subunits together with the membrane-bound subunits. In neurons, p47phox is activated by phosphorylation by PKCzeta (reprinted from Bedard and Krause, 2007, with permission). (B) Production of superoxide by NADPH oxidase requires glucose because glucose is the requisite substrate for the pentose phosphate shunt that regenerates NADPH from NADP+. By contrast, superoxide production from mitochondria can be fueled by pyruvate or other non-glucose substrates. (C) Neurons exposed to NMDA exhibit very attenuated superoxide production when deprived of glucose, indicating NADPH oxidase rather than mitochondria as the primary source. Data are means ± SEM; *p < 0.05 vs. no glucose. Redrawn from Brennan et al. (2009).
Several lines of evidence identify NOX2, rather than mitochondria, as the primary source of excitotoxic superoxide production. In cultured neurons, oxidation of the superoxide-sensitive fluorescent probe dihydroethidium is completely blocked by either pharmacological inhibition of NOX2 or genetic ablation of the p47phox subunit (Brennan et al., 2009; Reyes et al., 2012; Brennan-Minnella et al., 2013). Pharmacologic or genetic inhibition of NOX2 likewise prevents the lipid oxidation, DNA damage, and cell death caused by NMDA receptor stimulation in vitro or by brain ischemia in vivo (Brennan et al., 2009; Girouard et al., 2009; Raz et al., 2010; Guemez-Gamboa et al., 2011; Reyes et al., 2012; Clausen et al., 2013; Koriauli et al., 2015; Ma et al., 2017). Of note, superoxide production by NOX2 has an absolute requirement for glucose, which fuels regeneration of NADPH through the pentose–phosphate pathway (Decoursey and Ligeti, 2005). By contrast, mitochondrial superoxide production can be fueled by a variety of substrates (Figure 1). Neurons deprived of glucose (and metabolically supported by pyruvate) are unable to produce superoxide in response to NMDAr activation (Brennan et al., 2009), further supporting NOX2 rather than mitochondria as the superoxide source.
The question then arises, how can it be that blocking either mitochondrial Ca2+ uptake or NOX2 activity suppresses excitotoxic superoxide production? One possibility is that excitotoxic neuronal NOX2 activation induces mitochondrial superoxide production and vice versa, such that blocking either source of superoxide also reduces superoxide production by the other. This “ROS-induced ROS” production has been demonstrated in other cell types. In vascular cells, both increased mitochondrial superoxide production (Nazarewicz et al., 2013) and depletion of mitochondrial superoxide dismutase (Dikalova et al., 2010) increase NOX2 activity. Conversely, (putative) mitochondrial superoxide production induced by angiotensin II can be blocked by suppressing NOX2 activation (Brandes, 2005; Kimura et al., 2005; Doughan et al., 2008; Dikalov et al., 2014). Evidence also suggests that superoxide produced by mitochondria can elicit further superoxide release from the densely packed mitochondria of cardiac cells (Zorov et al., 2006). To our knowledge, definitive evidence of ROS-induced ROS production has not yet been demonstrated in neurons, but for the reasons outlined earlier, this process could be central to oxidative injury in excitotoxicity.
Superoxide is also generated by other enzymes, such as xanthine oxidase, lipoxygenases, and cyclooxygenases (Snezhkina et al., 2019). However, these have not borne out as major sources of superoxide production in excitotoxicity, and in some cases the observed suppression of superoxide formation by inhibitors of these enzymes is by indirect mechanisms (as further discussed in section “Signaling Pathways Underlying NOX2 Activation by NMDA Receptors”).
Significance of the Superoxide Production Source
Researchers working with neuronal cultures have long recognized that excitotoxicity has a spreading or “bad neighborhood” effect, whereby neurons usually die in clusters and survive better when located away from other neurons. Excitotoxic death in vivo similarly affects contiguous populations of cells, rather than scattered individual neurons. The underlying mechanism of this spreading effect has not been established, but one possibility is the cell-to-cell effects of extracellular superoxide release. This possibility was examined in a cell culture study in which only a small sub-population of cultured neurons expressed functional NOX2 (Reyes et al., 2012). After incubation with NMDA, neurons in the vicinity of the few NOX2-competent neurons exhibited oxidative stress (lipid peroxidation), whereas neurons at a distance from these neurons did not (Figure 2). Moreover, the NOX2-competent neurons did not themselves exhibit greater oxidative stress than the NOX2-deficient cells, and the oxidative stress in all the cells was reduced by addition of superoxide dismutase to the culture medium. These observations demonstrate a trans-cellular movement of superoxide from cell to cell during NMDAr activation, and are most consistent with superoxide generated by NOX2 at the neuronal plasma membrane where it is released directly into the extracellular space. The observations are more difficult to reconcile with superoxide production by mitochondria because mitochondrial superoxide is released into the cytosol from where it can access the extracellular space only by eluding the cytosolic superoxide and H2O2 scavenging systems and crossing the lipophilic cell membrane (Figure 2). The ability of NMDAr to activate NOX2 indicates an underlying signaling pathway between them, and thus the possibility of regulating this pathway to either amplify or suppress NMDAr induced superoxide production.
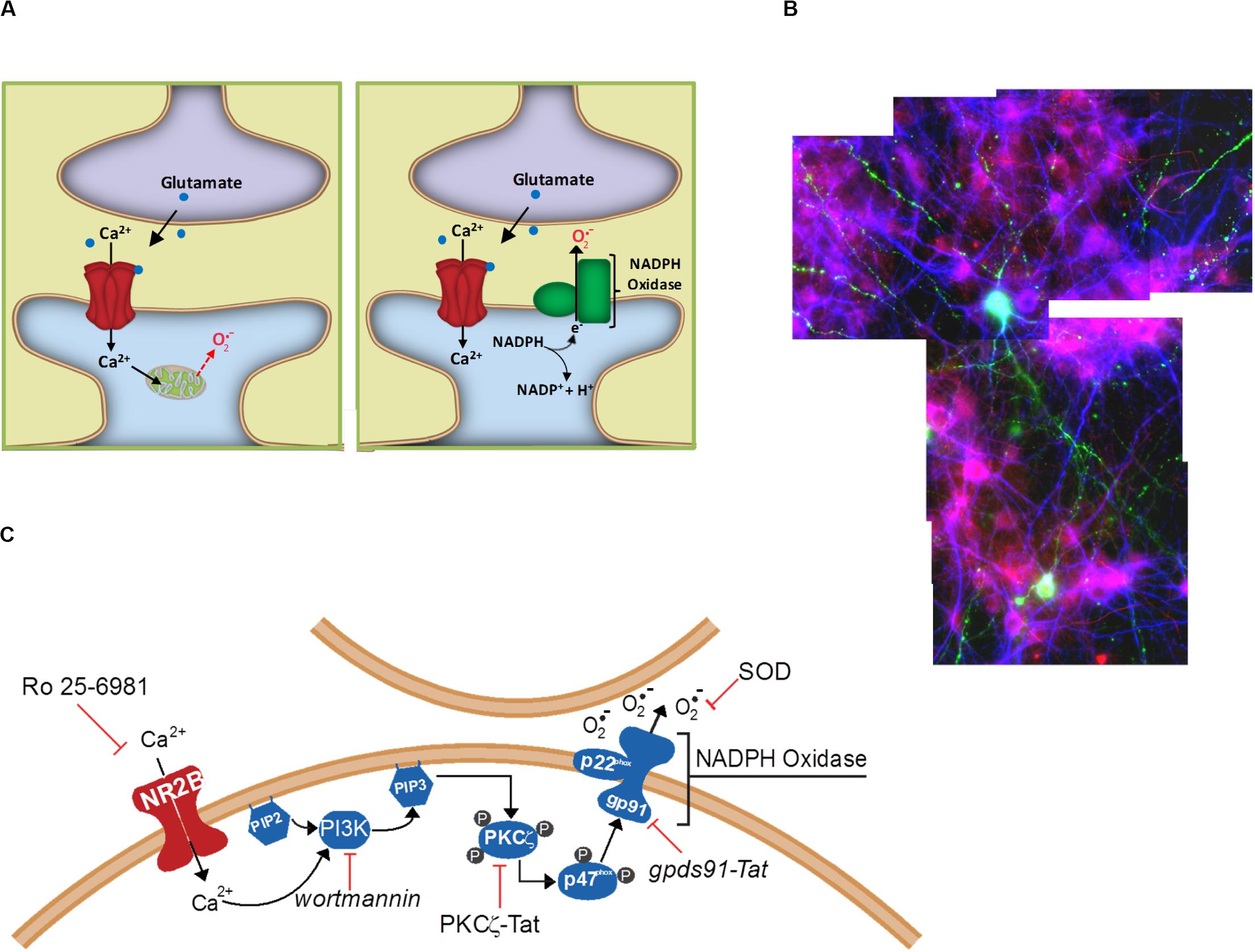
Figure 2. Extracellular release of superoxide by NMDA-induced NADPH oxidase activation. (A) Superoxide release from mitochondria will first encounter intracellular constituents of the cell in which it is enclosed, and can reach the extracellular space only be eluding cytosolic superoxide dismutase and crossing the lipid plasma membrane. By contrast, NADPH oxidase releases superoxide directly into the cytosol, where it can more readily interact with neighboring cells. (B) Cell-to-cell transmission of oxidative stress in neuronal cultures in which only a small subset of neurons (labeled green) contain functional NADPH oxidase. Application of NMDA to these cultures induces oxidative stress in neighboring neurons, as detected by the lipid peroxidation marker 4-hydroxynonenal (red). Cell nuclei are counterstained with DAPI (blue). Reprinted from Reyes et al. (2012). (C) Known steps in the signaling pathway linking NADPH oxidase to NMDAr activation. Reprinted from Minnella et al. (2018).
Signaling Pathways Underlying NOX2 Activation by NMDA Receptors
Several steps in this pathway have now been identified (Figure 2). NOX2 regulation is intrinsically complex, involving activation of the GTPase Rac1 and phospholipase A, permeability of a proton channel, proline isomerization, and phosphorylation of the p47phox organizing subunit, and phosphorylation of other NOX subunits (Levy et al., 2000; Bedard and Krause, 2007; El-Benna et al., 2009; Raad et al., 2019). Of these factors, phosphorylation of p47phox has been best characterized. p47phox-mediated assembly of the cytosolic and membrane subunits of NOX2 into a functional complex can occur only after p47phox is phosphorylated at several serine residues, most notably ser328 (El-Benna et al., 2009). In neurons, this phosphorylation is performed primarily by protein kinase C zeta (PKCζ), an atypical, calcium-independent protein kinase. Studies using a peptide inhibitor to PKCζ showed near-complete suppression of NMDAr-induced superoxide in vitro and in vivo (Brennan et al., 2009; Brennan-Minnella et al., 2013). Interestingly, PKCζ also has a fundamental role in memory formation (Sacktor et al., 1993), which like excitotoxicity also involves superoxide signaling (Massaad and Klann, 2011).
As an atypical protein kinase C, PKCζ is not activated by Ca2+ or diacyl glycerol, but instead by phosphoinositol-3-phosphate, the product of phosphoinositol-3-kinase (PI3K). Accordingly, NMDAr-induced superoxide production and cell death can be blocked with the PI3K inhibitor wortmannin, and this blockade can be circumvented with either exogenous phosphoinositol-3-phosphate or with a constitutively active form of PKCζ (Brennan-Minnella et al., 2013). A more granular description of which PI3K isoform is involved in this process, and how it is activated by NMDAr activation, remains to be attained. This information would be useful, as a targeted disruption of this interaction could then dissociate superoxide production and excitotoxic injury from other aspects of NMDAr activation.
This role of PI3K in excitotoxicity appears to contrast with the other, “pro-survival” effects associated with PI3K (Brunet et al., 2001). However, neurons express several different PI3K isoforms. PI3Ks are divided into several classes on the basis of structure, regulation, and function (Hawkins et al., 2006; Gross and Bassell, 2014). Class1A PI3Ks that contain the P110γ catalytic subunit are involved in NMDAr signaling pathways (Kim et al., 2011), suggesting this may be the isoform also involved in superoxide production. On the other hand, immunoprecipitation studies have instead identified the P110β subunit in association with the NMDAr complex (Wang et al., 2012). The association between PI3K and NMDAr may be via either an adaptor protein, APPL1, which links P110β to the PSD95 complex to which synaptic NMDAr are also bound (Wang et al., 2012), or via direct binding of the PI3K p85 regulatory subunit to GluN2B (Hisatsune et al., 1999).
NMDA receptors exist as hetero-tetramers, usually with two GluN1 subunits and two GluN2 subunits. The GluN2 subunits are in turn usually of the GluN2A or GluN2B isoforms (Watanabe et al., 1993; Traynelis et al., 2010). GluN2B-containing NMDA receptors are most clearly involved in excitotoxicity (Martel et al., 2012; McQueen et al., 2017), and three lines of evidence also indicate that GluN2B is likewise specifically involved in excitotoxic superoxide production. First, superoxide production is suppressed by the putative GluN2B specific antagonist Ro 25-6981 (Brennan-Minnella et al., 2013) and by depletion of GluN2B from the NMDAr complexes (Minnella et al., 2018). Second, replacement of the GluN2A intracellular C-terminal with the GluN2B intracellular C-terminal leads to superoxide formation from cells expressing these chimeric GluN2A/B subunits (Minnella et al., 2018). Third, a synthetic peptide that blocks GluN2B binding to the multiprotein PSD-95 complex also blocks excitotoxic superoxide production (Chen et al., 2015).
Of note, this peptide (termed “Tat-NR2B9c” or “NA-1”) was originally designed to block NMDAr-induced nitric oxide production by displacing GluN2B from the PSD95, with which neuronal nitric oxide synthase is associated (Sattler et al., 1999; Aarts et al., 2002). Measures of cGMP formation in the presence of Tat-NR2B9c confirm that it effectively reduces NMDAr-induced NO production, and the salutary effects on this peptide are widely attributed to this effect. However, given that the peptide also blocks NMDAr-induced NOX2 activation (also by displacing GluN2B from PSD95; Chen et al., 2015), and given that reductions in either nitric oxide or superoxide reduce peroxynitrite formation, it is equally possible that the neuroprotective effect of Tat-NR2B9c is attributable to instead to reduced superoxide production (or to the combined effects of reducing both superoxide and nitric oxide production). The Tat-NR2B9c peptide is now being used in clinical trials for ischemic stroke (Hill et al., 2020).
Route of Calcium Influx in Excitotoxic Superoxide Production
While several aspects of the signaling pathway linking NMDAr to NOX2 activation remain uncertain, the fundamental question of which step is Ca2+ dependent remains unresolved. Other Ca2+-dependent NMDAr signaling events have been shown to be mediated by calcium/calmodulin-dependent protein kinases (Bayer and Schulman, 2019), and in particular CaMKII, which binds to NMDAr. Inhibition of CaMKII has been shown to block excitotoxicity, although with a complex temporal pattern (Ashpole and Hudmon, 2011). Several GluN2B phosphorylation sites regulate NMDA receptor trafficking, including tyrosines 1070, 1472, and 1480 (Prybylowski et al., 2005; Sanz-Clemente et al., 2010; Lu et al., 2015; Chiu et al., 2019). Of these, deletion of the 1472 tyrosine residue has been shown to suppress NMDAr-induced superoxide production (Knox et al., 2014). It is also possible that Ca2+ influx is instead in involved in one of the other processes required for NOX2 activation, such as Rac1 activation (Puri, 2020), phospholipase activation, or p47phox proline isomerization.
A related issue is the longstanding controversy as to whether there is a “special” role for Ca2+ entering via NMDAr or whether only the magnitude of Ca2+ increase is important. Published data support both sides of this issue (e.g., Tymianski et al., 1993; Sattler et al., 1998; Stanika et al., 2009, 2012). This controversy may be resolved by a study from our laboratory that compared effects of Ca2+ influx induced by the calcium ionophore ionomycin with that of Ca2+ influx induced by NMDAr activation. Whereas NMDAr activation induced NOX2 activation, ionomycin did not, under conditions in which ionomycin was titrated to match the intracellular Ca2+ elevations induced by NMDAr activation (Minnella et al., 2018). Very similar effects were observed using activation of voltage-gated calcium channels to induce Ca2+ influx. However, ionomycin used at concentrations that raised intracellular Ca2+ well above the levels induced by NMDA did induce superoxide production, and this mode of superoxide production was not blocked by NOX2 inhibition. These observations suggest that Ca2+ influx by routes other than NMDAr can induce superoxide production only if the resulting Ca2+ elevations are far higher than achieved by NMDAr activation. They also provide support for a special effect of Ca2+ influx via NMDAr, as this induced superoxide production, oxidative stress, and cell death at intracellular Ca2+ levels that did none of these things when produced by ionomycin or voltage-gated calcium channels. However, as outlined later, these observations may alternatively be explained by concurrent, non-ionotropic effects of NMDAr stimulation that are not engaged during Ca2+ influx by other routes.
Non-Ionotropic NMDAr Signaling in Excitotoxicity
Ca2+ influx through NMDAr ion channels has long been established as a necessary event in excitotoxicity (Choi, 1985), as evidenced by the potent cytoprotective effects of drugs such as MK801 that block NMDAr ion channels. As noted earlier, Ca2+ elevations achieved by influx routes other than NMDAr (of comparable magnitude) do not produce nitric oxide production, superoxide production, or excitotoxic cell death (Tymianski et al., 1993; Sattler et al., 1998; Minnella et al., 2018). These observations are widely interpreted as evidence for a privileged access to certain local signaling pathways by Ca2+ influx specifically through NMDAr, however, it has recently been recognized that these observations can alternatively be explained by engagement of non-ionotropic NMDAr signaling in parallel to NMDAr-induced Ca2+ influx.
“Non-ionotropic” receptor signaling refers to signal transduction events triggered by transmembrane conformational changes rather than by ion flux. Non-ionotropic signaling by NMDAr was first identified by Westbrook and colleagues, who found that agonist binding independent of ion flux could downregulate GluN1/GluN2A receptor function through a process involving dephosphorylation of GluN1 tyrosine residues (Vissel et al., 2001). Subsequent studies showed that non-ionotropic NMDAr signaling also mediated long-term depression, p38 phosphorylation (Nabavi et al., 2013; Tamburri et al., 2013), and dendritic spine shrinkage (Stein et al., 2020). These studies identified non-ionotropic signaling by selectively blocking flux through the NMDAr ion channel while preserving agonist binding to the NMDAr itself (Figure 3). Using this same approach, Minnella and colleagues showed that NMDAr channel blockers prevented the production of superoxide (as expected), and that superoxide production and excitotoxicity were restored when Ca2+ influx was reconstituted through ionomycin or voltage-gated calcium channels during NMDAr stimulation (Minnella et al., 2018). Crucially, the Ca2+ influx induced by these routes failed to induce superoxide production in the absence of concurrent agonist binding to NMDAr, thus demonstrating a requisite role for non-ionotropic NMDAr binding in addition to Ca2+ influx (Figure 3). Ligand binding to NMDAr in the absence of Ca2+ influx increased PI3K association with GluN2B (as assessed by immunoprecipitation), thus suggesting a mechanism coupling non-ionotropic signaling to NOX2 activation.
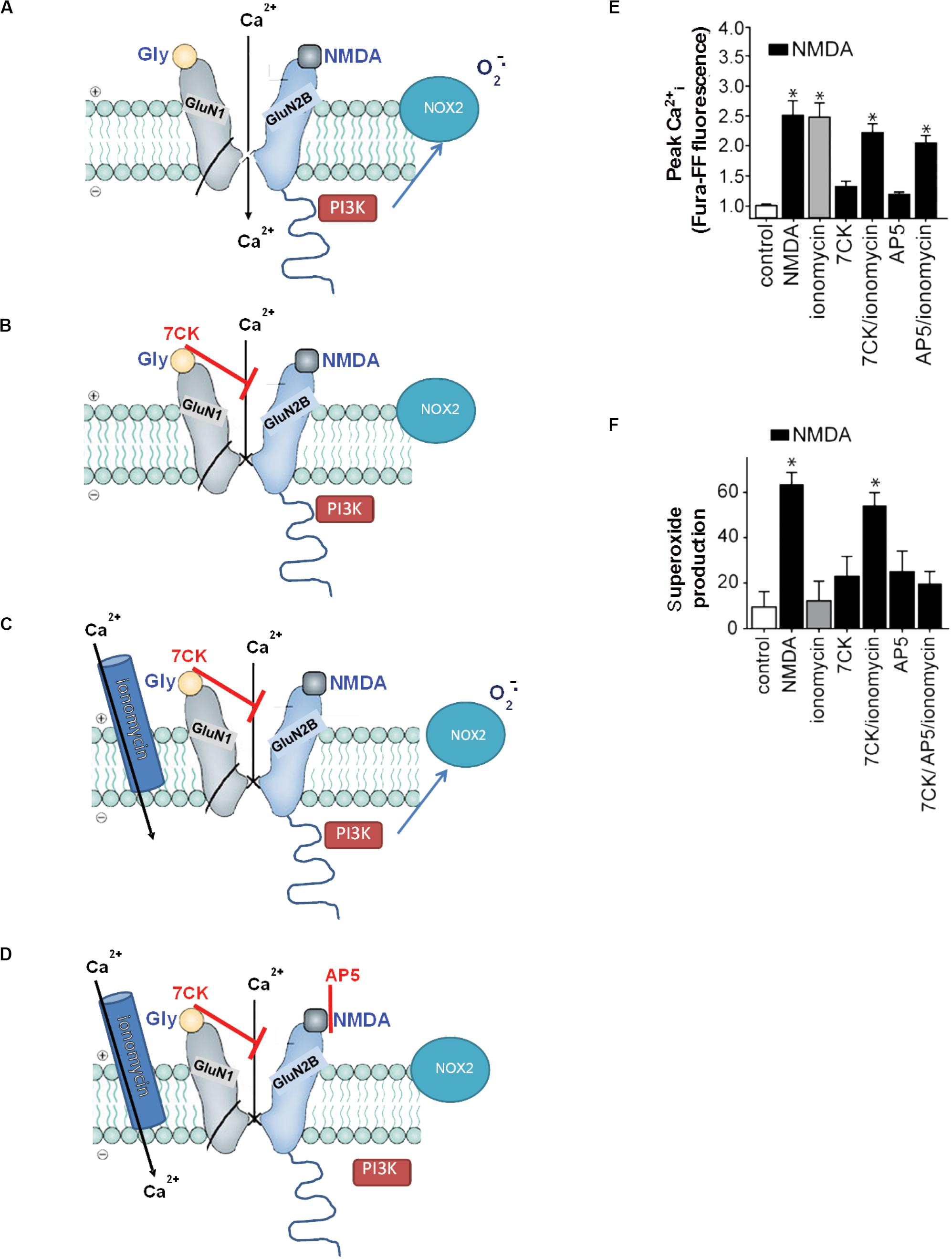
Figure 3. Non-ionotropic signaling in excitotoxicity. (A) Binding of the agonists glycine and NMDA to an NMDAr containing a GluN2B subunit triggers Ca2+ influx through the NMDAr channel and subsequent superoxide production by NOX2. (B) If the Ca2+ influx is blocked by the NMDAr glycine site antagonist 7-chlorokynurenic acid (7CK), there is no superoxide formation despite NMDA binding. (C) If, in addition, ionomycin is used to reconstitute the Ca2+ influx, superoxide is again produced by NOX2. (D) Ionomycin does not induce superoxide production when NMDA binding to GluN2B is blocked by (2R)-amino-5-phosphonopentanoate (AP5). Note the association of PI3K with the GluN2 subunit of the NMDAr requires ligand binding to the GluN2 subunit, but does not require Ca2+ influx. (E,F) Quantified measure of intracellular Ca2+ elevations and superoxide production under the conditions diagrammed in (A–D). Data are means ± SEM; *p < 0.05 vs. control. Reprinted from Minnella et al. (2018), with modifications.
A second and apparently independent role of metabotropic signaling in excitotoxicity involves pannexin channel opening. When activated, pannexin ion channels become permeable to Na+, Ca2+, and other ions and molecules (Yeung et al., 2020). Thompson and colleagues have shown that sustained NMDAr activation can induce pannexin-1 channel activation in the absence of ion flux through the NMDAr ion channel, with the resulting Ca2+ and Na+ influx essentially amplify the elevations caused by ion flux through the NMDAr itself. A synthetic peptide that prevents pannexin-1 channel activation by preventing its phosphorylation by Src kinase attenuates NMDAr-induced Ca2+ elevations and neuronal death (Weilinger et al., 2012, 2016). The relationship between this non-ionotropic signaling pathway and non-ionotropic NOX2 activation has not yet been resolved. However, NOX2 activation does not appear to be downstream of pannexin opening because the pannexin-1 inhibitor probenecid does not suppress NMDAr-induced superoxide production (J. Wang and R. Swanson, unpublished results).
Summary
The early observations that identified key roles for Ca2+, superoxide, and nitric oxide in excitotoxicity have stood the test of time, but subsequent observations have identified complexities that both expand our understanding of this process and open additional questions. Among these complexities is that elevations in either superoxide or nitric oxide levels can drive production of peroxynitrite, such that it is rarely possible to ascertain which one of these ROS is driving excitotoxic injury. It has also been demonstrated that superoxide production induced by NMDAr stimulation is generated primarily by NOX2, rather than mitochondria, but it remains uncertain whether this superoxide signal may be amplified by resultant mitochondrial dysfunction. The identification of NOX2 as the primary source of superoxide and provides an explanation for the extracellular actions of superoxide in excitoxicity, and also indicates the existence of a signaling cascade linking NMDAr to NADPH oxidase. This signaling cascade has been shown to include Ca2+ influx, PI3K activation, and PKCζ activation, but the nature of the Ca2+-dependent step remains to be established. Notably, Ca2+ influx alone is not sufficient to induce superoxide formation or excitotoxicity, and recent studies suggest that these processes also require concomitant non-ionotropic signaling induced by agonist binding to NMDAr.
Author Contributions
RS and JW both contributed to the literature research and writing for this review. Both authors contributed to the article and approved the submitted version.
Funding
RS received research support from the U.S. Department of Veterans Affairs and National Institutes of Health, but the preparation of this review was not supported by any specific grant from any funding agencies.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aarts, M., Liu, Y., Liu, L., Besshoh, S., Arundine, M., Gurd, J. W., et al. (2002). Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 298, 846–850. doi: 10.1126/science.1072873
Andreyev, A. Y., Kushnareva, Y. E., and Starkov, A. A. (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70, 200–214.
Angelova, P. R., Vinogradova, D., Neganova, M. E., Serkova, T. P., Sokolov, V. V., Bachurin, S. O., et al. (2019). Pharmacological sequestration of mitochondrial calcium uptake protects neurons against glutamate excitotoxicity. Mol. Neurobiol. 56, 2244–2255. doi: 10.1007/s12035-018-1204-8
Ashpole, N. M., and Hudmon, A. (2011). Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol. Cell Neurosci. 46, 720–730. doi: 10.1016/j.mcn.2011.02.003
Bayer, K. U., and Schulman, H. (2019). CaM kinase: still inspiring at 40. Neuron 103, 380–394. doi: 10.1016/j.neuron.2019.05.033
Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624. doi: 10.1073/pnas.87.4.1620
Bedard, K., and Krause, K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Bindokas, V. P., Jordan, J., Lee, C. C., and Miller, R. J. (1996). Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J. Neurosci. 16, 1324–1336. doi: 10.1523/jneurosci.16-04-01324.1996
Brandes, R. P. (2005). Triggering mitochondrial radical release: a new function for NADPH oxidases. Hypertension 45, 847–848. doi: 10.1161/01.hyp.0000165019.32059.b2
Brennan, A. M., Suh, S. W., Won, S. J., Narasimhan, P., Kauppinen, T. M., Lee, H., et al. (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863. doi: 10.1038/nn.2334
Brennan-Minnella, A. M., Shen, Y., El-Benna, J., and Swanson, R. A. (2013). Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis. 4:e580. doi: 10.1038/cddis.2013.111
Brunet, A., Datta, S. R., and Greenberg, M. E. (2001). Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 11, 297–305. doi: 10.1016/s0959-4388(00)00211-7
Campolo, N., Issoglio, F. M., Estrin, D. A., Bartesaghi, S., and Radi, R. (2020). 3-Nitrotyrosine and related derivatives in proteins: precursors, radical intermediates and impact in function. Essays Biochem. 64, 111–133. doi: 10.1042/ebc20190052
Chen, Y., Brennan-Minnella, A. M., Sheth, S., El-Benna, J., and Swanson, R. A. (2015). Tat-NR2B9c prevents excitotoxic neuronal superoxide production. J. Cereb. Blood Flow Metab. 35, 739–742. doi: 10.1038/jcbfm.2015.16
Chiu, A. M., Wang, J., Fiske, M. P., Hubalkova, P., Barse, L., Gray, J. A., et al. (2019). NMDAR-Activated PP1 dephosphorylates GluN2B to modulate NMDAR synaptic content. Cell Rep. 28, 332–341.e5..
Choi, D. W. (1985). Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 58, 293–297. doi: 10.1016/0304-3940(85)90069-2
Clausen, A., Mcclanahan, T., Ji, S. G., and Weiss, J. H. (2013). Mechanisms of rapid reactive oxygen species generation in response to cytosolic Ca2+ or Zn2+ loads in cortical neurons. PLoS One 8:e83347. doi: 10.1371/journal.pone.0083347
Dawson, V. L., Dawson, T. M., London, E. D., Bredt, D. S., and Snyder, S. H. (1991). Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. U.S.A. 88, 6368–6371. doi: 10.1073/pnas.88.14.6368
Dawson, V. L., Kizushi, V. M., Huang, P. L., Snyder, S. H., and Dawson, T. M. (1996). Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J. Neurosci. 16, 2479–2487. doi: 10.1523/jneurosci.16-08-02479.1996
Decoursey, T. E., and Ligeti, E. (2005). Regulation and termination of NADPH oxidase activity. Cell Mol. Life Sci. 62, 2173–2193. doi: 10.1007/s00018-005-5177-1
Dikalov, S. I., Nazarewicz, R. R., Bikineyeva, A., Hilenski, L., Lassegue, B., Griendling, K. K., et al. (2014). Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox. Signal 20, 281–294. doi: 10.1089/ars.2012.4918
Dikalova, A. E., Bikineyeva, A. T., Budzyn, K., Nazarewicz, R. R., Mccann, L., Lewis, W., et al. (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116. doi: 10.1161/circresaha.109.214601
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496. doi: 10.1161/circresaha.107.162800
Duan, Y., Gross, R. A., and Sheu, S. S. (2007). Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(ADP-ribose) polymerase-1 activation during glutamate excitotoxicity. J. Physiol. 585, 741–758. doi: 10.1113/jphysiol.2007.145409
Dugan, L. L., Sensi, S. L., Canzoniero, L. M., Handran, S. D., Rothman, S. M., Lin, T. S., et al. (1995). Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J. Neurosci. 15, 6377–6388. doi: 10.1523/jneurosci.15-10-06377.1995
El-Benna, J., Dang, P. M., Gougerot-Pocidalo, M. A., Marie, J. C., and Braut-Boucher, F. (2009). p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp. Mol. Med. 41, 217–225.
Girouard, H., Wang, G., Gallo, E. F., Anrather, J., Zhou, P., Pickel, V. M., et al. (2009). NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci. 29, 2545–2552. doi: 10.1523/jneurosci.0133-09.2009
Gross, C., and Bassell, G. J. (2014). Neuron-specific regulation of class I PI3K catalytic subunits and their dysfunction in brain disorders. Front. Mol. Neurosci. 7:12. doi: 10.3389/fnmol.2014.00012
Guemez-Gamboa, A., Estrada-Sanchez, A. M., Montiel, T., Paramo, B., Massieu, L., and Moran, J. (2011). Activation of NOX2 by the stimulation of ionotropic and metabotropic glutamate receptors contributes to glutamate neurotoxicity in vivo through the production of reactive oxygen species and calpain activation. J. Neuropathol. Exp. Neurol. 70, 1020–1035. doi: 10.1097/nen.0b013e3182358e4e
Hawkins, P. T., Anderson, K. E., Davidson, K., and Stephens, L. R. (2006). Signalling through Class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 34, 647–662. doi: 10.1042/bst0340647
Hill, M. D., Goyal, M., Menon, B. K., Nogueira, R. G., Mctaggart, R. A., Demchuk, A. M., et al. (2020). Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): a multicentre, double-blind, randomised controlled trial. Lancet 395, 878–887.
Hisatsune, C., Umemori, H., Mishina, M., and Yamamoto, T. (1999). Phosphorylation-dependent interaction of the N-methyl-D-aspartate receptor epsilon 2 subunit with phosphatidylinositol 3-kinase. Genes Cells 4, 657–666. doi: 10.1046/j.1365-2443.1999.00287.x
Johnson-Cadwell, L. I., Jekabsons, M. B., Wang, A., Polster, B. M., and Nicholls, D. G. (2007). ‘Mild Uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J. Neurochem. 101, 1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x
Kim, J. I., Lee, H. R., Sim, S. E., Baek, J., Yu, N. K., Choi, J. H., et al. (2011). PI3Kgamma is required for NMDA receptor-dependent long-term depression and behavioral flexibility. Nat. Neurosci. 14, 1447–1454.
Kimura, S., Zhang, G. X., Nishiyama, A., Shokoji, T., Yao, L., Fan, Y. Y., et al. (2005). Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 45, 860–866. doi: 10.1161/01.hyp.0000163462.98381.7f
Knox, R., Brennan-Minnella, A. M., Lu, F., Yang, D., Nakazawa, T., Yamamoto, T., et al. (2014). NR2B phosphorylation at tyrosine 1472 contributes to brain injury in a rodent model of neonatal hypoxia-ischemia. Stroke 45, 3040–3047. doi: 10.1161/strokeaha.114.006170
Koriauli, S., Natsvlishvili, N., Barbakadze, T., and Mikeladze, D. (2015). Knockdown of interleukin-10 induces the redistribution of sigma1-receptor and increases the glutamate-dependent NADPH-oxidase activity in mouse brain neurons. Biol. Res. 48, 55.
Lafon-Cazal, M., Pietri, S., Culcasi, M., and Bockaert, J. (1993). NMDA-dependent superoxide production and neurotoxicity. Nature 364, 535–537. doi: 10.1038/364535a0
Levy, R., Lowenthal, A., and Dana, R. (2000). Cytosolic phospholipase A2 is required for the activation of the NADPH oxidase associated H+ channel in phagocyte-like cells. Adv. Exp. Med. Biol. 479, 125–135. doi: 10.1007/0-306-46831-x_11
Lipton, S. A., Singel, D. J., and Stamler, J. S. (1994). Nitric oxide in the central nervous system. Prog. Brain Res. 103, 359–364.
Lu, W., Fang, W., Li, J., Zhang, B., Yang, Q., Yan, X., et al. (2015). Phosphorylation of Tyrosine 1070 at the GluN2B subunit is regulated by synaptic activity and critical for surface expression of N-Methyl-D-aspartate (n.d.) receptors. J. Biol. Chem. 290, 22945–22954. doi: 10.1074/jbc.m115.663450
Ma, M. W., Wang, J., Zhang, Q., Wang, R., Dhandapani, K. M., Vadlamudi, R. K., et al. (2017). NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 12, 7.
Martel, M. A., Ryan, T. J., Bell, K. F., Fowler, J. H., Mcmahon, A., Al-Mubarak, B., et al. (2012). The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron 74, 543–556. doi: 10.1016/j.neuron.2012.03.021
Massaad, C. A., and Klann, E. (2011). Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox. Signal 14, 2013–2054. doi: 10.1089/ars.2010.3208
McQueen, J., Ryan, T. J., Mckay, S., Marwick, K., Baxter, P., Carpanini, S. M., et al. (2017). Pro-death NMDA receptor signaling is promoted by the GluN2B C-terminus independently of Dapk1. Elife 6:e17161.
Minnella, A. M., Zhao, J. X., Jiang, X., Jakobsen, E., Lu, F., Wu, L., et al. (2018). Excitotoxic superoxide production and neuronal death require both ionotropic and non-ionotropic NMDA receptor signaling. Sci. Rep. 8:17522.
Nabavi, S., Kessels, H. W., Alfonso, S., Aow, J., Fox, R., and Malinow, R. (2013). Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. U.S.A. 110, 4027–4032. doi: 10.1073/pnas.1219454110
Nazarewicz, R. R., Dikalova, A. E., Bikineyeva, A., and Dikalov, S. I. (2013). Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 305, H1131–H1140.
Nguyen, D., Alavi, M. V., Kim, K. Y., Kang, T., Scott, R. T., Noh, Y. H., et al. (2011). A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2:e240. doi: 10.1038/cddis.2011.117
Nicholls, D. G. (2006). Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J. Biol. Chem. 281, 14864–14874. doi: 10.1074/jbc.m510916200
Nicholls, D. G., and Ward, M. W. (2000). Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 23, 166–174. doi: 10.1016/s0166-2236(99)01534-9
Olney, J. W., Ho, O. L., and Rhee, V. (1971). Cytotoxic effects of acidic and sulphur containing amino acids on the infant mouse central nervous system. Exp. Brain Res. 14, 61–76.
Pacher, P., Beckman, J. S., and Liaudet, L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424.
Patel, M., Day, B. J., Crapo, J. D., Fridovich, I., and Mcnamara, J. O. (1996). Requirement for superoxide in excitotoxic cell death. Neuron 16, 345–355. doi: 10.1016/s0896-6273(00)80052-5
Prybylowski, K., Chang, K., Sans, N., Kan, L., Vicini, S., and Wenthold, R. J. (2005). The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 47, 845–857. doi: 10.1016/j.neuron.2005.08.016
Puri, B. K. (2020). Calcium signaling and gene expression. Adv. Exp. Med. Biol. 1131, 537–545. doi: 10.1007/978-3-030-12457-1_22
Qiu, J., Tan, Y. W., Hagenston, A. M., Martel, M. A., Kneisel, N., Skehel, P. A., et al. (2013). Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 4:2034.
Raad, H., Derkawi, R. A., Tlili, A., Belambri, S. A., Dang, P. M., and El-Benna, J. (2019). Phosphorylation of gp91(phox)/NOX2 in Human Neutrophils. Methods Mol. Biol. 1982, 341–352.
Radi, R. (2018). Oxygen radicals, nitric oxide, and peroxynitrite: redox pathways in molecular medicine. Proc. Natl. Acad. Sci. U.S.A. 115, 5839–5848. doi: 10.1073/pnas.1804932115
Raz, L., Zhang, Q. G., Zhou, C. F., Han, D., Gulati, P., Yang, L. C., et al. (2010). Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS One 5:e12606. doi: 10.1371/journal.pone.0012606
Reyes, R. C., Brennan, A. M., Shen, Y., Baldwin, Y., and Swanson, R. A. (2012). Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. 32, 12973–12978. doi: 10.1523/jneurosci.1597-12.2012
Reynolds, I. J., and Hastings, T. G. (1995). Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 15, 3318–3327. doi: 10.1523/jneurosci.15-05-03318.1995
Sacktor, T. C., Osten, P., Valsamis, H., Jiang, X., Naik, M. U., and Sublette, E. (1993). Persistent activation of the zeta isoform of protein kinase C in the maintenance of long-term potentiation. Proc. Natl. Acad. Sci. U.S.A. 90, 8342–8346. doi: 10.1073/pnas.90.18.8342
Sanz-Clemente, A., Matta, J. A., Isaac, J. T., and Roche, K. W. (2010). Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 67, 984–996. doi: 10.1016/j.neuron.2010.08.011
Sattler, R., Charlton, M. P., Hafner, M., and Tymianski, M. (1998). Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J. Neurochem. 71, 2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x
Sattler, R., Xiong, Z., Lu, W. Y., Hafner, M., Macdonald, J. F., and Tymianski, M. (1999). Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284, 1845–1848. doi: 10.1126/science.284.5421.1845
Sengpiel, B., Preis, E., Krieglstein, J., and Prehn, J. H. (1998). NMDA-induced superoxide production and neurotoxicity in cultured rat hippocampal neurons: role of mitochondria. Eur. J. Neurosci. 10, 1903–1910. doi: 10.1046/j.1460-9568.1998.00202.x
Snezhkina, A. V., Kudryavtseva, A. V., Kardymon, O. L., Savvateeva, M. V., Melnikova, N. V., Krasnov, G. S., et al. (2019). ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell Longev. 2019:1–17. doi: 10.1155/2019/6175804
Stanika, R. I., Pivovarova, N. B., Brantner, C. A., Watts, C. A., Winters, C. A., and Andrews, S. B. (2009). Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9854–9859. doi: 10.1073/pnas.0903546106
Stanika, R. I., Villanueva, I., Kazanina, G., Andrews, S. B., and Pivovarova, N. B. (2012). Comparative impact of voltage-gated calcium channels and NMDA receptors on mitochondria-mediated neuronal injury. J. Neurosci. 32, 6642–6650. doi: 10.1523/jneurosci.6008-11.2012
Starkov, A. A., Chinopoulos, C., and Fiskum, G. (2004). Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 36, 257–264. doi: 10.1016/j.ceca.2004.02.012
Stein, I. S., Park, D. K., Flores, J. C., Jahncke, J. N., and Zito, K. (2020). Molecular mechanisms of non-ionotropic NMDA receptor signaling in dendritic spine shrinkage. J. Neurosci. 40, 3741–3750. doi: 10.1523/jneurosci.0046-20.2020
Tamburri, A., Dudilot, A., Licea, S., Bourgeois, C., and Boehm, J. (2013). NMDA-receptor activation but not ion flux is required for amyloid-beta induced synaptic depression. PLoS One 8:e65350. doi: 10.1371/journal.pone.0065350
Tejada-Simon, M. V., Serrano, F., Villasana, L. E., Kanterewicz, B. I., Wu, G. Y., Quinn, M. T., et al. (2005). Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol. Cell Neurosci. 29, 97–106. doi: 10.1016/j.mcn.2005.01.007
Traynelis, S. F., Wollmuth, L. P., Mcbain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496.
Tymianski, M., Charlton, M. P., Carlen, P. L., and Tator, C. H. (1993). Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J. Neurosci. 13, 2085–2104. doi: 10.1523/jneurosci.13-05-02085.1993
Vissel, B., Krupp, J. J., Heinemann, S. F., and Westbrook, G. L. (2001). A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat. Neurosci. 4, 587–596. doi: 10.1038/88404
Wang, G., Coleman, C. G., Chan, J., Faraco, G., Marques-Lopes, J., Milner, T. A., et al. (2013a). Angiotensin II slow-pressor hypertension enhances NMDA currents and NOX2-dependent superoxide production in hypothalamic paraventricular neurons. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R1096–R1106.
Wang, J. Q., Chen, Q., Wang, X., Wang, Q. C., Wang, Y., Cheng, H. P., et al. (2013b). Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 288, 3070–3084. doi: 10.1074/jbc.m112.407726
Wang, Y. B., Wang, J. J., Wang, S. H., Liu, S. S., Cao, J. Y., Li, X. M., et al. (2012). Adaptor protein APPL1 couples synaptic NMDA receptor with neuronal prosurvival phosphatidylinositol 3-kinase/Akt pathway. J. Neurosci. 32, 11919–11929. doi: 10.1523/jneurosci.3852-11.2012
Watanabe, M., Inoue, Y., Sakimura, K., and Mishina, M. (1993). Distinct distributions of five N-methyl-D-aspartate receptor channel subunit mRNAs in the forebrain. J. Comp. Neurol. 338, 377–390. doi: 10.1002/cne.903380305
Weilinger, N. L., Lohman, A. W., Rakai, B. D., Ma, E. M., Bialecki, J., Maslieieva, V., et al. (2016). Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat. Neurosci. 19, 432–442. doi: 10.1038/nn.4236
Weilinger, N. L., Tang, P. L., and Thompson, R. J. (2012). Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 32, 12579–12588. doi: 10.1523/jneurosci.1267-12.2012
Yeung, A. K., Patil, C. S., and Jackson, M. F. (2020). Pannexin-1 in the CNS: emerging concepts in health and disease. J. Neurochem. doi: 10.1111/jnc.15004 online ahead of print,
Keywords: glutamate, glucose, Glun2B, phosphoinositol-3-kinase, metabotropic, peroxynitrite, NADPH oxidase, calcium
Citation: Wang J and Swanson RA (2020) Superoxide and Non-ionotropic Signaling in Neuronal Excitotoxicity. Front. Neurosci. 4:861. doi: 10.3389/fnins.2020.00861
Received: 31 May 2020; Accepted: 24 July 2020;
Published: 03 September 2020.
Edited by:
Alberto Granzotto, Aging Sciences and Translational Medicine Center (CeSI-MeT), ItalyReviewed by:
Elias Aizenman, University of Pittsburgh, United StatesVeronica Perez De La Cruz, Manuel Velasco Suárez Instituto Nacional de Neurología y Neurocirugía, Mexico
Copyright © 2020 Wang and Swanson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raymond A. Swanson, cmF5bW9uZC5zd2Fuc29uQHVjc2YuZWR1; cmF0c3dhbnNvbkBnbWFpbC5jb20=