
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 12 November 2019
Sec. Neuropharmacology
Volume 13 - 2019 | https://doi.org/10.3389/fnins.2019.01186
This article is part of the Research Topic Sigma-1 Receptor: Neuronal Functions, Disorders of the Nervous System, and Therapeutic Potential View all 8 articles
 Olivier Soriani1*
Olivier Soriani1* Saïd Kourrich2,3,4,5*
Saïd Kourrich2,3,4,5*The sigma-1 receptor (σ1R) is an endoplasmic reticulum (ER)-resident chaperone protein that acts like an inter-organelle signaling modulator. Among its several functions such as ER lipid metabolisms/transports and indirect regulation of genes transcription, one of its most intriguing feature is the ability to regulate the function and trafficking of a variety of functional proteins. To date, and directly relevant to the present review, σ1R has been found to regulate both voltage-gated ion channels (VGICs) belonging to distinct superfamilies (i.e., sodium, Na+; potassium, K+; and calcium, Ca2+ channels) and non-voltage-gated ion channels. This regulatory function endows σ1R with a powerful capability to fine tune cells’ electrical activity and calcium homeostasis—a regulatory power that appears to favor cell survival in pathological contexts such as stroke or neurodegenerative diseases. In this review, we present the current state of knowledge on σ1R’s role in the regulation of cellular electrical activity, and how this seemingly adaptive function can shift cell homeostasis and contribute to the development of very distinct chronic pathologies such as psychostimulant abuse and tumor cell growth in cancers.
The sigma-1 receptor (σ1R, a.k.a., Sig-1R), also called sigma non-opioid intracellular receptor 1 and encoded by the gene SIGMAR1 in humans, is actually not a receptor but a poorly characterized endoplasmic reticulum (ER) chaperone protein. Indeed, upon its discovery, σ1R was originally thought to be a novel opioid receptor (Martin et al., 1976). However, further pharmacological characterization shows that σ1R does not bind classical opioid ligands but rather binds (+)-benzomorphans (Walker et al., 1990; see review Schmidt and Kruse, 2019). σ1R has no known homologs in the eukaryotic genome and is the only known chaperone protein whose activity is regulated by endogenous and synthetic compounds in a clear agonist-antagonist manner (Hayashi and Su, 2007; Hayashi et al., 2011). At resting state, σ1R is coupled to BiP (Binding immunoglobulin Protein), another ER-resident chaperone protein. Upon agonist binding and in contrast to antagonist action, σ1R dissociates from BiP and acts as an interorganelle signaling modulator. Importantly, σ1R has been associated with many and diverse chronic diseases ranging from amyotrophic lateral sclerosis to Alzheimer’s disease, cancer and drug addiction (Romieu et al., 2004; Maurice and Su, 2009; Katz et al., 2011; Kourrich et al., 2012b; Yasui and Su, 2016). Among several distinct functions of σ1R that may contribute to these chronic pathologies, its ability to regulate and traffic a variety of functional proteins to the plasma membrane is gaining attraction (Su et al., 2010, 2016; Crottès et al., 2013; Kourrich, 2017). Specifically, σ1R regulates cellular electrical activity, a mechanism that occurs through physical protein-protein interactions with several VGICs and non-VGICs (Su et al., 2016; Kourrich, 2017; Schmidt and Kruse, 2019). In contrast to typical auxiliary ion channel subunits (e.g., Kvβs, Cavβ), this ability to associate and regulate functions and surface expression of a myriad of client ion channels from distinct superfamilies (i.e., sodium, Na+; potassium, K+; and calcium, Ca2+ channels) posits σ1R as an atypical auxiliary subunit (Aydar et al., 2002; reviewed in Kourrich, 2017). However, the use of in vitro model systems, such as cell culture models, Xenopus oocytes (Aydar et al., 2002; Crottès et al., 2011; Kinoshita et al., 2012) and neuroendocrinal tissue (Lupardus et al., 2000; Aydar et al., 2002) has been a major impediment to our understanding of how σ1R exerts its auxiliary subunit functions in vivo. Nonetheless, recent advancement provides evidence that σ1R can bind to ion channels in the brain and that endogenous or exogenous stimuli promotes the formation of these protein complexes, which results in long-lasting changes in cellular electrical activity (Crottès et al., 2013; Kourrich, 2017). The physiological and behavioral consequences of these changes are still largely unknown; however, with the exception of stimulant addiction and cancer, activation of σ1R are typically associated with both physiological and behavioral positive outcomes. Indeed, agonist activation of σ1R ameliorates the negative symptoms in many models of chronic diseases. For example, evidence suggests that activation of σ1R exhibits anti-amnesic effect against Aβ neurotoxicity in a model of Alzheimer’s disease (Jin et al., 2015); and while σ1R knockout (KO) mice exhibit a depression-like phenotype, σ1 agonists exhibit antidepressant properties (Hayashi et al., 2011; Wang et al., 2016). In stark contrast, activation of σ1R promotes the development of tumor growth in cancers (i.e., prostate, colorectal and breast cancers, and leukemia (Renaudo et al., 2007; Crottès et al., 2016; Gueguinou et al., 2017; Thomas et al., 2017) and addiction-relevant behaviors (Katz et al., 2016).
Stimulant addiction and cancer are very distinct types of severe chronic diseases. In both cases, many factors can trigger or contribute to their development and maintenance. These factors range from external stimuli such as intake or exposure to biological agents to internal factors such as genetic background that may confer vulnerability to the development of the disease. A common adaptation between stimulant addiction and some cancers is that both implicate enduring changes in cellular electrical activity, i.e., cellular intrinsic plasticity (Prevarskaya et al., 2010; Huang and Jan, 2014; Pardo and Stuhmer, 2014; Kourrich et al., 2015). The mechanism by which these changes are initiated and their contributions to the development and maintenance of the disease remain elusive. Nonetheless, recent studies revealed that σ1R is a common molecular player involved in the development of cellular electrical plasticity that characterizes both cancer (reviewed in Crottès et al., 2013) and psychostimulant drug addiction (Kourrich et al., 2013; Delint-Ramirez et al., 2018). We speculate that a reason that explains this opposite negative outcomes of σ1R activity on the development of cancer and psychostimulant abuse lies is the fundamental role of σ1R in cell’s health and survival, and especially, its ability to regulate cellular electrical properties.
As such, in this review, we will present the state of knowledge on σ1R-dependent regulation of ion channels. For the sake of brevity, we will focus on channels that directly contribute to the intrinsic electrical properties of the cell, in both neuronal and non-neuronal cells. Then, we will speculate on how this ability for σ1R to regulate cellular electrical activity, a seemingly adaptive function, can be hijacked and lead to chronic pathological conditions such as cancer and stimulant addiction.
σ1R has been proposed to be considered a ligand-regulated auxiliary potassium channel subunit since early 2000s (Aydar et al., 2002). However, whether σ1R is a stricto sensu auxiliary subunit is unclear, as the concept of auxiliary subunits has not been clearly defined. Auxiliary subunits are non-conducting, modulatory components of the multi-protein ion channel complexes that underlie normal neuronal signaling. Typically, auxiliary subunits, including σ1R, are characterized by several features, including (1) directly modulating the biophysical properties of the α pore-forming subunits (Aydar et al., 2002; Zhang et al., 2009; Kinoshita et al., 2012), (2) participating to the assembly, trafficking and surface expression of the pore-forming subunits (Crottès et al., 2011; Kinoshita et al., 2012; Kourrich et al., 2013; Balasuriya et al., 2014), (3) regulating ion currents in ligand-dependent and independent manner (Aydar et al., 2002; Kinoshita et al., 2012), (4) altering pharmacological interactions or bind drugs directly (see reviews Hayashi et al., 2011; Kourrich et al., 2012b; Schmidt and Kruse, 2019), and (5) interacting directly with the pore-forming subunit (Balasuriya et al., 2012, 2013, 2014). However, and in contrast to typical auxiliary subunits, indirect evidence suggest that σ1R is not stably associated with the pore-forming α subunits or present in purified channel complexes, but rather its association with client proteins is dynamic (Kourrich et al., 2013; Delint-Ramirez et al., 2018). Furthermore, σ1R exhibits specific functional characteristics that are not shared with typical auxiliary subunits. For example, canonical VGIC auxiliary subunits are either cytoplasmic proteins located at the plasmalemma level, i.e., in close proximity of the plasma membrane, or fully integrated in the phospholipidic membrane (Vacher et al., 2008). Instead, σ1R exhibits a heterogeneous distribution and can be found, in addition to these typical subcellular locations, at the nuclear and associated ER membranes, mitochondrial membrane (Hayashi and Su, 2003a, 2007; Luty et al., 2010; Su et al., 2010; Shioda et al., 2012) and potentially in extracellular space (Hayashi and Su, 2003a). Furthermore, while emerging evidence suggest that some channel family-specific auxiliary subunits have the ability to regulate ion channels from other superfamilies, such as the Na+ channel auxiliary subunits Navβ modulating voltage-gated K+ channels (Marionneau et al., 2012; Calhoun and Isom, 2014), this feature is extended and even exacerbated in σ1R. In particular, σ1R regulates via direct protein-protein interactions the functions of all VGIC superfamilies (i.e., Na+, Ca2+, and K+ families) and classes (i.e., ligand-gated ion channels, e.g., NMDA receptors, NMDARs; and G protein-coupled receptors, e.g., dopamine receptors, DARs) (reviewed in Crottès et al., 2013; Kourrich, 2017; Soriani and Rapetti-Mauss, 2017; Schmidt and Kruse, 2019). This unique characteristic positions σ1R as a powerful regulator of various cellular functions and neuronal excitability.
A feature that differentiates neuronal from non-neuronal cells is their excitability and their unique shape, which together endow neurons with the capability to receive, integrate and propagate the information within brain circuits. As such, this section will refer mainly to voltage-gated Na+, Ca2+, and K+ channels, i.e., the non-synaptic factors that are responsible for the generation and propagation of action potentials (Hille, 2001). This process is made possible thanks to a tight control of ion channels’ maturation, their delivery at the plasma membrane and their functional regulation. While the role of common K+ channel auxiliary subunits (e.g., Kvβs and KChips) is these processes are extensively studied (Maffie and Rudy, 2008; Vacher et al., 2008), the regulatory power of σ1R, emerging as a new but atypical auxiliary subunit (Aydar et al., 2002; reviewed in Kourrich, 2017), is scarcely understood. On that matter, we know today that σ1R regulates neuronal intrinsic excitability via trafficking of VGICs (particularly Kv channels) to the plasma membrane (reviewed in Kourrich, 2017), a function that operate in ligand-dependent and -independent manner. Though σ1R regulates the trafficking of VGICs from the ER to the cell surface, the mechanism remains unclear. Interestingly, σ1R possesses a double-arginine ER retention signal (RR) at the N-terminus (Hayashi and Su, 2003b). Because arginine-based intracellular retention signals are used to regulate assembly and surface transport of several multimeric complexes including ion channels (Scott et al., 2003; Gassmann et al., 2005; Phartiyal et al., 2008), it is tempting to speculate that such a mechanism may also apply to the regulation of VGICs by σ1R.
Modulation of neuronal intrinsic excitability is a critical mechanism that controls a plethora of physiological and cognitive functions. These functions range from the generation and propagation of information within brain circuits to the control of the capability of neurons to undergo synaptic plasticity, and thereby lead to lasting changes in behavior such as learning and memory. Yet, the regulatory power of σ1R remains elusive, partly because σ1R’s regulatory functions have been investigated using mainly heterologous cell culture systems (Aydar et al., 2002; Crottès et al., 2011; Kinoshita et al., 2012; reviewed in Kourrich, 2017). Further, the use of promiscuous ligands when investigating the regulatory functions of σ1R has so far prevented reaching molecular specificity.
σ1R-dependent regulation of VGICs can be mediated through direct protein-protein interactions or indirectly through G proteins. As such, σ1R has the capability to regulate neuronal intrinsic excitability and exert a strong influence on the ability for a neuron to generate action potentials in response to synaptic inputs, fine tune firing frequency, and conduct action potentials along the axon. This section summarizes evidence supporting σ1R regulations of VGICs and its resulting effects on their functions (Table 1 and Figure 1), which appear to be influenced by several factors, including the physiological milieu, brain regions, experimental preparations and neuronal types. However, a critical factor that exerts a strong influence on functional outcomes is σ1R’s conformational and oligomerization states, which appear to be dependent on the class of ligands used (Gromek et al., 2014; Mishra et al., 2015; reviewed in Chu and Ruoho, 2016).
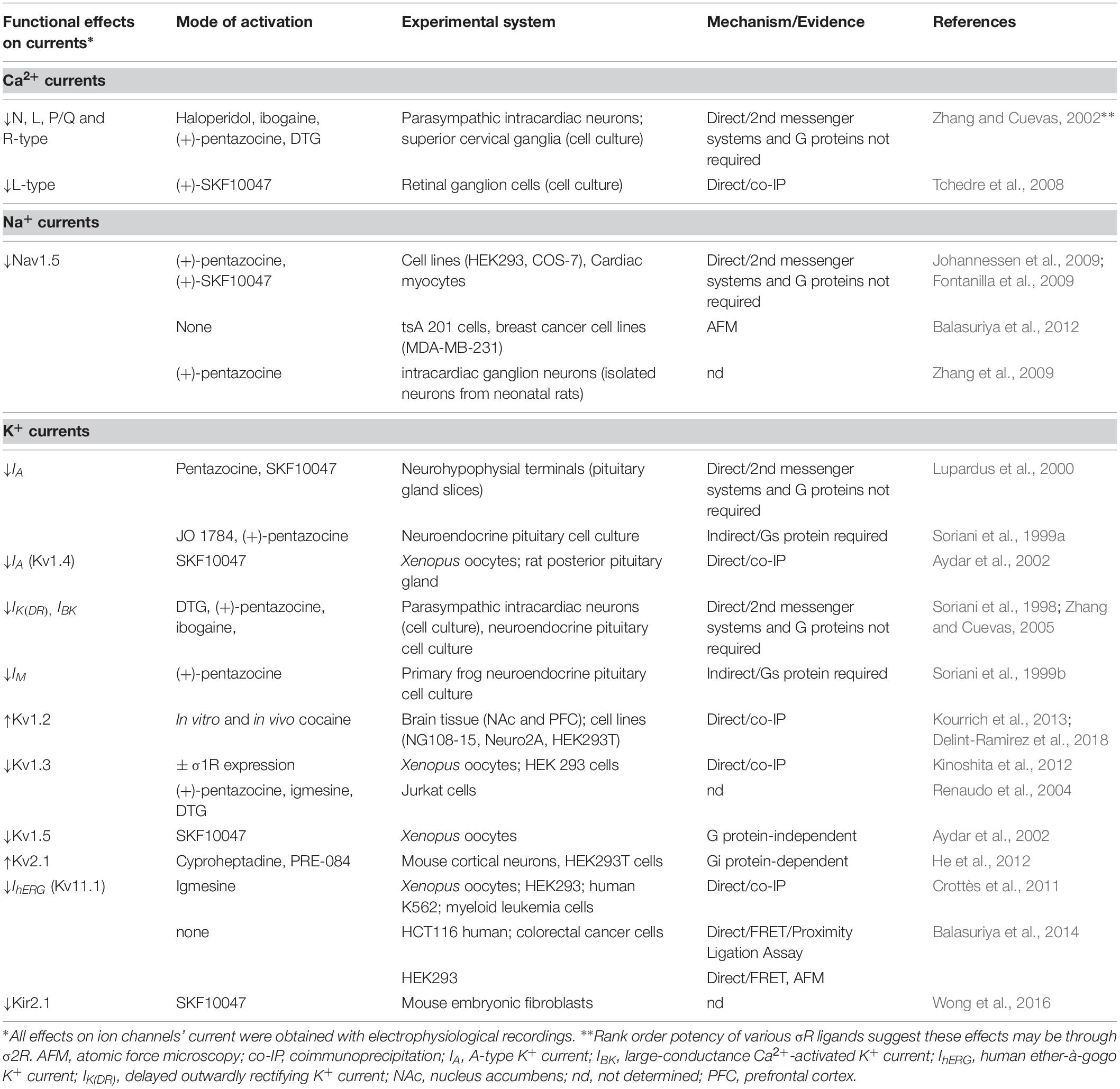
Table 1. Summary of σ1R activation effects on VGICs.
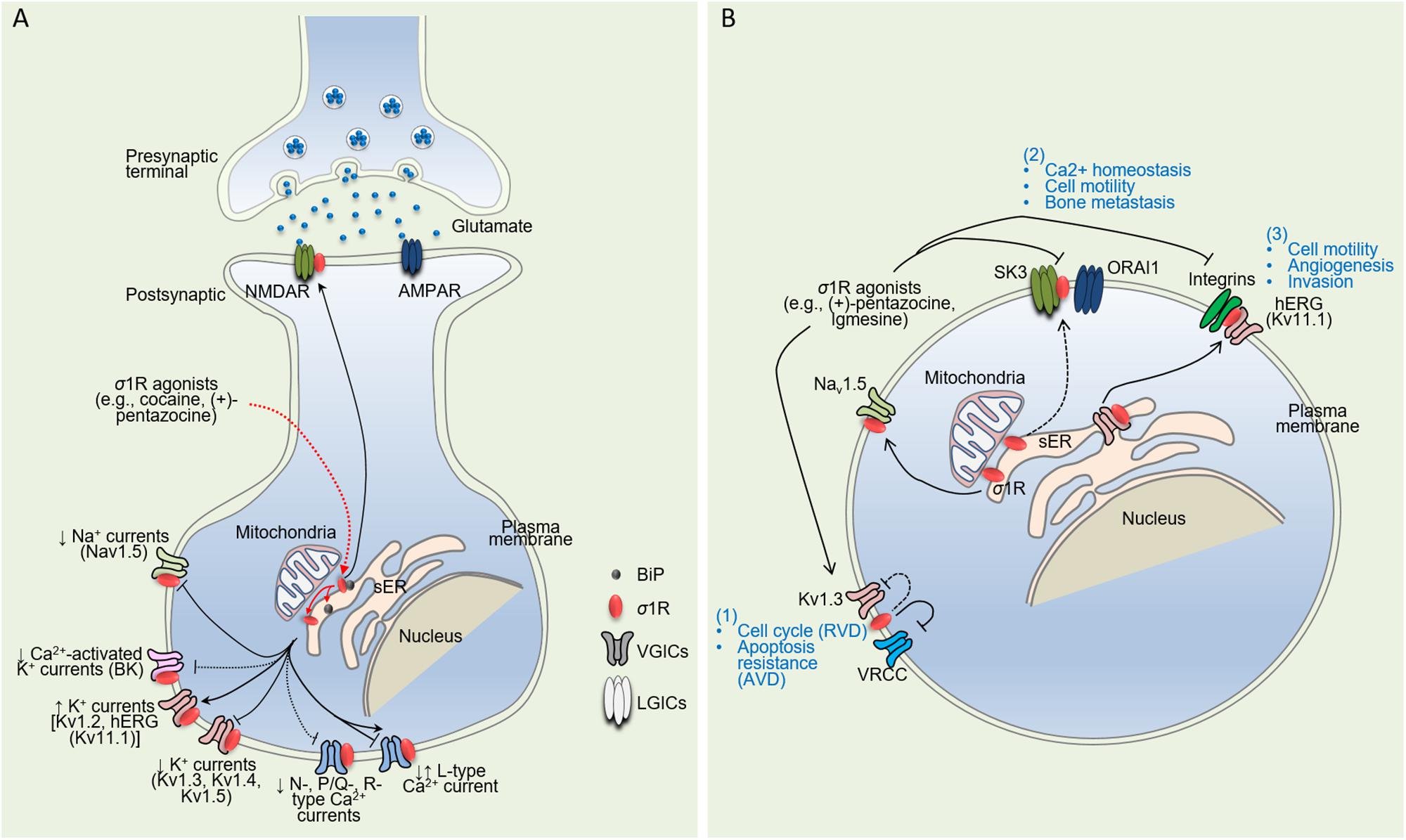
Figure 1. Schematic diagram illustrating direct σ1R-dependent regulation of ion channels in neuronal and cancer cells. (A) Upon ligand stimulation [e.g., cocaine, (+)-pentazocine, PRE-084], σ1R dissociates from binding immunoglobulin protein (BiP), another endoplasmic reticulum (ER) chaperone protein, and then translocates from the mitochondrion-associated ER membrane (MAM, interface between mitochondrion and ER) to the ER and plasmalemma. Acting as an interorganelle signaling modulator, σ1R regulates a variety of functional proteins, both directly and indirectly. Here are represented only the regulations mediated by direct interaction with the targets. Pointed and flathead arrows indicate positive and negative regulations respectively. Unbroken and dashed lines indicate direct and indirect evidence for σ1R:Ion channels physical interactions. On the one hand, σ1R upregulates ion channel expression at the plasma membrane either through the regulation of subunit trafficking activity (hERG) (Crottès et al., 2011) or a mechanism that remains unidentified (Kv1.2) (Kourrich et al., 2013; Delint-Ramirez et al., 2018). σ1R activation by (+)-SKF 10,047 enhances binding with NMDARs, a mechanism that may play a role in NMDAR subunits trafficking to the cell surface (Balasuriya et al., 2013; Pabba et al., 2014). On the other hand, σ1R inhibits ion currents through modulation of target’s biophysical properties (Kv1.3, Kv1.4) (Aydar et al., 2002; Kinoshita et al., 2012) and likely trafficking mechanisms (Nav1.5) (Johannessen et al., 2009; Balasuriya et al., 2012). This can occur through both ligand-independent (Kv1.3, Kv1.4, Kv1.5) (Aydar et al., 2002; Kinoshita et al., 2012) and ligand-dependent mechanisms (Kv1.4, Kv1.5) (Aydar et al., 2002). σ1R can both enhance (Sabeti et al., 2007) and inhibit (Tchedre et al., 2008) L-type Ca2+ currents. Adapted from Kourrich (2017). (B) By shaping cancer cell electrical signature, σ1R participates to cancer hallmarks. (1) σ1R functionally modulates VRCC and K+ channels restricting cell sensitivity to AVD without altering cell cycle progression; (2) σ1R binds SK3 channel and promotes the formation of SK3:ORAI1 complexes within cholesterol-rich nanodomains responsible for increased Ca2+ influx and migration potency; and (3) σ1R dynamically associates hERG channels to integrins upon cell stimulation by ECM triggering motility, angiogenesis and invasiveness.
Ca2+ influx through voltage-gated Ca2+ channels at the synapse play a key role in both fast synaptic transmission, i.e., allows the initiation of a mechanism necessary for neurotransmitter release, and in the regulation of intracellular signaling, thereby initiating slow and lasting changes in neuronal activity (Dolphin, 2009; Catterall, 2010). σ1R modulates intracellular Ca2+ concentration through both the regulation of membrane voltage-gated Ca2+ channels (Table 1 and Figure 1A) and Ca2+ mobilization from endoplasmic stores (Hayashi et al., 2000; Hayashi and Su, 2001; for reviews see Maurice and Su, 2009; Fishback et al., 2010; Su et al., 2010).
Although the subtype of σ receptors, σ1R or σ2R, was not identified yet, early indirect evidence linked σ receptors to modulations of Ca2+ channels’ functions (Rothman et al., 1991; Church and Fletcher, 1995; Brent et al., 1997). These studies, using binding, pharmacological and electrophysiological assays, showed that various σ receptor ligands inhibit intracellular Ca2+ dynamics. Although high concentrations of σ receptors’ ligands used in these studies did not exclude unspecific off-target effects, such as direct actions of ligands on Ca2+ channels rather than on σ receptors, the discovery of highly selective toxins for specific voltage-gated Ca2+ channels provided evidence that σ receptors associate with and regulate voltage-gated Ca2+ channels (Brent et al., 1997). Consistent with these findings, Cuevas and colleagues showed that σ receptor agonists, although the rank order potency of these ligands suggests these effects may be through σ2 rather than σ1 receptors, alters several Ca2+ channels’ biophysical properties on intracardiac ganglionic neurons (N-, L-, P/Q-, and R-type Ca2+ channels) (Table 1 and Figure 1A), all consistent with inhibition of Ca2+ influx. Interestingly, the mechanism through which σ receptors agonists exerted their inhibitory effects were independent on second messenger systems and G proteins (Zhang and Cuevas, 2002), suggesting that the putative σ receptor exerts its inhibitory action through protein-protein interactions with its client ion channel. To date, only the L-type voltage-gated Ca2+ channel has been identified as a direct target for σ1R (Tchedre et al., 2008). In particular, using both specific σ1R receptor ligands and co-immunoprecipitation assays, Tchedre et al. (2008) showed that σ1R activation with (+)-SKF 10,047 inhibits Ca2+ currents—an effect that is prevented by σ1R antagonist BD 1047 and that appears to be mediated via direct σ1R binding to L-type voltage-gated Ca2+ channel (Tchedre et al., 2008) (Table 1 and Figure 1A). In contrast, pregnenolone sulfate activation of σ1R increases L-type Ca2+ currents in the CA1 region of the hippocampus—a mechanism that promotes the induction of synaptic plasticity (i.e., long-term potentiation) (Sabeti et al., 2007). These findings suggest that the effects of σ1R ligands differ between preparations and/or regions of the nervous system. These discrepancies may also be due to the presence and type of ligands, which control σ1R’s conformational and oligomerization states (monomeric vs. oligomeric) (Gromek et al., 2014; Mishra et al., 2015; reviewed in Chu and Ruoho, 2016).
Besides voltage-gated Ca2+channels, σ1R also regulates non-voltage-gated Ca2+-permeable channels via protein-protein interactions, including IP3 receptors at the ER level (Hayashi and Su, 2007; reviewed in Maurice and Su, 2009; Fishback et al., 2010; Su et al., 2010), and plasma membrane acid-sensing ion channels 1a (ASIC1a) (Herrera et al., 2008; Carnally et al., 2010).
To date, and in contrast to bidirectional action of σ1R on Ca2+ currents, σ1R ligands exert inhibitory actions on Na+ currents (Table 1 and Figures 1A,B). Zhang et al. (2009) reported that σ1R ligand activation decreases neuronal intrinsic excitability, an effect mediated by hyperpolarization of Na+ channels’ steady-state inactivation and characterized by a decreased ability to initiate neuronal firing. In contrast, other studies found that σ1R ligand activation in various cell preparations [e.g., mouse cardiac myocytes, COS-7 and human embryonic kidney cells (HEK293)] inhibits Nav1.5-mediated currents without altering channel’s biophysical properties (Fontanilla et al., 2009; Johannessen et al., 2009). Since these effects were mediated in the absence of GTP or ATP, this mechanism is thought to be independent of G-proteins and protein kinases (Johannessen et al., 2009), suggesting direct interaction between σ1R and Nav1.5. Indeed, studies using atomic force microscopy (AFM) imaging provided direct evidence for σ1R-Nav1.5 protein-protein association (Balasuriya et al., 2012). However, since only 6% of the two proteins appears to interact, σ1R may be involved in Nav1.5 trafficking or maturation, a process that requires association-dissociation with Nav1.5. Strikingly, these authors also observed that the molecular silencing of σ1R in a breast cancer line reduced Nav1.5 current density and that (+)-pentazocine reduced interaction between σ1R and Nav1.5 alpha subunits (Balasuriya et al., 2012). Taken together, these results suggest that σ1R increases the number of channels at the plasma membrane, while σ1R ligands exerts inhibitory action through either decreasing Na+ current or rendering Na+ channels unavailable.
Early seminal studies found that the σ1R modulates K+ currents (Bartschat and Blaustein, 1988; Kennedy and Henderson, 1990; Wu et al., 1991). However, results from Morio et al. (1994) were likely the first to suggest that this modulatory ability may involve direct interactions with K+ channels. Specifically, authors showed that σ1R ligand activation inhibits outward K+ conductance in GTP- and second messenger systems-independent manner (e.g., protein kinase A, PKA; and protein kinase C, PKC) (Morio et al., 1994). Later studies conducted in neuronal cells [i.e., rodent neurohypophysial nerve terminals (Lupardus et al., 2000) and parasympathic intracardiac neurons (Zhang and Cuevas, 2005)] provided additional evidence that σ1R agonist activation inhibits various K+ currents independently of ATP- and GTP-dependent processes, including delayed outward rectifier K+ current (IKDR), large conductance Ca2+-sensitive K+ channels (IBK) and M-current (Table 1 and Figure 1A). By contrast, in frog pituitary melanotroph cells (neuroendocrine cells), sigma ligands including Igmesine and (+)-pentazocine, increase action potential firing through a Gs-dependent inhibition of voltage-dependent K+ currents. Indeed, sigma ligands decrease IKDR current density and accelerate IA inactivation, together with a leftward shift in voltage-dependent inactivation curve (Soriani et al., 1998, 1999a). Moreover, (+)-pentazocine was shown to accelerate deactivation rate of the M-current, together with rightward shift in the steady-state activation curve (Soriani et al., 1999b) (Table 1).
To date, several K+ channels have been found to be regulated by direct σ1R binding (Table 1 and Figures 1A,B). Interestingly, this regulatory mechanism can occur through σ1R ligand-dependent or -independent mechanisms. Specifically, studies in Xenopus oocytes showed that in the absence of ligand, σ1R accelerates Kv1.4 inactivation time constant—an effect that is dependent on σ1R:Kv1.4 ratio—and similar to that observed with Kv1.3 (Kinoshita et al., 2012). Instead, application of the σ1R agonist SKF10,047 dramatically reduces outward K+ current of both Kv1.4 and Kv1.5 by ∼75% (Aydar et al., 2002). Using both Xenopus oocytes and neuroendocrine tissue (i.e., posterior pituitary gland), Aydar et al. (2002) found that this modulatory effect is mediated through σ1R-Kv1.4 protein-protein interactions, which is also further supported by co-localization studies in CHO-K1 cells (Mavlyutov and Ruoho, 2007).
Human Ether-a-go-go Related Gene (hERG, also named Kv11.1), another member of the voltage-dependent K+ channel family, also directly interacts with σ1R, as shown by co-immunoprecipitation, fluorescence resonance energy transfer (FRET) and AFM experiments (Crottès et al., 2011; Balasuriya et al., 2014). hERG is mainly expressed in the heart, brain or neuroendocrine tissues where it regulates firing pattern (Vandenberg et al., 2012). hERG is also aberrantly expressed in leukemias and epithelial cancers and has been clearly involved in disease progression (Becchetti et al., 2019). In HEK cells, σ1R increases maturation rate and channel stability at the plasma membrane. Accordingly, σ1R molecular silencing reduces current density in colorectal cancer (CRC) and leukemia cell lines, demonstrating that, in the absence of any ligand, σ1R stimulates hERG function (Crottès et al., 2011, 2013, 2016), a result which is in contrast with those obtained with Kv1.5 channels in Xenopus oocytes where σ1R behaves as a negative modulator (Aydar et al., 2002; Crottès et al., 2013). Noteworthy, cell incubation with the σ1R agonist igmesine (JO1784) produces the same effect as protein silencing on hERG current density. In contrast to Nav1.5/σ1R interaction, hERG/σ1R was resistant to (+)-pentazocine (Balasuriya et al., 2014). Interestingly, σ1R is required for the fast recruitment of hERG by integrins at the plasma membrane following cancer cell activation by the extra cellular matrix (ECM), suggesting that σ1R dynamically regulates ion channel activity depending on external stimulations (Crottès et al., 2016).
While most of previous studies were performed in heterologous systems (Aydar et al., 2002; Kinoshita et al., 2012) or in neuroendocrinal tissue in vitro (Aydar et al., 2002), σ1R-dependent modulation of Kv currents has also been shown to occur in the brain in vivo (Kourrich et al., 2013). Here, authors found that σ1R forms a complex with Kv1.2 channels in the nucleus accumbens (NAc), a critical hub of motivational neural networks, and the prefrontal cortex (PFC), a brain region involved in decisional processes. These protein complexes undergo enduring cocaine-driven upregulation in NAc inhibitory medium spiny projection neurons (MSNs) and thereby lead to persistent decrease in neuronal intrinsic excitability that lasts beyond the detoxification period—a neuroadaptation that contributes to behavioral response to cocaine (Kourrich et al., 2013). Cocaine-induced increase in σ1R-Kv1.2 complexes, a mechanism that promotes Kv1.2 trafficking to the plasma membrane, was also evidence in heterologous expression systems (NG108-15, Neuro2A and HEK293T cell lines), suggesting that the association between Kv1.2 and σ1R is a conserved mechanism (Kourrich et al., 2013; Delint-Ramirez et al., 2018). In sum, these studies demonstrate that the ability for σ1R to bind and regulate Kv functions and surface expression levels can be hijacked by external stimuli and lead to sustained maladaptive changes in cells’ electrical signature and contribute to nervous system disorders. Interestingly, a recent study confirming basal interaction between σ1R and Kv1.2 in HEK293T cells also found that this mechanism influences Kv1.2’s biophysical properties such as bimodal activation gating (Abraham et al., 2019).
Taken together, the σ1R associates with several K+ channels, and these associations can modulate K+ channels functions (Aydar et al., 2002; Kinoshita et al., 2012) or regulate subunits maturation and trafficking to the plasma membrane (Crottès et al., 2011; Kourrich et al., 2013; Balasuriya et al., 2014; Delint-Ramirez et al., 2018).
σ1R not only binds voltage-gated channels. For example, AFM studies revealed that the chaperone also forms aggregates with Asic1a and NMDA receptors (Carnally et al., 2010; Balasuriya et al., 2013) and a recent work indicated a functional coupling with Kir2.1 channels (Wong et al., 2016). Last, in breast cancer cells, σ1R is tightly associated to SK3 (KCNN3) and promotes the functional association of the K+ channel to Orai1 Ca2+ channels. Interestingly, the coupling between the two channels is destroyed either in the absence of σ1R (molecular silencing) or by incubation with the sigma ligand igmesine (Gueguinou et al., 2017).
σ1R is associated with distinct types of chronic diseases such as nervous system disorders, including Alzheimer’s disease, multiple sclerosis, depression, stimulant addiction, and cancer types such as colorectal, lung, prostate, breast cancers and leukemia (Renaudo et al., 2004, 2007; Romieu et al., 2004; Maurice and Su, 2009; Crottès et al., 2013; Kourrich et al., 2013; Aydar et al., 2016; Thomas et al., 2017). Nonetheless, the mechanism through which σ1R operates in such diseases remains elusive. Over the last few years, cancer research has accumulated evidence suggesting that σ1R, particularly via modulation of voltage-gated K+ channels, may contribute to various cellular functions that result in tumor growth (Crottès et al., 2013). Regarding stimulant addiction, another chronic disease, recent discoveries identified novel and unconventional σ1R-driven mechanisms that contribute to the development and likely the maintenance of psychostimulant abuse, some of which implicate enduring changes in neuronal intrinsic excitability (Delint-Ramirez et al., 2018). In addition to cancer and stimulant addiction, these findings extend the putative role for σ1R-driven intrinsic plasticity to other chronic neurological and neuropsychiatric disorders where both changes in intrinsic excitability and σ1R are engaged, e.g., Alzheimer’s disease, multiple sclerosis, and neuropathic pain. For the sake of brevity, this section will focus on stimulant addiction and cancer, two distinct type of chronic diseases whose contributing mechanisms implicate enduring changes in σ1R-driven trafficking of VGICs.
Drugs of abuse, and especially psychostimulant drugs, enhance dopamine (DA) and/or other monoamines (e.g., serotonin, 5-HT; and noradrenaline, NA) signaling in brain reward circuits (Di Chiara and Imperato, 1988), including in the NAc shell and the prefrontal cortex. Overactivation of DA receptors triggers second messengers’ systems, involving protein kinases and phosphatases, leads to enduring changes in neuronal activity. To date, while the effects of psychostimulant drugs on glutamate synaptic strength has been extensively studied (Wolf, 2010, 2016; Luscher and Malenka, 2011; Wolf and Tseng, 2012), little is known about how they alter neuronal firing (Kourrich et al., 2015). While σ1R has long been associated with addiction to psychostimulant drugs [e.g., cocaine and methamphetamine (METH)] and alcohol (Romieu et al., 2004; Maurice and Su, 2009; Katz et al., 2011; Kourrich et al., 2012b; Yasui and Su, 2016), it is unclear whether and how σ1R contributes to these changes. Although several scenario have been proposed, a prominent hypothesis originates from σ1R’s ability to modulate monoaminergic systems, and thereby neuronal intrinsic excitability (Nichols and Nichols, 2008; Andrade, 2011; Ciranna and Catania, 2014). In this section, for the sake of brevity and due to the state of knowledge, we review evidence linking σ1R-driven plasticity of neuronal intrinsic excitability and addictive processes triggered by exposure to psychostimulant drugs; and when possible, we will discuss mechanisms by which monoamine signaling systems may also be involved.
Regarding psychostimulant drugs, although σR subtypes (σ1R vs. σ2R) were not identified yet, early studies from Ujike and colleagues found that σRs antagonists (BMY-14802, rimcazole or SR-31742A) block the development of psychomotor sensitization to cocaine and METH (Ujike et al., 1992a,b,c, 1996). Today, we know that this inhibitory effect of σR antagonists, and especially σ1R, extends to other addiction-relevant behaviors, including conditioned-place preference and drug self-administration, a model that closely mimic human condition (reviewed in Maurice and Su, 2009; Katz et al., 2011, 2016). While the conventional mechanism of action of psychostimulant drugs is to enhance DA (and other monoamines) signaling in the brain, accumulating evidence suggest that in addition to this mechanism, σ1R can exert its pro-addictive functions independently of DA signaling. An intriguing candidate mechanism originate from early studies that demonstrate that psychostimulant drugs such as cocaine and METH can directly activate σ1R in agonist-dependent manner (Sharkey et al., 1988; Kahoun and Ruoho, 1992; Nguyen et al., 2005; Chen et al., 2007; Hayashi and Su, 2007; Hayashi et al., 2011).
Rising evidence suggests that cocaine addiction also involves regional-specific alterations in intrinsic excitability (Nasif et al., 2005; Dong et al., 2006; Ishikawa et al., 2009; Kourrich and Thomas, 2009; for a review, see Kourrich et al., 2015). Specifically, it is well established that cocaine experience alters several VGICs, including Na+, Ca2+, and K+ conductances in NAc neurons (Zhang et al., 1998, 2002; Hu et al., 2004). Alterations of these channels are all consistent with decreased NAc MSNs intrinsic excitability (Dong et al., 2006; Ishikawa et al., 2009; Kourrich and Thomas, 2009; Mu et al., 2010). This adapatation is characterized by a decreased ability of neurons to trigger action potentials, and will be referred to as neuronal hypoactivity. However, evidence from both biophysical and pharmacological approaches found that the slowly inactivating A-type K+ current (also called D-type, ID) (Kourrich and Thomas, 2009; Kourrich et al., 2013) and the small conductance Ca2+-activated K+ current (SK type) (Ishikawa et al., 2009; Mu et al., 2010) play a significant role in the expression of cocaine-induced neuronal hypoactivity. Importantly, this neuroadaptation is observed after weeks of cocaine abstinence from both non-contingent intraperitoneal injection (Ishikawa et al., 2009; Kourrich and Thomas, 2009) and contingent intravenous cocaine self-administration (Segev et al., 2018; Wang et al., 2018).
Whereas enhanced DA signaling is accepted as the canonical mechanism initiating addictive processes, evidence indicates that cocaine also triggers non-canonical, DA-independent mechanism(s) that contribute to addiction (Sora et al., 1998), some of which involve σ1R (Garces-Ramirez et al., 2011; Katz et al., 2011; Hiranita et al., 2013; reviewed in Katz et al., 2011, 2016). These mechanisms are not excluding the critical role of DA in the development of stimulant addiction; however, unconventional DA-independent (or monoamine-independent) are still underestimated by the scientific community. Recent studies demonstrate that cocaine-induced neuronal hypoactivity in NAc shell MSNs is independent of DA, and potentially other monoamine signaling (Delint-Ramirez et al., 2018). In particular, upon both in vivo and in vitro cocaine exposure, σ1R associates with Kv1.2 channels, a channel that drives ID in striatal MSNs (Shen et al., 2004). This mechanism promotes Kv1.2 trafficking from the ER to the plasma membrane, which enhances ID, decreases neuronal firing, and thereby enhances behavioral response to cocaine (Kourrich et al., 2013; Delint-Ramirez et al., 2018) (Figure 1A). Consistent with a DA-independent mechanism, cocaine-induced σ1R-Kv1.2 protein-protein associations and enhanced membrane Kv1.2 also occurs in HEK293T cells that are devoid of DARs. Although other voltage- and Ca2+-gated ion channels are thought to contribute to cocaine-induced neuronal hypoactivity (Zhang et al., 1998, 2002; Hu et al., 2004; Ishikawa et al., 2009) and that σ1R can modulate all classes of VGICs (Figure 1A), it is still unknown whether changes in these channels after exposure to cocaine implicate σ1R-dependent trafficking mechanisms.
While conventional mechanisms of actions for abused substances involve extracellular targets, DA transporters (DAT) (and other monoamine transporters) in the case of psychostimulant drugs, work form Henry Lester’s group provide an alternative hypothesis. Here, drugs of abuse or psychiatric drugs in physiological milieu coexist with their deprotonoted form (membrane permeant). These neutral forms can freely diffuse through the plasma membrane and act directly on intracellular targets. σ1R is enriched in intracellular organelles, especially at the ER level. Binding assays carried out on cell homogenates — a preparation that nevertheless does not preserve plasma membrane integrity — showed that cocaine, at doses found to be rewarding in rodents, binds to σ1R (Kahoun and Ruoho, 1992; Chen et al., 2007). Using a combination of in vitro molecular and electrophysiological approaches in HEK293T cells and NAc shell brain slices, respectively, Delint-Ramirez et al. (2018) demonstrated that cocaine binding to intracellular σ1R initiates the mechanism responsible for MSNs hypoactivity. Together, this suggests that beyond canonical DA-driven mechanisms initiated by extracellular actions of cocaine, cocaine also exerts its addictive properties via intracellular actions triggered by direct binding to σ1R (Garces-Ramirez et al., 2011; Hiranita et al., 2013; reviewed in Katz et al., 2016). This is consistent with the work form Dr. Katz group demonstrating that animals with cocaine experience self-administer σ1R agonists (e.g., PRE-084 and (+)-Pentazocine) (Hiranita et al., 2013) at doses that do not induce DA release in the NAc shell (Garces-Ramirez et al., 2011; reviewed in Katz et al., 2016). This switch in reinforcement does not occur after experience with food reinforcement (reviewed in Katz et al., 2016).
Importantly, METH, a psychostimulant drug that is structurally distinct from cocaine and that exhibits high addiction liability, also depresses NAc shell neuronal excitability (Graves et al., 2015), an effect that may be triggered by METH binding to σ1R (Nguyen et al., 2005). Indeed, structural and crystallography studies found that σ1R ligand-binding cavity is capable of binding structurally different compounds (Su et al., 2010; Schmidt et al., 2016).
In regards to the implication of σ1R in the development of addictive processes, σ1R and DA 1 receptors (D1Rs) can form heteromers. Cocaine binding to this complex amplifies D1R-mediated increases in cyclic AMP (cAMP), a mechanism that appears to occur in mouse striatum (Navarro et al., 2010). Although the complex formed by the D1R and σ1R appears to amplify PKA signaling, the mechanisms by which this adaptation contributes to cocaine addiction has not been identified yet. A putative mechanism may be the activation of PKA and thereby the inhibition of striatal ID current, which is expected to enhance neuronal firing. Indeed, previous studies indicate that inhibiting PKA pathways decreases neuronal firing (Hopf et al., 2003; Perez et al., 2006), therefore excluding this scenario as a putative mechanism for cocaine-induced neuronal hypoactivity in NAc shell MSNs, a mechanism that occurs in D1R- but not in DA 2 receptors (D2R)-containing MSNs (Delint-Ramirez et al., 2018). An important consideration to take account is that although the complex formed by the D1R and the σ1R occurs, the ability for cocaine to activate this protein complex in vivo is yet to be demonstrated. Indeed, authors applied cocaine in vitro at 30–300 μM (Navarro et al., 2010), a concentration that exhibits off-target effects, especially on VGICs (Reith et al., 1986; Zimanyi et al., 1989; Wheeler et al., 1993; O’Leary and Chahine, 2002; Xiao et al., 2004; Chen et al., 2006). Furthermore, this concentration range is significantly beyond cocaine concentration found in the brain upon exposure to standard non-toxic rewarding dose (l–3 μM) (Pettit et al., 1990), and even higher than the total serum concentrations of cocaine (>10 μM) that produce toxicity in humans (Van Dyke et al., 1976; Paly et al., 1982; Escobedo et al., 1991). Nonetheless, assuming that σ1R-D1R complex plays a role in the behavioral effects of cocaine, the signaling pathway that is initiated does not seem to result in changes in intrinsic postsynaptic neuronal excitability. One possible mechanism by which this heteromeric complex may contribute to addictive processes may be by enhancing presynaptic glutamate release in prelimbic cortex (Dong et al., 2007), a region in brain reward circuits that is critical for initiating cocaine seeking and relapse to drug seeking behaviors (McFarland and Kalivas, 2001; Capriles et al., 2003; McFarland et al., 2003, 2004; McLaughlin and See, 2003; for reviews, see Kalivas and O’Brien, 2008; Peters et al., 2009).
An intriguing candidate mechanism that may also implicate DAR-activated kinases in the regulation of neuronal intrinsic excitability is the activation of the α-isoform of calcium/calmodulin-dependent protein kinase II (αCaMKII). Indeed, D1R activation enhances αCaMKII (CaMKII-pThr286) (Anderson et al., 2008), and besides its established role in glutamate AMPAR trafficking to the surface, αCaMKII regulates surface K+ channel density and currents directly, especially A-type K+ currents (Yao and Wu, 2001; Varga et al., 2004; Sergeant et al., 2005). A previous study found that mice that overexpress a constitutively active form of striatal neuron-specific αCaMKII (in which Thr286 is mutated to Asp) exhibit a decreased MSN firing capacity in the NAc shell in the absence of any detectable changes in glutamate transmission (Kourrich et al., 2012a). Interestingly, while σ1R also has the capability to increase αCaMKII-pThr286 (Moriguchi et al., 2011), whether this mechanism is downstream of D1R activation and results in modulation of neuronal firing is yet to be investigated.
From a behavioral viewpoint, neurobiological manipulations that promotes NAc neuronal hypoactivity (e.g., viral enhancement of K+ currents or via overexpression of αCaMKII-pThr286) enhances rewarding properties of cocaine (Kourrich et al., 2012a) and produce a hypersensitivity to cocaine’s psychomotor effects (Dong et al., 2006; Kourrich et al., 2012a)—a mechanism that is thought to contribute to the transition from recreational to compulsive drug use (Ferrario et al., 2005; Robinson and Berridge, 2008). At the circuit level, enhanced behavioral response to cocaine following inhibition of NAc shell neurons is thought to be driven by the disinhibition of downstream reward-related brain regions. In sum, the decreased neuronal intrinsic excitability in the NAc shell is an adaptation sufficient to promote addiction phenotype and is now considered as one of the hallmarks for cocaine addiction (reviewed in Kourrich et al., 2015). This adaptation is also consistent with the predominant tonic inhibition in NAc shell during short-access cocaine self-administration sessions (Peoples et al., 1998, 1999; Peoples and Cavanaugh, 2003) and the role of the NAc hypoactivity in cocaine behavioral effects (Peoples et al., 2007).
Together, accumulating evidence demonstrate that in addition to the established role of DA in initiating and shaping addiction-relevant behaviors, cocaine (and potentially other psychostimulant drugs) engages unidentified mechanisms that also contribute to shaping the addiction phenotype (Kuribara and Uchihashi, 1993; Mattingly et al., 1994; White et al., 1998; Prinssen et al., 2004). Accordingly, a recent review from David J. Nutt and colleagues discusses evidence that enhanced DA release in the brain may not be the sole mediator or initiator for addictive processes (Nutt et al., 2015). This underscores the high clinical significance for identifying novel mechanisms of actions for drugs of abuse, e.g., DA-independent, which would stimulate the development of novel and combinatorial pharmacotherapies to treat stimulant abuse or to provide alternatives for individuals resistant to conventional treatments.
In a larger extent, the discovery that σ1R can form complexes with specific VGICs and that these protein complexes can undergo enduring experience-dependent maladaptive plasticity suggests new mechanistic hypotheses for other neuropsychiatric disorders that are associated with alterations in σ1R and VGICs functions (e.g., Alzheimer’s’ disease and multiple sclerosis).
While the presence of ion channels has been observed for decades in cancer cell lines (Strobl et al., 1995), the understanding of their influence on cancer biology has only emerged recently. Instability in cancer cell genotypes induce several pathological features (hallmarks) including proliferation, resistance to apoptosis, epithelial to mesenchymal transition, tissue invasion and angiogenesis (Hanahan and Weinberg, 2011). Moreover, cancer cells entertain a dynamic bi-directional dialog with their local microenvironment, formed by cancer-activated fibroblasts, immune cell, blood vessels and proteins of the extra-cellular matrix, triggering signaling pathways shaping the development of the disease (Quail and Joyce, 2013). A general scheme has emerged in which ion channels shape an electrical signature that accompanies all steps of disease development, relapse and finally patient survival (Wulff et al., 2009; Pardo and Stuhmer, 2014; Arcangeli and Becchetti, 2017; Rapetti-Mauss et al., 2017; Prevarskaya et al., 2018; Becchetti et al., 2019).
The functional relationships between σ1R and ion channels in the central nervous system (CNS) raises the question of the function of the chaperone in cancers. In the 1990’s, several teams pointed out the presence of high densities of σ1R in cancer cell lines (i.e., glioblastoma, neuroblastoma, leukemia, breast cancer, melanoma, prostate or lung cancer cell lines) and suggested that it could be involved in cancer cell biology since sigma ligands (e.g., BD737) decreased cell proliferation or induced cell death in vitro (Vilner et al., 1995a, b). Despite the large number of publications dealing with σ1R and cancer, most of the knowledge until recently came from descriptive pharmacological studies with few answers regarding the innate function of the protein in disease progression as well as the molecular mechanisms involved. However, the evolution of the concept of σ1R from a classical receptor to a ligand-regulated chaperone protein has been accompanied by new information on its functions in cancer cell biology.
Spruce and collaborators showed that σ1R expression in breast and prostate cancer cells induced a break on Caspase-3-dependent apoptosis that could be attenuated by the σ1R antagonist rimcazole, in turn reducing tumor growth and metastatic proliferation in mice xenograft models. Accordingly, the effects of rimcazole could be attenuated by the σ1R agonists SKF10.047 and (+)-pentazocine (Spruce et al., 2004). In Jurkat T cell lines, it was observed that σ1R expression increased cell resistance to thapsigargin-induce apoptotic volume decrease (AVD), an early step in cell death process (Renaudo et al., 2007). In Jurkat and small cell lung cancer cells (NCI-H209), sigma ligands [igmesine and (+)-pentazocine] blocked cell cycle by inhibiting regulatory volume decrease (RVD), a mechanism related to AVD and that is required for G1/S transition (Renaudo et al., 2004). Overexpression of σ1R when compared to healthy tissues is also observed in colorectal (CRC) and myeloid leukemia (ML). In these cancers, σ1R promotes integrin-dependent invasive process in vitro and in vivo. In breast cancer patients, σ1R expression correlates with reduced overall survival; and at the cellular level, σ1R potentiates Ca2+-dependent migration, a mechanism also found in CRC cells (Crottès et al., 2016; Gueguinou et al., 2017). In prostate cancer, a recent study revealed that σ1R expression contributes to cell resistance to ER stress, and to the development of treatment-induced castration-resistant prostate cancer by promoting androgen receptor resurgence (Thomas et al., 2017).
Altogether, these results suggest that σ1R plays a significant role in cancer cell by regulating ion channel-related mechanisms (e.g., AVD, RVD, integrin signaling, Ca2+ homeostasis) (Aydar et al., 2016; Crottès et al., 2016; Guéguinou et al., 2016; Soriani and Rapetti-Mauss, 2017; Rapetti-Mauss et al., 2018).
Cancer cell growth is the result of a balance between cell division and cell apoptosis. Early works by Bowen and coworkers back in the 1990s revealed that incubation of cancer cell lines with sigma ligands such as haloperidol and BD737 impaired cell growth in vitro. Interestingly, cell growth arrest was accompanied by a cellular swelling (Vilner et al., 1995a). Further works revealed that sigma ligands such as (+)-pentazocine or igmesine induced a cell swelling in vitro by inhibiting Kv and volume regulated chloride channels (VRAC). In fact, these two channels are the main effectors of the RVD process, allowing, in response to hypo-osmotic challenge, cell volume recovery by allowing potassium chloride (KCl) and osmotic water efflux (through activation of K+ and chloride channels). RVD is required for cells to pursue their cell cycle through the G1/S phase transition (Rouzaire-Dubois and Dubois, 1998; Rouzaire-Dubois et al., 2000). Accordingly, in SCLC and leukemia cell lines, inhibition of Kv and VRAC channels by σ1R ligands provoked accumulation of the p27kip1, and a decrease in cyclin A contents, reflecting an arrest of cell cycle at the end of the G1 phase (Renaudo et al., 2004, 2007). Cell volume regulation is also an important feature in the context of cell death. Indeed, cell shrinking by water efflux following activation of VRAC and K+ channels, is an early event of apoptosis signaling (Maeno et al., 2000; Lambert et al., 2008). In the studies performed by Renaudo and colleagues (2004, 2007), several clues indicated that σ1R was also involved in cell response to apoptosis. In leukemia T cells, the sigma ligand igmesine reduced VRAC current density and delayed staurosporine-induced AVD. Experiments in HEK293 cells showed that σ1R overexpression decreased volume-regulated chloride channels (VRCC) activation rate and delayed stausporine-induced AVD.
Taken together, these data suggest that σ1R expression in cancer provides cells better resistance capacity to apoptotic signals. Indeed, the tonic brake exerted by σ1R on VRAC channel activation rate restricts AVD without impeding RVD, the latter being necessary for cell cycle progression (Renaudo et al., 2007). This idea, in which σ1R exerts membrane-channel dependent pro-survival function seems to mirror the model proposed by Hayashi and Su in which σ1R, by chaperoning IP3R at the mitochondrion-associated ER membrane (MAM), stimulates CHO cell resistance to ER stress-induced apoptosis (Hayashi and Su, 2007).
In cancer, mortality is mainly the consequence of metastasis spreading, a complex process requiring the capacity of cells to migrate from the primary site and disseminate. Among the cellular processes involved in cell migration, Ca2+ homeostasis is crucial. Indeed, a deep Ca2+ influx remodeling occurs during the development of the disease and recent findings have revealed the formation of gain-of-function ion channel platforms at the plasma membrane as key events for metastasis progression (Guéguinou et al., 2014; Monteith et al., 2017). In breast and colorectal cancers (BC and CRC), σ1R is required to trigger the physical and functional coupling between the Ca2+ channel Orai1 and the Ca2+-dependent K+ channel SK3 (KCNN3). The group of Vandier has shown that the coupling of these two channels within cholesterol-rich membrane nanodomains (labeled by the presence of Caveolin1) potentiated constitutive (in BC) or capacitive (in CRC) Ca2+ influx, which in turn stimulated cell migration and bone metastasis formation in BC (Chantôme et al., 2013; Guéguinou et al., 2016). Co-immunoprecipitation and FRET assays demonstrated that σ1R binds SK3. Furthermore, σ1R silencing abrogated SK3-dependent Ca2+ influx and migration by chasing both SK3 and Orai1 out of caveolae lipid nanodomains. Interestingly, the sigma ligand igmesine mimicked these effects on Ca2+ influx and migration by dissociating Orai1 from SK3, the former being excluded from lipid caveolae nanodomains in MDA MB 435s BC cells. Noteworthy, σ1R is overexpressed in CRC and BC human samples and is associated with higher-grade tumors in CRC and reduced overall survival in BC patients (Gueguinou et al., 2017).
Voltage-gated sodium channels (VGSC) have been extensively characterized for their electrogenic functions in cell excitability (action potential firing). During the last decade, an increasing number of studies have described the anomalous expression of VGSC in epithelial cancers where they are associated to motility or invasiveness, therefore increasing the risk of metastasis development. In particular, the neonatal form of Nav1.5 α-subunit (SCN5A) has been observed in breast cancers where it generates a tonic inward Na+ current (in other words a window current) (Roger et al., 2006). It was shown that Na+ entry through VGSC leads to the formation and activity of invadopodia with the polymerization of actin and increase in Na+-H+ exchanger type 1 (NHE1) activity. This mechanism contributes to the acidification of the extracellular surface of the plasma membrane, making a favorable milieu for the activity of acidic cysteine cathepsins (reviewed in Brisson et al., 2013). As previously mentioned, σ1R binds Nav1.5 with a fourfold symmetry (one σ1R per set of six transmembrane domains). At a more functional level, σ1R silencing in the metastatic breast cancer cell line MDA-MD-231 led to a significant decrease in Nav1.5 current density, suggesting that the presence of the chaperone in BC cells potentiates their invasive potency (Balasuriya et al., 2012). Interestingly, further studies showed in the same cell line that the functional coupling between σ1R and Nav1.5 was also involved in cell adhesion properties, but not in cell proliferation or migration (Aydar et al., 2016).
Integrin-mediated cell adhesion has a pivotal role on cell fate determination and is intimately associated with ion transport. Ion channels and integrins form signaling hubs that recruits membrane receptors involved in proliferation, migration, differentiation, invasion or angiogenesis. A growing number of studies indicates that ion channels regulate signaling pathways downstream integrin activation by the ECM. Remarkably, their association with integrins determines their open probability, which in turn influences downstream pathways (reviewed in Becchetti et al., 2019). hERG K+ channel has been pointed out as a partner of several integrins and is prognostic a marker in several solid tumors and leukemias. As stated above, σ1R binds hERG α-subunits and enhances hERG trafficking to the plasma membrane in K562 leukemia cells or transfected HEK293 cells, leading to increased current density (Crottès et al., 2013; Balasuriya et al., 2014). In myeloid leukemia and CRC, the rapid association of hERG with the β1 subunit of integrin upon ECM stimulation requires σ1R. The silencing of the chaperone abolishes both ECM-induced stimulation of hERG and PI3/AKT pathway downstream of integrin stimulation. Consequently, σ1R inhibition reduces migration, angiogenesis and metastasis spreading in vitro and in vivo using zebra fish and mice models (Balasuriya et al., 2014; Crottès et al., 2016). These data demonstrate that σ1R dynamically shapes cancer cell electrical signature in response to the tumor microenvironment. The role of σ1R in electrical plasticity within the cancer tissue obviously contributes to the signaling underlying the phenotypic adaptation of cancer cells to a highly stringent environment.
Altogether, these results suggest that the innate pro-adaptive function of σ1R is hijacked to the benefit of tumor development.
Due to their critical role in regulating trafficking and functions of VGICs, knocking-out classical ion channels’ auxiliary subunits alters a variety of cellular functions (Giese et al., 1998; Pongs et al., 1999; Vacher et al., 2008; Sun et al., 2011; Martinez-Espinosa et al., 2014), which can lead to various chronic diseases such as long QT syndrome (Schulze-Bahr et al., 1997; Splawski et al., 1997), epilepsy (Heilstedt et al., 2001) and even premature death (Arikkath and Campbell, 2003). In contrast, σ1R KO mice are viable and do not exhibit clear behavioral and physiological phenotypes (Langa et al., 2003), raising the hypothesis that σ1R becomes critical only when the system is challenged.
Indeed, σ1R KO mice exhibit behavioral deficits when confronted to novel situations that require behavioral adaptations, such as during learning and memory tasks (Entrena et al., 2009; Chevallier et al., 2011) and during stressful and anxiogenic situations that require coping mechanisms (Sabino et al., 2009; Chevallier et al., 2011). Similarly, in the context of neuronal electrical activity, preventing σ1R activation with various antagonists both in freely moving animals or in vitro does not alter Na+ currents (Zhang et al., 2009), Ca2+ dynamics (Tchedre et al., 2008; Cuevas et al., 2011; Pan et al., 2014) and K+ currents (Kourrich et al., 2013). In fact, these findings suggest that σ1R’s functions are not necessary in situations that are not threatening the biological integrity of the individual. This idea is further supported by studies demonstrating that σ1R ligands activation exhibit robust neuroprotective properties, such as reducing infarct volume after embolic stroke (Allahtavakoli and Jarrott, 2011), preventing apoptotic retinal ganglion cell death induced by glutamate and excitotoxic perinatal brain injury (Griesmaier et al., 2012). σ1R ligands also rescue various behavioral and biological deficits associated with amnesia, depression, neuropathic pain, cocaine-induced immune alteration and HIV expression, and Alzheimer’s disease (Maurice and Su, 2009). In stark contrast with these protective properties of σ1R activation, ligand activation of σ1R contributes to tumor growth (Crottès et al., 2013) and the development of psychostimulant addiction (Katz et al., 2011, 2016). Based on these findings, we speculate that primary σ1R’s functions, which are to preserve cellular health and survival, are hijacked to contribute to maladaptive cellular functions, thereby leading to chronic diseases such as cancer and stimulant addiction.
Besides their rewarding properties, cocaine and other psychostimulants drugs trigger neurotoxic mechanisms (Pereira et al., 2015). This toxicity originates from various drugs’ mechanisms of actions, such as supraphysiological elevation of extracellular DA. While enhanced DA signaling contributes to the rewarding properties of psychostimulant drugs, it also induces oxidative stress and associated neuronal apoptosis (Pereira et al., 2015). Indeed, increase of oxidative stress in brain regions associated with the brain reward circuits (NAc, frontal cortex, and hippocampus) (Dietrich et al., 2005; Muriach et al., 2010; Jang et al., 2015) upregulates pro-inflammatory mediators (e.g., cytokines and chemokines) or astroglia/microglia activation (Renthal et al., 2009; Piechota et al., 2010; Blanco-Calvo et al., 2014; Lopez-Pedrajas et al., 2015; see review Pereira et al., 2015). Interestingly, a physiologically relevant concentration of DA that does not cause apoptosis becomes toxic in σ1R knockdown cells (Mori et al., 2012), consistent with neuroprotective and other associated positive effects of σ1R ligands activation on various chronic neurodegenerative diseases such as Alzheimer’s (Ryskamp et al., 2019) and Huntington’s diseases (Bol’shakova et al., 2017; Ryskamp et al., 2017; see review Cai et al., 2017). Indeed, σ1R agonist PRE-084 reduces oxidative species, calcium flux and other inflammatory molecules [including interleukin (IL) IL-1β, IL-6, IL-8 and tumor necrosis factor alpha (TNFα)] in various cell types (Katnik et al., 2006; Szabo et al., 2014). As such, we speculate that identified cocaine-driven σ1R mechanism associated with the development of cocaine addiction may be initially a neuroprotective mechanism aimed to counteract deleterious effect of psychostimulant drugs of abuse on neuronal health. Here, we present a hypothesis that may provide clues onto the dual effect of σ1R activation, aimed primarily to be pro-survival but could be hijacked to promote the development of addiction to psychostimulant drugs.
Today, we know that repeated cocaine exposure leads to several long-lasting neuroadaptations in the brain reward circuits (Luscher and Malenka, 2011; Wolf and Tseng, 2012; Kourrich et al., 2015; Wolf, 2016). The mechanisms through which these neuroadaptations are initiated and their contributions to shaping lasting changes in behavior are active fields of research. A recent study provided direct evidence that persistent cocaine-induced neuronal hypoactivity in the NAc shell, now considered as one of the hallmarks for cocaine addiction, is initiated by σ1R activation (Delint-Ramirez et al., 2018). This adapatation of the NAc shell neurons is characterized by a decreased ability for neurons to trigger action potentials and is associated with enhanced hyperpolarizing and decreased depolarizing membrane ion channels. It is tempting to speculate that the primary function of this early σ1R-driven mechanism is to protect the neuron from cocaine-induced cytotoxic mechanisms, a scenario supported by indirect evidence from two lines of studies. Specifically, cocaine administration induces an early increase of membrane NMDA glutamate receptors (NMDARs) in the NAc shell, especially receptors that contain the GluN2B subunit. These GluN2B-containing NMDARs appear to contribute to the development of further lasting changes in glutamate neurotransmission in the reward circuits and thereby may participate to the development of cocaine addiction (Dong and Nestler, 2014). NMDARs are highly permeable to Ca2+ and inclusion of GluN2B subunits further prolongs Ca2+ entry (Monyer et al., 1994; Vicini et al., 1998; Rumbaugh and Vicini, 1999; Schilstrom et al., 2006). NMDAR-induced Ca2+ neurotoxicity is one of the primary factor that leads to cell damage (see review Pchitskaya et al., 2018). A recent study showed that D-cycloserine (DCS), an agonist of NMDARs, leads to decreased neuronal excitability, and disrupting GluN2B prevents both this mechanism and cocaine-induced neuronal hypoactivity in the NAc shell (Wang et al., 2018). Another study found that this same adaptation, i.e., cocaine-induced neuronal hypoactivity, is initiated by direct cocaine binding to σ1R (Delint-Ramirez et al., 2018).
Altogether, cocaine-induced neuronal hypoactivity in the NAc shell may originally be an adaptive mechanism aimed to counteract Ca2+-induced neurotoxicity. However, at the circuit level, decreased intrinsic excitability of MSNs, which are inhibitory projection neurons, is expected to disinhibit downstream reward-related brain regions and thereby promote addiction-relevant behaviors (reviewed in Kourrich et al., 2015). Consistent with this framework, Khoshbouei and colleagues found that σ1R activation decreases METH-stimulated DA efflux and prevents METH-induced, DAT-mediated increases in firing activity of DA neurons, which together may also play a role in limiting DA-induced neurotoxicity (Sambo et al., 2017). It is noteworthy to mention that METH is a psychostimulant drug that exhibits strong neurotoxic effects. Importantly, in both cocaine and METH studies, σ1R activation dampens neuronal firing. However, at the behavioral level, resulting effects of σ1R-driven inhibition of neuronal firing in the NAc shell MSNs or ventral tegmental area (VTA) DA neurons, which may be a mechanism limiting activity-dependent neurotoxicity, differentially affect rewarding properties of the drug. While this mechanism supports the role of σ1R in neuroprotection, its effects on the development or maintenance of stimulant abuse likely depends on its brain site of action. Taken together, this theory warrants further studies to elucidate brain region-specific mechanisms by which σ1R, likely depending on client proteins available, contributes to the development of stimulant abuse. This idea is further supported by a recent study demonstrating that cocaine administration induces a cascade of cellular mechanisms in the NAc that aim to counteract each other in homeostatic fashion (Wang et al., 2018), which ultimately heightens cocaine seeking after drug abstinence.
During tumor development, cancer cells are facing intrinsic (oncogene activation) and extrinsic (limiting nutrient or oxygen supply, inflammation) challenges, with which they must cope to survive. Moreover, imbalance between protein folding demand and capacity in the ER leads to a situation of ER stress that is often observed in highly proliferative tumor cells (reviewed in Papaioannou et al., 2018; Obacz et al., 2019). These challenges share a common point with those faced by neurons in many neurodegenerative diseases or stroke for which activation of σ1R revealed beneficial effects (Chu and Ruoho, 2016; Weng et al., 2017; Penke et al., 2018; Smith et al., 2018). The review of studies exploring the function of σ1R in cancers indicates that the protein is generally associated with enhanced invasive properties of cancer cells and a poorer diagnosis at the patient level. In fact, depending on the type of cancer considered, σ1R participates to many hallmark of cancers as defined by Hanahan and Weinberg (2011), including apoptosis resistance, migration, invasive potency, angiogenesis and cell response to the microenvironment. The contribution of σ1R to these hallmarks can be synthesized as follows:
σ1R expression lowers cell sensitivity to apoptosis by slowing activation of VRAC channels involved in AVD, but no drastically enough to impede RVD, the latter being required for cell cycle progression (Renaudo et al., 2004, 2007) (Figure 1B).
Enhanced migration in BC and CRC cancers is the consequence of a σ1R-dependent formation of the special coupling between a Ca2+-dependent channel (SK3) and a Ca2+ channel (Orai1) responsible for increased constitutive or capacitive Ca2+ entry, which finally triggers cell motility (Guéguinou et al., 2016; Gueguinou et al., 2017). In BC cells, σ1R expression increases Nav1.5 current density, probably through a direct association between the two proteins (Balasuriya et al., 2012). Since a Nav1.5-associated Na+ window current stimulates NHE1 activity and subsequent extracellular acidification, σ1R likely potentiates cell invasiveness potency through increased Nav1.5 activity (Roger et al., 2003, 2004, 2015; Brisson et al., 2011, 2013) (Figure 1B).
σ1R also deeply influences the dialog between tumor cells and the microenvironment by regulating ion channels. By shaping cancer cell electrical signature in response to ECM, σ1R orchestrates the formation of [channel:receptor] complexes at the plasma membrane, contributing to the integration of signals from the tumor microenvironment and the subsequent adaptive phenotype. Indeed, σ1R is a key actor of the formation of [integrin:hERG] signaling hub at the plasma membrane, triggering AKT-dependent pro-metastatic cell behavior in vivo by stimulating migration, vascular endothelial vascular factor (VEGF) secretion, finally leading to migration, extravasation and angiogenesis in chronic myeloid leukemia and CRC (Crottès et al., 2016; Becchetti et al., 2019) (Figure 1B).
Altogether, these results support the idea that the innate function of σ1R, aimed at a better cell survival in physiopathological conditions (Hayashi and Su, 2007; Chu and Ruoho, 2016), is hijacked to the benefit of tumor cell growth and invasive properties. It is therefore tempting to postulate that within cancer tissues, σ1R shapes tumor cell electrical signature (Figure 1B), thereby enhancing their adaptation potency to their environment.
The comprehension of the physiological significance of σ1R as well as the cellular and molecular mechanisms associated with this protein has deeply progressed during the past 10 years. The data accumulated describing the functional interactions between ion channels and σ1R have refined the picture of the contribution of σ1R to CNS diseases including stimulant addiction. The recent data described above demonstrate that contribution to hallmarks of cancer cells and stimulant addiction represent two groups of pathological contexts where [σ1R:ion channel] complexes may play central roles. One of the most striking finding supporting this idea is that a common mechanism, i.e., the chaperoning of voltage-gated K+ channels, is involved in both neuronal response to cocaine and cancer cell response to tumor microenvironment. In a unifying view, it is tempting to speculate that both cocaine exposure (Figure 1A) and oncogenic processes (Figure 1B) activate the protective function of σ1R that in turn contributes to a shift in cell homeostasis leading to deleterious behaviors.
OS and SK contributed to all sections in the review. OS and SK were entirely responsible for the sections related to cancer and drug addiction, respectively.
OS is supported by the Fondation ARC (N°PJA20161204740 and PJA20181207701), Fondation de France (FdF Eotp 535412), and Cancéropôle PACA. SK is supported by a grant from the National Institutes of Health (National Institute on Drug Abuse R01DA041390), the Université du Quebec à Montreal (UQAM), and by the Natural Sciences and Engineering Research Council of Canada (NSERC) (funding reference number RGPIN-2019-06666). SK is a Fonds de la Recherche du Québec - Santé (FRQS) Senior Research Scholar.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abraham, M. J., Fleming, K. L., Raymond, S., Wong, A. Y. C., and Bergeron, R. (2019). The sigma-1 receptor behaves as an atypical auxiliary subunit to modulate the functional characteristics of Kv1.2 channels expressed in HEK293 cells. Physiol. Rep. 7:e14147. doi: 10.14814/phy2.14147
Allahtavakoli, M., and Jarrott, B. (2011). Sigma-1 receptor ligand PRE-084 reduced infarct volume, neurological deficits, pro-inflammatory cytokines and enhanced anti-inflammatory cytokines after embolic stroke in rats. Brain Res. Bull. 85, 219–224. doi: 10.1016/j.brainresbull.2011.03.019
Anderson, S. M., Famous, K. R., Sadri-Vakili, G., Kumaresan, V., Schmidt, H. D., Bass, C. E., et al. (2008). CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat. Neurosci. 11, 344–353. doi: 10.1038/nn2054
Andrade, R. (2011). Serotonergic regulation of neuronal excitability in the prefrontal cortex. Neuropharmacology 61, 382–386. doi: 10.1016/j.neuropharm.2011.01.015
Arcangeli, A., and Becchetti, A. (2017). hERG channels: from antitargets to novel targets for cancer therapy. Clin. Cancer Res. 23, 3–5. doi: 10.1158/1078-0432.CCR-16-2322
Arikkath, J., and Campbell, K. P. (2003). Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr. Opin. Neurobiol. 13, 298–307.
Aydar, E., Palmer, C. P., Klyachko, V. A., and Jackson, M. B. (2002). The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 34, 399–410.
Aydar, E., Stratton, D., Fraser, S. P., Djamgoz, M. B., and Palmer, C. (2016). Sigma-1 receptors modulate neonatal na. Eur. Biophys. J. 45, 671–683.
Balasuriya, D., D’Sa, L., Talker, R., Dupuis, E., Maurin, F., Martin, P., et al. (2014). A direct interaction between the sigma-1 receptor and the hERG voltage-gated K+ channel revealed by atomic force microscopy and homogeneous time-resolved fluorescence (HTRF(R)). J. Biol. Chem. 289, 32353–32363. doi: 10.1074/jbc.M114.603506
Balasuriya, D., Stewart, A. P., Crottes, D., Borgese, F., Soriani, O., and Edwardson, J. M. (2012). The sigma-1 receptor binds to the Nav1.5 voltage-gated Na+ channel with 4-fold symmetry. J. Biol. Chem. 287, 37021–37029. doi: 10.1074/jbc.M112.382077
Balasuriya, D., Stewart, A. P., and Edwardson, J. M. (2013). The sigma-1 receptor interacts directly with GluN1 but not GluN2A in the GluN1/GluN2A NMDA receptor. J. Neurosci. 33, 18219–18224. doi: 10.1523/JNEUROSCI.3360-13.2013
Bartschat, D. K., and Blaustein, M. P. (1988). Psychotomimetic sigma-ligands, dexoxadrol and phencyclidine block the same presynaptic potassium channel in rat brain. J. Physiol. 403, 341–353.
Becchetti, A., Petroni, G., and Arcangeli, A. (2019). Ion channel conformations regulate integrin-dependent signaling. Trends Cell Biol. 29, 298–307. doi: 10.1016/j.tcb.2018.12.005
Blanco-Calvo, E., Rivera, P., Arrabal, S., Vargas, A., Pavon, F. J., Serrano, A., et al. (2014). Pharmacological blockade of either cannabinoid CB1 or CB2 receptors prevents both cocaine-induced conditioned locomotion and cocaine-induced reduction of cell proliferation in the hippocampus of adult male rat. Front. Integrat. Neurosci. 7:106. doi: 10.3389/fnint.2013.00106
Bol’shakova, A. V., Kraskovskaya, N. A., Gainullina, A. N., Kukanova, E. O., Vlasova, O. L., and Bezprozvanny, I. B. (2017). Neuroprotective effect of sigma1-receptors on the cell model of huntington’s disease. Bull. Exp. Biol. Med. 164, 252–258. doi: 10.1007/s10517-017-3968-7
Brent, P. J., Herd, L., Saunders, H., Sim, A. T., and Dunkley, P. R. (1997). Protein phosphorylation and calcium uptake into rat forebrain synaptosomes: modulation by the sigma ligand, 1,3-ditolylguanidine. J. Neurochem. 68, 2201–2211.
Brisson, L., Driffort, V., Benoist, L., Poet, M., Counillon, L., Antelmi, E., et al. (2013). NaV1.5 Na+2 channels allosterically regulate the NHE-1 exchanger and promote the activity of breast cancer cell invadopodia. J. Cell. Sci. 126, 4835–4842.
Brisson, L., Gillet, L., Calaghan, S., Besson, P., Le Guennec, J. Y., Roger, S., et al. (2011). Na(V)1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H(+) efflux in caveolae. Oncogene 30, 2070–2076. doi: 10.1038/onc.2010.574
Cai, Y., Yang, L., Niu, F., Liao, K., and Buch, S. (2017). Role of sigma-1 receptor in cocaine abuse and neurodegenerative disease. Adv. Exp. Med. Biol. 964, 163–175. doi: 10.1007/978-3-319-50174-1_12
Calhoun, J. D., and Isom, L. L. (2014). The role of non-pore-forming beta subunits in physiology and pathophysiology of voltage-gated sodium channels. Handb. Exp. Pharmacol. 221, 51–89. doi: 10.1007/978-3-642-41588-3_4
Capriles, N., Rodaros, D., Sorge, R. E., and Stewart, J. (2003). A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology 168, 66–74.
Carnally, S. M., Johannessen, M., Henderson, R. M., Jackson, M. B., and Edwardson, J. M. (2010). Demonstration of a direct interaction between sigma-1 receptors and acid-sensing ion channels. Biophys. J. 98, 1182–1191. doi: 10.1016/j.bpj.2009.12.4293
Catterall, W. A. (2010). Signaling complexes of voltage-gated sodium and calcium channels. Neurosci. Lett. 486, 107–116. doi: 10.1016/j.neulet.2010.08.085
Chantôme, A., Potier-Cartereau, M., Clarysse, L., Fromont, G., Marionneau-Lambot, S., Guéguinou, M., et al. (2013). Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 73, 4852–4861. doi: 10.1158/0008-5472.CAN-12-4572
Chen, Y., Hajipour, A. R., Sievert, M. K., Arbabian, M., and Ruoho, A. E. (2007). Characterization of the cocaine binding site on the sigma-1 receptor. Biochemistry 46, 3532–3542.
Chen, Y. H., Lin, C. H., Lin, P. L., and Tsai, M. C. (2006). Cocaine elicits action potential bursts in a central snail neuron: the role of delayed rectifying K+ current. Neuroscience 138, 257–280.
Chevallier, N., Keller, E., and Maurice, T. (2011). Behavioural phenotyping of knockout mice for the sigma-1 (sigma(1)) chaperone protein revealed gender-related anxiety, depressive-like and memory alterations. J. Psychopharmacol. 25, 960–975. doi: 10.1177/0269881111400648
Chu, U. B., and Ruoho, A. E. (2016). Biochemical pharmacology of the sigma-1 receptor. Mol. Pharmacol. 89, 142–153. doi: 10.1124/mol.115.101170
Church, J., and Fletcher, E. J. (1995). Blockade by sigma site ligands of high voltage-activated Ca2+ channels in rat and mouse cultured hippocampal pyramidal neurones. Br. J. Pharmacol. 116, 2801–2810.
Ciranna, L., and Catania, M. V. (2014). 5-HT7 receptors as modulators of neuronal excitability, synaptic transmission and plasticity: physiological role and possible implications in autism spectrum disorders. Front. Cell. Neurosci. 8:250. doi: 10.3389/fncel.2014.00250
Crottès, D., Guizouarn, H., Martin, P., Borgese, F., and Soriani, O. (2013). The sigma-1 receptor: a regulator of cancer cell electrical plasticity? Front. Physiol. 4:175. doi: 10.3389/fphys.2013.00175
Crottès, D., Martial, S., Rapetti-Mauss, R., Pisani, D. F., Loriol, C., Pellissier, B., et al. (2011). Sig1R protein regulates hERG channel expression through a post-translational mechanism in leukemic cells. J. Biol. Chem. 286, 27947–27958. doi: 10.1074/jbc.M111.226738
Crottès, D., Rapetti-Mauss, R., Alcaraz-Perez, F., Tichet, M., Gariano, G., Martial, S., et al. (2016). SIGMAR1 regulates membrane electrical activity in response to extracellular matrix stimulation to drive cancer cell invasiveness. Cancer Res. 76, 607–618. doi: 10.1158/0008-5472.CAN-15-1465
Cuevas, J., Behensky, A., Deng, W., and Katnik, C. (2011). Afobazole modulates neuronal response to ischemia and acidosis via activation of sigma-1 receptors. J. Pharmacol. Exp. Ther. 339, 152–160. doi: 10.1124/jpet.111.182774
Delint-Ramirez, I., Garcia-Oscos, F., Segev, A., and Kourrich, S. (2018). Cocaine engages a non-canonical, dopamine-independent, mechanism that controls neuronal excitability in the nucleus accumbens. Mol. Psychiatr. doi: 10.1038/s41380-018-0092-7
Di Chiara, G., and Imperato, A. (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. U.S.A. 85, 5274–5278.
Dietrich, J. B., Mangeol, A., Revel, M. O., Burgun, C., Aunis, D., and Zwiller, J. (2005). Acute or repeated cocaine administration generates reactive oxygen species and induces antioxidant enzyme activity in dopaminergic rat brain structures. Neuropharmacology 48, 965–974.
Dolphin, A. C. (2009). Calcium channel diversity: multiple roles of calcium channel subunits. Curr. Opin. Neurobiol. 19, 237–244. doi: 10.1016/j.conb.2009.06.006
Dong, L. Y., Cheng, Z. X., Fu, Y. M., Wang, Z. M., Zhu, Y. H., Sun, J. L., et al. (2007). Neurosteroid dehydroepiandrosterone sulfate enhances spontaneous glutamate release in rat prelimbic cortex through activation of dopamine D1 and sigma-1 receptor. Neuropharmacology 52, 966–974.
Dong, Y., Green, T., Saal, D., Marie, H., Neve, R., Nestler, E. J., et al. (2006). CREB modulates excitability of nucleus accumbens neurons. Nat. Neurosci. 9, 475–477.
Dong, Y., and Nestler, E. J. (2014). The neural rejuvenation hypothesis of cocaine addiction. Trends Pharmacol. Sci. 35, 374–383. doi: 10.1016/j.tips.2014.05.005
Entrena, J. M., Cobos, E. J., Nieto, F. R., Cendan, C. M., Gris, G., Del Pozo, E., et al. (2009). Sigma-1 receptors are essential for capsaicin-induced mechanical hypersensitivity: studies with selective sigma-1 ligands and sigma-1 knockout mice. Pain 143, 252–261. doi: 10.1016/j.pain.2009.03.011
Escobedo, L. G., Ruttenber, A. J., Agocs, M. M., Anda, R. F., and Wetli, C. V. (1991). Emerging patterns of cocaine use and the epidemic of cocaine overdose deaths in dade county. florida. Arch. Pathol. Laborator. Med. 115, 900–905.
Ferrario, C. R., Gorny, G., Crombag, H. S., Li, Y., Kolb, B., and Robinson, T. E. (2005). Neural and behavioral plasticity associated with the transition from controlled to escalated cocaine use. Biol. Psychiatry 58, 751–759.
Fishback, J. A., Robson, M. J., Xu, Y. T., and Matsumoto, R. R. (2010). Sigma receptors: potential targets for a new class of antidepressant drug. Pharmacol. Ther. 127, 271–282. doi: 10.1016/j.pharmthera.2010.04.003
Fontanilla, D., Johannessen, M., Hajipour, A. R., Cozzi, N. V., Jackson, M. B., and Ruoho, A. E. (2009). The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 323, 934–937. doi: 10.1126/science.1166127
Garces-Ramirez, L., Green, J. L., Hiranita, T., Kopajtic, T. A., Mereu, M., Thomas, A. M., et al. (2011). Sigma receptor agonists: receptor binding and effects on mesolimbic dopamine neurotransmission assessed by microdialysis. Biol. Psychiatry 69, 208–217. doi: 10.1016/j.biopsych.2010.07.026
Gassmann, M., Haller, C., Stoll, Y., Abdel Aziz, S., Biermann, B., Mosbacher, J., et al. (2005). The RXR-type endoplasmic reticulum-retention/retrieval signal of GABAB1 requires distant spacing from the membrane to function. Mol. Pharmacol. 68, 137–144.
Giese, K. P., Storm, J. F., Reuter, D., Fedorov, N. B., Shao, L. R., Leicher, T., et al. (1998). Reduced K+ channel inactivation, spike broadening, and after-hyperpolarization in Kvbeta1.1-deficient mice with impaired learning. Learn Mem. 5, 257–273.
Graves, S. M., Clark, M. J., Traynor, J. R., Hu, X. T., and Napier, T. C. (2015). Nucleus accumbens shell excitability is decreased by methamphetamine self-administration and increased by 5-HT2C receptor inverse agonism and agonism. Neuropharmacology 89, 113–121. doi: 10.1016/j.neuropharm.2014.09.001
Griesmaier, E., Posod, A., Gross, M., Neubauer, V., Wegleiter, K., Hermann, M., et al. (2012). Neuroprotective effects of the sigma-1 receptor ligand PRE-084 against excitotoxic perinatal brain injury in newborn mice. Exp. Neurol. 237, 388–395. doi: 10.1016/j.expneurol.2012.06.030
Gromek, K. A., Suchy, F. P., Meddaugh, H. R., Wrobel, R. L., LaPointe, L. M., Chu, U. B., et al. (2014). The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J. Biol. Chem. 289, 20333–20344. doi: 10.1074/jbc.M113.537993
Guéguinou, M., Chantôme, A., Fromont, G., Bougnoux, P., Vandier, C., and Potier-Cartereau, M. (2014). KCa and Ca(2+) channels: the complex thought. Biochim. Biophys. Acta 1843, 2322–2333. doi: 10.1016/j.bbamcr.2014.02.019
Gueguinou, M., Crottès, D., Chantôme, A., Rapetti-Mauss, R., Potier-Cartereau, M., Clarysse, L., et al. (2017). The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca. Oncogene 36, 3640–3647. doi: 10.1038/onc.2016.501
Guéguinou, M., Harnois, T., Crottes, D., Uguen, A., Deliot, N., Gambade, A., et al. (2016). SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: a novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 7, 36168–36184. doi: 10.18632/oncotarget.8786
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Hayashi, T., Maurice, T., and Su, T. P. (2000). Ca(2+) signaling via sigma(1)-receptors: novel regulatory mechanism affecting intracellular Ca(2+) concentration. J. Pharmacol. Exp. Ther. 293, 788–798.
Hayashi, T., and Su, T. P. (2001). Regulating ankyrin dynamics: roles of sigma-1 receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 491–496.
Hayashi, T., and Su, T. P. (2003a). Intracellular dynamics of sigma-1 receptors (sigma(1) binding sites) in NG108-15 cells. J. Pharmacol. Exp. Ther. 306, 726–733.
Hayashi, T., and Su, T. P. (2003b). Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 306, 718–725.
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610.
Hayashi, T., Tsai, S. Y., Mori, T., Fujimoto, M., and Su, T. P. (2011). Targeting ligand-operated chaperone sigma-1 receptors in the treatment of neuropsychiatric disorders. Expert Opin. Ther. Targets 15, 557–577. doi: 10.1517/14728222.2011.560837
He, Y.-L., Zhang, C.-L., Gao, X.-F., Yao, J.-J., Hu, C.-L., and Mei, Y.-A. (2012). Cyproheptadine enhances the IK of mouse cortical neurons through sigma-1 receptor-mediated intracellular signal pathway. PLoS One 7:e41303. doi: 10.1371/journal.pone.0041303
Heilstedt, H. A., Burgess, D. L., Anderson, A. E., Chedrawi, A., Tharp, B., Lee, O., et al. (2001). Loss of the potassium channel beta-subunit gene, KCNAB2, is associated with epilepsy in patients with 1p36 deletion syndrome. Epilepsia 42, 1103–1111.
Herrera, Y., Katnik, C., Rodriguez, J. D., Hall, A. A., Willing, A., Pennypacker, K. R., et al. (2008). sigma-1 receptor modulation of acid-sensing ion channel a (ASIC1a) and ASIC1a-induced Ca2+ influx in rat cortical neurons. J. Pharmacol. Exp. Ther. 327, 491–502. doi: 10.1124/jpet.108.143974
Hille, B. (2001). Ionic Channels of Excitable Membranes, 3rd Edn. Sunderland, MA: Sinauer Associates.
Hiranita, T., Mereu, M., Soto, P. L., Tanda, G., and Katz, J. L. (2013). Self-administration of cocaine induces dopamine-independent self-administration of sigma agonists. Neuropsychopharmacology 38, 605–615. doi: 10.1038/npp.2012.224
Hopf, F. W., Cascini, M. G., Gordon, A. S., Diamond, I., and Bonci, A. (2003). Cooperative activation of dopamine D1 and D2 receptors increases spike firing of nucleus accumbens neurons via G-protein betagamma subunits. J. Neurosci. 23, 5079–5087.
Hu, X. T., Basu, S., and White, F. J. (2004). Repeated cocaine administration suppresses HVA-Ca2+ potentials and enhances activity of K+ channels in rat nucleus accumbens neurons. J. Neurophysiol. 92, 1597–1607.
Huang, X., and Jan, L. Y. (2014). Targeting potassium channels in cancer. J. Cell Biol. 206, 151–162.
Ishikawa, M., Mu, P., Moyer, J. T., Wolf, J. A., Quock, R. M., Davies, N. M., et al. (2009). Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J. Neurosci. 29, 5820–5831. doi: 10.1523/JNEUROSCI.5703-08.2009
Jang, E. Y., Ryu, Y. H., Lee, B. H., Chang, S. C., Yeo, M. J., Kim, S. H., et al. (2015). Involvement of reactive oxygen species in cocaine-taking behaviors in rats. Addict. Biol. 20, 663–675. doi: 10.1111/adb.12159
Jin, J. L., Fang, M., Zhao, Y. X., and Liu, X. Y. (2015). Roles of sigma-1 receptors in Alzheimer’s disease. Int. J. Clin. Exp. Med. 8, 4808–4820.
Johannessen, M., Ramachandran, S., Riemer, L., Ramos-Serrano, A., Ruoho, A. E., and Jackson, M. B. (2009). Voltage-gated sodium channel modulation by sigma-receptors in cardiac myocytes and heterologous systems. Am. J. Physiol. Cell Physiol. 296, C1049–C1057. doi: 10.1152/ajpcell.00431.2008
Kahoun, J. R., and Ruoho, A. E. (1992). (125I)iodoazidococaine, a photoaffinity label for the haloperidol-sensitive sigma receptor. Proc. Natl. Acad. Sci. U.S.A. 89, 1393–1397.
Kalivas, P. W., and O’Brien, C. (2008). Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology 33, 166–180.
Katnik, C., Guerrero, W. R., Pennypacker, K. R., Herrera, Y., and Cuevas, J. (2006). Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical neurons during in vitro ischemia. J. Pharmacol. Exp. Ther. 319, 1355–1365.
Katz, J. L., Hong, W. C., Hiranita, T., and Su, T. P. (2016). A role for sigma receptors in stimulant self-administration and addiction. Behav. Pharmacol. 27, 100–115. doi: 10.1097/FBP.0000000000000209
Katz, J. L., Su, T. P., Hiranita, T., Hayashi, T., Tanda, G., Kopajtic, T., et al. (2011). A role for sigma receptors in stimulant self administration and addiction. Pharmaceuticals 4, 880–914.
Kennedy, C., and Henderson, G. (1990). Inhibition of potassium currents by the sigma receptor ligand (+)-3-(3-hydroxyphenyl)-N-(1-propyl)piperidine in sympathetic neurons of the mouse isolated hypogastric ganglion. Neuroscience 35, 725–733.
Kinoshita, M., Matsuoka, Y., Suzuki, T., Mirrielees, J., and Yang, J. (2012). Sigma-1 receptor alters the kinetics of Kv1.3 voltage gated potassium channels but not the sensitivity to receptor ligands. Brain Res. 1452, 1–9. doi: 10.1016/j.brainres.2012.02.070
Kourrich, S. (2017). Sigma-1 receptor and neuronal excitability. Handb. Exp. Pharmacol. 244, 109–130. doi: 10.1007/164_2017_8
Kourrich, S., Calu, D. J., and Bonci, A. (2015). Intrinsic plasticity: an emerging player in addiction. Nat. Rev. Neurosci. 16, 173–184. doi: 10.1038/nrn3877
Kourrich, S., Hayashi, T., Chuang, J. Y., Tsai, S. Y., Su, T. P., and Bonci, A. (2013). Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 152, 236–247. doi: 10.1016/j.cell.2012.12.004
Kourrich, S., Klug, J. R., Mayford, M., and Thomas, M. J. (2012a). AMPAR-independent effect of striatal CaMKII promotes the sensitization of cocaine reward. J. Neurosci. 32, 6578–6586. doi: 10.1523/jneurosci.6391-1511.2012
Kourrich, S., Su, T. P., Fujimoto, M., and Bonci, A. (2012b). The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 35, 762–771. doi: 10.1016/j.tins.2012.09.007
Kourrich, S., and Thomas, M. J. (2009). Similar neurons, opposite adaptations: psychostimulant experience differentially alters firing properties in accumbens core versus shell. J. Neurosci. 29, 12275–12283. doi: 10.1523/JNEUROSCI.3028-09.2009
Kuribara, H., and Uchihashi, Y. (1993). Dopamine antagonists can inhibit methamphetamine sensitization, but not cocaine sensitization, when assessed by ambulatory activity in mice. J. Pharm. Pharmacol. 45, 1042–1045.
Lambert, I. H., Hoffmann, E. K., and Pedersen, S. F. (2008). Cell volume regulation: physiology and pathophysiology. Acta Physiol. 194, 255–282. doi: 10.1111/j.1748-1716.2008.01910.x
Langa, F., Codony, X., Tovar, V., Lavado, A., Gimenez, E., Cozar, P., et al. (2003). Generation and phenotypic analysis of sigma receptor type I (sigma 1) knockout mice. Eur. J. Neurosci. 18, 2188–2196.
Lopez-Pedrajas, R., Ramirez-Lamelas, D. T., Muriach, B., Sanchez-Villarejo, M. V., Almansa, I., Vidal-Gil, L., et al. (2015). Cocaine promotes oxidative stress and microglial-macrophage activation in rat cerebellum. Front. Cell. Neurosci. 9:279. doi: 10.3389/fncel.2015.00279
Lupardus, P. J., Wilke, R. A., Aydar, E., Palmer, C. P., Chen, Y., Ruoho, A. E., et al. (2000). Membrane-delimited coupling between sigma receptors and K+ channels in rat neurohypophysial terminals requires neither G-protein nor ATP. J. Physiol. 526(Pt 3), 527–539.
Luscher, C., and Malenka, R. C. (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663. doi: 10.1016/j.neuron.2011.01.017
Luty, A. A., Kwok, J. B., Dobson-Stone, C., Loy, C. T., Coupland, K. G., Karlstrom, H., et al. (2010). Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann. Neurol. 68, 639–649. doi: 10.1002/ana.22274
Maeno, E., Ishizaki, Y., Kanaseki, T., Hazama, A., and Okada, Y. (2000). Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 9487–9492.
Maffie, J., and Rudy, B. (2008). Weighing the evidence for a ternary protein complex mediating A-type K+ currents in neurons. J. Physiol. 586, 5609–5623. doi: 10.1113/jphysiol.2008.161620
Marionneau, C., Carrasquillo, Y., Norris, A. J., Townsend, R. R., Isom, L. L., Link, A. J., et al. (2012). The sodium channel accessory subunit Navbeta1 regulates neuronal excitability through modulation of repolarizing voltage-gated K(+) channels. J. Neurosci. 32, 5716–5727. doi: 10.1523/JNEUROSCI.6450-11.2012
Martin, W. R., Eades, C. G., Thompson, J. A., Huppler, R. E., and Gilbert, P. E. (1976). The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 197, 517–532.
Martinez-Espinosa, P. L., Yang, C., Gonzalez-Perez, V., Xia, X. M., and Lingle, C. J. (2014). Knockout of the BK beta2 subunit abolishes inactivation of BK currents in mouse adrenal chromaffin cells and results in slow-wave burst activity. J. Gen. Physiol. 144, 275–295. doi: 10.1085/jgp.201411253
Mattingly, B. A., Hart, T. C., Lim, K., and Perkins, C. (1994). Selective antagonism of dopamine D1 and D2 receptors does not block the development of behavioral sensitization to cocaine. Psychopharmacology 114, 239–242.
Maurice, T., and Su, T. P. (2009). The pharmacology of sigma-1 receptors. Pharmacol. Ther. 124, 195–206. doi: 10.1016/j.pharmthera.2009.07.001
Mavlyutov, T. A., and Ruoho, A. E. (2007). Ligand-dependent localization and intracellular stability of sigma-1 receptors in CHO-K1 cells. J. Mol. Signal. 2:8.
McFarland, K., Davidge, S. B., Lapish, C. C., and Kalivas, P. W. (2004). Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J. Neurosci. 24, 1551–1560.
McFarland, K., and Kalivas, P. W. (2001). The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J. Neurosci. 21, 8655–8663.
McFarland, K., Lapish, C. C., and Kalivas, P. W. (2003). Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J. Neurosci. 23, 3531–3537.
McLaughlin, J., and See, R. E. (2003). Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology 168, 57–65. doi: 10.1007/s00213-002-1196-x
Mishra, A. K., Mavlyutov, T., Singh, D. R., Biener, G., Yang, J., Oliver, J. A., et al. (2015). The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem. J. 466, 263–271. doi: 10.1042/BJ20141321
Monteith, G. R., Prevarskaya, N., and Roberts-Thomson, S. J. (2017). The calcium-cancer signalling nexus. Nat. Rev. Cancer 17, 367–380. doi: 10.1038/nrc.2017.18
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540.
Mori, T., Hayashi, T., and Su, T. P. (2012). Compromising sigma-1 receptors at the endoplasmic reticulum render cytotoxicity to physiologically relevant concentrations of dopamine in a nuclear factor-kappaB/Bcl-2-dependent mechanism: potential relevance to Parkinson’s disease. J. Pharmacol. Exp. Ther. 341, 663–671. doi: 10.1124/jpet.111.190868
Moriguchi, S., Yamamoto, Y., Ikuno, T., and Fukunaga, K. (2011). Sigma-1 receptor stimulation by dehydroepiandrosterone ameliorates cognitive impairment through activation of CaM kinase II, protein kinase C and extracellular signal-regulated kinase in olfactory bulbectomized mice. J. Neurochem. 117, 879–891. doi: 10.1111/j.1471-4159.2011.07256.x
Morio, Y., Tanimoto, H., Yakushiji, T., and Morimoto, Y. (1994). Characterization of the currents induced by sigma ligands in NCB20 neuroblastoma cells. Brain Res. 637, 190–196.
Mu, P., Moyer, J. T., Ishikawa, M., Zhang, Y., Panksepp, J., Sorg, B. A., et al. (2010). Exposure to cocaine dynamically regulates the intrinsic membrane excitability of nucleus accumbens neurons. J. Neurosci. 30, 3689–3699. doi: 10.1523/JNEUROSCI.4063-09.2010
Muriach, M., Lopez-Pedrajas, R., Barcia, J. M., Sanchez-Villarejo, M. V., Almansa, I., and Romero, F. J. (2010). Cocaine causes memory and learning impairments in rats: involvement of nuclear factor kappa B and oxidative stress, and prevention by topiramate. J. Neurochem. 114, 675–684. doi: 10.1111/j.1471-4159.2010.06794.x
Nasif, F. J., Sidiropoulou, K., Hu, X. T., and White, F. J. (2005). Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J. Pharmacol. Exp. Ther. 312, 1305–1313.
Navarro, G., Moreno, E., Aymerich, M., Marcellino, D., McCormick, P. J., Mallol, J., et al. (2010). Direct involvement of sigma-1 receptors in the dopamine D1 receptor-mediated effects of cocaine. Proc. Natl. Acad. Sci. U.S.A. 107, 18676–18681. doi: 10.1073/pnas.1008911107
Nguyen, E. C., McCracken, K. A., Liu, Y., Pouw, B., and Matsumoto, R. R. (2005). Involvement of sigma (sigma) receptors in the acute actions of methamphetamine: receptor binding and behavioral studies. Neuropharmacology 49, 638–645.
Nutt, D. J., Lingford-Hughes, A., Erritzoe, D., and Stokes, P. R. (2015). The dopamine theory of addiction: 40 years of highs and lows. Nat. Rev. Neurosci. 16, 305–312. doi: 10.1038/nrn3939
Obacz, J., Avril, T., Rubio-Patiño, C., Bossowski, J. P., Igbaria, A., Ricci, J. E., et al. (2019). Regulation of tumor-stroma interactions by the unfolded protein response. FEBS J. 286, 279–296. doi: 10.1111/febs.14359
O’Leary, M. E., and Chahine, M. (2002). Cocaine binds to a common site on open and inactivated human heart (Na(v)1.5) sodium channels. J. Physiol. 541, 701–716.
Pabba, M., Wong, A. Y., Ahlskog, N., Hristova, E., Biscaro, D., Nassrallah, W., et al. (2014). NMDA receptors are upregulated and trafficked to the plasma membrane after sigma-1 receptor activation in the rat hippocampus. J. Neurosci. 34, 11325–11338. doi: 10.1523/JNEUROSCI.0458-14.2014
Paly, D., Jatlow, P., Van Dyke, C., Jeri, F. R., and Byck, R. (1982). Plasma cocaine concentrations during cocaine paste smoking. Life Sci. 30, 731–738.
Pan, B., Guo, Y., Kwok, W. M., Hogan, Q., and Wu, H. E. (2014). Sigma-1 receptor antagonism restores injury-induced decrease of voltage-gated Ca2+ current in sensory neurons. J. Pharmacol. Exp. Ther. 350, 290–300. doi: 10.1124/jpet.114.214320
Papaioannou, A., Higa, A., Jégou, G., Jouan, F., Pineau, R., Saas, L., et al. (2018). Alterations of EDEM1 functions enhance ATF6 pro-survival signaling. FEBS J. 285, 4146–4164. doi: 10.1111/febs.14669
Pardo, L. A., and Stuhmer, W. (2014). The roles of K(+) channels in cancer. Nat. Rev. Cancer 14, 39–48.
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. doi: 10.1016/j.ceca.2017.06.008
Penke, B., Fulop, L., Szucs, M., and Frecska, E. (2018). The role of sigma-1 receptor, an intracellular chaperone in neurodegenerative diseases. Curr. Neuropharmacol. 16, 97–116. doi: 10.2174/1570159X15666170529104323
Peoples, L. L., and Cavanaugh, D. (2003). Differential changes in signal and background firing of accumbal neurons during cocaine self-administration. J. Neurophysiol. 90, 993–1010.
Peoples, L. L., Kravitz, A. V., and Guillem, K. (2007). The role of accumbal hypoactivity in cocaine addiction. Sci. World J. 7, 22–45.
Peoples, L. L., Uzwiak, A. J., Gee, F., and West, M. O. (1999). Tonic firing of rat nucleus accumbens neurons: changes during the first 2 weeks of daily cocaine self-administration sessions. Brain Res. 822, 231–236.
Peoples, L. L., Uzwiak, A. J., Guyette, F. X., and West, M. O. (1998). Tonic inhibition of single nucleus accumbens neurons in the rat: a predominant but not exclusive firing pattern induced by cocaine self-administration sessions. Neuroscience 86, 13–22.
Pereira, R. B., Andrade, P. B., and Valentao, P. (2015). a comprehensive view of the neurotoxicity mechanisms of cocaine and ethanol. Neuro. Res. 28, 253–267. doi: 10.1007/s12640-015-9536-x
Perez, M. F., White, F. J., and Hu, X. T. (2006). Dopamine D(2) receptor modulation of K(+) channel activity regulates excitability of nucleus accumbens neurons at different membrane potentials. J. Neurophysiol. 96, 2217–2228.
Peters, J., Kalivas, P. W., and Quirk, G. J. (2009). Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn. Mem. 16, 279–288. doi: 10.1101/lm.1041309
Pettit, H. O., Pan, H. T., Parsons, L. H., and Justice, J. B. (1990). Extracellular concentrations of cocaine and dopamine are enhanced during chronic cocaine administration. J. Neurochem. 55, 798–804.
Phartiyal, P., Sale, H., Jones, E. M., and Robertson, G. A. (2008). Endoplasmic reticulum retention and rescue by heteromeric assembly regulate human ERG 1a/1b surface channel composition. J. Biol. Chem. 283, 3702–3707.
Piechota, M., Korostynski, M., Solecki, W., Gieryk, A., Slezak, M., Bilecki, W., et al. (2010). The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biol. 11:R48. doi: 10.1186/gb-2010-11-5-r48
Pongs, O., Leicher, T., Berger, M., Roeper, J., Bahring, R., Wray, D., et al. (1999). Functional and molecular aspects of voltage-gated K+ channel beta subunits. Ann. N. Y. Acad. Sci. 868, 344–355.
Prevarskaya, N., Skryma, R., and Shuba, Y. (2010). Ion channels and the hallmarks of cancer. Trends Mol. Med. 16, 107–121.
Prevarskaya, N., Skryma, R., and Shuba, Y. (2018). Ion channels in cancer: are cancer hallmarks oncochannelopathies? Physiol. Rev. 98, 559–621. doi: 10.1152/physrev.00044.2016
Prinssen, E. P., Colpaert, F. C., Kleven, M. S., and Koek, W. (2004). Ability of dopamine antagonists to inhibit the locomotor effects of cocaine in sensitized and non-sensitized C57BL/6 mice depends on the challenge dose. Psychopharmacology 172, 409–414.
Quail, D. F., and Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. doi: 10.1038/nm.3394
Rapetti-Mauss, R., Borgese, F., Harvey, B. J., and Soriani, O. (2018). [KCNQ1: a new regulator of the epithelio-mesenchymal transition in colorectal cancers]. Med. Sci. 34, 21–24.
Rapetti-Mauss, R., Bustos, V., Thomas, W., McBryan, J., Harvey, H., Lajczak, N., et al. (2017). Bidirectional KCNQ1:β-catenin interaction drives colorectal cancer cell differentiation. Proc. Natl. Acad. Sci. U.S.A. 114, 4159–4164.
Reith, M. E. A., Sershen, H., and Lajtha, A. (1986). Binding-sites for [H-3] cocaine in mouse striatum and cerebral-cortex have different dissociation kinetics. J. Neurochem. 46, 309–312.
Renaudo, A., L’Hoste, S., Guizouarn, H., Borgese, F., and Soriani, O. (2007). Cancer cell cycle modulated by a functional coupling between sigma-1 receptors and Cl- channels. J. Biol. Chem. 282, 2259–2267.
Renaudo, A., Watry, V., Chassot, A. A., Ponzio, G., Ehrenfeld, J., and Soriani, O. (2004). Inhibition of tumor cell proliferation by sigma ligands is associated with K+ Channel inhibition and p27kip1 accumulation. J. Pharmacol. Exp. Ther. 311, 1105–1114.
Renthal, W., Kumar, A., Xiao, G., Wilkinson, M., Covington, H. E. III, Maze, I., et al. (2009). Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 62, 335–348. doi: 10.1016/j.neuron.2009.03.026
Robinson, T. E., and Berridge, K. C. (2008). Review. The incentive sensitization theory of addiction: some current issues. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 3137–3146. doi: 10.1098/rstb.2008.0093
Roger, S., Besson, P., and Le Guennec, J. Y. (2003). Involvement of a novel fast inward sodium current in the invasion capacity of a breast cancer cell line. Biochim. Biophys. Acta 1616, 107–111.
Roger, S., Gillet, L., Le Guennec, J. Y., and Besson, P. (2015). Voltage-gated sodium channels and cancer: is excitability their primary role? Front. Pharmacol. 6:152. doi: 10.3389/fphar.2015.00152
Roger, S., Le Guennec, J. Y., and Besson, P. (2004). Particular sensitivity to calcium channel blockers of the fast inward voltage-dependent sodium current involved in the invasive properties of a metastastic breast cancer cell line. Br. J. Pharmacol. 141, 610–615.
Roger, S., Potier, M., Vandier, C., Besson, P., and Le Guennec, J. Y. (2006). Voltage-gated sodium channels: new targets in cancer therapy? Curr. Pharm. Des. 12, 3681–3695.
Romieu, P., Meunier, J., Garcia, D., Zozime, N., Martin-Fardon, R., Bowen, W. D., et al. (2004). The sigma1 (sigma1) receptor activation is a key step for the reactivation of cocaine conditioned place preference by drug priming. Psychopharmacology 175, 154–162.
Rothman, R. B., Reid, A., Mahboubi, A., Kim, C. H., De Costa, B. R., Jacobson, A. E., et al. (1991). Labeling by [3H]1,3-di(2-tolyl)guanidine of two high affinity binding sites in guinea pig brain: evidence for allosteric regulation by calcium channel antagonists and pseudoallosteric modulation by sigma ligands. Mol. Pharmacol. 39, 222–232.
Rouzaire-Dubois, B., and Dubois, J. M. (1998). K+ channel block-induced mammalian neuroblastoma cell swelling: a possible mechanism to influence proliferation. J. Physiol. 510(Pt 1), 93–102.
Rouzaire-Dubois, B., Milandri, J. B., Bostel, S., and Dubois, J. M. (2000). Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflugers. Arch. 440, 881–888.
Rumbaugh, G., and Vicini, S. (1999). Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J. Neurosci. 19, 10603–10610.
Ryskamp, D., Wu, J., Geva, M., Kusko, R., Grossman, I., Hayden, M., et al. (2017). The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of huntington disease. Neurobiol. Dis. 97, 46–59. doi: 10.1016/j.nbd.2016.10.006
Ryskamp, D., Wu, L., Wu, J., Kim, D., Rammes, G., Geva, M., et al. (2019). Pridopidine stabilizes mushroom spines in mouse models of Alzheimer’s disease by acting on the sigma-1 receptor. Neurobiol. Dis. 124, 489–504. doi: 10.1016/j.nbd.2018.12.022
Sabeti, J., Nelson, T. E., Purdy, R. H., and Gruol, D. L. (2007). Steroid pregnenolone sulfate enhances NMDA-receptor-independent long-term potentiation at hippocampal CA1 synapses: role for L-type calcium channels and sigma-receptors. Hippocampus 17, 349–369.
Sabino, V., Cottone, P., Parylak, S. L., Steardo, L., and Zorrilla, E. P. (2009). Sigma-1 receptor knockout mice display a depressive-like phenotype. Behav. Brain Res. 198, 472–476. doi: 10.1016/j.bbr.2008.11.036
Sambo, D. O., Lin, M., Owens, A., Lebowitz, J. J., Richardson, B., Jagnarine, D. A., et al. (2017). The sigma-1 receptor modulates methamphetamine dysregulation of dopamine neurotransmission. Nat. Commun. 8:2228. doi: 10.1038/s41467-017-02087-x
Schilstrom, B., Yaka, R., Argilli, E., Suvarna, N., Schumann, J., Chen, B. T., et al. (2006). Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J. Neurosci. 26, 8549–8558.
Schmidt, H. R., and Kruse, A. C. (2019). The molecular function of sigma receptors: past, present, and future. Trends Pharmacol. Sci. 40, 636–654. doi: 10.1016/j.tips.2019.07.006
Schmidt, H. R., Zheng, S., Gurpinar, E., Koehl, A., Manglik, A., and Kruse, A. C. (2016). Crystal structure of the human sigma1 receptor. Nature 532, 527–530. doi: 10.1038/nature17391
Schulze-Bahr, E., Wang, Q., Wedekind, H., Haverkamp, W., Chen, Q., Sun, Y., et al. (1997). KCNE1 mutations cause jervell and lange-nielsen syndrome. Nat. Genet. 17, 267–268.
Scott, D. B., Blanpied, T. A., and Ehlers, M. D. (2003). Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 45, 755–767.
Segev, A., Delint-Ramirez, I., and Kourrich, S. (2018). Contingent versus non-contingent cocaine administration differentially affects the persistence of neuronal intrinsic plasticity of medium spiny neurons in the nucleus accumbens shell. Soc. Neurosci. Abstract 419.17.
Sergeant, G. P., Ohya, S., Reihill, J. A., Perrino, B. A., Amberg, G. C., Imaizumi, Y., et al. (2005). Regulation of Kv4.3 currents by Ca2+/calmodulin-dependent protein kinase II. Am. J. Physiol. Cell Physiol. 288, C304–C313.
Sharkey, J., Glen, K. A., Wolfe, S., and Kuhar, M. J. (1988). Cocaine binding at sigma receptors. Eur. J. Pharmacol. 149, 171–174.
Shen, W., Hernandez-Lopez, S., Tkatch, T., Held, J. E., and Surmeier, D. J. (2004). Kv1.2-containing K+ channels regulate subthreshold excitability of striatal medium spiny neurons. J. Neurophysiol. 91, 1337–1349.
Shioda, N., Ishikawa, K., Tagashira, H., Ishizuka, T., Yawo, H., and Fukunaga, K. (2012). Expression of a truncated form of the endoplasmic reticulum chaperone protein, sigma1 receptor, promotes mitochondrial energy depletion and apoptosis. J. Biol. Chem. 287, 23318–23331. doi: 10.1074/jbc.M112.349142
Smith, S. B., Wang, J., Cui, X., Mysona, B. A., Zhao, J., and Bollinger, K. E. (2018). Sigma 1 receptor: a novel therapeutic target in retinal disease. Prog. Retin Eye Res. 67, 130–149. doi: 10.1016/j.preteyeres.2018.07.003
Sora, I., Wichems, C., Takahashi, N., Li, X. F., Zeng, Z., Revay, R., et al. (1998). Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc. Natl. Acad. Sci. U.S.A. 95, 7699–7704.
Soriani, O., Foll, F. L., Roman, F., Monnet, F. P., Vaudry, H., and Cazin, L. (1999a). A-Current down-modulated by sigma receptor in frog pituitary melanotrope cells through a G protein-dependent pathway. J. Pharmacol. Exp. Ther. 289, 321–328.
Soriani, O., Le Foll, F., Galas, L., Roman, F., Vaudry, H., and Cazin, L. (1999b). The sigma-ligand (+)-pentazocine depresses M current and enhances calcium conductances in frog melanotrophs. Am. J. Physiol. 277, E73–E80. doi: 10.1152/ajpendo.1999.277.1.E73
Soriani, O., and Rapetti-Mauss, R. (2017). Sigma 1 receptor and ion channel dynamics in cancer. Adv. Exp. Med. Biol. 964, 63–77. doi: 10.1007/978-3-319-50174-1_6
Soriani, O., Vaudry, H., Mei, Y. A., Roman, F., and Cazin, L. (1998). Sigma ligands stimulate the electrical activity of frog pituitary melanotrope cells through a G-protein-dependent inhibition of potassium conductances. J. Pharmacol. Exp. Ther. 286, 163–171.
Splawski, I., Tristani-Firouzi, M., Lehmann, M. H., Sanguinetti, M. C., and Keating, M. T. (1997). Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 17, 338–340.
Spruce, B. A., Campbell, L. A., McTavish, N., Cooper, M. A., Appleyard, M. V., O’Neill, M., et al. (2004). Small molecule antagonists of the sigma-1 receptor cause selective release of the death program in tumor and self-reliant cells and inhibit tumor growth in vitro and in vivo. Cancer Res. 64, 4875–4886.
Strobl, J. S., Wonderlin, W. F., and Flynn, D. C. (1995). Mitogenic signal transduction in human breast cancer cells. Gen. Pharmacol. JID 7602417, 1643–1649.
Su, T. P., Hayashi, T., Maurice, T., Buch, S., and Ruoho, A. E. (2010). The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 31, 557–566. doi: 10.1016/j.tips.2010.08.007
Su, T. P., Su, T. C., Nakamura, Y., and Tsai, S. Y. (2016). The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol. Sci. 37, 262–278. doi: 10.1016/j.tips.2016.01.003
Sun, W., Maffie, J. K., Lin, L., Petralia, R. S., Rudy, B., and Hoffman, D. A. (2011). DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron 71, 1102–1115. doi: 10.1016/j.neuron.2011.08.008
Szabo, A., Kovacs, A., Frecska, E., and Rajnavolgyi, E. (2014). Psychedelic N,N-dimethyltryptamine and 5-methoxy-N,N-dimethyltryptamine modulate innate and adaptive inflammatory responses through the sigma-1 receptor of human monocyte-derived dendritic cells. PLoS One 9:e106533. doi: 10.1371/journal.pone.0106533
Tchedre, K. T., Huang, R. Q., Dibas, A., Krishnamoorthy, R. R., Dillon, G. H., and Yorio, T. (2008). Sigma-1 receptor regulation of voltage-gated calcium channels involves a direct interaction. Invest. Ophthalmol. Visual Sci. 49, 4993–5002. doi: 10.1167/iovs.08-1867
Thomas, J. D., Longen, C. G., Oyer, H. M., Chen, N., Maher, C. M., Salvino, J. M., et al. (2017). Sigma1 targeting to suppress aberrant androgen receptor signaling in prostate cancer. Cancer Res. 77, 2439–2452. doi: 10.1158/0008-5472.CAN-16-1055
Ujike, H., Kanzaki, A., Okumura, K., Akiyama, K., and Otsuki, S. (1992a). Sigma (sigma) antagonist BMY 14802 prevents methamphetamine-induced sensitization. Life Sci. 50, L129–L134.
Ujike, H., Okumura, K., Zushi, Y., Akiyama, K., and Otsuki, S. (1992b). Persistent supersensitivity of sigma receptors develops during repeated methamphetamine treatment. Eur. J. Pharmacol. 211, 323–328.
Ujike, H., Tsuchida, K., Akiyama, K., and Otsuki, S. (1992c). Supersensitivity of sigma receptors after repeated administration of cocaine. Life Sci. 51, L31–L36.
Ujike, H., Kuroda, S., and Otsuki, S. (1996). sigma Receptor antagonists block the development of sensitization to cocaine. Eur. J. Pharmacol. 296, 123–128.
Vacher, H., Mohapatra, D. P., and Trimmer, J. S. (2008). Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 88, 1407–1447. doi: 10.1152/physrev.00002.2008
Van Dyke, C., Barash, P. G., Jatlow, P., and Byck, R. (1976). Cocaine: plasma concentrations after intranasal application in man. Science 191, 859–861.
Vandenberg, J. I., Perry, M. D., Perrin, M. J., Mann, S. A., Ke, Y., and Hill, A. P. (2012). hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478.
Varga, A. W., Yuan, L. L., Anderson, A. E., Schrader, L. A., Wu, G. Y., Gatchel, J. R., et al. (2004). Calcium-calmodulin-dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A-type potassium currents. J. Neurosci. 24, 3643–3654.
Vicini, S., Wang, J. F., Li, J. H., Zhu, W. J., Wang, Y. H., Luo, J. H., et al. (1998). Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 79, 555–566.
Vilner, B. J., Decosta, B. R., and Bowen, W. D. (1995a). Cytotoxic effects of sigma ligands: sigma receptor- mediated alterations in cellular morphology and viability. J. Neurosci. 15, 117–134.
Vilner, B. J., John, C. S., and Bowen, W. D. (1995b). Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 55, 408–413.
Walker, J. M., Bowen, W. D., Walker, F. O., Matsumoto, R. R., De Costa, B., and Rice, K. C. (1990). Sigma receptors: biology and function. Pharmacol. Rev. 42, 355–402.
Wang, J., Ishikawa, M., Yang, Y., Otaka, M., Kim, J. Y., Gardner, G. R., et al. (2018). Cascades of Homeostatic dysregulation promote incubation of cocaine craving. J. Neurosci. 38, 4316–4328. doi: 10.1523/JNEUROSCI.3291-17.2018
Wang, Y., Guo, L., Jiang, H. F., Zheng, L. T., Zhang, A., and Zhen, X. C. (2016). Allosteric modulation of sigma-1 receptors elicits rapid antidepressant activity. CNS Neurosci. Ther. 22, 368–377. doi: 10.1111/cns.12502
Weng, T. Y., Tsai, S. A., and Su, T. P. (2017). Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases. J. Biomed. Sci. 24:74. doi: 10.1186/s12929-017-0380-6
Wheeler, D. D., Edwards, A. M., and Ondo, J. G. (1993). The effect of cocaine on membrane-potential, on membrane depolarization by veratridine or elevated [K]O and on sodium-potassium permeability ratios in synaptosomes from the limbic cortex of the rat. Neuropharmacology 32, 195–204.
White, F. J., Joshi, A., Koeltzow, T. E., and Hu, X. T. (1998). Dopamine receptor antagonists fail to prevent induction of cocaine sensitization. Neuropsychopharmacology 18, 26–40.
Wolf, M. E. (2010). The bermuda triangle of cocaine-induced neuroadaptations. Trends Neurosci. 33, 391–398. doi: 10.1016/j.tins.2010.06.003
Wolf, M. E. (2016). Synaptic mechanisms underlying persistent cocaine craving. Nat. Rev. Neurosci. 17, 351–365. doi: 10.1038/nrn.2016.39
Wolf, M. E., and Tseng, K. Y. (2012). Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front. Mol. Neurosci. 5:72. doi: 10.3389/fnmol.2012.00072
Wong, A. Y., Hristova, E., Ahlskog, N., Tasse, L. A., Ngsee, J. K., Chudalayandi, P., et al. (2016). Aberrant subcellular dynamics of sigma-1 receptor mutants underlying neuromuscular diseases. Mol. Pharmacol. 90, 238–253. doi: 10.1124/mol.116.104018
Wu, X. Z., Bell, J. A., Spivak, C. E., London, E. D., and Su, T. P. (1991). Electrophysiological and binding studies on intact NCB-20 cells suggest presence of a low affinity sigma receptor. J. Pharmacol. Exp. Ther. 257, 351–359.
Wulff, H., Castle, N. A., and Pardo, L. A. (2009). Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 8, 982–1001. doi: 10.1038/nrd2983
Xiao, Y. F., Ke, Q., Wang, S. Y., Yang, Y., Chen, Y., Wang, G. K., et al. (2004). Electrophysiologic properties of lidocaine, cocaine, and n-3 fatty-acids block of cardiac Na+ channels. Eur. J. Pharmacol. 485, 31–41.
Yao, W. D., and Wu, C. F. (2001). Distinct roles of CaMKII and PKA in regulation of firing patterns and K(+) currents in Drosophila neurons. J. Neurophysiol. 85, 1384–1394.
Yasui, Y., and Su, T. P. (2016). Potential molecular mechanisms on the role of the sigma-1 receptor in the action of cocaine and methamphetamine. J. Drug Alcohol. Res. 5:235970.
Zhang, H., and Cuevas, J. (2002). Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J. Neurophysiol. 87, 2867–2879.
Zhang, H., and Cuevas, J. (2005). sigma receptor activation blocks potassium channels and depresses neuroexcitability in rat intracardiac neurons. J. Pharmacol. Exp. Ther. 313, 1387–1396.
Zhang, H., Katnik, C., and Cuevas, J. (2009). Sigma receptor activation inhibits voltage-gated sodium channels in rat intracardiac ganglion neurons. Int. J. Physiol. Pathophysiol. Pharmacol. 2, 1–11.
Zhang, X. F., Cooper, D. C., and White, F. J. (2002). Repeated cocaine treatment decreases whole-cell calcium current in rat nucleus accumbens neurons. J. Pharmacol. Exp. Ther. 301, 1119–1125.
Zhang, X. F., Hu, X. T., and White, F. J. (1998). Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J. Neurosci. 18, 488–498.
Keywords: sigma-1 receptor, chaperone protein, voltage-gated ion channels, intrinsic excitability, plasticity, nervous system disorders, cancer, drug addiction
Citation: Soriani O and Kourrich S (2019) The Sigma-1 Receptor: When Adaptive Regulation of Cell Electrical Activity Contributes to Stimulant Addiction and Cancer. Front. Neurosci. 13:1186. doi: 10.3389/fnins.2019.01186
Received: 30 May 2019; Accepted: 21 October 2019;
Published: 12 November 2019.
Edited by:
Juan J. Canales, University of Tasmania, AustraliaReviewed by:
Teruo Hayashi, Nishikawa Hospital, JapanCopyright © 2019 Soriani and Kourrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Soriani, b2xpdmllci5zb3JpYW5pQHVuaWNlLmZy; Saïd Kourrich, a291cnJpY2guc2FpZEB1cWFtLmNh; U2FpZC5Lb3VycmljaEBVVFNvdXRod2VzdGVybi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.