 Laís S. S. Ferreira
Laís S. S. Ferreira Caroline S. Fernandes1,2†
Caroline S. Fernandes1,2† Marcelo N. N. Vieira
Marcelo N. N. Vieira Fernanda G. De Felice
Fernanda G. De Felice
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci. , 13 November 2018
Sec. Neuroendocrine Science
Volume 12 - 2018 | https://doi.org/10.3389/fnins.2018.00830
This article is part of the Research Topic Insulin and The Brain View all 7 articles
The epidemiological connection between diabetes, obesity, and dementia represents an important public health challenge but also an opportunity to further understand these conditions. The key intersection among the three diseases is insulin resistance, which has been classically described to occur in peripheral tissues in diabetes and obesity and has recently been shown to develop in Alzheimer's disease (AD) brains. Here we review encouraging preclinical and clinical data indicating the potential of targeting impaired insulin signaling with antidiabetic drugs to treat dementia. We further discuss biological mechanisms through which peripheral metabolic dysregulation may lead to brain malfunction, providing possible explanations for the connection between diabetes, obesity, and AD. Finally, we briefly discuss how lifelong allostatic load may interact with aging to increase the risk of dementia in late life.
Alzheimer's disease (AD), type 2 diabetes (T2D), and obesity are among the most expensive and disabling disorders worldwide. For a long time, the correlation between cognitive impairment and metabolic diseases was undetected. Now, increasingly epidemiological evidence supports an important association among these conditions (Razay et al., 2006; Baker et al., 2011; Crane et al., 2013; Ikram et al., 2017). Accordingly, experimental observations are identifying that markers of metabolic dysregulation are also present in AD, the most remarkable being insulin resistance (Talbot et al., 2012; De Felice, 2013; Boles et al., 2017). However, the molecular mechanisms underlying this crosstalk are still elusive, as well as how central and peripheral insulin signaling operate in AD (Biessels and Despa, 2018).
Over the last decade, cumulative data have reinforced that the brain is an insulin-sensitive organ. The insulin receptor (IR) and related insulin-like growth factor receptors 1 and 2 (IGF1-R/IGF2-R) are expressed not only in rodent hypothalamus, the critical brain region for metabolic control, but also in cortex, hippocampus, thalamus, olfactory bulb, and in lower levels, in cerebellum, striatum, midbrain, and brainstem (Fernandez and Torres-Alemán, 2012; Kleinridders et al., 2014).
Beyond the canonical role of insulin in whole body metabolism regulation (Brüning et al., 2000), insulin also modifies neuronal activity promoting synaptic plasticity (Wan et al., 1997; Schmitz et al., 2018) and improves memory function in mammalian brain (Park et al., 2000; Benedict et al., 2007). Glial insulin signaling functions are gaining attention. Hypothalamic astrocyte IRs control glucose-induced activation of POMC neurons, central and peripheral response to glucose availability, and glucose transport through the blood-brain barrier (BBB, García-Cáceres et al., 2016). Additionally, insulin modulates proinflammatory cytokine secretion in microglia and astrocytes in vitro (Spielman et al., 2015; Kurochkin et al., 2018). Under brain injury, IGF1 can be produced by activated microglia and is required for vessel remodeling (Walter et al., 1997; Lopez-Lopez et al., 2004). Further studies are required to decipher the complex regulation of insulin signaling in distinct cell types, but these data suggest that IR/IGF-Rs play a role in central nervous system (CNS) physiology.
Remarkably, several studies show that insulin signaling is impaired in the brains of AD patients and AD experimental models (Boyt et al., 2000; Craft et al., 2003; Steen et al., 2005; Bomfim et al., 2012; Hiltunen et al., 2012; Talbot et al., 2012). Neuronal insulin resistance can be induced by Aβ-oligomers in primary cultures of hippocampal neurons and by intracerebroventricular injection of AβOs in mice and monkeys. It is mediated by TNF-α activation and IRS inhibition and has major impact on synaptic dysfunction, impaired synaptic plasticity, and synapse loss (Townsend et al., 2007; De Felice et al., 2009; Bomfim et al., 2012; Batista et al., 2018) Remarkably, our group found that icv injection of Aβ oligomers also induce peripheral glucose intolerance with classic hallmarks of peripheral insulin resistance, a process also observed in transgenic AD mice models(Clarke et al., 2015) and that may underlie increased risk for diabetes in AD (Janson et al., 2004). Additionally, anti-diabetic drugs exerts beneficial effects on cognition, synapse protection, insulin signaling deficits, and other AD-related pathological mechanisms, such as endoplasmic reticulum stress and chronic inflammation (Lourenco et al., 2013; Sebastião et al., 2014; Batista et al., 2018; Tai et al., 2018).
Here, we review evidence regarding similar mechanisms of insulin resistance shared by T2D, obesity, and AD. We will discuss recent findings that help explain the connection between peripheral metabolic deregulation and AD, the cause-consequence between these diseases and how boosting insulin pathway provide therapeutic alternatives for treating AD.
A key question to understand the connection between diabetes and obesity to AD is to learn how metabolic impairment in periphery may cause pathological alterations in the brain.
Fatty acids and lipids in general are well-established players in obesity, diabetes, and AD. A recent meta-analysis study revealed the potential mechanistic, diagnostic, and therapeutic implications of different classes of lipids in AD (Zarrouk et al., 2017). Obesity and diabetes present inflammatory components (Wang et al., 2018). Indeed, obese patients often display basal low-grade systemic inflammation in adipose tissue and increased susceptibility to immune-mediated diseases (Sun et al., 2012; Mraz and Haluzik, 2014). Free fatty acids (FFA) from feeding trigger an inflammatory cascade initiated by Toll-like receptor 4 (TLR4) stimulation, releasing pro-inflammatory cytokines such as TNF-α and interleukins IL-1β and IL-6 (Weisberg et al., 2003; Shu et al., 2012). High levels of FFA can further inhibit the anti-lipolytic action of insulin, increasing the rate of FFA release into the bloodstream (Guenther, 2009). FFA binding to BBB endothelial cell alters permeability, allowing FFA infiltration into the brain (Rapoport, 2001). Hence, the increase in cerebral FFA triggers detrimental events such as ceramide production, pattern recognition receptors activation, inflammation, and ER stress (Spiegel, 2005; Guenther, 2009; Groop et al., 2018). When the brain detects homeostatic disruption, microglia are activated leading to neuroinflammation (Jha et al., 2016). Saturated fatty acids and mono-unsaturated fatty acids were shown to activate microglial NF-kB pathway in a TLR4-dependent manner, leading to increased production of proinflammatory cytokines, and reactive oxygen species (ROS, Wang et al., 2012; Arnold et al., 2014; Button et al., 2014; Carroll et al., 2018). Saturated fatty acids were also shown to induce TLR4-dependent activation of astrocytes in culture leading to cytokine production (Gupta et al., 2012; Wang et al., 2012).
Advanced glycation end-products (AGEs) also represent a common feature in diabetes and AD, and may be involved in the crosstalk between periphery and CNS that underlies this connection. AGEs are the product of unspecific and uncontrolled reactions between proteins or lipids with sugars. AGEs increase during normal aging, but their formation is promoted in glucose-rich environments, such as in hyperglycemia. Interestingly, elevated AGEs levels are also observed in AD brains (Shuvaev et al., 2001; Choei et al., 2004; Takeuchi et al., 2007). Importantly, AGE-receptor (RAGE) was suggested as a possible receptor for Aβ (Yan et al., 1996) and to mediate Abeta toxic mechanisms such as induction of ER-stress (Chen et al., 2018). Moreover, RAGEs have been shown to promote Aβ production, tau hyperphosphorylation and tangle formation, synaptic impairment cognitive decline, and neurodegeneration (Cai et al., 2016). Downregulating RAGE signaling specifically in microglia prevented synaptic deficit and cognitive impairment and diminished the activation of stress-related kinases in a mouse model of AD (Criscuolo et al., 2017). Importantly, recent evidences indicate that RAGE mediates Abeta impact on BBB integrity and tight junction regulation as well APOE4-induced BBB anomalies (Park et al., 2014; Wan et al., 2014, 2015; Alata et al., 2015).
Collectively, these observations indicate that lipidemic dysregulation in metabolic disorders may lead to BBB permeabilization to FFAs, triggering a cascade of events leading to activation of glial cells, and neuroinflammation. In parallel, increased AGE levels in hyperglycemia may contribute to RAGE-mediated disruption of BBB integrity and further promote brain pathology leading to AD. Proinflammatory cytokines reaching the brain through compromised BBB form a toxic environment for neurons, leading to neuronal insulin resistance, and synaptic dysfunction (Bomfim et al., 2012; Gupta et al., 2012; Lourenco et al., 2013; Kiernan et al., 2016; Vieira et al., 2017, Figure 1). Consistent with this hypothesis, it has been shown that HFD activates inflammatory responses in the mouse hippocampus (Lu et al., 2011; Almeida-Suhett et al., 2017) and impairs insulin signaling (Arnold et al., 2014) whereas a high-AGE diet aggravates AD-like phenotypes in a mouse model of AD.
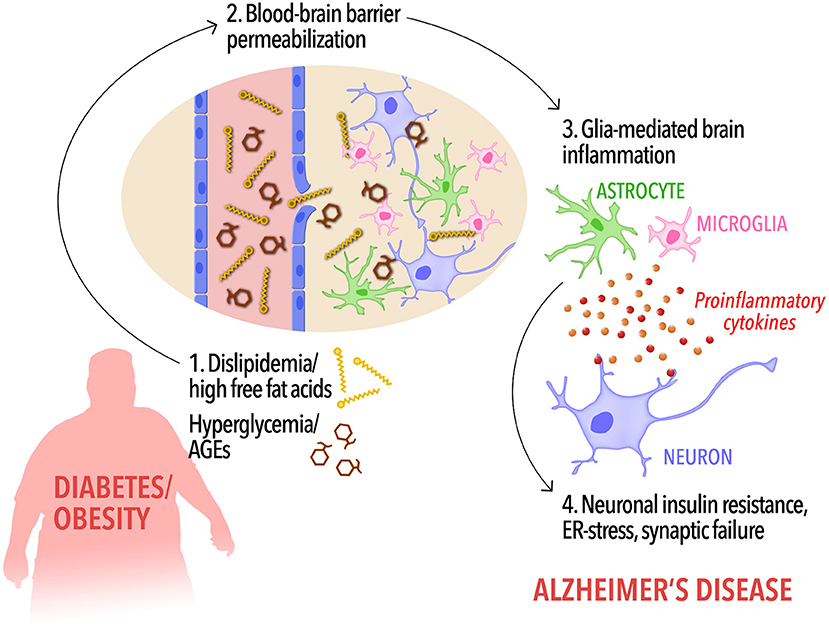
Figure 1. A possible cascade of events connecting peripheral metabolic dysregulation to dementia. In diabetic and/or obese subjects, dyslipidemia, and increased circulating free fat acids as well as hyperglycemia and elevated peripheral AGEs levels (1) may increase blood-brain barrier permeability, allowing the influx of FFAs into the brain (2). Disrupted BBB along with high levels of brain FFAs and AGEs, in turn, would cause activation of microglia and astrocytes and the release of proinflammatory cytokines (3). Low-grade, chronic brain inflammation leads to detrimental events in neurons, including insulin resistance (4), priming the brain to cognitive impairment and Alzheimer's disease.
Intriguingly, many studies suggest that the incidence of AD is higher in T2D patients and obese individuals, implying common mechanisms driving these disorders (Kivipelto et al., 2005; Razay et al., 2006; Whitmer et al., 2007; Baker et al., 2011; Crane et al., 2013; Ikram et al., 2017). A core feature shared among diabetes, obesity, and AD is insulin resistance (Kullmann et al., 2016). In AD patients brains, impaired insulin signaling is a risk factor but also aggravates the pathology (Matsuzaki et al., 2010; Arnold et al., 2018).
During the past decades, mounting studies correlate the alterations in IR/IGF-R signaling pathways and AD (Boyt et al., 2000; Craft et al., 2003; Bomfim et al., 2012; Hiltunen et al., 2012; Talbot et al., 2012; Pitt et al., 2017). In addition, alterations in other proteins involved in insulin signaling were also described. Insulin-degrading enzyme (IDE) is essential for insulin and Aβ clearance (Farris et al., 2003). Insulin resistance in AD and diabetes can lead to hyperinsulinemia, thereby, saturating IDE for insulin and Aβ degradation. Furthermore, IDE function declines with age, the major risk factor for sporadic AD (Kurochkin et al., 2018). Also, phosphatase and tensin homolog (PTEN), protein kinase B (Akt), and glycogen synthase kinase 3β (GSK3β) are recruited to the synaptic compartments impairing LTP after Aβ exposure (Knafo et al., 2016).
In diabetes and obesity, increased levels of pro-inflammatory cytokines, particularly tumor necrosis factor alpha (TNF-α) activates c-Jun terminal kinase (JNK), resulting in insulin receptor substrate 1 (IRS1) suppression by increasing the inhibitory phosphorylation of Ser312IRS1, Ser636IRS1, and decreasing activating phosphorylation of Tyr465IRS1 (Hotamisligil et al., 1993; Rui et al., 2001; Pedersen et al., 2003). These alterations were also identified in AD pathogenesis: JNK activation and IRS1 inhibition are present in the hippocampus of AD transgenic mice and cynomolgus monkeys after intracerebroventricular (icv) injections of Aβ oligomers (Ma et al., 2009; Forny-Germano et al., 2014). Of note, these alterations in insulin signaling pathway were also described in AD patients. Initial evidence comes from a study showing that AD brains display markedly reduced levels of both insulin and insulin-like growth factors (IGF)-I and –II, accompanied by decreased expression of IR mRNA, IRS-associated PI3K, and activated Akt (Steen et al., 2005). This observations were subsequently corroborated by studies demonstrating progressively increased phosphorylation of IRS-1 in inhibitory serine residues, accompanied by augmented levels of their activated serine-kinases GSK-3, IKK, JNK, mTOR, and PKCζ/λ, as patients evolve from non-demented to MCI to AD (Bomfim et al., 2012; Talbot et al., 2012). Increased IRS-1 serine phosphorylation was later found to co-localize with neurons presenting tau-pathology and tangles (Yarchoan et al., 2014). Additionally, it was observed increased phosphorylation of Ser312IRS1 in neural-derived blood exosomes of AD, frontal dementia, and T2D patients. The most interest point of this study is that increased p-Ser312IRS1 manifested in prodromal AD patients that sustained these alterations 10 years later, as AD patients (Kapogiannis et al., 2015), suggesting that insulin resistance in AD develops years before clinical manifestations and that neural-derived exosomes carries potential for early AD diagnosis. Exosomal biomarkers for insulin resistance were further associated with morphometric changes in AD brains (Mullins et al., 2017). Notably, the inhibitory phosphorylation of IRS-1 and IRS-2 described in AD patients brains and transgenic mice are correlated to memory deficits and leads to insulin resistant states (Steen et al., 2005; Bomfim et al., 2012; Talbot et al., 2012).
In peripheral tissues, such as the adipose tissue, of diabetes and obesity models, TNF-α release also activates stress kinases IκB kinase (IKK) and double-stranded RNA-dependent protein kinase (PKR), promoting inflammation and inducing endoplasmic reticulum stress besides insulin signaling deregulation (Hotamisligil et al., 1993; Wellen and Hotamisligil, 2005; Yang et al., 2009). Conversely, TNF-α depletion protects mice from obesity-induced insulin resistance (Uysal et al., 1997). Similarly, Aβ oligomers also induce TNF-α secretion and the consequent induction of IKK and PKR activation in AD animal models (Bomfim et al., 2012; Lourenco et al., 2013), suggesting a common mechanism promoting insulin resistance.
Interestingly, icv injections of Aβ oligomers in mice provokes peripheral glucose intolerance, insulin resistance, and inflammation characterized by activation of JNK and IKK and IRS-1 inhibition, connecting AD pathology progression to the development of diabetes (Clarke et al., 2015). In AD patients, it was proposed an association between peripheral and central insulin resistance by correlating [18F]-fln AD patients, i positron emission tomography (FDG-PET) scans in the brain and the homeostasis model assessment of insulin resistance (HOMA-IR) in the blood. The study evaluated AD-vulnerable regions of interest (ROIs) and predicted higher HOMA-IR and lower FDG in all ROIs, indicating that central hypoglycemia and peripheral insulin resistance are related. Additionally, both were associated with immediate and delayed memory deficits in the medial temporal lobe (Willette et al., 2015).
Although not completely elucidated, it is now acknowledged that the alterations of insulin signaling in AD and diabetes are associated. On that account, a great effort has been made to distinguish the selective insulin responses in cellular types and contexts. Most of insulin effects were thought to be mediated by neurons, but recent findings indicate insulin actions on glial cells affecting their functions and whole-body response (Bélanger et al., 2011; García-Cáceres et al., 2016; Fernandez et al., 2017). Astrocytic IR-knockout mice present moderate glucose intolerance and depressive-like behavior. Furthermore, astrocytic-mediated ATP traffic to neurons is impaired, affecting purinergic signaling and, consequently, decreasing dopamine release. Curiously, these phenotypes were only observed in male mice, suggesting a possible sex-specific role for IR in astrocytes (Cai et al., 2018). This could represent a new molecular link between AD and other psychiatric disorders, since clinical evidence has linked depression and AD (Ownby et al., 2006; Modrego, 2010). In line with these observations, knocking out astrocytic IGF1-R in mice debilitates mitochondrial function, elevating ROS production, and impairs working memory. In addition, astrocytic IGF1-R knockout decreases glucose and Aβ uptake by astrocytes, contributing to Aβ accumulation in the brain (Logan et al., 2018).
A neurotoxic astrocyte phenotype induced by activated microglia was recently described. This A1-like astrocyte is associated with aging and aggravation of several neurodegenerative disorders (Liddelow et al., 2017; Clarke et al., 2018). Conversely, targeting microglia-mediated conversion of astrocytes into A1 type with NLYP01, a novel glucagon like peptide 1 receptor (GLP1R) agonist, increases lifespan and reverses the loss of dopaminergic neurons, and behavioral deficits in mouse models of Parkinson's disease (Yun et al., 2018). In the future, it will be interesting to investigate if the beneficial effects of targeting microglia GLP1 pathway offer any improvement to other neurodegenerative diseases, such as AD (Liddelow and Barres, 2017).
The strongest genetic risk factor for late-onset Alzheimer is the isoform ApoE4 of apolipoprotein E (ApoE4) protein, involved in cholesterol metabolism (Strittmatter et al., 1993a,b). Alongside with ApoE4 risk, AD patients also present abnormal levels of 24-hydroxycholesterol (24-OHC), a cholesterol oxide derivative which is important for learning and memory, in the plasma and cerebrospinal fluid (CSF) (Zarrouk et al., 2017). Interestingly, ApoE4 appears to exacerbate AD neuropathology in individuals with T2D (Malek-Ahmadi et al., 2013). It was also observed that insulin resistance in ApoE4 carriers is correlated with higher levels of phosphorylated tau in the CSF (Starks et al., 2015). Increasingly reports are trying to figure out the connection between ApoE4 carriers, development of AD, and metabolic changes associated with obesity and diabetes (Peila et al., 2002; Reiman et al., 2005; Moser and Pike, 2017; Zhao et al., 2017). It was described that ApoE4 genotype aggravates weight gain, impaired glucose metabolism, augmented Aβ plaque load, and gliosis in mice under high fat and sugar diet (Moser and Pike, 2017). Also, human ApoE4 variant interacts with insulin receptor trapping it on endosomes in neurons, reducing IRs availability in neuronal surface and diminishing insulin sensitivity. Remarkably, these effects are age-dependent and accelerated under high fat diet (HFD, Zhao et al., 2017). It was also described that the TREM2-ApoE pathway regulates the switch to neurodegenerative microglia induced by phagocytosis of apoptotic neurons, playing a detrimental role in AD (Krasemann et al., 2017). However, if and how insulin signaling participates in this process remains to be determined. Importantly, TREM2 polymorphisms also determines high risk for late-onset AD, but their role in neurodegenerative diseases are controversial (Jay et al., 2015; Wang et al., 2015). Therefore, the current challenge is to identify distinct roles of insulin signaling in multiple genetic and environmental backgrounds comprising AD risk and development to selectively modulate insulin signaling.
Drugs currently in use for AD treatment target acetylcholinergic, NMDA-type glutamatergic, and glutaminergic pathways. These therapies offer symptomatic relief of memory defects but do not combat the pathological mechanisms underlying AD progression (Graham et al., 2017). Therapies based on the amyloid hypothesis, aimed to reduce Aβ production, and toxic Aβ aggregates in the brain (Barage and Sonawane, 2015), have proven unsuccessful due to severe collateral effects (Searfoss et al., 2003; Wong et al., 2004), unfavorable pharmacological properties (Vassar, 2014), or fail to improve cognition of AD patients in clinical trials (Salloway et al., 2014; Lasser et al., 2015; Kennedy et al., 2016; Egan et al., 2018; Honig et al., 2018). In this context, it is important to explore alternative targets that may provide symptom relief and ameliorate pathology with minor collateral effects.
On this matter, mounting data indicate that anti-diabetes drugs could be neuroprotective in AD models and clinical studies. Our group has contributed investigating the modulation of insulin signaling by clinically used anti-diabetic drugs in diverse AD models. We identified that insulin prevents Aβ oligomers-induced synapse loss and surface IR reduction in vitro (De Felice et al., 2009) and also rescued the PKR-mediated endoplasmic reticulum stress in AD models (Lourenco et al., 2013). Also, we investigated the beneficial effects of GLP1-R agonists, such as exendin-4 and liraglutide, in hippocampal cultures exposed to Aβ oligomers, transgenic AD mice, and cynomolgus monkeys injected icv with Aβ oligomers. These drugs activate insulin-related pathways through G-protein dependent signaling, regardless of IR, and IGF-R (Andersen et al., 2018). Exendin-4 acted decreasing the inhibitory phosphorylation of Ser312IRS1, Ser636IRS1, and of JNK, while restoring activating Tyr465IRS1 phosphorylation, then counteracting insulin signaling impairment, memory deficits and diminishing amyloid plaque load in APP/PS1 transgenic AD mice (Bomfim et al., 2012). Liraglutide reduced tau phosphorylation and prevented IR reduction and synapse loss in a c-AMP dependent manner in cynomolgus monkeys injected icv with Aβ oligomers (Batista et al., 2018). Other groups also showed that liraglutide can reduce inflammation and enhance LTP in AD transgenic mice (McClean et al., 2011; McClean and Hölscher, 2014), that exendin-4 prevents neuronal excitotoxicity in a neurodegenerative rat model (Perry, 2002) and icv insulin injections enhance cognitive performance (Park et al., 2000, Table 1).
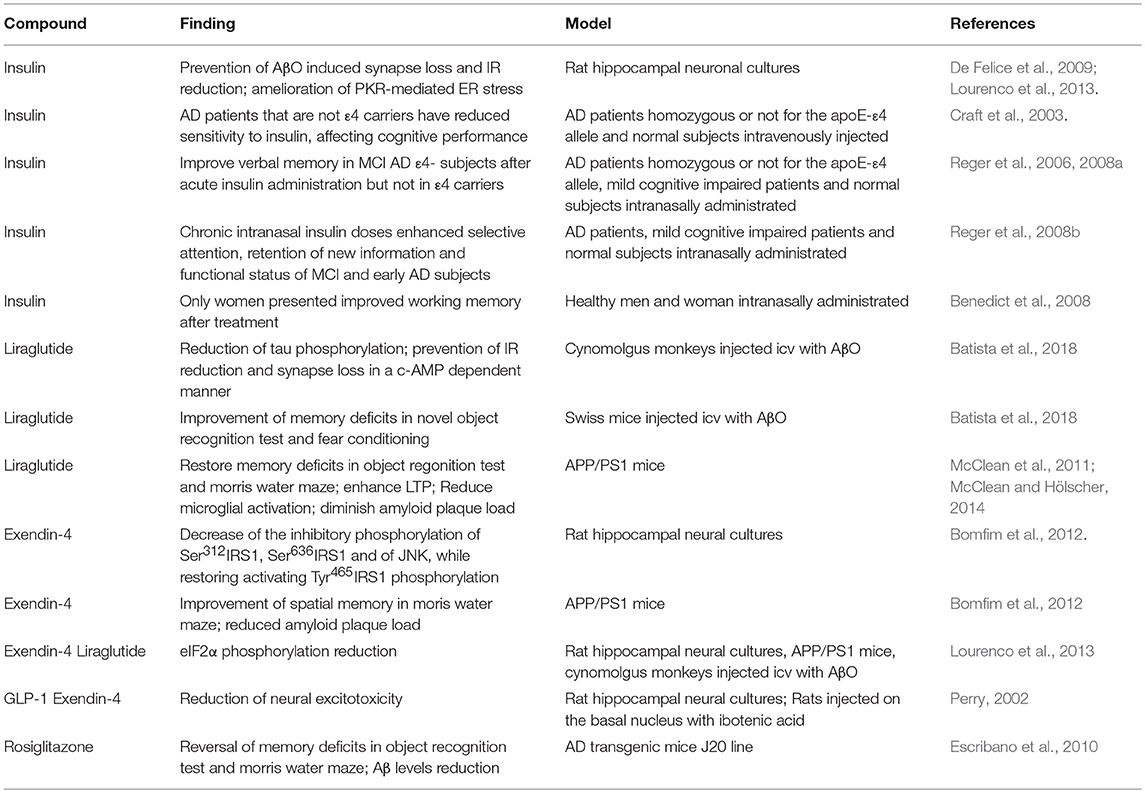
Table 1. Summary of preclinical and clinical studies on the efficacy of anti-diabetic, insulin-sensitizing drugs on multiple aspects of AD pathology in human patients and animal models.
Observations taken from preclinical and epidemiological data encouraged clinical trials repurposing insulin as a prospect treatment to AD patients (Table 1). Anti-diabetics exerts neuroprotective effects by mitigating Aβ toxicity, reducing inflammation, and improving memory deficits (Bomfim et al., 2012; Lourenco et al., 2013). In this context, anti-diabetic therapy offers a multitarget approach covering several aspects of AD pathology progression. However, concerns about off-targeting consequences have been raised, as insulin signaling systemically controls various cellular processes. To bypass this problem, one propitious solution is the intranasal delivery, restricting it to CNS and avoiding major peripheral effects such as hypoglicemia, besides being more effective than oral administration (Born et al., 2002; Spetter and Hallschmid, 2015; Schmid et al., 2018). In mild cognitive impairment (MCI) and early AD patients, acute intranasal insulin administration facilitates verbal memory recall. Nevertheless, ApoE4 carriers presented poorer verbal memory recall after the treatment, suggesting a role for ApoE4 genotype in central effects of insulin (Craft et al., 2003; Reger et al., 2006, 2008a). The same group described that chronic intranasal insulin doses enhanced selective attention, retention of new information, and functional status of MCI and early AD subjects (Reger et al., 2008b). Importantly, selective effects of intranasal insulin treatment were observed. It was reported that women show better scores of cognitive improvement than men after the intranasal insulin treatment (Benedict et al., 2008). Conversely, in obese individuals, intranasal insulin have no effect on body weight but improved declarative memory and mood (Schneider et al., 2014). At this moment, there are ten ongoing unpublished clinical trials accessing intranasal insulin beneficial effects on AD patients (NCT00581867, NCT02010476, NCT01436045, NCT01636596, NCT03038282, NCT01767909, NCT00018382, NCT01595646, NCT01547169, NCT02462161).
Besides insulin, GLP1-R agonists are also an alternative to be invested on clinical trials, considering prior positive results in preclinical models (Duarte et al., 2013; Tramutola et al., 2017). One pilot study with few patients noted that subcutaneous liraglutide prevented the decline of brain glucose consumption, but had no effect on Aβ load or cognition (Gejl et al., 2016). Currently, there are two ongoing larger clinical trials accessing liraglutide neuroprotective effects in AD (NCT01469351, NCT01843075). Other anti-diabetic drugs targeting alternative pathways are also considered to address AD, such as peroxisome-proliferator activated receptor γ (PPAR γ) agonists rosiglitazone and pioglitazone (Miller et al., 2011). Pre-clinical evidence showed that rosiglitazone improved memory and decreased phosphorylated tau (Escribano et al., 2010). However, these studies are also incipient and reported no beneficial effects so far (Harrington et al., 2011; Miller et al., 2011). In the next years, the results of the ongoing larger clinical trials will provide further information about how metabolic status of AD patients interfere on treatment success and also the potential of anti-diabetic therapies in dementia, specially, insulin intranasal use as a possible AD therapy (Femminella et al., 2017).
Some puzzles of AD progression are now assembled, however, the sequence of biological alterations that elicits disease development needs clarification. One of the most accepted hypothesis to explain AD is that pathology develops as consequence of the build up of Aβ oligomers and amyloid plaques in the brain (Hardy and Higgins, 1992; Haass and Selkoe, 2007). In familial AD, many mutations are associated with Aβ processing, such as APP and PSEN, supporting this idea. However, it is still elusive the mechanism of Aβ oligomers accumulation and how other mutations apparently non-related to Aβ increase sporadic AD risk (Rao et al., 2014). The understanding that AD can be triggered by inflammation and insulin resistance brought a new perspective. It is possible that allostatic load, multiple environmental effects accumulated during life, accelerates AD pathogenesis. A crosstalk between brain and whole-body metabolic homeostasis is thought to be one of the drivers of sporadic AD (De Felice, 2013; Mattson and Arumugam, 2018).
Environmental factors interfere on cognitive performance even in first years of life, when traumatic brain injury, maternal separation, and psychological trauma might increase the risk of neurodegenerative disease (Barlow, 2005). This correlation is also present in brain injury suffered during adulthood (McKee and Robinson, 2014). Other psychiatric disorders, such as depression, can increase the susceptibility of AD (Ownby et al., 2006). In this context, the brain is subordinate to many types of insults and can also reflect peripheral tissue abnormal function. Sleep disordered breathing is associated with glucose intolerance and insulin resistance that may lead to T2D (Punjabi et al., 2004). Moreover, sleep deprivation also play a role in AD pathogenesis modulating Aβ accumulation (Bliwise, 2004; Kang et al., 2009) and AD-like pathology in mice (Kincheski et al., 2017).
An unhealth lifestyle accelerates the detrimental effects of aging, such as loss of endocrinological control leading to insulin resistance, declines in growth hormone, IGF-1, and sex steroids (Barzilai et al., 2012). A recent study showed that western diet (composed majority by sugar and fat) is associated with hippocampal atrophy (Jacka et al., 2015) and was reported to be associated with a significant rise of AD incidence in Japan (Grant, 2014). Smoking, lack of physical activity, and alcohol abuse are some of other unhealthy practices that increase the risk of AD (Baumgart et al., 2015).
Importantly, older women show faster deterioration of cognition than men, present higher risk for developing AD and abnormal insulin signaling (Li and Singh, 2014; Duarte et al., 2018). Gene network analysis in mice revealed that age-related transcriptional changes happen differently and earlier in female compared to male brain (Zhao et al., 2016). Furthermore, abdominal obesity is correlated with increased death risk in older women and contributes to insulin resistance (Folsom et al., 1993). Besides, other treatments can increase the risk for AD. The Woman Health Initiative observed that hormone replacement therapy combining estrogen plus progestin could upregulate several age related markers, augmenting the risks for cognitive decline, and cardiovascular disease (Writing Group for the Women's Health Initiative Investigators, 2002; Duarte et al., 2018).
In this scenario, it is essential to identify modifiable risks that reduce AD incidence and/or slow down sporadic AD progression. In accordance with the notion that metabolic changes triggers aging, it is possible that AD occurs as a consequence of metabolic dysfunction, that can be caused by cumulative lifelong impacts of lifestyle and other conditions.
The clinical/epidemiological evidence associating metabolic disorders with dementia has encouraged scientists to search for the biological mechanisms underlying this connection. The pivotal finding that insulin signaling is impaired in AD brains represents a major advance in our current understanding of AD physiopathology. Furthermore, compelling evidence demonstrate that the molecular mechanisms leading to brain insulin resistance in AD share striking similarity to those involved in peripheral insulin resistance in diabetes and obesity. Those include chronic, low-grade inflammation, TNF-α-mediated inhibition of IRS-1, and endoplasmic-reticulum stress. Importantly, these findings also led to the proposition that drugs currently used to overcome peripheral insulin resistance in diabetes may be repurposed to rescue brain insulin signaling in AD. Since insulin signaling is neurotrophic, neuroprotective and plays a key role in synaptic plasticity and cognitive processes, boosting neuronal insulin signaling may come to be a disease modifying therapeutic approach in AD. Indeed, preclinical studies and clinical trials have been performed to test the efficacy of anti-diabetic drugs, including insulin itself, in AD, with some promising results (Table 1). In addition, dissection of the actions of insulin in neuronal and glial cells is likely to foster knowledge on the roles of insulin and related signaling pathways in CNS physiology. Finally, a better comprehension of the crosstalk between peripheral energy metabolism and brain function may also help identifying therapeutic targets and modifiable risk factors for AD and other brain disorders, improving public health policies and public awareness.
LF, CF, and MV wrote the manuscript. MV and FDF planned and reviewed the final manuscript.
FDF laboratory is funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), National Institute for Translational Neuroscience (INNT, Brazil), Canadian Institutes of Health Research (CIHR) and the Weston Brain Institute. LF is a master's student fellow of CNPq, CF is a Ph.D. student fellow of CNPq, MV is a postdoctoral fellow of FAPERJ and awardee of the International Society for Neurochemistry (ISN) Career Interruption, Re-entry Grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alata, W., Ye, Y., St-Amour, I., Vandal, M., and Calon, F. (2015). Human apolipoprotein e ε4 expression impairs cerebral vascularization and blood—brain barrier function in mice. J. Cereb. Blood Flow Metab. 35, 86–94. doi: 10.1038/jcbfm.2014.172
Almeida-Suhett, C. P., Graham, A., Chen, Y., and Deuster, P. (2017). Behavioral changes in male mice fed a high-fat diet are associated with IL-1β expression in specific brain regions. Physiol. Behav. 169, 130–140. doi: 10.1016/j.physbeh.2016.11.016
Andersen, A., Lund, A., Knop, F. K., and Vilsbøll, T. (2018). Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 14, 390–403. doi: 10.1038/s41574-018-0016-2
Arnold, S. E., Arvanitakis, Z., Macauley-Rambach, S. L., Koenig, A. M., Wang, H. Y., Ahima, R. S., et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 14, 168–181. doi: 10.1038/nrneurol.2017.185
Arnold, S. E., Lucki, I., Brookshire, B. R., Carlson, G. C., Browne, C. A., Kazi, H., et al. (2014). High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 67, 79–87. doi: 10.1016/j.nbd.2014.03.011
Baker, L. D., Cross, D. J., Minoshima, S., Belongia, D., Stennis Watson, G., and Craft, S. (2011). Insulin resistance and alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 68, 51–57. doi: 10.1001/archneurol.2010.225
Barage, S. H., and Sonawane, K. D. (2015). Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer's disease. Neuropeptides 52, 1–18. doi: 10.1016/j.npep.2015.06.008
Barlow, K. M. (2005). Late neurologic and cognitive sequelae of inflicted traumatic brain injury in infancy. Pediatrics 116:e174–e185. doi: 10.1542/peds.2004-2739
Barzilai, N., Huffman, D. M., Muzumdar, R. H., and Bartke, A. (2012). The critical role of metabolic pathways in aging. Diabetes 61, 1315–1322. doi: 10.2337/db11-1300
Batista, A. F., Forny-Germano, L., Clarke, J. R., Lyra e Silva, N. M., Brito-Moreira, J., Boehnke, S. E., et al. (2018). The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer's disease. J. Pathol. 245, 85–100. doi: 10.1002/path.5056
Baumgart, M., Snyder, H. M., Carrillo, M. C., Fazio, S., Kim, H., and Johns, H. (2015). Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimer's Dement. 11, 718–726. doi: 10.1016/j.jalz.2015.05.016
Bélanger, M., Allaman, I., and Magistretti, P. J. (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. doi: 10.1016/j.cmet.2011.08.016
Benedict, C., Hallschmid, M., Schmitz, K., Schultes, B., Ratter, F., Fehm, H. L., et al. (2007). Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology 32, 239–243. doi: 10.1038/sj.npp.1301193
Benedict, C., Kern, W., Schultes, B., Born, J., and Hallschmid, M. (2008). Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 93, 1339–1344. doi: 10.1210/jc.2007-2606
Biessels, G. J., and Despa, F. (2018). Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat. Rev. Endocrinol. 14, 591–604. doi: 10.1038/s41574-018-0048-7
Bliwise, D. L. (2004). Sleep disorders in Alzheimer's disease and other dementias. Clin. Cornerstone 6, S16–S28. doi: 10.1016/S1098-3597(04)90014-2
Boles, A., Kandimalla, R., and Reddy, P. H. (2017). Dynamics of diabetes and obesity: epidemiological perspective. Biochim. Biophys. Acta Mol. Basis. Dis. 1863, 1026–1036. doi: 10.1016/j.bbadis.2017.01.016
Bomfim, T. R., Forny-Germano, L., Sathler, L. B., Brito-Moreira, J., Houzel, J. C., Decker, H., et al. (2012). An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease- associated Aβ oligomers. J. Clin Invest. 122, 1339–1353. doi: 10.1172/JCI57256DS1
Born, J., Lange, T., Kern, W., McGregor, G. P., Bickel, U., and Fehm, H. L. (2002). Sniffing neuropeptides: a transnasal approach to the human brain. Nat. Neurosci. 5, 514–516. doi: 10.1038/nn0602-849
Boyt, A. A., Taddei, T. K., Hallmayer, J., Helmerhorst, E., Gandy, S. E., Craft, S., et al. (2000). The effect of insulin and glucose on the plasma concentration of Alzheimer's amyloid precursor protein. Neuroscience 95, 727–734. doi: 10.1016/S0306-4522(99)00458-3
Brüning, J. C., Gautam, D., Burks, D. J., Gillette, J., Schubert, M., Orban, P. C., et al. (2000). Role of brain insulin receptor in control of body weight and reproduction. Science 289, 2122–2125. doi: 10.1126/science.289.5487.2122
Button, E. B., Mitchell, A. S., Domingos, M. M., Chung, J. H., Bradley, R. M., Hashemi, A., et al. (2014). Microglial cell activation increases saturated and decreases monounsaturated fatty acid content, but both lipid species are proinflammatory. Lipids 49, 305–316. doi: 10.1007/s11745-014-3882-y
Cai, W., Xue, C., Sakaguchi, M., Konishi, M., Shirazian, A., Ferris, H. A., et al. (2018). Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Invest. 128, 2914–2926. doi: 10.1172/JCI99366
Cai, Z., Liu, N., Wang, C., Qin, B., Zhou, Y., Xiao, M., et al. (2016). Role of RAGE in Alzheimer's disease. Cell Mol. Neurobiol. 36, 483–495. doi: 10.1007/s10571-015-0233-3
Carroll, R. G., Zasłona, Z., Galván-Peña, S., Koppe, E. L., Sévin, D. C., Angiari, S., et al. (2018). An unexpected link between fatty acid synthase and cholesterol synthesis in proinflammatory macrophage activation. J. Biol. Chem. 293, 5509–5521. doi: 10.1074/jbc.RA118.001921
Chen, H., Guo, S., Xia, Y., Yuan, L., Lu, M., Zhou, M., et al. (2018). The role of rho-GEF trio in regulating tooth root development through the p38 MAPK pathway. Exp. Cell Res. 372, 158–167. doi: 10.1016/j.yexcr.2018.09.022
Choei, H., Sasaki, N., Takeuchi, M., Yoshida, T., Ukai, W., Yamagishi, S. I., et al. (2004). Glyceraldehyde-derived advanced glycation end products in Alzheimer's disease. Acta Neuropathol. 108, 189–193. doi: 10.1007/s00401-004-0871-x
Clarke, J. R., Lyra e Silva, N. M., Figueiredo, C. P., Frozza, R. L., Ledo, J. H., Beckman, D., et al. (2015). Alzheimer-associated Aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 7, 190–210. doi: 10.15252/emmm.201404183
Clarke, L. E., Liddelow, S. A., Chakraborty, C., Münch, A. E., Heiman, M., and Barres, B. A. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U.S.A. 115, E1896–E1905. doi: 10.1073/pnas.1800165115
Craft, S., Asthana, S., Cook, D. G., Baker, L. D., Cherrier, M., Purganan, K., et al. (2003). Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer's disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology 28, 809–822. doi: 10.1016/S0306-4530(02)00087-2
Crane, P. K., Walker, R., Hubbard, R. A., Li, G., Nathan, D. M., Zheng, H., et al. (2013). Glucose levels and risk of dementia. N. Engl. J. Med. 369, 540–548. doi: 10.1056/NEJMoa1215740
Criscuolo, C., Fontebasso, V., Middei, S., Stazi, M., Ammassari-Teule, M., Yan, S. S., et al. (2017). Entorhinal cortex dysfunction can be rescued by inhibition of microglial RAGE in an Alzheimer's disease mouse model. Sci. Rep. 7:42370. doi: 10.1038/srep42370
De Felice, F., Vieira, N. N. M., Bomfim, T., Decker, H., Velasco, P., Lambert, M., et al. (2009). Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 1971–1976. doi: 10.1073/pnas.0901917106
De Felice, F. G. (2013). Alzheimer's disease and insulin resistance: translating basic science into clinical applications. J. Clin. Invest. 123, 531–539. doi: 10.1172/JCI64595.
Duarte, A. I., Candeias, E., Correia, S. C., Santos, R. X., Carvalho, C., Cardoso, S., et al. (2013). Crosstalk between diabetes and brain: glucagon-like peptide-1 mimetics as a promising therapy against neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 1832, 527–541. doi: 10.1016/j.bbadis.2013.01.008
Duarte, A. I., Santos, M. S., Oliveira, C. R., and Moreira, P. I. (2018). Brain insulin signalling, glucose metabolism and females' reproductive aging: a dangerous triad in Alzheimer's disease. Neuropharmacology 136, 223–242. doi: 10.1016/j.neuropharm.2018.01.044
Egan, M. F., Kost, J., Tariot, P. N., Aisen, P. S., Cummings, J. L., Vellas, B., et al. (2018). Randomized trial of verubecestat for mild-to-moderate Alzheimer's disease. N. Engl. J. Med. 378, 1691–1703. doi: 10.1056/NEJMoa1706441
Escribano, L., Simón, A. M., Gimeno, E., Cuadrado-Tejedor, M., De Maturana, R. L., García-Osta, A., et al. (2010). Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 35, 1593–1604. doi: 10.1038/npp.2010.32
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci.U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
Femminella, G. D., Bencivenga, L., Petraglia, L., Visaggi, L., Gioia, L., Grieco, F. V., et al. (2017). Antidiabetic drugs in Alzheimer's disease: mechanisms of action and future perspectives. J. Diabetes Res. 2017:7420796. doi: 10.1155/2017/7420796
Fernandez, A. M., Hernandez-Garzón, E., Perez-Domper, P., Perez-Alvarez, A., Mederos, S., Matsui, T., et al. (2017). Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes 66, 64–74. doi: 10.2337/db16-0861
Fernandez, A. M., and Torres-Alemán, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Folsom, A. R., Kaye, S. A., Sellers, T. A., Hong, C. P., Cerhan, J. R., Potter, J. D., et al. (1993). Body fat distribution and 5-year risk of death in older women. JAMA J. Am. Med. Assoc. 269, 483–487. doi: 10.1001/jama.1993.03500040049035
Forny-Germano, L., Lyra e Silva, N. M., Batista, A. F., Brito-Moreira, J., Gralle, M., Boehnke, S. E., et al. (2014). Alzheimer's disease-like pathology induced by amyloid- oligomers in nonhuman primates. J. Neurosci. 34, 13629–13643. doi: 10.1523/JNEUROSCI.1353-14.2014
García-Cáceres, C., Quarta, C., Varela, L., Gao, Y., Gruber, T., Legutko, B., et al. (2016). Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166, 867–880. doi: 10.1016/j.cell.2016.07.028
Gejl, M., Gjedde, A., Egefjord, L., Moller, A., Hansen, S. B., Vang, K., et al. (2016). In Alzheimer's disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 8:108. doi: 10.3389/fnagi.2016.00108
Graham, W. V., Bonito-Oliva, A., and Sakmar, T. P. (2017). Update on Alzheimer's disease therapy and prevention strategies. Annu. Rev. Med. 68, 413–430. doi: 10.1146/annurev-med-042915-103753
Grant, W. B. (2014). Trends in diet and Alzheimer's disease during the nutrition transition in Japan and developing countries. J. Alzheimer's Dis. 38, 611–620. doi: 10.3233/JAD-130719
Groop, L. C., Petrides, S., Bonadonna, C., Shank, M., Simonson, C., Defronzo, R. A., et al. (2018). Effect of insulin on oxidative of free fatty acid metabolism and nonoxidative in human obesity pathways. Am. J. Physiol. 263, 79–84. doi: 10.1152/ajpendo.1992.263.1.E79
Guenther, B. (2009). Obesity and free fatty acids (FFA). Endocrinol. Metab. Clin. North Am. 185, 351–356. doi: 10.1016/j.ecl.2008.06.007
Gupta, S., Knight, A. G., Gupta, S., Keller, J. N., and Bruce-Keller, A. J. (2012). Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 120, 1060–1071. doi: 10.1111/j.1471-4159.2012.07660
Haass Selkoe, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Harrington, C., Sawchak, S., Chiang, C., Davies, J., Donovan, C. M., Saunders, A., et al. (2011). Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimers disease: two phase 3 studies. Curr. Alzheimer Res. 8, 592–606. doi: 10.2174/156720511796391935
Hiltunen, M., Khandelwal, V. K. M., Yaluri, N., Tiilikainen, T., Tusa, M., Koivisto, H., et al. (2012). Contribution of genetic and dietary insulin resistance to Alzheimer phenotype in APP/PS1 transgenic mice. J. Cell Mol. Med. 16, 1206–1222. doi: 10.1111/j.1582-4934.2011.01384
Honig, L. S., Vellas, B., Woodward, M., Boada, M., Bullock, R., Borrie, M., et al. (2018). Trial of solanezumab for mild dementia due to Alzheimer's disease. N. Engl. J. Med. 378, 321–330. doi: 10.1056/NEJMoa1705971
Hotamisligil, G. S., Shargill, N., and Spiegelman, B. M. (1993). Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259, 87–91. doi: 10.1126/science.7678183
Ikram, M. A., Brusselle, G. G. O., Murad, S. D., van Duijn, C. M., Franco, O. H., Goedegebure, A., et al. (2017). The rotterdam study: 2018 update on objectives, design and main results. Eur. J. Epidemiol. 32, 807–850. doi: 10.1007/s10654-017-0321-4
Jacka, F. N., Cherbuin, N., Anstey, K. J., Sachdev, P., and Butterworth, P. (2015). Western diet is associated with a smaller hippocampus: a longitudinal investigation. BMC Med. 13:215. doi: 10.1186/s12916-015-0461-x
Janson, J., Laedtke, T., Parisi, J. E., O'Brien, P., Petersen, R. C., and Butler, P. C. (2004). Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53, 474–481. doi: 10.2337/diabetes.53.2.474
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., Broihier, M. L., et al. (2015). TREM2 deficiency eliminates TREM2 + inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295. doi: 10.1084/jem.20142322
Jha, M. K., Lee, W. H., and Suk, K. (2016). Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem. Pharmacol. 103, 1–16. doi: 10.1016/j.bcp.2015.11.003
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kapogiannis, D., Boxer, A., Schwartz, J. B., Abner, E. L., Biragyn, A., Masharani, U., et al. (2015). Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer's disease. FASEB J. 29, 589–596. doi: 10.1096/fj.14-262048
Kennedy, M. E., Stamford, A. W., Chen, X., Cox, K., Cumming, J. N., Dockendorf, M. F., et al. (2016). The BACE1 inhibitor verubecestat (MK-8931) reduces CNS b-Amyloid in animal models and in Alzheimer's disease patients. Sci. Transl. Med. 8, 1–14. doi: 10.1126/scitranslmed.aad9704
Kiernan, E. A., Smith, S. M. C., Mitchell, G. S., and Watters, J. J. (2016). Mechanisms of microglial activation in models of inflammation and hypoxia: implications for chronic intermittent hypoxia. J. Physiol. 594, 1563–1577. doi: 10.1113/JP271502
Kincheski, G. C., Valentim, I. S., Clarke, J. R., Cozachenco, D., Castelo-Branco, M. T. L., Ramos-Lobo, A. M., et al. (2017). Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-β oligomers in mice. Brain Behav. Immun. 64, 140–151. doi: 10.1016/j.bbi.2017.04.007
Kivipelto, M., Ngandu, T., Fratiglioni, L., Viitanen, M., Kåreholt, I., Winblad, B., et al. (2005). Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 62, 1556–1560. doi: 10.1001/archneur.62.10.1556
Kleinridders, A., Ferris, H. A., Cai, W., and Kahn, C. R. (2014). Insulin action in brain regulates systemic metabolism and brain function. Diabetes 63, 2232–2243. doi: 10.2337/db14-0568
Knafo, S., Sánchez-Puelles, C., Palomer, E., Delgado, I., Draffin, J. E., Mingo, J., et al. (2016). PTEN recruitment controls synaptic and cognitive function in Alzheimer's models. Nat. Neurosci. 19, 443–453. doi: 10.1038/nn.4225
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566.e9–581.e9. doi: 10.1016/j.immuni.2017.08.008
Kullmann, S., Heni, M., Hallschmid, M., Fritsche, A., Preissl, H., and Häring, H. U. (2016). Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol. Rev. 96, 1169–1209. doi: 10.1152/physrev.00032.2015
Kurochkin, I. V., Guarnera, E., and Berezovsky, I. N. (2018). Insulin-degrading enzyme in the fight against Alzheimer's disease. Trends Pharmacol. Sci. 39, 49–58. doi: 10.1016/j.tips.2017.10.008
Lasser, R., Ostrowitzki, S., Scheltens, P., Boada, M., Dubois, B., Dorflinger, E., et al. (2015). Efficacy and safety of gantenerumab in prodromal Alzheimer's disease: results from scarlet road—a global, multicenter trial. Alzheimer's Dement. 11, P331–P332. doi: 10.1016/j.jalz.2015.08.153
Li, R., and Singh, M. (2014). Sex differences in cognitive impairment and Alzheimer's disease. Front. Neuroendocrinol. 35, 385–403. doi: 10.1016/j.yfrne.2014.01.002
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Logan, S., Pharaoh, G. A., Marlin, M. C., Masser, D. R., Matsuzaki, S., Wronowski, B., et al. (2018). Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-β uptake in astrocytes. Mol. Metab. 9, 141–155. doi: 10.1016/j.molmet.2018.01.013
Lopez-Lopez, C., LeRoith, D., and Torres-Aleman, I. (2004). Insulin-like growth factor I is required for vessel remodeling in the adult brain. Proc. Natl. Acad. Sci. U.S.A. 101, 9833–9838. doi: 10.1073/pnas.0400337101
Lourenco, M. V., Clarke, J. R., Frozza, R. L., Bomfim, T. R., Forny-Germano, L., Batista, A. F., et al. (2013). TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer's β-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843. doi: 10.1016/j.cmet.2013.11.002
Lu, J., Wu, D., Zheng, Y. L., Hu, B., Cheng, W., Zhang, Z., et al. (2011). Ursolic acid improves high fat diet-induced cognitive impairments by blocking endoplasmic reticulum stress and IκB kinase β/nuclear factor-κB-mediated inflammatory pathways in mice. Brain Behav. Immun. 25, 1658–1667. doi: 10.1016/j.bbi.2011.06.009
Ma, Q. L., Yang, F., Rosario, E. R., Ubeda, O. J., Beech, W., Gant, D. J., et al. (2009). Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J. Neurosci. 29, 9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009
Malek-Ahmadi, M., Beach, T., Obradov, A., Sue, L., Belden, C., Davis, K., et al. (2013). Increased Alzheimer's disease neuropathology is associated with type 2 diabetes and ApoE ε.4 carrier status. Curr. Alzheimer Res. 10, 654–659. doi: 10.2174/15672050113109990006
Matsuzaki, T., Sasaki, K., Tanizaki, Y., Hata, J., Fujimi, K., Matsui, Y., et al. (2010). Insulin resistance is associated with the pathology of Alzheimer disease: the hisayama study. Neurology 75, 764–770. doi: 10.1212/WNL.0b013e3181eee25f
Mattson, M. P., and Arumugam, T. V. (2018). Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 27, 1176–1199. doi: 10.1016/j.cmet.2018.05.011
McClean, P. L., and Hölscher, C. (2014). Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer's disease. Neuropharmacology 76, 57–67. doi: 10.1016/j.neuropharm.2013.08.005
McClean, P. L., Parthsarathy, V., Faivre, E., and Hölscher, C. (2011). The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J. Neurosci. 31, 6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011
McKee Robinson, A. C., and Robinson, M. E. (2014). Military-related traumatic brain injury and neurodegeneration. Alzheimer's Dement. 10, 242–253. doi: 10.1111/j.1743-6109.2008.01122
Miller, B. W., Willett, K. C., and Desilets, A. R. (2011). Rosiglitazone and pioglitazone for the treatment of Alzheimer's disease. Ann. Pharmacother. 45, 1416–1424. doi: 10.1345/aph.1Q238
Modrego, P. J. (2010). Depression in Alzheimer's disease. Pathophysiol. Diag. Treat. J. Alzheimer's Dis. 21, 1077–1087. doi: 10.3233/JAD-2010-100153
Moser Pike, V. A., and Pike, C. J. (2017). Obesity accelerates Alzheimer-related pathology in APOE4 but not APOE3 mice. Eneuro. 4:ENEURO.0077–17.2017. doi: 10.1523/ENEURO.0077-17.2017
Mraz, M., and Haluzik, M. (2014). The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 222, 113–127. doi: 10.1530/JOE-14-0283
Mullins, R. J., Mustapic, M., Goetz, E. J., and Kapogiannis, D. (2017). Exosomal biomarkers of brain insulin resistance associated with regional atrophy in Alzheimer's disease. Hum. Brain Mapp. 38, 1933–1940. doi: 10.1002/hbm.23494
Ownby, R. L., Crocco, E., Acevedo, A., John, V., and Loewenstein, D. (2006). Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch. Gen. Psychiatry 63, 530–538. doi: 10.1001/archpsyc.63.5.530
Park, C. R., Seeley, R. J., Craft, S., and Woods, S. C. (2000). Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol. Behav. 68, 509–514. doi: 10.1016/S0031-9384(99)00220-6
Park, R., Kook, S. Y., Park, J. C., and Mook-Jung, I. (2014). Aβ1-42reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-κB signaling. Cell Death Dis. 5:e1299. doi: 10.1038/cddis.2014.258
Pedersen, M., Bruunsgaard, H., Weis, N., Hendel, H. W., Andreassen, B. U., Eldrup, E., et al. (2003). Circulating levels of TNF-alpha and IL-6-relation to truncal fat mass and muscle mass in healthy elderly individuals and in patients with type-2 diabetes. Mech. Ageing Dev. 124, 495–502. doi: 10.1016/S0047-6374(03)00027-7
Peila, R., Rodriguez, B. L., and Launer, L. J. (2002). Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the honolulu-asia aging study. Diabetes 51, 1256–1262. doi: 10.2337/diabetes.51.4.1256
Perry, T. (2002). Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J. Pharmacol. Exp. Ther. 302, 881–888. doi: 10.1124/jpet.102.037481
Pitt, J., Wilcox, K. C., Tortelli, V., Diniz, L. P., Oliveira, M. S., Dobbins, C., et al. (2017). Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Aβ oligomers. Mol. Biol. Cell. 28, 2623–2636. doi: 10.1091/mbc.E17-06-0416
Punjabi, N. M., Shahar, E., Redline, S., Gottlieb, D. J., Givelber, R., and Resnick, H. E. (2004). Sleep-disordered breathing, glucose intolerance, and insulin resistance: the sleep heart health study. Am. J. Epidemiol. 160, 521–530. doi: 10.1093/aje/kwh261
Rao, A. T., Degnan, A. J., and Levy, L. M. (2014). Genetics of Alzheimer disease. Am. J. Neuroradiol. 35, 457–458. doi: 10.3174/ajnr.A3545
Rapoport, S. I. (2001). In vivo fatty acid incorporation into brain phospholipids in relation to plasma availability, signal transduction and membrane remodeling. J. Mol. Neurosci. 16, 243–261. doi: 10.1385/JMN:16,2-3,243
Razay, G., Vreugdenhil, A., and Wilcock, G. (2006). Obesity, abdominal obesity and Alzheimer disease. Dement. Geriatr. Cogn. Disord. 22, 173–176. doi: 10.1159/000094586
Reger, M. A., Watson, G. S., Frey, W. H., Baker, L. D., Cholerton, B., Keeling, M. L., et al. (2006). Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol. Aging 27, 451–458. doi: 10.1016/j.neurobiolaging.2005.03.016
Reger, M. A., Watson, G. S., Green, P. S., Baker, L. D., Cholerton, B., Fishel, M. A., et al. (2008a). Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-β in memory-impaired older adults. J. Alzheimer's Dis. 13, 323–331. doi: 10.3233/JAD-2008-13309
Reger, M. A., Watson, G. S., Green, P. S., Wilkinson, C. W., Baker, L. D., Cholerton, B., et al. (2008b). Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 70, 440–448. doi: 10.1212/01.WNL.0000265401.62434.36
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2005). Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc. Natl. Acad. Sci. U.S.A. 102, 8299–8302. doi: 10.1073/pnas.0500579102
Rui, L., Aguirre, V., Kim, J. K., Shulman, G. I., Lee, A., Corbould, A., et al. (2001). Insulin/IGF-1 and TNF-α stimulate phosphorylation of IRS-1 at inhibitory Ser307via distinct pathways. J. Clin. Invest. 107, 181–189. doi: 10.1172/JCI10934
Salloway, S., Sperling, R., Fox, N. C., Blennow, K., Klunk, W., Raskind, M., et al. (2014). Two Phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N. Engl. J. Med. 370, 322–333. doi: 10.1056/NEJMoa1304839
Schmid, V., Kullmann, S., Gfrörer, W., Hund, V., Hallschmid, M., Lipp, H. P., et al. (2018). Safety of intranasal human insulin: a review. Diabetes Obes. Metab. 20, 1563–1577. doi: 10.1111/dom.13279
Schmitz, L., Kuglin, R., Bae-Gartz, I., Janoschek, R., Appel, S., Mesaros, A., et al. (2018). Hippocampal insulin resistance links maternal obesity with impaired neuronal plasticity in adult offspring. Psychoneuroendocrinology 89, 46–52. doi: 10.1016/j.psyneuen.2017.12.023
Schneider, L. S., Mangialasche, F., Andreasen, N., Feldman, H., Giacobini, E., Jones, R., et al. (2014). Clinical trials and late-stage drug development for Alzheimer's disease: an appraisal from 1984 to 2014. J. Intern. Med. 275, 251–283. doi: 10.1111/joim.12191
Searfoss, G. H., Jordan, W. H., Calligaro, D. O., Galbreath, E. J., Schirtzinger, L. M., Berridge, B. R., et al. (2003). Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional γ-secretase inhibitor. J. Biol. Chem. 278, 46107–46116. doi: 10.1074/jbc.M307757200
Sebastião, I., Candeias, E., Santos, M. S., de Oliveira, C. R., Moreira, P. I., and Duarte, A. I. (2014). Insulin as a bridge between type 2 diabetes and alzheimer disease - how anti-diabetics could be a solution for dementia. Front. Endocrinol. 5:110. doi: 10.3389/fendo.2014.00110
Shu, C. J., Benoist, C., and Mathis, D. (2012). The immune system's involvement in obesity-driven type 2 diabetes. Semin. Immunol. 24, 436–442. doi: 10.1016/j.smim.2012.12.001
Shuvaev, V. V., Laffont, I., Serot, J., and Fujii, J. (2001). Increased protein glycation in cerebrospinal fluid of Alzheimer' s disease. Electrophoresis 22, 397–402. doi: 10.1016/S0197-4580(00)00253-0
Spetter, M. S., and Hallschmid, M. (2015). Intranasal neuropeptide administration to target the human brain in health and disease. Mol. Pharm. 12, 2767–2780. doi: 10.1021/acs.molpharmaceut.5b00047
Spielman, L. J., Bahniwal, M., Little, J. P., Walker, D. G., and Klegeris, A. (2015). Insulin modulates in vitro secretion of cytokines and cytotoxins by human glial cells. Curr. Alzheimer Res. 12, 684–693. doi: 10.2174/1567205012666150710104428
Starks, E. J., Patrick O'Grady, J., Hoscheidt, S. M., Racine, A. M., Carlsson, C. M., Zetterberg, H., et al. (2015). Insulin resistance is associated with higher cerebrospinal fluid tau levels in asymptomatic APOE-4 carriers. J. Alzheimers Dis. 46, 525–533. doi: 10.3233/JAD-150072
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease - Is this type 3 diabetes? J. Alzheimer's Dis. 7, 63–80. doi: 10.3233/JAD-2005-7107
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993a). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981. doi: 10.1073/pnas.90.5.1977
Strittmatter, W. J., Weisgraber, K. H., Huang, D. Y., Dong, L. M., Salvesen, G. S., Pericak-Vance, M., et al. (1993b). Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 8098–8102. doi: 10.1073/pnas.90.17.8098
Sun, S., Ji, Y., Kersten, S., and Qi, L. (2012). Mechanisms of inflammatory responses in obese adipose tissue. Annu. Rev. Nutr. 32, 261–286. doi: 10.1146/annurev-nutr-071811-150623
Tai, J., Liu, W., Li, Y., Li, L., and Hölscher, C. (2018). Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer's disease. Brain Res. 1678, 64–74. doi: 10.1016/j.brainres.2017.10.012
Takeuchi, M., Sato, T., Takino, J., Kobayashi, Y., Furuno, S., and Kikuchi, S. (2007). Diagnostic utility of serum or cerebrospinal fluid levels of toxic advanced glycation end-products (TAGE) in early detection of Alzheimer's disease. Med. Hypotheses 69, 1358–1366. doi: 10.1016/j.mehy.2006.12.017
Talbot, K., Wang, H., Kazi, H., Han, L., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer' s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/JCI59903DS1
Townsend, M., Mehta, T., and Selkoe, D. J. (2007). Soluble Aβ inhibits specific signal transduction cascades common to the insulin receptor pathway. J. Biol. Chem. 282, 33305–33312. doi: 10.1074/jbc.M610390200
Tramutola, A., Arena, A., Cini, C., Butterfield, D. A., and Barone, E. (2017). Modulation of GLP-1 signaling as a novel therapeutic approach in the treatment of Alzheimer's disease pathology. Expert Rev. Neurother. 17, 59–75. doi: 10.1080/14737175.2017.1246183
Uysal, K. T., Wiesbrock, S. M., Marino, M. W., and Hotamisligil, G. S. (1997). Protection from obesity-induced insulin resistance in mice lacking TNF- α function. Nature 389, 610–614. doi: 10.1038/39335
Vassar, R. (2014). BACE1 inhibitor drugs in clinical trials for Alzheimer's disease. Alzheimers Res. Ther. 6, 1–14. doi: 10.1186/s13195-014-0089-7
Vieira, M. N. N., Lima-Filho, R. A. S., and De Felice, F. G. (2017). Connecting Alzheimer's disease to diabetes: underlying mechanisms and potential therapeutic targets. Neuropharmacology 136, 160–171. doi: 10.1016/j.neuropharm.2017.11.014
Walter, H. J., Berry, M., Hill, D. J., and Logan, A. (1997). Spatial and temporal changes in the insulin-like growth factor (IGF) axis indicate autocrine/paracrine actions of IGF-I within wounds of the rat brain. Endocrinology 138, 3024–3034. doi: 10.1210/endo.138.7.5284
Wan, Q., Xiong, Z. G., Man, H. Y., Ackerley, C. A., Braunton, J., Lu, W. Y., et al. (1997). Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature 388, 686–690. doi: 10.1038/41792
Wan, W., Cao, L., Liu, L., Zhang, C., Kalionis, B., Tai, X., et al. (2015). Aβ1-42oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J. Neurochem. 134, 382–393. doi: 10.1111/jnc.13122
Wan, W., Chen, H., and Li, Y. (2014). The potential mechanisms of Aβ-receptor for advanced glycation end-products interaction disrupting tight junctions of the blood-brain barrier in Alzheimer's disease. Int. J. Neurosci. 124, 75–81. doi: 10.3109/00207454.2013.825258
Wang, M., Chen, F., Wang, J., Zeng, Z., Yang, Q., and Shao, S. (2018). Th17 and Treg lymphocytes in obesity and Type 2 diabetic patients. Clin. Immunol. 197, 77–85. doi: 10.1016/j.clim.2018.09.005
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wang, Z., Liu, D., Wang, F., Liu, S., Zhao, S., Ling, E. A., et al. (2012). Saturated fatty acids activate microglia via Toll-like receptor 4/NF-κB signalling. Br. J. Nutr. 107, 229–241. doi: 10.1017/S0007114511002868
Weisberg, S. P., McCann, D., Desai, M., Rosenbaum, M., Leibel, R. L., and Ferrante, A. W. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808. doi: 10.1172/JCI200319246
Wellen Hotamisligil, K. E., and Hotamisligil, G. S. (2005). Inflammation, stress, and diabetes. J. Clin. Invest. 115, 1111–1119. doi: 10.1172/JCI200525102
Whitmer, R., Gunderson, E., Quesenberry, C., Zhou, J., and Yaffe, K. (2007). Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr. Alzheimer Res. 4, 103–109. doi: 10.2174/156720507780362047
Willette, A. A., Modanlo, N., and Kapogiannis, D. (2015). Insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease. Diabetes 64, 1933–1940. doi: 10.2337/db14-1507
Wong, G. T., Manfra, D., Poulet, F. M., Zhang, Q., Josien, H., Bara, T., et al. (2004). Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits γ-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 279, 12876–12882. doi: 10.1074/jbc.M311652200
Writing Group for the Women's Health Initiative Investigators. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women's health initiative randomized controlled trial. JAMA J. Am. Med. Assoc. 288, 321–333. doi: 10.1001/jama.288.3.321
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., et al. (1996). RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 382, 685–691. doi: 10.1038/382685a0
Yang, J., Park, Y., Zhang, H., Xu, X., Laine, G. A, Dellsperger, K. C., et al. (2009). Feed-forward signaling of TNF-alpha and NF-kappaB via IKK-beta pathway contributes to insulin resistance and coronary arteriolar dysfunction in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 296, H1850–H1858. doi: 10.1152/ajpheart.01199.2008
Yarchoan, M., Toledo, J. B., Lee, E. B., Arvanitakis, Z., Kazi, H., Han, L. Y., et al. (2014). Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer's disease and tauopathies. Acta Neuropathol. 128, 679–689. doi: 10.1007/s00401-014-1328-5
Yun, S. P., Kam, T., Panicker, N., Kim, S., Oh, Y., Park, J., et al. (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson' s disease. Nat. Med. 24, 931–938. doi: 10.1038/s41591-018-0051-5
Zarrouk, A., Debbabi, M., Bezine, M., Karym, E. M., Badreddine, A., Rouaud, O., et al. (2017). Lipid biomarkers in Alzheimer's disease. Curr. Alzheimer Res. 14, 303–312. doi: 10.2174/1567205014666170505101426
Zhao, L., Mao, Z., Woody, S. K., and Brinton, R. D. (2016). Sex differences in metabolic aging of the brain: insights into female susceptibility to Alzheimer's disease. Neurobiol. Aging 42, 69–79. doi: 10.1515/jci-2013-0007
Keywords: insulin signaling, insulin resistance, Alzheimer's disease, obesity, type 2 diabetes, metabolic dysregulation, blood-brain barrier
Citation: Ferreira LSS, Fernandes CS, Vieira MNN and De Felice FG (2018) Insulin Resistance in Alzheimer's Disease. Front. Neurosci. 12:830. doi: 10.3389/fnins.2018.00830
Received: 31 July 2018; Accepted: 23 October 2018;
Published: 13 November 2018.
Edited by:
Mohammed Taouis, UMR9197 Institut des Neurosciences Paris Saclay (Neuro-PSI), FranceReviewed by:
Nils Lambrecht, University of California, Irvine, United StatesCopyright © 2018 Ferreira, Fernandes, Vieira and De Felice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo N. N. Vieira, bW51bmVzQGJpb3FtZWQudWZyai5icg==
Fernanda G. De Felice, ZmVsaWNlQGJpb3FtZWQudWZyai5icg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.