 Lindsay M. Lueptow
Lindsay M. Lueptow Amanda K. Fakira
Amanda K. Fakira Erin N. Bobeck
Erin N. Bobeck- 1Department of Psychiatry, Semel Institute for Neuroscience and Human Behavior UCLA, Los Angeles, CA, United States
- 2Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 3Department of Biology, Utah State University, Logan, UT, United States
Opioids remain among the most effective pain-relieving therapeutics. However, their long-term use is limited due to the development of tolerance and potential for addiction. For many years, researchers have explored the underlying mechanisms that lead to this decreased effectiveness of opioids after repeated use, and numerous theories have been proposed to explain these changes. The most widely studied theories involve alterations in receptor trafficking and intracellular signaling. Other possible mechanisms include the recruitment of new structural neuronal and microglia networks. While many of these theories have been developed using molecular and cellular techniques, more recent behavioral data also supports these findings. In this review, we focus on the mechanisms that underlie tolerance within the descending pain modulatory pathway, including alterations in intracellular signaling, neural-glial interactions, and neurotransmission following opioid exposure. Developing a better understanding of the relationship between these various mechanisms, within different parts of this pathway, is vital for the identification of more efficacious, novel therapeutics to treat chronic pain.
Descending Pain Pathway in Opioid Functions
Opioids are widely used pain therapeutics; however, the development of tolerance limits the long-term use of opioids due to the need for dose escalation over time in order to maintain analgesia. While there are four main types of opioid receptors, most pain therapeutics, including morphine, methadone, fentanyl, and oxycodone, target the mu opioid receptor (MOPr). The MOPr is a G-protein coupled receptor that couples to inhibitory heterotrimeric G-proteins (Gi/o) producing subsequent intracellular signaling and ion conductance (Goode and Raffa, 1997; Gintzler and Chakrabarti, 2004). MOPr expression within the descending pain modulatory pathway, which includes the ventrolateral periaqueductal gray (PAG), rostral ventromedial medulla (RVM), and the dorsal horn (DH) of the spinal cord, contribute to opioid-induced antinociception and the development of opioid tolerance (Fang et al., 1989; Fairbanks and Wilcox, 1997; Tortorici et al., 2001; Morgan et al., 2006; Bobeck et al., 2009).
GABAergic neurons within the PAG are a critical site of action by opioids. Under normal conditions, these neurons have tonic activity (Figure 1, naive); however, upon binding of opioids to MOPr, the activity of these neurons is decreased, disinhibiting PAG projections to the RVM (Figure 1, Acute Morphine) (Stiller et al., 1996; Vaughan et al., 1997; Bobeck et al., 2014). In vitro electrophysiology studies have shown that opioids reduce the frequency of spontaneous mIPSCs in PAG (Vaughan et al., 1997; Bobeck et al., 2014), which indicates a reduction in the probability of GABA release. This is also supported by in vivo studies. Microinjection of bicuculline (GABAA agonist) into the PAG produces antinociception, which suggests that GABA is being tonically released (Bobeck et al., 2014). Furthermore, microdialysis in the PAG reveals a reduction in extracellular GABA following administration of morphine (Stiller et al., 1996).
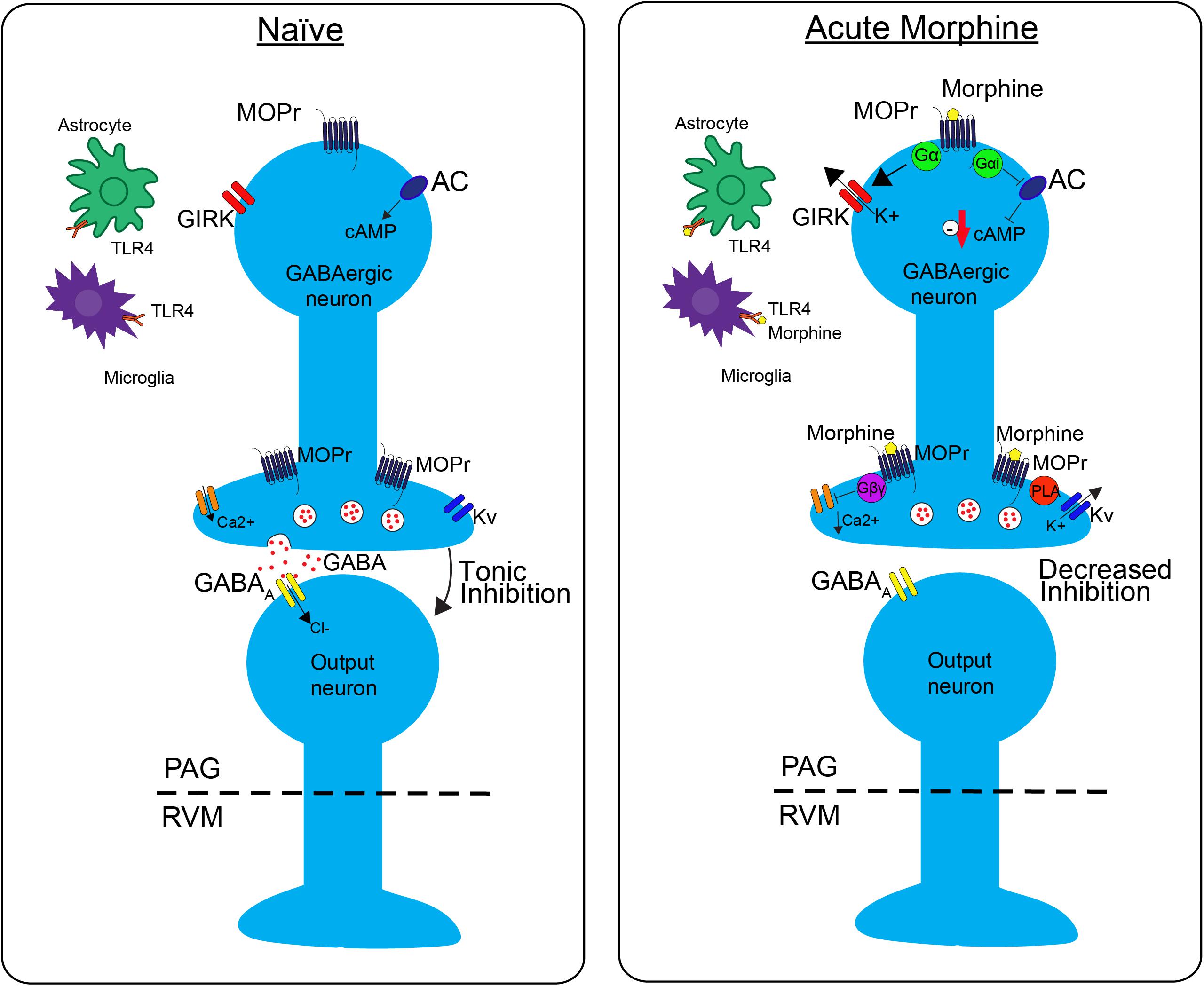
FIGURE 1. The effects of morphine on neuronal transmission in the descending pain pathway. In the naïve state, GABAergic interneurons in the periaqueductal gray (PAG) fire tonically, thereby producing a steady release of GABA and inhibition of PAG output neurons. Upon administration of acute morphine, postsynaptic mu opioid receptor (MOPr) activate GIRK channels via Gα proteins resulting in K+ release and hyperpolarization of the neuron. Additionally, MOPr activate Gi/o proteins, which result in the inhibition of adenylyl cyclase (AC) and decrease cAMP production. Morphine binding of presynaptic MOPr inhibits voltage dependent calcium (Ca2+) conductances via Gβγ proteins and activated voltage dependent potassium conductances (Kv) via Phospholipase A (PLA). Overall, these two mechanisms block release of the neurotransmitter GABA, therefore suppressing inhibition, increasing output, of the PAG neurons projecting to the rostral ventromedial medulla (RVM). Acute morphine treatment also activates toll-like receptor 4 (TLR4) receptors on astrocytes and microglia in the PAG inducing several signaling cascades.
Opioids activate different signaling cascades depending on whether the MOPr is expressed at pre- or post- synaptic sites. Opioid binding to postsynaptic MOPr results in the activation of a G-protein inwardly rectifying potassium channels (GIRK) that hyperpolarize GABAergic neurons in the PAG producing an overall decrease in GABAergic neuron activity (Figure 1; Acute Moprhine) (North and Williams, 1983; Pan et al., 1990). Alternatively, when MOPr are expressed at presynaptic sites they produce an inhibition of voltage gated calcium channels and voltage gated potassium channels (Kv) resulting in the inhibition of GABA release (Figure 1; Acute Morphine) (Wilding et al., 1995; Vaughan et al., 1997; Connor et al., 1999; Williams et al., 2001). Overall, the combined action of MOPr binding by opioids is a decrease in GABAergic neuronal activity, and therefore an increase in output from the PAG to the RVM (Figure 1; Moreau and Fields, 1986; Depaulis et al., 1987; Jacquet, 1988; Osborne et al., 1996). Recent studies support the hypothesis that this increase in PAG output to the RVM is a main contributor to the opioid-induced antinociception by demonstrating that selective inhibition of GABAergic neurons or activation of glutamatergic output neurons in the PAG mimics the antinociceptive effects of opioids (Samineni et al., 2017). In summary, these findings support the notion that analgesia is produced by disinhibition of excitatory outputs from the PAG.
The overall effect of MOPr activation in the PAG is an increase in output to the two distinct cell types within the RVM: off-cells and on-cells (Fields et al., 1983; Fields and Heinricher, 1985). The activity of off-cells pauses just prior to the response to a painful stimulus, while the activity of on-cells increases during this response, and both of these activities are blocked during the administration of opioids. There is conflicting evidence regarding the excitatory versus inhibitory nature of the PAG projections to the on- and off-cells in the RVM. Studies in GAD67-GFP mice, a marker for GABAergic neurons, show that retrogradely labeled neurons from the RVM do no co-localize with GAD67 in the PAG (Park et al., 2010), indicating that the PAG to RVM projection is glutamatergic. In contrast, studies in rats demonstrate that PAG to RVM projections are a mix of GABAergic and glutamatergic neurons (Morgan et al., 2008). Furthermore, these studies demonstrate that GABAergic neurons project from PAG and target on-cells and glutamatergic neurons project from the PAG and target off-cells (Morgan et al., 2008). Despite these differences, both studies support the notion that opioids inhibit GABA release from interneurons in the PAG, which disinhibit (i.e., excite) glutamate projections to off-cells. Given that the off-cells in the RVM are GABAergic, they subsequently inhibit pain responses in the DH (Fields et al., 1983; Moreau and Fields, 1986; Morgan et al., 2008). Overall, these studies support the concept that opioid-induced antinociception is mediated by direct excitation of off-cells and subsequent inhibition of pain in the spinal cord.
At each level of this pathway, a myriad of cellular effects drives the physiological changes mentioned above, and are highly correlated with the development of opioid tolerance. One of the most studied mechanisms involves regulation and signaling at the MOPr. Current research demonstrates that while MOPr is a key player in the development of antinociceptive tolerance, mechanisms beyond simple receptor desensitization, including alterations in neurotransmission and β-arrestin dependent signaling, are also critical. Furthermore, MOPr expression in non-neuronal cells, specifically on microglia and astrocytes within the spinal cord, and more recently within the PAG, greatly contributes to the development of opioid tolerance.
Opioid Tolerance and Neurotransmission in the Descending Pain Pathway
Evidence suggests that tolerance is due to changes in the properties of GABAergic neurons in the PAG (Morgan et al., 2003). First of all, while microinjection of morphine into the PAG or RVM produces antinociception, repeated microinjection into the PAG and not the RVM results in tolerance (Morgan et al., 2005; Campion et al., 2016). Secondly, inhibition of MOPrs within the PAG blocks tolerance to systemic morphine (Lane et al., 2005). Furthermore, inactivation of RVM by a GABA agonist during direct administration of morphine into the PAG still leads to tolerance development (Lane et al., 2005). Therefore, MOPr within the PAG are necessary and sufficient in the development of opioid tolerance.
However, the development of tolerance within the PAG produces downstream effects along the descending pain pathway. This is evidenced by the fact that direct injection of morphine into the PAG affects RVM signaling, suggesting that their activity is in fact coupled (Tortorici et al., 2001). While acute administration of opioids into the PAG disrupts the activity of on- and off- cells in response to painful stimuli, these cells respond normally following chronic infusion that is associated with tolerance (Lane et al., 2004). Another side effect of chronic morphine treatment is hyperalgesia, or the increased sensitivity to pain following chronic morphine treatment. One theory is that hyperalgesia may manifest as opioid-induced tolerance since increased sensitivity to pain would counteract the pain-relieving effects of opioids. Some studies suggest that increased activation of the descending pain pathway by chronic morphine produces neuroadaptations with in the RVM that result in hyperalgesia (Vanderah et al., 2001). In support of this, one study demonstrated that chronic morphine produced an increase in the number of active on-cells, likely increasing sensitivity to noxious stimuli (Meng and Harasawa, 2007), which may be responsible for morphine-induced hyperalgesia. While RVM plays a role in opioid-induced tolerance, direct injections into the RVM leads to a lesser development of tolerance compared to PAG administration (Morgan et al., 2005), indicating that activation of the entire descending pain circuit is essential.
The neurophysiological mechanisms of tolerance in the PAG are mediated by MOPr uncoupling from downstream G-protein mediated signaling (Figure 2). One key study demonstrated that chronic morphine decreases opioid-mediated GIRK currents in the PAG (Bagley et al., 2005), supporting the notion that morphine tolerance is associated with uncoupling of G-protein mediated signaling. Since GIRK channels regulate neuronal excitability, this mechanism would result in a reduction in the ability of MOPr activation to suppress GABAergic neuron activity. Additionally, morphine tolerance is also associated with decreased efficacy of other MOPr agonists ability to reduce voltage gated calcium currents in the PAG (Bagley et al., 2005). The net effect of the uncoupling of MOPr activation from voltage gated calcium channels would be the attenuation of MOPr mediated inhibition of GABA release. However, the precise mechanisms underlying MOPr uncoupling from voltage gated calcium channels are complex, as cellular tolerance associated with this effect was not observed in β-arrestin two knockout mice (Connor et al., 2015), suggesting that β-arrestin two also plays a role in this interaction.
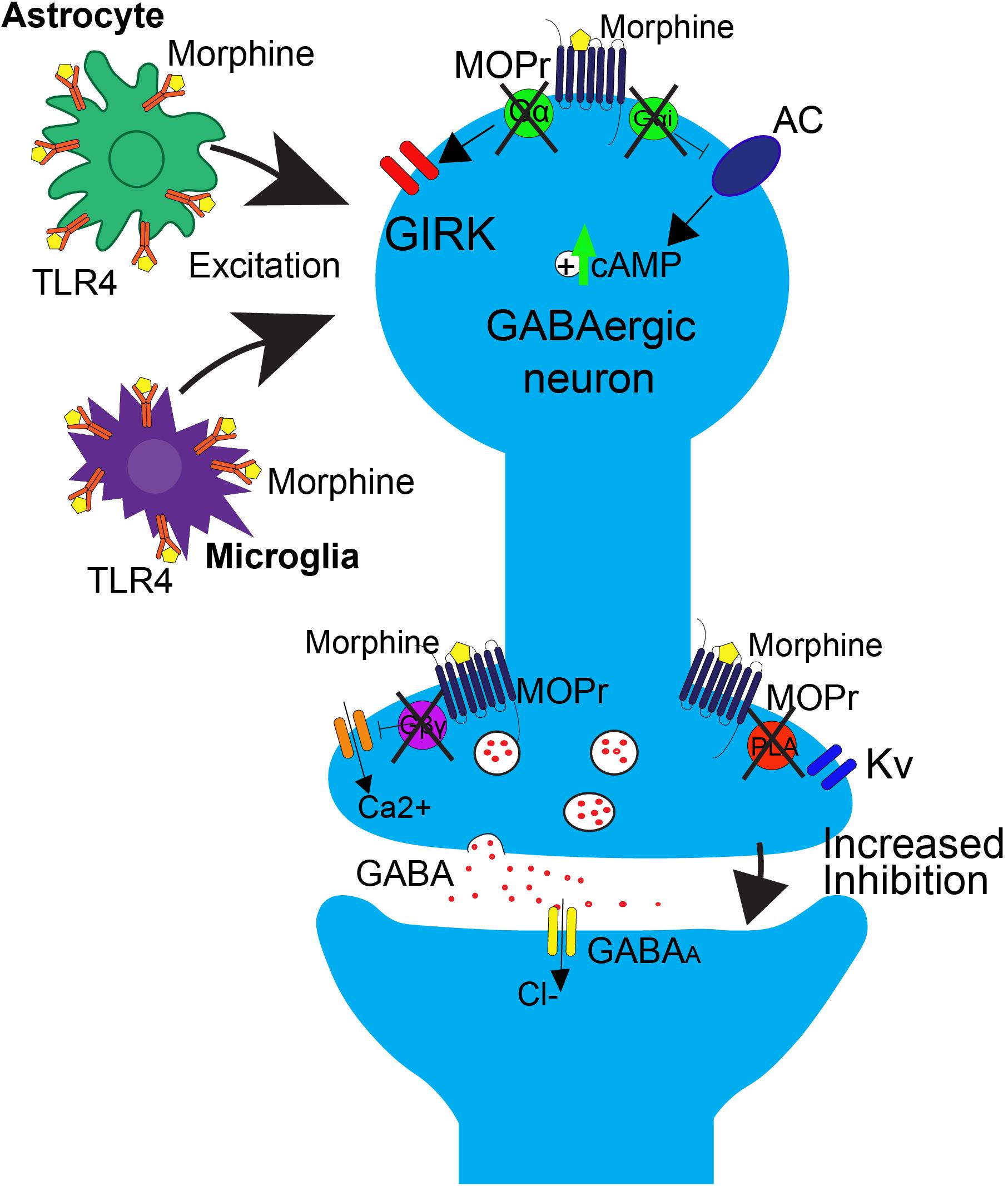
FIGURE 2. Effects of repeated morphine treatment on glial and neuronal signaling in the PAG. Chronic morphine treatment induces several side effects that block opioid-induced decreases in GABAergic interneuron activity at the neuronal level, postsynaptically. This includes the uncoupling of MOPr from G-protein mediated effects on GIRK channels and AC. This results in an upregulation of cAMP. Uncoupling also occurs in the presynaptic region, blocking the Gβγ mediated inhibition of calcium channels and PLA mediated activation of Kv channels. In this state, binding of opioids to MOPr no longer results in suppression of GABA release. At the level of glial signaling, upon repeated treatment with morphine, there is a rapid upregulation of TLR4 on astrocytes and microglia within the PAG, resulting in an increase in excitatory cytokine release, as well as a switch from Gi/o to Gs coupling at MOPr, resulting in an overall increase in excitatory tone that is correlated with opioid tolerance.
GABA release is also regulated by signaling through phospholipase A2-mediated activation of voltage gated potassium channels (Figure 1; Wimpey and Chavkin, 1991; Vaughan et al., 1997). This signaling pathway is differentially affected by morphine tolerance versus withdrawal. Morphine tolerance is associated with a decrease in opioid-mediated inhibition of GABA release (Figure 2) that is not a result of MOPr desensitization (Fyfe et al., 2010). However, during naloxone-precipitated withdrawal following chronic morphine, GABA release is enhanced via an increase in adenylyl cyclase (AC) signaling (Sharma et al., 1975; Ingram et al., 1998; Hack et al., 2003). These two outcomes may be related as studies have demonstrated that inhibition of AC in the PAG prevents the development and expression of morphine tolerance (Bobeck et al., 2014). Moreover, AC activation is sufficient to increase GABA release from PAG neurons (Bobeck et al., 2014).
Intracellular Signaling Changes in the PAG-RVM-DH Pathway in Opioid Tolerance
Direct activation of the MOPr results in the Gα subunit-mediated inhibition of the AC-cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) pathway (Figure 1; Sharma et al., 1975; Guitart and Nestler, 1989; Hirst and Lambert, 1995). However, opioid binding activates other signaling proteins, such as protein kinase C (PKC) and extracellular signal-regulated kinase 1/2 (ERK1/2) via β-arrestin pathways, which are independent of G-protein signaling. As mentioned previously, downstream of G-protein mediated signaling, there is an inhibition of calcium channels and activation of potassium channels that leads to hyperpolarization and a reduction in neurotransmitter release in the PAG that produces antinociception (Bourinet et al., 1996; Ippolito et al., 2002; Torrecilla et al., 2002). Chronic morphine produces adaptations that contribute to opioid-tolerance within all these downstream signaling pathways.
Long-term opioid treatment leads to adaptations in many signaling proteins within the PAG-RVM-DH pathway, which have been proposed as mechanisms of opioid tolerance. In contrast to the acute inhibitory effect of opioids on cAMP production, chronic morphine treatment upregulates cAMP (Figure 2; Guitart and Nestler, 1989; Gintzler and Chakrabarti, 2004). It has been proposed that this upregulation in cAMP is caused by compensatory changes in intracellular signaling, or an uncoupling of Gi/o-proteins from the receptor and a switch to coupling with Gs-proteins (Gintzler and Chakrabarti, 2004). Very few in vivo studies have evaluated the role of the AC pathway in morphine tolerance, but inhibition of the AC pathway, via either intracerebroventricular (ICV) or intra-PAG injection, during morphine pretreatment has been shown to block the development of morphine tolerance (Smith et al., 2006; Gabra et al., 2008; Bobeck et al., 2014). In the DH, administration of morphine results in no change or even an increase in MOPr expression, but a significant down-regulation of the G-protein activation in the DH, as measured by [35S]-GTPyS (Maher et al., 2001; Ray et al., 2004). The loss of G-protein signaling is likely a switch in MOPr G-protein coupling, from Gi/o to Gs coupling (Gintzler and Chakrabarti, 2004). Recently, adrenomedullin, a pronociceptive peptide from the CGRP family, has been implicated in mediating this G-protein switch in the DH. Following chronic morphine, adrenomedullin is significantly upregulated in the DH and dorsal root ganglia, and inhibition of its receptor prevents or reverses morphine tolerance and blocks the MOPr Gi/o to Gs switch in coupling (Wang et al., 2016).
Behavioral studies suggest that the mechanisms underlying tolerance are dependent on the specific MOPr agonist being studied. Some agonists, such as morphine, do not readily recruit β-arrestin or internalize the receptor, as compared to high efficacy agonists, such as DAMGO or fentanyl, which readily do both. This difference in signaling suggests differences in tolerance mechanisms, where morphine-mediated tolerance utilizes a G-protein dependent mechanism, and other MOPr agonists, such as DAMGO or fentanyl, use a β-arrestin dependent mechanism (Hull et al., 2010; Melief et al., 2010; Bobeck et al., 2014, 2016; Morgan et al., 2014). For example, inhibition of ERK1/2 within the PAG during the development of tolerance enhances morphine tolerance (Macey et al., 2009), but reduces tolerance to DAMGO and has no effect on fentanyl tolerance (Bobeck et al., 2016). Furthermore, inhibition of the G-protein dependent pathway (i.e., c-Jun N-terminal kinase) blocks development of tolerance to morphine, but not fentanyl. However, inhibition of β-arrestin dependent signaling (i.e., G protein-coupled receptor kinase) blocks expression of fentanyl tolerance (Morgan et al., 2014).
Neuropeptides within the descending pain pathway have also been shown to regulate opioid tolerance. One such neuropeptide, cholecystokinin (CCK), is particularly enriched in supraspinal midbrain regions known to regulate spinal nociception (King et al., 2005). There is evidence that CCK acting within the PAG-RVM-DH pathway regulates morphine tolerance (Xie et al., 2005; Thomas et al., 2015). A CCK receptor antagonist, directly injected into the PAG, is able to block morphine tolerance (Xie et al., 2005). In the RVM, injection of CCK blocks opioid activation of off-cells that mediate descending antinociception, resulting in a blockade of the analgesic effects of morphine (Xu et al., 2014; Thomas et al., 2015).
N-methyl D-aspartate receptors (NMDArs) have been heavily implicated in the development of both spinal-mediated hyperalgesia and opioid tolerance. NMDAr antagonists or the targeted disruption of the NR2 subunits, NR2a and NR2b, attenuates opioid tolerance (Price et al., 2000; Zhao et al., 2012). Deletion of PSD-93, the anchoring protein for NR2a and NR2b in the synapse, leads to a DH site-specific down-regulation of both subunits from the plasma membrane into the cytosolic compartment, and a reduction in the development of morphine tolerance (Liaw et al., 2008). This is a region-specific effect, as other portions of the descending pain pathway did not see changes in the NR2 subunit localization (Kozela and Popik, 2007). Interestingly, NMDArs in the PAG have not been implicated in tolerance (Morgan et al., 2009).
A few other main signaling targets have been implicated in DH-mediated opioid tolerance, as well. Mammalian target of rapamycin (mTOR) is found to be upregulated following repeated intrathecal morphine administration, and this effect is mediated by activation of PI3K/AKT following MOPr activation (Xu et al., 2015). Administration of mTOR inhibitors is able to both prevent and reverse morphine tolerance (Xu et al., 2014, 2015; Jiang et al., 2016; Chen et al., 2017). Calcium/calmodulin-dependent protein kinase IIα has also been implicated in the development of tolerance (Brüggemann et al., 2000). It has been shown to colocalize with MOPr, in the DH specifically, following opioid administration, possibly resulting in increased MOPr phosphorylation and desensitization (Brüggemann et al., 2000).
Impact of Opioid-Induced Neuroinflammation on the Development of Tolerance
Over the past few decades, researchers have discovered that opioids are potent activators of immune cells within the CNS, and this inflammation is a strong contributor to the development of opioid tolerance (Giron et al., 2015; Cahill and Taylor, 2017). Specifically, repeated administration of opioids, which leads to activation of glia within the PAG and spinal cord of the descending pain pathway, results in alterations in both intracellular signaling cascades and signaling properties of neurons. Furthermore, microglial inhibitors have been shown to attenuate morphine-induced tolerance (Song and Zhao, 2001; Raghavendra et al., 2002, 2004; Cui et al., 2008; Eidson and Murphy, 2013; Harada et al., 2013). Though the precise mechanisms that underlie these changes are only beginning to be uncovered, a few notable pathways have emerged that are likely significant contributors to the development of opioid tolerance.
One prominent pro-inflammatory signaling cascade that has been implicated in opioid tolerance involves the immune receptor, toll-like receptor 4 (TLR4, Figure 2). Upon agonist binding to TRL4, sphingomyelinase produces ceramide, which allows for interaction of the receptor with its co-activators myeloid differentiation factor-2 (MD-2) and CD14, resulting in subsequent activation of 3 parallel pathways: the p38-MAPK pathway, the PI3K/AKT pathway (cell survival/apoptosis), and the NFκB pathway (proinflammatory cytokine release) (Rönnbäck and Hansson, 1988; Watkins et al., 2009; Nakamoto et al., 2012; Eidson and Murphy, 2013). In the spinal cord, TLR4 is primarily expressed on microglial cells and is shown to be upregulated (Figure 2) along with its cofactor MD-2 following morphine treatment (Wang et al., 2012), and activation of TLR4 signaling can induce “naïve tolerance” to opioids (Eidson and Murphy, 2013; Grace et al., 2015). Furthermore, inhibition of TLR4, co-activators MD-2 or CD14, or inhibition of ceramide biosynthesis, leads to attenuation of morphine tolerance, as well as decreased microglial activation, suggesting a prominent role for the TLR4 pathway in the development of opioid tolerance, at the level of the spinal cord (Ndengele et al., 2009; Hutchinson et al., 2010, 2011; Muscoli et al., 2010; Thomas et al., 2015).
Interestingly, it is also thought that TLR4 is directly activated by opioids (Figures 1, 2; Hutchinson et al., 2011; Wang et al., 2012; Grace et al., 2015), and, perhaps more importantly, the accessory protein MD-2 is able to non-stereoselectively bind opioids and signal through TRL4 (Grace et al., 2015). Since the classic opioid receptors only bind the (−)-opioid isomer, the (+)-opioid isomer antagonists could be used to block TLR4-mediated microglial activation and pro-inflammatory cytokine production. In fact, studies have demonstrated that (+)-naloxone is able to attenuate morphine-induced analgesia, specifically at the level of the spinal cord (Hutchinson et al., 2010; Lewis et al., 2010). This non-stereoselectivity at the TLR4 receptor complex could potentially be leveraged for the enhancement of the therapeutic efficacy of opioids, including enhancing analgesic effects and reducing tolerance.
How does the activation of glial cells lead to alterations in neuronal signaling? One possibility is through the alteration of neuronal excitability via increased release of glially-derived pro-inflammatory cytokines, including TNF (tumor necrosis factor) and IL-1β, which are known to increase neuronal AMPA and NMDA receptors, as well as down regulate GABA receptors (Viviani et al., 2003; Stellwagen, 2005). Within the PAG, repeated morphine administration results in an upregulation of TLR4, which subsequently leads to an increase in release of TNF and IL-1β (Eidson and Murphy, 2013; Eidson et al., 2017). This upregulation is concurrent with a downregulation of astrocyte glutamate transporters GLT-1 and GLAST, which are responsible for synaptic glutamate uptake. The overall effect is an increase in neuronal excitability, thereby lowering the ability of opioids to hyperpolarize mu-containing GABAergic neurons (Figure 2). Within the PAG to RVM circuitry, this results in an inability for morphine to disinhibit output neurons to RVM (Eidson and Murphy, 2013; Eidson et al., 2017).
Another potential point of cross talk is via purinergic receptors, specifically P2X4 and P2X7, which are primarily expressed on microglia. These receptors are also capable of upregulating pro-inflammatory cytokines, and blocking their activity in the spinal cord attenuates morphine tolerance (Horvath et al., 2010; Zhou et al., 2010; Xiao et al., 2015). P2X4 activates the p38-MAPK pathway, resulting in the release of IL-1β, TNF-α, and BDNF, which, as mentioned above, are known to alter neuronal excitability and contribute to pain hypersensitivity, but no direct connection has been made to opioid tolerance (Ferrini et al., 2013; Grace et al., 2015; Thomas et al., 2015). However, P2X7 mediated release of IL-18 from microglia induces activation of the IL-18 receptor on astrocytes, thereby increasing the release of D-serine, which is able to activate NMDA receptors in neurons. Activation of both receptors is able to alter glial activation and neuronal excitability, suggesting a complicated crosstalk between cell types in the spinal cord that is correlated with morphine tolerance (Chen et al., 2012).
Conclusion
The descending pain pathway is a critical modulator of nociception and plays an important role in mediating endogenous and exogenous opioid-induced analgesia. Because of this, it is highly implicated in allostatic cellular and molecular changes following repeated opioid use that lead to the development of tolerance. While this review has touched on a number of those changes at each level of the descending pain pathway, including desensitization of MOPr, altered cellular excitability and signaling, and induction of immune-competent cells, we do not yet have a complete understanding of all the factors that might be contributing to opioid tolerance.
Much of the literature on opioid tolerance has focused the effects of morphine on this system. Future research must expand to include other commonly used opioids, especially in light of the increasing use of oxycodone and fentanyl, as each of these has widely different pharmacokinetic and signaling profiles, and may have differential effects on each level of the PAG-RVM-DH pathway. Indeed, studies looking at cross-tolerance between opioid analgesics suggest that differences in the distribution of the drug within the pain pathway may be differentially contributing to the development of tolerance. Furthermore, the cellular signaling pathways initiated within these spinal and supraspinal regions following administration of different opioids are known to vary.
Finally, the research design of the studies related to opioids and tolerance has varied widely in terms of not only the drugs used, but also routes of administration, length of exposure, and use of biological systems. Also, the majority of studies on opioid tolerance have focused on males and have largely excluded females. Given that males show greater morphine potency, tolerance, and activation of neurons from PAG to RVM following morphine, as compared to females (Loyd et al., 2008), it is imperative to further explore these differences. Overall, these variations in research design have resulted in a myriad of observed cellular changes that correlate with tolerance, but with no definite conclusions or unifying theories of tolerance. While no one specific etiology may exist, future researchers must be careful in designing these studies, in order to make meaningful conclusions regarding the cellular impact of opioids in the development of tolerance.
Author Contributions
LL, AF, and EB contributed to the writing and editing of this manuscript.
Funding
This work was supported by a Young Investigator Grant from the Brain and Behavior Research Foundation to EB, National Institute Health Award T32 DA007135 to LL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bagley, E. E., Chieng, B. C. H., Christie, M. J., and Connor, M. (2005). Opioid tolerance in periaqueductal gray neurons isolated from mice chronically treated with morphine. Br. J. Pharmacol. 146, 68–76. doi: 10.1038/sj.bjp.0706315
Bobeck, E. N., Chen, Q., Morgan, M. M., and Ingram, S. L. (2014). Contribution of adenylyl cyclase modulation of pre- and postsynaptic GABA neurotransmission to morphine antinociception and tolerance. Neuropsychopharmacology 39, 2142–2152. doi: 10.1038/npp.2014.62
Bobeck, E. N., Ingram, S. L., Hermes, S. M., Aicher, S. A., and Morgan, M. M. (2016). Ligand-biased activation of extracellular signal-regulated kinase 1/2 leads to differences in opioid induced antinociception and tolerance. Behav. Brain Res. 298, 17–24. doi: 10.1016/j.bbr.2015.10.032
Bobeck, E. N., McNeal, A. L., and Morgan, M. M. (2009). Drug dependent sex-differences in periaqueducatal gray mediated antinociception in the rat. Pain 147, 210–216. doi: 10.1016/j.pain.2009.09.008
Bourinet, E., Soong, T. W., Stea, A., and Snutch, T. P. (1996). Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc. Natl. Acad. Sci. U.S.A. 93, 1486–1491. doi: 10.1073/pnas.93.4.1486
Brüggemann, I., Schulz, S., Wiborny, D., and Höllt, V. (2000). Colocalization of the μ-opioid receptor and calcium/calmodulin-dependent kinase II in distinct pain-processing brain regions. Mol. Brain Res. 85, 239–250. doi: 10.1016/S0169-328X(00)00265-5
Cahill, C. M., and Taylor, A. M. (2017). Neuroinflammation—a co-occurring phenomenon linking chronic pain and opioid dependence. Curr. Opin. Behav. Sci. 13, 171–177. doi: 10.1016/j.cobeha.2016.12.003
Campion, K. N., Saville, K. A., and Morgan, M. M. (2016). Relative contribution of the dorsal raphe nucleus and ventrolateral periaqueductal gray to morphine antinociception and tolerance in the rat. Eur. J. Neurosci. 44, 2667–2672. doi: 10.1111/ejn.13378
Chen, M. L., Cao, H., Chu, Y. X., Cheng, L. Z., Liang, L. L., Zhang, Y. Q., et al. (2012). Role of P2X7 receptor-mediated IL-18/IL-18R signaling in morphine tolerance: Multiple glial-neuronal dialogues in the rat spinal cord. J. Pain 13, 945–958. doi: 10.1016/j.jpain.2012.06.007
Chen, S.-P., Zhou, Y.-Q., Liu, D.-Q., Zhang, W., Manyande, A., Guan, X.-H., et al. (2017). PI3K/Akt pathway: a potential therapeutic target for chronic pain. Curr. Pharm. Des. 23, 1860–1868. doi: 10.2174/1381612823666170210150147
Connor, M., Bagley, E. E., Chieng, B. C., and Christie, M. J. (2015). β-Arrestin-2 knockout prevents development of cellular μ-opioid receptor tolerance but does not affect opioid-withdrawal-related adaptations in single PAG neurons. Br. J. Pharmacol. 172, 492–500. doi: 10.1111/bph.12673
Connor, M., Schuller, A., Pintar, J. E., and Christie, M. J. (1999). μ-opioid receptor modulation of calcium channel current in periaqueductal grey neurons from C57B16/J mice and mutant mice lacking MOR-1. Br. J. Pharmacol. 126, 1553–1558. doi: 10.1038/sj.bjp.0702457
Cui, Y., Liao, X. X., Liu, W., Guo, R. X., Wu, Z. Z., Zhao, C. M., et al. (2008). A novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain. Behav. Immun. 22, 114–123. doi: 10.1016/j.bbi.2007.07.014
Depaulis, A., Morgan, M. M., and Liebeskind, J. C. (1987). GABAergic modulation of the analgesic effects of morphine microinjected in the ventral periaqueductal gray matter of the rat. Brain Res. 436, 223–228. doi: 10.1016/0006-8993(87)91665-9
Eidson, L. N., Inoue, K., Young, L. J., Tansey, M. G., and Murphy, A. Z. (2017). Toll-like receptor 4 mediates morphine-induced neuroinflammation and tolerance via soluble tumor necrosis factor signaling. Neuropsychopharmacology 42, 661–670. doi: 10.1038/npp.2016.131
Eidson, L. N., and Murphy, A. Z. (2013). Blockade of toll-like receptor 4 attenuates morphine tolerance and facilitates the pain relieving properties of morphine. J. Neurosci. 33, 15952–15963. doi: 10.1523/JNEUROSCI.1609-13.2013
Fairbanks, C. A., and Wilcox, G. L. (1997). Acute tolerance to spinally administered morphine compares mechanistically with chronically induced morphine tolerance. J. Pharmacol. Exp. Ther. 282, 1408–1417.
Fang, F. G., Haws, C. M., Drasner, K., Williamson, A., and Fields, H. L. (1989). Opioid peptides (DAGO-enkephalin, dynorphin A(1-13), BAM 22P) microinjected into the rat brainstem: comparison of their antinociceptive effect and their effect on neuronal firing in the rostral ventromedial medulla. Brain Res. 501, 116–128. doi: 10.1016/0006-8993(89)91033-0
Ferrini, F., Trang, T., Mattioli, T. A. M., Laffray, S., Del’Guidice, T., Lorenzo, L. E., et al. (2013). Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl-homeostasis. Nat. Neurosci. 16, 183–192. doi: 10.1038/nn.3295
Fields, H. L., Bry, J., Hentall, I., and Zorman, G. (1983). The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J. Neurosci. 3, 2545–2552. doi: 10.1523/JNEUROSCI.03-12-02545.1983
Fields, H. L., and Heinricher, M. M. (1985). Anatomy and physiology of a nociceptive modulatory system. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 308, 361–374. doi: 10.1098/rstb.1985.0037
Fyfe, L. W., Cleary, D. R., Macey, T. A., Morgan, M. M., and Ingram, S. L. (2010). Tolerance to the antinociceptive effect of morphine in the absence of short-term presynaptic desensitization in rat periaqueductal gray neurons. J. Pharmacol. Exp. Ther. 335, 674–680. doi: 10.1124/jpet.110.172643
Gabra, B. H., Bailey, C. P., Kelly, E., Smith, F. L., Henderson, G., and Dewey, W. L. (2008). Pre-treatment with a PKC or PKA inhibitor prevents the development of morphine tolerance but not physical dependence in mice. Brain Res. 1217, 70–77. doi: 10.1016/j.brainres.2008.04.036
Gintzler, A. R., and Chakrabarti, S. (2004). Chronic morphine-induced plasticity among signalling molecules. Novartis Found. Symp. 261, 191–193. doi: 10.1002/0470869127.ch13
Giron, S. E., Griffis, C. A., and Burkard, J. F. (2015). Chronic pain and decreased opioid efficacy: an inflammatory link. Pain Manag. Nurs. 16, 819–831. doi: 10.1016/j.pmn.2015.04.001
Goode, T. L., and Raffa, R. B. (1997). An examination of the relationship between mu-opioid antinociceptive efficacy and G-protein coupling using pertussis and cholera toxins. Life Sci. 60, l107–l113. doi: 10.1016/S0024-3205(96)00684-4
Grace, P. M., Maier, S. F., and Watkins, L. R. (2015). Opioid-Induced central immune signaling: implications for opioid analgesia. Headache 55, 475–489. doi: 10.1111/head.12552
Guitart, X., and Nestler, E. J. (1989). Identification of morphine- and cyclic AMP-regulated phosphoproteins (MARPPs) in the locus coeruleus and other regions of rat brain: regulation by acute and chronic morphine. J. Neurosci. 9, 4371–4387. doi: 10.1523/JNEUROSCI.09-12-04371.1989
Hack, S. P., Vaughan, C. W., and Christie, M. J. (2003). Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology 45, 575–584. doi: 10.1016/S0028-3908(03)00205-3
Harada, S., Nakamoto, K., and Tokuyama, S. (2013). The involvement of midbrain astrocyte in the development of morphine tolerance. Life Sci. 93, 573–578. doi: 10.1016/j.lfs.2013.08.009
Hirst, R. A., and Lambert, D. G. (1995). Adenylyl cyclase in SH-SY5Y human neuroblastoma cells is regulated by intra- and extracellular calcium. Biochem. Pharmacol. 49, 1633–1640. doi: 10.1016/0006-2952(95)00075-B
Horvath, R. J., Romero-Sandoval, E. A., and Leo, J. A. D. (2010). Inhibition of microglial P2X4receptors attenuates morphine tolerance, Iba1, GFAP and μ opioid receptor protein expression while enhancing perivascular microglial ED2. Pain 150, 401–413. doi: 10.1016/j.pain.2010.02.042
Hull, L. C., Llorente, J., Gabra, B. H., Smith, F. L., Kelly, E., Bailey, C., et al. (2010). The effect of protein kinase C and G protein-coupled receptor kinase inhibition on tolerance induced by mu-opioid agonists of different efficacy. J. Pharmacol. Exp. Ther. 332, 1127–1135. doi: 10.1124/jpet.109.161455
Hutchinson, M. R., Shavit, Y., Grace, P. M., Rice, K. C., Maier, S. F., and Watkins, L. R. (2011). Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol. Rev. 63, 772–810. doi: 10.1124/pr.110.004135
Hutchinson, M. R., Zhang, Y., Shridhar, M., Evans, J. H., Buchman, M. M., Zhao, T. X., et al. (2010). Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav. Immun. 24, 83–95. doi: 10.1016/j.bbi.2009.08.004
Ingram, S. L., Vaughan, C. W., Bagley, E. E., Connor, M., and Christie, M. J. (1998). Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J. Neurosci. 18, 10269–10276. doi: 10.1523/JNEUROSCI.18-24-10269.1998
Ippolito, D. L., Temkin, P. A., Rogalski, S. L., and Chavkin, C. (2002). N-terminal tyrosine residues within the potassium channel Kir3 modulate GTPase activity of Galphai. J. Biol. Chem. 277, 32692–32696. doi: 10.1074/jbc.M204407200
Jacquet, Y. F. (1988). The NMDA receptor: central role in pain inhibition in rat periaqueductal gray. Eur. J. Pharmacol. 154, 271–276. doi: 10.1016/0014-2999(88)90201-4
Jiang, Z., Wu, S., Wu, X., Zhong, J., Lv, A., Jiao, J., et al. (2016). Blocking mammalian target of rapamycin alleviates bone cancer pain and morphine tolerance via μ-opioid receptor. Int. J. Cancer 138, 2013–2020. doi: 10.1002/ijc.29927
King, T., Ossipov, M. H., Vanderah, T. W., Porreca, F., and Lai, J. (2005). Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals 14, 194–205. doi: 10.1159/000087658
Kozela, E., and Popik, P. (2007). A complete analysis of NMDA receptor subunits in periaqueductal grey and ventromedial medulla of morphine tolerant mice. Drug Alcohol Depend. 86, 290–293. doi: 10.1016/j.drugalcdep.2006.06.018
Lane, D. A., Patel, P. A., and Morgan, M. M. (2005). Evidence for an intrinsic mechanism of antinociceptive tolerance within the ventrolateral periaqueductal gray of rats. Neuroscience 135, 227–234. doi: 10.1016/J.NEUROSCIENCE.2005.06.014
Lane, D. A., Tortorici, V., and Morgan, M. M. (2004). Behavioral and electrophysiological evidence for tolerance to continuous morphine administration into the ventrolateral periaqueductal gray. Neuroscience 125, 63–69. doi: 10.1016/j.neuroscience.2004.01.023
Lewis, S. S., Hutchinson, M. R., Rezvani, N., Loram, L. C., Zhang, Y., Maier, S. F., et al. (2010). Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1β. Neuroscience 165, 569–583. doi: 10.1016/j.neuroscience.2009.10.011
Liaw, W. J., Zhu, X. G., Yaster, M., Johns, R. A., Gauda, E. B., and Tao, Y. X. (2008). Distinct expression of synaptic NR2A and NR2B in the central nervous system and impaired morphine tolerance and physical dependence in mice deficient in postsynaptic density-93 protein. Mol. Pain 4:45. doi: 10.1186/1744-8069-4-45
Loyd, D. R., Morgan, M. M., and Murphy, A. Z. (2008). Sexually dimorphic activation of the periaqueductal gray-rostral ventromedial medullary circuit during the development of tolerance to morphine in the rat. Eur. J. Neurosci. 27, 1517–1524. doi: 10.1111/j.1460-9568.2008.06100.x
Macey, T. A., Bobeck, E. N., Hegarty, D. M., Aicher, S. A., Ingram, S. L., and Morgan, M. M. (2009). Extracellular signal-regulated kinase 1/2 activation counteracts morphine tolerance in the periaqueductal gray of the rat. J. Pharmacol. Exp. Ther. 331, 412–418. doi: 10.1124/jpet.109.152157
Maher, C. E., Eisenach, J. C., Pan, H. L., Xiao, R., and Childers, S. R. (2001). Chronic intrathecal morphine administration produces homologous mu receptor/G-protein desensitization specifically in spinal cord. Brain Res. 895, 1–8. doi: 10.1016/S0006-8993(00)03093-6
Melief, E. J., Miyatake, M., Bruchas, M. R., and Chavkin, C. (2010). Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 11608–11613. doi: 10.1073/pnas.1000751107
Meng, I. D., and Harasawa, I. (2007). Chronic morphine exposure increases the proportion of on-cells in the rostral ventromedial medulla in rats. Life Sci. 80, 1915–1920. doi: 10.1016/j.lfs.2007.02.022
Moreau, J. L., and Fields, H. L. (1986). Evidence for GABA involvement in midbrain control of medullary neurons that modulate nociceptive transmission. Brain Res. 397, 37–46. doi: 10.1016/0006-8993(86)91367-3
Morgan, M. M., Bobeck, E. N., and Ingram, S. L. (2009). Glutamate modulation of antinociception, but not tolerance, produced by morphine microinjection into the periaqueductal gray of the rat. Brain Res. 1295, 59–66. doi: 10.1016/j.brainres.2009.07.100
Morgan, M. M., Clayton, C. C., and Boyer-Quick, J. S. (2005). Differential susceptibility of the PAG and RVM to tolerance to the antinociceptive effect of morphine in the rat. Pain 113, 91–98. doi: 10.1016/j.pain.2004.09.039
Morgan, M. M., Clayton, C. C., and Lane, D. A. (2003). Behavioral evidence linking opioid-sensitive GABAergic neurons in the ventrolateral periaqueductal gray to morphine tolerance. Neuroscience 118, 227–232. doi: 10.1016/S0306-4522(02)00822-9
Morgan, M. M., Fossum, E. N., Levine, C. S., and Ingram, S. L. (2006). Antinociceptive tolerance revealed by cumulative intracranial microinjections of morphine into the periaqueductal gray in the rat. Pharmacol. Biochem. Behav. 85, 214–219. doi: 10.1016/j.pbb.2006.08.003
Morgan, M. M., Reid, R. A., and Saville, K. A. (2014). Functionally selective signaling for morphine and fentanyl antinociception and tolerance mediated by the rat periaqueductal gray. PLoS One 9:e114269. doi: 10.1371/journal.pone.0114269
Morgan, M. M., Whittier, K. L., Hegarty, D. M., and Aicher, S. A. (2008). Periaqueductal gray neurons project to spinally projecting GABAergic neurons in the rostral ventromedial medulla. Pain 140, 376–386. doi: 10.1016/j.pain.2008.09.009
Muscoli, C., Doyle, T., Dagostino, C., Bryant, L., Chen, Z., Watkins, L. R., et al. (2010). Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J. Neurosci. 30, 15400–15408. doi: 10.1523/JNEUROSCI.2391-10.2010
Nakamoto, K., Kawasaki, S., Kobori, T., Fujita-Hamabe, W., Mizoguchi, H., Yamada, K., et al. (2012). Involvement of matrix metalloproteinase-9 in the development of morphine tolerance. Eur. J. Pharmacol. 683, 86–92. doi: 10.1016/j.ejphar.2012.03.006
Ndengele, M. M., Cuzzocrea, S., Masini, E., Vinci, M. C., Esposito, E., Muscoli, C., et al. (2009). Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J. Pharmacol. Exp. Ther. 329, 64–75. doi: 10.1124/jpet.108.146290
North, R. A., and Williams, J. T. (1983). Opiate activation of potassium conductance inhibits calcium action potentials in rat locus coeruleus neurones. Br. J. Pharmacol. 80, 225–228. doi: 10.1111/j.1476-5381.1983.tb10023.x
Osborne, P. B., Vaughan, C. W., Wilson, H. I., and Christie, M. J. (1996). Opioid inhibition of rat periaqueductal grey neurones with identified projections to rostral ventromedial medulla in vitro. J. Physiol. 490, 383–389. doi: 10.1113/jphysiol.1996.sp021152
Pan, Z. Z., Williams, J. T., and Osborne, P. B. (1990). Opioid actions on single nucleus raphe magnus neurons from rat and guinea? pig in vitro. J. Physiol. 427, 519–532. doi: 10.1113/jphysiol.1990.sp018185
Park, C., Kim, J.-H., Yoon, B.-E., Choi, E.-J., Lee, C. J., and Shin, H.-S. (2010). T-type channels control the opioidergic descending analgesia at the low threshold-spiking GABAergic neurons in the periaqueductal gray. Proc. Natl. Acad. Sci. U.S.A. 107, 14857–14862. doi: 10.1073/pnas.1009532107
Price, D. D., Mayer, D. J., Mao, J., and Caruso, F. S. (2000). NMDA-receptor antagonists and opioid receptor interactions as related to analgesia and tolerance. J. Pain Symptom Manage. 19(1 Suppl.), S7–S11. doi: 10.1016/S0885-3924(99)00121-9
Raghavendra, V., Rutkowski, M. D., and DeLeo, J. A. (2002). The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J. Neurosci. 22, 9980–9989. doi: 10.1523/jneurosci.1850-04.2004
Raghavendra, V., Tanga, F. Y., and DeLeo, J. A. (2004). Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammtory immune responses by propentofylline in rats. Neuropsychopharmacology 29, 327–334. doi: 10.1038/sj.npp.1300315
Ray, S. B., Gupta, H., and Gupta, Y. K. (2004). Up-regulation of mu-opioid receptors in the spinal cord of morphine-tolerant rats. J. Biosci. 29, 51–56. doi: 10.1007/BF02702561
Rönnbäck, L., and Hansson, E. (1988). Are astroglial cells involved in morphine tolerance? Neurochem. Res. 13, 87–103.
Samineni, V. K., Grajales-Reyes, J. G., Copits, B. A., O’Brien, D. E., Trigg, S. L., Gomez, A. M., et al. (2017). Divergent modulation of nociception by glutamatergic and gabaergic neuronal subpopulations in the periaqueductal gray. Eneuro 4:ENEURO.0129-16.2017. doi: 10.1523/ENEURO.0129-16.2017
Sharma, S. K., Klee, W. A., and Nirenberg, M. (1975). Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc. Natl. Acad. Sci. U.S.A. 72, 3092–3096. doi: 10.1073/pnas.72.8.3092
Smith, F. L., Javed, R. R., Smith, P. A., Dewey, W. L., and Gabra, B. H. (2006). PKC and PKA inhibitors reinstate morphine-induced behaviors in morphine tolerant mice. Pharmacol. Res. 54, 474–480. doi: 10.1016/j.phrs.2006.09.007
Song, P., and Zhao, Z.-Q. (2001). The involvement of glial cells in the development of morphine tolerance. Neurosci. Res. 39, 281–286. doi: 10.1016/S0168-0102(00)00226-1
Stellwagen, D. (2005). Differential Regulation of AMPA Receptor and GABA receptor trafficking by tumor necrosis factor-. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Stiller, C. O., Bergquist, J., Beck, O., Ekman, R., and Brodin, E. (1996). Local administration of morphine decreases the extracellular level of GABA in the periaqueductal gray matter of freely moving rats. Neurosci. Lett. 209, 165–168. doi: 10.1016/0304-3940(96)12638-0
Thomas, J., Mustafa, S., Johnson, J., Nicotra, L., and Hutchinson, M. (2015). The relationship between opioids and immune signaling in the spinal cord. Handb. Exp. Pharmacol. 227, 207–238. doi: 10.1007/978-3-662-46450-2_11
Torrecilla, M., Marker, C. L., Cintora, S. C., Stoffel, M., Williams, J. T., and Wickman, K. (2002). G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J. Neurosci. 22, 4328–4334. doi: 10.1523/JNEUROSCI.22-11-04328.2002
Tortorici, V., Morgan, M. M., and Vanegas, H. (2001). Tolerance to repeated microinjection of morphine into the periaqueductal gray is associated with changes in the behavior of off- and on-cells in the rostral ventromedial medulla of rats. Pain 89, 237–244. doi: 10.1016/S0304-3959(00)00367-5
Vanderah, T. W., Suenaga, N. M., Ossipov, M. H., Malan, T. P., Lai, J., and Porreca, F. (2001). Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J. Neurosci. 21, 279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001
Vaughan, C. W., Ingram, S. L., Connor, M. A., and Christie, M. J. (1997). How opioids inhibit GABA-mediated neurotransmission. Nature 390, 611–614. doi: 10.1038/37610
Viviani, B., Bartesaghi, S., Gardoni, F., Vezzani, A., Behrens, M. M., Bartfai, T., et al. (2003). Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
Wang, D., Zeng, J., Li, Q., Huang, J., Couture, R., and Hong, Y. (2016). Contribution of adrenomedullin to the switch of G protein-coupled μ-opioid receptors from Gi to Gs in the spinal dorsal horn following chronic morphine exposure in rats. Br. J. Pharmacol. 173, 1196–1207. doi: 10.1111/bph.13419
Wang, X., Loram, L. C., Ramos, K., de Jesus, A. J., Thomas, J., Cheng, K., et al. (2012). Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. U.S.A. 109, 6325–6330. doi: 10.1073/pnas.1200130109
Watkins, L. R., Hutchinson, M. R., Rice, K. C., and Maier, S. F. (2009). The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol. Sci. 30, 581–591. doi: 10.1016/j.tips.2009.08.002
Wilding, T. J., Womack, M. D., and McCleskey, E. W. (1995). Fast, local signal transduction between the mu opioid receptor and Ca2+ channels. J. Neurosci. 15(5 Pt 2), 4124–4132. doi: 10.1523/JNEUROSCI.15-05-04124.1995
Williams, J. T., Christie, M. J., and Manzoni, O. (2001). Cellular and synaptic adaptations mediating opioid dependence. Physiol. Rev. 81, 299–343. doi: 10.1152/physrev.2001.81.1.299
Wimpey, T. L., and Chavkin, C. (1991). Opioids activate both an inward rectifier and a novel voltage-gated potassium conductance in the hippocampal formation. Neuron 6, 281–289. doi: 10.1016/0896-6273(91)90363-5
Xiao, Z., Li, Y. Y., and Sun, M. J. (2015). Activation of P2X7 receptors in the midbrain periaqueductal gray of rats facilitates morphine tolerance. Pharmacol. Biochem. Behav. 135, 145–153. doi: 10.1016/j.pbb.2015.06.002
Xie, J. Y., Herman, D. S., Stiller, C.-O., Gardell, L. R., Ossipov, M. H., Lai, J., et al. (2005). Cholecystokinin in the rostral ventromedial medulla mediates opioid-induced hyperalgesia and antinociceptive tolerance. J. Neurosci. 25, 409–416. doi: 10.1523/JNEUROSCI.4054-04.2005
Xu, J., Sun, L., Lutz, B., Bekker, A. T., and Tao, Y. (2015). Intrathecal rapamycin attenuates morphine-induced analgesic tolerance and hyperalgesia in rats with neuropathic pain. Transl. Perioper. Pain Med. 2, 27–34.
Xu, J.-T. T., Yaster, M., Tao, Y.-X. X., Zhao, J.-Y. Y., Zhao, X., Ligons, D., et al. (2014). Opioid receptor-triggered spinal mTORC1 activation contributes to morphine tolerance and hyperalgesia. J. Clin. Invest 124, 592–603. doi: 10.1172/JCI70236
Zhao, Y. L., Chen, S. R., Chen, H., and Pan, H. L. (2012). Chronic opioid potentiates presynaptic but impairs postsynaptic N-methyl-D-aspartic acid receptor activity in spinal cords: implications for opioid hyperalgesia and tolerance. J. Biol. Chem. 287, 25073–25085. doi: 10.1074/jbc.M112.378737
Keywords: opioid, tolerance, periaqueductal gray (PAG), RVM, dorsal horn
Citation: Lueptow LM, Fakira AK and Bobeck EN (2018) The Contribution of the Descending Pain Modulatory Pathway in Opioid Tolerance. Front. Neurosci. 12:886. doi: 10.3389/fnins.2018.00886
Received: 03 July 2018; Accepted: 13 November 2018;
Published: 27 November 2018.
Edited by:
Lawrence Toll, Florida Atlantic University, United StatesReviewed by:
Mary M. Heinricher, Oregon Health & Science University, United StatesElyssa Margolis, University of California, San Francisco, United States
Copyright © 2018 Lueptow, Fakira and Bobeck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erin N. Bobeck, ZXJpbi5ib2JlY2tAdXN1LmVkdQ==