 Alireza Majdi
Alireza Majdi Farzin Kamari
Farzin Kamari Saeed Sadigh-Eteghad
Saeed Sadigh-Eteghad Albert Gjedde
Albert Gjedde- 1Neurosciences Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
- 2Department of Clinical Research, University of Southern Denmark, Odense, Denmark
- 3Department of Neuroscience, University of Copenhagen, Copenhagen, Denmark
- 4Department of Neurology and Neurosurgery, McGill University, Montreal, QC, Canada
- 5Department of Radiology and Radiological Science, Johns Hopkins University, Baltimore, MD, United States
Background: The alleged procognitive effects of nicotine and its metabolites in brain are controversial.
Objective: Here, we review the pharmacologically active metabolites of nicotine in brain and their effects on neuronal mechanisms involving two main cognitive domains, i.e., learning and memory.
Methods: We searched Embase, Medline via PubMed, Scopus, and Web of Science databases for entries no later than May 2018, and restricted the search to articles about nicotine metabolites and cognitive behavior or cognitive mechanisms.
Results: The initial search yielded 425 articles, of which 17 were eligible for inclusion after application of exclusion criteria. Of these, 13 were experimental, two were clinical, and two were conference papers.
Conclusions: The results revealed three pharmacologically active biotransformations of nicotine in the brain, including cotinine, norcotinine, and nornicotine, among which cotinine and nornicotine both had a procognitive impact without adverse effects. The observed effect was significant only for cotinine.
Introduction
The procognitive effects of nicotine are controversial. Some studies have shown positive effects of nicotine on learning and memory impairment in specific neurological disorders (López-Hidalgo et al., 2012; Newhouse et al., 2012; Allison and Shoaib, 2013; Majdi et al., 2018), while others reported negative effects of nicotine on cognitive abilities (Mundy and Iwamoto, 1988; Park et al., 2000).
After systemic administration, nicotine is extensively metabolized by the liver. Nicotine and some of its metabolites are biotransformed in the brain where they affect cognitive outcomes (Benowitz et al., 2009). Different metabolites of nicotine mediate different molecular and behavioral effects (Barreto et al., 2014), reported in studies of the influence of the metabolites on specific brain functions.
As the target of nicotine and its metabolites, nicotinic acetylcholine receptors (nAChR) modulate specific aspects of learning and memory (Majdi et al., 2017). Among different subtypes of nAChR, the α7 subtype may be mainly responsible for the procognitive and neuroprotective properties of acetylcholine (Sadigh-Eteghad et al., 2014; Wong et al., 2018). Although the metabolites of nicotine have lower affinity than nicotine to nAChR, the products interact with the receptors most likely as type 1 positive allosteric modulators (PAM) (Takeshima et al., 2007). The biotransformed metabolite cotinine long has been held to be responsible for memory supportive effects of nicotine, without the adverse effects (Green et al., 2000; Echeverria et al., 2011; Patel et al., 2014). In contrast, nornicotine has been found to possess the same addictive characteristics as nicotine (Green et al., 2000). Prevention of apoptosis, oxidative stress, and neuroinflammation, as well as augmentation of synaptic plasticity, modulation of glutamate release, and blockade of amyloid-beta or tau protein production pathways, are among the procognitive mechanisms proposed to underlie the effects of the metabolites (Soto-Otero et al., 2002; Hooper et al., 2008; Rehani et al., 2008; Echeverria et al., 2011; Moran, 2012), but the details of the molecular and behavioral mechanisms are incompletely understood.
Systematic reviews are tools that find relevant and unbiased answers to a research question (Sena et al., 2014). Due to the methodological strength, systematic reviews are reference standards for topics of controversy (Moher et al., 2015). The primary aim of this study was to identify known biotransformed products of nicotine in the brain, and the secondary aim was to reveal the known impacts on learning and memory and the mechanisms mediating the effects. First, we searched for specific biotransformed metabolites of nicotine in the brain, and second, we attempted to resolve the known effects on cognitive performance, including learning and memory and the mechanisms that mediate these brain functions.
Methods
Search Strategy
We electronically searched Embase, ISI Web of Science, MEDLINE via PubMed, and SCOPUS for studies that had investigated (1) nicotine metabolites in the brain as follows: [(nicotine)] AND [(metabolite)] AND [(brain) OR (central nervous system) OR (CNS)] and (2) the effects of nicotine metabolites on cognitive impairment as follows: [(memory) OR (learning) OR (cognition)] AND [(cotinine) OR (nicotine metabolite) OR (nornicotine) OR (nor-nicotine) OR (norcotinine) OR (nor-cotinine)]. Two investigators independently screened title, abstract and, where necessary, the full text, based on the inclusion and exclusion criteria. Where there were disagreements, the third investigator resolved the controversy. There was no date (all studies until May 2018) or species restriction in the search, but the search was limited to texts in English and original articles.
Inclusion and Exclusion Criteria
We included all experimental and clinical studies reporting the effects of nicotine metabolites (i.e., cotinine, nornicotine, and norcotinine) as opposed to placebo or vehicle on learning and memory. Because cognition is a broad topic, and because evaluation of each domain requires comprehensive review, we focused on learning and memory in this systematic review, regardless of type or assessment task. All other domains of cognition were not investigated in this review. We excluded every study of the effects of smoking cigarettes, cigars, or pipe, or of ingesting tobacco in any form, on cognitive abilities. We also excluded studies that evaluated the effects of nicotine (rather than its metabolites) on the cognitive function. We examined the effects of nicotine in a previous publication (Majdi et al., 2017).
Study Outcomes
The primary outcome of this review was evidence of specific biotransformed metabolites of nicotine in the brain, and the secondary outcome was evidence of effects on learning and memory and the mechanisms that mediate these brain functions.
Data Extraction
From the included articles, we extracted data of the metabolites, the type of studies (clinical or experimental), the nature of the condition in which metabolites had effects, the actual effect(s) (positive or negative), and the mechanism, dose, duration, and route of metabolite administration. We also noted study quality measures to evaluate the risk of bias (see below).
Quality of Selected Studies
A modified version of the CAMARADES' study quality checklist (Sadigh-Eteghad et al., 2017) was used to evaluate the methods used in the selected animal studies. The checklist provides the tools for assessment of the internal validity of the included studies (e.g., selection, performance, detection, and attrition bias) and other study quality measures (e.g., reporting quality and power). The items in the list include publication in a peer-reviewed journal, randomization to treatment or control, allocation concealment, blinded assessment of outcome, statement of inclusion and exclusion of animals from the study, sample-size calculation, statement of compliance with regulatory requirements and statement regarding possible conflicts of interest. The Cochrane risk of bias tool (Higgins et al., 2011) was used for human studies to determine different forms of bias, such as selection, performance, detection, attrition, and reporting.
Results and Discussion
Study Selection
The electronic search of the mentioned databases identified 426 articles of which 17 studies met the inclusion criteria (Figure 1). Fifteen articles reported animal experiments, and two articles reported studies of humans. The search identified five nicotine metabolites in the brain including cotinine, nornicotine, norcotinine, and two unnamed minor metabolites that have not been characterized fully yet. All included articles addressed the effects of cotinine on learning and memory, and no study addressed the impact of other nicotine metabolites on cognition.
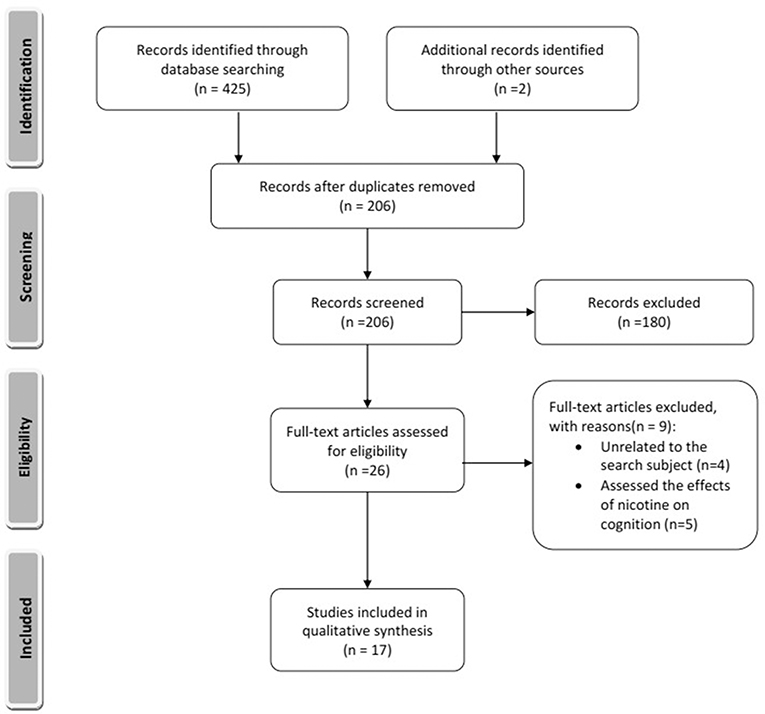
Figure 1. Summary of included and excluded articles. The style was adopted from Moher et al. (2009).
Study Quality
Low methodological quality of studies leads to overvaluation of effect sizes (Sadigh-Eteghad et al., 2017). We included 13 out of 17 publications into CAMARADES assessment. Two articles were human studies, assessed by the Cochrane tool, and two articles were conference papers that could not be evaluated by the checklists. The assessment showed that the quality of the animal studies included in the systematic review was modest (3.37 out of 8 items) (Figure 2). Some items on the checklist, such as reporting of animal exclusions, sample size calculation, and blinded induction of the model, usually were not reported. In contrast, the two human studies included in the review both had a low risk of bias. Considering the bias items in the design of future studies will reduce the risk of bias.
Figure 2. Quality assessment of the included animal studies according to modified CAMARADES' study quality checklist.
Nicotine
Nicotine Metabolism
Upon delivery to the systemic circulation, nicotine is distributed throughout the body as ionized (69%) and unionized (31%) forms, and its binding to proteins is insignificant (Benowitz et al., 1982). The main organ of nicotine metabolism is liver, followed by kidney, spleen, and lungs. The metabolism of nicotine is also substantial in the brain, and due to the upregulation of nAChR, the metabolism in the brain is higher in smokers than in non-smokers (Hukkanen et al., 2005).
After distribution throughout the body, including the liver, nicotine is extensively metabolized by the liver, and the metabolites or the remaining nicotine are then excreted in the urine. A main first pass pathway of nicotine metabolism in the human liver is C-terminal oxidation to cotinine by cytochrome P450 2A6 (CYP2A6) which is the predominant metabolite (70–80%) of nicotine in mammals (Nakajima and Yokoi, 2005). Other metabolites of nicotine are nicotine N′-oxide (4–7%), nicotine glucuronide (3–5%), 4-oxo-4-(3-pyridyl) butanoic acid (1–2%), nicotine isomethonium ion (0.4–1%), and nornicotine (0.4–0.8%) (Byrd et al., 1992; Hukkanen et al., 2005). Cotinine is further metabolized by cytochrome P450 2A5 (CYP2A5), mainly to trans-3′-hydroxycotinine and its glucuronides (Ghosheh and Hawes, 2002; Kuehl and Murphy, 2003). Other metabolites of cotinine are 5′-hydroxycotinine, cotinine N-oxide, cotinine methonium ion, cotinine glucuronide, and norcotinine (Hukkanen et al., 2005). Nicotine, cotinine, and their metabolites are then excreted in the urine by kidneys (Meger et al., 2002).
Nicotine Metabolism in the Brain
Nicotine distributes to the brain shortly after peripheral administration (whether intraperitoneal, intravenous, oral, or subcutaneous) with maximum between 30 and 60 min, and can be detected in the CNS as late as 4 h after injection (Crooks and Dwoskin, 1997). In contrast to distribution after peripheral administration, smoking causes nicotine to massively distribute to the bloodstream and from there to the brain in 10–20 s (Majdi et al., 2017). Due to the prominence of the base and associated lipid solubility, nicotine readily penetrates the blood-brain barrier (BBB) at physiological pH (Oldendorf et al., 1979; Tega et al., 2013). In addition, nicotine is transported through the BBB as a monoprotonated cation by organic cationic transport systems (Tega et al., 2013). Upon administration of a single dose of nicotine (0.54 mg/kg) in rats, the following quantities of metabolites were found in the brain at 4 h post-injection: cotinine (44.6 ng/g brain), nornicotine (11.7 ng/g brain), and norcotinine (3.1 ng/g brain) (Crooks and Dwoskin, 1997).
Until recently, little attention has been paid to nicotine's metabolism in the central nervous system (CNS). The current urge to study nicotine and its metabolites in the brain arose from the evidence that the metabolites are pharmacologically active and may mediate nicotine's apparent effects in the brain (Crooks et al., 1995).
Besides nicotine, five metabolites of nicotine can be identified in the brain, including cotinine, norcotinine, nornicotine, and two minor N-demethylated metabolites that as yet have not been fully elucidated (Crooks et al., 1997; Ghosheh et al., 2001). They are either transported from the periphery, or they are the biotransformation products of nicotine in the brain (Crooks and Dwoskin, 1997; Ghosheh et al., 2001). The half-lives of the main metabolites (i.e., cotinine, norcotinine, nornicotine) significantly exceed their precursor's sojourn in the brain, and their concentrations are 6, 4, and 3 times higher than that of nicotine, respectively (Ghosheh et al., 2001). It has been shown that repeated peripheral administration of nicotine can cause significant accumulation of the metabolites in the brain that may contribute to the neuropharmacological effects of nicotine in the brain (Crooks et al., 1997; Dwoskin et al., 1999).
Although a large body of evidence supports the procognitive effects of nicotine, there is insufficient knowledge of the metabolites and their impact in the brain (White and Levin, 1999, 2004; Rezvani and Levin, 2001; Grizzell and Echeverria, 2015; Majdi et al., 2017). There is evidence that nicotine metabolites play a role in the positive neuropharmacological effects of nicotine (e.g., on memory and learning) in the brain (Crooks and Dwoskin, 1997), and the metabolites, and especially cotinine, do not show the common cardiovascular and addictive effects of nicotine in the tested subjects (Moran, 2012). Therefore, studies of the role of nicotine metabolites in the treatment of cognitive impairment have gained considerable attention.
Cotinine
Cotinine Properties
Cotinine [(S)-1-methyl-5-(3-pyridinyl)-2-pyrrolidinone] is believed to be the main proximate metabolite of nicotine in the brain (Crooks et al., 1997). Structurally, it differs from nicotine only by an acetyl group (Fox et al., 2015). The accumulation of cotinine in the brain and its passage through the BBB are much slower than those of nicotine. Nicotine has been found to be present in the brain five min after subcutaneous injection, compared to cotinine's 30–60 min. The concentration peaks in 4 h and is detectable in the brain until 18 h after nicotine injection. As a result, its residence in blood and brain tissue is much longer than that of nicotine, and it may be responsible for nicotine's more prolonged pharmacological effects in the brain (Ghosheh et al., 1999; Buccafusco and Terry, 2003; Terry et al., 2005). Besides redistribution from the systemic circulation by passage through the BBB, some cotinine in the brain can also stem from local transformation of nicotine (Crooks and Dwoskin, 1997).
Cotinine does not cause tachyphylaxis, addiction, or nicotine-like withdrawal symptoms, and it has no negative cardiovascular effects as opposed to nicotine (Terry et al., 2005; Benowitz et al., 2009; Zeitlin et al., 2012). On the other hand, cotinine has positive effects on cognition and enhances learning, memory, and attention (Terry et al., 2005; Zeitlin et al., 2012). Therefore, as a pharmacologically active metabolite of nicotine, it may be a promising therapeutic option in the treatment of cognitive disorders (Terry et al., 2005).
Receptor Interactions
As a type 1 PAM of nAChR, cotinine's affinity is low compared to that of nicotine (Riah et al., 1999; Vainio and Tuominen, 2001; Takeshima et al., 2007). However, the affinity is high enough to trigger nicotinic responses in the brain (Vainio and Tuominen, 2001). Cotinine may enhance the effectiveness of endogenous ligands, such as acetylcholine but is unlikely to have agonist effects or to change the receptors' expression. In contrast to nicotine, cotinine does not interfere with receptor desensitization (Wildeboer-Andrud et al., 2014).
The findings cited above are not universally replicated, and some studies yielded opposite results. Rezvani and Levin (2001) showed that cotinine administration has the same effects as nicotine on the trafficking and assembly of nAChR and can up- or downregulate their expression, but at higher concentrations of cotinine, this effect appears to be lost. Although a majority of cotinine effects are mediated via α7 subtype, a recent study showed that chronic cotinine administration increases α4β2 subtype expression and the trafficking of receptors to the plasma membrane at doses around 1 μM, which equals its average blood concentration in a typical smoker. On the other hand, the highest doses (10 μM) was found to induce endocytosis and decrease α4β2 expression (Fox et al., 2015). More studies are needed to resolve the exact interactions between cotinine and nAChR fully.
Cognition
A growing body of evidence supports a procognitive effect of cotinine in animals (Herzig et al., 1998; Grizzell et al., 2014a; Grizzell and Echeverria, 2015). However, the two studies of cognition in humans included here failed to replicate the positive effects of cotinine on the cognitive performance of animals (Hatsukami et al., 1997; Herzig et al., 1998) (Table 1). The discrepancy may stem from the fact that neither human study examined the effects of chronic cotinine administration on human subjects, with cotinine administered for either 1 or 3 days. Thus, chronic cotinine administration in clinical studies deserves further investigation. Also, interspecies differences between rodents and humans may justify the observed differences among studies. The limited qualities of experimental studies and the lack of vigorous designs may also play a role in this regard. Figure 3 illustrates the major pathways found to mediate procognitive effects of cotinine. The material discussed in the following sections is based on evidence from animal studies.
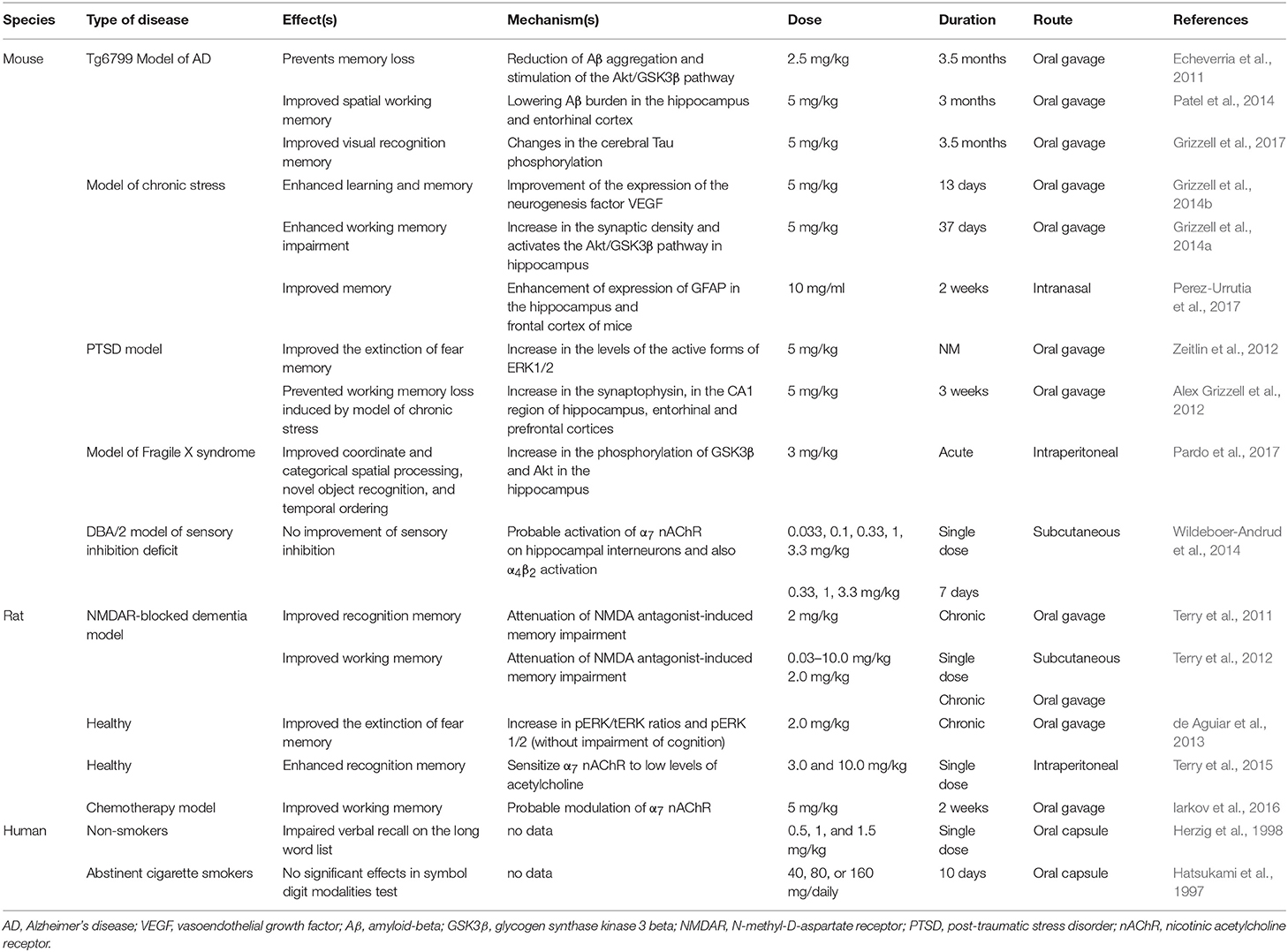
Table 1. Selected studies investigating the effects of cotinine on cognitive performance in various neurological disorders.
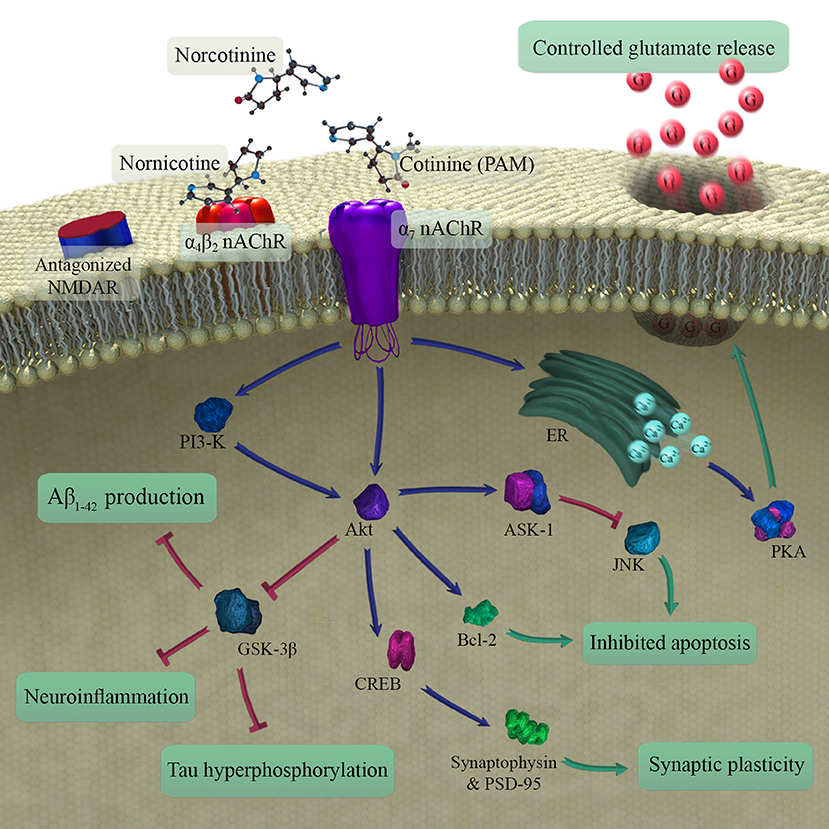
Figure 3. Schematic illustration of metabolites of nicotine in brain and mechanisms involved in the procognitive effects of cotinine (the main procognitive metabolite). As a type 1 PAM, cotinine modulates the function of α7 nAChR that in turn leads to reduced Aβ1−42 production and decreased neuroinflammation, tau hyperphosphorylation, and apoptosis. It also improves synaptic plasticity. In the end, the changes may contribute to the reduction of age-related cognitive impairment. PAM, positive allosteric modulator; nAChR, nicotinic acetylcholine receptor; NMDAR, N-methyl-D-aspartic acid receptor; ER, endoplasmic reticulum; PKA, protein kinase A; Aβ, amyloid-beta.
Apoptosis and neuronal survival
Apoptosis, a programmed form of cell death, has been implicated in the pathogenesis of memory disorders, such as AD (Majdi et al., 2016). This process is controlled by a variety of pro- and anti-apoptotic signals inside neurons (Kim et al., 2001). Akt is a family of serine-threonine-specific protein kinases that inhibit programmed cell death and promote neuronal survival by phosphorylation and inhibition of proapoptotic proteins, such as glycogen synthase kinase 3 (GSK3) (Dudek et al., 1997; Kim et al., 2001). Cotinine, by its positive allosteric effects on α7 nAChR, activates the Akt pathway that subsequently raises the expression of anti-apoptotic proteins, such as the cAMP response element binding (CREB) protein and B-cell lymphoma protein 2 (Bcl-2). Akt stimulation also decreases the activity of pro-apoptotic factors including c-Jun N-terminal kinase (JNK) by triggering apoptosis signal-regulating kinase 1 (Ask-1) that ultimately promotes neuronal survival (Kim et al., 2001; Moran, 2012).
Synaptic plasticity and density
Synaptic plasticity and density are of central importance to learning and memory (Silva, 2003). Studies prove that synaptic dysfunction happens before neuronal degeneration in neurodegenerative disorders and age-related cognitive decline (Selkoe, 2002; VanGuilder et al., 2011; Phan et al., 2017). A marker of synaptic density, synaptophysin is detected in synaptic vesicles (Valtorta et al., 2004). Cotinine has been shown to remarkably increase the expression of synaptophysin, and with it, synaptic density in the prefrontal cortex and hippocampus and thus to improve learning and memory (Grizzell et al., 2014a). An increase in the expression of post-synaptic density protein-95 (PSD-95), which also promotes synaptic plasticity, has been reported with cotinine treatment. The mechanism of both of these changes is the cotinine-induced modulation of α7 nAChR that subsequently stimulates protein kinases phosphoinositide-3 kinase (PI3K). PI3K then induces Akt phosphorylation, leading to increase in the CREB protein transcriptional activity. The increase raises the expression of the synaptic proteins and improves cognitive performance (Zeitlin et al., 2012; Grizzell et al., 2014b).
Amyloid-beta production and aggregation
As the main neurotoxic forms of Aβ, amyloid-beta1−42 (Aβ1−42) oligomers are believed by some to cause the cognitive dysfunction of AD (Resende et al., 2008; Sadigh-Eteghad et al., 2015). Cotinine blocks Aβ1−42 aggregation and oligomerisation, reduces number and size of plaques, decreases the Aβ42/Aβ40 ratio. Protection of neurons against Aβ1−42-induced neurotoxicity and possible subsequent improvement of cognition (Burgess et al., 2011; Echeverria et al., 2011) are not explained by interaction with nAChR, as the effects are not eliminated by blockade of the receptors (Burgess et al., 2011). The mechanism of cotinine's effects on the Aβ clearance, therefore, remains unclear, although cotinine inhibits activation of GSK3β and may reduce Aβ1−42 production by Akt activation in both cortex and hippocampus (Echeverria et al., 2011). GSK3β is a proline-directed serine-threonine kinase, and excessive activation may impair memory by increase of Aβ production and hyperphosphorylation of tau (Hooper et al., 2008).
Tau hyperphosphorylation and NFT formation
Hyperphosphorylated tau is the major component of neurofibrillary tangles (NFT) that are a key pathological finding in AD and other cognitive disorders (Mitchell et al., 2002). Tau accumulation in the temporal lobe correlates better with cognitive dysfunction than Aβ deposition in any region of the brain (Brier et al., 2016). Tau is phosphorylated by GSK3β, and this enzyme's activity is closely associated with NFT burden in AD brains (Baum et al., 1996; Plattner et al., 2006). As discussed above, cotinine inhibits tau hyperphosphorylation through activation of the Akt pathway and subsequent blockade of GSK3β in a concentration-dependent manner. Evidence suggests that α7 nAChR may mediate these effects of cotinine on the brain (Burgess et al., 2008; Echeverria et al., 2011).
Modulation of glutamate release
Controlled release of glutamate in the cortex regulates high cortical functions, such as learning and memory (Rahn et al., 2012), and disruption of glutamatergic neurotransmission has been implicated in the pathogenesis of cognitive decline (Tsai and Coyle, 2002). It has been shown that cotinine administration enhances attention and executive function in glutamate antagonist-induced cognitive impairment in the rat, possibly due to the activation of α7 nAChR (Terry et al., 2012).
Activation of α7 receptors stimulates calcium release from intracellular sources (Dajas-Bailador et al., 2002). Also, α7 nAChR enhance depolarization of nerve terminals, opening voltage-gated calcium channels with calcium entry into the cell. Increased calcium levels then, directly and indirectly, raise glutamate release from synapses through activation of cAMP-PKA-dependent pathways (Girod et al., 2000; Cheng and Yakel, 2015). The α7 nAChR induced glutamate surge also plays a role in presynaptic facilitation and synaptic plasticity (Livingstone et al., 2010).
Neuroinflammation
The anti-inflammatory properties of nAChR, especially the α7 subtype, are well-known from numerous studies (Metz and Tracey, 2005; de Jonge and Ulloa, 2007; Egea et al., 2015). Neuroinflammation is a hallmark both of normal brain aging and of pathological aging with cognitive disorders, such as AD (Ownby, 2010; Sadigh-Eteghad et al., 2016), and elevation of inflammatory markers is directly linked to the degree of cognitive impairment (Ownby, 2010). Through an nAChR and NF-κB-dependent pathway, cotinine lowers the levels of pro-inflammatory molecules, such as TNF-α, IL-1β as well as IL-6 and enforces anti-inflammatory cytokines including IL-10 production (Rehani et al., 2008). Also, cotinine exerts its anti-inflammatory effects via regulation of PI3K-Akt and inhibition of the GSK3β pathways that provoke neuroinflammation (Rehani et al., 2008; Echeverria et al., 2016). This action makes cotinine a potential candidate for the treatment of the neuroinflammatory disorders, e.g., as seen in AD.
Oxidative Stress
Under controlled circumstances, cotinine blocks Fenton's reaction and prevents free radical production in the brain (Soto-Otero et al., 2002). Evidence suggests that addition of cotinine and iron to the media before H2O2 blocks free radical formation and reduces oxidative stress. This can be partly explained by the fact that addition of nicotine or cotinine chelates iron and halts Fenton's reaction (Nakajima et al., 1996; Soto-Otero et al., 2002). It appears that cotinine also lowers lipid peroxidation in a manner that cannot be explained by the effects on Fenton's reaction. The reduction can result from chain-breaking antioxidant properties of cotinine (Soto-Otero et al., 2002). Oxidative stress and lipid peroxidation are of crucial importance to brain aging and neurodegeneration and the accompanying cognitive decline (Sadigh-Eteghad et al., 2015; Pourmemar et al., 2017). Thus, treatment with anti-oxidant effect is a top priority in these conditions (Fukui et al., 2002; Mecocci, 2004; Williams et al., 2006; Schrag et al., 2013).
Nornicotine
Nornicotine or demethylcotinine is a major pharmacologically active metabolite of nicotine in the brain which possibly acts via nAChR (Dwoskin et al., 2001). Oxidative N-demethylation of nicotine is the major pathway by which nornicotine is produced in the CNS (Crooks et al., 1997; Ghosheh et al., 2001). Compared with the periphery where nornicotine is considered to be a minor metabolite (0.8%), its concentration in the brain is higher for several reasons, including the longer half-life in comparison to nicotine, its superior partitioning as well as active transport to the CNS and transformation of nicotine to nornicotine in the brain (Ghosheh et al., 2001).
Although nornicotine is as potent as nicotine, it is less desensitizing at the major nAChR subtypes in the brain, and nornicotine's presence leads to the activation of α7 nAChR. Nornicotine's potency and efficacy differ by several folds, but it has been shown that peak currents caused by nornicotine acting at α7 nAChR are equal to those of acetylcholine. Considering nornicotine's durable presence in the brain, the molecule may mediate some of the neuroprotective effects of nicotine. A study showed that α7 receptors are responsive to nornicotine, and the action at the receptors of this nicotine metabolite leads to improved cognition and attention (Papke, 2006; Papke et al., 2007). Nornicotine may also alter Aβ's aggregation, possibly via reduced plaque formation or altered clearance of the peptide, or both, as well as by attenuated toxicity of soluble Aβ aggregates (Dickerson and Janda, 2003). More studies are needed to better define nornicotine effects on brain function, learning, and memory.
Norcotinine
In addition to the major metabolites mentioned above, there are minor CNS biotransformation products of nicotine, including norcotinine. After peripheral injection of nicotine, norcotinine is detected in the brain, and it is likely produced by 5′-C-oxidation of brain nornicotine. This fate is different from the processing in the periphery where N-demethylation of cotinine produces norcotinine. It has been shown that only 0.16% of cotinine is metabolized into norcotinine (Li et al., 2015). In vivo, the metabolite neither evoked the release of dopamine from rat striatal slices nor inhibited dopamine uptake into rat striatal synaptosomes (Crooks et al., 1995), suggesting that this minor metabolite, in fact, may be pharmacologically inactive (Crooks et al., 1997). Thus, there is no information on norcotinine's effects on cognitive performance, but possible effects are under investigation because of the pharmacological and therapeutic potentials of cotinine in cognitive disorders, such as AD (Li et al., 2012).
Conclusion
Nicotine lowers learning and memory impairment in some neurological disorders. However, its adverse cardiovascular and addictive effects limit the application in the clinical setting. Possible biological effects of nicotine in the human brain in principle could be mediated by nicotine itself or by its metabolites, but there is a considerable lack of evidence of the mechanistic effects of specific compounds in humans. This shortage of evidence can be rectified only by focused research in the future. On the other hand, evidence suggests that the biotransformation product cotinine is pharmacologically active in the brain of animal models with no adverse effects. Accumulating evidence makes it likely that this metabolite mediates the memory supportive effects of nicotine in the brain. Thus, a great deal of effort has been exerted to clinically apply cotinine as a treatment of learning and memory impairment and its underlying disorders. Taken together, we claim that this biologically active metabolite is more than just a biomarker of nicotine consumption and has potentially novel therapeutic value in the treatment of learning and memory declines.
Author Contributions
AM, FK, and SS-E performed the searches, interpreted the results and wrote the manuscript. AG, SS-E, and AM designed the study. AG critically interpreted data and critically revised and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by a grant from Neurosciences Research Centre—Tabriz University of Medical Sciences (grant number: 61017) to SS-E, and publication grants from Danish Alzheimer Foundation and University of Southern Denmark to AG.
References
Alex Grizzell, J., Iarkov, A., Holmes, R., Mori, T., and Moran, V. E. (2012). “Cotinine prevents working memory loss in a mouse model of Posttraumatic stress-induced cognitive impairment,” in Society for Neuroscience (New Orleans).
Allison, C., and Shoaib, M. (2013). Nicotine improves performance in an attentional set shifting task in rats. Neuropharmacology 64, 314–320. doi: 10.1016/j.neuropharm.2012.06.055
Barreto, G. E., Iarkov, A., and Moran, V. E. (2014). Beneficial effects of nicotine, cotinine and its metabolites as potential agents for Parkinson's disease. Front. Aging Neurosci. 6, 340. doi: 10.3389/fnagi.2014.00340
Baum, L., Hansen, L., Masliah, E., and Saitoh, T. (1996). Glycogen synthase kinase 3 alteration in Alzheimer disease is related to neurofibrillary tangle formation. Mol. Chem. Neuropathol. 29, 253–261. doi: 10.1007/BF02815006
Benowitz, N. L., Hukkanen, J., and Jacob, I. I. I.P (2009). “Nicotine chemistry, metabolism, kinetics and biomarkers,” in Nicotine Psychopharmacology (Berlin; Heidelberg: Springer-Verlag), 29–60.
Benowitz, N. L., Jacob, P., Jones, R. T., and Rosenberg, J. (1982). Interindividual variability in the metabolism and cardiovascular effects of nicotine in man. J. Pharmacol. Exp. Therap. 221, 368–372.
Brier, M. R., Gordon, B., Friedrichsen, K., McCarthy, J., Stern, A., Christensen, J., et al. (2016). Tau and Aβ imaging, CSF measures, and cognition in Alzheimer's disease. Sci. Trans. Med. 8:338ra66. doi: 10.1126/scitranslmed.aaf2362
Buccafusco, J. J., and Terry, A. V. (2003). The potential role of cotinine in the cognitive and neuroprotective actions of nicotine. Life Sci. 72, 2931–2942. doi: 10.1016/S0024-3205(03)00226-1
Burgess, S., Zeitlin, R., and Echeverria, V. (2011). Cotinine inhibits amyloid-β peptide neurotoxicity and oligomerization. J. Clin. Toxicol. S6:003. doi: 10.4172/2161-0495.S6-003
Burgess, S., Zeitlin, R., Gamble-George, J., and Echeverria Moran, V. (2008). P2-319: Cotinine is neuroprotective against beta-amyloid toxicity. Alzheimers Dement. 4:T466. doi: 10.1016/j.jalz.2008.05.1396
Byrd, G. D., Chang, K. M., Greene, J. M., and deBethizy, J. D. (1992). Evidence for urinary excretion of glucuronide conjugates of nicotine, cotinine, and trans-3′-hydroxycotinine in smokers. Drug Metab. Dispos. 20, 192–197.
Cheng, Q., and Yakel, J. L. (2015). The effect of α7 nicotinic receptor activation on glutamatergic transmission in the hippocampus. Biochem. Pharmacol. 97, 439–444. doi: 10.1016/j.bcp.2015.07.015
Crooks, P. A., and Dwoskin, L. P. (1997). Contribution of CNS nicotine metabolites to the neuropharmacological effects of nicotine and tobacco smoking. Biochem. Pharmacol. 54, 743–753. doi: 10.1016/S0006-2952(97)00117-2
Crooks, P. A., Li, M., and Dwoskin, L. P. (1995). Determination of nicotine metabolites in rat brain after peripheral radiolabeled nicotine administration: detection of nornicotine. Drug Metab. Dispos. 23, 1175–1177.
Crooks, P. A., Li, M., and Dwoskin, L. P. (1997). Metabolites of nicotine in rat brain after peripheral nicotine administration. Drug Metab. Dispos. 25, 47–54.
Dajas-Bailador, F. A., Mogg, A. J., and Wonnacott, S. (2002). Intracellular Ca2+ signals evoked by stimulation of nicotinic acetylcholine receptors in SH-SY5Y cells: contribution of voltage-operated Ca2+ channels and Ca2+ stores. J. Neurochem. 81, 606–614. doi: 10.1046/j.1471-4159.2002.00846.x
de Aguiar, R. B., Parfitt, G. M., Jaboinski, J., and Barros, D. M. (2013). Neuroactive effects of cotinine on the hippocampus: behavioral and biochemical parameters. Neuropharmacology 71, 292–298. doi: 10.1016/j.neuropharm.2013.03.032
de Jonge, W., and Ulloa, L. (2007). The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 151, 915–929. doi: 10.1038/sj.bjp.0707264
Dickerson, T. J., and Janda, K. D. (2003). Glycation of the amyloid β-protein by a nicotine metabolite: a fortuitous chemical dynamic between smoking and Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 8182–8187. doi: 10.1073/pnas.1332847100
Dudek, H., Datta, S. R., Franke, T. F., Birnbaum, M. J., Yao, R., Cooper, G. M., et al. (1997). Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661–665. doi: 10.1126/science.275.5300.661
Dwoskin, L. P., Crooks, P. A., Teng, L., Green, T. A., and Bardo, M. T. (1999). Acute and chronic effects of nornicotine on locomotor activity in rats: altered response to nicotine. Psychopharmacology 145, 442–451. doi: 10.1007/s002130051079
Dwoskin, L. P., Teng, L. H., and Crooks, P. A. (2001). Nornicotine, a nicotine metabolite and tobacco alkaloid: desensitization of nicotinic receptor-stimulated dopamine release from rat striatum. Eur. J. Pharmacol. 428, 69–79. doi: 10.1016/S0014-2999(01)01283-3
Echeverria, V., Alex Grizzell, J., and Barreto, G. E. (2016). Neuroinflammation: a therapeutic target of cotinine for the treatment of psychiatric disorders? Curr. Pharm. Des. 22, 1324–1333. doi: 10.2174/138161282210160304112511
Echeverria, V., Zeitlin, R., Burgess, S., Patel, S., Barman, A., Thakur, G., et al. (2011). Cotinine reduces amyloid-β aggregation and improves memory in Alzheimer's disease mice. J. Alzheimers Dis. 24, 817–835. doi: 10.3233/JAD-2011-102136
Egea, J., Buendia, I., Parada, E., Navarro, E., León, R., and Lopez, M. G. (2015). Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem. Pharmacol. 97, 463–472. doi: 10.1016/j.bcp.2015.07.032
Fox, A. M., Moonschi, F. H., and Richards, C. I. (2015). The nicotine metabolite, cotinine, alters the assembly and trafficking of a subset of nicotinic acetylcholine receptors. J. Biol. Chem. 290, 24403–24412. doi: 10.1074/jbc.M115.661827
Fukui, K., Omoi, N. O., Hayasaka, T., Shinnkai, T., Suzuki, S., Abe, K., et al. (2002). Cognitive impairment of rats caused by oxidative stress and aging, and its prevention by vitamin E. Ann. N. Y. Acad. Sci. 959, 275–284. doi: 10.1111/j.1749-6632.2002.tb02099.x
Ghosheh, O., Dwoskin, L. P., Li, W. K., and Crooks, P. A. (1999). Residence times and half-lives of nicotine metabolites in rat brain after acute peripheral administration of [2′-14C] nicotine. Drug Metab. Dispos. 27, 1448–1455.
Ghosheh, O., and Hawes, E. M. (2002). N-glucuronidation of nicotine and cotinine in human: formation of cotinine glucuronide in liver microsomes and lack of catalysis by 10 examined UDP-glucuronosyltransferases. Drug Metab. Dispos. 30, 991–996. doi: 10.1124/dmd.30.9.991
Ghosheh, O. A., Dwoskin, L. P., Miller, D. K., and Crooks, P. A. (2001). Accumulation of nicotine and its metabolites in rat brain after intermittent or continuous peripheral administration of [2′-14C] nicotine. Drug Metab. Dispos. 29, 645–651.
Girod, R., Barazangi, N., McGehee, D., and Role, L. W. (2000). Facilitation of glutamatergic neurotransmission by presynaptic nicotinic acetylcholine receptors. Neuropharmacology 39, 2715–2725. doi: 10.1016/S0028-3908(00)00145-3
Green, T. A., Phillips, S. B., Crooks, P. A., Dwoskin, L. P., and Bardo, M. T. (2000). Nornicotine pretreatment decreases intravenous nicotine self-administration in rats. Psychopharmacology (Berl). 152, 289–294. doi: 10.1007/s002130000524
Grizzell, J. A., and Echeverria, V. (2015). New insights into the mechanisms of action of cotinine and its distinctive effects from nicotine. Neurochem. Res. 40, 2032–2046. doi: 10.1007/s11064-014-1359-2
Grizzell, J. A., Iarkov, A., Holmes, R., Mori, T., and Echeverria, V. (2014a). Cotinine reduces depressive-like behavior, working memory deficits, and synaptic loss associated with chronic stress in mice. Behav. Brain Res. 268, 55–65. doi: 10.1016/j.bbr.2014.03.047
Grizzell, J. A., Mullins, M., Iarkov, A., Rohani, A., Charry, L. C., and Echeverria, V. (2014b). Cotinine reduces depressive-like behavior and hippocampal vascular endothelial growth factor downregulation after forced swim stress in mice. Behav. Neurosci. 128:713. doi: 10.1037/bne0000021
Grizzell, J. A., Patel, S., Barreto, G. E., and Echeverria, V. (2017). Cotinine improves visual recognition memory and decreases cortical Tau phosphorylation in the Tg6799 mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 78, 75–81. doi: 10.1016/j.pnpbp.2017.05.010
Hatsukami, D. K., Grillo, M., Pentel, P. R., Oncken, C., and Bliss, R. (1997). Safety of cotinine in humans: physiologic, subjective, and cognitive effects. Pharmacol. Biochem. Behav. 57, 643–650. doi: 10.1016/S0091-3057(97)80001-9
Herzig, K. E., Callaway, E., Halliday, R., Naylor, H., and Benowitz, N. L. (1998). Effects of cotinine on information processing in nonsmokers. Psychopharmacology 135, 127–132. doi: 10.1007/s002130050493
Higgins, J. P., Altman, D. G., Gøtzsche, P. C., Jüni, P., Moher, D., Oxman, A. D., et al. (2011). The cochrane collaboration's tool for assessing risk of bias in randomised trials. BMJ 343:d5928. doi: 10.1136/bmj.d5928
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer's disease. J. Neurochem. 104, 1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x
Hukkanen, J., Jacob, P., and Benowitz, N. L. (2005). Metabolism and disposition kinetics of nicotine. Pharmacol. Rev. 57, 79–115. doi: 10.1124/pr.57.1.3
Iarkov, A., Appunn, D., and Echeverria, V. (2016). Post-treatment with cotinine improved memory and decreased depressive-like behavior after chemotherapy in rats. Cancer Chemother. Pharmacol. 78, 1033–1039. doi: 10.1007/s00280-016-3161-0
Kim, A. H., Khursigara, G., Sun, X., Franke, T. F., and Chao, M. V. (2001). Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 21, 893–901. doi: 10.1128/MCB.21.3.893-901.2001
Kuehl, G. E., and Murphy, S. E. (2003). N-glucuronidation of trans-3′-hydroxycotinine by human liver microsomes. Chem. Res. Toxicol. 16, 1502–1506. doi: 10.1021/tx034173o
Li, P., Beck, W. D., Callahan, P. M., Terry, A. V., and Bartlett, M. G. (2012). Quantitation of cotinine and its metabolites in rat plasma and brain tissue by hydrophilic interaction chromatography tandem mass spectrometry (HILIC–MS/MS). J. Chromatogr. B 907, 117–125. doi: 10.1016/j.jchromb.2012.09.018
Li, P., Beck, W. D., Callahan, P. M., Terry, A. V., and Bartlett, M. G. (2015). Pharmacokinetics of cotinine in rats: a potential therapeutic agent for disorders of cognitive function. Pharmacol. Rep. 67, 494–500. doi: 10.1016/j.pharep.2014.12.004
Livingstone, P. D., Dickinson, J. A., Srinivasan, J., Kew, J. N., and Wonnacott, S. (2010). Glutamate-dopamine crosstalk in the rat prefrontal cortex is modulated by Alpha7 nicotinic receptors and potentiated by PNU-120596. J. Mol. Neurosci. 40, 172–176. doi: 10.1007/s12031-009-9232-5
López-Hidalgo, M., Salgado-Puga, K., Alvarado-Martínez, R., Medina, A. C., Prado-Alcalá, R. A., and García-Colunga, J. (2012). Nicotine uses neuron-glia communication to enhance hippocampal synaptic transmission and long-term memory. PLoS ONE 7:e49998. doi: 10.1371/journal.pone.0049998
Majdi, A., Kamari, F., Vafaee, M. S., and Sadigh-Eteghad, S. (2017). Revisiting nicotine's role in the ageing brain and cognitive impairment. Rev. Neurosci. 28, 767–781. doi: 10.1515/revneuro-2017-0008
Majdi, A., Mahmoudi, J., Sadigh-Eteghad, S., Golzari, S. E., Sabermarouf, B., and Reyhani-Rad, S. (2016). Permissive role of cytosolic pH acidification in neurodegeneration: a closer look at its causes and consequences. J. Neurosci. Res. 94, 879–887. doi: 10.1002/jnr.23757
Majdi, A., Sadigh-Eteghad, S., Talebi, M., Farajdokht, F., Erfani, M., Mahmoudi, J., et al. (2018). Nicotine modulates cognitive function in D-galactose-induced senescence in mice. Front. Aging Neurosci. 10:194. doi: 10.3389/fnagi.2018.00194
Mecocci, P. (2004). Oxidative stress in mild cognitive impairment and Alzheimer disease: a continuum. J. Alzheimers Dis. 6, 159–163. doi: 10.3233/JAD-2004-6207
Meger, M., Meger-Kossien, I., Schuler-Metz, A., Janket, D., and Scherer, G. (2002). Simultaneous determination of nicotine and eight nicotine metabolites in urine of smokers using liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 778, 251–261. doi: 10.1016/S0378-4347(01)00451-0
Metz, C. N., and Tracey, K. J. (2005). It takes nerve to dampen inflammation. Nat. Immunol. 6, 756–757. doi: 10.1038/ni0805-756
Mitchell, T. W., Mufson, E. J., Schneider, J. A., Cochran, E. J., Nissanov, J., Han, L. Y., et al. (2002). Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer's disease. Ann. Neurol. 51, 182–189. doi: 10.1002/ana.10086
Moher, D., Liberati, A., Tetzlaff, J., Altman, D. G., and The PRISMA Group. (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 6:e1000097. doi: 10.1371/journal.pmed1000097
Moher, D., Shamseer, L., Clarke, M., Ghersi, D., Liberati, A., Petticrew, M., et al. (2015). Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 4:1. doi: 10.1186/2046-4053-4-1
Moran, V. E.Moran, V. E. (2012). Cotinine: beyond that expected, more than a biomarker of tobacco consumption. Front. Pharmacol. 3:173. doi: 10.3389/fphar.2012.00173
Mundy, W. R., and Iwamoto, E. T. (1988). Nicotine impairs acquisition of radial maze performance in rats. Pharmacol. Biochem. Behav. 30, 119–122. doi: 10.1016/0091-3057(88)90433-9
Nakajima, M., Yamamoto, T., Nunoya, K., Yokoi, T., Nagashima, K., Inoue, K., et al. (1996). Role of human cytochrome P4502A6 in C-oxidation of nicotine. Drug Metab. Dispos. 24, 1212–1217.
Nakajima, M., and Yokoi, T. (2005). Interindividual variability in nicotine metabolism: C-oxidation and glucuronidation. Drug Metab. Pharmacokinet. 20, 227–235. doi: 10.2133/dmpk.20.227
Newhouse, P., Kellar, K., Aisen, P., White, H., Wesnes, K., Coderre, E., et al. (2012). Nicotine treatment of mild cognitive impairment: a 6-month double-blind pilot clinical trial. Neurology 78, 91–101. doi: 10.1212/WNL.0b013e31823efcbb
Oldendorf, W., Braun, L., and Cornford, E. (1979). pH dependence of blood-brain barrier permeability to lactate and nicotine. Stroke 10, 577–581. doi: 10.1161/01.STR.10.5.577
Ownby, R. L. (2010). Neuroinflammation and cognitive aging. Curr. Psychiatry Rep. 12, 39–45. doi: 10.1007/s11920-009-0082-1
Papke, R. L. (2006). Estimation of both the potency and efficacy of α7 nAChR agonists from single-concentration responses. Life Sci. 78, 2812–2819. doi: 10.1016/j.lfs.2005.11.009
Papke, R. L., Dwoskin, L. P., and Crooks, P. A. (2007). The pharmacological activity of nicotine and nornicotine on nAChRs subtypes: relevance to nicotine dependence and drug discovery. J. Neurochem. 101, 160–167. doi: 10.1111/j.1471-4159.2006.04355.x
Pardo, M., Beurel, E., and Jope, R. S. (2017). Cotinine administration improves impaired cognition in the mouse model of Fragile X syndrome. Eur. J. Neurosci. 45, 490–498. doi: 10.1111/ejn.13446
Park, S., Knopick, C., McGurk, S., and Meltzer, H. Y. (2000). Nicotine impairs spatial working memory while leaving spatial attention intact. Neuropsychopharmacology 22, 200–209. doi: 10.1016/S0893-133X(99)00098-6
Patel, S., Grizzell, J. A., Holmes, R., Zeitlin, R., Solomon, R., Sutton, T. L., et al. (2014). Cotinine halts the advance of Alzheimer's disease-like pathology and associated depressive-like behavior in Tg6799 mice. Front. Aging Neurosci. 6:162. doi: 10.3389/fnagi.2014.00162
Perez-Urrutia, N., Mendoza, C., Alvarez-Ricartes, N., Oliveros-Matus, P., Echeverria, F., Grizzell, J. A., et al. (2017). Intranasal cotinine improves memory, and reduces depressive-like behavior, and GFAP+ cells loss induced by restraint stress in mice. Exp. Neurol. 295, 211–221. doi: 10.1016/j.expneurol.2017.06.016
Phan, J. A., Stokholm, K., Zareba-Paslawska, J., Jakobsen, S., Vang, K., Gjedde, A., et al. (2017). Early synaptic dysfunction induced by α-synuclein in a rat model of Parkinson's disease. Sci. Rep. 7:6363. doi: 10.1038/s41598-017-06724-9
Plattner, F., Angelo, M., and Giese, K. P. (2006). The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 281, 25457–25465. doi: 10.1074/jbc.M603469200
Pourmemar, E., Majdi, A., Haramshahi, M., Talebi, M., Karimi, P., and Sadigh-Eteghad, S. (2017). Intranasal cerebrolysin attenuates learning and memory impairments in D-galactose-induced senescence in mice. Exp. Gerontol. 87, 16–22. doi: 10.1016/j.exger.2016.11.011
Rahn, K. A., Slusher, B. S., and Kaplin, A. I. (2012). Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition. Curr. Med. Chem. 19, 1335–1345. doi: 10.2174/092986712799462649
Rehani, K., Scott, D. A., Renaud, D., Hamza, H., Williams, L. R., Wang, H., et al. (2008). Cotinine-induced convergence of the cholinergic and PI3 kinase-dependent anti-inflammatory pathways in innate immune cells. Biochim. Biophys. Acta Mol. Cell Res. 1783, 375–382. doi: 10.1016/j.bbamcr.2007.12.003
Resende, R., Ferreiro, E., Pereira, C., and Resende de Oliveira, C. (2008). Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 155, 725–737. doi: 10.1016/j.neuroscience.2008.06.036
Rezvani, A. H., and Levin, E. D. (2001). Cognitive effects of nicotine. Biol. Psychiatry 49, 258–267. doi: 10.1016/S0006-3223(00)01094-5
Riah, O., Dousset, J. C., Courriere, P., Stigliani, J. L., Baziard-Mouysset, G., and Belahsen, Y. (1999). Evidence that nicotine acetylcholine receptors are not the main targets of cotinine toxicity. Toxicol. Lett. 109, 21–29. doi: 10.1016/S0378-4274(99)00070-3
Sadigh-Eteghad, S., Majdi, A., Mahmoudi, J., Golzari, S. E., and Talebi, M. (2016). Astrocytic and microglial nicotinic acetylcholine receptors: an overlooked issue in Alzheimer's disease. J. Neural Transm. 123, 1359–1367. doi: 10.1007/s00702-016-1580-z
Sadigh-Eteghad, S., Majdi, A., McCann, S. K., Mahmoudi, J., Vafaee, M. S., and Macleod, M. R. (2017). D-galactose-induced brain ageing model: a systematic review and meta-analysis on cognitive outcomes and oxidative stress indices. PLoS ONE 12:e0184122. doi: 10.1371/journal.pone.0184122
Sadigh-Eteghad, S., Sabermarouf, B., Majdi, A., Talebi, M., Farhoudi, M., and Mahmoudi, J. (2015). Amyloid-beta: a crucial factor in Alzheimer's disease. Med. Princ. Pract. 24, 1–10. doi: 10.1159/000369101
Sadigh-Eteghad, S., Talebi, M., Farhoudi, M., Golzari, S. E. J., Sabermarouf, B., and Mahmoudi, J. (2014). Beta-amyloid exhibits antagonistic effects on alpha 7 nicotinic acetylcholine receptors in orchestrated manner. J. Med. Hypotheses Ideas 8, 49–52. doi: 10.1016/j.jmhi.2014.01.001
Schrag, M., Mueller, C., Zabel, M., Crofton, A., Kirsch, W. M., Ghribi, O., et al. (2013). Oxidative stress in blood in Alzheimer's disease and mild cognitive impairment: a meta-analysis. Neurobiol. Dis. 59, 100–110. doi: 10.1016/j.nbd.2013.07.005
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sena, E. S., Currie, G. L., McCann, S. K., Macleod, M. R., and Howells, D. W. (2014). Systematic reviews and meta-analysis of preclinical studies: why perform them and how to appraise them critically. J. Cereb. Blood Flow Metab. 34, 737–742. doi: 10.1038/jcbfm.2014.28
Silva, A. J. (2003). Molecular and cellular cognitive studies of the role of synaptic plasticity in memory. Dev. Neurobiol. 54, 224–237. doi: 10.1002/neu.10169
Soto-Otero, R., Méndez-Alvarez, E., Hermida-Ameijeiras, A., López-Real, A. M., and Labandeira-García, J. L. (2002). Effects of (–)-nicotine and (–)-cotinine on 6-hydroxydopamine-induced oxidative stress and neurotoxicity: relevance for Parkinson's disease. Biochem. Pharmacol. 64, 125–135.
Takeshima, T., Hiroshi, T., Toru, E., and Shigehiro, K. (2007). Molecular structure of cotinine studied by gas electron diffraction combined with theoretical calculations. J. Mol. Struct. 841, 13–21. doi: 10.1016/j.molstruc.2006.11.041
Tega, Y., Akanuma, S., Kubo, Y., Terasaki, T., and Hosoya, K. (2013). Blood-to-brain influx transport of nicotine at the rat blood–brain barrier: involvement of a pyrilamine-sensitive organic cation transport process. Neurochem. Int. 62, 173–181. doi: 10.1016/j.neuint.2012.11.014
Terry, Jr A. V, Schade, R., Callachan, P. M., Chapman, J. M., and Bartlett, M. G. (2011). Chronic treatment with nicotine metabolite, cotinine, improves sustained attention and recognition memory in rats and attenuates glutamate (NMDA) antagonist-related impairments. Biochem. Pharmacol. 82, 1041–1042. doi: 10.1016/j.bcp.2011.07.048
Terry, A. V., Buccafusco, J. J., Schade, R. F., Vandenhuerk, L., Callahan, P. M., Beck, W. D., et al. (2012). The nicotine metabolite, cotinine, attenuates glutamate (NMDA) antagonist-related effects on the performance of the five choice serial reaction time task (5C-SRTT) in rats. Biochem. Pharmacol. 83, 941–951. doi: 10.1016/j.bcp.2011.12.043
Terry, A. V., Callahan, P. M., and Bertrand, D. (2015). R-(+) and S-(–) isomers of cotinine augment cholinergic responses in vitro and in vivo. J. Pharmacol. Exp. Therap. 352, 405–418. doi: 10.1124/jpet.114.219881
Terry, A. V., Hernandez, C. M., Hohnadel, E. J., Bouchard, K. P., and Buccafusco, J. J. (2005). Cotinine, a neuroactive metabolite of nicotine: potential for treating disorders of impaired cognition. CNS Drug Rev. 11, 229–252. doi: 10.1111/j.1527-3458.2005.tb00045.x
Tsai, G., and Coyle, J. T. (2002). Glutamatergic mechanisms in schizophrenia. Annu. Rev. Pharmacol. Toxicol. 42, 165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735
Vainio, P. J., and Tuominen, R. K. (2001). Cotinine binding to nicotinic acetylcholine receptors in bovine chromaffin cell and rat brain membranes. Nicotine Tob. Res. 3, 177–182. doi: 10.1080/14622200110043095
Valtorta, F., Pennuto, M., Bonanomi, D., and Benfenati, F. (2004). Synaptophysin: leading actor or walk-on role in synaptic vesicle exocytosis? Bioessays 26, 445–453. doi: 10.1002/bies.20012
VanGuilder, H. D., Farley, J. A., Yan, H., Van Kirk, C. A., Mitschelen, M., Sonntag, W. E., et al. (2011). Hippocampal dysregulation of synaptic plasticity-associated proteins with age-related cognitive decline. Neurobiol. Dis. 43, 201–212. doi: 10.1016/j.nbd.2011.03.012
White, H. K., and Levin, E. D. (1999). Four-week nicotine skin patch treatment effects on cognitive performance in Alzheimer's disease. Psychopharmacology 143, 158–165.
White, H. K., and Levin, E. D. (2004). Chronic transdermal nicotine patch treatment effects on cognitive performance in age-associated memory impairment. Psychopharmacology 171, 465–471. doi: 10.1007/s00213-003-1614-8
Wildeboer-Andrud, K. M., Zheng, L., Choo, K. S., and Stevens, K. E. (2014). Cotinine impacts sensory processing in DBA/2 mice through changes in the conditioning amplitude. Pharmacol. Biochem. Behav. 117, 144–150. doi: 10.1016/j.pbb.2013.12.005
Williams, T. I., Lynn, B. C., Markesbery, W. R., and Lovell, M. A. (2006). Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in mild cognitive impairment and early Alzheimer's disease. Neurobiol. Aging 27, 1094–1099. doi: 10.1016/j.neurobiolaging.2005.06.004
Wong, D. F., Kuwabara, H., Horti, A. G., Roberts, J. M., Nandi, A., Cascella, N., et al. (2018). PET Brain imaging of α7-nAChR with [18F] ASEM: Reproducibility, occupancy, receptor density, and changes in schizophrenia. Int. J. Neuropsychopharmacol. 21, 656–667. doi: 10.1093/ijnp/pyy021
Keywords: nicotine, metabolite, cotinine, norcotinine, nornicotine, cognition, systematic review
Citation: Majdi A, Kamari F, Sadigh-Eteghad S and Gjedde A (2019) Molecular Insights Into Memory-Enhancing Metabolites of Nicotine in Brain: A Systematic Review. Front. Neurosci. 12:1002. doi: 10.3389/fnins.2018.01002
Received: 22 August 2018; Accepted: 12 December 2018;
Published: 15 January 2019.
Edited by:
Alfredo Meneses, Centro de Investigación y de Estudios Avanzados (CINVESTAV), MexicoReviewed by:
Massimo Grilli, Università di Genova, ItalyEddy A. Van Der Zee, University of Groningen, Netherlands
Copyright © 2019 Majdi, Kamari, Sadigh-Eteghad and Gjedde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saeed Sadigh-Eteghad, c2FlZWQuc2FkaWdldGVnYWRAZ21haWwuY29t