 Karina Joppe
Karina Joppe Anna-Elisa Roser
Anna-Elisa Roser Fabian Maass
Fabian Maass Paul Lingor
Paul Lingor- 1Department of Neurology, University Medical Center Göttingen, Göttingen, Germany
- 2Center for Biostructural Imaging of Neurodegeneration, Göttingen, Germany
- 3German Center for Neurodegenerative Diseases, Göttingen, Germany
- 4Rechts der Isar Hospital, Technical University of Munich, Munich, Germany
The homeostasis of iron is of fundamental importance in the central nervous system (CNS) to ensure biological processes such as oxygen transport, mitochondrial respiration or myelin synthesis. Dyshomeostasis and accumulation of iron can be observed during aging and both are shared characteristics of several neurodegenerative diseases. Iron-mediated generation of reactive oxygen species (ROS) may lead to protein aggregation and cellular toxicity. The process of misfolding and aggregation of neuronal proteins such as α-synuclein, Tau, amyloid beta (Aβ), TDP-43 or SOD1 is a common hallmark of many neurodegenerative disorders and iron has been shown to facilitate protein aggregation. Thus, both, iron and aggregating proteins are proposed to amplify their detrimental effects in the disease state. In this review, we give an overview on effects of iron on aggregation of different proteins involved in neurodegeneration. Furthermore, we discuss the proposed mechanisms of iron-mediated toxicity and protein aggregation emphasizing the red-ox chemistry and protein-binding properties of iron. Finally, we address current therapeutic approaches harnessing iron chelation as a disease-modifying intervention in neurodegenerative disorders, such as Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis.
Introduction
Neurodegenerative disorders (NDDs) rapidly gain importance due to their age-related prevalence and the resulting socio-economic burden (Hindle, 2010; Abbott, 2011). Aging is one of the main risk factors for NDDs (Ashraf et al., 2018) and the constantly growing life expectancy will result in their increased prevalence (Oeppen and Vaupel, 2002). Although different NDDs present a variety of symptoms ranging from cognitive, motor, sensory and/or autonomic failure, neuronal loss is the shared characteristic. Therefore, it is of great relevance to identify common pathophysiological features that are present in multiple NDDs to elucidate general mechanisms of neurodegeneration and potential pathways for intervention.
Iron does not only play a main role in cellular senescence but also in NDDs such as Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS) or prion diseases (PrD) (Ashraf et al., 2018). As protein aggregation is another shared hallmark among many NDDs (Iadanza et al., 2018), it is suggestive to assume a mutual interplay of iron and protein aggregation amplifying their detrimental effects. In addition to iron, other transition metals such as copper and manganese are also considered in the pathogenesis of NDDs. This review, however, is focusing on the role of iron in protein aggregation disorders and the reader is referred to alternative publications on the role of other metals (e.g., Carboni and Lingor, 2015; Kim et al., 2018).
In this review, we discuss the mode of action of iron in NDDs and proposed mechanisms of iron-mediated protein aggregation. Finally, we outline recent therapeutic approaches targeting iron as promising treatment option for NDDs.
Iron Dyshomeostasis and Accumulation
Iron is the most abundant trace metal in the human brain, present in neuronal and glial cells. It acts as a catalytic center for multiple enzymes and supports the synthesis of DNA, neurotransmitters and myelin in the brain. Furthermore, it participates in oxygen transport, neurotransmitter metabolism and mitochondrial respiration (Ward R.J. et al., 2014; Ashraf et al., 2018).
Iron metabolism is tightly regulated by iron responsive elements (IREs). In the presence of iron, binding of the iron regulatory protein (IRP) to IREs controls the translation of mRNAs related to proteins of iron metabolism, e.g., iron import, export and storage proteins. Under physiological conditions, extracellular ferric iron (Fe3+) is mainly bound to the glycoprotein transferrin (Tf), being delivered to cells by transferrin receptors (TfR). Non-transferrin-bound ferrous iron (Fe2+) is mainly transported into the cell via the divalent metal-ion transporter 1 (DMT1) (Hider and Kong, 2013; Jiang et al., 2017). DMT1 is also an important player in mitochondrial uptake of Fe2+ (Wolff et al., 2018). Intracellularly, ferritin is the major iron storage protein complex. Before storage in both H- or L-ferritins, Fe2+ is oxidized by H-ferritin to Fe3+. Under normal conditions, the labile Fe2+ pool and ferritin molecules are at an equilibrium. The export of Fe2+ is regulated by ferroportin-1 (FPN1) being controlled by hepcidin (Hider and Kong, 2013; Ashraf et al., 2018).
Impaired iron metabolism coupled with its accumulation in various brain regions are hallmarks of physiological aging. H-and L-ferritins also are more abundant with age (Zecca et al., 2001). During life, both ferritin subunits increase in their concentration within the SN but stay constant within the locus coeruleus. Both regions are important target areas for PD whereas the locus coeruleus is also affected in AD (Zecca et al., 2004a). Brains of patients suffering from NDDs, e.g., PD and AD, are lacking the age-associated rise of both ferritins. In PD, reduced ferritin levels in SN and pathological iron accumulation were found (Dexter et al., 1991; Connor et al., 1995).
Excessive ROS production resulting in oxidative stress is a common feature of NDDs and accumulated redox-active iron triggers ROS formation by the Fenton and Haber-Weiss reactions, providing the basis for catalyzed oxidation processes. Accordingly, iron reacts with hydrogen peroxide, which is a by-product of the mitochondrial respiration and intracellularly abundant, resulting in hydroxyl free radicals (HO•). Therefore, iron fosters the formation of ROS that lead to oxidative stress, inducing mitochondrial dysfunction and cell death (Zecca et al., 2004b).
This said, the reasons for iron accumulation and its precise effects on pathomechanisms in neurodegeneration remain still incompletely understood. Its contribution to the aggregation of disease-relevant proteins may be a major effector of its toxicity in NDDs.
Protein Aggregation
A shared hallmark of numerous NDDs is protein aggregation. For example, α-synuclein aggregates are the main components of Lewy bodies in PD (Spillantini et al., 1998), whereas neurofibrillary tangles and plaques in AD are composed of Tau and Amyloid beta (Aβ), respectively (Glenner and Wong, 1984; Brion, 1998). Aggregation of TDP-43 or SOD1 are observed in ALS (Brown, 1998; Neumann et al., 2006). Recent data demonstrate, however, that aggregation of one particular protein is not specific for one disease (e.g., Cisbani et al., 2017; Trist et al., 2018). Under physiological conditions, the ubiquitin proteasome system (UPS), autophagosomes and chaperone activity ensure the clearance of protein aggregates (Stroo et al., 2017). However, genetic or environmental factors can disturb the balance of aggregate formation and clearance, so that native soluble proteins or peptides start misfolding and assemble into insoluble beta-sheet oligomers and protofibrils. This filamentous aggregation results in amyloid fibrils and protein inclusion formation. For different disease-dependent proteins this aggregation process is likely to follow similar pathways (Soto and Pritzkow, 2018).
Whereas for protein inclusions a possible neuroprotective role is still discussed, oligomers and protofibrils of the above-mentioned species are very likely neurotoxic. Amyloid structures are believed to impair axonal transport, DNA transcription and the UPS, and trigger mitochondrial dysfunction, synaptic dysfunction and oxidative stress (Dhouafli et al., 2018; Iadanza et al., 2018). Furthermore, oligomers increase the lipid bilayer conductance and, therefore, induce calcium dyshomeostasis (Verma et al., 2015). Altogether, these mechanisms contribute to cellular dysfunction and cytotoxicity.
Iron and Protein Aggregation
Via interaction with redox-active metal ions, amyloidogenic forms of, e.g., Aβ or α-synuclein triggered ROS production and oxidative cytotoxicity (Liu et al., 2011; Deas et al., 2016). Especially iron was shown to enhance aggregation processes of α-synuclein (Ostrerova-Golts et al., 2000), Aβ (Rottkamp et al., 2001) or Tau (Sayre et al., 2000). How iron enhances protein aggregation is not fully understood, but two distinct mechanisms are considered as relevant. First, the direct binding of iron to amyloidogenic proteins, and second, an indirect iron-mediated process, where the above-mentioned Fenton and Haber-Weiss reaction of Fe2+ triggers aggregation by ROS production and resulting oxidative stress. An overview on relevant interactions of iron and below-mentioned proteins is shown in Figure 1.
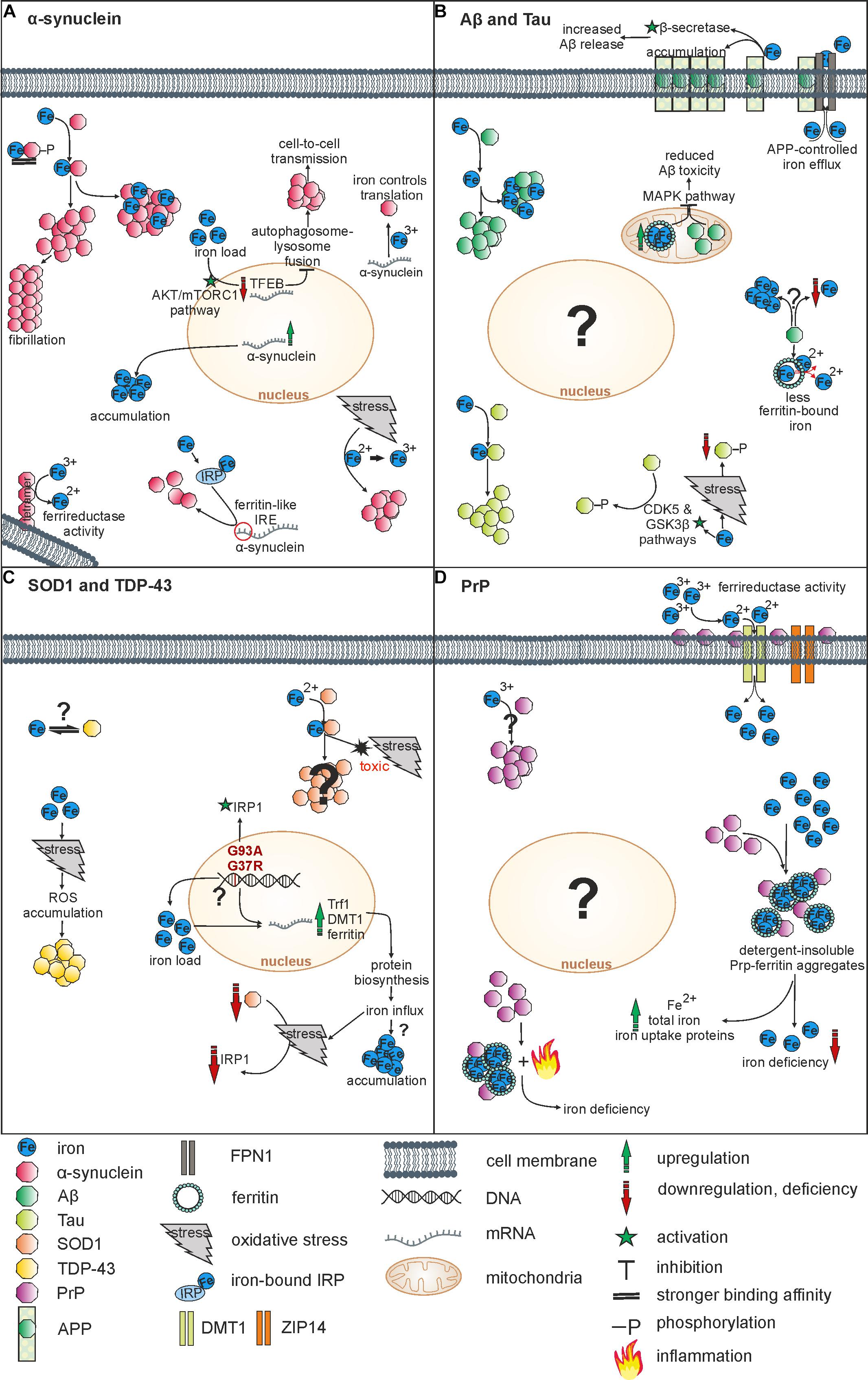
Figure 1. Overview on relevant interactions of iron and NDDs-associated proteins. (A) Iron induces α-synuclein aggregation by direct binding or via oxidation. Indirectly, iron also influences α-synuclein on its transcriptional and translational level. α-synuclein acts as a ferrireductase and can induce iron accumulation by overexpression. (B) Iron fosters aggregation of both Aβ und Tau by binding. Whereas Aβ reduces levels of ferritin-bound iron, an overexpression of mitochondrial ferritin reduces Aβ toxicity. APP controls iron efflux and together with iron it affects the Aβ release. Furthermore, there is evidence for both, Aβ-induced iron accumulation and Aβ-induced iron depletion. Whereas iron increases Tau-phosphorylation via CDK5 and GSK3ß pathways, iron-induced oxidative stress reduces Tau-phosphorylation. (C) Iron binds SOD1, inducing oxidative stress and toxicity. Mutations of SOD1 lead to an upregulation of iron metabolism proteins followed by iron influx. Iron is suggested to affect TDP-43 aggregation indirectly via oxidative stress-mediated ROS accumulation. An interaction of iron and TDP-43 has not been objectified so far. (D) PrP operates as a ferrireductase partner of ZIP14 and DMT1 increasing Fe3+ uptake. Furthermore, PrP-ferritin aggregates induce iron deficiency and an upregulation of total iron, Fe2+ and iron uptake proteins. Inflammation processes may contribute to iron deficiency. Vice versa, Fe3+ triggers PrP accumulation within the cell.
α-Synuclein
α-Synuclein is a 140 amino acid protein expressed in neuronal cytosol and presynaptic terminals that is thought to participate in vesicle packaging, release and trafficking as well as in membrane remodeling. Furthermore, interactions of α-synuclein with histones as well as with nuclear DNA are suspected, but its concrete function in the nucleus and presynaptic terminals needs to be further investigated (Bendor et al., 2013; Rocha et al., 2018). α-Synuclein is intensively studied in regard to the pathophysiology of PD, since some inherited forms of PD can result from point mutations and from overproduction of α-synuclein through multiplications of the SNCA gene encoding the human α-synuclein protein. In total, six point mutations (A30P, E46K, H50Q, G51D, A53T, and A53E) of the SNCA gene were identified so far leading to a biophysical change of amino acid substitutions (Uchihara and Giasson, 2016). Therefore, models overexpressing α-synuclein or a mutated form are used to understand the role of α-synuclein and its interaction with iron (e.g., Zhu et al., 2016; Carboni et al., 2017).
Analyzing the interaction of iron and α-synuclein, different studies could show that α-synuclein fibrillation can be induced by iron (Uversky et al., 2001; Golts et al., 2002; Kostka et al., 2008). In vitro, using BE-M17 neuroblastoma cells iron had stronger effects on aggregation of A53T and A30P mutant compared to wild-type α-synuclein (Ostrerova-Golts et al., 2000). In vivo, iron treatment showed reduced survival of α-synuclein mutant (A53T, A30P) and α-synuclein wild-type flies compared to w1118 wild-type controls, but only the mutant flies showed a strong motor decline (Zhu et al., 2016). Transmission electron microscopy resolved that ex vivo Fe3+ addition to wild-type and mutant (A53T, A30P, E46K) α-synuclein generates fibrils, resembling fibril conformations formed without iron incubation, whereas copper addition to mutant α-synuclein led to the formation of amorphous aggregates. These results indicate that the fibril morphology is metal-specific (Bharathi et al., 2007).
Even micromolar concentrations of Fe3+ increased α-synuclein aggregation and produced larger SDS-resistant oligomers. Since H2O2 treatment did not lead to the same effects as iron treatment, oxidation of α-synuclein per se cannot be the reason for the oligomerization, showing that trivalent ions play a relevant role (Kostka et al., 2008). However, further in vitro studies showed that Fe2+ also promoted α-synuclein aggregation, transmission and affected viability of SK-N-SH and SN4741 cells (Li et al., 2011; Xiao et al., 2018). Specifically, in SK-N-SH cells α-synuclein aggregation was increased around the nucleus (Li et al., 2011). Under aerobic conditions in vitro, Fe2+ treatment caused a polymerization into antiparallel soluble α-synuclein oligomers, whereas under anaerobic conditions both Fe2+ and Fe3+ induced parallel ß-sheet aggregates (Abeyawardhane et al., 2018).
How iron influences α-synuclein aggregation is not completely understood. However, α-synuclein has a high metal binding affinity and it is known that both Fe2+ and Fe3+ can bind α-synuclein, revealing a binding constant of 1.2 x 1013 M−1 for Fe3+ and 5.8 x 103 M−1 for Fe2+ (Peng et al., 2010). Fe2+ binds at the C-terminus, specifically at Asp-121, Asn-122, and Glu-123 (Binolfi et al., 2006) and the binding affinity is increased in phosphorylated (pY125 or pS129) α-synuclein (Lu et al., 2011). Additionally, Fe3+ can bind α-synuclein, having two binding sites, which are likely at the C-terminus (Davies et al., 2011).
Interestingly, there is a close homology in the 5′-UTR of human α-synuclein mRNA to the IRE of the ferritin mRNA, which could explain the regulation of α-synuclein levels by intracellular iron (Friedlich et al., 2007). Fe3+ was suggested to control the translation of α-synuclein mRNA since the iron chelator deferoxamine (DFO) highly decreased the polysome-associated endogenous α-synuclein mRNA in HEK293 cells (Febbraro et al., 2012). Furthermore, a knockdown of the IRP in SK-N-SH cells enhanced α-synuclein aggregation by upregulation of α-synuclein transcription, indicating that iron partially controls the aggregation process through the IRE/IRP system (Li et al., 2011). In addition to its direct binding there is thus evidence that iron also influences α-synuclein on a transcriptional and translational level.
Iron also promotes α-synuclein aggregation indirectly by regulating the nuclear transcription factor EB (TFEB), which is a transcriptional regulator of the autophagosome-lysosome pathway. Iron enrichment decreased the TFEB expression and inhibited its nuclear translocation through the activation of the Akt/mTORC1 pathway resulting in increased α-synuclein aggregation in cell lysates by the inhibition of TFEB-mediated autophagosome-lysosome fusion. Furthermore, iron increased α-synuclein cell-to-cell transmission, which was attenuated by TFEB overexpression (Xiao et al., 2018). As an indirect pathway, oxidative stress contributes to the iron-induced aggregation. This was further supported by the finding that supplementation of the antioxidative vitamin E attenuated aggregation in SK-N-SH cells (Li et al., 2011). Additionally, an ex vivo study indicated that oxidative stress enhances α-synuclein aggregation indirectly via oxidation of iron from Fe2+ to Fe3+ (Levin et al., 2011).
Vice versa, iron levels were increased by the overexpression of α-synuclein itself in primary midbrain neurons and PC12 cells which were analyzed with particle induced X-ray emission. Iron accumulation quantified by X-ray fluorescence was specifically observed within the perinuclear regions of PC12 cells (Ortega et al., 2016).
Furthermore, α-synuclein was also found to act as a ferrireductase, reducing Fe3+ to Fe2+ (Davies et al., 2011). The ferrireductase active form of α-synuclein is suggested to be a membrane-associated helical-rich tetramer (McDowall et al., 2017; Angelova and Brown, 2018). Previous studies suggest a resistance of the tetramer to fibril and aggregate formation, making the tetramer to an interesting subspecies in vivo (Bartels et al., 2011; Wang W. et al., 2011; Dettmer et al., 2015).
Regarding treatment strategies to reduce or prevent α-synuclein aggregation, iron seems to be a promising target. Iron recycling by Nramp1 (Soe-Lin et al., 2009) was shown to degrade microglial α-synuclein oligomers in vitro and in vivo, highlighting natural defense mechanisms in iron overload conditions (Wu et al., 2017). Furthermore, different iron chelators showed various beneficial effects in preclinical cell or animal PD models (e.g., Sangchot et al., 2002; Mandel et al., 2004; Billings et al., 2016; Finkelstein et al., 2016, 2017; Carboni et al., 2017; Das et al., 2017).
Iron and α-synuclein thus influence each other mutually: whereas iron contributes to α-synuclein aggregation by direct binding and indirectly via oxidative stress and transcription factors, α-synuclein shows ferrireductase activity influencing iron homeostasis.
Aβ and Tau
Aβ is a metalloprotein consisting of 39–43 residues that is derived from the transmembrane amyloid precursor protein (APP) by proteolytic cleavage. Aβ aggregates are found as amyloid plaques in AD and Aβ plays a role in metal sequestration and homeostasis, synaptic activity and neuronal plasticity (Smith et al., 2007; Rajasekhar et al., 2015). Aβ was found to be strongly colocalized with brain iron in risk patients for AD measured by magnetic resonance imaging and positron emission tomography (Van Bergen et al., 2016). X-ray studies analyzing Aβ plaques in cortex tissue of transgenic mice and Aβ plaque cores of AD patients confirmed a direct correlation of iron and Aβ localization, suggesting the formation of an iron-Aβ complex (Telling et al., 2017; Everett et al., 2018). Ex vivo, Fe3+ was found to promote aggregation of Aβ1-40 and Aβ1-42 visualized by fluorescence spectroscopy and atomic force microscopy (Tahmasebinia and Emadi, 2017). Fe3+ was also shown to bind Aβ using the phenolic oxygen of tyrosine 10 as binding site (Miura et al., 2001). Fe3+-mediated generation of Aβ aggregates was also reported in vitro. However, these aggregates were shorter and less ordered than in iron-free Aβ incubation (Liu et al., 2011). In drosophila, the affinity of Aβ for iron is mediated by three N-terminal histidines enhancing Aβ dimerization and leading to histidine-dependent oxidative damage (Ott et al., 2015).
Furthermore, treatment with iron chelators (clioquinol, YM-F24) increased survival and locomotor function of flies expressing Aβ1-42 compared to control wild-type flies, highlighting the relevance of oxidative stress for neurotoxicity of Aβ (Rival et al., 2009; Liu et al., 2011). Iron chelation also showed beneficial effects in mammalian models (Fine et al., 2015; Zhao et al., 2017) and reduced Aβ1-42 aggregation in an ex vivo study (Tahmasebinia and Emadi, 2017). Other studies showed that the presence of the important iron storage protein ferritin has beneficial effects on Aβ pathology. Accordingly, in vitro and in vivo, mitochondrial ferritin reduced neurotoxic effects exerted by Aβ (Wu et al., 2013; Wang P. et al., 2017). The overexpression of mitochondrial ferritin in SHSY5Y cells prevented the activation of the MAPK signaling pathway, which is related to oxidative stress-induced cell death (Wu et al., 2013).
Iron also strongly influences APP. Treating SHSY5Y cells with Fe3+ caused APP accumulation in membrane-enriched cellular fraction and increased activity of β-secretase that both triggered increased release of Aβ1-42 (Banerjee et al., 2014). Another study using SHSY5Y cells confirmed increased APP steady state levels and APP production after iron treatment (Rogers et al., 2016).
Furthermore, APP influences iron export by controlling the persistence of FPN1 on the neuronal surface, even if it does not function as a ferroxidase (Wong et al., 2014). There is evidence that Aβ induces iron accumulation in a cell-free ex vivo study (Everett et al., 2014b). Other studies suggest that Aβ controls the redox-activity of iron and reduces iron chemically in vivo (Everett et al., 2018). Accordingly, Aβ was able to inhibit ascorbate-dependent hydroxyl radical generation by free Fe3+ (Nakamura et al., 2007). Furthermore, interaction with Aβ led to a reduction of ferrihydrite (ferritin-bound iron) to pure redox-active Fe2+ (Everett et al., 2014a).
Iron was not only shown to modulate the aggregation of Aβ but also of Tau (Kim et al., 2018). Tau is a microtubule-associated protein that is the main component of neurofibrillary tangles in AD. Mostly, it is located in axons and sometimes in dendrites (Nisbet et al., 2015). So far, little is known about the interplay of iron and Tau and present studies are not consistent. Iron treatment enhanced Tau aggregation in iron-enriched hippocampal regions and iron was shown to bind Tau (Sayre et al., 2000). Some studies showed that only trivalent metal ions, as Fe3+, trigger Tau aggregation but not divalent ions (Yamamoto et al., 2002; Bader et al., 2011). Fe3+-generated Tau oligomers were even more stable than DMSO-generated ones (Nübling et al., 2012). However, a recent electrochemical study showed that both, Fe3+ and Fe2+, bind Tau at different binding sites, inducing a structural change, which was more pronounced with Fe2+ (Ahmadi et al., 2017).
Iron-induced oxidative stress reduced Tau phosphorylation by interfering with the function of the CDK5/p25 system of hippocampal neurons (Egaña et al., 2003). Another study using primary cultures of rat cortical neurons showed iron-induced Tau phosphorylation by activation of GSK3 (Lovell et al., 2004). Ebselen, an organo-selenium compound with antioxidant activity, inhibits the CDK5 and GSK3β pathway leading to less Tau phosphorylation. These effects were not only caused by the antioxidant effects of Ebselen, but rather by the inhibition of DMT1 in SHSY5Y cells (Xie et al., 2012). The same effects on CDK5 and GSK3β as well as on Tau phosphorylation were shown with the iron chelator DFO analyzing APP/PS1 transgenic mouse brains (Guo et al., 2013).
In summary, iron binds both, Aβ and Tau, partially modifying their structure and fostering their aggregation process. Furthermore, the Aβ and Tau phenotypes can be partially rescued by iron chelation.
SOD1 and TDP-43
SOD1 is an abundant antioxidant protein predominantly located within the cytosol. Its aggregation leads to, e.g., neuronal degeneration and changes in DNA/RNA metabolism, neurofilament and axonal transport (Pasinelli and Brown, 2006). An NMR study showed binding of iron to SOD1 with binding sites closely located to Cu/Zn binding pockets leading to Fe2+-bound SOD1 complexes, which are likely toxic (Lim and Song, 2015). Even if there is no direct evidence for iron-induced SOD1 aggregation, several studies emphasize a coherence of iron load and SOD1 pathology.
Therapeutic approaches showed that in SOD1 mouse models (G93A, G37R, G86R) iron chelators [VK-28, M30, HLA20, VAR-ced, deferiprone (DFP)] extended lifespan, increased locomotor function and motoneuron survival and decreased oxygen free radicals, iron levels and TfR expression (Jeong et al., 2009; Kupershmidt et al., 2009; Wang Q. et al., 2011; Golko-Perez et al., 2016; Moreau et al., 2018).
Furthermore, several studies observed an impact of SOD1 on iron metabolism. Accordingly, in vivo studies with SOD1.G93A mutant mice showed increased mRNA expression of ferritin, TfR1 and DMT1. These results indicate enhanced iron load, since iron is known to regulate ferritin expression (Jeong et al., 2009; Wang Q. et al., 2011). In vitro studies analyzing cell lysates showed an increased iron content and altered iron metabolism mediated by an impaired Akt signaling pathway. Accordingly, SOD1.G93A overexpression triggered an inactivation of Akt, activation of the transcription factor FOXO3a and subsequently increased ferritin synthesis and iron accumulation (Hadzhieva et al., 2013; Halon-Golabek et al., 2018). SOD1.G37R mutants showed in vivo and in vitro increased mRNA levels of TfR1, ferritin, DMT1 but also of mitochondrial ferritin (Jeong et al., 2009). The above-mentioned results of an increased iron content and increased TfR levels suggest an iron dyshomeostasis, which is not primarily controlled by IRE/IRP1 mechanisms (Lovejoy and Guillemin, 2014). Other studies showed diverse effects of SOD1 on the cytosolic iron sensor IRP1. SOD1 activation in G93A mutant mice showed no changes of IRP1 expression but more activated IRP1 (Jeong et al., 2009; Gajowiak et al., 2016). In vivo, SOD1 deficiency and the resulting oxidative stress caused IRP1 downregulation (Milczarek et al., 2017). Furthermore, in SOD1.G93A mutant mice ROS induction led to an upregulated iron import, triggering oxidative stress (Hadzhieva et al., 2013).
SOD1 is also thought to interact with the ALS-relevant protein TDP-43 (Higashi et al., 2010). Aggregates of the RNA-binding protein TDP-43 are incorporated in ubiquitinated inclusions within the neuronal cytoplasm found in ALS and in syndromes jointly named ‘neurodegeneration with brain iron accumulation’ (NBIA) (Neumann et al., 2006; Haraguchi et al., 2011). SOD1 was shown to initiate modification and accumulation of TDP-43 (Zeineddine et al., 2017; Jeon et al., 2018). In SOD1 mutant mice, iron chelators reduced TDP-43 aggregation, whereas vehicle-treated animals showed TDP-43 aggregates located in the cytoplasm of motor neurons (Wang Q. et al., 2011). Since oxidative stress-mediated accumulation of ROS fosters the TDP-43 aggregation in vitro (Cohen et al., 2012), iron chelator effects on TDP-43 aggregation suggest an indirect effect of iron by oxidative stress induction.
In conclusion, proteins of the iron metabolism are altered in SOD1 mutants, which partially could explain enhanced iron loading. Iron chelator effects in SOD1 models indicate an impact of iron on SOD1 pathology. Even if iron accumulation is a common feature of ALS (e.g., Moreau et al., 2018), a contribution of iron to aggregation of SOD1 or TDP-43 is yet unproven.
Prion Protein
The prion protein (PrP) is located intracellularly and is also an important membrane-bound protein at the cell surface. Therefore, PrP is involved in exocytotic and endocytic synaptic vesicle processing as well as in signaling pathways, but also in myelination and neurogenesis (Liebert et al., 2014; Legname, 2017; Watts et al., 2018).
By using PrP knockout or PrP-overexpressing mouse models, studies showed that upregulation or downregulation of PrP levels, respectively, affect the iron homeostasis (Singh et al., 2009a,b; Pushie et al., 2011; Ashok et al., 2018). Accordingly, iron deficiency is a crucial characteristic in brains of humans, hamster and mice affected with prion pathology. Analyzing pathological brain tissue as well as scrapie-infected ScN2a and SMB cells, iron dyshomeostasis was supposedly caused by the iron sequestration in detergent-insoluble PrP-scrapie-ferritin aggregates, resulting in a decreased bio-available iron pool and a state of cellular iron deficiency. These results also explain increased amounts of total iron and Fe2+ as well as iron uptake proteins (Singh et al., 2009a). Another study investigated PrP-mediated iron deficiency in retinas of scrapie-injected hamsters and demonstrated an accumulation of detergent-insoluble ferritin. Furthermore, they showed a correlation of the ferritin accumulation with microglia activity. These results suggested a contribution of chronic inflammation as a side effect of PrP accumulation to functional iron deficiency (Asthana et al., 2017).
Another explanation for the influence of PrP on the iron homeostasis is that PrP operates as a plasma membrane ferrireductase. Accordingly, PrP-expressing neuroblastoma cells showed a significant increase in ferrireductase activity compared to non-transfected cells. Furthermore, mutant PrP forms showed that for an optimal ferrireductase function of PrP the presence of NADH, the copper binding octa-peptide repeat region and the linkage to the plasma membrane is needed (Singh et al., 2013). Also in HepG2-cells, expressing PrP, ferrireductase activity was indicated by an increased uptake of Fe3+ but not of Fe2+. Since Fe3+ uptake was significantly increased after a co-expression of PrP with metal transporter ZIP14 and DMT1, PrP is suggested to be the ferrireductase partner of both metal transporter (Tripathi et al., 2015).
So far, there is not much evidence for the influence of iron on the PrP pathology. One study using PrP-deficient HpL3-4 cells showed that Fe3+ but not Fe2+ induced accumulation of internalized PrP after iron exposure and PrP treatment of cells (Choi et al., 2013). However, it is not clarified if a direct binding to the PrP protein or indirect mechanisms triggered by iron foster the aggregation of PrP.
Vice versa, PrP was shown to influence the iron homeostasis via its ferrireductase activity and PrP-ferritin aggregates but it needs further investigation to explain if these two modes of action or indirect mechanisms such as the activation of inflammation processes lead to iron deficiency.
Clinical Application
Iron accumulation in NDDs argues for a clinical evaluation of iron chelators as symptomatic or neuroprotective agents. Iron chelation by DFO, DFP or deferasirox (DFX) is clinically approved by the FDA for systemic conditions like acute iron intoxication and chronic iron overload1. DFO application is problematic due to the required continuous injection (short plasma half-life period) and dose-dependent neurotoxicity. DFX and DFP can be administered orally. DFX must be especially monitored for renal failure and DFP for agranulocytosis and neutropenia (Mobarra et al., 2016). With its ability to relocate iron and to cross the blood brain barrier, DFP presents the most promising candidate for targeting iron load in the CNS, being tested in NDDs with regional iron overload, e.g., Friedreich ataxia (e.g., Velasco-Sánchez et al., 2011; Pandolfo et al., 2014) and NBIA (e.g., Lim et al., 2018; Rohani et al., 2018).
In PD patients (FAIR-PARK-I, NCT00943748), DFP treatment showed benefits in motor performance and reduced MR-quantified substantia nigra iron content (Devos et al., 2014). In another trial (DeferipronPD, NCT01539837), administration of a lower dosage resulted in a reduced T2∗ MRI iron content in the dentate and caudate nucleus (Martin-Bastida et al., 2017). Based on these results, a European multicenter, parallel-group, placebo-controlled, randomized phase III trial is ongoing to assess disease-modifying effects of DFP in PD patients (FAIRPARKII, NCT02655315).
In a pilot trial (SAFEFAIRALS, NCT02164253), DFP treatment of ALS patients reduced R2∗ iron content in the cervical spinal cord, medulla oblongata and motor cortex. Additionally, patients showed a smaller decrease in the ALS function rating scale and in the body mass index in the first 3 months of treatment compared to the first treatment-free period (Moreau et al., 2018). Based on these results, a phase II/phase III study with a larger sample size has been initiated testing DFP (FAIR-ALSII, NCT03293069).
In AD, intramuscular application of DFO significantly reduced the decline of daily living skills (Crapper McLachlan et al., 1991). Besides, cognition of AD patients benefited from treatment with the metal-attenuating compound clioquinol or its derivate (Ritchie et al., 2003; Lannfelt et al., 2008). Now, AD patients are recruited for an ongoing phase II study investigating effects of DFP (The 3D Study, NCT03234686).
Details of the mentioned studies can be found in Supplementary Table 1. Summing up, iron chelators already showed promising results in smaller trials and this advocates for further assessment in larger studies.
Conclusion
Current investigations focus on iron accumulation and protein aggregation as two pathological hallmarks of multiple NDDs. The interaction of both features appears to be an essential part of common neurodegeneration mechanisms. So far, iron was found to bind amyloidogenic proteins like α-synuclein, Aβ and Tau fostering their aggregation. Indirectly, iron influences disease-related proteins via oxidative stress or manipulation of their transcription and translation. On the other hand, α-synuclein, Aβ and SOD1 were found to affect iron metabolism and iron accumulation, proposing a mutual interplay and an amplification of their detrimental effects in the disease state. Even if precise effects of iron on neurodegenerative pathomechanisms remain incompletely understood, translational trials in human patients already showed beneficial effects of iron chelation as a treatment strategy for NDDs. Thus, iron may be a substantial contributor to neurodegeneration and merits further investigation as molecular and therapeutic target.
Author Contributions
KJ developed the idea under the lead of PL, performed literature research, wrote and finalized the manuscript, and prepared the figure. A-ER and FM were involved in literature research and writing of the manuscript. PL developed the idea for this review and revised the manuscript. All authors have seen and approved the final version.
Funding
We acknowledge support by the German Research Foundation and the Open Access Publication Funds of the Göttingen University.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The support of the SFB 1286 “Quantitative Synaptology” was greatly acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.00015/full#supplementary-material
Footnote
References
Abeyawardhane, D. L., Fernández, R. D., Murgas, C. J., Heitger, D. R., Forney, A. K., Crozier, M. K., et al. (2018). Iron redox chemistry promotes antiparallel oligomerization of α-synuclein. J. Am. Chem. Soc. 140, 5028–5032. doi: 10.1021/jacs.8b02013
Ahmadi, S., Ebralidze, I. I., She, Z., and Kraatz, H.-B. (2017). Electrochemical studies of tau protein-iron interactions—Potential implications for Alzheimer’s disease. Electrochim. Acta 236, 384–393. doi: 10.1016/J.ELECTACTA.2017.03.175
Angelova, D. M., and Brown, D. R. (2018). Model senescent microglia induce disease related changes in α-synuclein expression and activity. Biomolecules 8:67. doi: 10.3390/biom8030067
Ashok, A., Karmakar, S., Chandel, R., Ravikumar, R., Dalal, S., Kong, Q., et al. (2018). Prion protein modulates iron transport in the anterior segment: implications for ocular iron homeostasis and prion transmission. Exp. Eye Res. 175, 1–13. doi: 10.1016/j.exer.2018.05.031
Ashraf, A., Clark, M., and So, P.-W. (2018). The aging of iron man. Front. Aging Neurosci. 10:65. doi: 10.3389/fnagi.2018.00065
Asthana, A., Baksi, S., Ashok, A., Karmakar, S., Mammadova, N., Kokemuller, R., et al. (2017). Prion protein facilitates retinal iron uptake and is cleaved at the β-site: implications for retinal iron homeostasis in prion disorders. Sci. Rep. 7, 1–14. doi: 10.1038/s41598-017-08821-1
Bader, B., Nübling, G., Mehle, A., Nobile, S., Kretzschmar, H., and Giese, A. (2011). Single particle analysis of tau oligomer formation induced by metal ions and organic solvents. Biochem. Biophys. Res. Commun. 411, 190–196. doi: 10.1016/j.bbrc.2011.06.135
Banerjee, P., Sahoo, A., Anand, S., Ganguly, A., Righi, G., Bovicelli, P., et al. (2014). Multiple mechanisms of iron-induced amyloid beta-peptide accumulation in SHSY5Y cells: protective action of negletein. Neuro Mol. Med. 16, 787–798. doi: 10.1007/s12017-014-8328-4
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–111. doi: 10.1038/nature10324
Bendor, J. T., Logan, T. P., and Edwards, R. H. (2013). The function of α-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
Bharathi, Indi, S. S., and Rao, K. S. J. (2007). Copper- and iron-induced differential fibril formation in α-synuclein: TEM study. Neurosci. Lett. 424, 78–82. doi: 10.1016/j.neulet.2007.06.052
Billings, J. L., Hare, D. J., Nurjono, M., Volitakis, I., Cherny, R. A., Bush, A. I., et al. (2016). Effects of neonatal iron feeding and chronic clioquinol administration on the parkinsonian human A53T transgenic mouse. ACS Chem. Neurosci. 7, 360–366. doi: 10.1021/acschemneuro.5b00305
Binolfi, A., Rasia, R. M., Bertoncini, C. W., Ceolin, M., Zweckstetter, M., Griesinger, C., et al. (2006). Interaction of α-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 128, 9893–9901. doi: 10.1021/ja0618649
Brion, J.-P. (1998). Neurofibrillary tangles and alzheimer’s disease. Eur. Neurol. 40, 130–140. doi: 10.1159/000007969
Brown, R. H. (1998). SOD1 aggregates in ALS: cause, correlate or consequence? Nat. Med. 4, 1362–1364. doi: 10.1038/3945
Carboni, E., and Lingor, P. (2015). Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics 7, 395–404. doi: 10.1039/C4MT00339J
Carboni, E., Tatenhorst, L., Tönges, L., Barski, E., Dambeck, V., Bähr, M., et al. (2017). Deferiprone rescues behavioral deficits induced by mild iron exposure in a mouse model of alpha-synuclein aggregation. Neuro Mol. Med. 19, 309–321. doi: 10.1007/s12017-017-8447-9
Choi, B. R., Lee, J., Kim, S. Y., Yim, I., Kim, E. H., and Woo, H. J. (2013). Prion protein conversion induced by trivalent iron in vesicular trafficking. Biochem. Biophys. Res. Commun. 432, 539–544. doi: 10.1016/j.bbrc.2013.02.021
Cisbani, G., Maxan, A., Kordower, J. H., Planel, E., Freeman, T. B., and Cicchetti, F. (2017). Presence of tau pathology within foetal neural allografts in patients with Huntington’s and Parkinson’s disease. Brain 140, 2982–2992. doi: 10.1093/brain/awx255
Cohen, T. J., Hwang, A. W., Unger, T., Trojanowski, J. Q., and Lee, V. M. (2012). Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 31, 1241–1252. doi: 10.1038/emboj.2011.471
Connor, J. R., Snyder, B. S., Arosio, P., Loeffler, D. A., and Lewitt, P. (1995). A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer’s diseased brains. J. Neurochem. 65, 717–724. doi: 10.1046/j.1471-4159.1995.65020717.x
Crapper, McLachlan, D. R., Dalton, A. J., Kruck, T. P., Bell, M. Y., Smith, W. L., et al. (1991). Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet (London, England) 337, 1304–1308. doi: 10.1016/0140-6736(91)92978-B
Das, B., Rajagopalan, S., Joshi, G. S., Xu, L., Luo, D., Andersen, J. K., et al. (2017). A novel iron (II) preferring dopamine agonist chelator D-607 significantly suppresses α-syn- and MPTP-induced toxicities in vivo. Neuropharmacology 123, 88–99. doi: 10.1016/J.NEUROPHARM.2017.05.019
Davies, P., Moualla, D., and Brown, D. R. (2011). Alpha-synuclein is a cellular ferrireductase. PLoS One 6:e015814. doi: 10.1371/journal.pone.0015814
Deas, E., Cremades, N., Angelova, P. R., Ludtmann, M. H. R., Yao, Z., Chen, S., et al. (2016). Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid. Redox Signal. 24, 376–391. doi: 10.1089/ars.2015.6343
Dettmer, U., Newman, A. J., von Saucken, V. E., Bartels, T., and Selkoe, D. (2015). KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 112, 9596–9601. doi: 10.1073/pnas.1505953112
Devos, D., Moreau, C., Devedjian, J. C., Kluza, J., Petrault, M., Laloux, C., et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 21, 195–210. doi: 10.1089/ars.2013.5593
Dexter, D. T., Carayon, A., Javoy-Agid, F., Agid, Y., Wells, F. R., Daniel, S. E., et al. (1991). Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative. Brain 114, 1953–1975. doi: 10.1093/brain/114.4.1953
Dhouafli, Z., Cuanalo-Contreras, K., Hayouni, E. A., Mays, C. E., Soto, C., and Moreno-Gonzalez, I. (2018). Inhibition of protein misfolding and aggregation by natural phenolic compounds. Cell. Mol Life Sci. 75, 3521–3538. doi: 10.1007/s00018-018-2872-2
Egaña, J. T., Zambrano, C., Nuñez, M. T., Gonzalez-Billault, C., and Maccioni, R. B. (2003). Iron-induced oxidative stress modify tau phosphorylation patterns in hippocampal cell cultures. BioMetals 16, 215–223. doi: 10.1023/A:1020727218493
Everett, J., Céspedes, E., Shelford, L. R., Exley, C., Collingwood, J. F., Dobson, J., et al. (2014a). Evidence of redox-active iron formation following aggregation of ferrihydrite and the Alzheimer’s disease peptide β-amyloid. Inorg. Chem. 53, 2803–2809. doi: 10.1021/ic402406g
Everett, J., Céspedes, E., Shelford, L. R., Exley, C., Collingwood, J. F., Dobson, J., et al. (2014b). Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide-amyloid (1-42). J. R. Soc. Interface 11:20140165. doi: 10.1098/rsif.2014.0165
Everett, J., Collingwood, J. F., Tjendana-Tjhin, V., Brooks, J., Lermyte, F., Plascencia-Villa, G., et al. (2018). Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale 10, 11782–11796. doi: 10.1039/C7NR06794A
Febbraro, F., Giorgi, M., Caldarola, S., Loreni, F., and Romero-ramos, M. (2012). α-Synuclein expression is modulated at the translational level by iron. Neuroreport 23, 576–580. doi: 10.1097/WNR.0b013e328354a1f0
Fine, J. M., Renner, D. B., Forsberg, A. C., Cameron, R. A., Galick, B. T., Le, C., et al. (2015). Intranasal deferoxamine engages multiple pathways to decrease memory loss in the APP/PS1 model of amyloid accumulation. Neurosci. Lett. 584, 362–367. doi: 10.1016/j.neulet.2014.11.013
Finkelstein, D. I., Billings, J. L., Adlard, P. A., Ayton, S., Sedjahtera, A., Masters, C. L., et al. (2017). The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 5:53. doi: 10.1186/s40478-017-0456-2
Finkelstein, D. I., Hare, D. J., Billings, J. L., Sedjahtera, A., Nurjono, M., Arthofer, E., et al. (2016). Clioquinol improves cognitive, motor function, and microanatomy of the alpha-synuclein hA53T transgenic mice. ACS Chem. Neurosci. 7, 119–129. doi: 10.1021/acschemneuro.5b00253
Friedlich, A. L., Tanzi, R. E., and Rogers, J. T. (2007). The 5′-untranslated region of Parkinson’s disease α-synuclein messenger RNA contains a predicted iron responsive element. Mol. Psychiatry 12, 222–223. doi: 10.1038/sj.mp.4001937
Gajowiak, A., Styś, A., Starzyński, R. R., Bednarz, A., Lenartowicz, M., Staroń, R., et al. (2016). Mice overexpressing both non-mutated human SOD1 and mutated SOD1G93A genes: a competent experimental model for studying iron metabolism in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 8:82. doi: 10.3389/fnmol.2015.00082
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/S0006-291X(84)80190-4
Golko-Perez, S., Amit, T., Youdim, M. B., and Weinreb, O. (2016). Beneficial effects of multitarget iron chelator on central nervous system and gastrocnemius muscle in SOD1(G93A) transgenic ALS mice. J. Mol. Neurosci. 59, 504–510. doi: 10.1007/s12031-016-0763-2
Golts, N., Snyder, H., Frasier, M., Theisler, C., Choi, P., and Wolozin, B. (2002). Magnesium inhibits spontaneous and iron-induced aggregation of α-synuclein. J. Biol. Chem. 277, 16116–16123. doi: 10.1074/jbc.M107866200
Guo, C., Wang, P., Zhong, M. L., Wang, T., Huang, X. S., Li, J. Y., et al. (2013). Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 62, 165–172. doi: 10.1016/j.neuint.2012.12.005
Hadzhieva, M., Kirches, E., Wilisch-Neumann, A., Pachow, D., Wallesch, M., Schoenfeld, P., et al. (2013). Dysregulation of iron protein expression in the G93A model of amyotrophic lateral sclerosis. Neuroscience 230, 94–101. doi: 10.1016/j.neuroscience.2012.11.021
Halon-Golabek, M., Borkowska, A., Kaczor, J. J., Ziolkowski, W., Flis, D. J., Knap, N., et al. (2018). hmSOD1 gene mutation-induced disturbance in iron metabolism is mediated by impairment of Akt signalling pathway. J. Cachexia Sarcopenia Muscle 9, 557–569. doi: 10.1002/jcsm.12283
Haraguchi, T., Terada, S., Ishizu, H., Yokota, O., Yoshida, H., Takeda, N., et al. (2011). Coexistence of TDP-43 and tau pathology in neurodegeneration with brain iron accumulation type 1 (NBIA-1, formerly Hallervorden-Spatz syndrome). Neuropathology 31, 531–539. doi: 10.1111/j.1440-1789.2010.01186.x
Hider, R. C., and Kong, X. (2013). “Iron: effect of overload and deficiency,” in Interrelations between Essential Metal Ions and Human Diseases, eds A. Sigel, H. Sigel, and R. K. O. Sigel (Dordrecht: Springer Netherlands), 1–535.
Higashi, S., Tsuchiya, Y., Araki, T., Wada, K., and Kabuta, T. (2010). TDP-43 physically interacts with amyotrophic lateral sclerosis-linked mutant CuZn superoxide dismutase. Neurochem. Int. 57, 906–913. doi: 10.1016/J.NEUINT.2010.09.010
Hindle, J. V. (2010). Ageing, neurodegeneration and Parkinson’s disease. Age Ageing 39, 156–161. doi: 10.1093/ageing/afp223
Iadanza, M. G., Jackson, M. P., Hewitt, E. W., Ranson, N. A., and Radford, S. E. (2018). A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 19, 755–773. doi: 10.1038/s41580-018-0060-8
Jeon, G. S., Shim, Y. M., Lee, D. Y., Kim, J. S., Kang, M. J., Ahn, S. H., et al. (2018). Pathological modification of TDP-43 in amyotrophic lateral sclerosis with SOD1 mutations. Mol. Neurobiol. doi: 10.1007/s12035-018-1218-2 [Epub ahead of print].
Jeong, S. Y., Rathore, K. I., Schulz, K., Ponka, P., Arosio, P., and David, S. (2009). Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 29, 610–619. doi: 10.1523/JNEUROSCI.5443-08.2009
Jiang, H., Wang, J., Rogers, J., and Xie, J. (2017). Brain iron metabolism dysfunction in Parkinson’s disease. Mol. Neurobiol. 54, 3078–3101. doi: 10.1007/s12035-016-9879-1
Kim, A. C., Lim, S., and Kim, Y. K. (2018). Metal ion effects on A β and tau aggregation. Int. J. Mol. Sci. 19:128. doi: 10.3390/ijms19010128
Kostka, M., Högen, T., Danzer, K. M., Levin, J., Habeck, M., Wirth, A., et al. (2008). Single particle characterization of iron-induced pore-forming α-synuclein oligomers. J. Biol. Chem. 283, 10992–11003. doi: 10.1074/jbc.M709634200
Kupershmidt, L., Weinreb, O., Amit, T., Mandel, S., Carri, M. T., and Youdim, M. B. (2009). Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 23, 3766–3779. doi: 10.1096/fj.09-130047
Lannfelt, L., Blennow, K., Zetterberg, H., Batsman, S., Ames, D., Harrison, J., et al. (2008). Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7, 779–786. doi: 10.1016/S1474-4422(08)70167-4
Legname, G. (2017). Elucidating the function of the prion protein. PLoS Pathog. 13:e1006458. doi: 10.1371/journal.ppat.1006458
Levin, J., Högen, T., Hillmer, A. S., Bader, B., Schmidt, F., Kamp, F., et al. (2011). Generation of ferric iron links oxidative stress to α-synuclein oligomer formation. J. Parkinsons. Dis. 1, 205–216. doi: 10.3233/JPD-2011-11040
Li, W., Jiang, H., Song, N., and Xie, J. (2011). Oxidative stress partially contributes to iron-induced alpha-synuclein aggregation in SK-N-SH cells. Neurotox. Res. 19, 435–442. doi: 10.1007/s12640-010-9187-x
Liebert, A., Bicknell, B., and Adams, R. (2014). Prion protein signaling in the nervous system—a review and perspective. Sign. Transduct. Insights 3:STI.S12319. doi: 10.4137/STI.S12319
Lim, L. Z., and Song, J. (2015). A novel SOD1-dependent mechanism for the iron-induced production of toxic SOD1 and oxidative stress that initiates ALS. bioRxiv [Preprint]. doi: 10.1101/018846
Lim, S.-Y., Tan, A. H., Ahmad-Annuar, A., Schneider, S. A., Bee, P. C., Lim, J. L., et al. (2018). A patient with beta-propeller protein-associated neurodegeneration: treatment with iron chelation therapy. J. Mov. Disord. 11, 89–92. doi: 10.14802/jmd.17082
Liu, B., Moloney, A., Meehan, S., Morris, K., Thomas, S. E., Serpell, L. C., et al. (2011). Iron promotes the toxicity of amyloid β peptide by impeding its ordered aggregation. J. Biol. Chem. 286, 4248–4256. doi: 10.1074/jbc.M110.158980
Lovejoy, D. B., and Guillemin, G. J. (2014). The potential for transition metal-mediated neurodegeneration in amyotrophic lateral sclerosis. Front. Aging Neurosci. 6:173. doi: 10.3389/fnagi.2014.00173
Lovell, M. A., Xiong, S., Xie, C., Davies, P., and Markesbery, W. R. (2004). Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. JAD 6, 659–671. doi: 10.3233/JAD-2004-6610
Lu, Y., Prudent, M., Fauvet, B., Lashuel, H. A., and Girault, H. H. (2011). Phosphorylation of alpha-synuclein at Y125 and S129 alters its metal binding properties: implications for understanding the role of alpha-synuclein in the pathogenesis of Parkinson’s disease and related disorders. ACS Chem. Neurosci. 2, 667–675. doi: 10.1021/cn200074d
Mandel, S., Maor, G., and Youdim, M. B. H. (2004). Iron and α-synuclein in the substantia nigra of MPTP-treated mice. J. Mol. Neurosci. 24, 401–416. doi: 10.1385/JMN:24:3:401
Martin-Bastida, A., Ward, R. J., Newbould, R., Piccini, P., Sharp, D., Kabba, C., et al. (2017). Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 7, 1–9. doi: 10.1038/s41598-017-01402-2
McDowall, J. S., Ntai, I., Hake, J., Whitley, P. R., Mason, J. M., Pudney, C. R., et al. (2017). Steady-state kinetics of α-synuclein ferrireductase activity identifies the catalytically competent species. Biochemistry 56, 2497–2505. doi: 10.1021/acs.biochem.7b00257
Milczarek, A., Starzyński, R. R., Styś, A., Jończy, A., Staroń, R., Grzelak, A., et al. (2017). A drastic superoxide-dependent oxidative stress is prerequisite for the down-regulation of IRP1: insights from studies on SOD1-deficient mice and macrophages treated with paraquat. PLoS One 12:e0176800. doi: 10.1371/journal.pone.0176800
Miura, T., Suzuki, K., and Takeuchi, H. (2001). Binding of iron (III) to the single tyrosine residue of amyloid b-peptide probed by Raman spectroscopy. J. Mol. Liq. 598, 79–84. doi: 10.1016/S0022-2860(01)00807-9
Mobarra, N., Shanaki, M., Ehteram, H., Nasiri, H., Sahmani, M., Saeidi, M., et al. (2016). A review on iron chelators in treatment of iron overload syndromes. Int. J. Hematol. Stem Cell Res. 10, 239–247.
Moreau, C., Danel, V., Devedjian, J. C., Grolez, G., Timmerman, K., Laloux, C., et al. (2018). Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid. Redox Signal. 29, 742–748. doi: 10.1089/ars.2017.7493
Nakamura, M., Shishido, N., Nunomura, A., Smith, M. A., Perry, G., Hayashi, Y., et al. (2007). Three histidine residues of amyloid-β peptide control the redox activity of copper and iron. Biochemistry 46, 12737–12743. doi: 10.1021/bi701079z
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (80-) 314, 130–134. doi: 10.1126/science.1134108
Nisbet, R. M., Polanco, J. C., Ittner, L. M., and Götz, J. (2015). Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 129, 207–220. doi: 10.1007/s00401-014-1371-2
Nübling, G., Bader, B., Levin, J., Hildebrandt, J., Kretzschmar, H., and Giese, A. (2012). Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with α-synuclein at the single molecule level. Mol. Neurodegener. 7, 1–13. doi: 10.1186/1750-1326-7-35
Oeppen, J., and Vaupel, J. W. (2002). Demography: broken limits to life expectancy. Science (80-) 296, 1029–1031. doi: 10.1126/science.1069675
Ortega, R., Carmona, A., Roudeau, S., Perrin, L., Duèiæ, T., Carboni, E., et al. (2016). α-Synuclein over-expression induces increased iron accumulation and redistribution in iron-exposed neurons. Mol. Neurobiol. 53, 1925–1934. doi: 10.1007/s12035-015-9146-x
Ostrerova-Golts, N., Petrucelli, L., Hardy, J., Lee, J. M., Farer, M., and Wolozin, B. (2000). The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 20, 6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000
Ott, S., Dziadulewicz, N., and Crowther, D. C. (2015). Iron is a specific cofactor for distinct oxidation- and aggregation- dependent Aβ toxicity mechanisms in a Drosophila model. Dis. Model. Mech. 8, 657–667. doi: 10.1242/dmm.019042
Pandolfo, M., Arpa, J., Delatycki, M. B., Le Quan, Sang, K. H., Mariotti, C., et al. (2014). Deferiprone in Friedreich ataxia: a 6-month randomized controlled trial. Ann. Neurol. 76, 509–521. doi: 10.1002/ana.24248
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723. doi: 10.1038/nrn1971
Peng, Y., Wang, C., Xu, H., Liu, Y.-N., and Zhou, F. (2010). Binding of α-synuclein with Fe(III) and with Fe(II) and biological implications of the resultant complexes. J. Inorg. Biochem. 104, 365–370. doi: 10.1016/j.jinorgbio.2009.11.005.Binding
Pushie, M. J., Pickering, I. J., Martin, G. R., Tsutsui, S., Jirik, F. R., and George, G. N. (2011). Prion protein expression level alters regional copper, iron and zinc content in the mouse brain. Metallomics 3, 206–214. doi: 10.1039/c0mt00037j
Rajasekhar, K., Chakrabarti, M., and Govindaraju, T. (2015). Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem. Commun. 51, 13434–13450. doi: 10.1039/c5cc05264e
Ritchie, C., Bush, A. L., Mackinnon, A., Macfarlane, S., Mastwyk, MacGregor L, M., et al. (2003). Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting aβ amyloid deposition and toxicity in alzheimer disease: a pilot phase 2 clinical trial. Arch. Neurol. 60, 1685–1691. doi: 10.1001/archneur.60.12.1685
Rival, T., Page, R. M., Chandraratna, D. S., Sendall, T. J., Ryder, E., Liu, B., et al. (2009). Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer’s disease. Eur. J. Neurosci. 29, 1335–1347. doi: 10.1111/j.1460-9568.2009.06701.x
Rocha, E. M., De Miranda, B., and Sanders, L. H. (2018). Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109, 249–257. doi: 10.1016/j.nbd.2017.04.004
Rogers, J. T., Venkataramani, V., Washburn, C., Liu, Y., Tummala, V., Jiang, H., et al. (2016). A role for amyloid precursor protein translation to restore iron homeostasis and ameliorate lead (Pb) neurotoxicity. J. Neurochem. 138, 479–494. doi: 10.1111/jnc.13671
Rohani, M., Razmeh, S., Shahidi, G. A., and Orooji, M. (2018). A pilot trial of deferiprone in pantothenate kinase-associated neurodegeneration patients. Neurol. Int. 9, 79–81. doi: 10.4081/ni.2017.7279
Rottkamp, C. A., Raina, A. K., Zhu, X., Gaier, E., Bush, A. I., Atwood, C. S., et al. (2001). Redox-active iron mediates amyloid-beta toxicity. Free Radic. Biol. Med. 30, 447–450. doi: 10.1016/S0891-5849(00)00494-9
Sangchot, P., Sharma, S., Chetsawang, B., Porter, J., Govitrapong, P., and Ebadi, M. (2002). Deferoxamine attenuates iron-induced oxidative stress and prevents mitochondrial aggregation and α-synuclein translocation in SK-N-SH cells in culture. Dev. Neurosci. 24, 143–153. doi: 10.1159/000065700
Sayre, L. M., Perry, G., Harris, P. L. R., Liu, Y., Schubert, K. A., and Smith, M. A. (2000). In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease. J. Neurochem. 74, 270–279. doi: 10.1046/j.1471-4159.2000.0740270.x
Singh, A., Haldar, S., Horback, K., Tom, C., Zhou, L., Meyerson, H., et al. (2013). Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimer’s Dis. 35, 541–552. doi: 10.3233/JAD-130218
Singh, A., Isaac, A. O., Luo, X., Mohan, M. L., Cohen, M. L., Chen, F., et al. (2009a). Abnormal brain iron homeostasis in human and animal prion disorders. PLoS Pathog. 5:e1000336. doi: 10.1371/journal.ppat.1000336
Singh, A., Kong, Q., Luo, X., Petersen, R. B., Meyerson, H., and Singh, N. (2009b). Prion protein (PrP) knock-out mice show altered iron metabolism: a functional role for PrP in iron uptake and transport. PLoS One 4:e6115. doi: 10.1371/journal.pone.0006115
Smith, D. G., Cappai, R., and Barnham, K. J. (2007). The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta – Biomembr. 1768, 1976–1990. doi: 10.1016/j.bbamem.2007.02.002
Soe-Lin, S., Apte, S. S., Andriopoulos, B., Andrews, M. C., Schranzhofer, M., Kahawita, T., et al. (2009). Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 5960–5965. doi: 10.1073/pnas.0900808106
Soto, C., and Pritzkow, S. (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 21, 1332–1340. doi: 10.1038/s41593-018-0235-9
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’ s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Stroo, E., Koopman, M., Nollen, E. A. A., and Mata-Cabana, A. (2017). Cellular regulation of amyloid formation in aging and disease. Front. Neurosci. 11:64. doi: 10.3389/fnins.2017.00064
Tahmasebinia, F., and Emadi, S. (2017). Effect of metal chelators on the aggregation of beta-amyloid peptides in the presence of copper and iron. BioMetals 30, 285–293. doi: 10.1007/s10534-017-0005-2
Telling, N. D., Everett, J., Collingwood, J. F., Gallagher, J. J., Wang, J., Hitchcock, A. P., et al. (2017). Iron Biochemistry is correlated with amyloid plaque morphology in an established mouse model of alzheimer’ s disease article iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of alzheimer’ s disease. Cell Chem. Biol. 24, 1205.e3–1215.e3. doi: 10.1016/j.chembiol.2017.07.014
Tripathi, A. K., Haldar, S., Qian, J., Beserra, A., Suda, S., Singh, A., et al. (2015). Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic. Biol. Med. 84, 322–330. doi: 10.2144/000114329.Functional
Trist, B. G., Fifita, J. A., Freckleton, S. E., Hare, D. J., Lewis, S. J. G., Halliday, G. M., et al. (2018). Accumulation of dysfunctional SOD1 protein in Parkinson’s disease is not associated with mutations in the SOD1 gene. Acta Neuropathol. 135, 155–156. doi: 10.1007/s00401-017-1779-6
Uchihara, T., and Giasson, B. I. (2016). Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 131, 49–73. doi: 10.1007/s00401-015-1485-1
Uversky, V. N., Li, J., and Fink, A. L. (2001). Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein: a possible molecular link between parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296. doi: 10.1074/jbc.M105343200
Van Bergen, J. M. G., Li, X., Hua, J., Schreiner, S. J., Steininger, S. C., Quevenco, F. C., et al. (2016). Colocalization of cerebral iron with amyloid beta in mild cognitive impairment. Sci. Rep. 6, 1–9. doi: 10.1038/srep35514
Velasco-Sánchez, D., Aracil, A., Montero, R., Mas, A., Jiménez, L., O’Callaghan, M., et al. (2011). Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 10, 1–8. doi: 10.1007/s12311-010-0212-7
Verma, M., Vats, A., and Taneja, V. (2015). Toxic species in amyloid disorders: oligomers or mature fibrils. Ann. Indian Acad. Neurol. 18, 138–145. doi: 10.4103/0972-2327.144284
Wang, P., Wu, Q., Wu, W., Li, H., Guo, Y., Yu, P., et al. (2017). Mitochondrial ferritin deletion exacerbates ß-amyloid-induced neurotoxicity in mice. Oxid. Med. Cell Longev. 2017:1020357. doi: 10.1155/2017/1020357
Wang, Q., Zhang, X., Chen, S., Zhang, X., Zhang, S., Youdium, M., et al. (2011). Prevention of motor neuron degeneration by novel iron chelators in SOD1(G93A) transgenic mice of amyotrophic lateral sclerosis. Neurodegener. Dis. 8, 310–321. doi: 10.1159/000323469
Wang, W., Perovic, I., Chittuluru, J., Kaganovich, A., Nguyen, L. T. T., Liao, J., et al. (2011). A soluble -synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U.S.A. 108, 17797–17802. doi: 10.1073/pnas.1113260108
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/j.cogdev.2010.08.003.Personal
Watts, J. C., Bourkas, M. E. C., and Arshad, H. (2018). The function of the cellular prion protein in health and disease. Acta Neuropathol. 135, 159–178. doi: 10.1007/s00401-017-1790-y
Wolff, N. A., Garrick, M. D., Zhao, L., Garrick, L. M., Ghio, A. J., and Thévenod, F. (2018). A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 8, 1–12. doi: 10.1038/s41598-017-18584-4
Wong, B. X., Tsatsanis, A., Lim, L. Q., Adlard, P. A., Bush, A. I., and Duce, J. A. (2014). B-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One 9:e114174. doi: 10.1371/journal.pone.0114174
Wu, K. C., Liou, H. H., Kao, Y. H., Lee, C. Y., and Lin, C. J. (2017). The critical role of Nramp1 in degrading α-synuclein oligomers in microglia under iron overload condition. Neurobiol. Dis. 104, 61–72. doi: 10.1016/j.nbd.2017.05.001
Wu, W.-S., Zhao, Y.-S., Shi, Z.-H., Chang, S.-Y., Nie, G.-J., Duan, X.-L., et al. (2013). Mitochondrial ferritin attenuates β-Amyloid-induced neurotoxicity: reduction in oxidative damage through the Erk/P38 mitogen-activated protein kinase pathways. Antioxid. Redox Signal. 18, 158–169. doi: 10.1089/ars.2011.4285
Xiao, Y., Chen, X., Huang, S., Li, G., Mo, M., Zhang, L., et al. (2018). Iron promotes α-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. J. Neurochem. 145, 34–50. doi: 10.1111/jnc.14312
Xie, L., Zheng, W., Xin, N., Xie, J. W., Wang, T., and Wang, Z. Y. (2012). Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem. Int. 61, 334–340. doi: 10.1016/j.neuint.2012.05.016
Yamamoto, A., Shin, R., and Hasegawa, K. (2002). Iron (III) induces aggregation of hyperphosphorylated s and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’ s disease. J. Neurochem. 82, 1137–1147. doi: 10.1046/j.1471-4159.2003.02066.x
Zecca, L., Gallorini, M., Schünemann, V., Trautwein, A. X., Gerlach, M., Riederer, P., et al. (2001). Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: consequences for iron storage and neurodegenerative processes. J. Neurochem. 76, 1766–1773. doi: 10.1046/j.1471-4159.2001.00186.x
Zecca, L., Stroppolo, A., Gatti, A., Tampellini, D., Toscani, M., Gallorini, M., et al. (2004a). The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. U.S.A. 101, 9843–9848. doi: 10.1073/pnas.0403495101
Zecca, L., Youdim, M. B. H., Riederer, P., Connor, J. R., and Crichton, R. R. (2004b). Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 5, 863–873. doi: 10.1038/nrn1537
Zeineddine, R., Farrawell, N. E., Lambert-Smith, I. A., and Yerbury, J. J. (2017). Addition of exogenous SOD1 aggregates causes TDP-43 mislocalisation and aggregation. Cell Stress Chaperones 22, 893–902. doi: 10.1007/s12192-017-0804-y
Zhao, Y., Sun, P., Chen, B., Lun, P., Dou, Y., and Cao, W. W. (2017). A study on the effect of ion chelating agent on Alzheimer disease. Biomed. Res. 28,8022–8026.
Keywords: iron, protein aggregation, neurodegeneration, disease mechanism, iron chelator
Citation: Joppe K, Roser A-E, Maass F and Lingor P (2019) The Contribution of Iron to Protein Aggregation Disorders in the Central Nervous System. Front. Neurosci. 13:15. doi: 10.3389/fnins.2019.00015
Received: 29 October 2018; Accepted: 08 January 2019;
Published: 22 January 2019.
Edited by:
Isabella Zanella, Università degli Studi di Brescia, ItalyReviewed by:
Neena Singh, Case Western Reserve University, United StatesLuigi Zecca, Italian National Research Council, Italy
Heather R. Lucas, Virginia Commonwealth University, United States
Copyright © 2019 Joppe, Roser, Maass and Lingor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karina Joppe, S2FyaW5hLkpvcHBlQG1lZC51bmktZ29ldHRpbmdlbi5kZQ== Paul Lingor, cGxpbmdvckBnd2RnLmRl