 Liu Liu1,2†
Liu Liu1,2† Zi-Rong Chen1,3†Hai-Qing Xu4De-Tian Liu1Yong Mao5Han-Kui Liu5
Zi-Rong Chen1,3†Hai-Qing Xu4De-Tian Liu1Yong Mao5Han-Kui Liu5 Xiao-Rong Liu1Peng Zhou1Si-Mei Lin1Bin Li1Na He1Tao Su1Qiong-Xiang Zhai6Heng Meng7Wei-Ping Liao1*
Xiao-Rong Liu1Peng Zhou1Si-Mei Lin1Bin Li1Na He1Tao Su1Qiong-Xiang Zhai6Heng Meng7Wei-Ping Liao1* Yong-Hong Yi1*
Yong-Hong Yi1*- 1Institute of Neuroscience, Department of Neurology of the Second Affiliated Hospital of Guangzhou Medical University, Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, Guangzhou, China
- 2Department of Neurology, Xiaoshan First People’s Hospital, Hangzhou, China
- 3Department of Neurology, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
- 4Department of Neurology, Xuzhou Central Hospital, Affiliated Hospital of Southeast University, Xuzhou, China
- 5BGI-Shenzhen, Shenzhen, China
- 6Department of Pediatrics, Guangdong General Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China
- 7Department of Neurology of the First Affiliated Hospital of Jinan University and Clinical Neuroscience Institute of Jinan University, Guangzhou, China
To explore the phenotype spectrum of DEPDC5 variants and the possible mechanisms underlying phenotypical variation, we performed targeted next-generation sequencing in 305 patients with focal epilepsies and 91 patients with generalized epilepsies. Protein modeling was performed to predict the effects of missense mutations. All previously reported epilepsy-related DEPDC5 variants were reviewed. The genotype–phenotype correlations with molecular sub-regional implications were analyzed. We identified a homozygous DEPDC5 mutation (p.Pro1031His) in a case with focal cortical dysplasia and eight heterozygous mutations in 11 families with mild focal epilepsies, including 13 patients in eight families with focal epilepsy with febrile seizures plus/febrile seizures (FEFS + /FS). The mutations included one termination codon mutation (p.Ser1601_Ter1604del_ext133), three truncating mutations (p.Val151Serfs∗27, p.Arg239∗, and p.Arg838∗), and four missense mutations (p.Tyr7Cys, p.Tyr836Cys, p.Pro1031His, and p.Gly1545Ser) that were predicted to affect hydrogen bonds and protein stability. Analysis on epilepsy-related DEPDC5 variants revealed that malformations of cortical development (MCDs) had a tendency of higher frequency of null mutations than those without MCD. MCD-associated heterozygous missense mutations were clustered in structural axis for binding arrangement (SABA) domain and close to the binding sites to NPRL2/NPRL3 complex, whereas those associated with FEFS + /FS were a distance away from the binding sites. Evidence from four aspects and one possible evidence from sub-regional implication suggested MCD and FEFS + /FS as phenotypes of DEPDC5 variants. This study suggested that the phenotypes of DEPDC5 variants vary from mild FEFS + /FS to severe MCD. Heterozygous DEPDC5 mutations are generally less pathogenic and commonly associated with mild phenotypes. Bi-allelic mutations and second hit of somatic mutations, together with the genotype–phenotype correlation and sub-regional implication of DEPDC5 variants, explain severe phenotypes.
Introduction
DEPDC5 gene (OMIM∗614191) encodes disheveled Egl-10 and pleckstrin (DEP) domain containing protein 5, which forms part of the GTPase-activating protein activity toward RAG complex 1, a repressor of the mammalian target of rapamycin (mTOR) signaling pathway that is closely related to epilepsies (Bar-Peled et al., 2013; Baldassari et al., 2019). The mTOR pathway is involved in a myriad of biological processes, including cell growth, proliferation, and protein synthesis (Fingar and Blenis, 2004; Sarbassov et al., 2005; Marsan and Baulac, 2018). DEPDC5 mutations have been demonstrated to be the most common cause of familial focal epilepsies (Dibbens et al., 2013). However, the phenotypes of DEPDC5 mutations included not only familial epilepsies such as familial focal epilepsy with variable foci (Dibbens et al., 2013), autosomal dominant (AD) nocturnal frontal epilepsy (Ishida et al., 2013; Picard et al., 2014), and familial temporal lobe epilepsy (TLE) (Ishida et al., 2013; Striano et al., 2015) but also non-familial focal epilepsies, such as childhood epilepsy with centrotemporal spikes (rolandic epilepsy) (Lal et al., 2014) and epilepsy with continuous spikes and waves during slow-wave sleep (Ricos et al., 2016). In addition, cases of focal cortical dysplasia (FCD) (Scerri et al., 2015) and hemimegalencephaly (D’Gama et al., 2015) were reported. On the other hand, DEPDC5 mutations have been occasionally identified in cases of epileptic spasms (Carvill et al., 2015); asymptomatic carriers were also common across the DEPDC5 mutation-related families (Tsai et al., 2017). Therefore, the phenotypic spectrum of DEPDC5 mutations requires further verification, and it is unknown whether the phenotypic variation is correlated with the genotypes of DEPDC5 mutations.
Here, we performed targeted next-generation sequencing approach in a cohort of patients with focal epilepsies or generalized epilepsies. Eight DEPDC5 variants were identified in 12 unrelated families with phenotypic heterogeneity, including eight families with 13 individuals with focal epilepsy with febrile seizures plus/febrile seizures (FEFS + /FS) and a homozygous mutation in a case with FCD. We systematically reviewed the DEPDC5 variants and analyzed the genotype–phenotype correlation, with special attention on the molecular sub-regional implications of mutations, which was suggested to be a critical consideration in evaluating the pathogenicity of sequence variants (Gussow et al., 2016; Tang et al., 2019). To determine the association between DEPDC5 variants and malformations of cortical development (MCDs) or FEFS + /FS, evidence from five clinical-genetic aspects was analyzed.
Materials and Methods
Patients
We recruited a cohort of patients with epilepsies, which consisted of 305 patients with focal epilepsies and 91 patients with generalized epilepsies. The patients were from the Epilepsy Center of the Second Affiliated Hospital of Guangzhou Medical University and Guangdong General Hospital from July 2015 to November 2019. Patients with focal epilepsies included 20 cases with FCD, considering that mutations of several genes were associated with focal epilepsies and cortical malformation (Guerrini, 2005). Patients with focal epilepsies caused by acquired etiologies, such as tumor, trauma, and stroke, were excluded. All individuals enrolled were unrelated ethnic Han Chinese who lived in southern China. None of the biological grandparents of the participants were from other races.
The collected clinical data included seizure onset age, seizure type and frequency, response to antiepileptic drugs, general and neurological examination results, and detailed family history. Magnetic resonance imaging (MRI) scans were performed to detect any brain structure abnormalities on a 3.0-T Magnet including three-dimensional T1 and T2 weighted, T2 fluid-attenuation inversion recovery (FLAIR) in a horizontal position, T1 weighted and T2 FLAIR in a coronary position, T2 weighted in a sagittal position, 3.0-mm-thick contiguous slices, and sequences. Long-term (24-h) video electroencephalography (EEG) monitoring records that included hyperventilation, intermittent photic stimulation, open–close eyes test, and sleeping recording were obtained. Epileptic seizures and epilepsies were diagnosed according to the criteria of the Commission on Classification and Terminology of the International League Against Epilepsy (ILAE) (1981, 1989, 2001, 2010, and 2017). Focal epilepsies were diagnosed based on focally originated seizures and/or focal discharges of EEG. Generalized epilepsies were diagnosed based on a range of seizure types including absence, spasms, myoclonic, clonic, atonic, tonic, and tonic–clonic seizures, supported by the finding of typically generalized discharges on EEG.
This study adhered to the guidelines of the International Committee of Medical Journal Editors regarding patient consent for research or participation. Written informed consent was obtained from the individuals or legal guardians. This study was approved by institutional review board and ethics committee of the Second Affiliated Hospital of Guangzhou Medical University (approval number: 2014004).
Targeted Sequencing
Whole blood samples were collected from the probands, their parents, and other available relatives to ascertain if the variants were inherited or de novo and for cosegregation analysis. Genomic DNA was extracted from whole blood using the Qiagen Flexi Gene DNA Kit (Qiagen, Hilden, Germany).
A gene panel was designed for targeted sequencing of 483 genes that are possibly associated with epilepsy to uncover disease-causing variants (Supplementary Table S1) (Zhou et al., 2018). Genes potentially associated with focal epilepsies in the panel included CHRNA2, CHRNA4, CHRNB2, CNTNAP2, DEPDC5, FLNA, GABRG2, GRIN2A, KCNQ2, KCNQ3, KCNT1, LGI1, MECP2, NPRL2, NPRL3, PCDH19, POLG, PRIMA1, PRRT2, RELN, SCN1A, SCN1B, SCN2A, SLC2A1, SRPX2, SYN1, TBC1D24, TSC1, and TSC2. The sequencing method and filtering criteria were as those described previously (Zhou et al., 2018). All candidate pathogenic variants were validated by Sanger sequencing. Paternity and maternity of the probands were confirmed by alignment of the segregated sequence variants.
Molecular Structural Analysis
Protein modeling was performed to predict the effects of missense variants on molecular structure by using Iterative Threading ASSEmbly Refinement (I-TASSER) software (Yang et al., 2015), based on the updated template of 6CES.pdb (chain D)1 (Shen et al., 2018). PyMOL 1.7 was used for three-dimensional protein structure visualization and analysis. We used mCSM to predict protein stability, which is indicated by free energy change (ΔΔG) (Pires et al., 2014). Mutations were discriminated into two classes: destabilizing mutations (ΔΔG < 0 kcal/mol) and stabilizing mutations (ΔΔG > 0 kcal/mol). Free SASA is used to calculate solvent-accessible surface area (SASA) (Mitternacht, 2016).
Genotype–Phenotype Correlation
We retrieved all DEPDC5 mutations from the PubMed2 and the HGMD3 up to November 2018 (Supplementary Table S2). All DEPDC5 mutations were validated by direct sequencing in the original reports. We rechecked all the mutations with nucleotide and amino acid numbering according to DEPDC5 reference transcript NM_001242896.1 (reference protein NP_001229825.1). To avoid duplicate recruitment, mutations were cross-referenced on their genetic and clinical information.
To facilitate analyzing the correlation between genotype and phenotype, gene mutations are classified into null and missense mutations. Null mutations are those causing gross protein malformations, including truncating mutations (nonsense and frameshifting), splice-site mutations, and mutations at initiation codon or with single/multiple exon deletion, which mainly lead to complete loss of function and haploinsufficiency (Richards et al., 2015).
Phenotypes of the DEPDC5 mutations were listed according to the original reports. Familial case was defined by existence of at least two members carrying the same DEPDC5 mutation. Families with only one affected individual (due to incomplete penetrance) were indicated. An affected family was considered a single case in data analysis. For our analysis, cases with MCD were separated from those without MCD. A familial case of MCD was defined by existence of at least one affected member presented MCD; and the detailed number of affected members with MCD or other phenotypes was indicated (Supplementary Table S2). The cases without MCD were further classified into cases with focal epilepsy and cases with FEFS + /FS. FS + was used to denote the individuals with FS extending outside the age range of 3 months to 6 years, or with afebrile seizures. It was observed in several mutations that the same mutation was reported to be associated with different phenotypes (Supplementary Table S2). The penetrance of DEPDC5 mutation was defined as the proportion of affected individuals with the mutation, that is, the number of affected individuals with the mutation divided by the total number of individuals with the mutation. A family, or sub-branches within a family, was recruited for penetrance analysis when all individuals in the family or sub-branches of the family were tested for DEPDC5 mutations (Meng et al., 2015).
Evaluating a Phenotype of DEPDC5 Variants
To determine the association between DEPDC5 variants and MCD or FEFS + /FS, evidence from five clinical-genetic aspects was analyzed. These include (1) whether variants were recurrently identified in unrelated cases of homogenous phenotype, or significantly high frequency, or hotspot in patient group (repetition); (2) for heterogeneous phenotypes, whether a phenotype was within a spectrum that was correlated with genotype (genotype–phenotype correlation); (3) defined inheritance pattern, for example, cosegregation in families with AD/autosomal recessive (AR) inheritance pattern, or mainly de novo origination (inheritance pattern); (4) correlation between genetic impairment and phenotype severity (genetic quantitative correlation); and (5) defined sub-regional (functional domains) or sub-molecular implications of the variants (molecular sub-regional implication), or distinct pathogenic functional alteration/mechanism.
Statistical Analysis
Statistical analyses were performed with the SPSS version 23.0 (SPSS, Inc., Chicago, IL, United States). Chi-square test or Fisher’s exact test was applied to compare the frequencies of null/de novo mutations and penetrance between different genotype groups. The relationship between phenotype and the occurrence of null/de novo mutations was analyzed by Spearman’s correlation test. Values of p < 0.05 (two-sided) were considered significant.
Results
Identification of Novel DEPDC5 Variants
Among the 305 patients with focal epilepsies, eight DEPDC5 mutations were identified in 12 unrelated families (Figure 1 and Table 1, sequencing graph, see Supplementary Figure S1). A heterozygous truncating mutations (p.Val151Serfs∗27), a termination codon mutation (p.Ser1601_Ter1604del_ext133), and two missense mutations (p.Tyr836Cys and p.Gly1545Ser) have not been reported previously and were novel findings. Truncating mutation p.Arg239∗ has been reported in cases of FCD and focal epilepsy (Ishida et al., 2013; Baulac et al., 2015; Baldassari et al., 2019), whereas mutation p.Arg838∗ has been identified in cases of sleep-related hypermotor epilepsy, frontal lobe epilepsy (FLE), or focal epilepsy (Baldassari et al., 2019). Mutation p.Tyr7Cys has previously been reported in a case of TLE (Tsai et al., 2017). Mutation p.Pro1031His, which has been described previously as a de novo mutation in a patient with late-onset epileptic spasms with focal discharges (Carvill et al., 2015), appeared as a heterozygous variant in four families with FEFS + /FS or rolandic epilepsy and as homozygous variant in a case of FLE with FCD. In contrast, no DEPDC5 mutation was identified in any of the 91 patients with generalized epilepsies.
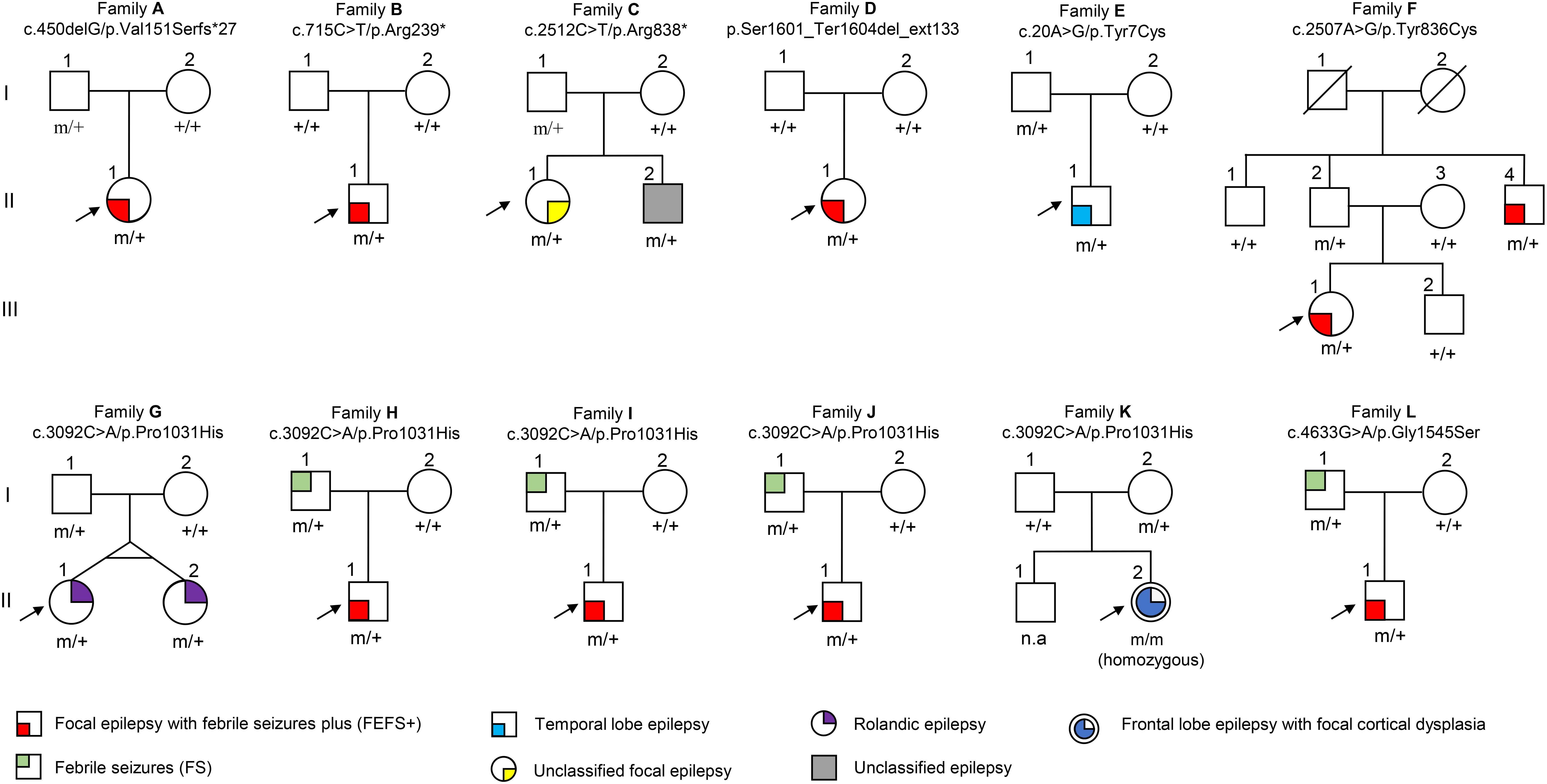
Figure 1. Pedigrees of the families with DEPDC5 mutations. Individuals with heterozygous mutation are indicated by m/+, those with homozygous mutation are indicated by m/m, and those negative for mutation are indicated by +/+. The probands are indicated by arrows. n.a., not available. The phenotype of each case is indicated by different symbols in the figure.

Table 1. Clinical feature of patients with DEPDC5 mutations.
We analyzed the potential molecular effects of the variants. A recent study demonstrated that DEPDC5 contains five functional domains, including N-terminal domain (NTD), structural axis for binding arrangement (SABA) domain, steric hindrance for enhancement of nucleotidase activity (SHEN) domain, DEP domain, and C-terminal domain (CTD) (Shen et al., 2018). Locations of the eight mutations identified in this study are indicated in Figure 2A.
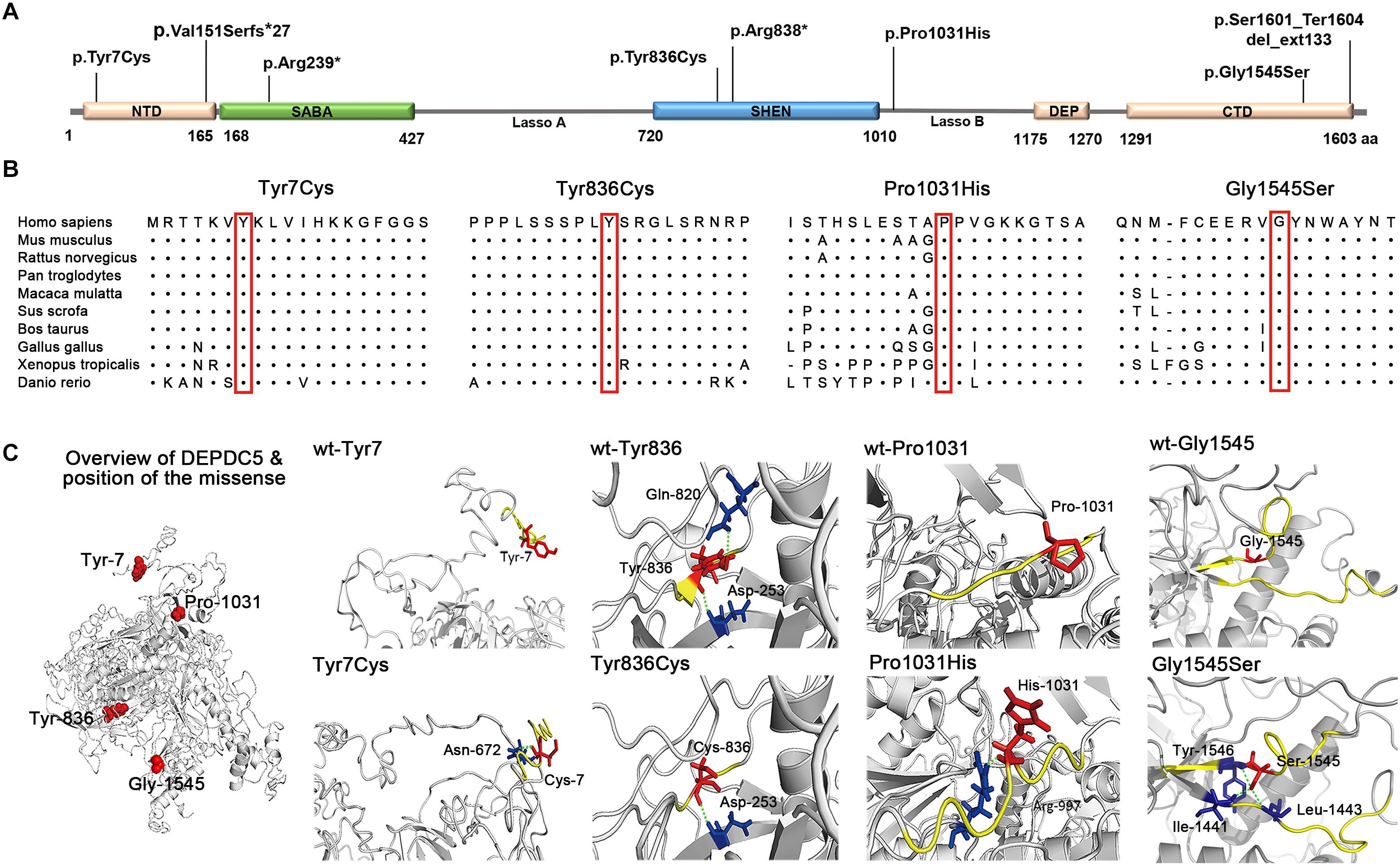
Figure 2. Schematic presentation of DEPDC5 structure. (A) The positions of the mutations identified in this study are indicated relative to functional domains. (B) All four missense mutations identified in this study affected amino acid residues that are highly conserved in various species. (C) Overview of DEPDC5 and focal structures of wild type and mutants. The residues where the mutations occurred are shown as red rods. The hydrogen-bonded residues are colored blue. The hydrogen bonds are shown as green spheres. The yellow-colored structure where Tyr836 located is a short β sheet. NTD, N-terminal domain; SABA, structural axis for binding arrangement; SHEN, steric hindrance for enhancement of nucleotidase activity; DEP, disheveled, Egl-10 and pleckstrin; CTD, C-terminal domain.
Truncating mutations (p.Val151Serfs∗27, p.Arg239∗, and p.Arg838∗) and termination codon mutation (p.Ser1601_Ter1604del_ext133) were not present in ExAC, 1000 Genomes, or gnomAD database. They could cause gross protein malformations and lead to functional haploinsufficiency. The four missense mutations substituted evolutionarily conserved amino acid residues (Figure 2B). These missense mutations presented in ExAC, 1000 Genomes, and gnomAD databases as a minor allele frequency of < 0.005 and were suggested to be damaging or possibly damaging by the web-based prediction tools (SIFT, PolyPhen-2, and Mutation Taster) (Table 2). However, p.Tyr7Cys, p.Tyr836Cys, and p.Pro1031His present at higher frequencies in East Asian population than that in general populations in ExAC database (0.0007 vs. 0.00005, 0.0013 vs. 0.0001, and 0.0076 vs. 0.0005, respectively). With the use of the standards and guidelines for the interpretation of sequence variants by the American College of Medical Genetics and Genomics, evaluation of pathogenicity of the variants showed that p.Arg239∗ and p.Ser1601_Ter1604del_ext133 were pathogenic; p.Arg838∗, p.Val151Serfs∗27, and p.Pro1031His were likely pathogenic; and p.Tyr7Cys, p.Tyr836Cys, and p.Gly1545Ser were of uncertain significance (Table 2).
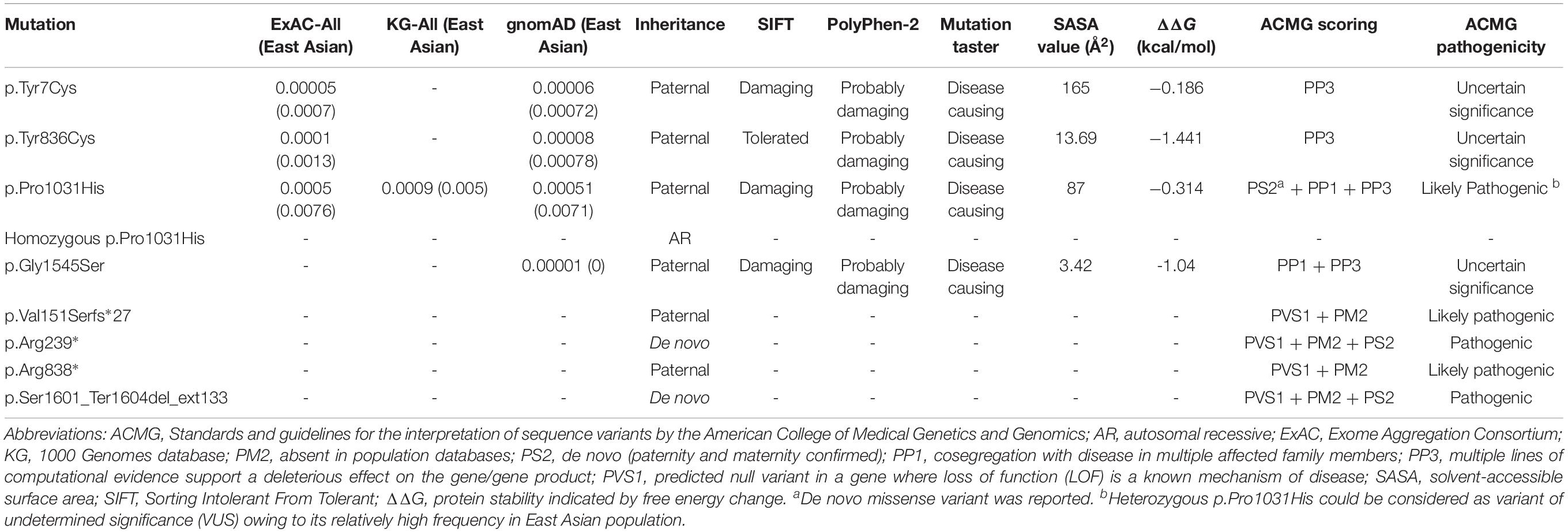
Table 2. In silico analysis and ACMG scoring of DEPDC5 variants.
The molecular effects of the missense variants were further predicted by protein modeling using I-TASSER. As shown in Figure 2C, Tyr7 and Pro1031 are located in protein surface, with the SASA values of 165 and 87 Å2, respectively. In contrast, Tyr836 and Gly1545 are deeper in protein cores, with the SASA value of 13.69 and 3.42 Å2, respectively. In the wild type of DEPDC5 monomer, Tyr7 residue is located in a loop region with no hydrogen bond to near residues. On the contrary, p.Tyr7Cys formed a new bond with residue Asn672 and would have mild influence on protein folding (the ΔΔG was −0.186 kcal/mol). Under normal conditions, Tyr836 residue forms two hydrogen bonds with Asp253 and Gln820. Conversely, p.Tyr836Cys destroyed the hydrogen bond with Gln820, which resulted in an alteration from short β sheet to loop structure. The difference of biochemical property of the amino acids, as the replacement of a ring-contained aromatic amino acid tyrosine by an aliphatic amino acid cysteine, would have strong influence on protein folding and affect the protein stability (ΔΔG of −1.441 kcal/mol). Normally, the Pro1031 residue is located in the loop region. When proline was replaced by histidine (p.Pro1031His), a new bond with Arg997 was formed and would lead to decreased structural stability (ΔΔG of −0.314 kcal/mol). Gly1545 is located in a loop region close to β sheet with no hydrogen bond to near residues. Three new hydrogen bonds generated when the Gly1545 is replaced by Ser1545, which decreases the structural stability (ΔΔG of −1.04 kcal/mol). These data suggested that the four missense mutations changed the hydrogen bonds and potentially affected the protein stability.
Phenotype of DEPDC5 Variants
In this cohort, eight DEPDC5 mutations were identified in 12 unrelated families featured by focal epilepsies. All probands had focally originated seizures or focal discharges on EEG recordings. Eight of the probands and another affected individual were diagnosed as focal (partial) epilepsy with FS +. Four of their parents had FS. Other phenotypes included rolandic epilepsy, TLE, unclassified focal epilepsy, and FLE with FCD (homozygous mutation). All cases with heterozygous mutations, including the cases with truncating mutations, presented mild phenotype with good responses to antiepileptic therapy. The case with homozygous mutation and MCD also became seizure free after 1.5 years of frequent seizures. The clinical information of patients with DEPDC5 mutations is summarized in Table 1, and their representative EEG and imaging data are shown in Figures 3A–J.
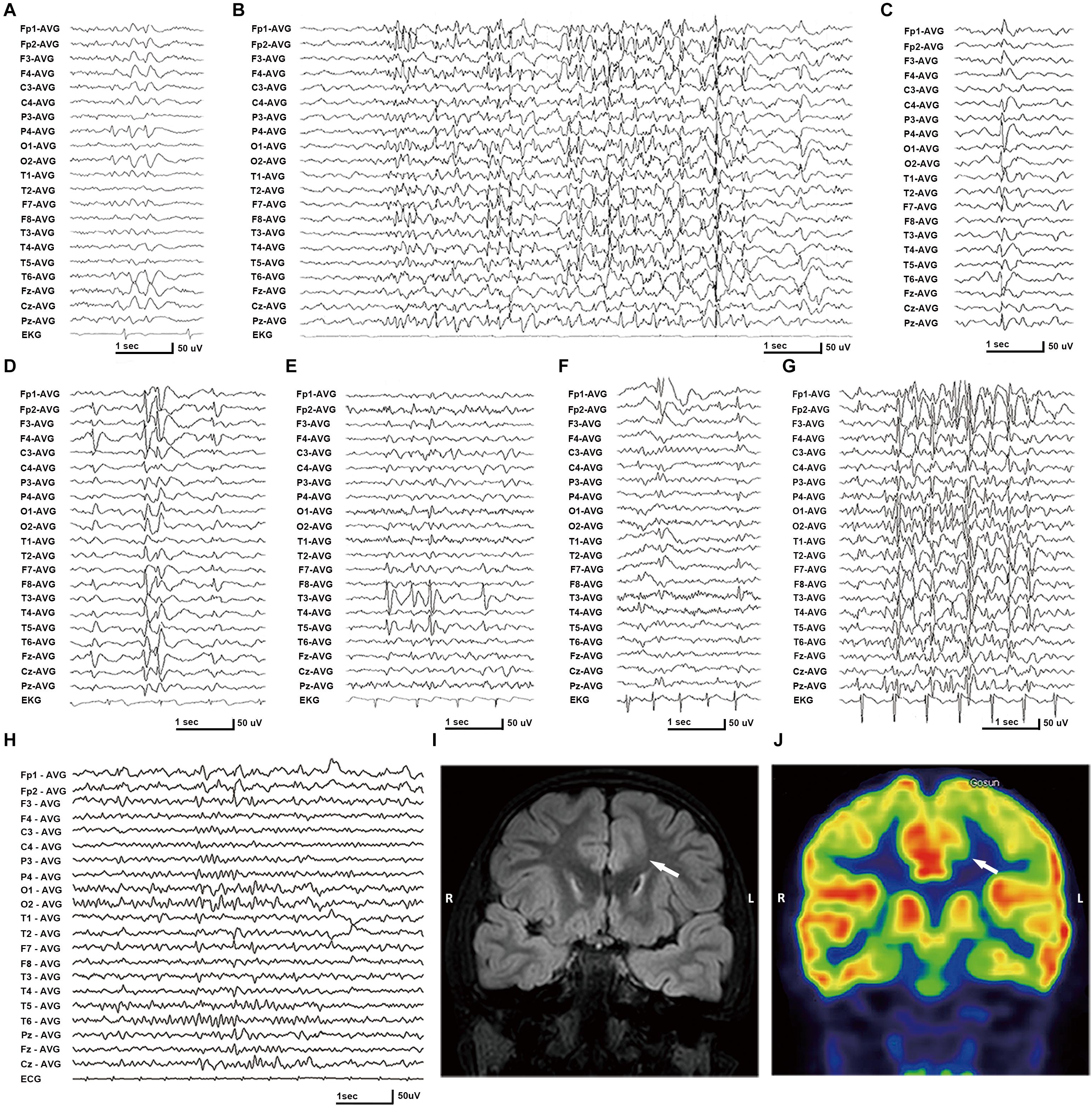
Figure 3. Electroencephalography (EEG) and imaging data of the patients with DEPDC5 mutations. (A) EEG of proband in family A (II-1) shows spikes and waves predominantly in the right parietal region. (B,D) Proband in family C (II-1) shows generalized polyspikes and slow waves (B) and multifocal discharges (C,D). (E) Proband in family E (II-1) shows spikes or spikes and waves in the left temporal lobe. (F,G) Proband in family H (II-1) shows both focal spikes and waves or spikes in the left frontal or temporal region (F) and generalized discharges (G). (H) The case with homozygous p.Pro1031His in family K (II-2) shows spikes or slow waves predominantly in the left frontal lobe; MRI shows focal cortical dysplasia in the anterior cingulate region of frontal lobe (I), which shows hypometabolic signal in PET (J).
The patient carrying the truncating mutation p.Val151Serfs∗27 (Figure 1, Family A: II-1) had a single FS at the age of 4 years. She presented with episodic vertigo since the age of 9, which usually lasted for 30–60 s without precipitating factor. Interictal EEG showed spikes and waves predominantly in the right parietal region (Figure 3A). A diagnosis of simple sensory seizure was considered. The attacks occurred at about three times per day and were controlled by levetiracetam monotherapy.
The patient with the truncating mutation p.Arg239∗ (Figure 1, Family B: II-1) had first febrile seizure at the age of 15 years. It was generalized tonic–clonic seizures (GTCSs), which lasted for 3–5 min and occurred four times per year during fever. Interictal EEGs showed sharp waves or spikes and slow waves predominantly in the left temporal area. The seizures were controlled by levetiracetam monotherapy.
The proband of the family with p.Arg838∗ had focal epilepsy (Figure 1, Family C: II-1). She was a 31-year-old woman who had her first seizure at 7 years of age. She presented with seizures that started with blank staring, automatism, and then limb jerks, lasting 30–90 s, one of which was recorded as a complex partial seizure during EEG monitoring. Interictal EEGs revealed generalized polyspikes and slow waves (Figure 3B), as well as multifocal discharges that tended to be generalized (Figures 3C,D). Her seizures responded well to valproate. Her younger brother (Figure 1, Family C: II-2) had occasional secondarily GTCS (sGTCS).
The patient with termination codon mutation p.Ser1601_Ter1604del_ext133 (Figure 1, Family D: II-1) had both febrile seizures and afebrile seizures that were complex partial seizures and occurred three to five times per year. Interictal EEGs recorded focal-sharp slow-wave discharges predominantly in the frontal and central areas. Seizure was controlled by levetiracetam monotherapy.
The affected individual with p.Tyr7Cys (Figure 1, Family E: II-1) had TLE with left hippocampal atrophy and discharges in the left temporal lobe on EEGs (Figure 3E), which was consistent with the phenotypic characteristics of the case reported previously (Tsai et al., 2017). He became seizure free after the application of oxcarbazepine.
In the family with p.Tyr836Cys, the two affected family members (Figure 1, Family F: II-4, III-1) had epilepsy with antecedent FS. The seizure was sGTCS. Interictal EEGs showed bilateral spike-and-wave discharges predominantly in the frontal area.
In this cohort, heterozygous p.Pro1031His was identified in four unrelated families with eight individuals involved, including a pair of monozygotic twins (Figure 1, Family G: II-1, II-2) with rolandic epilepsy and three families with FEFS + /FS (Figure 1, Family H to J, Table 1). An affected individual with FEFS + (Figure 1, Family H: II-1) showed both focal and generalized discharges on EEGs (Figures 3F,G). The girl with homozygous p.Pro1031His (Figure 1, Family K: II-2) had seizures since she was 3 years old, which usually started with a feeling of fear and followed by loss of consciousness that lasted for around 10 s. She had frequent seizures up to eight times per day for 1.5 years and became seizure free in recently for 1 year with a combination of levetiracetam (50 mg/kg/day), oxcarbazepine (15 mg/kg/day), and clonazepam (1 mg/night). EEG and neuroimaging confirmed the diagnosis of FLE with FCD (Figures 3H–J). One of the heterozygous p.Pro1031His mutations was from her mother, and the other one possibly originated de novo. Her paternity was confirmed by alignment of the segregated sequence variants, and all other variants detected were either maternal or paternal. p.Pro1031His has also been reported previously as a de novo mutation (Carvill et al., 2015).
The proband of the family with p.Gly1545Ser mutation (Figure 1, Family L: II-1) had two febrile seizures (sGTCS) at the age of 3 and 7 years. His brain imaging and EEG were normal.
Genotype–Phenotype Correlation
In the present study, six DEPDC5 mutations were identified in eight unrelated familial cases with FEFS + /FS. These mutations included one termination codon mutation (de novo), two truncating mutations (one of de novo), and three missense mutations (10 individuals who cosegregated with the mutations in each family). Previously, three DEPDC5 mutations associated with FS and one mutation associated with FEFS + have been reported (Martin et al., 2014; Pippucci et al., 2015; Ricos et al., 2016; Baldassari et al., 2019). These findings suggested that DEPDC5 variants were potentially associated with FEFS + /FS. On the other hand, homozygous p.Pro1031His was detected in a case with FLE and FCD. DEPDC5 variants have been reported to be associated with MCD (including FCD), and a second hit caused by mosaic somatic mutations was suggested (Ribierre et al., 2018). To explore the association between DEPDC5 variants and MCD or FS/FS +, we systematically reviewed all DEPDC5 mutations (Supplementary Table S2).
To date, 123 DEPDC5 mutations, including 87 null mutations and 35 missense mutations, two insertion–deletion mutations, have been identified in 170 unrelated cases. Most of the cases were featured by focal epilepsies (96.4%), except six cases with unclassified epilepsy. Thirty-nine cases were associated with MCD (22.9%). Among the cases without MCD, 113 cases presented as focal epilepsy and 12 cases (nine families and three de novo) presented as FEFS + /FS.
When we compared the mutation type of different groups, it was found that MCD group had a tendency of higher frequency of null mutation than the patients with FEFS + /FS (excluding p.Pro1031His, Figure 4A). Considering that the patients with MCD commonly presented frequent and refractory seizures (Baulac et al., 2015), a correlation between phenotype severity and null mutation was suggested. DEPDC5 mutations were originally reported in familial focal epilepsies, but asymptomatic carriers were common. We therefore compared the penetrance of different genotypes. The familial cases with truncating mutations had a penetrance of 66.9%. No statistical significance in penetrance was found among the families with truncating mutations, splice-site mutations, and missense mutations (Figure 4B). The overall penetrance of DEPDC5 mutations was 70.3% (142/202).
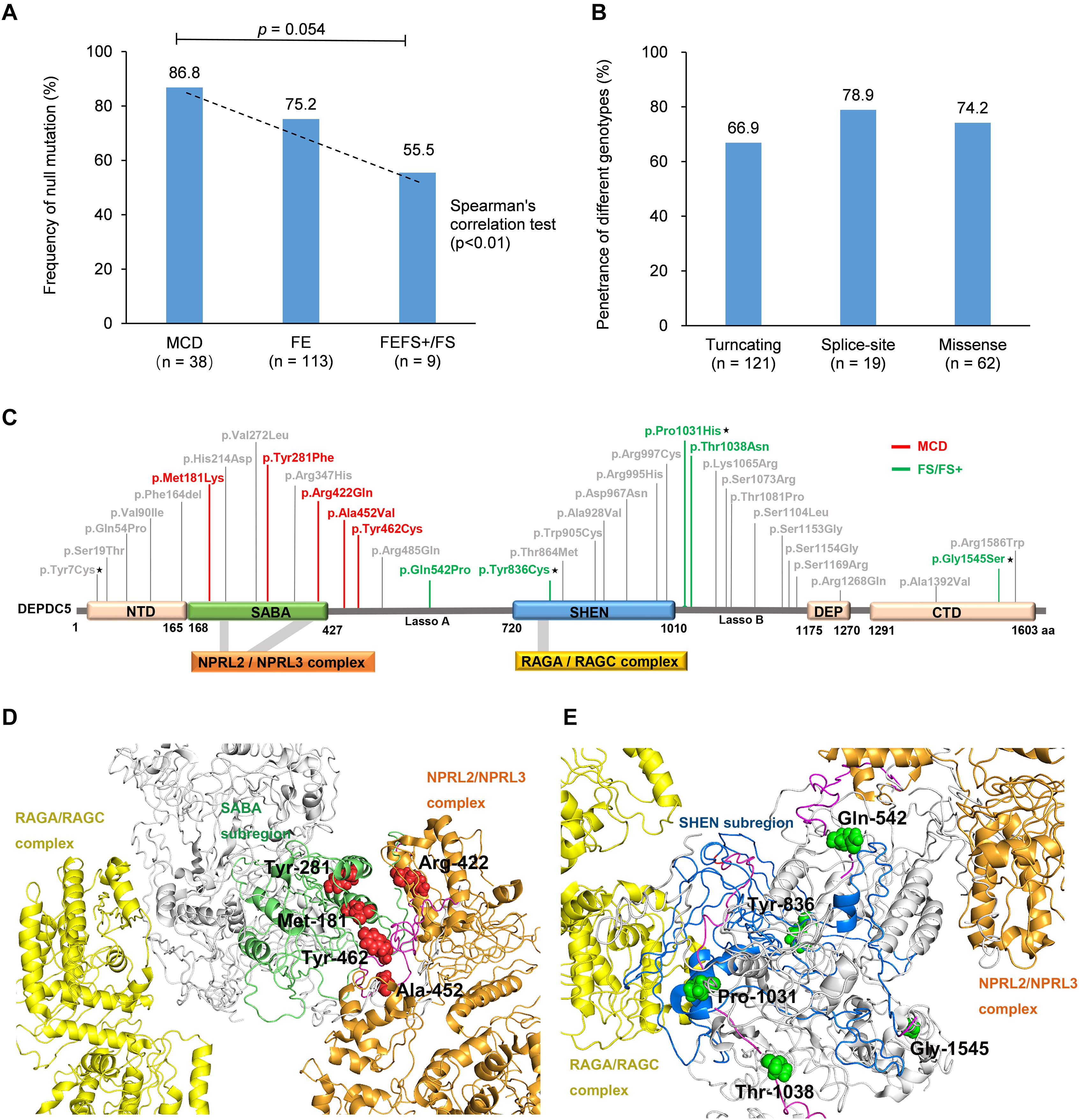
Figure 4. Genotype–phenotype correlations of DEPDC5 mutations. (A) The frequency of null mutations in DEPDC5 for each phenotype. The values are expressed as the percentage of cases with null mutations (cases with null mutations/total cases) in each group. (B) The penetrance of different genotypes of DEPDC5 mutations. Chi-square test or Fisher’s exact test was used for statistical analysis of the differences. Spearman’s correlation test was used for correlation analysis. (C) Schematic diagram of DEPDC5 domain and position of the missense mutations. Malformation of cortical development (MCD) and focal epilepsy with febrile seizures plus/febrile seizures (FEFS+/FS) associated mutations were colored red and green, respectively. The mutations reported in this study are indicated by black stars; the others were from literature. (D,E) The spatial location of MCD and FEFS+/FS associated mutations in 3D protein structure. The colors indicated in D,E for domains are the same as those in (C).
We further mapped the locations of the heterozygous missense variants on DEPDC5 (Figure 4C). The variants distributed over all DEPDC5. However, all MCD-associated heterozygous missense variants clustered around SABA domain and were close to the binding sites to NPRL2/NPRL3 complex (Figure 4D). MCD-related missense mutation identified in this study (p.Pro1031His) was not included for sub-regional analysis owing to its homozygous nature. In contrast, the FEFS + /FS-associated variants were located around SHEN domain and a distance away from NPRL2/NPRL3 complex or RAGA/RAGC complex (Figure 4E). The other three SABA domain-located missense mutations were associated with focal epilepsies (c.640C > G/p.His214Asp and c.814G > T/p.Val272Leu) or SUDEP (c.1040G > A/p.Arg347His), and whether these mutations were associated with FCD was unknown owing to the lack of neuroimaging data (Supplementary Table S2). The other five mutations in SHEN domain were excluded from the association with FCD by neuroimaging examinations (Supplementary Table S2). The three-dimensional protein modeling demonstrated more clearly the location of mutations in DEPDC5 (Supplementary Figures S2a,b4).
We analyzed evidence from five clinical-genetic aspects that potentially disclose the association between DEPDC5 variants and MCD or FEFS + /FS. Evidence from all five clinical-genetic aspects suggested MCD as a phenotype of DEPDC5 variants; and evidence from four aspects and one possible evidence from sub-regional implication aspect suggested FEFS + /FS as a phenotype of DEPDC5 variants (Table 3).

Table 3. Five-dimensional evaluation of phenotypes of DEPDC5 variants.
Discussion
In the present study, we identified eight DEPDC5 mutations in 12 unrelated families from a cohort of 305 patients affected by focal epilepsy (3.9%), including homozygous mutation in a case with FCD. In contrast, no DEPDC5 mutation was identified in the 91 patients with generalized epilepsies. Thirteen of the 19 affected individuals (68.4%) in this study had FEFS + /FS, suggesting a potential role of DEPDC5 mutations in FEFS + /FS. Our further analysis revealed potential genotype–phenotype correlations and sub-regional implications of DEPDC5 variants, which would help understanding the mechanism underlying phenotypical variation.
DEPDC5 is located on chromosome 22q12.3 and encodes a ubiquitous protein that inhibits the mTOR pathway (Bar-Peled et al., 2013; Marsan and Baulac, 2018). Homozygous Depdc5–/– embryos of rats died in utero owing to global growth delay. In contrast, heterozygous Depdc5± rats had altered cortical neuron excitability and firing patterns but without developmental abnormalities or spontaneous electro-clinical seizures (Marsan et al., 2016). These findings suggested a potential quantitative correlation between genetic impairment and phenotype severity and that heterozygous mutations would potentially cause mild phenotype or susceptibility alterations. All cases with heterozygous mutations in this study presented mild phenotype with good responses to antiepileptic therapy, and most individuals have become seizure free. Heterozygous mutations also presented an overall penetrance of 70.3%, which was lower than that in genes of high pathogenic potential like SCN1A (Meng et al., 2015). These findings suggested that heterozygous DEPDC5 mutations were less pathogenic, coincident with the evidence from heterozygous knockout.
In this study, 13 individuals in eight families with DEPDC5 mutations had FEFS + /FS. Previously, four FS-related families with DEPDC5 mutations, including two truncating mutations and two missense mutations, have been reported (Supplementary Table S2) (Martin et al., 2014; Pippucci et al., 2015; Ricos et al., 2016; Baldassari et al., 2019). Mutations in 11 of the 12 families inherited in a dominant pattern or originated de novo. Further analysis revealed that FEFS + /FS had a lower frequency of null mutation than MCD or other focal epilepsies; and missense mutations associated with FEFS + /FS were located away from the binding sites to NPRL2/NPRL3 or RAGA/RAGC. To define FEFS + /FS as a phenotype of DEPDC5 variants, we tried to evaluate evidence from five clinical-genetic aspects. Evidence from four aspects, including repetition, genotype–phenotype correlation, inheritance pattern, and genetic quantitative correlation, suggested FEFS + /FS as a phenotype of DEPDC5 variants. FEFS + /FS associated heterozygous missense variants located away from NPRL2/NPRL3 complex or RAGA/RAGC complex, which was a possible evidence in molecular sub-regional implication aspect (Table 3).
Previous studies have showed that DEPDC5 mutations are associated with diverse focal epileptic phenotypes, ranging from mild rolandic epilepsy to severe MCD-associated epilepsies (Baulac, 2016). Mechanisms underlying the phenotypic variation were unclear, especially for the severe phenotype like MCD. Previously, brain somatic DEPDC5 mutations, in addition to the germline mutations, have been identified in two patients with MCD, suggesting a second-hit mechanism (Baulac et al., 2015; Ribierre et al., 2018). The present study identified a homozygous DEPDC5 mutation (p.Pro1031His) that was associated with FCD. To our knowledge, this was the first report on homozygous DEPDC5 mutation in patients with FCD, which provided direct evidence on association between bi-allelic DEPDC5 mutation and MCD. Our further analysis revealed that MCD was more frequently associated with null mutations (Figure 4A); and MCD-associated heterozygous missense mutations were located on SABA domain and were close to the binding sites to NPRL2/NPRL3 complex (Figure 4D), provided additional possible explanations for the association between DEPDC5 mutations and MCD. DEPDC5 exerts inhibitory effect on mTOR pathway through binding with NPRL2/NPRL3 complex (Shen et al., 2018). It is therefore possible that mutations closer to the binding site of DEPDC5 to NPRL2/NPRL3 may lead to more severe phenotype like MCD. Evidence from four clinical-genetic aspects, including repetition, genotype–phenotype correlation, inheritance pattern, and genetic quantitative correlation, suggested MCD as a phenotype of DEPDC5 variants. MCD associated heterozygous missense variants clustered in SABA domain and close to the binding sites of NPRL2/NPRL3 complex, which was a possible evidence in molecular sub-regional implication aspect (Table 3).
In the present study, we identified four deleterious DEPDC5 mutations that would cause gene haploinsufficiency in four cases. Three of the four cases had FEFS + /FS. Additionally, two of the three missense mutations, excluding p.Pro1031His mutation, had FEFS + /FS. These cases suggest a potential role of DEPDC5 mutations in FEFS + /FS. However, the pathogenicity of missense mutations warrants further verification, especially on variant p.Pro1031His. Heterozygous p.Pro1031His was identified in four families with mild phenotype, including six individuals with FEFS + /FS that cosegregated with the variant in three small families (Figure 1 and Table 1). Previously, heterozygous p.Pro1031His has been identified as a de novo mutation in a patient with late-onset epileptic spasms with focal discharges (Carvill et al., 2015). It could be evaluated to be likely pathogenic by ACMG (Table 2). However, heterozygous p.Pro1031His presents at minor allele fractions (MAFs) of 0.00051 in the general populations and at 0.0071 in East Asian population in gnomAD (Table 2). The pathogenicity of p.Pro1031His could therefore be suspected and was reclassified as likely benign (Baldassari et al., 2019) or variant of undetermined significance. Evaluation of the pathogenicity of a variant is currently challenging (Tang et al., 2019), even for genes with de novo mutations (He et al., 2019). Generally, the MAFs are closely related to the prevalence of the phenotypes (Richards et al., 2015), among which mild phenotypes potentially have higher prevalence than severe ones. It was noted that a prevalence of febrile seizures as high as 6.9–8.2% has been reported in East Asian population (Tsuboi and Okada, 1984; Byeon et al., 2018). Our recent study has demonstrated that the damaging effects of variants usually vary and potential present a continuing distribution with overlaps between pathogenic variants and benign variants (Tang et al., 2019). It was therefore possible that heterozygous p.Pro1031His may be less pathogenic and even overlapped with rare variants in general populations. On the other hand, homozygous p.Pro1031His did not present in any populations in gnomAD (Table 2) and was potentially associated with MCD. Previous study showed that homozygous Depdc5–/– knockout was lethal, whereas heterozygous Depdc5± led to subclinical change of neuron excitability (Marsan et al., 2016), suggesting a potential quantitative correlation between genetic impairment and phenotype severity, which would help in understanding the difference in the pathogenicity between homogenous and heterogeneous DEPDC5 missense variants.
This study has several limitations. Previously, effects of DEPDC5 variants on protein expressions and interactions have been studied. Mutation Y281F presented slightly decreased protein expression, but the four missense mutations tested, including H214D, Y281F, 542P, and S1154F, did not show impacts on other parameters (Dawson et al., 2020). In a similar study, mutations S442F, A452V, T864M, S1073R, and K1088R presented slightly decreased protein expression; mutations A452V and R485Q led to slight increased activity of target of rapamycin complex 1 (Van Kranenburg et al., 2015). Missense mutations in families with cosegregation have been reported, which provided clinical genetic evidence on the pathogenicity of missense DEPDC5 variants (Dibbens et al., 2013; Ricos et al., 2016). However, further studies on direct functional impact of DEPDC5 variants are required, especially for missense variants. The present study suggests that the phenotypical spectrum of DEPDC5 variants potentially includes FEFS + /FS, which warrants validation on a large cohort of FS-related epilepsies.
Epilepsy comprises a huge group of heterogeneous phenotypes with heterogeneous genetic etiologies. So far, approximately 1,000 genes have been reported to be associated with epilepsy (Wang et al., 2017). Defining the association between a phenotype and a gene is practical challenging. Previously, we summarized the evidence required to define the associations between epileptic encephalopathies and genes with de novo mutations (He et al., 2019). In a more general sense, we now summarized the clinical-genetic aspects that potentially disclose the association between a phenotype and the genetic variants, which is expected to enable the formulation of guideline for defining a phenotype of genetic variants. This is also the first report on the molecular sub-regional effect of DEPDC5 variants, which may help in evaluating the pathogenicity of DEPDC5 variants and development of individualized predictive algorithms, as suggested in our recent study (Tang et al., 2019).
Data Availability Statement
The datasets are included in the Supplementary Material and uploaded to a public database on Figshare, https://figshare.com/s/17679354408cdca72fd7.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
YH-Y and WP-L designed and conceptualized the study. LL, Z-RC, H-QX, D-TL, X-RL, PZ, S-ML, BL, NH, and Q-XZ collected and analyzed the data. LL and Z-RC drafted the manuscript for intellectual content. LL, Z-RC, H-QX, D-TL, TS, HM, W-PL, and Y-HY revised the manuscript for intellectual content. H-KL, YM, and D-TL provided software support for 3D structure model. LL and D-TL prepared the figures. All authors have read and approved the final draft of the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant nos. 81870903, 81571273, 81471149, and 81501125), Omics-based precision medicine of epilepsy being entrusted by the Key Research Project of the Ministry of Science and Technology of China (Grant no. 2016YFC0904400), Science and Technology Project of Guangdong Province (Grant nos. 2017B090904036 and 2017B030314159), Science and Technology Project of Guangzhou (Grant nos. 201607010002, 20181A011076, and 201904020028), and Collaborative Innovation Center for Neurogenetics and Channelopathies. The funders had no role in study design, data collection, and analysis or in the decision to publish or the preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are deeply grateful to the families who participated in this research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2020.00821/full#supplementary-material
Footnotes
- ^ http://www.rcsb.org
- ^ http://www.ncbi.nlm.nih.gov/pubmed/
- ^ http://www.hgmd.cf.ac.uk/ac/index.php
- ^ https://figshare.com/s/17679354408cdca72fd7
References
Baldassari, S., Picard, F., Verbeek, N. E., Van Kempen, M., Brilstra, E. H., Lesca, G., et al. (2019). The landscape of epilepsy-related GATOR1 variants. Genet. Med. 21, 398–408. doi: 10.1038/s41436-018-0060-2
Bar-Peled, L., Chantranupong, L., Cherniack, A. D., Chen, W. W., Ottina, K. A., Grabiner, B. C., et al. (2013). A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106. doi: 10.1126/science.1232044
Baulac, S. (2016). mTOR signaling pathway genes in focal epilepsies. Prog. Brain Res. 226, 61–79. doi: 10.1016/bs.pbr.2016.04.013
Baulac, S., Ishida, S., Marsan, E., Miquel, C., Biraben, A., Nguyen, D. K., et al. (2015). Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann. Neurol. 77, 675–683. doi: 10.1002/ana.24368
Byeon, J. H., Kim, G. H., and Eun, B. L. (2018). Prevalence, incidence, and recurrence of febrile seizures in korean children based on national registry data. J. Clin. Neurol. 14, 43–47. doi: 10.3988/jcn.2018.14.1.43
Carvill, G. L., Crompton, D. E., Regan, B. M., Mcmahon, J. M., Saykally, J., Zemel, M., et al. (2015). Epileptic spasms are a feature of DEPDC5 mTORopathy. Neurol. Genet. 1:e17. doi: 10.1212/NXG.0000000000000016
Dawson, R. E., Nieto Guil, A. F., Robertson, L. J., Piltz, S. G., Hughes, J. N., and Thomas, P. Q. (2020). Functional screening of GATOR1 complex variants reveals a role for mTORC1 deregulation in FCD and focal epilepsy. Neurobiol. Dis. 134:104640. doi: 10.1016/j.nbd.2019.104640
D’Gama, A. M., Geng, Y., Couto, J. A., Martin, B., Boyle, E. A., Lacoursiere, C. M., et al. (2015). Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann. Neurol. 77, 720–725. doi: 10.1002/ana.24357
Dibbens, L. M., De Vries, B., Donatello, S., Heron, S. E., Hodgson, B. L., Chintawar, S., et al. (2013). Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat. Genet. 45, 546–551. doi: 10.1038/ng.2599
Fingar, D. C., and Blenis, J. (2004). Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23, 3151–3171. doi: 10.1038/sj.onc.1207542
Guerrini, R. (2005). Genetic malformations of the cerebral cortex and epilepsy. Epilepsia 46(Suppl. 1), 32–37. doi: 10.1111/j.0013-9580.2005.461010.x
Gussow, A. B., Petrovski, S., Wang, Q., Allen, A. S., and Goldstein, D. B. (2016). The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biol. 17:9. doi: 10.1186/s13059-016-0869-4
He, N., Lin, Z. J., Wang, J., Wei, F., Meng, H., Liu, X. R., et al. (2019). Evaluating the pathogenic potential of genes with de novo variants in epileptic encephalopathies. Genet. Med. 21, 17–27. doi: 10.1038/s41436-018-0011-y
Ishida, S., Picard, F., Rudolf, G., Noe, E., Achaz, G., Thomas, P., et al. (2013). Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat. Genet. 45, 552–555. doi: 10.1038/ng.2601
Lal, D., Reinthaler, E. M., Schubert, J., Muhle, H., Riesch, E., Kluger, G., et al. (2014). DEPDC5 mutations in genetic focal epilepsies of childhood. Ann. Neurol. 75, 788–792. doi: 10.1002/ana.24127
Marsan, E., and Baulac, S. (2018). Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol. Appl. Neurobiol. 44, 6–17. doi: 10.1111/nan.12463
Marsan, E., Ishida, S., Schramm, A., Weckhuysen, S., Muraca, G., Lecas, S., et al. (2016). Depdc5 knockout rat: a novel model of mTORopathy. Neurobiol. Dis. 89, 180–189. doi: 10.1016/j.nbd.2016.02.010
Martin, C., Meloche, C., Rioux, M. F., Nguyen, D. K., Carmant, L., Andermann, E., et al. (2014). A recurrent mutation in DEPDC5 predisposes to focal epilepsies in the French-Canadian population. Clin. Genet. 86, 570–574. doi: 10.1111/cge.12311
Meng, H., Xu, H. Q., Yu, L., Lin, G. W., He, N., Su, T., et al. (2015). The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum. Mutat. 36, 573–580. doi: 10.1002/humu.22782
Mitternacht, S. (2016). FreeSASA: an open source C library for solvent accessible surface area calculations. F1000Research 5:189. doi: 10.12688/f1000research.7931.1
Picard, F., Makrythanasis, P., Navarro, V., Ishida, S., De Bellescize, J., Ville, D., et al. (2014). DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology 82, 2101–2106. doi: 10.1212/wnl.0000000000000488
Pippucci, T., Licchetta, L., Baldassari, S., Palombo, F., Menghi, V., D’aurizio, R., et al. (2015). Epilepsy with auditory features: a heterogeneous clinico-molecular disease. Neurol. Genet. 1:e5. doi: 10.1212/NXG.0000000000000005
Pires, D. E., Ascher, D. B., and Blundell, T. L. (2014). mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342. doi: 10.1093/bioinformatics/btt691
Ribierre, T., Deleuze, C., Bacq, A., Baldassari, S., Marsan, E., Chipaux, M., et al. (2018). Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Investig. 128, 2452–2458. doi: 10.1172/JCI99384
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi: 10.1038/gim.2015.30
Ricos, M. G., Hodgson, B. L., Pippucci, T., Saidin, A., Ong, Y. S., Heron, S. E., et al. (2016). Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann. Neurol. 79, 120–131. doi: 10.1002/ana.24547
Sarbassov, D. D., Ali, S. M., and Sabatini, D. M. (2005). Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 17, 596–603. doi: 10.1016/j.ceb.2005.09.009
Scerri, T., Riseley, J. R., Gillies, G., Pope, K., Burgess, R., Mandelstam, S. A., et al. (2015). Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann. Clin. Transl. Neurol. 2, 575–580. doi: 10.1002/acn3.191
Shen, K., Huang, R. K., Brignole, E. J., Condon, K. J., Valenstein, M. L., Chantranupong, L., et al. (2018). Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 556, 64–69. doi: 10.1038/nature26158
Striano, P., Serioli, E., Santulli, L., Manna, I., Labate, A., Dazzo, E., et al. (2015). DEPDC5 mutations are not a frequent cause of familial temporal lobe epilepsy. Epilepsia 56, e168–e171. doi: 10.1111/epi.13094
Tang, B., Li, B., Gao, L. D., He, N., Liu, X. R., Long, Y. S., et al. (2019). Optimization of in silico tools for predicting genetic variants: individualizing for genes with molecular sub-regional stratification. Brief Bioinform. [Epub ahead of print]. doi: 10.1093/bib/bbz115
Tsai, M. H., Chan, C. K., Chang, Y. C., Yu, Y. T., Chuang, S. T., Fan, W. L., et al. (2017). DEPDC5 mutations in familial and sporadic focal epilepsy. Clin. Genet. 92, 397–404. doi: 10.1111/cge.12992
Tsuboi, T., and Okada, S. (1984). Seasonal variation of febrile convulsion in Japan. Acta Neurol. Scand. 69, 285–292. doi: 10.1111/j.1600-0404.1984.tb07814.x
Van Kranenburg, M., Hoogeveen-Westerveld, M., and Nellist, M. (2015). Preliminary functional assessment and classification of DEPDC5 variants associated with focal epilepsy. Hum. Mutat. 36, 200–209. doi: 10.1002/humu.22723
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Keywords: DEPDC5, focal epilepsy, febrile seizures, genotype–phenotype correlation, molecular sub-regional effect
Citation: Liu L, Chen Z-R, Xu H-Q, Liu D-T, Mao Y, Liu H-K, Liu X-R, Zhou P, Lin S-M, Li B, He N, Su T, Zhai Q-X, Meng H, Liao W-P and Yi Y-H (2020) DEPDC5 Variants Associated Malformations of Cortical Development and Focal Epilepsy With Febrile Seizure Plus/Febrile Seizures: The Role of Molecular Sub-Regional Effect. Front. Neurosci. 14:821. doi: 10.3389/fnins.2020.00821
Received: 01 April 2020; Accepted: 14 July 2020;
Published: 11 August 2020.
Edited by:
Paul Pavlidis, University of British Columbia, CanadaReviewed by:
Gemma Louise Carvill, Northwestern University, United StatesMichael John Gambello, Emory University, United States
Copyright © 2020 Liu, Chen, Xu, Liu, Mao, Liu, Liu, Zhou, Lin, Li, He, Su, Zhai, Meng, Liao and Yi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Ping Liao, d3BsaWFvQDE2My5uZXQ=; Yong-Hong Yi, eXloMTY4QHNpbmEuY29t
†These authors have contributed equally to this work