 Danielle N. Edwards
Danielle N. Edwards Gregory J. Bix
Gregory J. Bix- 1Sanders–Brown Center on Aging, University of Kentucky, Lexington, KY, United States
- 2Department of Neuroscience, University of Kentucky, Lexington, KY, United States
- 3Department of Neurology, University of Kentucky, Lexington, KY, United States
- 4Department of Neurosurgery, University of Kentucky, Lexington, KY, United States
Ischemic stroke is a leading cause of death and disability with limited therapeutic options. Resulting inflammatory mechanisms after reperfusion (removal of the thrombus) result in cytokine activation, calcium influx, and leukocytic infiltration to the area of ischemia. In particular, leukocytes migrate toward areas of inflammation by use of integrins, particularly integrins β1 and β2. Integrins have been shown to be necessary for leukocyte adhesion and migration, and thus are of immediate interest in many inflammatory diseases, including ischemic stroke. In this review, we identify the main integrins involved in leukocytic migration following stroke (αLβ2, αDβ2, α4β1, and α5β1) and targeted clinical therapeutic interventions.
Introduction
Ischemic stroke is a leading cause of death and disability in the United States with limited therapeutic interventions available, including tissue plasminogen activator (t-PA) and endovascular mechanical thrombectomy (Rao et al., 2014; Benjamin et al., 2017; Rai et al., 2017). These interventions are focused on the removal of the thrombus, restoring blood flow, oxygen and glucose to hypoperfused areas, but are unable to affect the inflammatory, necrotic, and blood-brain barrier (BBB) mechanism that follow. In particular, the initial inflammatory cascade is initiated by the decrease in ATP production, release of cytokines, influx of intracellular calcium, reactive oxygen species, etc., that develops during occlusion and continues for days afterward (Sandoval and Witt, 2008). Using shear forces from cerebral blood flow, marrow-derived leukocytes (including polymorphonuclear leukocytes (PMNs), neutrophils, lymphocytes and monocytes) are recruited to the site of injury (Dereski et al., 1993; del Zoppo, 1994; Stefanidakis and Koivunen, 2006).
For the purpose of this review, we will focus on the recruitment and rolling of leukocytes under the direction of integrins, as well as some of their ligands following reperfusion after ischemic stroke. We will then introduce recent β2 and β1 integrin-specific stroke clinical trials, and, finally, discuss potential future directions for the field.
Leukocytic Infiltration Following Ischemic Stroke
Leukocyte Recruitment
The initial endothelial response upregulates endothelial selectins, particularly P-selectin and E-selectin, translocating them from an intracellular, inactive state, to the available extracellular matrix for leukocytic binding, while the upregulation of L-selectin on the leukocyte is essential for recruitment to the site of injury (Bargatze et al., 1994). Both are acutely regulated, P-selectin at 15 min and E-selectin at 2 h post ischemia. These extracellularly located selectins then facilitate the recruitment and activation of leukocytes to the area of ischemia (Zhang et al., 1998). Leukocytes then undergo a conformational change, facilitating polarization and the development of certain cellular characteristics: a leading edge, main body, and rear-uropod protrusion. The uropod, or posterior protrusion, in fast moving leukocytes promotes mobility, while the leading protrusions (lamellipodia and filopodia, small leg-like projections) are less likely to be used due to the rate limiting interaction with actin filaments (Ridley et al., 2003) (summarized in Figure 1). Though this mechanism is less obvious in the highly mobile leukocytes. Once leukocytes are bound to selectins, additional binding to integrins and adhesion molecules (intracellular adhesion molecule (ICAMs and vascular adhesion molecule-1 (VCAM) occurs, permitting leukocytic rolling (del Pozo et al., 1995; Lorant et al., 1995; Kindzelskii et al., 1996; Becker, 2002). Additional damage can occur once at the site of ischemia, as infiltration into the brain parenchyma across the BBB destroys surrounding vasculature (Clark et al., 1993; Chou et al., 2004), and leukocytes continually release additional factors (reactive oxygen species, cytokines, and proteases) that enhance leukocytic recruitment (Wang et al., 2008).
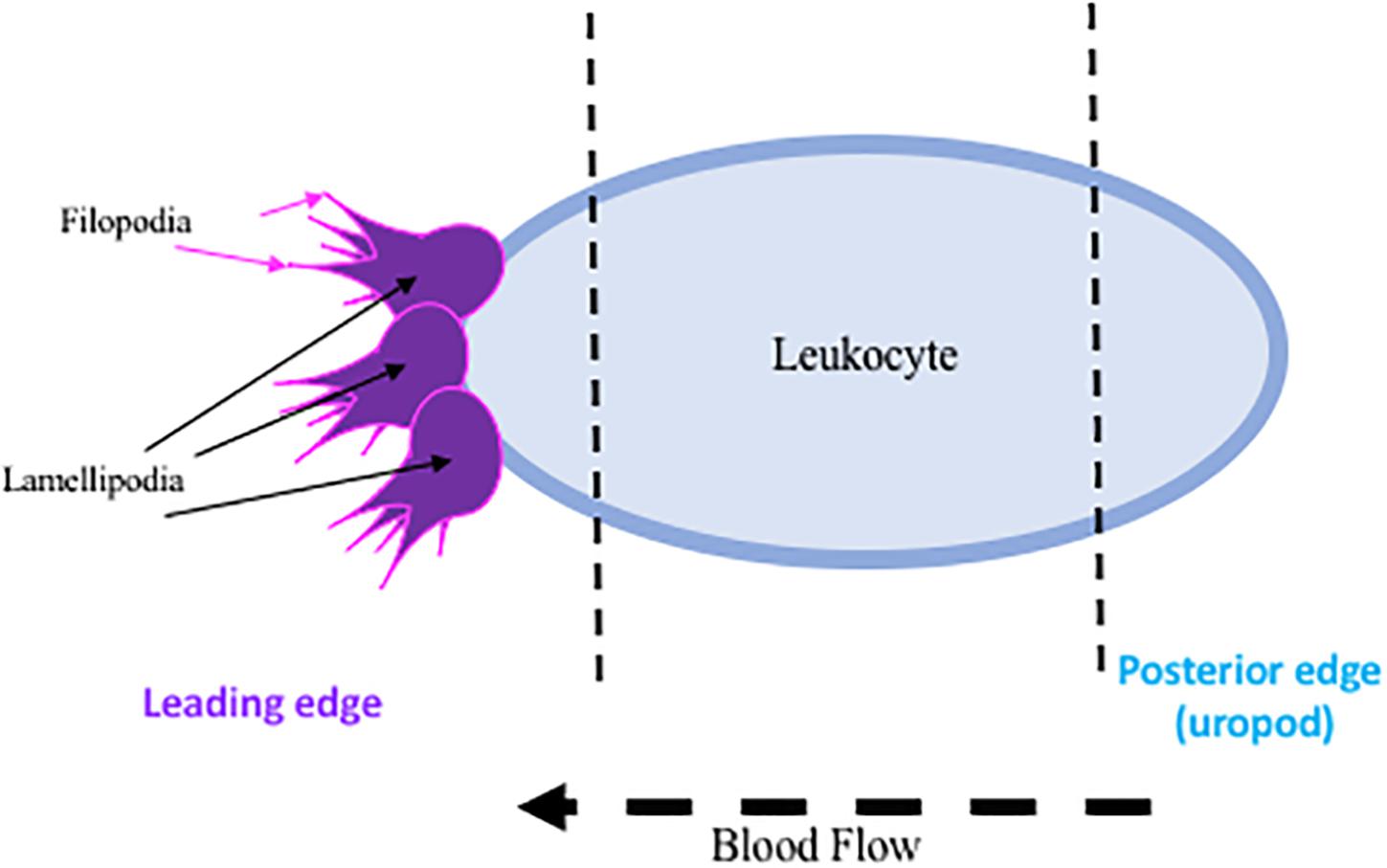
Figure 1. Representative diagram of regions of interest on activated leukocytes.
Leukocyte Infiltration
At the site of injury, leukocytes continue to increase binding on cerebrovasculature up to 48 h following ischemic stroke, and use transendothelial migration as a method for infiltration from the cerebrovasculature into the surrounding brain parenchyma. Early adhesion, prior to 24 h following reperfusion, is attributed to neutrophils. Within 30 min to a few hours following reperfusion, neutrophils arrive at the site of injury, peaking at maximum expression around 1–3 days, though expression can still be detected 7–15 days later in preclinical stroke models (Weston et al., 2007). This upregulation is also seen in ischemic stroke patients, where neutrophils have been detected beginning at 6 h, with radiolabeled imaging, and continue to be detected up to 72 h (Aspey et al., 1989). The early recruitment and infiltration of neutrophils across the BBB appears to be highly significant in stroke, as high neutrophilic infiltration is associated with damaged cerebrovasculature (Enzmann et al., 2013), while depletion reduces infarct volume and dysfunction in stroke models (Chou et al., 2004).
As reperfusion injury continues, the circulating leukocytes switch from neutrophils to mononucleuar leukocytes (monocytes/lymphocytes) which dominate the adherent culture from 24 h to 7 days post reperfusion (Schroeter et al., 1994; Stevens et al., 2002; Ishikawa et al., 2005). Of the two types of lymphocytes, B- and T-, T cells have emerged as the dominant, damage-inducing lymphocyte in ischemic stroke (Brait et al., 2011). Preclinical studies have shown that the inhibition of all lymphocytes results in smaller infarct and improved neurological outcomes, but only the reintroduction of T-lymphocytes to mice reversed any benefits (Yilmaz et al., 2006; Kleinschnitz et al., 2010). Activated T-lymphocytes, not B-, have been detected in patients up to 60 days post-stroke, and are correlated with an increased risk of stroke reoccurrence and death (Tarkowski et al., 1995; Nadareishvili et al., 2004).
Clinical Importance of Leukocytes
Multiple studies have established the importance of leukocytic adherence and infiltration into the brain parenchyma following ischemic stroke, but targeting the leukocytes has a high degree of risk. This is evident as inhibition of leukocytic cells increase the occurrence of bacterial infection and mortality as evident by Leukocytic Adhesion Deficiency (LAD-1) (Stefanidakis and Koivunen, 2006). Because these cells are necessary for bacterial resistance, systemic inhibition following ischemic stroke is exceptionally risky. However, studies in stroke patients show a strong correlation between increased levels of peripheral leukocytes and neutrophils, and increased infarct volume (Price et al., 2004; Buck et al., 2008). Thus, some current therapeutic strategies have focused on the inflammatory cascade have focused on blocking the adhesion and infiltration of cells at the site of injury, primarily endothelial expressed ICAMs and VCAM. This method has shown success in preclinical studies, but has failed to translate to the clinic. Thus, the focus has switched to directly targeting the integrins, a primary mediator of leukocyte adhesion, rather than their ligands, as discussed above. This review is focused on integrins β2 and β1 that have shown promise in therapeutically targeting the ischemic stroke inflammatory cascade.
Role of Integrins Post-Stroke: an Overview
Integrins are a diverse group of heterodimers composed of 18 different α and β subunits, creating 24 unique combinations. Integrins exist on every cell type, while exhibiting a high diversity of ligands and grouped into four different receptor groups: RGD (Arg-Gly-Asp), laminin receptors, collagen receptors, and leukocyte-specific receptors. Within these groups, integrins can have a variety of ligands and roles following ischemic stroke (reviewed in Edwards and Bix, 2019). Under normal cerebrovascular conditions, integrins are in a highly inactive state, typically in a bent conformation (Takagi et al., 2002; Nishida et al., 2006). Following ischemic stroke, activation signals are sent. Chemokines are translocated to the lumen, on the apical side of endothelial cells, to induce “inside-out” signaling (Chavakis, 2012). Integrins then undergo a conformational change to increase integrin affinity for potential ligands while enhancing detection by localizing to the leading or rear-facing edge of the leukocyte’s cell wall for ligand detection (Ridley et al., 2003; Hyun et al., 2009). Activated integrins then bind to available ligands, permitting leukocytic rolling and intracellular signaling. This is termed “outside-in” signaling (Hato et al., 1998; Tominaga et al., 1998; Ley et al., 2007). Leukocytes continue movement to the site of injury, looking for areas to cross the endothelial cell barrier, and eventually coming to a halt. Aggregation/clustering of integrins increases binding avidity (strength of binding), preventing flow conditions from detaching leukocytes from the endothelial cells (Ley et al., 2007). Using transmigration, leukocytes will infiltrate into the cerebral parenchyma using these integrin-ligand connections.
β2 Integrins
β2 integrins are the only group of integrins exclusively expressed on leukocytes (derived from hematopoietic cells) (Schenkel et al., 2004), and like most integrins, are highly conserved across species (Schittenhelm et al., 2017). They are also the most highly expressed integrin on circulating blood leukocytes, tending to cluster at the retraction area of the cell (the rear), in both an active and inactive state, compared to other β1, β4, β3, and β7 integrins found on circulating leukocytes (Pierini et al., 2000; Lindbom and Werr, 2002). Genetic leukocyte adhesion changes (LAD-1, as discussed above) has been attributed to mutations in the β2 subunit, reducing β2 expression. Thus, leukocytic movement is reduced on the cell surface with less movement toward the site of inflammation (Arnaout, 1990; Scharffetter-Kochanek et al., 1998). Importantly, in β2 inhibited mice, there is not total arrest of leukocytic recruitment or infiltration (Pierini et al., 2000), suggesting that other factors likely play a role. There are 4 identified heterodimers of β2 integrins, and of these, the most highly studied are αLβ2 and αMβ2 in ischemic stroke, and will be reviewed in more detail below. The other β2 integrins, αXβ2 and αDβ2, have not been individually studied in the context of stroke as have αLβ2 and αMβ2 integrins, though CD18 (β2) inhibition in addition to t-PA has been shown to increase the time window of t-PA administration without an increase in hemorrhagic transformation in a rat embolic stroke model (Zhang et al., 1999). Furthermore, Figure 2 summarizes the results in this section.
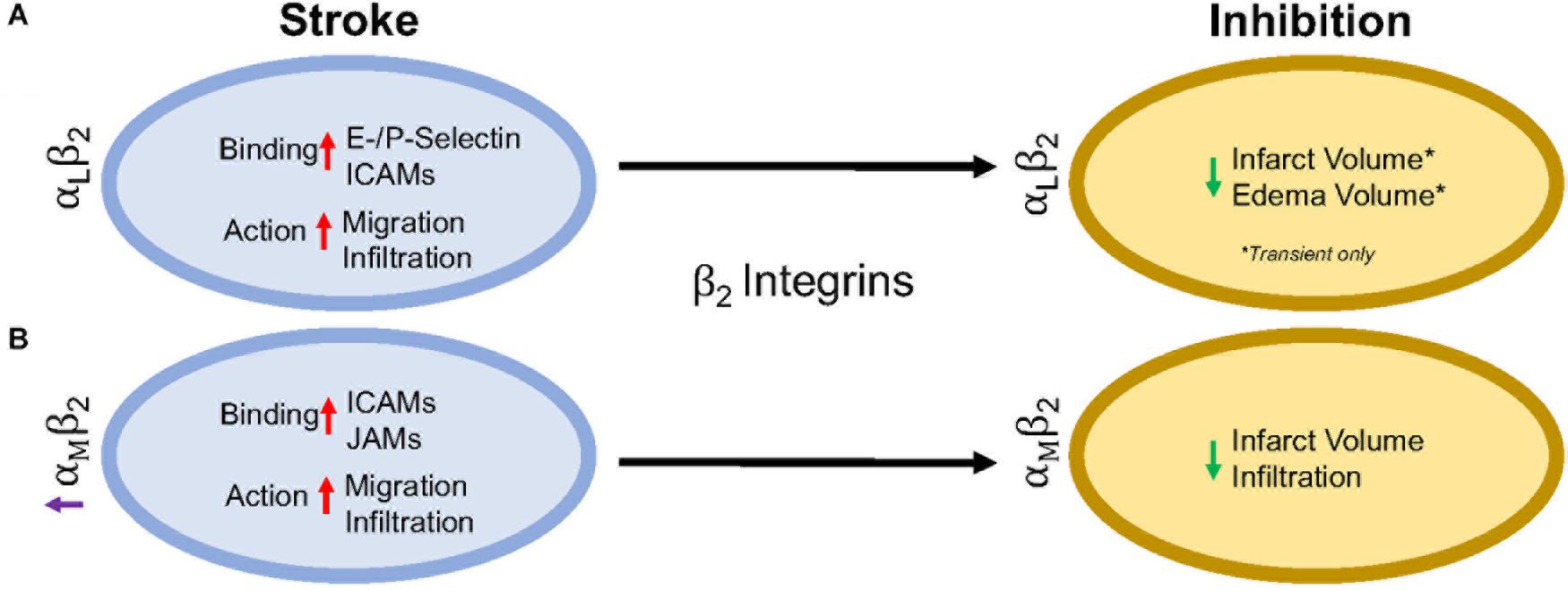
Figure 2. Representative image of the β2 integrin response following experimental stroke and inhibitory antibody treatment in preclinical trials. Inhibition of (A) αLβ2 and (B) αMβ2 integrins post-stroke responses and effects.
αLβ2 Integrin
Integrin αLβ2 is also referred to as CD11a/CD18 and LFA-1 (lymphocyte functional-associating antigen-1). αLβ2 integrin acutely increases in ischemic stroke patients, with detectable amounts through 72 h associated around the area of ischemia (Gerhard et al., 2000; Zhao et al., 2002). This suggests a correlation between αLβ2 integrin expression and inflammatory damage following ischemia. αLβ2 is expressed on all leukocytes (Soriano et al., 1999), though at particularly high levels on T-lymphocytes (Hammond et al., 2014; Walling and Kim, 2018). In healthy individuals, extracted blood analysis revealed that αLβ2 activation requires leukocytic rolling on P- or E-selectins, inducing an active conformational change (Kuwano et al., 2010), but it is the binding of chemokines g-protein coupled receptors (GPCR) and Rap-1 activation that induces the high-affinity conformational state of αLβ2 (Steffen et al., 1994; Greenwood et al., 1995; Ghandour et al., 2007). In this state, αLβ2 has many possible ligands, ICAM-1, ICAM-2, ICAM-3, ICAM-4, ICAM-5, and junctional adhesion molecule-1 (JAM-1) (Marlin and Springer, 1987; de Fougerolles et al., 1991, 1994; Tian et al., 2000), though ICAM-1 is preferentially bound (Walling and Kim, 2018). The high avidity αLβ2-ICAM-1 complex, once formed, allows t-lymphocytes to move against circulatory flow and the shear forces, resulting in the high-speed movement of leukocytes (Katakai et al., 2013; Dominguez et al., 2015).
In an intraluminal model of experimental ischemic stroke, αLβ2 inhibition with the use of transgenic mice results in reduced infarct volume, edema volume and mortality. However, this phenomenon is evident in transient, but not permanent middle cerebral artery occlusion (Arumugam et al., 2004). This may be due to the previously mentioned high avidity of αLβ2-ICAM-1 bonds, and is evident in an in vitro study using αLβ2 (LFA-1) knock-in mice that experience high avidity through binding of lymphocytes mediated through ICAM-1 binding, but are unable to continue movement due to a non-polarized uropod (Park et al., 2010). An explanation for this phenomenon may be that the recycling process within the leukocyte is overwhelmed (Shaw et al., 2004). By enhancing αLβ2 expression, recycling may not be able to allow for dislocation of αLβ2-ICAM-1 complexes, preventing movement from the loss of high adhesion bonds. Enhanced αLβ2 expression could be a potential new avenue for therapy, especially if no enhanced mortality, infection, etc., are observed.
Independently, ICAMs play a significant role in inflammation following ischemic stroke. ICAM-1, in particular, is acutely increased in both cultured human endothelial cells undergoing hypoxia and following intraluminal suture middle cerebral artery occlusion, while expression remains sustained for up to a week post-injury (Hess et al., 1994a,b; Zhang et al., 1995). ICAM-2, another possible ligand, does not change in expression following cytokine stimulation (de Fougerolles et al., 1991; Nortamo et al., 1991a,b). Furthermore, serum of ischemic stroke patients contains soluble ICAM-1, but not ICAM-2 in addition to being a risk factor (Kaplanski et al., 1994; Shyu et al., 1997). Antibodies targeting ICAM-1 in rodents and humans have shown contradictory results. An intraluminal suture middle cerebral artery occlusion model in mice and rats showed a decrease in leukocyte infiltration and infarct volume (Connolly et al., 1996; Kitagawa et al., 1998; Vemuganti et al., 2004), while one study reported opposing effects (Furuya et al., 2001). ICAM-1 inhibition was translated to the clinic through testing of the murine ICAM-1 antibody, Enlimomab in ischemic stroke. Unfortunately, the study was halted early due to increased rate of infection, infarct volumes, neurological scores and mortality for patients (Furuya et al., 2001).
αMβ2 Integrin
Integrin αMβ2, also known as CD11b/CD18 and Mac-1 (macrophage-1 antigen), exhibits many similarities to αLβ2 through its expression on all leukocytes (Springer et al., 1979), and common ligand binding partners such as the family of ICAMs and JAMs (von Andrian et al., 1991). Additional ligands are fibrinogen, heparin (von Andrian et al., 1991), elastase (Cai and Wright, 1996), complement C3 fragment (C3bi) (Micklem and Sim, 1985), kinogen components, and urokinase and its receptor (Chavakis et al., 1999). Just as αLβ2, hypoxia induced factors (cytokines, chemokines, etc.) induce conformational change of αMβ2 to a high affinity ligand-binding state (Stanimirovic et al., 1997). Binding assays with ICAM-1 as a ligand and both αLβ2 and αMβ2 as receptors show αLβ2 integrin is preferably bound (Lub et al., 1996). This suggests that the binding sites on both αLβ2 and αMβ2 compete for ICAM-1 binding.
Following experimental ischemic stroke in rats, integrin αMβ2 is upregulated (Campanella et al., 2002), and has shown benefit when inhibited. Antibodies against both CD11b/CD18 reduce infarct volume and reestablish cerebral blood flow as a result of decreased neutrophil infiltration following intraluminal stroke surgery (Chen et al., 1994; Bowes et al., 1995; Zhang et al., 1995). In a different approach, the addition of recombinant neutrophil inhibitory factor (rNIF) inhibits a binding domain on Mac-1 and yields similar results in the same intraluminal occlusion model (Jiang et al., 1998). Furthermore, and similarly to αLβ2 integrin inhibition, inhibition of αMβ2 is also effective in transient, but not permanent experimental ischemic stroke in an embolic occlusion model (Zhang et al., 2003).
β1 Integrins
β1 integrins are a diverse set of integrins, with laminin-binding, collagen-binding, RGD-binding and leukocyte heterodimers. β1 integrins are not as highly expressed on leukocytes as β2 integrins, but they do play a major role in leukocyte adhesion and migration following ischemic stroke. The activity of β1 integrins is similar to β2 integrins. They undergo a conformational change to induce “inside-out” and “outside-in” cellular signaling (Campanero et al., 1994). As the cells migrate, the β1 integrins are most commonly clustered around the uropod, but will be located in any area of the leukocyte that is in contact with the endothelial cell or extracellular matrix (Campanero et al., 1994; Caimi et al., 2001). Inhibition of the β1 integrin, just as with β2 integrin inhibition, does not fully stop leukocyte rolling. However, when both β1 and β2 integrins are inhibited, complete leukocyte arrest occurs (Lobb and Hemler, 1994; Pierini et al., 2000). This suggests that both β1 and β2 integrins are necessary for leukocyte migration, regardless of expression load. Of all the β1 integrins, both α4β1 and α5β1 appear to be the most highly expressed and the most studied in post-stroke inflammation. The other β1 integrin expressed on leukocytes, α9β1, has not been studied in the context of stroke as its expression and role has not yet been fully elucidated in the brain. Figure 3 summarizes the results discussed in this section.
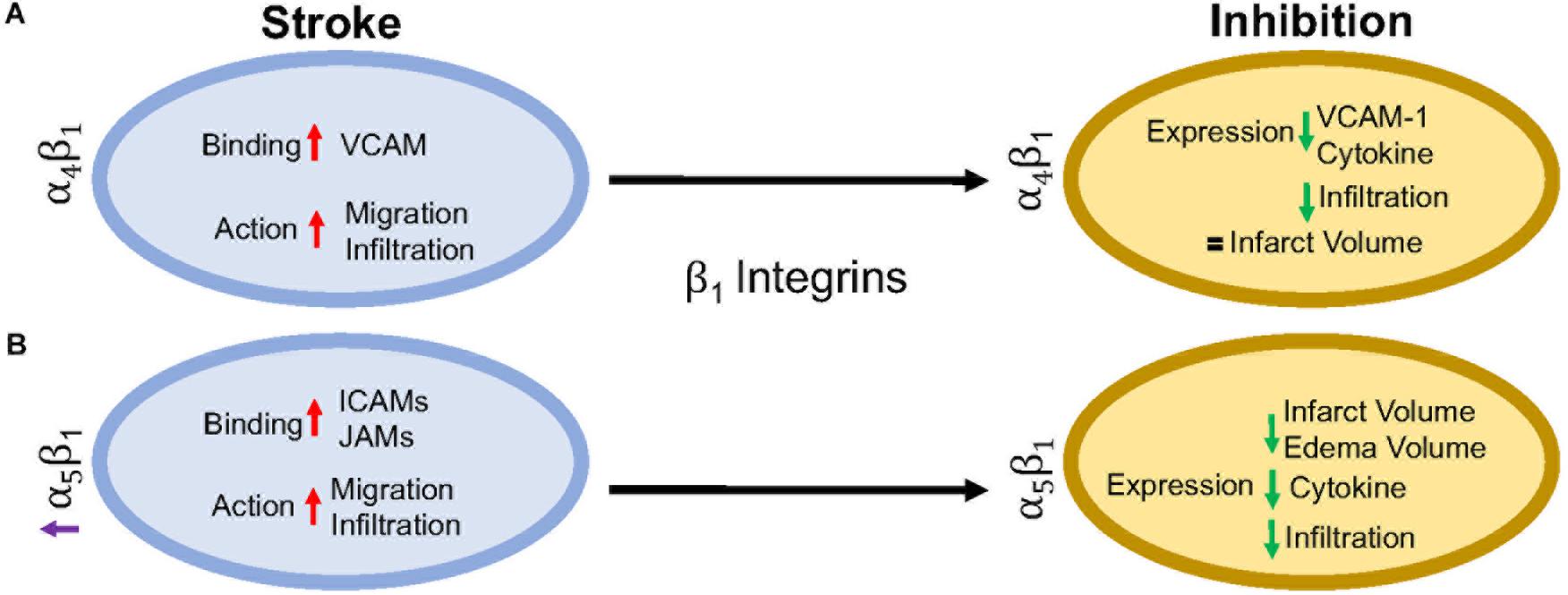
Figure 3. Representative image of the β1 integrin response following experimental stroke and inhibitory antibody treatment in preclinical trials. Inhibition of (A) α4β1 and (B) α5β1 integrins post-stroke responses and effects.
α4β1 Integrin
α4β1, also known as CD49d/CD29 VLA-4 (very late antigen-4), is localized primarily to leukocytes (neutrophils, monocytes, lymphocytes, macrophages, etc.) and microglia as a leukocyte-specific receptor. Additionally, α4 will also dimerize with β4, which is found in gut endothelium (Hammond et al., 2014). Activation of α4β1 integrin results from the binding of upregulated chemokines to GPCRs in the same manner as αLβ2 as discussed above (Vajkoczy et al., 2001). This stimulates binding to α4β1’s preferred ligand, VCAM-1, but experiments have shown some preference for paxillin ICAM-1 (Steffen et al., 1994; Ghandour et al., 2007), and fibronectin (Hart and Greaves, 2010) as well. Interestingly, instead of using the β1 submit of the heterodimer for binding, integrin α4β1 uses its α subunit of α4β1 to mediate binding to VCAM-1 (Luo et al., 2007).
Preclinical ischemic stroke studies targeting α4β1 have shown increasingly varied results. Most researchers reported a decrease in VCAM-1 expression, cytokine production, and infiltrating leukocytes (Liesz et al., 2011; Langhauser et al., 2014; Llovera et al., 2015), but this reduction in inflammation did not result in reduced infarct volumes or functional deficit following analysis of a randomized preclinical trial involving six different centers (Llovera et al., 2015). Langhauser et al went one step further and found that no treatment paradigm (prophylactic or therapeutic) and no model (transient or permanent) showed efficacy (Langhauser et al., 2014). On the other hand, both Becker, 2002 and Relton et al., 2000 found that inhibition of α4 improved both infarct volumes and functional deficits. When a preclinical randomized control trial was implemented at multiple centers, researchers found efficacy only in patients with small infarct volumes (Llovera et al., 2015). Collectively, these contradictory results may be caused by a couple of scenarios, 1) the varying expression of integrin α4β1 expression following ischemic stroke resulting in continued leukocyte infiltration, or 2) integrin α4β1 is not a primary driver of post-stroke pathophysiology, but other factors, including other integrins, promote leukocyte migration (Hammond et al., 2014).
α5β1 Integrin
α5β1, also known as CD49e/CD29 and VLA-5, plays an as yet largely undetermined role in inflammation, with studies primarily limited to cell culture. What is known is that leukocytes express different β1 integrins with α5β1 composing around 50% of all β1 –integrins expressed on neutrophils (Pierini et al., 2000) and monocytes (Pacifici et al., 1994). Additionally, α5β1 is necessary for leukocyte adhesion. Only inhibition of both α5β1 and β2 integrins completely blocks adhesion in vitro (Pierini et al., 2000), while inhibition of α5β1 alone prevents transmigration across the BBB (Labus et al., 2018) in vitro. As an RGD receptor, fibronectin has been shown to be the primary and preferred [over other potential ligands such as fibrinogen (Suehiro et al., 1997)] ligand for α5β1 on endothelial cells and leukocytes (Schaffner et al., 2013; Bharadwaj et al., 2017). Importantly, in the presence of activated αLβ2, leukocyte α5β1 binding to fibronectin is enhanced (Bohnsack, 1992; Loike et al., 1999; Gronholm et al., 2016). α5β1 integrin expression is induced by cytokines, particularly TNFα (Li et al., 2011) toward the leading edge of the cell in contrast with other integrins at the uropod (Pierini et al., 2000). Furthermore, α5β1 integrin appears to be highly sensitive to calcium (Pierini et al., 2000), an ion that is increased rapidly following reperfusion (Sandoval and Witt, 2008). Upon calcium buffering, α5β1 expression moves from the front of the cell to the uropod and the leukocyte becomes elongated. The change in expression localization and morphology is attributed to non-movement as the leukocyte cannot detach α5β1 from the vascular wall (Pierini et al., 2000). Recently, Edwards et al. (2019) found that inhibition of α5β1 integrin by the small peptide ATN-161 prevented CD45+ leukocytes from infiltrating the brain parenchyma following the tandem/transient common carotid artery/middle cerebral artery occlusion model. Additionally, mice were observed to have reduced BBB permeability, functional deficits, edema, and infarct volume following middle cerebral artery occlusion (Roberts et al., 2015; Edwards et al., 2019). Thus, targeting α5β1 after ischemic stroke could be a new avenue for reduction of inflammation following ischemic stroke.
Clinical Implications
The preclinical studies discussed here point towards the potential of targeting β2 and β1 integrins in the treatment of post-stroke inflammation. Though their potential has not fully been elucidated, many clinical trials, not just limited to stroke, have been approved in the last 10 years targeting these integrins.
The most common target for post-stroke inflammation are the β2 integrins. Though some efficacy has been reported, no clinical stroke trials to date have targeted the αL subunit in stroke patients. However, one clinical trial with the monoclonal antibody, Efalizumab, has shown promise in decreasing T-lymphocyte rolling in patients with moderate-severe plaque psoriasis (Lebwhohl et al., 2003).
In preclinical studies targeting αMβ2, a hookworm isolated recombinant glycoprotein targeting rNIF (UK279276) (Zhang et al., 2003) and humanized Hu23F2G (Leukarrest) (Yenari et al., 1998), were both shown to decrease infarct volume and increase functional recovery following reperfusion. Both therapies had negligible side effects in Phase 1 studies and thus were continued to a Phase II study, respectively, before the trials were halted due to no observed efficacy (Becker, 2002; Krams et al., 2003). The failure to target αMβ2 integrin may be due to the observation that human ischemic stroke patients do not experience the increase in αMβ2 expression as seen in rodent stroke models (Caimi et al., 2001). Interestingly, when given in conjunction with United Kingdom279276, patients experienced a slight improvement (Krams et al., 2003), but no follow-up has been conducted. This interesting effect may be worth additional investigation in future clinical trials.
Clinical inhibition of β1 integrins, on the other hand, is small and varied. Of the current clinical trials, one trial has emerged targeting α4β1 in the context of ischemic stroke. The monoclonal antibody targeting the α4 subunit (Natalizumab) has been successful in protecting patients from relapses in multiple sclerosis (Polman et al., 2006) and Crohn’s disease (Sandborn et al., 2015). However, in a Phase II ischemic stroke study, patients receiving Natalizumab showed no improvement in infarct growth or neurological scores over 30 days. Furthermore, two patients (out of 79) died from serious infections attributed to Natalizumab treatment (Elkins et al., 2017). At this time, there are no further clinical trials planned.
Future Considerations
As discussed in this review, targeting leukocytic integrins has had limited to no efficacy in clinical trials. Importantly, these studies have collectively employed only three different therapeutics and two targets; there are still significant areas that can be investigated. Though not discussed here, most preclinical investigations have focused on the ligands themselves rather than the integrin as the therapeutic target, highlighting the continued importance of integrins in stroke.
It is also important to note that preclinical studies carried out in rodents inadequately model the post-stroke pathophysiology that patients experience. Preclinical stroke research is also typically limited, focusing on one species, sex, and age that do not necessarily match the demographic of stroke patients (see Kahle and Bix, 2012 for a review of this topic). Furthermore, as the changes following stroke and/or reperfusion are inadequately understood, identifying appropriate therapeutic targets that translate from the lab to clinical trials, has been particularly challenging.
However, this does not suggest abandoning therapeutic trials for ischemic stroke. As mentioned above, stroke is a leading cause of death and disability, separate from cardiovascular disease. This will not improve without intervention with our aging and obese population. Fortunately, with the advent of stroke mortality-altering therapies, i.e., t-PA and endovascular mechanical thrombectomy, our financial burden has shifted to aftercare. When we review the amount of trials performed for thrombolytic agents (Multicentre Acute Stroke Trial–Italy (MAST-I) Group, 1995; National Institute of Neurological Disorders and Stroke rt-Pa Stroke Study Group, 1995; The Multicenter Acute Stroke Trial–Europe Study Group, 1996) and endovascular thrombectomy [MR CLEAN (Berkhemer et al., 2015), ESCAPE (Goyal et al., 2015), EXTEND IA (Campbell et al., 2015), SWIFT PRIME (Saver et al., 2015), and REVASCAT (Jovin et al., 2015)] as potential treatments of ischemic stroke, it is obvious that the complexities of stroke affect the outcome of the clinical trial. This includes, but is not limited to, the time a patient takes to arrive at an ER, time to treatment, location of the stroke, amount of surrounding collaterals, current medications and co-morbidities (diabetes, cancer, etc.), and if the patient has experienced multiple strokes.
Based on current advances, as well as previous failures, a focus on integrins as a therapeutic target for stroke is emerging. A significant reason for this focus may be the complex, multi-dimensional role that integrins appear to play in brain pathophysiology. Integrins are diverse, existing on all cell types with varying roles depending upon expression and activation. This complexity can represent a significant challenge to integrin-targeted therapies inasmuch as such therapies could have diverse, even unintended off-target effects. However, we believe that this can be overcome by a better understanding of how integrin function and expression is altered after stroke, with the potential to exploit stroke-dependent integrin changes to therapeutic effect. For example, identifying a specific integrin to be upregulated in select cells in the post-stroke brain or brain-targeting cells, but not in other organs, could render it a viable therapeutic target. This emphasizes the need and importance of preclinical stroke research to discover and unravel the complexities of integrin biology. We are confident that such studies will result in viable new stroke therapies.
Conclusion
In this review, we have implicated integrins as an area of research for limiting inflammation following ischemic stroke. To date, therapeutic inhibition of αLβ2, αMβ2, and α4β1 has shown promising results in preclinical studies, but translation to the clinic has been disappointing. Going forward, more targeted antibodies to all reactive β1 and β2 integrins after ischemic stroke may prove more beneficial, but more research needs to be done to completely understand the human inflammatory response and how that relates to changes in preclinical models.
Author Contributions
DE wrote the manuscript. DE and GB edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arnaout, M. A. (1990). Leukocyte adhesion molecules deficiency: its structural basis, pathophysiology and implications for modulating the inflammatory response. Immunol. Rev. 114, 145–180. doi: 10.1111/j.1600-065x.1990.tb00564.x
Arumugam, T. V., Salter, J. W., Chidlow, J. H., Ballantyne, C. M., Kevil, C. G., and Granger, D. N. (2004). Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 287, H2555–H2560. doi: 10.1152/ajpheart.00588.2004
Aspey, B., Jessimer, C., Pereira, S., and Harrison, M. (1989). Do leukocytes have a role in the cerebral no-reflow phenomenon? J. Neurol. Neurosurg. Psychiatry 52, 526–528. doi: 10.1136/jnnp.52.4.526
Bargatze, R., Kurk, S., Butcher, E., and Jutila, M. (1994). Neutrophils roll on adherent neutrophils bound to cytokine-induced endothelial cells via L-selectin on the rolling cells. J. Exp. Med. 180, 1785–1792. doi: 10.1084/jem.180.5.1785
Becker, K. (2002). Anti-leukocyte antibodies: leukarrest (Hu23F2G) and enlimomab (R6.5) in acute stroke. Curr. Med. Res. Opin. 18(Suppl. 2), 18–22.
Benjamin, E. J., Blaha, M. J., Chiuve, S. E., Cushman, M., Das, S. R., Deo, R., et al. (2017). Heart disease and stroke statistics—2017 update: a report from the american heart association. Stroke 137, 1–459.
Berkhemer, O. A., Fransen, P. S., Beumer, D., van den Berg, L. A., Lingsma, H. F., Yoo, A. J., et al. (2015). A randomized trial of intraarterial treatment for acute ischemic stroke. N. Engl. J. Med. 372, 11–20. doi: 10.1056/NEJMoa1411587
Bharadwaj, M., Strohmeyer, N., Colo, G. P., Helenius, J., Beerenwinkel, N., Schiller, H. B., et al. (2017). alphaV-class integrins exert dual roles on alpha5beta1 integrins to strengthen adhesion to fibronectin. Nat. Commun. 8:14348. doi: 10.1038/ncomms14348
Bohnsack, J. (1992). CD18-independent neutrophil adherence to laminin is mediated by the integrin VLA-6. Blood 79, 1545–1552.
Bowes, M., Rothlein, R., Fagan, S., and Zivin, J. (1995). Monoclonal antibodies preventing leukocyte activation reduce experimental neurologic injury and enhance efficacy of thrombolytic therapy. Neurology 45, 815–819. doi: 10.1212/WNL.45.4.815
Brait, V. H., Rivera, J., Broughton, B. R., Lee, S., Drummond, G. R., and Sobey, C. G. (2011). Chemokine-related gene expression in the brain following ischemic stroke: no role for CXCR2 in outcome. Brain Res. 1372, 169–179. doi: 10.1016/j.brainres.2010.11.087
Buck, B. H., Liebeskind, D. S., Saver, J. L., Bang, O. Y., Yun, S. W., Starkman, S., et al. (2008). Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 39, 355–360. doi: 10.1161/STROKEAHA.107.490128
Cai, T., and Wright, S. (1996). Human leukocyte elastase is an endogenous ligand for the integrin CR3 (CDllb/CD18, Mac-l, alphaM beta2) and modulates polymorphonuclear leukocyte adhesion. J. Exp. Med. 184, 1213–1223. doi: 10.1084/jem.184.4.1213
Caimi, G., Canino, B., Ferrara, F., Montana, M., Musso, M., Porretto, F., et al. (2001). Granulocyte integrins before and after activation in acute ischaemic stroke. J. Neurol. Sci. 186, 23–26. doi: 10.1016/s0022-510x(01)00495-6
Campanella, M., Sciorati, C., Tarozzo, G., and Beltramo, M. (2002). Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke 33, 586–592. doi: 10.1161/hs0202.103399
Campanero, M., Sanchez-Mateos, P., del Pozo, M., and Sanchez-Madrid, F. (1994). ICAM-3 regulates lymphocyte morphology and integrin-mediated T cell interaction with endothelial cell and extracellular matrix ligands. J. Cell Biol. 127, 867–878. doi: 10.1083/jcb.127.3.867
Campbell, B. C., Mitchell, P. J., Kleinig, T. J., Dewey, H. M., Churilov, L., Yassi, N., et al. (2015). Endovascular therapy for ischemic stroke with perfusion-imaging selection. N. Engl. J. Med. 372, 1009–1018. doi: 10.1056/NEJMoa1414792
Chavakis, T. (2012). Leucocyte recruitment in inflammation and novel endogenous negative regulators thereof. Eur. J. Clin. Invest. 42, 686–691. doi: 10.1111/j.1365-2362.2012.02677
Chavakis, T., May, A., Preissner, K., and Kanse, S. (1999). Molecular mechanisms of zinc-dependent leukocyte adhesion involving the urokinase receptor and B2-integrins. Blood 93, 2976–2983.
Chen, H., Chopp, M., Zhang, R., Bodzin, G., Chen, Q., Rusche, J., et al. (1994). Anti-CD11b monoclonal antibody reduces ischemic cell damage after transient focal cerebral ischemia in rat. Ann. Neurol. 35, 458–463. doi: 10.1002/ana.410350414
Chou, W., Choi, D., Zhang, H., Mu, D., McMahon, T., Kharazia, V., et al. (2004). Neutrophil protein kinase Cδ as a mediator of stroke-reperfusion injury. J. Clin. Invest. 114, 49–56. doi: 10.1172/JCI200421655
Clark, R., Lee, E., Fish, C., White, R., Price, W. J., Jonak, Z., et al. (1993). Development of tissue damage, inflammation, and resolution following stroke: an immunohistochemical and quantitative planimetric study. Brain Res. Bull. 31, 565–572. doi: 10.1016/0361-9230(93)90124-t
Connolly, E., Winfree, C., Springer, T. A., Naka, Y., Liao, H., Yan, S., et al. (1996). Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. J. Clin. Invest. 97, 209–216. doi: 10.1172/jci118392
de Fougerolles, A., Qin, X., and Springer, T. (1994). Characterization of the function of intercellular adhesion molecule (ICAM)-3 and comparison with ICAM-1 and ICAM-2 in immune responses. J. Exp. Med. 179, 619–629. doi: 10.1084/jem.179.2.619
de Fougerolles, A., Stacker, S., Schwarting, R., and Springer, T. (1991). Characterization of ICAM-2 and evidence for a third counter-receptor for LFA-1. J. Exp. Med. 174, 253–267. doi: 10.1084/jem.174.1.253
del Pozo, M. A., Sánchez-Mateos, P., Nieto, M., and Sánchez-Madrid, F. (1995). Chemokines regulate cellular polarization and adhesion receptor redistribution during lymphocyte interaction with endothelium and extracellular matrix. Involvement of cAMP signaling pathway. J. Cell Biol. 131, 495–508. doi: 10.1083/jcb.131.2.495
del Zoppo, G. (1994). Microvascular changes during cerebral ischemia and reperfusion. Cerebrovasc. Brain Metab. Rev. 6, 47–96.
Dereski, M., Chopp, M., Knight, R., Rodolosi, L., and Garcia, J. (1993). The heterogeneous temporal evolution of focal ischemic neuronal damage in the rat. Acta Neuropathol. 85, 327–333.
Dominguez, G. A., Anderson, N. R., and Hammer, D. A. (2015). The direction of migration of T-lymphocytes under flow depends upon which adhesion receptors are engaged. Integr. Biol. 7, 345–355. doi: 10.1039/c4ib00201f
Edwards, D., and Bix, G. (2019). Roles of integrins and extracellular matrix proteins after ischemic stroke. Am. J. Physiol. Cell Physiol. 316, C252–C263. doi: 10.1152/ajpcell.00151.2018
Edwards, D. N., Salmeron, K., Lukins, D., Trout, A., Fraser, J. F., and Bix, G. J. (2019). “Integrin a5b1 inhibition by ATN-161 reduces blood-brain barrier permeability, neuroinflammation, and is neuroprotective,” in Proceedings of the Abstract, International Stroke Conference 2019, Honolulu.
Elkins, J., Vetkamp, R., Montaner, J., Johnston, S., Singhal, A., Becker, K., et al. (2017). Safety and efficacy of natalizumab in patients with acute ischemic stroke (ACTION): a randomized, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 16, 217–226. doi: 10.1016/S1474-4422(16)30357-X
Enzmann, G., Mysiorek, C., Gorina, R., Cheng, Y. J., Ghavampour, S., Hannocks, M. J., et al. (2013). The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol. 125, 395–412. doi: 10.1007/s00401-012-1076-3
Furuya, K., Takeda, H., Azhar, S., McCarron, R., Chen, Y., Ruetzler, C., et al. (2001). Examination of several potential mechanisms for the negative outcome in a clinical stroke trial of enlimomab, a murine anti-human intercellular adhesion molecule-1 antibody: a bedside-to-bench study. Stroke 32, 2665–2674. doi: 10.1161/hs3211.098535
Gerhard, A., Neumaier, B., Elitok, E., Glatting, G., Tomczak, R., Ludolph, A., et al. (2000). In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport 11, 2957–2960. doi: 10.1097/00001756-200009110-00025
Ghandour, H., Cultere, X., Alvarez, A., Luscinskas, F., and Mayadas, T. (2007). Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin mediated human T-cell adhesion. Blood 110, 3682–3690. doi: 10.1182/blood-2007-
Goyal, M., Demchuk, A. M., Menon, B. K., Eesa, M., Rempel, J. L., Thornton, J., et al. (2015). Randomized assessment of rapid endovascular treatment of ischemic stroke. N. Engl. J. Med. 372, 1019–1030. doi: 10.1056/NEJMoa1414905
Greenwood, J., Wang, Y., and Calder, V. (1995). Lymphocyte adhesion and transendothelial migration in the central nervous system: the role of LFA-1, ICAM-1, VLA-4 and VCAM-1. off. Immunology 86, 408–415.
Gronholm, M., Jahan, F., Bryushkova, E. A., Madhavan, S., Aglialoro, F., Soto Hinojosa, L., et al. (2016). LFA-1 integrin antibodies inhibit leukocyte alpha4beta1-mediated adhesion by intracellular signaling. Blood 128,1270–1281. doi: 10.1182/blood-2016-03-705160
Hammond, M. D., Ambler, W. G., Ai, Y., and Sansing, L. H. (2014). alpha4 integrin is a regulator of leukocyte recruitment after experimental intracerebral hemorrhage. Stroke 45, 2485–2487. doi: 10.1161/STROKEAHA.114.005551
Hart, R., and Greaves, D. (2010). Chemerin contributes to inflammation by promoting macrophage adhesion to VCAM-1 and fibronectin through clustering of VLA-1 and VLA-5. J. Immunol. 185, 3728–3739. doi: 10.4049/jimmunol.0902154
Hato, T., Pampori, N., and Shattil, S. J. (1998). Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin aIIb b3. J. Cell Biol. 141, 1685–1695. doi: 10.1083/jcb.141.7.1685
Hess, D., Bhutwala, T., Sheppard, J., Zhao, W., and Smith, J. (1994a). ICAM-1 expression on human brain microvascular endothelial cells. Neurosci. Lett. 168, 201–204. doi: 10.1016/0304-3940(94)90450-2
Hess, D., Zhao, W., Carroll, J., McEachin, M., and Buchanan, K. (1994b). Increased expression of ICAM-1 during reoxygenation in brain endothelial cells. Stroke 25, 1463–1468.
Hyun, Y., Lefort, C., and Kim, M. (2009). Leukocyte integrins and their ligand interactions. Immunol. Res. 45, 195–208. doi: 10.1007/s12026-009-8101-1
Ishikawa, M., Vowinkel, T., Stokes, K. Y., Arumugam, T. V., Yilmaz, G., Nanda, A., et al. (2005). CD40/CD40 ligand signaling in mouse cerebral microvasculature after focal ischemia/reperfusion. Circulation 111, 1690–1696. doi: 10.1161/01.CIR.0000160349.42665.0C
Jiang, N., Chopp, M., and Chahwala, S. (1998). Neutrophil inhibitory factor treatment of focal cerebral ischemia in the rat. Brain Res. 788, 25–34. doi: 10.1016/s0006-8993(97)01503-5
Jovin, T. G., Chamorro, A., Cobo, E., de Miquel, M. A., Molina, C. A., Rovira, A., et al. (2015). Thrombectomy within 8 hours after symptom onset in ischemic stroke. N. Engl. J. Med. 372, 2296–2306. doi: 10.1056/NEJMoa1503780
Kahle, M. P., and Bix, G. J. (2012). Successfully climbing the “STAIRs”: surmounting failed translation of experimental ischemic stroke treatments. Stroke Res. Treat. 2012, 1–8. doi: 10.1155/2012/374098
Kaplanski, G., Farnaier, C., Benoliel, A. M., Foa, C., Kaplanski, S., and Bongrand, P. (1994). A novel role for E- and P-selectins: shape control of endothelial cell monolayers. J. Cell Sci. 107, 2449–2457.
Katakai, T., Habiro, K., and Kinashi, T. (2013). Dendritic cells regulate high-speed interstitial T cell migration in the lymph node via LFA-1/ICAM-1. J. Immunol. 191, 1188–1199. doi: 10.4049/jimmunol.1300739
Kitagawa, K., Matsumoto, M., Mabuchi, T., Yagita, Y., O’htsuki, T., Hori, M., et al. (1998). Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. JCBFM 18, 1336–1345. doi: 10.1097/00004647-199812000-00008
Kindzelskii, A. L., Laska, Z. O., Todd, R. F., and Petyy, H. R. (1996). Urokinase-type plasminogen activator receptor reversibly dissociates from complement receptor type 3 (alpha M beta 2’ CD11b/CD18) during neutrophil polarization. J. Immunol. 156, 297–309.
Kleinschnitz, C., Schwab, N., Kraft, P., Hagedorn, I., Dreykluft, A., Schwarz, T., et al. (2010). Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 115, 3835–3842. doi: 10.1182/blood-2009-10-249078
Krams, M., Lees, K. R., Hacke, W., Grieve, A. P., Orgogozo, J. M., Ford, G. A., et al. (2003). Acute stroke therapy by inhibition of neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke 34, 2543–2548. doi: 10.1161/01.STR
Kuwano, Y., Spelten, O., Zhang, H., Ley, K., and Zarbock, A. (2010). Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood 116, 617–624. doi: 10.1182/blood-2010-01-266122
Labus, J., Woltje, K., Stolte, K. N., Hackel, S., Kim, K. S., Hildmann, A., et al. (2018). IL-1beta promotes transendothelial migration of PBMCs by upregulation of the FN/alpha5beta1 signaling pathway in immortalized human brain microvascular endothelial cells. Exp. Cell Res. 373, 99–111. doi: 10.1016/j.yexcr.2018.10.002
Langhauser, F., Kraft, P., Gob, E., Leinweber, J., Schuhmann, M. K., Lorenz, K., et al. (2014). Blocking of alpha4 integrin does not protect from acute ischemic stroke in mice. Stroke 45, 1799–1806. doi: 10.1161/STROKEAHA.114.005000
Lebwhohl, M., Tyring, S., Hamilton, T., Toth, D., Glazer, S., Tawflk, N., et al. (2003). A novel targeted T-cell modulator, efalizumab, for plaque psoriasis. N. Eng. J. Med. 349, 2004–2013. doi: 10.1056/NEJMoa030002
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689. doi: 10.1038/nri2156
Li, B., Pozzi, A., and Young, P. P. (2011). TNFalpha accelerates monocyte to endothelial transdifferentiation in tumors by the induction of integrin alpha5 expression and adhesion to fibronectin. Mol. Cancer Res. 9, 702–711. doi: 10.1158/1541-7786.MCR-10-0484
Liesz, A., Zhou, W., Mracsko, E., Karcher, S., Bauer, H., Schwarting, S., et al. (2011). Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain 134(Pt 3), 704–720. doi: 10.1093/brain/awr008
Lindbom, L., and Werr, J. (2002). Integrin-dependent neutrophil migration in extravascular tissue. Semin. Immunol. 14, 115–121. doi: 10.1006/smim.2001.0348
Llovera, G., Hofmann, K., Roth, S., Salas-Perdomo, A., Ferrer-Ferrer, M., Perego, C., et al. (2015). Results of a preclinical randomized controlled multicenter trial (pRCT): anti-CD49d treatment for acute brain ischemia. Sci. Transl. Med. 7:299ra121. doi: 10.1126/scitranslmed.aaa9853
Lobb, R. R., and Hemler, M. E. (1994). The pathophysiologic role of alpha 4 integrins in vivo. J. Clin. Invest. 94, 1722–1728. doi: 10.1172/JCI117519
Loike, J., Cao, L., Budhu, S., Maracantonio, E., Khoury, J., Hoffman, S., et al. (1999). Differential regulation of b1 integrins by chemoattractants regulates neutrophil migration through fibrin. J. Cell Biol. 144, 1047–1056. doi: 10.1083/jcb.144.5.1047
Lorant, D., McEver, R., McIntyre, T. M., Moore, K., Prescott, S. M., and Zimmerman, G. A. (1995). Activation of polymorphonuclear leukocytes reduces their adhesion to P-selectin and causes redistribution of ligands for P-selectin on their surfaces. J. Clin. Invest. 96, 171–182. doi: 10.1172/jci118018
Lub, M., van Koyk, Y., and Figdor, C. (1996). Competition between lymphocyte function-associated antigen 1 (CD11a/CD18) and Mac-1 (CD11b/CD18) for binding to intercellular adhesion molecule-1 (CD54). J. Leukoc. Biol. 59, 648–655. doi: 10.1002/jlb.59.5.648
Luo, B. H., Carman, C. V., and Springer, T. A. (2007). Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647. doi: 10.1146/annurev.immunol.25.022106.141618
Marlin, S., and Springer, T. (1987). Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell 51, 813–819. doi: 10.1016/0092-8674(87)90104-8
Micklem, K., and Sim, R. (1985). Isolation of complement-fragment-iC3b-binding proteins by affinity chromatography. Biochemistry 231, 233–236. doi: 10.1042/bj2310233
Multicenter Acute Stroke Trial–Europe Study Group (1996). Thrombolytic therapy with streptokinase in acute ischemic stroke. N. Engl. J. Med. 335, 145–150. doi: 10.1056/nejm199607183350301
Multicentre Acute Stroke Trial–Italy (Mast-. (I)) Group (1995). Randomised controlled trial of streptokinase, aspirin, and combination of both in treatment of acute ischaemic stroke. Lancet 346, 1509–1514. doi: 10.1016/s0140-6736(95)92049-8
Nadareishvili, Z., Li, H., Wright, V., Maric, D., Warach, S., Hallenbeck, J., et al. (2004). Elevated pro-inflammatory CD4+CD28+ lymphocytes and stroke recurrence and death. Neurology 63, 1446–1451. doi: 10.1212/01.wnl.0000142260.61443.7c
National Institute of Neurological Disorders and Stroke rt-Pa Stroke Study Group (1995). Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 333, 1581–1587.
Nishida, N., Xie, C., Shimaoka, M., Cheng, Y., Walz, T., and Springer, T. A. (2006). Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity 25, 583–594. doi: 10.1016/j.immuni.2006.07.016
Nortamo, P., Li, R., Renkonen, R., Timonen, T., Prieto, J., Patarroyo, M., et al. (1991a). The expression of human intercellular adhesion molecule-2 is refractory to inflammatory cytokines. Eur. J. Immunol. 21, 2629–2632. doi: 10.1002/eji.1830211049
Nortamo, P., Salcedo, R., Timonen, T., Patarroyo, M., and Gahmber, C. (1991b). A monoclonal antibody to the human leukocyte adhesion molecule intercellular adhesion molecule-2. Cellular distribution and molecular characterization of the antigen. J. Immunol. 146, 2530–2535.
Pacifici, R., Roman, J., Kimble, R., Civitelli, R., Brownfield, C., and Bizzarri, C. (1994). Ligand binding to monocyte alpha 5 beta 1 integrin activates the alpha 2 beta 1 receptor via the alpha 5 subunit cytoplasmic domain and protein kinase C. J. Immunol. 153, 2222–2233.
Park, E. J., Peixoto, A., Imai, Y., Goodarzi, A., Cheng, G., Carman, C. V., et al. (2010). Distinct roles for LFA-1 affinity regulation during T-cell, diapedesis, and interstitial migration in lymph nodes. Immunology 115, 1572–1581. doi: 10.1182/blood-2009-08-237917
Pierini, L. M., Lawson, M. A., Eddy, R. J., Hendey, B., and Maxfield, F. R. (2000). Oriented endocytic recycling of a5b1 in motile neutrophils. Blood 95, 2471–2480.
Polman, C., O”Conner, P., Havrdova, E., Hutchinson, M., Kappos, L., Miller, D., et al. (2006). A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Eng. J. Med. 354, 899–910. doi: 10.1056/NEJMoa044397
Price, C. J., Menon, D. K., Peters, A. M., Ballinger, J. R., Barber, R. W., Balan, K. K., et al. (2004). Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke 35, 1659–1664. doi: 10.1161/01.STR.0000130592.71028.92
Rai, A. T., Seldon, A. E., Boo, S., Link, P. S., Domico, J. R., Tarabishy, A. R., et al. (2017). A population-based incidence of acute large vessel occlusions and thrombectomy eligible patients indicates significant potential for growth of endovascular stroke therapy in the USA. J. Neurointerv. Surg. 9, 722–726. doi: 10.1136/neurintsurg-2016-012515
Rao, N. M., Levine, S. R., Gornbein, J. A., and Saver, J. L. (2014). Defining clinically relevant cerebral hemorrhage after thrombolytic therapy for stroke: analysis of the National Institute of Neurological Disorders and Stroke tissue-type plasminogen activator trials. Stroke 45, 2728–2733. doi: 10.1161/STROKEAHA.114.005135
Relton, J., Sloan, K., Frew, E., Whalley, E., Adams, S., and Lobb, R. (2000). Inhibition of alpha4 integrins protects against transient focal cerebral ischemia in normotensive and hypertensive rats. Stroke 32, 199–205. doi: 10.1161/01.str.32.1.199
Ridley, A., Schwartz, M., Burridge, K., Firtel, R., Ginsberg, M., Borisy, G., et al. (2003). Cell migration: integrating signals from front to back. Science 302, 1704–1709. doi: 10.1126/science.1092053
Roberts, J., de Hoog, L., and Bix, G. J. (2015). Mice deficient in endothelial alpha5 integrin are profoundly resistant to experimental ischemic stroke. J. Cereb. Blood Flow Metab. 37, 85–96. doi: 10.1177/0271678X15616979
Sandborn, W., Colombel, J., Enns, R., Feagen, B., Hanauer, S., Lawrence, I., et al. (2015). Natalizumab induction and maintenance therapy for crohn’s disease. N. Eng. J. Med. 372:2074. doi: 10.1056/NEJMx140055
Sandoval, K. E., and Witt, K. A. (2008). Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 32, 200–219. doi: 10.1016/j.nbd.2008.08.005
Saver, J. L., Goyal, M., Bonafe, A., Diener, H. C., Levy, E. I., Pereira, V. M., et al. (2015). Stent-retriever thrombectomy after intravenous t-PA vs. t-PA alone in stroke. N. Engl. J. Med. 372, 2285–2295. doi: 10.1056/NEJMoa1415061
Schaffner, F., Ray, A. M., and Dontenwill, M. (2013). Integrin alpha5beta1, the fibronectin receptor, as a pertinent therapeutic target in solid tumors. Cancers 5, 27–47. doi: 10.3390/cancers5010027
Scharffetter-Kochanek, K., Lu, H., Norman, K., van Nood, N., Munoz, F., Grabbe, S., et al. (1998). Spontaneous skin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 188, 119–131. doi: 10.1084/jem.188.1.119
Schenkel, A. R., Mamdouh, Z., and Muller, W. A. (2004). Locomotion of monocytes on endothelium is a critical step during extravasation. Nat. Immunol. 5,393–400. doi: 10.1038/ni1051
Schittenhelm, L., Hilkens, C. M., and Morrison, V. L. (2017). beta2 integrins as regulators of dendritic cell, monocyte, and macrophage function. Front. Immunol. 8:1866. doi: 10.3389/fimmu.2017.01866
Schroeter, M., Jander, S., Witte, O., and Stoll, G. (1994). Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J. Neuroimmunol. 55, 195–203. doi: 10.1016/0165-5728(94)90010-8
Shaw, S. K., Ma, S., Kim, M. B., Rao, R. M., Hartman, C. U., Froio, R. M., et al. (2004). Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J. Exp. Med. 200, 1571–1580. doi: 10.1084/jem.20040965
Shyu, K., Chang, H., and Lin, C. (1997). Serum levels of intercellular adhesion molecule-1 and E-selectin in patients with acute ischaemic stroke. J. Neurosci. 244, 90–93. doi: 10.1007/s004150050055
Soriano, S., Coxon, A., Wang, Y., Frosch, M., Lipton, S., Hickey, P., et al. (1999). Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke 30, 134–139. doi: 10.1161/01.str.30.1.134
Springer, T. A., Galfre, G., Secher, D., and Milstein, C. (1979). Mac-1: a macrophage differentiation antigen identified by monoclonal antibody. Eur. J. Immunol. 9, 301–306. doi: 10.1002/eji.1830090410
Stanimirovic, D., Wong, J., Shapiro, A., and Durkin, J. (1997). Increase in surface expression of ICAM-1, VCAM-1 and E-selectin in human cerebromicrovascular endothelial cells subjected to ischemia-like insults. Acta Neurochir. Suppl. 70, 12–16. doi: 10.1007/978-3-7091-6837-0_4
Stefanidakis, M., and Koivunen, E. (2006). Cell-surface association between matrix metalloproteinases and integrins: role of the complexes in leukocyte migration and cancer progression. Blood 108, 1441–1450. doi: 10.1182/blood-2006-02-005363
Steffen, B., Butcher, E., and Engelhardt, B. (1994). Evidence for involvement of ICAM-1 and VCAM-1 in lymphocyte interaction with endothelium in experimental autoimmune encephalomyelitis in the central nervous system in the SJL/J mouse. Am. J. Pathol. 145, 189–201.
Stevens, S., Bao, J., Hollis, J., Lessov, N., Clark, W., and Stenzel-Poore, M. (2002). The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 932, 110–119. doi: 10.1016/s0006-8993(02)02292-8
Suehiro, K., Gailit, J., and Plow, E. F. (1997). Fibrinogen is a ligand for integrin alpha5beta1 on endothelial cells. J. Biol. Chem. 272, 5360–5366. doi: 10.1074/jbc.272.8.5360
Takagi, J., Petre, B., Walz, T., and Springer, T. A. (2002). Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599–611. doi: 10.1016/s0092-8674(02)00935-2
Tarkowski, E., Naver, H., Walling, B., Blomstrand, C., and Tarkowski, A. (1995). Lateralization of T-lymphocyte responses in patients with stroke. Effect of sympathetic dysfunction? Stroke 26, 57–62. doi: 10.1161/01.str.26.1.57
Tian, L., Kilgannon, P., Yoshihara, Y., Mori, K., Gallatin, W., Carpen, O., et al. (2000). Binding of T lymphocytes to hippocampal neurons through ICAM-5 (telencephalin) and characterization of its interaction with the leukocyte integrin CD11a/CD18. Eur. J. Immunol. 30, 810–818. doi: 10.1002/1521-4141(200003)30:3<810::aid-immu810>3.3.co;2-o
Tominaga, Y., Kita, T., Satoh, A., Asai, S., Kato, K., Ishikawa, K., et al. (1998). Affinity and kintetic analysis of the molecular interaction of ICAM-1 and leukocyte function-associated antigen-1. J. Immunol. 161, 4016–4022.
Vajkoczy, P., Laschinger, M., and Engelhardt, B. (2001). α4-integrin-VCAM-1 binding mediates G protein–independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J. Clin. Invest. 108, 557–565. doi: 10.1172/JCI200112440
Vemuganti, R., Dempsey, R. J., and Bowen, K. K. (2004). Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke 35, 179–184. doi: 10.1161/01.STR
von Andrian, U., Chambers, J., McEvoy, L., Bargatze, R., Arfors, K., and Butcher, E. (1991). Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta2 integrins in vivo. Proc. Natl. Acad. Sci. U.S.A. 88, 7538–7542. doi: 10.1073/pnas.88.17.7538
Walling, B. L., and Kim, M. (2018). LFA-1 in T cell migration and differentiation. Front. Immunol. 9:952. doi: 10.3389/fimmu.2018.00952
Wang, W. Z., Fang, X. H., Stephenson, L. L., Khiabani, K. T., and Zamboni, W. A. (2008). Ischemia/reperfusion-induced necrosis and apoptosis in the cells isolated from rat skeletal muscle. J. Orthop. Res. 26, 351–356. doi: 10.1002/jor.20493
Weston, R. M., Jones, N. M., Jarrott, B., and Callaway, J. K. (2007). Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. JCBFM 27, 100–114. doi: 10.1038/sj.jcbfm.9600324
Yenari, M. A., Kunis, D., Sun, G., Onley, D., Watson, L., Turner, S., et al. (1998). Hu23F2G, an antibody recognizing the leukocyte CD11/CD18 integrin, reduces injury in a rabbit model of transient focal cerebral ischemia. Exp. Neurol. 153, 223–233. doi: 10.1006/exnr.1998.6876
Yilmaz, G., Arumugam, T. V., Stokes, K. Y., and Granger, D. N. (2006). Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 113, 2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046
Zhang, L., Zhang, Z. G., Zhang, R. L., Lu, M., Krams, M., and Chopp, M. (2003). Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke 34, 1790–1795. doi: 10.1161/01.STR.0000077016.55891.2E
Zhang, R., Chopp, M., Zhang, Z., Jiang, N., and Powers, C. (1998). The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 785, 207–214. doi: 10.1016/s0006-8993(97)01343-7
Zhang, R., Zhang, Z., and Chopp, M. (1999). Increased therapeutic efficacy with rt-PA and anti-CD18 antibody treatment of stroke in the rat. Neurology 52, 273–279.
Zhang, Z., Chopp, M., Tang, W., Jiang, N., and Zhang, R. (1995). Postischemic treatment (2-4 h) with anti-CDllb and anti-CD18 monoclonal antibodies are neuroprotective after transient (2 h) focal cerebral ischemia in the rat. Brain Res. 698, 79–85. doi: 10.1016/0006-8993(95)00830-j
Keywords: ischemic stroke, integrins, inflammation, leukocytes, clinical trial results
Citation: Edwards DN and Bix GJ (2019) The Inflammatory Response After Ischemic Stroke: Targeting β2 and β1 Integrins. Front. Neurosci. 13:540. doi: 10.3389/fnins.2019.00540
Received: 07 March 2019; Accepted: 09 May 2019;
Published: 28 May 2019.
Edited by:
Xin Qi, Case Western Reserve University, United StatesReviewed by:
Jan Rossaint, University of Münster, GermanyShan Huang, University of California, Los Angeles, United States
Copyright © 2019 Edwards and Bix. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregory J. Bix, Z3JlZ29yeWJpeEB1a3kuZWR1