 Thomas J. Hedl1
Thomas J. Hedl1 Rebecca San Gil1
Rebecca San Gil1 Flora Cheng2
Flora Cheng2 Stephanie L. Rayner2
Stephanie L. Rayner2 Jennilee M. Davidson2
Jennilee M. Davidson2 Alana De Luca2
Alana De Luca2 Maria D. Villalva2
Maria D. Villalva2 Heath Ecroyd3,4
Heath Ecroyd3,4 Adam K. Walker1,2*
Adam K. Walker1,2* Albert Lee2*
Albert Lee2*- 1Neurodegeneration Pathobiology Laboratory, Queensland Brain Institute, The University of Queensland, St Lucia, QLD, Australia
- 2Centre for Motor Neuron Disease Research, Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University, North Ryde, NSW, Australia
- 3School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, NSW, Australia
- 4Illawarra Health and Medical Research Institute, Wollongong, NSW, Australia
Neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are increasing in prevalence but lack targeted therapeutics. Although the pathological mechanisms behind these diseases remain unclear, both ALS and FTD are characterized pathologically by aberrant protein aggregation and inclusion formation within neurons, which correlates with neurodegeneration. Notably, aggregation of several key proteins, including TAR DNA binding protein of 43 kDa (TDP-43), superoxide dismutase 1 (SOD1), and tau, have been implicated in these diseases. Proteomics methods are being increasingly applied to better understand disease-related mechanisms and to identify biomarkers of disease, using model systems as well as human samples. Proteomics-based approaches offer unbiased, high-throughput, and quantitative results with numerous applications for investigating proteins of interest. Here, we review recent advances in the understanding of ALS and FTD pathophysiology obtained using proteomics approaches, and we assess technical and experimental limitations. We compare findings from various mass spectrometry (MS) approaches including quantitative proteomics methods such as stable isotope labeling by amino acids in cell culture (SILAC) and tandem mass tagging (TMT) to approaches such as label-free quantitation (LFQ) and sequential windowed acquisition of all theoretical fragment ion mass spectra (SWATH-MS) in studies of ALS and FTD. Similarly, we describe disease-related protein-protein interaction (PPI) studies using approaches including immunoprecipitation mass spectrometry (IP-MS) and proximity-dependent biotin identification (BioID) and discuss future application of new techniques including proximity-dependent ascorbic acid peroxidase labeling (APEX), and biotinylation by antibody recognition (BAR). Furthermore, we explore the use of MS to detect post-translational modifications (PTMs), such as ubiquitination and phosphorylation, of disease-relevant proteins in ALS and FTD. We also discuss upstream technologies that enable enrichment of proteins of interest, highlighting the contributions of new techniques to isolate disease-relevant protein inclusions including flow cytometric analysis of inclusions and trafficking (FloIT). These recently developed approaches, as well as related advances yet to be applied to studies of these neurodegenerative diseases, offer numerous opportunities for discovery of potential therapeutic targets and biomarkers for ALS and FTD.
Introduction
Neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), demonstrate dysfunctional protein clearance and accumulation of protein-rich inclusions in neuronal cells. Resolving whether these inclusions are a cause of cellular degeneration or a symptom of homeostatic dysfunction has proven difficult, and the pathological mechanisms underlying their formation are still largely unknown. However, through attempting to answer these questions, the fundamental roles of protein aggregation and protein clearance in the pathology of these diseases have been established. Recent advances in proteomics technologies have advanced our understanding of both the primary pathological proteins as well as other associated proteins that may play downstream roles in disease mechanisms. The purpose of this review is to evaluate the current proteomic tools and techniques being used to understand these diseases, to summarize the state of knowledge gained from proteomic studies on ALS and FTD, and to discuss development of more effective disease-modifying treatments and biomarkers for clinical assessment driven by new advances in proteomic technologies.
Pathology and Disease Mechanisms of ALS and FTD
ALS and FTD are both debilitating neurodegenerative diseases caused by selective loss of subsets of neurons. In ALS, disease pathology primarily affects the motor neurons of the primary motor cortex and spinal cord (Foster and Salajegheh, 2019), and in FTD, neurons of the frontal and temporal lobe are the primary targets of degeneration (Olney et al., 2017). Characteristic of these neurodegenerative diseases is the accumulation of protein inclusions in the cytoplasm of neurons (Ling et al., 2013). These pathological proteins, which may be affected by inherited mutations in familial disease or undergo unclear pathological changes in sporadic cases of disease, include but are not limited to TAR DNA binding protein of 43 kDa (TDP-43), superoxide dismutase 1 (SOD1) and microtubule-associated protein tau (Ling et al., 2013). Numerous mutations in these and other proteins have been shown to individually cause either ALS or FTD, or indeed both diseases in the same patient or within the same family; however, the vast majority of ALS cases and approximately half of all FTD cases have no known underlying dominant mechanism of inheritance (Ling et al., 2013; Nguyen et al., 2018).
Prominent amongst these pathological proteins is TDP-43, which is a highly conserved predominantly nuclear protein with functional roles in RNA metabolism (Ayala et al., 2008; Weskamp and Barmada, 2018). TDP-43 has been reproducibly identified as one of the components of cytosolic protein inclusions, a pathological hallmark in almost all ALS and some FTD cases (known pathologically as frontotemporal lobar degeneration with TDP-43 pathology, FTLD-TDP), irrespective of genetic inheritance or mutation (Neumann et al., 2006, 2007; Wang et al., 2008). Aberrant cytoplasmic TDP-43 has been reported in neurons and glial cells of the central nervous system in patients with either ALS or FTD (Arai et al., 2006; Neumann et al., 2006), and TDP-43 is therefore a primary target for studies of mechanisms of these diseases. Indeed, mutations in numerous other ALS-linked genes, including C9ORF72, are also characterized by the presence of TDP-43 pathology (Chew et al., 2015).
Although TDP-43 is the main pathological protein in almost all ALS cases, the first known causative mutations linked to ALS were identified in the protein SOD1 (Rosen et al., 1993), and these account for approximately 20% of all familial ALS cases (Pasinelli and Brown, 2006). SOD1 is a cytosolic protein responsible for catalyzing the breakdown of harmful superoxide radicals (Field et al., 2003; Sea et al., 2015). However, disease-causative mutations in the SOD1 protein variably affect protein function, and knockout of SOD1 in animals does not result in an ALS-like disease, such that the primary mechanism of toxicity of mutant SOD1 is considered to be a gain-of-function even if alterations in SOD1 function may modify disease presentation (Valentine et al., 2005; Saccon et al., 2013). Mutant SOD1 forms large intraneuronal inclusions in people with ALS-linked SOD1 mutations as well as in cellular and animal model systems (Bruijn et al., 1998; Kato et al., 2000; Ayers et al., 2017).
Unlike SOD1, tau is a primarily neuronal axoplasmic protein that stabilizes microtubules (Kadavath et al., 2015). Numerous other roles for tau have been also been described, such as inhibition of HDAC6, a protein involved in tubulin acetylation (Perez et al., 2009), and in intraneuronal transport (Bodea et al., 2016). Importantly, phosphorylation of tau has been shown to correlate with the formation of tau inclusions that are present in tissue from people with FTD (Vega et al., 2005), and mutations within the MAPT gene encoding tau are a prominent cause of non-TDP-43-associated cases of FTD (Rademakers et al., 2004). Indeed, aggregation of tau and alterations in tau function are prominent in FTLD-tau as well as other neurodegenerative diseases, including Alzheimer’s disease (Frost et al., 2015).
Overall, numerous mechanisms have been implicated in the pathogenesis of these diseases, related to mutations and/or dysfunctions which impact on neuronal viability via changes in numerous pathways including intracellular transport, cellular stress responses, RNA metabolism and protein clearance machinery (Walker and Atkin, 2011; Ling et al., 2013; Zhang et al., 2015; Tank et al., 2018). However, despite the diversity of possible upstream causes of disease, the prominence of protein aggregation suggests that this plays a key role in driving neurodegeneration in ALS and FTD.
Proteostasis and Protein Aggregation in ALS and FTD
Proteins are the functional components that drive the majority of cellular processes. Protein homeostasis or “proteostasis” describes a network of constitutively expressed housekeeping and cellular stress-inducible molecular pathways that maintain proteins in a biologically active conformation, or degrade them, to ensure that cell viability is not compromised (Balch et al., 2008; Hipp et al., 2014). The proteostasis network can be clustered into several pathways including the heat shock response, unfolded protein response, ubiquitin-proteasome system (UPS), and autophagy machinery (Webster et al., 2017). Under physiological conditions, the mechanisms of proteostasis function sufficiently to maintain cell viability. However, if proteostasis deteriorates or becomes overwhelmed, for example in the context of ALS and FTD, aberrant protein accumulation and aggregation can occur, and cell viability may be threatened.
Under normal cellular conditions, proteins exist in their native conformation, consisting of external hydrophilic surfaces and an internal hydrophobic core. Apart from the folding that occurs for nascent polypeptides as they are synthesized on the ribosome, protein folding and unfolding occurs at other important times during the lifespan of many proteins. For example, proteins unfold and are refolded during trafficking across intracellular membranes, cellular secretion, and during times of cellular stress (Kincaid and Cooper, 2007; Gregersen and Bross, 2010). When proteins are subjected to cellular stresses, such as oxidative stress or increased burden to mitochondria or the endoplasmic reticulum, they may unfold and form partially folded protein intermediates that expose the hydrophobic regions of the polypeptide to the cytosol, which are otherwise buried within the protein (Hipp et al., 2014). Exposed hydrophobic regions are attracted to similar hydrophobic regions on adjacent partially folded protein intermediates, which may aggregate together and enter thermodynamically favorable pathways that lead to the formation of higher-order oligomers (Stefani, 2008). These oligomers may be toxic and also form the building blocks of larger aggregates and protein inclusions in neurodegenerative diseases (Lasagna-Reeves et al., 2012; Blair et al., 2013; Ait-Bouziad et al., 2017; Shafiei et al., 2017).
The maintenance of functional proteostasis to ameliorate protein aggregation is particularly important in post-mitotic cells such as neurons, since disrupted proteostasis cannot be simply counteracted by apoptosis and replacement with new healthy neurons, unlike most other cell types (Morimoto, 2008). A recent review has discussed evidence that cellular stress in the spinal cord of the SOD1G93A mouse, the most widely used model of ALS, does not result in the induction of the anti-aggregation heat shock response, which may suggest that this pathway is impaired in disease (San Gil et al., 2017). Impairment of proteasomal degradation likely also contributes to the accumulation of ubiquitinated proteins, including SOD1 and TDP-43 (Cheroni et al., 2005, 2009; Scotter et al., 2014). Numerous factors, such as activation of cellular stress responses and dysfunctions in proteasome activity and autophagy, may contribute to varying degrees to the accumulation of proteins in disease, although the underlying mechanisms of inclusion formation and associated pathophysiology remain unclear (Tanaka and Matsuda, 2014). However, since protein aggregation and inclusion formation are hallmarks of these diseases, understanding these processes, and how protein aggregation perturbs neuronal function, is likely to reveal new ways to delay or prevent neurodegeneration.
Proteins that are supersaturated, meaning that their concentration in the cell is greater than their predicted solubility, also have a higher propensity to aggregate (Ciryam et al., 2017). In ALS and FTD, proteins such as TDP-43 have been shown to be supersaturated due to their unusually high concentration (Ciryam et al., 2017). Phase transitions may result in proteins precipitating out in the cytoplasm, a liquid-liquid separation, and proteins with low complexity domains such as TDP-43 may be more prone to this transition (Li et al., 2018). Indeed, recently RNA has been proposed as a regulator of protein phase separation, suggesting that dysregulation of RNA: protein interactions may contribute to the formation of protein aggregates (Maharana et al., 2018; Zacco et al., 2019).
Analyzing how disease-related proteins aggregate, identification of the interaction partners of native and aggregated pathological proteins, and determination of the consequences of protein aggregation within neurons will likely elucidate the patho-mechanisms involved in ALS and FTD. The remainder of this review will focus on the methods and outcomes of previous and potential future studies using proteomics technologies to address these fundamental issues. By developing an understanding of the numerous tools available for these experiments and devising the most relevant biological questions, these studies offer the potential to elucidate the mechanisms of pathogenesis of ALS/FTD, and to identify biomarkers for these diseases. Together, this information may allow the future development of better treatments for people living with ALS and FTD.
Proteomic Approaches to Study ALS and FTD
Proteomic-based platforms are becoming increasingly powerful to identify both potential disease mechanisms as well as disease biomarkers. Proteomics involves using highly complex protein screening technology for biological understanding on a wide-scale level. This information can then be used in combination with genomic data to provide an understanding of the fundamental biological mechanisms underlying neurodegenerative diseases. The emergence of newer sophisticated mass spectrometry (MS) technology in the past decade, with higher resolution and faster scan rates, has enabled smoother and quicker identification of highly complex proteomes with shorter analysis periods (Chapman et al., 2014). The typical sample preparation procedures for proteomics analysis involves digesting proteins into peptides using a protease (e.g., trypsin and/or Lys-C) followed by reverse phase C18 liquid chromatographic separation and analysis by mass spectrometry (LC-MS/MS) (Tholey and Becker, 2017). The peptides that are eluted from the C18 column are directly ionized into the mass spectrometer for analysis, where they can be fragmented (MS/MS) further for peptide identification. The data generated by MS can be searched using algorithms that incorporate protein databases to generate lists of identified proteins. The two major streams for quantitative proteomics that are widely used are label-free and labeled quantitation (Figure 1). Both methods are used for the identification and quantitation of protein components between altered physiological states such as those in ALS and FTD, to differentiate their cellular and molecular mechanisms.
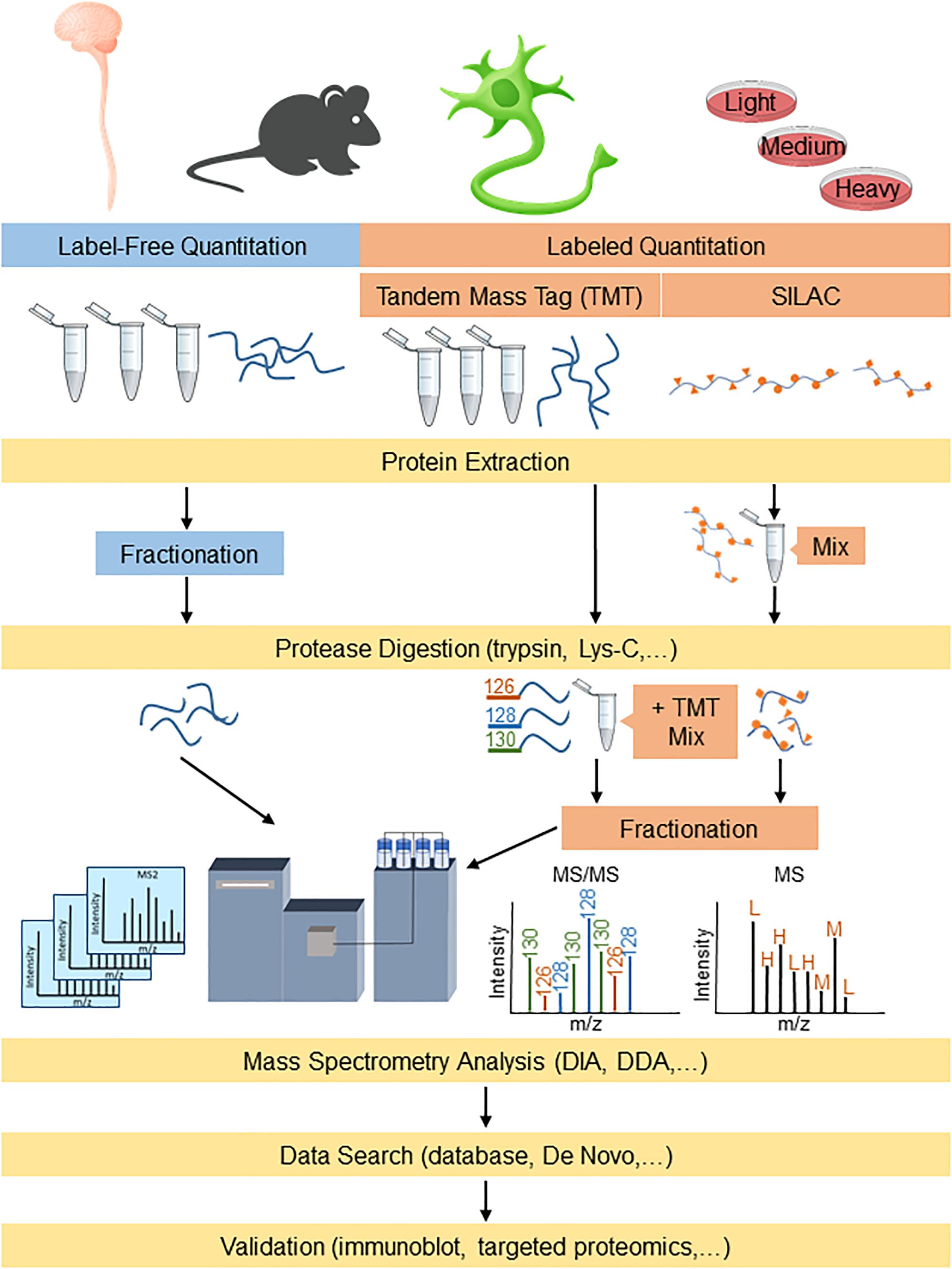
Figure 1. Proteomics workflow for label-free and labeling quantitation of proteins from complex mixtures relevant to understanding ALS and FTD. Protein samples derived from models/human tissue can either be labeled for targeted proteomics or analyzed label-free for broader detection. DDA, data-dependent acquisition; DIA, data-independent acquisition; and SILAC, stable isotope labeling by amino acids in cell culture.
Label-Free Quantitation Proteomics Studies of ALS and FTD
Label-free quantitation (LFQ) can use either data-dependent or data-independent acquisition (DIA) analyses. Data-dependent acquisition quantitation can be performed on either spectral counts or spectrometric signal intensity of product ions from selected precursors generated by the mass spectrometer (Yu F. et al., 2016). Recently, LFQ has been more frequently adopted for biomarker discovery studies since it is less expensive than labeled methods and allows comparative analysis of large groups of samples. A comprehensive review of the technical aspects of label-free quantitative proteomic approaches is available (Neilson et al., 2011). LFQ proteomics can also be used as an unbiased approach to characterize changes to a human proteome at a pathway level. As an example, amongst the identification of hundreds of proteins, overlapping mitochondrial and metabolic pathway alterations have been identified from samples of both human ALS and FTD brain and spinal cord samples, highlighting the dysfunctional similarities between these diseases (Iridoy et al., 2018). Similar analysis of brain tissues has provided insight into the various subtypes of ALS and FTD, distinguishing between them by highlighting differences in levels of numerous proteins as well as differences in protein aggregate assembly, distribution and morphology (Umoh et al., 2018; Laferriere et al., 2019). A sequential biochemical extraction technique to purify detergent-insoluble aggregated proteins resulted in enrichment of phosphorylated TDP-43 and identification of low-solubility proteins associated with TDP-43 pathology (Laferriere et al., 2019). Subsequent MS of these enriched proteins determined distinct patterns of enrichment amongst ALS (23 proteins) and subtypes of FTLD (FTLD-A: 8 proteins; FTLD-C: 10 proteins), providing insights into potential causes of disease heterogeneity (Laferriere et al., 2019). A summary of studies to investigate the mechanisms of ALS and FTD that have used human brain and spinal cord samples for LFQ are presented in Table 1.
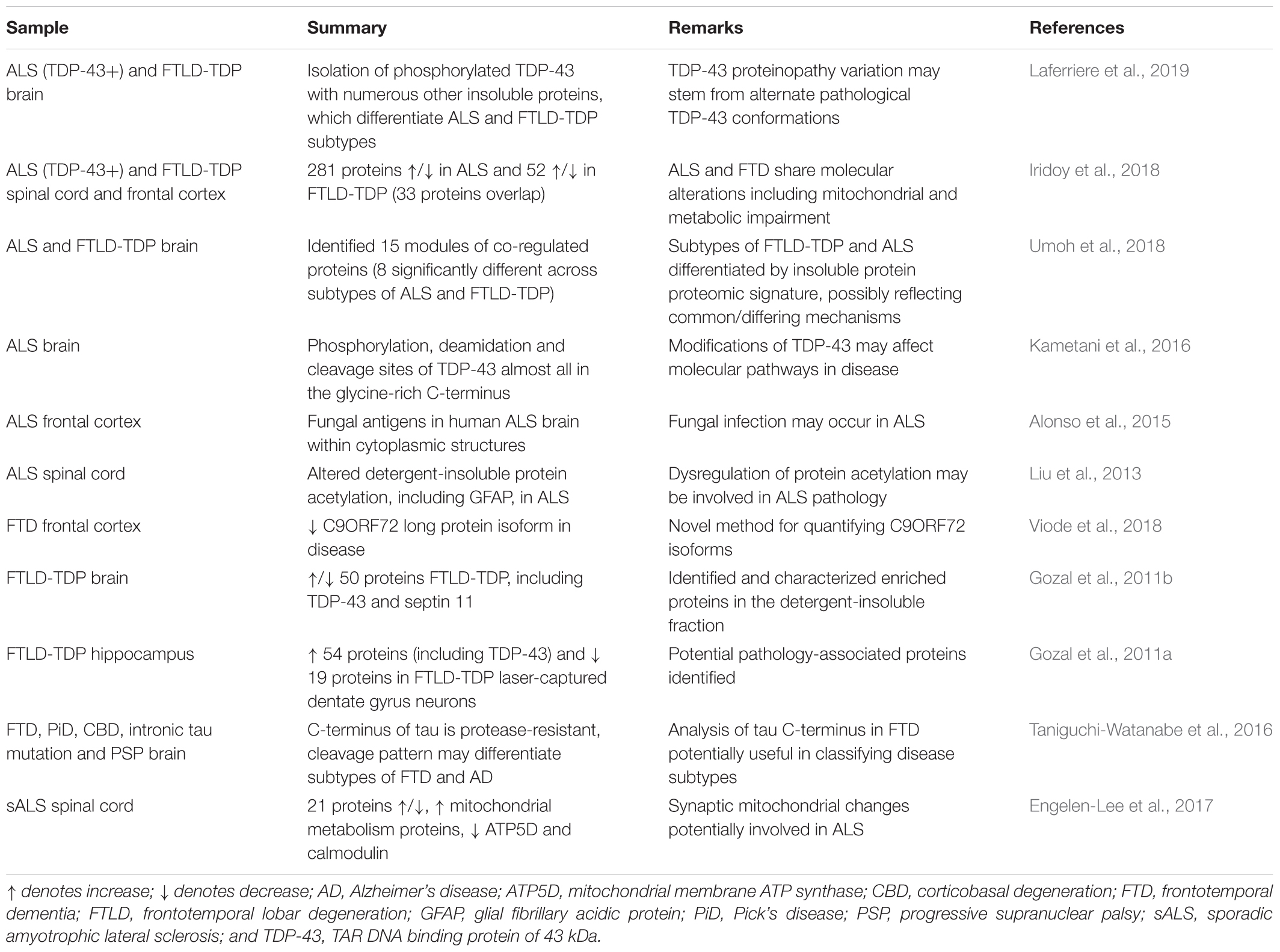
Table 1. Human tissue proteomics studies using label-free techniques for mechanistic insight into ALS and FTD.
LFQ proteomics has also been applied to animal models, for example identifying interactors of misfolded SOD1 in the spinal cord of SOD1G93A mice over three pre-symptomatic time points in disease (Ruegsegger et al., 2016). Only 5 identified proteins were common across these groups with high confidence, including HSPA8. The bulk of proteins found to interact with misfolded SOD1 were identified at the latest time point (Ruegsegger et al., 2016). Of all mutant SOD1 interactors, the activity of Na+/K+ATPase-α3 was decreased and exhibited higher levels of protein expression in high-vulnerability motor neurons (Ruegsegger et al., 2016).
Mutations in cyclin F (CCNFS621G), which cause rare familial cases of ALS/FTD, have also been studied by overexpression in neuronal cell lines and zebrafish to investigate the effects and mechanisms of ALS/FTD (Williams et al., 2016; Hogan et al., 2017; Lee A. et al., 2018). Hundreds of proteins were increased or decreased upon expression of disease-linked mutant cyclin F protein (Lee A. et al., 2018). The differentially expressed proteins clustered to cell pathways involved with cellular survival and toxicity, and predicted activation of caspase-3 mediated cell death (Hogan et al., 2017). Studies such as these that combine analysis of different models, such as cells and in vivo systems, using both proteomics and complementary validation approaches offer the best approach to identify biologically relevant changes. The selection of proteins for validation is often based on the statistically significant findings from the proteomic analysis as well as previously published work in the literature, however, pathway-level approaches for analysis of large datasets are often more informative than extensive analysis of individual proteins. A summary of ALS and FTD studies that have used animal models for LFQ are presented in Table 2.
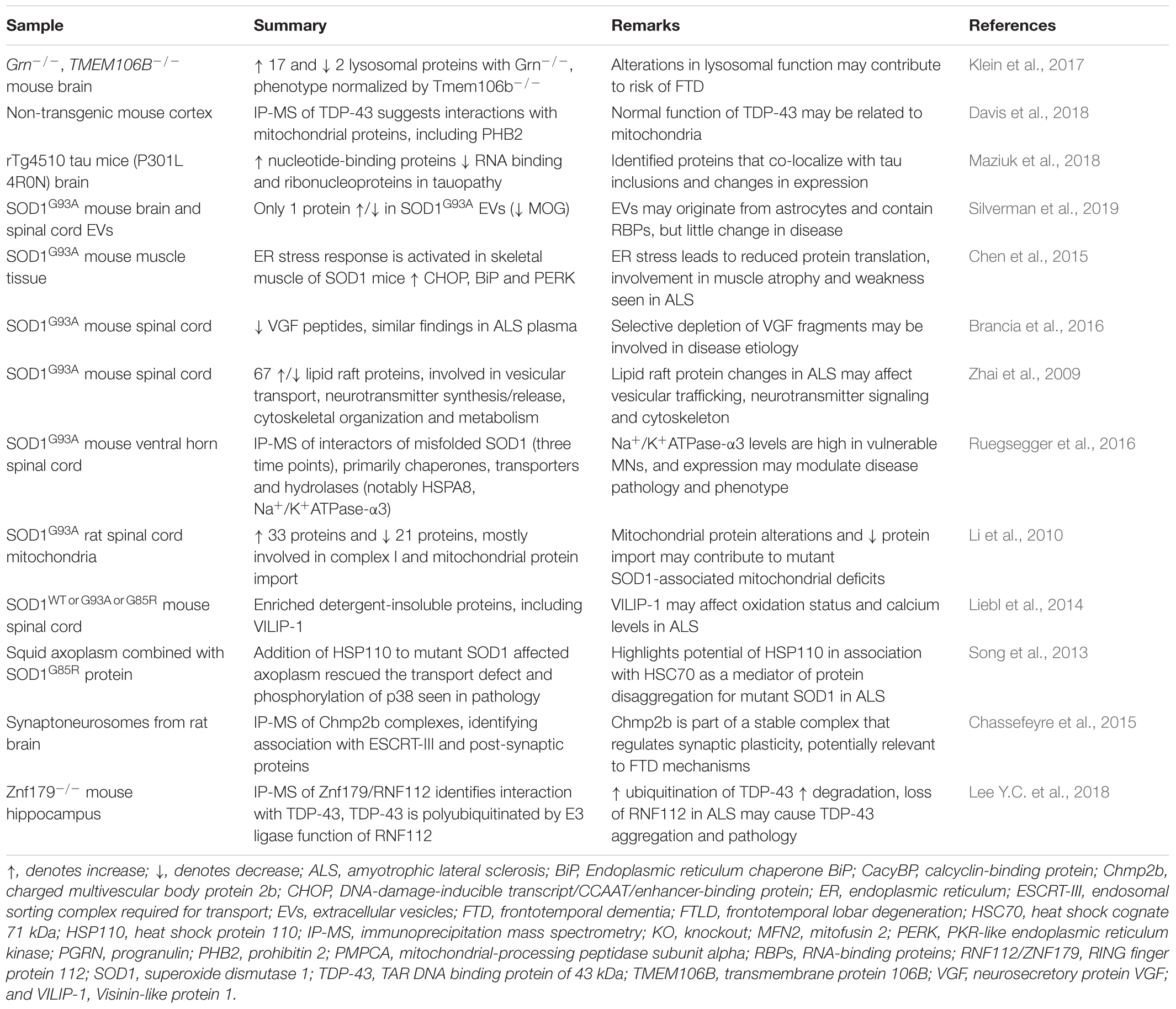
Table 2. Animal model proteomics studies using label-free techniques for mechanistic insight into ALS and FTD.
The majority of the previous investigative studies have used heterogeneous samples, which poses a limitation. Rather than representing proteomes from only a specific cell type of interest, they largely consist of populations of homogenized and variable cell types. To account for this, reemergence of laser-capture microdissection of tissues has been used to isolate distinct cell-types, and with recent advances in MS technologies this can now be successfully applied to proteomics studies of neurons from brain samples (Aring et al., 2018). Newer technology that is highly sensitive and specific can thus generate a greater number of protein hits from small amounts of extracted sample, and this offers opportunities for future studies of both model systems and human ALS and FTD samples.
In addition to studies of tissue samples, much proteomics research has been applied to understand ALS and FTD pathogenesis using cell culture models. This approach, which usually makes use of homogenous populations of cells such as immortalized cell lines, removes the confound of studying mixed samples that is often inherent using tissue samples and biofluids. Expression of disease-associated proteins, including mutant SOD1, TDP-43, and tau, recapitulate some features of disease in cell models, such as protein aggregation, inclusion formation and cellular toxicity (Gauthier-Kemper et al., 2011; Cohen et al., 2015; Celona et al., 2017; Monahan et al., 2017; Wang et al., 2017). Proteomics studies of these models have been used to identify proteins that co-aggregate or interact with the known pathological proteins, which may provide insight into disease pathogenesis. Identification of proteins associated with disease pathology may also help reveal proteins of potential therapeutic use if, for example, those proteins are able to modulate protein aggregation or toxicity (Celona et al., 2017; Mallik et al., 2018). A summary of ALS and FTD studies that have used cell models for LFQ are presented in Table 3.
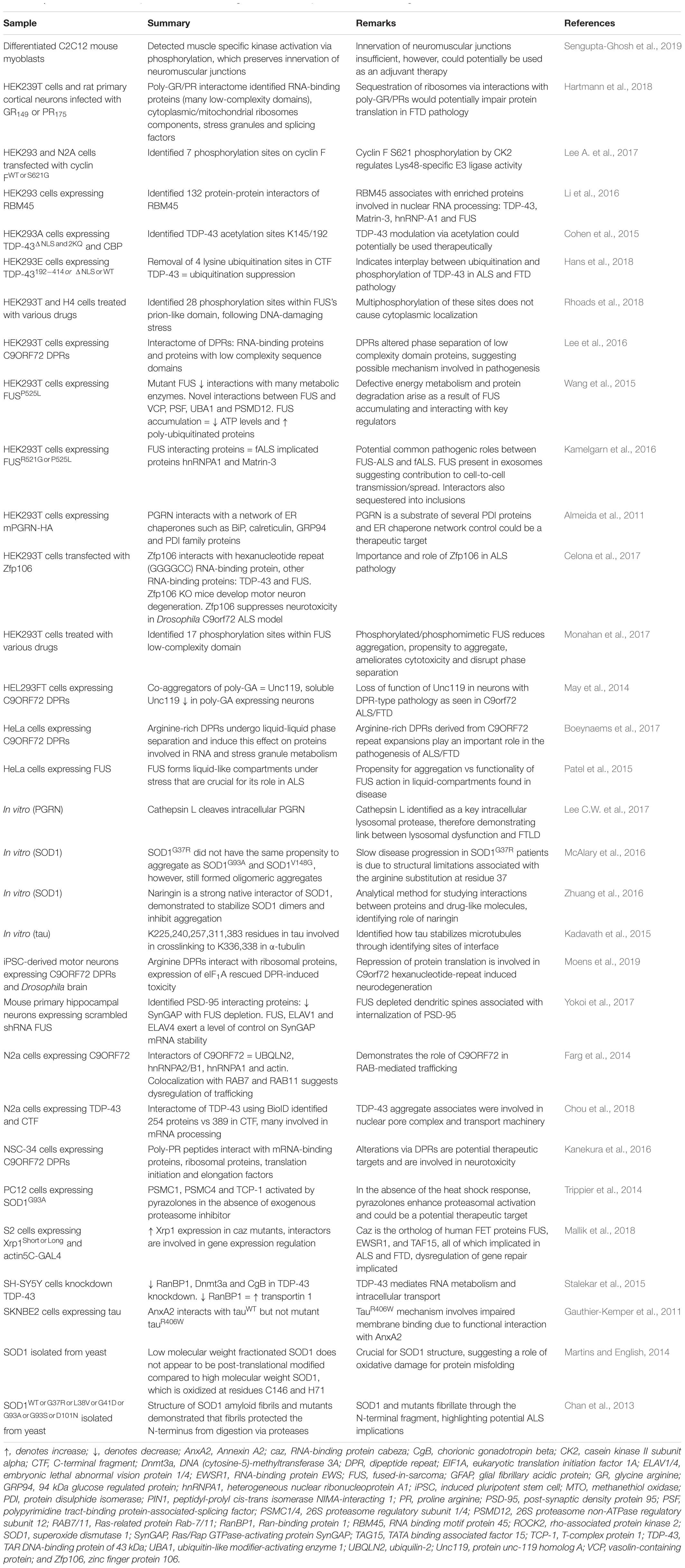
Table 3. Cell and in vitro model proteomics studies using label-free techniques for mechanistic insight into ALS and FTD.
The main limitations of LFQ using data-dependent acquisition is the generally low proteome coverage and low sensitivity, since many low intensity ions (often from low abundance proteins or poorly ionized peptides) are missed in precursor ion selection. In order to improve this coverage, additional sample prefractionation steps (such as strong cation exchange or basic reverse phase chromatography) can be used to reduce the complexity of the sample to be analyzed by MS. A recent LFQ approach that is being more widely adopted relies on data independent quantitation with methods such as Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH) that attempt to circumvent some of these issues associated with analyzing complex samples by data-dependent acquisition.
SWATH, or data-independent acquisition (DIA), acquires data by cycling through predefined sequential windows of a chromatographic elution range generating a larger number of identified proteins from a complex mixture (Gillet et al., 2012). Recently, SWATH has been used to identify differences in blood samples between 42 ALS patients and 18 healthy controls, which revealed a panel of novel potential biomarkers for diagnosis and use in clinical trials (Xu et al., 2018). This study found significant differences in the expression of 30 proteins that varied in ALS patients with or without cognitive impairments. This study highlights the potential of DIA methodologies, such as SWATH, to discover markers in biofluids that may have further utility for clinical use that offers promise to provide further advances in ALS and FTD studies. A comprehensive review of the technical aspects of SWATH is discussed elsewhere (Ludwig et al., 2018).
Matrix-assisted laser desorption ionization (MALDI) is an alternative method to introduce a sample into a MS. This differs from electro-spray ionization (ESI) through focusing laser energy at a matrix-embedded sample for low fragmentation and reduced multi-charged ion states (Nadler et al., 2017). Typically ESI dominates the literature as the intermediate in LC-MS, used in all the studies presented in Tables 1–3. There are limitations to each technique, which are discussed elsewhere (Nadler et al., 2017), and the choice of technique generally depends on the biological question. For example, in ALS and FTD studies, MALDI-MS has been successfully applied to identify increased levels of ubiquitin carboxy-terminal hydrolase L1 (UCHL1) in FTLD-tau cortex (Schweitzer et al., 2006) as well as increased carbonylation of UCHL1 in the spinal cord of SOD1 mice (Poon et al., 2005). Together these findings have highlighted the roles of the oxidative stress response and ubiquitin-mediated degradation in ALS and FTD. MALDI-MS has also been used to identify increased levels of proprotein convertase 1 inhibitor (ProSAAS) in FTD CSF (Davidsson et al., 2002) and potentially pathologically involved ProSAAS N-terminal fragments in the temporal lobe of FTD samples (Kikuchi et al., 2003). More recently, MALDI-MS has been used to identify interactions between Staufen1 and dynein, mediated via protein phosphatase 1-beta, implicating a role of Staufen1 in regulating mRNA localization in ALS (Gershoni-Emek et al., 2016). This has been reinforced through a described link between Staufen1 RNA stress granules and autophagy through interaction with ataxin-2 (Paul et al., 2018). A summary of ALS and FTD studies that have used MALDI-MS is presented in Table 4.

Table 4. MALDI-MS proteomics studies for mechanistic insight into ALS and FTD.
Quantitation via Labeling Proteomics Approaches for ALS and FTD
Labeled-based approaches have considerably higher quantitation accuracy in exchange for lower proteome coverage compared to LFQ (Megger et al., 2014). The labeling techniques involve the introduction of stable isotope labels on the proteins or peptides, which allow the mass spectrometer to distinguish between identical peptides from different samples within a mixture. Quantitative labeling methods can also be used for protein-protein interactions (PPIs) and post-translational modifications (PTM) analyses which are discussed below (Sullivan et al., 2016; Russell et al., 2017). A comprehensive review of the technical aspects of labeled-based proteomic approaches is available (Lindemann et al., 2017).
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)
Stable Isotope Labeling by Amino acids in Cell culture (SILAC) labeling is achieved by growing cells in culture with growth medium containing different isotopically labeled amino acids that are incorporated during protein synthesis (Ong et al., 2002). The protein lysates from different isotope-labeled cell culture are pooled together in equal amounts and prepared as one grouped sample to decrease sample preparation variability. High resolution mass spectrometers can analyze and resolve the different precursor (peptide) ions within an experiment and detect the signal intensities of the labeled peptides. The intensities of the precursor ions are used as a measure of the relative abundance of the protein within each sample. One limitation of SILAC, however, is the relatively small number of labeled amino acid combinations [maximum reported to date is 6-plex (Wang et al., 2013)] that can be used in one experiment, which limits the number of comparisons.
The versatility of SILAC can extend to the generation of an internal standard in a cell-line to quantify low abundance proteins in tissue (known as “Super SILAC”). This was applied to investigate the accumulated proteins in detergent-insoluble brain lysates from four control and four FTLD brain samples, with comparison to a HEK293 standard (Seyfried et al., 2012). A summary of ALS and FTD studies that have used SILAC is presented with other labeled methods in Table 5.
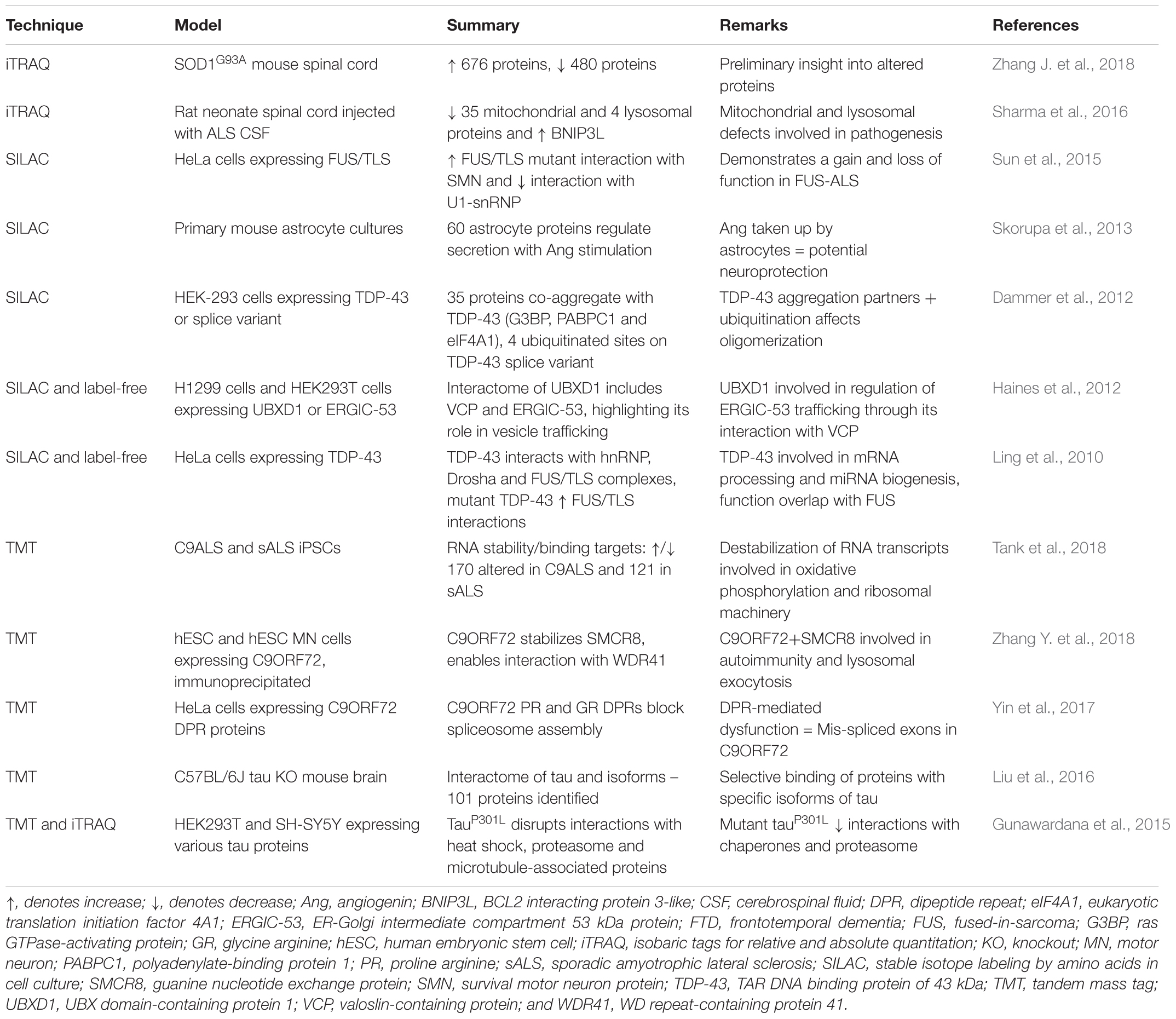
Table 5. Labeled-MS proteomics studies for mechanistic insight into ALS and FTD.
Tandem Mass Tags (TMT)
Unlike SILAC, which incorporates isotopically labeled amino acids at the protein level, the Tandem Mass Tag (TMT) system is used to label peptides following proteolytic digestions (Thompson et al., 2003). TMT tags are covalently coupled to both N-terminal α-amino groups and 𝜀-amino groups of lysine residues (Thompson et al., 2003). Once labeled, peptide samples are pooled (commonly up to 10-plex), and subsequently fractionated and analyzed by a high-resolution mass spectrometer. The secondary detection of reporter ions (MS2) allow the peptides to be quantitated based on the TMT signal intensities, which can be extended to a third round (MS3) to decrease ratio distortion (Ting et al., 2011).
TMT-based quantitative proteomics is being increasingly applied in neuroscience, including in several investigations of proteome-wide nucleocytoplasmic changes in cell-based models of ALS. In mouse motor neuron-like NSC-34 cells overexpressing mutant hSOD1G93A, TMT-labeling of peptides combined with the results of RNA-Seq demonstrated impairments to nuclear and cytoplasmic transport (Kim et al., 2017). Specifically, proteins enriched in the nuclear fraction of mutant SOD1-expressing cells were related to RNA transport/processing and known Huntington’s disease/Alzheimer’s disease pathways, whilst proteins enriched in cytoplasmic fractions were involved in protein folding, aminoacyl-tRNA biosynthesis, Wnt signaling, synaptic vesicle cycle and Hippo signaling pathways (Kim et al., 2017). In another study, TMT 10-plex was used to analyze ALS patient fibroblast-derived iPSC cells to validate genome-wide RNA instability in ALS and FTD patients (Tank et al., 2018). Bromouridine tagging of RNA transcripts identified profound destabilization of ribosomal and mitochondrial transcripts, which was verified by TMT-quantitative proteomics and revealed corresponding decreases in mitochondrial components and compensatory increases in protein synthesis (Tank et al., 2018). This approach suggested that RNA instability could be a targeted effect of TDP-43 accumulation, disrupting energy production and protein synthesis, culminating in cell death (Tank et al., 2018). A summary of studies that have used various labeling-based MS techniques such as TMT for investigation of ALS and FTD is presented in Table 5.
Protein-Protein Interactions
Interrogating PPIs in cultured cells, animal models of disease and from human tissue can provide valuable insight into mechanisms that underlie neurodegeneration. Characterizing the interactome of proteins of interest in both healthy and disease states not only reveals normal protein function but also sheds light on pathogenic changes. Characterizing PPIs also has therapeutic value since specific interactions may be amenable to therapeutic modulation.
MS-based methods enable high-throughput identification of PPIs and have been extensively used to characterize the interactomes of many proteins implicated in neurodegeneration (Hosp et al., 2015). Standard methods used to identify PPIs are based on an immunoprecipitation (IP) followed by MS; however, in recent years, proximity-ligation methods have emerged which complement standard IPs and provide additional insight into protein interactomes. These methods include proximity-dependent biotin identification (BioID) and ascorbate peroxidase (APEX)-based proximity tagging (Roux et al., 2012, 2013; Chu et al., 2017). BioID, APEX and related techniques can also be used to study interaction partners of insoluble proteins, a feature which makes these methods particularly useful when analyzing neurodegenerative diseases characterized by protein aggregation (Roux et al., 2013).
Immunoprecipitation and MS (IP-MS)
A standard method that is used to identify PPIs relies on maintaining the physical interaction between the interaction partners. Prior to IPs, cells are lysed in non-denaturing buffers which can maintain stable but not transient interactions. A typical process for IP-MS studies is based on antibody recognition of a protein of interest within a lysate, followed by specific isolation of the antibody (and associated proteins) using protein A or G conjugated to beads (Markham et al., 2007). Of note, this method requires that PPIs remain stable after cell lysis and that the protein complex of interest remains soluble in the chosen lysis buffers. In ALS and FTD studies, IP-MS has been used to identify interacting partners of TDP-43, revealing many proteins involved in RNA metabolism (Freibaum et al., 2010). Recently, identified interactors of RNA-binding protein 45 (RBM45), a protein that colocalizes with ALS and FTD inclusions, were associated with multiple hnRNP and EIF proteins involved in RNA metabolism, suggesting disturbance of these processes upon pathology formation in disease (Li et al., 2016). As IP-MS studies generally incorporate label-free MS they have been included amongst studies previously discussed in Tables 1–3.
Proximity-Ligation Methods
BioID and APEX are proximity-ligation methods which facilitate the covalent attachment of biotin onto proteins in close proximity to a protein of interest. The application of these methods for interrogating PPIs of proteins is based on the strong interaction between biotin and streptavidin (Green, 1990) and the stability of streptavidin in a large range of conditions. This includes stability in denaturing buffers containing ionic detergents such as sodium dodecylsulfate (SDS) (Sano et al., 1998) and chaotropic reagents such as urea (Kurzban et al., 1991), such that these approaches are applicable to studies of both detergent-soluble and -insoluble proteins from cells and tissues.
Proximity-dependent biotin identification (BioID)
Proximity-dependent biotin identification or BioID is based on the use of an engineered biotin ligase which carries an R118G mutation within its active site, effectively nullifying self-association and DNA binding (Roux et al., 2012). Normally, biotin ligase (BirA) works by converting biotin to a highly reactive biotinoyl-AMP intermediate in an ATP-dependent manner. This intermediate is then deposited onto lysine residues within the natural substrate of BirA, Biotin Carboxyl Carrier Protein (BCCP) (Cronan, 1990). Engineered biotin ligase (BirA∗) is also able to convert biotin to a highly reactive intermediate, however, due to this R118G mutation, the intermediate is prematurely released from the active site of BirA∗ and diffuses away leading to biotinylation of nearby lysine residues (Kwon et al., 2000). In this way BirA∗ can biotinylate lysine residues of proteins in close proximity to the protein of interest. In a typical BioID workflow, a construct encoding BirA∗ in frame with a protein of interest is first generated and expressed in live cells. Exogenous biotin is then added to cell culture media such that biotin can be processed by BirA∗ and deposited onto proteins within proximity to the fusion protein. Here, labeling typically occurs over 12–24 h to generate enough material for analysis. In addition, the half-life of the biotin intermediate is in the minutes range which results in a large labeling radius (Rhee et al., 2013). Cells can then be lysed before streptavidin-conjugated beads are used to isolate biotinylated proteins from the complex mixture. These isolated biotinylated proteins can then be identified using standard MS-based workflows. BioID approaches have only recently begun to be applied to studies to understand mechanisms of ALS and FTD and discussion continues into their use for neurodegenerative diseases (Rayner et al., 2019). Most notably, a BioID approach was used to characterize the interactome of TDP-43 and a C-terminal fragment which revealed a correlation between C-terminal fragments and nuclear pore defects (Chou et al., 2018).
In recent years, a smaller, more efficient form of the biotin ligase, termed BioID2, has been established (Kim et al., 2016). BirA∗ has also been modified using directed evolution which has improved the labeling kinetics of biotin ligase such that labeling can be completed in 10 min. This variant of BirA∗, termed “TurboID” has been implemented in live organisms including Caenorhabditis elegans and Drosophila melanogaster (Branon et al., 2018), demonstrating the versatility of this method to interrogate the interactome of proteins in both live cells and organisms. These improvements make this system a key new technology for future application to the understanding of protein interactions in neurodegeneration, using both cultured cells and animal models of disease.
Ascorbate peroxidase (APEX)-based proximity tagging
APEX is a 28 kDa engineered peroxidase that is derived from dimeric pea or soybean ascorbate peroxidases (Martell et al., 2012; Rhee et al., 2013). APEX can be used in a similar manner to BioID for the biotinylation of proteins in the vicinity of a protein of interest. Like BioID, constructs are first generated to encode engineered soybean peroxidase in frame with a protein of interest before the fusion protein is expressed in live cells. Notably, APEX can be used for temporally resolved proteomics as well as high-resolution microscopy, making this a flexible technique for studies of protein interactions.
Once the APEX fusion protein is expressed in live cells, exogenous biotin is added to cell culture media. For labeling of proximal proteins, hydrogen peroxide (H2O2) and biotin phenol is added to media such that APEX can catalyze a one-electron oxidization of biotin phenol to a biotinphenoxyl radical which subsequently biotinylates proteins in proximity (Lam et al., 2015). Notably, labeling kinetics of APEX is fast. The produced biotin-phenoxyl radical lasts for <1 ms which enables labeling times of 1 min and limits the labeling radius to 20 nm (Chu et al., 2017). After 1 min, the reaction is stopped with quenching buffer to prevent further biotinylation post-lysis. Cells may then be lysed before biotinylated proteins are isolated by streptavidin conjugated to beads and identified using MS-based workflows. An advantage of the short labeling timeframe is that APEX can be used to capture dynamic changes in PPIs. This may become advantageous for time course studies where protein interactions may dynamically change in response to various stimuli such as cell stress or drug treatments.
APEX can also be applied to analysis by electron microscopy (EM) (Martell et al., 2012; Lam et al., 2015), thereby enabling the interrogation of protein localization at high resolution. Here, fusion proteins are expressed in cells before cells are fixed and treated with diaminobenzidine (DAB) and H2O2 (Lam et al., 2015). APEX catalyzes the polymerization and local deposition of DAB in the vicinity of the fusion protein, enabling subsequent recruitment of electron dense osmium for EM applications (Lam et al., 2015).
By applying APEX for both proteomics and EM workflows, APEX can be used to characterize dynamic changes in the protein interactome of live cells whilst also accurately defining protein localization at high resolution. In recent years, variations of APEX have emerged. This includes APEX2 (Lam et al., 2015) which has improved catalytic efficiency and split APEX (sAPEX) which enables the interrogation of two known protein interaction partners or proteins known to be in close proximity (Han et al., 2018). Recently, the APEX technique has been applied to profile the components of stress granules, which are enriched in RNA-binding proteins including the ALS/FTD-linked proteins TDP-43 and FUS (Markmiller et al., 2018). Notably, these studies revealed previously unrecognized stress granules (SGs) proteins that also displayed alterations in induced pluripotent stem cell-derived motor neurons from ALS patients and for which modulation of expression was protective in Drosophila ALS models, suggesting disturbances in SGs may be related to disease pathogenesis (Markmiller et al., 2018).
Future Applications of Proximity Labeling Techniques
Biotinylation by Antibody Recognition (BAR) is another proximity-labeling technique which has recently been established for investigating PPIs in fixed samples (Bar et al., 2018). Here, cultured cells or tissue are fixed and permeabilized before a primary antibody is used to target a protein of interest. A secondary antibody conjugated to horseradish peroxidase (HRP) is then used to recognize the primary antibody, and labeling of proximal proteins is achieved by HRP in the presence of H2O2 and biotin phenol. The conjugation of biotin enables the isolation of these proximal proteins. Although this method has not yet been applied for the study of neurodegenerative diseases, it is a promising technique for future analysis of proteins involved in ALS and FTD and may be particularly useful for analysis of detergent-insoluble protein aggregates and inclusions.
Analysis of Protein Post-translational Modifications in ALS and FTD
Post-translational modifications provide the basis of biological diversity in the proteome by enabling the same protein to carry out diverse functions through characteristic variations of modifications and their temporal regulation. PTMs are small, covalent, amino acid modifications added onto a protein; they are highly dynamic and play key roles in selectively regulating protein function in cells, tissue, and biofluids, such as CSF (Guldbrandsen et al., 2014). PTMs regulate most aspects of intracellular function involving but not limited to, DNA repair, proliferation, subcellular localization, transport, and proteolytic cleavage of functional units or protein degradation. On a larger scale PTMs also modulate intercellular functions like cell signaling and adhesion (Minguez et al., 2012; Theillet et al., 2012; Olsen and Mann, 2013). Various types of PTMs exist (Prabakaran et al., 2012), with arguably the most studied in proteostasis and neurodegenerative diseases being: ubiquitination, phosphorylation, nitration, acetylation, oxidation, glycosylation, methylation, and sumoylation (Minguez et al., 2012).
Pathological phosphorylation of proteins such as tau, TDP-43, α-synuclein or FUS contribute to the formation of protein inclusions in neurodegenerative disease (Hasegawa et al., 2008; Despres et al., 2017; Monahan et al., 2017; Cykowski et al., 2018). For example, tauopathies caused by the phosphorylation of tau in FTD and AD are comprised of aggregated proteins in fibrillary tangles (Ferrer et al., 2002). While the underlying mechanisms of misfolded protein aggregation and neurotoxicity in neurodegeneration is not yet understood, PTMs such as ubiquitination are of interest given their regulatory role in protein degradation. For example, the temporal regulation of phosphorylation and ubiquitination in protein signaling cascades, and hyperubiquitination by mutant cyclin F cause defects to protein degradation pathways that are associated with ALS/FTD (Lee A. et al., 2017). Furthermore, other examples of aberrant PTMs include acetylation of glial fibrillary acidic protein in ALS (Liu et al., 2013), acetylated TDP-43 in ALS (Wang et al., 2017) and sialylation of amyloid precursor protein in AD (Nakagawa et al., 2006).
Detecting PTMs by Mass Spectrometry
Many PTMs can be mapped by tandem MS (MS/MS) (Dephoure et al., 2013), which offers an unbiased approach that can be verified by alternative biochemical methods such as immunoblotting and immunofluorescence microscopy. To date, the presence of PTMs such as ubiquitination, phosphorylation, acetylation, and nitration, and their stoichiometric analysis have been enabled by qualitative and quantitative proteomics, respectively, on a global proteomic scale in ALS-linked proteins (Sacksteder et al., 2006; Liu et al., 2013; Shi et al., 2013a; Lee A. et al., 2017; Wang et al., 2017). Despite the capabilities of MS to identify PTMs, they are often difficult to detect as they vary in physiochemical properties, can exist as transient modifications, be present in sub-stoichiometric concentrations and are sensitive to sample preparative steps that include high and low pH buffers, trypsinization, and de-salting steps (Wei and Li, 2008; Olsen and Mann, 2013). To circumvent many of the issues associated with analyzing PTMs such as ubiquitination and phosphorylation, enrichment strategies to isolate peptides containing these modifications together with quantitative proteomic approaches can provide a more in-depth analysis of the sub-proteome.
Ubiquitination
A popular enrichment strategy for identifying ubiquitination sites on peptides called diglycine (diGLY) enrichment exploits the cleavage sites (K-𝜀-GG) of ubiquitinated proteins following trypsin digestion (Kim et al., 2011). The covalent attachment of ubiquitin on lysine residues on proteins allows trypsin to cleave both the C-terminus of ubiquitin and the C-terminus of the lysine amino acid on the ubiquitinated protein exposing two glycine residues. Ubiquitin remnant motif (K-𝜀-GG) antibodies are used to enrich peptides containing the di-Gly-Gly motif, followed by elution and analysis by LC-MS/MS in which the presence of the GlyGly residue on lysine (+m/z 114.04) in the MS spectra is an indication of a ubiquitinated peptide (Peng et al., 2003; Xu et al., 2010; Fulzele and Bennett, 2018). However, ubiquitin-like proteins also exist that may be identified using this method such as SUMO (sumoylation), NEDD8 (neddylation) and interferon-stimulated gene 15 (ISG15; isg15ylation) also known as ubiquitin cross-reactive protein (UCRP) (Hemelaar et al., 2004). Quantitative assessment of the stoichiometry of site-specific ubiquitination can be achieved using the recently reported “isotopically balanced quantification of ubiquitination” (IBAQ-Ub) method which employs an amine-reactive chemical tag (AcGG-NHS) that is structurally homologous to a tryptically cleaved ubiquitinated peptide containing a GG remnant of ubiquitin on the modified lysine residue. These AcGG-NHS tagged peptides allows the generation of structurally identical peptides from ubiquitinated and unmodified lysine residues that can be further labeled using a secondary stable isotope (Li et al., 2019). Other areas which are generating interest are investigating the specific poly-ubiquitin linkages, in which isotope-labeling of the lysine residues in ubiquitin can be used, such as “UB-AQUA” and detected by high resolution MS via narrow window extracted ion chromatogram or by selected reaction monitoring (SRM-MS) using a triple quadrupole MS (Phu et al., 2011). Overall, these various methods enable stoichiometric analysis of the poly-ubiquitin modifications on target proteins, which could provide insights into the ubiquitin code underlying altered protein degradation in neurodegeneration.
Phosphorylation
Protein aggregates containing phosphorylated proteins is a key pathological feature in many neurodegenerative diseases, such as phospho-tau in Alzheimer’s disease and FTD (Sjogren et al., 2001), phospho-TDP-43 in ALS and FTD (Hasegawa et al., 2008) and phospho-alpha-synuclein in Parkinson’s disease (Wang et al., 2012). Phosphorylation of serine, threonine, and tyrosine residues are generally the most common sites in mammals, which are estimated to compromise of approximately 90, 10, and <1% of the total phosphoproteome, respectively (Nita-Lazar et al., 2008). The main challenges with characterizing phosphorylated proteins using MS is the direct detection of low stoichiometric concentrations of phosphopeptides resulting from trypsin digestion of complex samples. This is further complicated by ion suppression of phosphopeptides due to the negative charge and lability of the phosphate moiety. Therefore, many approaches have been developed to enrich phosphopeptides prior to MS-based analysis, including titanium dioxide (TiO2), immobilized metal affinity chromatography (IMAC), and hydrophilic interaction chromatography (HILIC), discussed elsewhere (Arrington et al., 2017). The detection of a phosphopeptide can be interpreted by the neutral loss of H3PO4 (-m/z 98) in the mass spectra. Many algorithms have been developed for interpreting MS/MS fragmentation spectra and phosphorylation site localization such as Mascot Delta Score (Savitski et al., 2011), PhosphoRS localization (Taus et al., 2011) and PTM score of MaxQuant (Cox and Mann, 2008). These algorithms match the spectra generated to a protein sequence database of choice (e.g., UniProtKB/Swiss-Prot) to assign the probabilities of potential phosphorylation sites.
Previously, sarkosyl-insoluble pathological TDP-43 from brains of two ALS patients were purified and subjected to LC-MS/MS analysis, identifying several novel phosphorylation sites, deamidation sites, and cleavage sites (Kametani et al., 2016). Seventeen phosphorylation sites were identified from both ALS patients which were predominately located in the Gly-rich C-terminal region on TDP-43, while most of the cleavage sites were located in N-terminal half, suggesting that these sites may be more accessible to proteolytic enzymes (Kametani et al., 2016). Immunoblot analysis using the phospho-specific TDP-43 S409/410 antibody, which recognizes the abnormal phosphorylation of Ser409 and 410, verified the presence of pathological phospho-forms of TDP-43 and additional fragments of 18∼24 kDa. These findings indicate that phosphorylation plays an important role in the mechanism of TDP-43 pathogenesis, and suggests that enrichment strategies to comprehensively characterize the phosphoproteome are highly relevant to understanding neurodegenerative diseases.
Bioinformatics
Tandem mass spectra generated by MS are analyzed using specialized software and algorithms to identify and quantitate peptides and proteins using two main approaches: database search and de novo search. Database searching follows the “Exact Pattern-Matching rule,” which consists of only selecting spectra masses that exactly match a sequence in a multi-species- or species-specific database. The most frequently used programs with a database search implemented are SEQUEST (Eng et al., 1994), Mascot (Perkins et al., 1999), and X!Tandem (Craig and Beavis, 2004). These programs not only extract candidate proteins using a combination of the open access database and their own in-built database but also score them using algorithms based on the ion signals observed in the spectra. Some limitations of the database search method include that a large portion of acquired spectra will be rejected because of the “Exact Pattern-Matching rule” and not all organisms have a complete protein sequence reference.
De novo peptide sequencing generates a list of all the highest scored peptides from the MS/MS spectra and the mass values given without the need of a reference database. With high resolution mass spectrometers producing quality data, the performance of de novo sequencing has remarkably improved. However, a significant caveat pertains to resolving amino acids with or without modifications, due to either identical mass or near-identical masses. In addition, there is an inverse relationship between the accuracy of this method and peptide length (Muth and Renard, 2018). New strategies are being created in this space to overcome these limitations, such as post novo, which post-processes de novo sequence filter prediction (Miller et al., 2018).
Commonly used bioinformatics programs such as STRING (Szklarczyk et al., 2019), BioGrid (Oughtred et al., 2019), Ingenuity Pathway Analysis (IPA) (Yu J. et al., 2016), and DAVID (Huang da et al., 2009) are frequently used to interrogate the data obtained from large scale quantitative proteomic experiments to get a snapshot of the expressional changes that occur within a sub-proteome. Notably, depending on the biological question, these informatics tools can assist in quickly identifying networks, pathways and functions to facilitate further experiments and generate new hypotheses. For example, proteomics data generated from IP of TDP-43 from HEK293 lysates were analyzed by STRING, which demonstrated clear nuclear and cytoplasmic interaction networks (Freibaum et al., 2010). Assigning protein functions using bioinformatics software therefore provides a higher-level overview into changes and perturbations occurring during disease, for example allowing the identification of disease relevant biochemical pathways. A more detailed review of the bioinformatics tools used for proteomics is discussed elsewhere (Schmidt et al., 2014; Martens and Vizcaino, 2017).
Flow Cytometric Purification of Inclusion Bodies Associated With ALS and FTD
Determination of the composition of insoluble inclusions formed in many neurodegenerative diseases is key to understanding pathogenesis. The use of proteomics will allow greater understanding of the global protein composition of these inclusions and allow us to identify proteins and cellular pathways that are either involved in the assembly or attempted disassembly of the inclusion, and distinguish these from proteins that were non-specifically sequestered. Comparing the protein composition of inclusions formed from a variety of pathogenic proteins (e.g., TDP-43- or SOD1-positive inclusions in ALS) could also help to establish common pathways involved in this process across different types of neurodegenerative diseases.
Fluorescence activated cell sorting (FACS) is one technique that can be used to purify insoluble protein or inclusions from cell culture or tissues. Regarding cell-based models of neurodegenerative disease-associated protein aggregation, gentle plasma membrane permeabilization in combination with FACS could be employed to purify inclusions for proteomic analyses. A method for this was recently described, whereby cells are permeabilized with 0.5% (v/v) TritonX-100 in PBS supplemented with protease/phosphatase inhibitors to quantify and characterize inclusions comprised of fluorescently tagged proteins of interest by flow cytometry (Whiten et al., 2016). This work also demonstrated that fluorescent aggregates and nuclei can be sorted by FACS (Whiten et al., 2016). Alternatively, cells can be treated with 0.03% (w/v) saponin to selectively permeabilize the plasma membrane, allowing soluble proteins to diffuse out of the cytoplasm, while retaining the nucleus and inclusions intact (Farrawell et al., 2018; Pokrishevsky et al., 2018). These treated cells can be washed to remove the soluble proteins that have diffused out of the cells and subsequently purify cells with inclusions by FACS. It follows that inclusions comprised of fluorescently tagged proteins, or fluorescent antibody-labeled inclusions can be purified by FACS from cells and tissues and subjected to proteomic analysis to determine their composition.
Purification of inclusions by FACS for proteomic analysis has previously been adopted to investigate the protein composition of Huntington’s disease-associated polyglutamine inclusions (Mitsui et al., 2002, 2006). Using this strategy, heat shock protein 84 and elongation factor 1α were identified as novel inclusion-interacting proteins and their over-expression in cell-based assays demonstrated cytoprotective activities (Mitsui et al., 2002, 2006). Future experiments could employ this strategy to purify insoluble protein and inclusions for proteomic analysis from ALS/FTD patient samples, rodent tissues, or cell-based models of ALS/FTLD.
Biomarker Studies of ALS and FTD
As high-throughput technologies have advanced, many proteomic-based biomarker studies have presented an abundance of potential candidates; however, it is often not feasible to properly validate each individual protein considering the heterogeneity amongst different patient cohorts, which can hinder the level of accuracy and reproducibility. Selection of which candidates to validate is not clearly defined, uniform, or standardized. This selection can be made based on a combination of statistics, current literature as well as assessed potential based on PPIs, both predicted and real.
One of the major issues that arises as a result of subjective validation is the variation of proteins identified across parallel studies, rarely reinforcing the previously presented data. For example, measurements of HeLa cells prepared identically and analyzed across different laboratories using the same instrumentation revealed a high level of biological heterogeneity on both transcriptomic and proteomic levels (Liu et al., 2019). This occurs for a number of reasons, including the sample composition and sample cohorts, environmental conditions affecting instrument parameters, as well as technical variation due to extraction/digestion procedures (Piehowski et al., 2013). Alternatively, the same protein could be reported with differing results across samples, such as decreased levels of galectin-3 in the plasma of SOD1 mice (Zubiri et al., 2018) and increased levels in human CSF (Zhou et al., 2010). These results are not necessarily conflicting but highlight the importance of validation across multiple large cohorts for biological relevance.
In recent years, an increased number of small and large-scale biomarker studies have been performed on human ALS samples by LFQ, as recently reviewed (Barschke et al., 2017). CSF from ALS, primary lateral sclerosis (PLS), cross-sectional healthy and disease controls (Parkinsons’ disease and ALS mimic disorders) were analyzed via LFQ for novel biomarker discovery (Thompson et al., 2018). Elevation in levels of chitotriosidase, chitinase-3-like protein 1 and chitinase-3-like protein 2 in a cohort of 43 patients with ALS has been revealed, indicating neuroinflammation due to increased microglial activation (Thompson et al., 2018). No direct validation was performed to confirm the proteomic data, although reference to previous ELISA studies of these chitinase proteins in ALS patient samples (likely including some from the same cohort), suggested a similar trend (Varghese et al., 2013).
Biofluid sample usage is prominent due to the need for a directly translatable and easily retrievable marker for rapid and accurate diagnosis. Alongside disease diagnosis, the ability to distinguish between subtypes of a disease would prove instrumental in developing personalized treatments. Whilst many studies have identified altered proteins across cohorts of patients, others focus on teasing apart unclear differences such as those found in subtypes of FTD (Teunissen et al., 2016). Recently, a CSF-wide proteomic analysis was performed to highlight the usefulness of biofluid MS for biomarker elucidation, identifying relevant disease-related proteins such as tau (Macron et al., 2018). In the context of biomarker discovery, newer labeled methods are being utilized to narrow down on protein hits of interest due to the greater quantitation accuracy compared to label-free methods (Megger et al., 2014). A summary of ALS and FTD biomarker elucidation studies in the last decade that have used both label-free and labeled methods is presented in Table 6. Ultimately, it is crucial to have a clear biological question to pursue for selection of sample, technique and subsequent validation approaches, although unbiased screening of potential disease-related biofluid alterations may help identify previously unrecognized research directions.
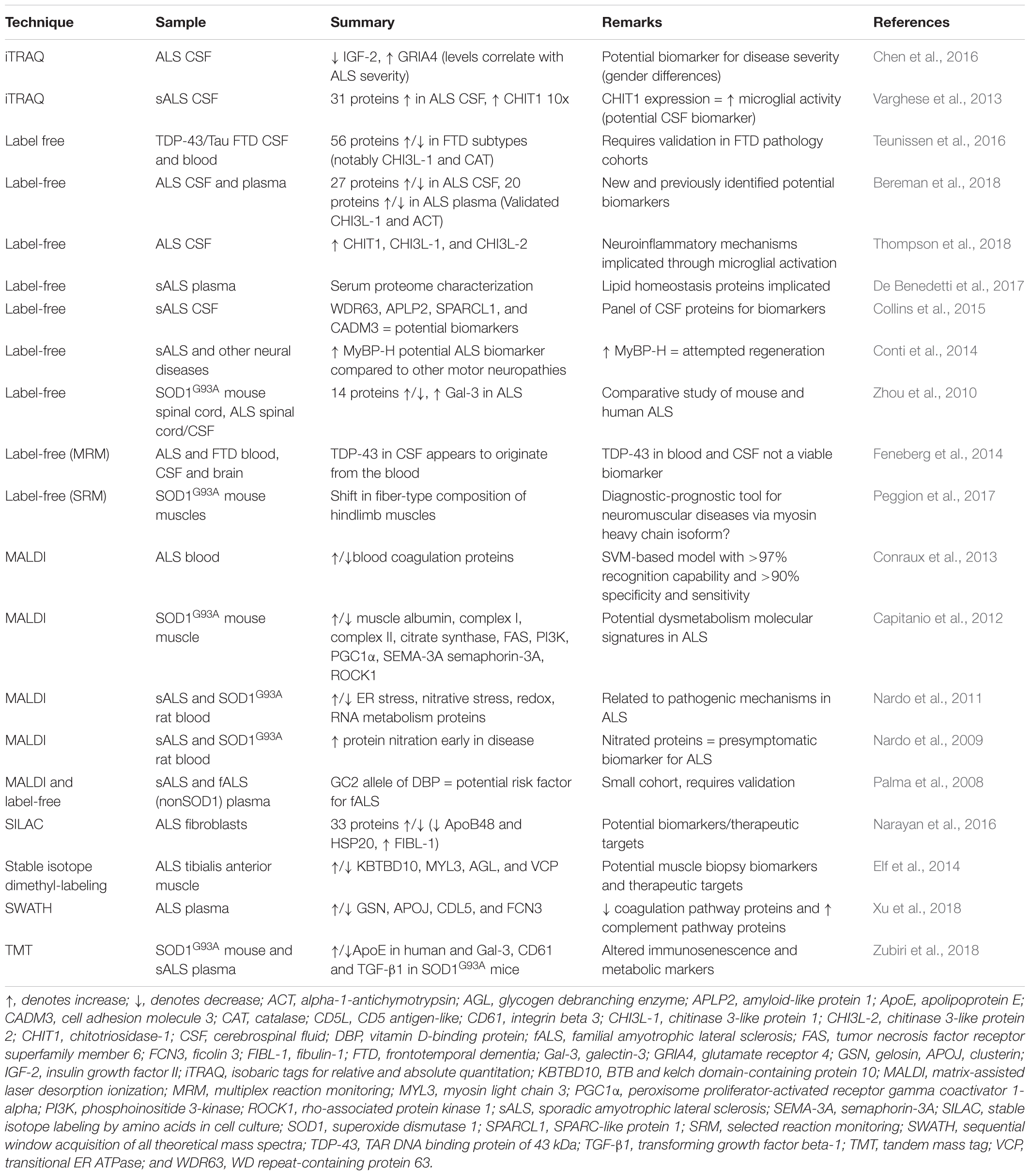
Table 6. Biomarker discovery studies using proteomics for ALS and FTD.
Future Directions and Conclusion
The technologies presented in this review have demonstrated their utility when applied to neurodegenerative diseases such as ALS and FTD. However, there needs to be a focus on validating the research laterally, solidifying findings to progress with ensuing biological questions. Incorporation of parallel approaches such as transcriptomics using RNASeq alongside results from proteomics can reinforce potential hits of interest. Studies have successfully combined transcriptomic and proteomics findings to provide mechanistic insights, such as the finding of lysosomal changes in Grn-/- mice that are modulated by the FTD-risk protein TMEM106B (Klein et al., 2017). Additionally, recent advances in this area have included correlation of transcriptomic findings with underlying TDP-43 pathology burden in a study of ALS spinal cord laser-capture dissected motor neurons (Krach et al., 2018). This was one of the first studies to utilize overlapping distinct datasets to stratify patient cohorts to identify the more relevant and important proteins involved in disease. Previously, careful patient sub-type selection has led to the identification of genetic contributors to disease, such as the finding that TMEM106b genotype is a risk modifier of FTD by GWAS of a highly specific and well-defined subgroup of FTLD samples with confirmed TDP-43 histopathology (Van Deerlin et al., 2010). These findings suggest that planning sample selection to allow multiple comparisons of disease subtypes, alongside the combined use of new protein- and RNA-profiling technologies, will lead to further advances in the field of ALS and FTD research.
Each technique discussed in this review has advantages and limitations, described above and in additional detail elsewhere (Johnson et al., 2005; Parker et al., 2009; Neilson et al., 2011; Megger et al., 2014; Arrington et al., 2017; Lindemann et al., 2017; Martens and Vizcaino, 2017; Nadler et al., 2017; Tholey and Becker, 2017). Importantly, issues of reproducibility present in less frequently used and outdated techniques such as surface-enhanced laser desorption/ionization (SELDI) have been addressed by the introduction of newer technologies. Whole proteome screening at high resolutions has also replaced the need for “precision” techniques such as two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) coupled to MS. The labeling of proximal proteins to investigate PPIs using techniques such as BAR will allow for more complex investigation that is not currently provided by standard affinity chromatography methods. The implementation of novel and high-throughput techniques in ALS/FTD can be used to overcome current issues in variability between studies as well as the low correlation of findings between differing studies. Furthermore, the application of different techniques is context-dependent, and choice of technology must take into account both biological and technical considerations. In addition, it is crucial that both experimental approaches and data interpretation are considered in study design.
In this review we have identified proteomics papers relevant to ALS/FTD and presented them alongside discussion of current technologies. The consistency amongst proteins identified strictly depend on the disease type/model used, however, in ALS research, TDP-43 as well as hnRNPs appear to reoccur (Ling et al., 2013). An assessment as to whether these commonalities are consistent has not been carried out as these would have to account for differences in stress (oxidative/heat) as well as cell type (neuron/cell-line/species).
In the context of ALS and FTD, greater focus should be given to comparisons between new and existing proteomics datasets as the growing number of studies are released using a variety of available techniques. Further comparisons between ALS and FTD datasets and those obtained from other neurodegenerative diseases should also be performed, since there are likely to be overlap in mechanisms which may be broadly involved in neurodegeneration. This may also provide information on how disease-specific alterations arise and why certain cell populations or CNS regions are susceptible to different neurodegenerative disease processes. For example, recent studies have applied TMT to identify changes in various Alzheimer’s disease mice, identifying hundreds of differentially expressed proteins (Kim et al., 2018). Previously, these comparisons have also been used for biomarker elucidation, attempting to highlight differences between similar neurodegenerative diseases such as FTD and Alzheimer’s disease (Teunissen et al., 2016).
Finally, it will also be informative to use these proteomic techniques to more closely investigate biochemical changes longitudinally in disease, particularly in studies of model systems from which brain and spinal cord samples can be collected at pre-defined time points. Strategies such as this are most likely to reveal the earliest pathogenic changes in disease, which may be most amenable to therapeutic intervention, but which may not be identified by studies at later disease time points or by using end-stage human tissue samples alone. The use of comparative studies and recent advances in new technologies and labeling techniques offers great hope for future understanding of ALS and FTD, and development of clinical tests and therapeutics based on these findings.
Author Contributions
TH, AW, and AL conceived of the manuscript and drafted the tables. All authors were responsible for drafting, writing and editing, and read and approved the final manuscript.
Funding
This work and related studies was supported by the Australian Government Department of Education and Training RTP, QBI Research Higher Degree Top Up Scholarship, the MND Research Institute of Australia (GIA1628, 1715, and 1727), the Australian National Health and Medical Research Council (Project Grant Nos. GNT1124005, 1095215, and 1107644 and RD Wright Career Development Fellowship GNT1140386), the Ross Maclean Fellowship, and the Brazil Family Program for Neurology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Nick Valmas for assistance with graphic design.
References
Acquadro, E., Caron, I., Tortarolo, M., Bucci, E. M., Bendotti, C., and Corpillo, D. (2014). Human SOD1-G93A specific distribution evidenced in murine brain of a transgenic model for amyotrophic lateral sclerosis by MALDI imaging mass spectrometry. J. Proteome Res. 13, 1800–1809. doi: 10.1021/pr400942n
Ait-Bouziad, N., Lv, G., Mahul-Mellier, A.-L., Xiao, S., Zorludemir, G., Eliezer, D., et al. (2017). Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun. 8:1678. doi: 10.1038/s41467-017-01575-4
Almeida, S., Zhou, L., and Gao, F.-B. (2011). Progranulin, a glycoprotein deficient in frontotemporal dementia, is a novel substrate of several protein disulfide isomerase family proteins. PLoS One 6:e26454. doi: 10.1371/journal.pone.0026454
Alonso, R., Pisa, D., Marina, A. I., Morato, E., Rabano, A., Rodal, I., et al. (2015). Evidence for fungal infection in cerebrospinal fluid and brain tissue from patients with amyotrophic lateral sclerosis. Int. J. Biol. Sci. 11, 546–558. doi: 10.7150/ijbs.11084
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611.
Aring, L., Steinbach, S., Marcus, K., and May, C. (2018). Isolation of distinct types of neurons from fresh brain tissue using laser microdissection in combination with high-performance liquid chromatography-mass spectrometry. Methods Mol. Biol. 1723, 247–260. doi: 10.1007/978-1-4939-7558-7_14
Arrington, J. V., Hsu, C.-C., Elder, S. G., and Andy Tao, W. (2017). Recent advances in phosphoproteomics and application to neurological diseases. Analyst 142, 4373–4387. doi: 10.1039/C7AN00985B
Ayala, Y. M., Zago, P., D’Ambrogio, A., Xu, Y. F., Petrucelli, L., Buratti, E., et al. (2008). Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121(Pt 22), 3778–3785. doi: 10.1242/jcs.038950
Ayers, J. I., McMahon, B., Gill, S., Lelie, H. L., Fromholt, S., Brown, H., et al. (2017). Relationship between mutant Cu/Zn superoxide dismutase 1 maturation and inclusion formation in cell models. J. Neurochem. 140, 140–150. doi: 10.1111/jnc.13864
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Bar, D. Z., Atkatsh, K., Tavarez, U., Erdos, M. R., Gruenbaum, Y., and Collins, F. S. (2018). Biotinylation by antibody recognition-a method for proximity labeling. Nat. Methods 15, 127–133. doi: 10.1038/nmeth.4533
Barschke, P., Oeckl, P., Steinacker, P., Ludolph, A., and Otto, M. (2017). Proteomic studies in the discovery of cerebrospinal fluid biomarkers for amyotrophic lateral sclerosis. Exp. Rev. Proteom. 14, 769–777. doi: 10.1080/14789450.2017.1365602
Basso, M., Samengo, G., Nardo, G., Massignan, T., D’Alessandro, G., Tartari, S., et al. (2009). Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One 4:e8130. doi: 10.1371/journal.pone.0008130
Bereman, M. S., Beri, J., Enders, J. R., and Nash, T. (2018). Machine learning reveals protein signatures in CSF and plasma fluids of clinical value for ALS. Sci. Rep. 8:16334. doi: 10.1038/s41598-018-34642-x
Bergemalm, D., Forsberg, K., Jonsson, P. A., Graffmo, K. S., Brannstrom, T., Andersen, P. M., et al. (2009). Changes in the spinal cord proteome of an amyotrophic lateral sclerosis murine model determined by differential in-gel electrophoresis. Mol. Cell Proteom. 8, 1306–1317. doi: 10.1074/mcp.M900046-MCP200
Blair, L. J., Nordhues, B. A., Hill, S. E., Scaglione, K. M., O’Leary, J. C. III, Fontaine, S. N., et al. (2013). Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 123, 4158–4169. doi: 10.1172/JCI69003
Blokhuis, A. M., Koppers, M., Groen, E. J., van den Heuvel, D. M., Dini Modigliani, S., Anink, J. J., et al. (2016). Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta Neuropathol. 132, 175–196. doi: 10.1007/s00401-016-1575-8
Bodea, L. G., Eckert, A., Ittner, L. M., Piguet, O., and Gotz, J. (2016). Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J. Neurochem. 138(Suppl. 1), 71–94. doi: 10.1111/jnc.13600
Boeynaems, S., Bogaert, E., Kovacs, D., Konijnenberg, A., Timmerman, E., Volkov, A., et al. (2017). Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell 65, 1044.e5–1055.e5. doi: 10.1016/j.molcel.2017.02.013
Brancia, C., Noli, B., Boido, M., Boi, A., Puddu, R., Borghero, G., et al. (2016). VGF protein and Its C-terminal derived peptides in amyotrophic lateral sclerosis: human and animal model studies. PLoS One 11:e0164689. doi: 10.1371/journal.pone.0164689
Branon, T. C., Bosch, J. A., Sanchez, A. D., Udeshi, N. D., Svinkina, T., Carr, S. A., et al. (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887. doi: 10.1038/nbt.4201
Bruijn, L. I., Houseweart, M. K., Kato, S., Anderson, K. L., Anderson, S. D., Ohama, E., et al. (1998). Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851. doi: 10.1126/science.281.5384.1851
Capitanio, D., Vasso, M., Ratti, A., Grignaschi, G., Volta, M., Moriggi, M., et al. (2012). Molecular signatures of amyotrophic lateral sclerosis disease progression in hind and forelimb muscles of an SOD1(G93A) mouse model. Antioxid. Redox Signal. 17, 1333–1350. doi: 10.1089/ars.2012.4524
Celona, B., Dollen, J. V., Vatsavayai, S. C., Kashima, R., Johnson, J. R., Tang, A. A., et al. (2017). Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife 6:e19032. doi: 10.7554/eLife.19032
Chan, P. K., Chattopadhyay, M., Sharma, S., Souda, P., Gralla, E. B., Borchelt, D. R., et al. (2013). Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 110, 10934–10939. doi: 10.1073/pnas.1309613110
Chapman, J. D., Goodlett, D. R., and Masselon, C. D. (2014). Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 33, 452–470. doi: 10.1002/mas.21400
Chassefeyre, R., Martínez-Hernández, J., Bertaso, F., Bouquier, N., Blot, B., Laporte, M., et al. (2015). Regulation of postsynaptic function by the dementia-related ESCRT-III subunit CHMP2B. J. Neurosci. 35, 3155–3173. doi: 10.1523/JNEUROSCI.0586-14.2015
Chen, D., Wang, Y., and Chin, E. R. (2015). Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A∗SOD1 amyotrophic lateral sclerosis mice. Front. Cell. Neurosci. 9:170. doi: 10.3389/fncel.2015.00170
Chen, Y., Liu, X. H., Wu, J. J., Ren, H. M., Wang, J., Ding, Z. T., et al. (2016). Proteomic analysis of cerebrospinal fluid in amyotrophic lateral sclerosis. Exp. Ther. Med. 11, 2095–2106. doi: 10.3892/etm.2016.3210
Cheroni, C., Marino, M., Tortarolo, M., Veglianese, P., De Biasi, S., Fontana, E., et al. (2009). Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 82–96. doi: 10.1093/hmg/ddn319
Cheroni, C., Peviani, M., Cascio, P., Debiasi, S., Monti, C., and Bendotti, C. (2005). Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol. Dis. 18, 509–522. doi: 10.1016/j.nbd.2004.12.007
Chew, J., Gendron, T. F., Prudencio, M., Sasaguri, H., Zhang, Y.-J., Castanedes-Casey, M., et al. (2015). C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154. doi: 10.1126/science.aaa9344
Chou, C. C., Zhang, Y., Umoh, M. E., Vaughan, S. W., Lorenzini, I., Liu, F., et al. (2018). TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 21, 228–239. doi: 10.1038/s41593-017-0047-3
Chu, Q., Rathore, A., Diedrich, J. K., Donaldson, C. J., Yates, J. R. III, and Saghatelian, A. (2017). Identification of microprotein-protein interactions via APEX tagging. Biochemistry 56, 3299–3306. doi: 10.1021/acs.biochem.7b00265
Ciryam, P., Lambert-Smith, I. A., Bean, D. M., Freer, R., Cid, F., Tartaglia, G. G., et al. (2017). Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. U.S.A. 114, E3935–E3943. doi: 10.1073/pnas.1613854114
Cohen, T. J., Hwang, A. W., Restrepo, C. R., Yuan, C. X., Trojanowski, J. Q., and Lee, V. M. (2015). An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 6:5845. doi: 10.1038/ncomms6845
Collins, M. A., An, J., Hood, B. L., Conrads, T. P., and Bowser, R. P. (2015). Label-Free LC-MS/MS proteomic analysis of cerebrospinal fluid identifies protein/pathway alterations and candidate biomarkers for amyotrophic lateral sclerosis. J. Proteome Res. 14, 4486–4501. doi: 10.1021/acs.jproteome.5b00804
Conraux, L., Pech, C., Guerraoui, H., Loyaux, D., Ferrara, P., Guillemot, J. C., et al. (2013). Plasma peptide biomarker discovery for amyotrophic lateral sclerosis by MALDI-TOF mass spectrometry profiling. PLoS One 8:e79733. doi: 10.1371/journal.pone.0079733
Conti, A., Riva, N., Pesca, M., Iannaccone, S., Cannistraci, C. V., Corbo, M., et al. (2014). Increased expression of myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim. Biophys. Acta 1842, 99–106. doi: 10.1016/j.bbadis.2013.10.013
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi: 10.1038/nbt.1511
Craig, R., and Beavis, R. C. (2004). TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467. doi: 10.1093/bioinformatics/bth092
Cronan, J. E. (1990). Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J. Biol. Chem. 265, 10327–10333.
Cykowski, M. D., Powell, S. Z., Appel, J. W., Arumanayagam, A. S., Rivera, A. L., and Appel, S. H. (2018). Phosphorylated TDP-43 (pTDP-43) aggregates in the axial skeletal muscle of patients with sporadic and familial amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 6:28. doi: 10.1186/s40478-018-0528-y
Dammer, E. B., Fallini, C., Gozal, Y. M., Duong, D. M., Rossoll, W., Xu, P., et al. (2012). Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PLoS One 7:e38658. doi: 10.1371/journal.pone.0038658
David, D. C., Hauptmann, S., Scherping, I., Schuessel, K., Keil, U., Rizzu, P., et al. (2005). Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 280, 23802–23814. doi: 10.1074/jbc.M500356200
Davidsson, P., Sjogren, M., Andreasen, N., Lindbjer, M., Nilsson, C. L., Westman-Brinkmalm, A., et al. (2002). Studies of the pathophysiological mechanisms in frontotemporal dementia by proteome analysis of CSF proteins. Brain Res. Mol. Brain Res. 109, 128–133.
Davis, S. A., Itaman, S., Khalid-Janney, C. M., Sherard, J. A., Dowell, J. A., Cairns, N. J., et al. (2018). TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 678, 8–15. doi: 10.1016/j.neulet.2018.04.053
De Benedetti, S., Gianazza, E., Banfi, C., Marocchi, A., Lunetta, C., Penco, S., et al. (2017). Serum proteome in a sporadic amyotrophic lateral sclerosis geographical cluster. Proteom. Clin. Appl. 11:1700043. doi: 10.1002/prca.201700043
Dephoure, N., Gould, K. L., Gygi, S. P., Kellogg, D. R., and Drubin, D. G. (2013). Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Mol. Biol. Cell 24, 535–542. doi: 10.1091/mbc.e12-09-0677
Despres, C., Byrne, C., Qi, H., Cantrelle, F.-X., Huvent, I., Chambraud, B., et al. (2017). Identification of the Tau phosphorylation pattern that drives its aggregation. Proc. Natl. Acad. Sci. U. S. A. 114, 9080–9085. doi: 10.1073/pnas.1708448114
Duplan, L., Bernard, N., Casseron, W., Dudley, K., Thouvenot, E., Honnorat, J., et al. (2010). Collapsin response mediator protein 4a (CRMP4a) is upregulated in motoneurons of mutant SOD1 mice and can trigger motoneuron axonal degeneration and cell death. J. Neurosci. 30, 785–796. doi: 10.1523/jneurosci.5411-09.2010
Ekegren, T., Hanrieder, J., Aquilonius, S. M., and Bergquist, J. (2006). Focused proteomics in post-mortem human spinal cord. J. Proteome Res. 5, 2364–2371. doi: 10.1021/pr060237f
Elf, K., Shevchenko, G., Nygren, I., Larsson, L., Bergquist, J., Askmark, H., et al. (2014). Alterations in muscle proteome of patients diagnosed with amyotrophic lateral sclerosis. J. Proteom. 108, 55–64. doi: 10.1016/j.jprot.2014.05.004
Eng, J. K., McCormack, A. L., and Yates, J. R. (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989. doi: 10.1016/1044-0305(94)80016-2
Engelen-Lee, J., Blokhuis, A. M., Spliet, W. G. M., Pasterkamp, R. J., Aronica, E., Demmers, J. A. A., et al. (2017). Proteomic profiling of the spinal cord in ALS: decreased ATP5D levels suggest synaptic dysfunction in ALS pathogenesis. Amyotroph. Lateral Scler. Frontotemporal. Degener. 18, 210–220. doi: 10.1080/21678421.2016.1245757
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A. K., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
Farrawell, N. E., Yerbury, M. R., Plotkin, S. S., McAlary, L., and Yerbury, J. J. (2018). CuATSM protects against the in vitro cytotoxicity of wild type-like SOD1 mutants but not mutants that disrupt metal binding. ACS Chem. Neurosci. 10, 1555-1564 doi: 10.1021/acschemneuro.8b00527
Feneberg, E., Steinacker, P., Lehnert, S., Schneider, A., Walther, P., Thal, D. R., et al. (2014). Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Frontotemporal. Degener. 15, 351–356. doi: 10.3109/21678421.2014.905606
Ferrer, I., Barrachina, M., and Puig, B. (2002). Anti-tau phospho-specific Ser262 antibody recognizes a variety of abnormal hyper-phosphorylated tau deposits in tauopathies including Pick bodies and argyrophilic grains. Acta Neuropathol. 104, 658–664. doi: 10.1007/s00401-002-0600-2
Field, L. S., Furukawa, Y., O’Halloran, T. V., and Culotta, V. C. (2003). Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J. Biol. Chem. 278, 28052–28059. doi: 10.1074/jbc.M304296200
Foster, L. A., and Salajegheh, M. K. (2019). Motor neuron disease: pathophysiology, diagnosis, and management. Am. J. Med. 132, 32–37. doi: 10.1016/j.amjmed.2018.07.012
Freibaum, B. D., Chitta, R. K., High, A. A., and Taylor, J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9, 1104–1120. doi: 10.1021/pr901076y
Frost, B., Gotz, J., and Feany, M. B. (2015). Connecting the dots between tau dysfunction and neurodegeneration. Trends Cell. Biol. 25, 46–53. doi: 10.1016/j.tcb.2014.07.005
Fukada, K., Zhang, F., Vien, A., Cashman, N. R., and Zhu, H. (2004). Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis. Mol. Cell. Proteom. 3, 1211–1223. doi: 10.1074/mcp.M400094-MCP200
Fulzele, A., and Bennett, E. J. (2018). Ubiquitin diGLY proteomics as an approach to identify and quantify the ubiquitin-modified proteome. Methods Mol. Biol. 1844, 363–384. doi: 10.1007/978-1-4939-8706-1_23
Gauthier-Kemper, A., Weissmann, C., Golovyashkina, N., Sebo-Lemke, Z., Drewes, G., Gerke, V., et al. (2011). The frontotemporal dementia mutation R406W blocks tau’s interaction with the membrane in an annexin A2-dependent manner. J. Cell Biol. 192, 647–661. doi: 10.1083/jcb.201007161
Gershoni-Emek, N., Mazza, A., Chein, M., Gradus-Pery, T., Xiang, X., Li, K. W., et al. (2016). Proteomic analysis of dynein-interacting proteins in amyotrophic lateral sclerosis synaptosomes reveals alterations in the RNA-binding protein staufen1. Mol. Cell. Proteom. 15, 506–522. doi: 10.1074/mcp.M115.049965
Gillet, L. C., Navarro, P., Tate, S., Rost, H., Selevsek, N., Reiter, L., et al. (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 11:O111016717. doi: 10.1074/mcp.O111.016717
Gozal, Y. M., Dammer, E. B., Duong, D. M., Cheng, D., Gearing, M., Rees, H. D., et al. (2011a). Proteomic analysis of hippocampal dentate granule cells in frontotemporal lobar degeneration: application of laser capture technology. Front. Neurol. 2:24. doi: 10.3389/fneur.2011.00024
Gozal, Y. M., Seyfried, N. T., Gearing, M., Glass, J. D., Heilman, C. J., Wuu, J., et al. (2011b). Aberrant septin 11 is associated with sporadic frontotemporal lobar degeneration. Mol. Neurodegener. 6:82. doi: 10.1186/1750-1326-6-82
Gregersen, N., and Bross, P. (2010). Protein misfolding and cellular stress: an overview. Methods Mol. Biol. 648, 3–23. doi: 10.1007/978-1-60761-756-3_1
Guldbrandsen, A., Vethe, H., Farag, Y., Oveland, E., Garberg, H., Berle, M., et al. (2014). In-depth characterization of the cerebrospinal fluid proteome displayed through the CSF proteome resource (CSF-PR). Mol. Cell. Proteom. 13, 3152–3163. doi: 10.1074/mcp.M114.038554
Gunawardana, C. G., Mehrabian, M., Wang, X., Mueller, I., Lubambo, I. B., Jonkman, J. E. N., et al. (2015). The human tau interactome: binding to the ribonucleoproteome, and impaired binding of the proline-to-leucine mutant at position 301 (P301L) to chaperones and the proteasome. Mol. Cell. Proteom. 14, 3000–3014. doi: 10.1074/mcp.M115.050724
Haines, D. S., Lee, J. E., Beauparlant, S. L., Kyle, D. B., den Besten, W., Sweredoski, M. J., et al. (2012). Protein interaction profiling of the p97 adaptor UBXD1 points to a role for the complex in modulating ERGIC-53 trafficking. Mol. Cell. Proteom. 11:M111016444. doi: 10.1074/mcp.M111.016444
Han, Y., Martell, J., Branon, T., Boassa, D., Shechner, D., Ellisman, M., et al. (2018). Directed evolution of split APEX peroxidase. bioRxiv doi: 10.1101/452888
Hans, F., Eckert, M., von Zweydorf, F., Gloeckner, C. J., and Kahle, P. J. (2018). Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 293, 16083–16099. doi: 10.1074/jbc.RA118.003440
Hartmann, H., Hornburg, D., Czuppa, M., Bader, J., Michaelsen, M., Farny, D., et al. (2018). Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 1:e201800070. doi: 10.26508/lsa.201800070
Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70. doi: 10.1002/ana.21425
Hemelaar, J., Borodovsky, A., Kessler, B. M., Reverter, D., Cook, J., Kolli, N., et al. (2004). Specific and covalent targeting of conjugating and deconjugating enzymes of ubiquitin-like proteins. Mol. Cell. Biol. 24, 84–95. doi: 10.1128/mcb.24.1.84-95.2004
Hipp, M. S., Park, S.-H., and Hartl, F. U. (2014). Proteostasis impairment in protein-misfolding and-aggregation diseases. Trends Cell Biol. 24, 506–514. doi: 10.1016/j.tcb.2014.05.003
Hogan, A. L., Don, E. K., Rayner, S. L., Lee, A., Laird, A. S., Watchon, M., et al. (2017). Expression of ALS/FTD-linked mutant CCNF in zebrafish leads to increased cell death in the spinal cord and an aberrant motor phenotype. Hum. Mol. Genet. 26, 2616–2626. doi: 10.1093/hmg/ddx136
Hosp, F., Vossfeldt, H., Heinig, M., Vasiljevic, D., Arumughan, A., Wyler, E., et al. (2015). Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 11, 1134–1146. doi: 10.1016/j.celrep.2015.04.030
Huang, Y. H., Shih, C. M., Huang, C. J., Lin, C. M., Chou, C. M., Tsai, M. L., et al. (2006). Effects of cadmium on structure and enzymatic activity of Cu, Zn-SOD and oxidative status in neural cells. J. Cell Biochem. 98, 577–589. doi: 10.1002/jcb.20772
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Iridoy, O. M., Zubiri, I., Zelaya, V. M., Martinez, L., Ausín, K., Lachen-Montes, M., et al. (2018). Neuroanatomical quantitative proteomics reveals common pathogenic biological routes between amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Int. J. Mol. Sci. 20:E4. doi: 10.3390/ijms20010004
Johnson, R. S., Davis, M. T., Taylor, J. A., and Patterson, S. D. (2005). Informatics for protein identification by mass spectrometry. Methods 35, 223–236. doi: 10.1016/j.ymeth.2004.08.014
Kadavath, H., Hofele, R. V., Biernat, J., Kumar, S., Tepper, K., Urlaub, H., et al. (2015). Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. U.S.A. 112, 7501–7506. doi: 10.1073/pnas.1504081112
Kamelgarn, M., Chen, J., Kuang, L., Arenas, A., Zhai, J., Zhu, H., et al. (2016). Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta 1862, 2004–2014. doi: 10.1016/j.bbadis.2016.07.015
Kametani, F., Obi, T., Shishido, T., Akatsu, H., Murayama, S., Saito, Y., et al. (2016). Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci. Rep. 6:23281. doi: 10.1038/srep23281
Kanekura, K., Yagi, T., Cammack, A. J., Mahadevan, J., Kuroda, M., Harms, M. B., et al. (2016). Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet. 25, 1803–1813. doi: 10.1093/hmg/ddw052
Kato, S., Horiuchi, S., Liu, J., Cleveland, D. W., Shibata, N., Nakashima, K., et al. (2000). Advanced glycation endproduct-modified superoxide dismutase-1 (SOD1)-positive inclusions are common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutations and transgenic mice expressing human SOD1 with a G85R mutation. Acta Neuropathol. 100, 490–505. doi: 10.1007/s004010000226
Kikuchi, K., Arawaka, S., Koyama, S., Kimura, H., Ren, C. H., Wada, M., et al. (2003). An N-terminal fragment of ProSAAS (a granin-like neuroendocrine peptide precursor) is associated with tau inclusions in Pick’s disease. Biochem. Biophys. Res. Commun. 308, 646–654.
Kim, D. I., Jensen, S. C., Noble, K. A., Kc, B., Roux, K. H., Motamedchaboki, K., et al. (2016). An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 27, 1188–1196. doi: 10.1091/mbc.E15-12-0844
Kim, D. K., Park, J., Han, D., Yang, J., Kim, A., Woo, J., et al. (2018). Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener. 13:2. doi: 10.1186/s13024-017-0234-4
Kim, J. E., Hong, Y. H., Kim, J. Y., Jeon, G. S., Jung, J. H., Yoon, B. N., et al. (2017). Altered nucleocytoplasmic proteome and transcriptome distributions in an in vitro model of amyotrophic lateral sclerosis. PLoS One 12:e0176462. doi: 10.1371/journal.pone.0176462
Kim, W., Bennett, E. J., Huttlin, E. L., Guo, A., Li, J., Possemato, A., et al. (2011). Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340. doi: 10.1016/j.molcel.2011.08.025
Kincaid, M. M., and Cooper, A. A. (2007). Misfolded proteins traffic from the endoplasmic reticulum (ER) due to ER export signals. Mol. Biol. Cell 18, 455–463. doi: 10.1091/mbc.e06-08-0696
Klein, Z. A., Takahashi, H., Ma, M., Stagi, M., Zhou, M., Lam, T. T., et al. (2017). Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 95, 281.e6–296.e6. doi: 10.1016/j.neuron.2017.06.026
Krach, F., Batra, R., Wheeler, E. C., Vu, A. Q., Wang, R., Hutt, K., et al. (2018). Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol. 136, 405–423. doi: 10.1007/s00401-018-1870-7
Kurzban, G. P., Bayer, E. A., Wilchek, M., and Horowitz, P. M. (1991). The quaternary structure of streptavidin in urea. J. Biol. Chem. 266, 14470–14477.
Kwon, K., Streaker, E. D., Ruparelia, S., and Beckett, D. (2000). Multiple disordered loops function in corepressor-induced dimerization of the biotin repressor. J. Mol. Biol. 304, 821–833. doi: 10.1006/jmbi.2000.4249
Laferriere, F., Maniecka, Z., Perez-Berlanga, M., Hruska-Plochan, M., Gilhespy, L., Hock, E. M., et al. (2019). TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci. 22, 65–77. doi: 10.1038/s41593-018-0294-y
Lam, S. S., Martell, J. D., Kamer, K. J., Deerinck, T. J., Ellisman, M. H., Mootha, V. K., et al. (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54. doi: 10.1038/nmeth.3179
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Sarmiento, J., Troncoso, J., Jackson, G. R., et al. (2012). Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 26, 1946–1959. doi: 10.1096/fj.11-199851
Lee, K. H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774.e17–788.e17. doi: 10.1016/j.cell.2016.10.002
Lee, A., Rayner Stephanie, L., De Luca, A., Gwee Serene, S. L., Morsch, M., Sundaramoorthy, V., et al. (2017). Casein kinase II phosphorylation of cyclin F at serine 621 regulates the Lys48-ubiquitylation E3 ligase activity of the SCF(cyclin F) complex. Open Biol. 7:170058. doi: 10.1098/rsob.170058
Lee, C. W., Stankowski, J. N., Chew, J., Cook, C. N., Lam, Y. W., Almeida, S., et al. (2017). The lysosomal protein cathepsin L is a progranulin protease. Mol Neurodegener. 12:55. doi: 10.1186/s13024-017-0196-6
Lee, A., Rayner, S. L., Gwee, S. S. L., De Luca, A., Shahheydari, H., Sundaramoorthy, V., et al. (2018). Pathogenic mutation in the ALS/FTD gene, CCNF, causes elevated Lys48-linked ubiquitylation and defective autophagy. Cell Mol. Life Sci. 75, 335–354. doi: 10.1007/s00018-017-2632-8
Lee, Y. C., Huang, W. C., Lin, J. H., Kao, T. J., Lin, H. C., Lee, K. H., et al. (2018). Znf179 E3 ligase-mediated TDP-43 polyubiquitination is involved in TDP-43- ubiquitinated inclusions (UBI) (+)-related neurodegenerative pathology. J. Biomed. Sci. 25:76. doi: 10.1186/s12929-018-0479-4
Li, H.-R., Chiang, W.-C., Chou, P.-C., Wang, W.-J., and Huang, J.-R. (2018). TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 293, 6090–6098. doi: 10.1074/jbc.AC117.001037
Li, Q., Vande Velde, C., Israelson, A., Xie, J., Bailey, A. O., Dong, M. Q., et al. (2010). ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. U.S.A. 107, 21146–21151. doi: 10.1073/pnas.1014862107
Li, Y., Collins, M., An, J., Geiser, R., Tegeler, T., Tsantilas, K., et al. (2016). Immunoprecipitation and mass spectrometry defines an extensive RBM45 protein-protein interaction network. Brain Res. 1647, 79–93. doi: 10.1016/j.brainres.2016.02.047
Li, Y., Evers, J., Luo, A., Erber, L., Postler, Z., and Chen, Y. (2019). A quantitative chemical proteomics approach for site-specific stoichiometry analysis of ubiquitination. Angew. Chem. Int. Ed. Engl. 58, 537–541. doi: 10.1002/anie.201810569
Liebl, M. P., Kaya, A. M., Tenzer, S., Mittenzwei, R., Koziollek-Drechsler, I., Schild, H., et al. (2014). Dimerization of visinin-like protein 1 is regulated by oxidative stress and calcium and is a pathological hallmark of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 72, 41–54. doi: 10.1016/j.freeradbiomed.2014.04.008
Lin, P. Y., Simon, S. M., Koh, W. K., Folorunso, O., Umbaugh, C. S., and Pierce, A. (2013). Heat shock factor 1 over-expression protects against exposure of hydrophobic residues on mutant SOD1 and early mortality in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 8:43. doi: 10.1186/1750-1326-8-43
Lindemann, C., Thomanek, N., Hundt, F., Lerari, T., Meyer, H. E., Wolters, D., et al. (2017). Strategies in relative and absolute quantitative mass spectrometry based proteomics. Biol. Chem. 398, 687–699. doi: 10.1515/hsz-2017-0104
Ling, S. C., Albuquerque, C. P., Han, J. S., Lagier-Tourenne, C., Tokunaga, S., Zhou, H., et al. (2010). ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. U.S.A. 107, 13318–13323. doi: 10.1073/pnas.1008227107
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, C., Song, X., Nisbet, R., and Gotz, J. (2016). Co-immunoprecipitation with tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J. Biol. Chem. 291, 8173–8188. doi: 10.1074/jbc.M115.641902
Liu, D., Liu, C., Li, J., Azadzoi, K., Yang, Y., Fei, Z., et al. (2013). Proteomic analysis reveals differentially regulated protein acetylation in human amyotrophic lateral sclerosis spinal cord. PLoS One 8:e80779. doi: 10.1371/journal.pone.0080779
Liu, J., Akhavan, A., Lu, M., Gruzman, A., Lingappa, V. R., An, J., et al. (2010). Carbonic anhydrase I is recognized by an SOD1 antibody upon biotinylation of human spinal cord extracts. Int. J. Mol. Sci. 11, 4051–4062. doi: 10.3390/ijms11104051
Liu, Y., Mi, Y., Mueller, T., Kreibich, S., Williams, E. G., Van Drogen, A., et al. (2019). Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat. Biotechnol. 37, 314–322. doi: 10.1038/s41587-019-0037-y
Ludwig, C., Gillet, L., Rosenberger, G., Amon, S., Collins, B. C., and Aebersold, R. (2018). Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 14:e8126. doi: 10.15252/msb.20178126
Macron, C., Lane, L., Nunez Galindo, A., and Dayon, L. (2018). Deep dive on the proteome of human cerebrospinal fluid: a valuable data resource for biomarker discovery and missing protein identification. J. Proteome Res. 17, 4113–4126. doi: 10.1021/acs.jproteome.8b00300
Maharana, S., Wang, J., Papadopoulos, D. K., Richter, D., Pozniakovsky, A., Poser, I., et al. (2018). RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921. doi: 10.1126/science.aar7366
Mallik, M., Catinozzi, M., Hug, C. B., Zhang, L., Wagner, M., Bussmann, J., et al. (2018). Xrp1 genetically interacts with the ALS-associated FUS orthologue caz and mediates its toxicity. J. Cell Biol. 217, 3947–3964. doi: 10.1083/jcb.201802151
Markham, K., Bai, Y., and Schmitt-Ulms, G. (2007). Co-immunoprecipitations revisited: an update on experimental concepts and their implementation for sensitive interactome investigations of endogenous proteins. Anal. Bioanal. Chem. 389, 461–473. doi: 10.1007/s00216-007-1385-x
Markmiller, S., Soltanieh, S., Server, K. L., Mak, R., Jin, W., Fang, M. Y., et al. (2018). Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590.e13–604.e13. doi: 10.1016/j.cell.2017.12.032
Martell, J. D., Deerinck, T. J., Sancak, Y., Poulos, T. L., Mootha, V. K., Sosinsky, G. E., et al. (2012). Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 30, 1143–1148. doi: 10.1038/nbt.2375
Martens, L., and Vizcaino, J. A. (2017). A golden age for working with public proteomics data. Trends Biochem. Sci. 42, 333–341. doi: 10.1016/j.tibs.2017.01.001
Martins, D., and English, A. M. (2014). SOD1 oxidation and formation of soluble aggregates in yeast: relevance to sporadic ALS development. Redox. Biol. 2, 632–639. doi: 10.1016/j.redox.2014.03.005
Massignan, T., Casoni, F., Basso, M., Stefanazzi, P., Biasini, E., Tortarolo, M., et al. (2007). Proteomic analysis of spinal cord of presymptomatic amyotrophic lateral sclerosis G93A SOD1 mouse. Biochem. Biophys. Res. Commun. 353, 719–725. doi: 10.1016/j.bbrc.2006.12.075
May, S., Hornburg, D., Schludi, M. H., Arzberger, T., Rentzsch, K., Schwenk, B. M., et al. (2014). C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 128, 485–503. doi: 10.1007/s00401-014-1329-4
Maziuk, B. F., Apicco, D. J., Cruz, A. L., Jiang, L., Ash, P. E. A., da Rocha, E. L., et al. (2018). RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 6:71. doi: 10.1186/s40478-018-0574-5
McAlary, L., Aquilina, J. A., and Yerbury, J. J. (2016). Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not in vitro aggregation propensity. Front. Neurosci. 10:499. doi: 10.3389/fnins.2016.00499
Megger, D. A., Pott, L. L., Ahrens, M., Padden, J., Bracht, T., Kuhlmann, K., et al. (2014). Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim. Biophys. Acta 1844, 967–976. doi: 10.1016/j.bbapap.2013.07.017
Miller, S. E., Rizzo, A. I., and Waldbauer, J. R. (2018). Postnovo: postprocessing enables accurate and FDR-controlled de novo peptide sequencing. J. Proteome Res. 17, 3671–3680. doi: 10.1021/acs.jproteome.8b00278
Min, S.-W., Cho, S.-H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Minguez, P., Parca, L., Diella, F., Mende, D. R., Kumar, R., Helmer-Citterich, M., et al. (2012). Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 8:599. doi: 10.1038/msb.2012.31
Mitsui, K., Doi, H., and Nukina, N. (2006). “Proteomics of polyglutamine aggregates,” in Methods in Enzymology, ed. S. P. Colowick (Cambridge, MA: Academic Press), 63–76.
Mitsui, K., Nakayama, H., Akagi, T., Nekooki, M., Ohtawa, K., Takio, K., et al. (2002). Purification of polyglutamine aggregates and identification of elongation factor-1α and heat shock protein 84 as aggregate-interacting proteins. J. Neurosci. 22, 9267–9277. doi: 10.1523/jneurosci.22-21-09267.2002
Moens, T. G., Niccoli, T., Wilson, K. M., Atilano, M. L., Birsa, N., Gittings, L. M., et al. (2019). C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 137, 487–500. doi: 10.1007/s00401-018-1946-4
Monahan, Z., Ryan, V. H., Janke, A. M., Burke, K. A., Rhoads, S. N., Zerze, G. H., et al. (2017). Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 36, 2951–2967. doi: 10.15252/embj.201696394
Morimoto, R. I. (2008). Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22, 1427–1438. doi: 10.1101/gad.1657108
Muntane, G., Dalfo, E., Martinez, A., Rey, M. J., Avila, J., Perez, M., et al. (2006). Glial fibrillary acidic protein is a major target of glycoxidative and lipoxidative damage in pick’s disease. J. Neurochem. 99, 177–185. doi: 10.1111/j.1471-4159.2006.04032.x
Muth, T., and Renard, B. Y. (2018). Evaluating de novo sequencing in proteomics: already an accurate alternative to database-driven peptide identification? Brief. Bioinform. 19, 954–970. doi: 10.1093/bib/bbx033
Nadler, W. M., Waidelich, D., Kerner, A., Hanke, S., Berg, R., Trumpp, A., et al. (2017). MALDI versus ESI: the Impact of the Ion Source on Peptide Identification. J. Proteome Res. 16, 1207–1215. doi: 10.1021/acs.jproteome.6b00805
Nakagawa, K., Kitazume, S., Oka, R., Maruyama, K., Saido, T. C., Sato, Y., et al. (2006). Sialylation enhances the secretion of neurotoxic amyloid-β peptides. J. Neurochem. 96, 924–933. doi: 10.1111/j.1471-4159.2005.03595.x
Narayan, M., Seeley, K. W., and Jinwal, U. K. (2016). Identification of Apo B48 and other novel biomarkers in amyotrophic lateral sclerosis patient fibroblasts. Biomark. Med. 10, 453–462. doi: 10.2217/bmm-2016-0025
Nardo, G., Pozzi, S., Mantovani, S., Garbelli, S., Marinou, K., Basso, M., et al. (2009). Nitroproteomics of peripheral blood mononuclear cells from patients and a rat model of ALS. Antioxid. Redox. Signal. 11, 1559–1567. doi: 10.1089/ars.2009.2548
Nardo, G., Pozzi, S., Pignataro, M., Lauranzano, E., Spano, G., Garbelli, S., et al. (2011). Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One 6:e25545. doi: 10.1371/journal.pone.0025545
Neilson, K. A., Ali, N. A., Muralidharan, S., Mirzaei, M., Mariani, M., Assadourian, G., et al. (2011). Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553. doi: 10.1002/pmic.201000553
Neumann, M., Kwong, L. K., Sampathu, D. M., Trojanowski, J. Q., and Lee, V. M.-Y. (2007). TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch. Neurol. 64, 1388–1394.
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133.
Nguyen, H. P., Van Broeckhoven, C., and van der Zee, J. (2018). ALS genes in the genomic era and their implications for FTD. Trends Genet. 34, 404–423. doi: 10.1016/j.tig.2018.03.001
Nita-Lazar, A., Saito-Benz, H., and White, F. M. (2008). Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics 8, 4433–4443. doi: 10.1002/pmic.200800231
Olney, N. T., Spina, S., and Miller, B. L. (2017). Frontotemporal dementia. Neurol. Clin. 35, 339–374. doi: 10.1016/j.ncl.2017.01.008
Olsen, J. V., and Mann, M. (2013). Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteom. 12, 3444–3452. doi: 10.1074/mcp.O113.034181
Ong, S. E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A., et al. (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 1, 376–386.
Oughtred, R., Stark, C., Breitkreutz, B. J., Rust, J., Boucher, L., Chang, C., et al. (2019). The BioGRID interaction database: 2019 update. Nucleic Acids Res. 47, D529–D541. doi: 10.1093/nar/gky1079
Palma, A. S., De Carvalho, M., Grammel, N., Pinto, S., Barata, N., Conradt, H. S., et al. (2008). Proteomic analysis of plasma from portuguese patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 9, 339–349. doi: 10.1080/17482960801934239
Parker, C., Mocanu, V., Mocanu, M., Dicheva, N., and Warren, M. (2009). Mass Spectrometry for Post-Translational Modifications. Boca Raton, FL: CRC Press.
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7:710. doi: 10.1038/nrn1971
Patel, A., Lee, H. O., Jawerth, L., Maharana, S., Jahnel, M., Hein, M. Y., et al. (2015). A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077. doi: 10.1016/j.cell.2015.07.047
Paul, S., Dansithong, W., Figueroa, K. P., Scoles, D. R., and Pulst, S. M. (2018). Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun. 9:3648. doi: 10.1038/s41467-018-06041-3
Peggion, C., Massimino, M. L., Biancotto, G., Angeletti, R., Reggiani, C., Sorgato, M. C., et al. (2017). Absolute quantification of myosin heavy chain isoforms by selected reaction monitoring can underscore skeletal muscle changes in a mouse model of amyotrophic lateral sclerosis. Anal. Bioanal. Chem. 409, 2143–2153. doi: 10.1007/s00216-016-0160-2
Peng, J., Schwartz, D., Elias, J. E., Thoreen, C. C., Cheng, D., Marsischky, G., et al. (2003). A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21:921. doi: 10.1038/nbt849
Perez, M., Santa-Maria, I., Gomez de Barreda, E., Zhu, X., Cuadros, R., Cabrero, J. R., et al. (2009). Tau–an inhibitor of deacetylase HDAC6 function. J. Neurochem. 109, 1756–1766. doi: 10.1111/j.1471-4159.2009.06102.x
Perkins, D. N., Pappin, D. J., Creasy, D. M., and Cottrell, J. S. (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567.
Perluigi, M., Fai Poon, H., Hensley, K., Pierce, W. M., Klein, J. B., Calabrese, V., et al. (2005). Proteomic analysis of 4-hydroxy-2-nonenal-modified proteins in G93A-SOD1 transgenic mice-A model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 38, 960–968. doi: 10.1016/j.freeradbiomed.2004.12.021
Phu, L., Izrael-Tomasevic, A., Matsumoto, M. L., Bustos, D., Dynek, J. N., Fedorova, A. V., et al. (2011). Improved quantitative mass spectrometry methods for characterizing complex ubiquitin signals. Mol. Cell. Proteom. 10:M110003756. doi: 10.1074/mcp.M110.003756
Piehowski, P. D., Petyuk, V. A., Orton, D. J., Xie, F., Moore, R. J., Ramirez-Restrepo, M., et al. (2013). Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. J. Proteome Res. 12, 2128–2137. doi: 10.1021/pr301146m
Pokrishevsky, E., McAlary, L., Farrawell, N. E., Zhao, B., Sher, M., Yerbury, J. J., et al. (2018). Tryptophan 32-mediated SOD1 aggregation is attenuated by pyrimidine-like compounds in living cells. Sci. Rep. 8, 15590–15590. doi: 10.1038/s41598-018-32835-y
Poon, H. F., Hensley, K., Thongboonkerd, V., Merchant, M. L., Lynn, B. C., Pierce, W. M., et al. (2005). Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice–a model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 39, 453–462. doi: 10.1016/j.freeradbiomed.2005.03.030
Prabakaran, S., Lippens, G., Steen, H., and Gunawardena, J. (2012). Post-translational modification: nature’s escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 565–583. doi: 10.1002/wsbm.1185
Rademakers, R., Cruts, M., and van Broeckhoven, C. (2004). The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 24, 277–295. doi: 10.1002/humu.20086
Rayner, S. L., Morsch, M., Molloy, M. P., Shi, B., Chung, R., and Lee, A. (2019). Using proteomics to identify ubiquitin ligase–substrate pairs: how novel methods may unveil therapeutic targets for neurodegenerative diseases. Cell. Mol. Life Sci. doi: 10.1007/s00018-019-03082-9
Rhee, H. W., Zou, P., Udeshi, N. D., Martell, J. D., Mootha, V. K., Carr, S. A., et al. (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331. doi: 10.1126/science.1230593
Rhoads, S. N., Monahan, Z. T., Yee, D. S., Leung, A. Y., Newcombe, C. G., O’Meally, R. N., et al. (2018). The prionlike domain of FUS is multiphosphorylated following DNA damage without altering nuclear localization. Mol. Biol. Cell 29, 1786–1797. doi: 10.1091/mbc.E17-12-0735
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rotunno, M. S., Auclair, J. R., Maniatis, S., Shaffer, S. A., Agar, J., and Bosco, D. A. (2014). Identification of a misfolded region in superoxide dismutase 1 that is exposed in amyotrophic lateral sclerosis. J. Biol. Chem. 289, 28527–28538. doi: 10.1074/jbc.M114.581801
Roux, K. J., Kim, D. I., and Burke, B. (2013). BioID: a screen for protein-protein interactions. Curr. Protoc. Protein Sci. 74, 19.23.1–19.23.14. doi: 10.1002/0471140864.ps1923s74
Roux, K. J., Kim, D. I., Raida, M., and Burke, B. (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810. doi: 10.1083/jcb.201112098
Ruegsegger, C., Maharjan, N., Goswami, A., Filezac de L’Etang, A., Weis, J., Troost, D., et al. (2016). Aberrant association of misfolded SOD1 with Na(+)/K(+)ATPase-alpha3 impairs its activity and contributes to motor neuron vulnerability in ALS. Acta Neuropathol. 131, 427–451. doi: 10.1007/s00401-015-1510-4
Russell, C. L., Mitra, V., Hansson, K., Blennow, K., Gobom, J., Zetterberg, H., et al. (2017). Comprehensive quantitative profiling of tau and phosphorylated tau peptides in cerebrospinal fluid by mass spectrometry provides new biomarker candidates. J. Alzheimers Dis. 55, 303–313. doi: 10.3233/JAD-160633
Saccon, R. A., Bunton-Stasyshyn, R. K., Fisher, E. M., and Fratta, P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136(Pt 8), 2342–2358. doi: 10.1093/brain/awt097
Sacksteder, C. A., Qian, W.-J., Knyushko, T. V., Wang, H., Chin, M. H., Lacan, G., et al. (2006). Endogenously nitrated proteins in mouse brain: links to neurodegenerative disease. Biochemistry 45, 8009–8022. doi: 10.1021/bi060474w
San Gil, R., Ooi, L., Yerbury, J. J., and Ecroyd, H. (2017). The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol. Neurodegener. 12:65. doi: 10.1186/s13024-017-0208-6
Sano, T., Vajda, S., and Cantor, C. R. (1998). Genetic engineering of streptavidin, a versatile affinity tag. J. Chromatogr. B Biomed. Sci. Appl. 715, 85–91.
Savitski, M. M., Lemeer, S., Boesche, M., Lang, M., Mathieson, T., Bantscheff, M., et al. (2011). Confident phosphorylation site localization using the mascot delta score. Mol. Cell. Proteom. 10:M110003830. doi: 10.1074/mcp.M110.003830
Schmidt, A., Forne, I., and Imhof, A. (2014). Bioinformatic analysis of proteomics data. BMC Syst. Biol. 8(Suppl. 2):S3. doi: 10.1186/1752-0509-8-s2-s3
Schweitzer, K., Decker, E., Zhu, L., Miller, R. E., Mirra, S. S., Spina, S., et al. (2006). Aberrantly regulated proteins in frontotemporal dementia. Biochem. Biophys. Res. Commun. 348, 465–472. doi: 10.1016/j.bbrc.2006.07.113
Scotter, E. L., Vance, C., Nishimura, A. L., Lee, Y. B., Chen, H. J., Urwin, H., et al. (2014). Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 127(Pt 6), 1263–1278. doi: 10.1242/jcs.140087
Sea, K., Sohn, S. H., Durazo, A., Sheng, Y., Shaw, B. F., Cao, X., et al. (2015). Insights into the role of the unusual disulfide bond in copper-zinc superoxide dismutase. J. Biol. Chem. 290, 2405–2418. doi: 10.1074/jbc.M114.588798
Sengupta-Ghosh, A., Dominguez, S. L., Xie, L., Barck, K. H., Jiang, Z., Earr, T., et al. (2019). Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1G93A mouse model of ALS. Neurobiol. Dis. 124, 340–352. doi: 10.1016/j.nbd.2018.12.002
Seyfried, N. T., Gozal, Y. M., Donovan, L. E., Herskowitz, J. H., Dammer, E. B., Xia, Q., et al. (2012). Quantitative analysis of the detergent-insoluble brain proteome in frontotemporal lobar degeneration using SILAC internal standards. J. Proteome Res. 11, 2721–2738. doi: 10.1021/pr2010814
Shafiei, S. S., Guerrero-Muñoz, M. J., and Castillo-Carranza, D. L. (2017). Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 9:83. doi: 10.3389/fnagi.2017.00083
Sharma, A., Varghese, A. M., Vijaylakshmi, K., Sumitha, R., Prasanna, V. K., Shruthi, S., et al. (2016). Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces mitochondrial and lysosomal dysfunction. Neurochem. Res. 41, 965–984. doi: 10.1007/s11064-015-1779-7
Shi, Y., Mowery, R. A., and Shaw, B. F. (2013a). Effect of metal loading and subcellular pH on net charge of superoxide dismutase-1. J. Mol. Biol. 425, 4388–4404. doi: 10.1016/j.jmb.2013.07.018
Shi, Y., Rhodes, N. R., Abdolvahabi, A., Kohn, T., Cook, N. P., Marti, A. A., et al. (2013b). Deamidation of asparagine to aspartate destabilizes Cu, Zn superoxide dismutase, accelerates fibrillization, and mirrors ALS-linked mutations. J. Am. Chem. Soc. 135, 15897–15908. doi: 10.1021/ja407801x
Silverman, J. M., Christy, D., Shyu, C. C., Moon, K.-M., Fernando, S., Gidden, Z., et al. (2019). CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 294, 3744–3759. doi: 10.1074/jbc.RA118.004825
Sjogren, M., Davidsson, P., Tullberg, M., Minthon, L., Wallin, A., Wikkelso, C., et al. (2001). Both total and phosphorylated tau are increased in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 70, 624–630.
Skorupa, A., Urbach, S., Vigy, O., King, M. A., Chaumont-Dubel, S., Prehn, J. H., et al. (2013). Angiogenin induces modifications in the astrocyte secretome: relevance to amyotrophic lateral sclerosis. J. Proteom. 91, 274–285. doi: 10.1016/j.jprot.2013.07.028
Song, Y., Nagy, M., Ni, W., Tyagi, N. K., Fenton, W. A., Lopez-Giraldez, F., et al. (2013). Molecular chaperone Hsp110 rescues a vesicle transport defect produced by an ALS-associated mutant SOD1 protein in squid axoplasm. Proc. Natl. Acad. Sci. U.S.A. 110, 5428–5433. doi: 10.1073/pnas.1303279110
Songsrirote, K., Li, Z., Ashford, D., Bateman, A., and Thomas-Oates, J. (2010). Development and application of mass spectrometric methods for the analysis of progranulin N-glycosylation. J. Proteom. 73, 1479–1490. doi: 10.1016/j.jprot.2010.02.013
Stalekar, M., Yin, X., Rebolj, K., Darovic, S., Troakes, C., Mayr, M., et al. (2015). Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience 293, 157–170. doi: 10.1016/j.neuroscience.2015.02.046
Stefani, M. (2008). Protein folding and misfolding on surfaces. Int. J. Mol. Sci. 9, 2515–2542. doi: 10.3390/ijms9122515
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Sun, S., Ling, S. C., Qiu, J., Albuquerque, C. P., Zhou, Y., Tokunaga, S., et al. (2015). ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 6:6171. doi: 10.1038/ncomms7171
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Tanaka, K., and Matsuda, N. (2014). Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta Mol. Cell Res. 1843, 197–204. doi: 10.1016/j.bbamcr.2013.03.012
Taniguchi-Watanabe, S., Arai, T., Kametani, F., Nonaka, T., Masuda-Suzukake, M., Tarutani, A., et al. (2016). Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 131, 267–280. doi: 10.1007/s00401-015-1503-3
Tank, E. M., Figueroa-Romero, C., Hinder, L. M., Bedi, K., Archbold, H. C., Li, X., et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 9:2845. doi: 10.1038/s41467-018-05049-z
Taus, T., Kocher, T., Pichler, P., Paschke, C., Schmidt, A., Henrich, C., et al. (2011). Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362. doi: 10.1021/pr200611n
Teunissen, C. E., Elias, N., Koel-Simmelink, M. J., Durieux-Lu, S., Malekzadeh, A., Pham, T. V., et al. (2016). Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimers Dement. 2, 86–94. doi: 10.1016/j.dadm.2015.12.004
Theillet, F. X., Smet-Nocca, C., Liokatis, S., Thongwichian, R., Kosten, J., Yoon, M. K., et al. (2012). Cell signaling, post-translational protein modifications and NMR spectroscopy. J. Biomol. NMR 54, 217–236. doi: 10.1007/s10858-012-9674-x
Tholey, A., and Becker, A. (2017). Top-down proteomics for the analysis of proteolytic events - Methods, applications and perspectives. Biochim. Biophys. Acta Mol. Cell Res. 1864, 2191–2199. doi: 10.1016/j.bbamcr.2017.07.002
Thompson, A., Schäfer, J., Kuhn, K., Kienle, S., Schwarz, J., Schmidt, G., et al. (2003). Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904. doi: 10.1021/ac0262560
Thompson, A. G., Gray, E., Thezenas, M. L., Charles, P. D., Evetts, S., Hu, M. T., et al. (2018). Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann. Neurol. 83, 258–268. doi: 10.1002/ana.25143
Ting, L., Rad, R., Gygi, S. P., and Haas, W. (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. methods 8, 937–940. doi: 10.1038/nmeth.1714
Trippier, P. C., Zhao, K. T., Fox, S. G., Schiefer, I. T., Benmohamed, R., Moran, J., et al. (2014). Proteasome activation is a mechanism for pyrazolone small molecules displaying therapeutic potential in amyotrophic lateral sclerosis. ACS Chem. Neurosci. 5, 823–829. doi: 10.1021/cn500147v
Umoh, M. E., Dammer, E. B., Dai, J., Duong, D. M., Lah, J. J., Levey, A. I., et al. (2018). A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 10, 48–62. doi: 10.15252/emmm.201708202
Valentine, J. S., Doucette, P. A., and Zittin Potter, S. (2005). Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593. doi: 10.1146/annurev.biochem.72.121801.161647
Van Deerlin, V. M., Sleiman, P. M., Martinez-Lage, M., Chen-Plotkin, A., Wang, L. S., Graff-Radford, N. R., et al. (2010). Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239. doi: 10.1038/ng.536
Varghese, A. M., Sharma, A., Mishra, P., Vijayalakshmi, K., Harsha, H. C., Sathyaprabha, T. N., et al. (2013). Chitotriosidase - a putative biomarker for sporadic amyotrophic lateral sclerosis. Clin. Proteom. 10:19. doi: 10.1186/1559-0275-10-19
Vega, I. E., Cui, L., Propst, J. A., Hutton, M. L., Lee, G., and Yen, S. H. (2005). Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Brain Res. Mol. Brain Res. 138, 135–144. doi: 10.1016/j.molbrainres.2005.04.015
Viode, A., Fournier, C., Camuzat, A., Fenaille, F., Latouche, M., Elahi, F., et al. (2018). New antibody-free mass spectrometry-based quantification reveals That C9ORF72 long protein isoform is reduced in the frontal cortex of hexanucleotide-repeat expansion carriers. Front. Neurosci. 12:589. doi: 10.3389/fnins.2018.00589
Walker, A. K., and Atkin, J. D. (2011). Stress signaling from the endoplasmic reticulum: a central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life 63, 754–763. doi: 10.1002/iub.520
Wang, F., Cheng, K., Wei, X., Qin, H., Chen, R., Liu, J., et al. (2013). A six-plex proteome quantification strategy reveals the dynamics of protein turnover. Sci. Rep. 3, 1827–1827. doi: 10.1038/srep01827
Wang, I. F., Wu, L. S., and Shen, C. K. (2008). TDP-43: an emerging new player in neurodegenerative diseases. Trends Mol. Med. 14, 479–485. doi: 10.1016/j.molmed.2008.09.001
Wang, P., Wander, C. M., Yuan, C.-X., Bereman, M. S., and Cohen, T. J. (2017). Acetylation-induced TDP-43 pathology is suppressed by an HSF1-dependent chaperone program. Nat. Commun. 8:82. doi: 10.1038/s41467-017-00088-4
Wang, T., Jiang, X., Chen, G., and Xu, J. (2015). Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration–associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiol. Aging 36, 527–535. doi: 10.1016/j.neurobiolaging.2014.07.044
Wang, Y., Shi, M., Chung, K. A., Zabetian, C. P., Leverenz, J. B., Berg, D., et al. (2012). Phosphorylated alpha-synuclein in parkinson’s disease. Sci. Transl. Med. 4:121ra120. doi: 10.1126/scitranslmed.3002566
Webster, C. P., Smith, E. F., Shaw, P. J., and De Vos, K. J. (2017). Protein homeostasis in amyotrophic lateral sclerosis: therapeutic opportunities? Front. Mol. Neurosci. 10:123. doi: 10.3389/fnmol.2017.00123
Wei, X., and Li, L. (2008). Mass spectrometry-based proteomics and peptidomics for biomarker discovery in neurodegenerative diseases. Int. J. Clin. Exp. Pathol. 2, 132–148.
Weskamp, K., and Barmada, S. J. (2018). TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res. 1693(Pt A), 67–74. doi: 10.1016/j.brainres.2018.01.015
Whiten, D. R., San Gil, R., McAlary, L., Yerbury, J. J., Ecroyd, H., and Wilson, M. R. (2016). Rapid flow cytometric measurement of protein inclusions and nuclear trafficking. Sci. Rep. 6:31138. doi: 10.1038/srep31138
Williams, K. L., Topp, S., Yang, S., Smith, B., Fifita, J. A., Warraich, S. T., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 7:11253. doi: 10.1038/ncomms11253
Xu, G., Paige, J. S., and Jaffrey, S. R. (2010). Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 28, 868–873. doi: 10.1038/nbt.1654
Xu, Z., Lee, A., Nouwens, A., Henderson, R. D., and McCombe, P. A. (2018). Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph. Lateral Scler. Frontotemporal. Degener. 19, 362–376. doi: 10.1080/21678421.2018.1433689
Yin, S., Lopez-Gonzalez, R., Kunz, R. C., Gangopadhyay, J., Borufka, C., Gygi, S. P., et al. (2017). Evidence that C9ORF72 dipeptide repeat proteins associate with U2 snRNP to cause Mis-splicing in ALS/FTD Patients. Cell Rep. 19, 2244–2256. doi: 10.1016/j.celrep.2017.05.056
Yokoi, S., Udagawa, T., Fujioka, Y., Honda, D., Okado, H., Watanabe, H., et al. (2017). 3’UTR length-dependent control of SynGAP isoform alpha2 mRNA by FUS and ELAV-like proteins promotes dendritic spine maturation and cognitive function. Cell Rep. 20, 3071–3084. doi: 10.1016/j.celrep.2017.08.100
Yu, F., Qiu, F., and Meza, J. (2016). “12 - design and statistical analysis of mass-spectrometry-based quantitative proteomics data,” in Proteomic Profiling and Analytical Chemistry, 2nd Edn, eds P. Ciborowski and J. Silberring (Boston, MA: Elsevier), 211–237.
Yu, J., Gu, X., and Yi, S. (2016). Ingenuity pathway analysis of gene expression profiles in distal nerve stump following nerve injury: insights into wallerian degeneration. Front. Cell. Neurosci. 10:274. doi: 10.3389/fncel.2016.00274
Zacco, E., Graña-Montes, R., Martin, S. R., de Groot, N. S., Alfano, C., Tartaglia, G. G., et al. (2019). RNA as a key factor in driving or preventing self-assembly of the TAR DNA-binding protein 43. J. Mol. Biol. 431, 1671–1688. doi: 10.1016/j.jmb.2019.01.028
Zetterstrom, P., Graffmo, K. S., Andersen, P. M., Brannstrom, T., and Marklund, S. L. (2011). Proteins that bind to misfolded mutant superoxide dismutase-1 in spinal cords from transgenic amyotrophic lateral sclerosis (ALS) model mice. J. Biol. Chem. 286, 20130–20136. doi: 10.1074/jbc.M111.218842
Zhai, J., Strom, A. L., Kilty, R., Venkatakrishnan, P., White, J., Everson, W. V., et al. (2009). Proteomic characterization of lipid raft proteins in amyotrophic lateral sclerosis mouse spinal cord. FEBS J. 276, 3308–3323. doi: 10.1111/j.1742-4658.2009.07057.x
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Zhang, J., Huang, P., Wu, C., Liang, H., Li, Y., Zhu, L., et al. (2018). Preliminary observation about alteration of proteins and their potential functions in spinal Cord of SOD1 G93A transgenic mice. Int. J. Biol. Sci. 14, 1306–1320. doi: 10.7150/ijbs.26829
Zhang, Y., Burberry, A., Wang, J. Y., Sandoe, J., Ghosh, S., Udeshi, N. D., et al. (2018). The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev. 32, 929–943. doi: 10.1101/gad.313932.118
Zhou, J. Y., Afjehi-Sadat, L., Asress, S., Duong, D. M., Cudkowicz, M., Glass, J. D., et al. (2010). Galectin-3 is a candidate biomarker for amyotrophic lateral sclerosis: discovery by a proteomics approach. J. Proteome Res. 9, 5133–5141. doi: 10.1021/pr100409r
Zhuang, X., Zhao, B., Liu, S., Song, F., Cui, F., Liu, Z., et al. (2016). Noncovalent interactions between superoxide dismutase and flavonoids studied by native mass spectrometry combined with molecular simulations. Anal. Chem. 88, 11720–11726. doi: 10.1021/acs.analchem.6b03359
Keywords: amyotrophic lateral sclerosis, frontotemporal dementia, proteomics, mass spectrometry, protein aggregation
Citation: Hedl TJ, San Gil R, Cheng F, Rayner SL, Davidson JM, De Luca A, Villalva MD, Ecroyd H, Walker AK and Lee A (2019) Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front. Neurosci. 13:548. doi: 10.3389/fnins.2019.00548
Received: 14 March 2019; Accepted: 13 May 2019;
Published: 11 June 2019.
Edited by:
Olivia Belbin, Sant Pau Institute of Biomedical Research, SpainReviewed by:
Fuyuki Kametani, Tokyo Metropolitan Institute of Medical Science, JapanValentina Bonetto, Istituto Di Ricerche Farmacologiche Mario Negri, Italy
Copyright © 2019 Hedl, San Gil, Cheng, Rayner, Davidson, De Luca, Villalva, Ecroyd, Walker and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Albert Lee, YWxiZXJ0LmxlZUBtcS5lZHUuYXU=; Adam K. Walker, YWRhbS53YWxrZXJAdXEuZWR1LmF1