 Jie Zhang1,2
Jie Zhang1,2 Haoliang Huang
Haoliang Huang Yang Hu
Yang Hu- 1Department of Ophthalmology, Stanford University School of Medicine, Palo Alto, CA, United States
- 2Department of Ophthalmology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
In the nearly 10 years since PTEN was identified as a prominent intrinsic inhibitor of CNS axon regeneration, the PTEN negatively regulated PI3K-AKT-mTOR pathway has been intensively explored in diverse models of axon injury and diseases and its mechanism for axon regeneration is becoming clearer. It is therefore timely to summarize current knowledge and discuss future directions of translational regenerative research for neural injury and neurodegenerative diseases. Using mouse optic nerve crush as an in vivo retinal ganglion cell axon injury model, we have conducted an extensive molecular dissection of the PI3K-AKT pathway to illuminate the cross-regulating mechanisms in axon regeneration. AKT is the nodal point that coordinates both positive and negative signals to regulate adult CNS axon regeneration through two parallel pathways, activating mTORC1 and inhibiting GSK3ββ. Activation of mTORC1 or its effector S6K1 alone can only slightly promote axon regeneration, whereas blocking mTORC1 significantly prevent axon regeneration, suggesting the necessary role of mTORC1 in axon regeneration. However, mTORC1/S6K1-mediated feedback inhibition prevents potent AKT activation, which suggests a key permissive signal from an unidentified AKT-independent pathway is required for stimulating the neuron-intrinsic growth machinery. Future studies into this complex neuron-intrinsic balancing mechanism involving necessary and permissive signals for axon regeneration is likely to lead eventually to safe and effective regenerative strategies for CNS repair.
Introduction
Axon injury is a frequent consequence of trauma and a common early feature of CNS degenerative diseases causing life-long neurological deficits. Injuries of CNS axons often result in loss of vital functions because CNS axons fail to regenerate in adult mammals (Schwab and Bartholdi, 1996; Goldberg et al., 2002b; Fitch and Silver, 2008). Both the diminished intrinsic regenerative capacity of mature neurons (Park et al., 2010) and the inhibitory environment of the adult CNS (Yiu and He, 2006) contribute to the growth failure. Neutralizing extracellular inhibitory molecules genetically or pharmacologically yields only limited regeneration and functional recovery (Lee et al., 2010), highlighting the critical importance of neuron-intrinsic factors (Benowitz et al., 2017). To explore the intrinsic regenerative signaling molecules, mouse retinal ganglion cell (RGC) and optic nerve (ON) provide a valuable in vivo neural injury system that is relatively simple but robustly replicates CNS traumatic injury and permits straightforward interpretation (Figure 1).
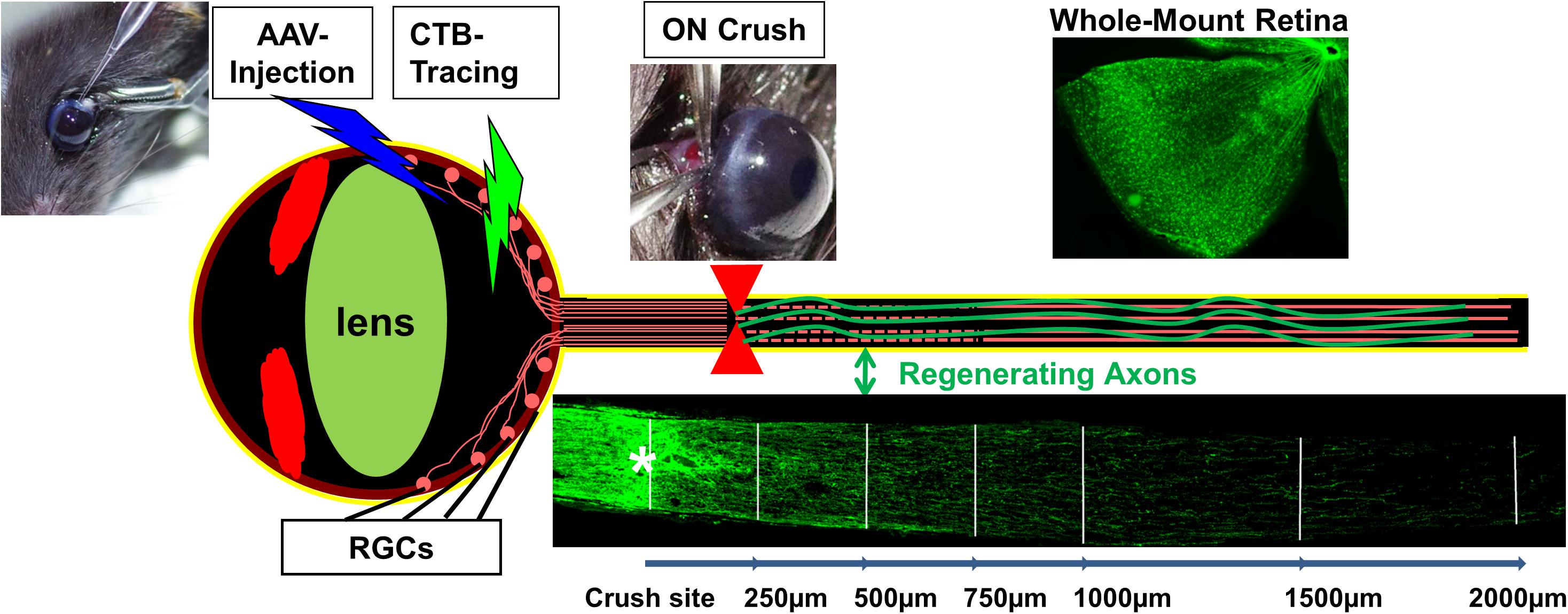
FIGURE 1. RGC/ON crush provides a straightforward in vivo CNS axon injury/regeneration model with clear readout, easy access and amenable to genetic manipulation. Mouse eye can be injected with AAV for genetic manipulation of RGCs and fluorescence-labeled CTB for regenerating axon tracing. Whole-mount retina is for RGC detection and ON longitudinal cryostat section is for visualizing regenerating axons.
Retinal ganglion cells are the only projection neurons in retina to relay visual information from retina to brain. The ON is formed by the projection axons sent exclusively from RGCs; it has the simplicity of an unidirectional axon pathway, which ensures that any nerve fibers passing through the complete crush site are regenerated and do not represent spared axons that underwent collateral sprouting. Adeno-associated viruses (AAV) can be injected directly into the vitreous chamber of the eye to express transgenes specifically and efficiently in adult RGCs. This spatially and temporally controlled genetic manipulation allows us to overcome developmental issues associated with germ line manipulation and to test interventions that can potentially be translated to therapies (Hu, 2015). Exploiting the anatomical and technical advantages of the RGC/ON crush model, multiple signal transduction pathways and transcriptional factors have been linked with CNS axon regeneration (Benowitz et al., 2017; Mahar and Cavalli, 2018). Here we focused on the PTEN/mTOR pathway that we and others have conducted an extensive molecular dissection of the cross-regulating mechanisms in axon regeneration that involve the downstream effectors of PTEN and PI3K to understand the intrinsic mechanisms of CNS axon regeneration (Park et al., 2008; Yang et al., 2014; Guo et al., 2016; Miao et al., 2016; Al-Ali et al., 2017).
PTEN Deletion Promotes Significant CNS Axon Regeneration
The initial efforts to understand the intrinsic mechanisms of regenerative failure have led us to postulate that CNS neurons tightly regulate the evolutionarily conserved molecular pathways that control cell growth (Weinberg, 2007) to prevent overgrowth when development is complete. We subsequently used the mouse ON crush model to screen multiple tumor suppressor genes and discovered that deletion of phosphatase and tensin homolog (PTEN), but not Rb (retinoblastoma), P53, Smad4, or LKB1 (liver kinase B1), promotes significant ON regeneration and RGC survival (Park et al., 2008). PTEN, a lipid phosphatase, is a major negative regulator of the phosphatidylinositol 3-kinase (PI3K)-mammalian target of rapamycin complex 1 (mTORC1) pathway (Figure 2). Similar axon regeneration phenotypes after PTEN deletion have been reported for mouse cortical motor neurons (Liu et al., 2010; Jin et al., 2015), drosophila sensory neurons (Song Y. et al., 2012) and Caenorhabditis elegans motor neurons (Byrne et al., 2014), presumably through activating PI3K-mTORC1-controlled cell growth. Direct activation of mTORC1 also promotes axon regeneration in dopaminergic neurons (Kim S.R. et al., 2011) and RGCs (Duan et al., 2015; Bei et al., 2016; Lim et al., 2016) and peripheral nerves (Abe et al., 2010), providing further support for the critical role of PI3K-mTORC1 in axon regeneration. However, deregulated PTEN/mTOR activities have been implicated in various disorders including metabolic diseases, tumor formation and even senescence (Zoncu et al., 2011). Presumably, uncontrolled or non-specific activation of mTOR and protein synthesis may result in severe negative consequences, such as tumor formation or cognitive impairment. However, not all tumor suppressor genes involved in axon regeneration in our initial screen (Park et al., 2008), indicating the unique role of PTEN/mTOR in determining the neuronal intrinsic regenerative ability. It is scientifically intriguing and clinically important to carry out a molecular dissection of the PTEN/mTOR pathway to acquire unambiguous understanding of the roles of their downstream signaling molecules in axon regeneration, in hope of differentiating to their tumorigenesis roles.
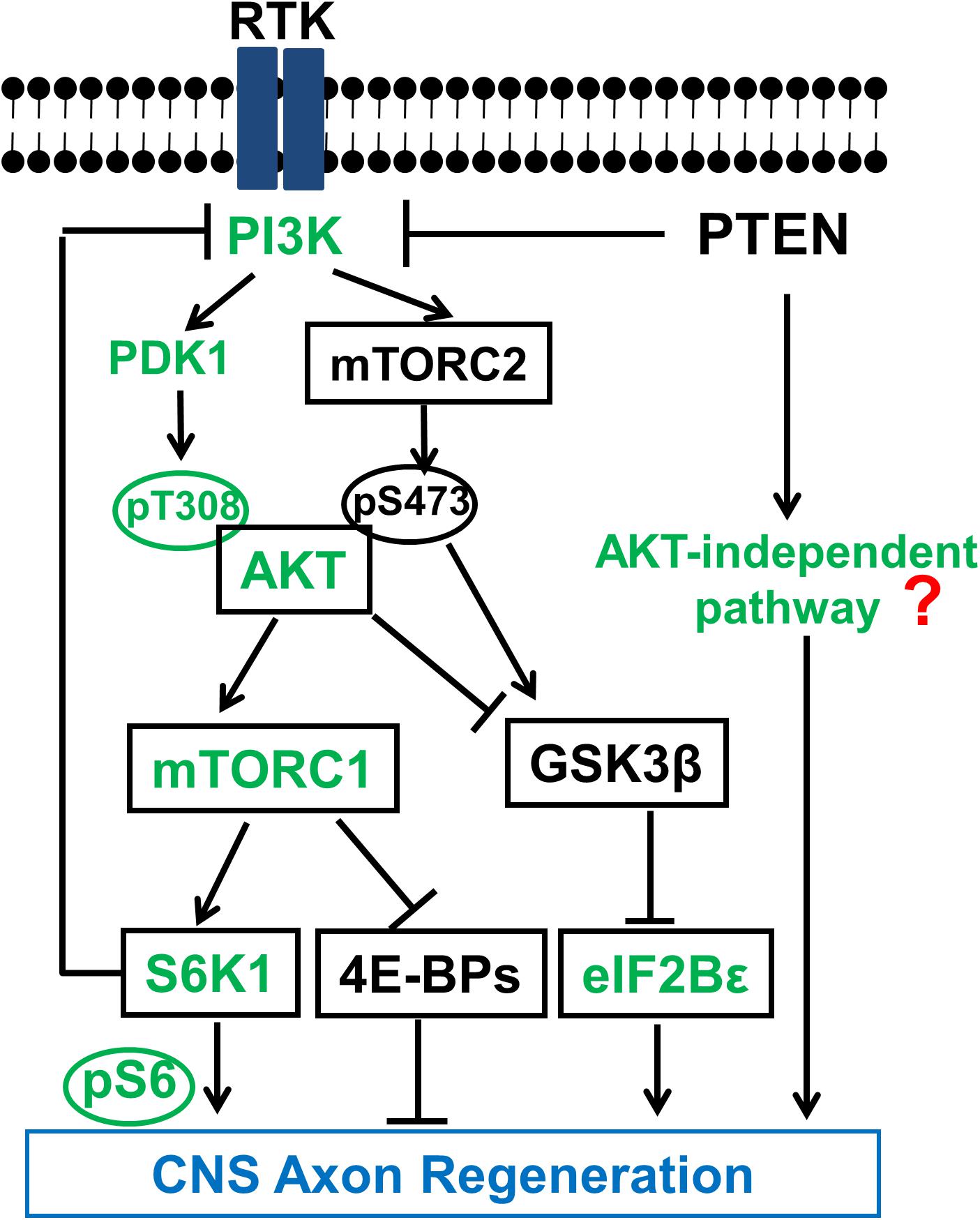
FIGURE 2. Schematic illustration of the PTEN regulated PI3K-AKT-mTOR signaling pathways in CNS axon regeneration. AKT is positively regulated by the PI3K/PDK1 pathway through phosphorylation of T308, and negatively regulated by the PI3K/mTORC2 pathway through phosphorylation of S473, through at least partially regulation of GSK3β phosphorylation and inhibition. Both AKT downstream effectors, activation of mTORC1 and phosphorylation/inhibition of GSK3β, synergistically promote axon regeneration; inhibition of GSK3β alone is also sufficient for axon regeneration, at least partially through eIF2B𝜀. The activation of mTORC1 and its substrates 4E-BP and S6K is necessary but not sufficient for potent axon regeneration. The question mark represents unknown effectors downstream of PTEN that are AKT-independent and sufficient to initiate CNS axon regeneration, and which potentially interact with the translational targets of mTORC1 to promote potent axon regeneration. Green color-coated molecules are pro-axon regeneration and dark color-coated molecules are anti-axon regeneration.
The Necessary Role of mTORC1 in CNS Axon Regeneration
PI3K is a lipid kinase which can be activated by growth factors, such as insulin and insulin-like growth factor-1 (IGF1), through receptor tyrosine kinase (RTK) (Figure 2). PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to produce phosphatidylinositol (3,4,5)-triphosphate (PIP3) in the lipid membrane. PIP3 in turn recruits AKT to the membrane to be phosphorylated at T308 and activated by phosphoinositide-dependent kinase-1 (PDK1) (Manning and Cantley, 2007). One of the multiple AKT downstream effectors is the complex formed by tuberous sclerosis 1 and 2 (TSC1/TSC2) heterodimer, the negative regulator of mTORC1. AKT activation removes the inhibition of TSC and activates mTORC1. PTEN converts PIP3 to PIP2 and thus inhibits the activation of AKT and its downstream effectors. PTEN deletion therefore results in constitutive activation of the PI3K-AKT-mTORC1 pathway, suggesting an important role of mTORC1 in the intrinsic regenerative ability of injured adult CNS neurons. Consistently, rapamycin, an inhibitor of mTORC1, blocked PTEN knockout (KO)-induced ON regeneration (Park et al., 2008), indicating mTORC1 activation is required for axon regeneration.
The functional complex mTORC1 regulates cell growth, proliferation, metabolism, motility and survival (Ma and Blenis, 2009; Laplante and Sabatini, 2012) and its downstream effectors are potential targets for promoting axon regeneration and functional recovery after CNS injury. Unfortunately, the clinical usefulness of mTORC1 activation is limited by the threat of deleterious side effects such as malignancy and cognitive deficits due to uncontrolled protein synthesis and cell proliferation (Laplante and Sabatini, 2012; Song M.S. et al., 2012). We studied the two best-characterized downstream signaling molecules of mTORC1, ribosomal protein S6 kinase (S6K) and eukaryotic translation initiation factor 4E (eIF4E)-binding protein (4E-BP) (Hay and Sonenberg, 2004) in ON regeneration (Yang et al., 2014). Phosphorylation of 4E-BP by mTORC1 releases its binding and inhibition from eIF4E, thus to initiate cap-dependent translation. Through different mechanisms, S6K also promotes protein and lipid synthesis (Hannan et al., 2003; Duvel et al., 2010). Although they are both involved in protein synthesis, previous studies suggested that S6K regulates cell size but not cell division (Ohanna et al., 2005), whereas 4E-BP controls cell proliferation but not cell size (Dowling et al., 2010). In RGC neurons, we found that over-expression of the constitutively active mutant of S6K1 significantly increases RGC cell size after ON crush but only promote axon regeneration to a small degree (Yang et al., 2014). Together with the similar effect of TSC deletion, which activates mTORC1 to a greater extent than PTEN deletion but results in very little axon regeneration (Park et al., 2008), we conclude that mTORC1 activation itself is necessary for axon regeneration but has only minimal effect on initiating axon regeneration. Another evidence to support the necessary role of mTORC1 in axon regeneration comes from 4E-BP1-4A mutant, which cannot be inhibited by mTORC1 since it cannot be phosphorylated, as it largely blocks PTEN KO-induced axon regeneration (Yang et al., 2014). However, double deletion of 4E-BP1 and 4E-BP2 in RGCs does not promote axon regeneration, indicating the necessary but insufficient role of 4E-BP inhibition by mTORC1 in axon regeneration. The possibility that other substrates of mTORC1 in addition to S6K1 and 4E-BP may contribute to axon regeneration cannot be rule out, however, we do favor the idea that the mTORC1 pathway essentially plays a necessary role in axon regeneration which requires a key permissive signal from unidentified effectors downstream of PTEN to trigger the neuron-intrinsic growth machinery.
AKT1 and AKT3 Are the Predominant Isoforms of AKT in RGCs and Display Different Effects on On Regeneration
AKT is downstream of PTEN/PI3K but upstream of mTORC1. AKT1 and AKT2 are widely expressed in almost any tissues, however, AKT3 is the predominant isoform in brain (Easton et al., 2005) and retina (Miao et al., 2016). Consistently, AKT1 deletion reduces whole body size and AKT2 deletion results in diabetes-like syndrome (Cho et al., 2001a,b). Deletion only of AKT3 reduces brain size (Easton et al., 2005), indicating a specific role of AKT3 in CNS growth control. We determined quantitatively the expression levels of the three AKT isoforms in RGCs and their distinct roles in axon regeneration, which provides strong evidence of the unique properties of AKT3 in retina: AKT1 and AKT3 are the major isoforms of AKTs in RGCs and activation of AKT3 promotes significantly greater RGC survival and ON regeneration than AKT1, presumably through its unique ability to activate mTORC1 (higher pS6) in retina (Miao et al., 2016). This is consistent with the results in brain, as AKT3 but not AKT1 deletion, decreases pS6 significantly (Easton et al., 2005). AKT3 may also selectively activate neuronal-specific signaling molecules that are currently unidentified.
AKT Coordinates Positive Signals From PI3K-PDK1 and Negative Signals From mTORC2 in Regulating mTORC1 Activation and GSK3β Phosphorylation for Axon Regeneration
In addition to T308 phosphorylation by PI3K-PDK1, AKT is also phosphorylated at S473 by mTORC2 (Hresko and Mueckler, 2005; Sarbassov et al., 2005; Guertin et al., 2006). mTORC2 is activated by PI3K in an uncharacterized way but depends on ribosome (Zinzalla et al., 2011) and its activation promotes cell survival and actin cytoskeleton dynamics (Jacinto et al., 2004). Like mTORC1, mTORC2 also plays a role in lipogenesis (Lamming and Sabatini, 2013; Yao et al., 2013). It is not clear how mTORC1 and mTORC2 interact to determine multiple downstream cellular events. AKT-pS473 enhances AKT-T308 phosphorylation (Scheid et al., 2002; Yang et al., 2002) and blocking S473 phosphorylation decreases AKT-T308 phosphorylation (Hresko and Mueckler, 2005; Sarbassov et al., 2005; Guertin et al., 2009; Yuan et al., 2012; Carson et al., 2013).
We confirmed the phosphorylation of AKT-T308 and the kinase activity of AKT are essential for axon regeneration by overexpression kinase dead mutant or T308A mutant of AKT in RGCs; whereas AKT-S473A mutant results in even more axon regeneration than wild type AKT, indicating the negative role of AKT-S473 phosphorylation in axon regeneration (Miao et al., 2016). This surprising finding implies that pT308 and pS473 of AKT may have different substrates or regulate the same substrates differentially, to allow their opposite roles in axon regeneration. Interestingly, pAKT-S473 regulates β-cell proliferation whereas pAKT-T308 controls β-cell (Hashimoto et al., 2006; Gu et al., 2011), possibly through different downstream effectors (Jacinto et al., 2004, 2006; Guertin et al., 2006; Yang et al., 2006; Gu et al., 2011). Phosphorylation of glycogen synthase kinase 3β-S9 (GSK3β-S9) by AKT inhibits GSK3β activity, which is critical for neuronal polarization, axon branching and axon growth (Kim Y.T. et al., 2011). The significantly increased pGSK3β-S9 after blocking mTORC2 or overexpression of AKT3-S472A mutant suggests that GSK3β is one of the AKT effectors that are differentially regulated by pAKT-T308 and pAKT-S473 (Miao et al., 2016). The results of our studies using GSK3β-S9A mutant and GSK3β KO mice further proved the inhibitory role of GSK3β in axon regeneration (Miao et al., 2016), thus to definitively resolve the contradictory results in the literature regarding the role of GSK3β in CNS axon regeneration (Dill et al., 2008; Abe et al., 2010; Christie et al., 2010; Saijilafu Hur et al., 2013; Gobrecht et al., 2014). More interestingly, Guo et al identified eIF2B𝜀 as an important downstream effector of GSK3β for axon regeneration (Guo et al., 2016), indicating the critical role of translation regulatory machinery and protein synthesis in axon regeneration.
mTORC2 and pAKT-S473 are necessary for PTEN deletion-induced tissue overgrowth in prostate cancer of mice (Guertin et al., 2009) and eyes of drosophila (Hietakangas and Cohen, 2007). Thus blocking mTORC2 and pAKT-S473 in PTEN KO mice may allow us to minimize their deleterious tumorigenic effect but boost PTEN/AKT’s regeneration-promoting effect. mTORC1 inhibition (deletion of RPTOR or mTOR, over-expression of dominant negative mutant S6K1-DN or 4E-BP1-4A) decreased AKT3-induced ON regeneration (Miao et al., 2016), consistent with our conclusion that mTORC1 is necessary for AKT-induced axon regeneration. In summary, mTORC1 activation and GSK3β inhibition act in parallel and synergistically downstream of AKT to promote CNS axon regeneration as we and others have shown (Guo et al., 2016; Miao et al., 2016).
Feedback Inhibition of PI3K-AKT Mediated by mTORC1-S6K1 and AKT-Independent Pathways
Proper translational control is crucial for normal cell growth. The increased protein synthesis induced by PI3K-mTORC1 activation needs to be balanced by an antagonistic mechanism. It has previously been shown that a negative feedback loop involved with S6K1 and insulin receptor substrate 1 (IRS-1) reduces activities of PI3K and its downstream effectors (Laplante and Sabatini, 2012). Indeed we detected decreased AKT phosphorylation and axon regeneration in PTEN KO mice after overexpression of S6K1 (Yang et al., 2014). Possibly through a similar mechanism, S6K inhibits axon regeneration in C. elegans (Hubert et al., 2014) and inhibition of S6K1 promotes corticospinal tract regeneration in mice (Al-Ali et al., 2017). It would not be surprising if additional balancing mechanisms can fine-tune the growth control loop of PI3K-AKT-mTORC1-PI3K. This feedback inhibition keeps AKT activation at minimum even after PTEN deletion (Laplante and Sabatini, 2012; Yang et al., 2014), suggesting AKT-independent signals downstream of PTEN for axon regeneration. PTEN deletion-induced PIP3-dependent signaling includes many AKT-independent pathways (Lien et al., 2017). In addition, PTEN can dephosphorylate focal adhesion kinase (FAK) and Shc, and deletion of PTEN activates FAK, RAS, and ERK (Godena and Ning, 2017). Furthermore, PTEN is also present in the nucleus to play a non-catalytic role in chromosomal instability and DNA repair (Shen et al., 2007; Song et al., 2011). Elucidation of these PTEN-dependent but AKT-independent pathways in axon regeneration will be an important future direction for the field.
Neuronal Survival and Axon Regeneration
Neuronal survival is an obvious prerequisite for axon regeneration. But our observation is that the increased neuron survival is not invariably linked with proportionately greater axon regeneration and this is consistent with others findings (Benowitz et al., 2015). For example, ON crush injured RGC survival can be increased significantly by inhibition of apoptosis, deleting tumor suppressor genes or by manipulating ER stress, but these manipulations do not induce more ON regeneration (Goldberg et al., 2002a; Park et al., 2008; Hu et al., 2012). On the other hand, spinal cord injury does not cause significant death of corticospinal neurons (Nielson et al., 2010, 2011), but they fail to regenerate axons (Schwab and Bartholdi, 1996; Goldberg et al., 2002b; Fitch and Silver, 2008). These results indicate that the neuronal intrinsic growth signals for axon regeneration is different to the signals for neuron survival. But no convincing evidence proves a direct causative relationship between these two events cannot totally exclude the possibility that more RGC survival contributes to more potent axon regeneration. PTEN deletion or AKT activation and their signaling effectors are normally related to both intrinsic growth control and cell survival, suggesting partially overlapping functionalities of these two events. Currently we can only allow a small percentage of surviving RGCs to regenerate their axons and different subtypes of RGCs have different regeneration abilities (Duan et al., 2015). Elucidating the mechanisms caused this difference will be a hot topic in the field to maximize RGC axon regeneration.
Axonal mRNA Translation and Axon Regeneration
The importance of localized protein synthesis in peripheral and central axon regeneration has been demonstrated in vitro and in vivo (Willis and Twiss, 2006; Jung et al., 2012; Baleriola et al., 2014; Perry and Fainzilber, 2014). And certain components of translation machinery including pS6 and 4E-BP1 have been detected in rat regenerating spinal cord (Kalinski et al., 2015). We also found that wildtype AKT and AKT-S473A mutant were localized in RGC axons whereas AKT mutants that cannot promote axon regeneration were excluded from RGC axons (Miao et al., 2016). Is the axonal AKT related with axonal mRNA translation? If so, does the regeneration phenotype caused by AKT activation rely on local protein synthesis in axons? These are very intriguing questions for future studies to investigate the significance of axonal protein synthesis and axonal signal transduction in axon regeneration.
Conclusion and Future Perspective
In summary, genetic manipulations specifically in RGCs provide a molecular dissection of the PTEN, PI3K-AKT-mTORC1/GSK3β, and PI3K-mTORC2-AKT-mTORC1/GSK3β pathways and definitively determine the linear and parallel signals that contribute to CNS axon regeneration (Figure 2). The balance between mTORC1 and mTORC2’s activities after PT3K activation converges on AKT phosphorylation of T308 and S473, which in turn control the activation of mTORC1 and inhibition of GSK3β that act in parallel and synergistically downstream of AKT to promote potent CNS axon regeneration. mTORC1/S6K also functions as feedback inhibition of PI3K signaling (Laplante and Sabatini, 2012) to keep AKT and mTORC1 on check (Yang et al., 2014), which suggests that another proactive signal originating from PTEN deletion may trigger the neuron-intrinsic growth capability (Hu, 2015). It is extremely intriguing scientifically and critical clinically to identify these permissive signals of axon regeneration and elucidate the mechanisms by which they are cross regulated with the necessary mTORC1 signals. The increased understanding of the complicated cross-regulation and feedback-control mechanisms involved in PTNE-PI3K-AKT-mTORC1/2 will certainly inspire more studies on these critical growth control mechanisms, which will eventually lead to safe and effective therapeutic strategies for CNS injury, and to isolate them from targets that mediate deleterious effects.
Author Contributions
YH and JZ contributed to bibliographical search, writing, and figure design. HH, DY, and YS wrote the paper.
Funding
This work was supported by grants from NIH NEI (EY024932 and EY023295 to YH and EY022058 and EY025295 to YS) and VA merit CX001298 to YS, and partially supported by Research for Prevention of Blindness Unrestricted grant and NEI P30-EY026877.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abe, N., Borson, S. H., Gambello, M. J., Wang, F., and Cavalli, V. (2010). Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J. Biol. Chem. 285, 28034–28043. doi: 10.1074/jbc.M110.125336
Al-Ali, H., Ding, Y., Slepak, T., Wu, W., Sun, Y., Martinez, Y., et al. (2017). The mTOR substrate S6 kinase 1 (S6K1) is a negative regulator of axon regeneration and a potential drug target for central nervous system injury. J. Neurosci. 37, 7079–7095. doi: 10.1523/JNEUROSCI.0931-17.2017
Baleriola, J., Walker, C. A., Jean, Y. Y., Crary, J. F., Troy, C. M., Nagy, P. L., et al. (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. doi: 10.1016/j.cell.2014.07.001
Bei, F., Lee, H. H., Liu, X., Gunner, G., Jin, H., Ma, L., et al. (2016). Restoration of visual function by enhancing conduction in regenerated axons. Cell 164, 219–232. doi: 10.1016/j.cell.2015.11.036
Benowitz, L. I., He, Z., and Goldberg, J. L. (2015). Reaching the brain: advances in optic nerve regeneration. Exp. Neurol. 15, 30141–30142.
Benowitz, L. I., He, Z., and Goldberg, J. L. (2017). Reaching the brain: advances in optic nerve regeneration. Exp. Neurol. 287, 365–373. doi: 10.1016/j.expneurol.2015.12.015
Byrne, A. B., Walradt, T., Gardner, K. E., Hubbert, A., Reinke, V., and Hammarlund, M. (2014). Insulin/IGF1 signaling inhibits age-dependent axon regeneration. Neuron 81, 561–573. doi: 10.1016/j.neuron.2013.11.019
Carson, R. P., Fu, C., Winzenburger, P., and Ess, K. C. (2013). Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum. Mol. Genet. 22, 140–152. doi: 10.1093/hmg/dds414
Cho, H., Mu, J., Kim, J. K., Thorvaldsen, J. L., Chu, Q., Crenshaw, E. B., et al. (2001a). Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292, 1728–1731. doi: 10.1126/science.292.5522.1728
Cho, H., Thorvaldsen, J. L., Chu, Q., Feng, F., and Birnbaum, M. J. (2001b). Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 276, 38349–38352. doi: 10.1074/jbc.C100462200
Christie, K. J., Webber, C. A., Martinez, J. A., Singh, B., and Zochodne, D. W. (2010). PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J. Neurosci. 30, 9306–9315. doi: 10.1523/JNEUROSCI.6271-09.2010
Dill, J., Wang, H., Zhou, F., and Li, S. (2008). Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J. Neurosci. 28, 8914–8928. doi: 10.1523/JNEUROSCI.1178-08.2008
Dowling, R. J., Topisirovic, I., Alain, T., Bidinosti, M., Fonseca, B. D., Petroulakis, E., et al. (2010). mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328, 1172–1176. doi: 10.1126/science.1187532
Duan, X., Qiao, M., Bei, F., Kim, I. J., He, Z., and Sanes, J. R. (2015). Subtype-specific regeneration of retinal ganglion cells following axotomy: effects of osteopontin and mTOR signaling. Neuron 85, 1244–1256. doi: 10.1016/j.neuron.2015.02.017
Duvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell. 39, 171–183. doi: 10.1016/j.molcel.2010.06.022
Easton, R. M., Cho, H., Roovers, K., Shineman, D. W., Mizrahi, M., Forman, M. S., et al. (2005). Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell. Biol. 25, 1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005
Fitch, M. T., and Silver, J. (2008). CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp. Neurol. 209, 294–301. doi: 10.1016/j.expneurol.2007.05.014
Gobrecht, P., Leibinger, M., Andreadaki, A., and Fischer, D. (2014). Sustained GSK3 activity markedly facilitates nerve regeneration. Nat. Commun. 5:4561. doi: 10.1038/ncomms5561
Godena, V. K., and Ning, K. (2017). Phosphatase and tensin homologue: a therapeutic target for SMA. Signal. Trans. Target Ther. 2:17038. doi: 10.1038/sigtrans.2017.38
Goldberg, J. L., Espinosa, J. S., Xu, Y., Davidson, N., Kovacs, G. T., and Barres, B. A. (2002a). Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron 33, 689–702. doi: 10.1016/S0896-6273(02)00602-5
Goldberg, J. L., Klassen, M. P., Hua, Y., and Barres, B. A. (2002b). Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science 296, 1860–1864. doi: 10.1126/science.1068428
Gu, Y., Lindner, J., Kumar, A., Yuan, W., and Magnuson, M. A. (2011). Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes Metab. Res. Rev. 60, 827–837.
Guertin, D. A., Stevens, D. M., Saitoh, M., Kinkel, S., Crosby, K., Sheen, J. H., et al. (2009). mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159. doi: 10.1016/j.ccr.2008.12.017
Guertin, D. A., Stevens, D. M., Thoreen, C. C., Burds, A. A., Kalaany, N. Y., Moffat, J., et al. (2006). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 11, 859–871. doi: 10.1016/j.devcel.2006.10.007
Guo, X., Snider, W. D., and Chen, B. (2016). GSK3beta regulates AKT-induced central nervous system axon regeneration via an eIF2Bepsilon-dependent, mTORC1-independent pathway. Elife 5:e11903. doi: 10.7554/eLife.11903
Hannan, K. M., Brandenburger, Y., Jenkins, A., Sharkey, K., Cavanaugh, A., Rothblum, L., et al. (2003). mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell. Biol. 23, 8862–8877. doi: 10.1128/MCB.23.23.8862-8877.2003
Hashimoto, N., Kido, Y., Uchida, T., Asahara, S., Shigeyama, Y., Matsuda, T., et al. (2006). Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat. Genet. 38, 589–593. doi: 10.1038/ng1774
Hay, N., and Sonenberg, N. (2004). Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. doi: 10.1101/gad.1212704
Hietakangas, V., and Cohen, S. M. (2007). Re-evaluating AKT regulation: role of TOR complex 2 in tissue growth. Genes Dev. 21, 632–637. doi: 10.1101/gad.416307
Hresko, R. C., and Mueckler, M. (2005). mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem. 280, 40406–40416. doi: 10.1074/jbc.M508361200
Hu, Y. (2015). The necessary role of mTORC1 in central nervous system axon regeneration. Neural Regen Res 10, 186–188. doi: 10.4103/1673-5374.152363
Hu, Y., Park, K. K., Yang, L., Wei, X., Yang, Q., Cho, K. S., et al. (2012). Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron 73, 445–452. doi: 10.1016/j.neuron.2011.11.026
Hubert, T., Wu, Z., Chisholm, A. D., and Jin, Y. (2014). S6 kinase inhibits intrinsic axon regeneration capacity via AMP kinase in Caenorhabditis elegans. J. Neurosci. 34, 758–763. doi: 10.1523/JNEUROSCI.2886-13.2014
Jacinto, E., Facchinetti, V., Liu, D., Soto, N., Wei, S., Jung, S. Y., et al. (2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137. doi: 10.1016/j.cell.2006.08.033
Jacinto, E., Loewith, R., Schmidt, A., Lin, S., Ruegg, M. A., Hall, A., et al. (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128. doi: 10.1038/ncb1183
Jin, D., Liu, Y., Sun, F., Wang, X., Liu, X., and He, Z. (2015). Restoration of skilled locomotion by sprouting corticospinal axons induced by co-deletion of PTEN and SOCS3. Nat. Commun. 6:8074. doi: 10.1038/ncomms9074
Jung, H., Yoon, B. C., and Holt, C. E. (2012). Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci. 13, 308–324. doi: 10.1038/nrn3210
Kalinski, A. L., Sachdeva, R., Gomes, C., Lee, S. J., Shah, Z., Houle, J. D., et al. (2015). mRNAs and protein synthetic machinery localize into regenerating spinal cord axons when they are provided a substrate that supports growth. J. Neurosci. 35, 10357–10370. doi: 10.1523/JNEUROSCI.1249-15.2015
Kim, S. R., Chen, X., Oo, T. F., Kareva, T., Yarygina, O., Wang, C., et al. (2011). Dopaminergic pathway reconstruction by Akt/Rheb-induced axon regeneration. Ann. Neurol. 70, 110–120. doi: 10.1002/ana.22383
Kim, Y. T., Hur, E. M., Snider, W. D., and Zhou, F. Q. (2011). Role of GSK3 signaling in neuronal morphogenesis. Front. Mol Neurosci. 4:48. doi: 10.3389/fnmol.2011.00048
Lamming, D. W., and Sabatini, D. M. (2013). A central role for mTOR in lipid homeostasis. Cell Metab. 18, 465–469. doi: 10.1016/j.cmet.2013.08.002
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Lee, J. K., Geoffroy, C. G., Chan, A. F., Tolentino, K. E., Crawford, M. J., Leal, M. A., et al. (2010). Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron 66, 663–670. doi: 10.1016/j.neuron.2010.05.002
Lien, E. C., Dibble, C. C., and Toker, A. (2017). PI3K signaling in cancer: beyond AKT. Curr. Opin. Cell Biol. 45, 62–71. doi: 10.1016/j.ceb.2017.02.007
Lim, J. H., Stafford, B. K., Nguyen, P. L., Lien, B. V., Wang, C., Zukor, K., et al. (2016). Neural activity promotes long-distance, target-specific regeneration of adult retinal axons. Nat. Neurosci. 19, 1073–1084. doi: 10.1038/nn.4340
Liu, K., Lu, Y., Lee, J. K., Samara, R., Willenberg, R., Sears-Kraxberger, I., et al. (2010). PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 13, 1075–1081. doi: 10.1038/nn.2603
Ma, X. M., and Blenis, J. (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318. doi: 10.1038/nrm2672
Mahar, M., and Cavalli, V. (2018). Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 19, 323–337. doi: 10.1038/s41583-018-0001-8
Manning, B. D., and Cantley, L. C. (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. doi: 10.1016/j.cell.2007.06.009
Miao, L., Yang, L., Huang, H., Liang, F., Ling, C., and Hu, Y. (2016). mTORC1 is necessary but mTORC2 and GSK3beta are inhibitory for AKT3-induced axon regeneration in the central nervous system. Elife 5:e14908. doi: 10.7554/eLife.14908
Nielson, J. L., Sears-Kraxberger, I., Strong, M. K., Wong, J. K., Willenberg, R., and Steward, O. (2010). Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J. Neurosci. 30, 11516–11528. doi: 10.1523/JNEUROSCI.1433-10.2010
Nielson, J. L., Strong, M. K., and Steward, O. (2011). A reassessment of whether cortical motor neurons die following spinal cord injury. J. Comp. Neurol. 519, 2852–2869. doi: 10.1002/cne.22661
Ohanna, M., Sobering, A. K., Lapointe, T., Lorenzo, L., Praud, C., Petroulakis, E., et al. (2005). Atrophy of S6K1(-/-) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat. Cell Biol. 7, 286–294. doi: 10.1038/ncb1231
Park, K. K., Liu, K., Hu, Y., Kanter, J. L., and He, Z. (2010). PTEN/mTOR and axon regeneration. Exp. Neurol. 223, 45–50. doi: 10.1016/j.expneurol.2009.12.032
Park, K. K., Liu, K., Hu, Y., Smith, P. D., Wang, C., Cai, B., et al. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966. doi: 10.1126/science.1161566
Perry, R. B., and Fainzilber, M. (2014). Local translation in neuronal processes–in vivo tests of a “heretical hypothesis”. Dev. Neurobiol. 74, 210–217. doi: 10.1002/dneu.22115
Saijilafu Hur, E. M., Liu, C. M., Jiao, Z., Xu, W. L., and Zhou, F. Q. (2013). PI3K-GSK3 signalling regulates mammalian axon regeneration by inducing the expression of Smad1. Nat. Commun. 4:2690. doi: 10.1038/ncomms3690
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. doi: 10.1126/science.1106148
Scheid, M. P., Marignani, P. A., and Woodgett, J. R. (2002). Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol. Cell. Biol. 22, 6247–6260. doi: 10.1128/MCB.22.17.6247-6260.2002
Schwab, M. E., and Bartholdi, D. (1996). Degeneration and regeneration of axons in the lesioned spinal cord. Physiol. Rev. 76, 319–370. doi: 10.1152/physrev.1996.76.2.319
Shen, W. H., Balajee, A. S., Wang, J., Wu, H., Eng, C., Pandolfi, P. P., et al. (2007). Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170. doi: 10.1016/j.cell.2006.11.042
Song, M. S., Carracedo, A., Salmena, L., Song, S. J., Egia, A., Malumbres, M., et al. (2011). Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199. doi: 10.1016/j.cell.2010.12.020
Song, M. S., Salmena, L., and Pandolfi, P. P. (2012). The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296. doi: 10.1038/nrm3330
Song, Y., Ori-Mckenney, K. M., Zheng, Y., Han, C., Jan, L. Y., and Jan, Y. N. (2012). Regeneration of Drosophila sensory neuron axons and dendrites is regulated by the Akt pathway involving Pten and microRNA bantam. Genes Dev. 26, 1612–1625. doi: 10.1101/gad.193243.112
Willis, D. E., and Twiss, J. L. (2006). The evolving roles of axonally synthesized proteins in regeneration. Curr. Opin. Neurobiol. 16, 111–118. doi: 10.1016/j.conb.2006.01.002
Yang, J., Cron, P., Thompson, V., Good, V. M., Hess, D., Hemmings, B. A., et al. (2002). Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol. Cell. 9, 1227–1240. doi: 10.1016/S1097-2765(02)00550-6
Yang, L., Miao, L., Liang, F., Huang, H., Teng, X., Li, S., et al. (2014). The mTORC1 effectors S6K1 and 4E-BP play different roles in CNS axon regeneration. Nat. Commun. 5:5416. doi: 10.1038/ncomms6416
Yang, Q., Inoki, K., Ikenoue, T., and Guan, K. L. (2006). Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 20, 2820–2832. doi: 10.1101/gad.1461206
Yao, Y., Suraokar, M., Darnay, B. G., Hollier, B. G., Shaiken, T. E., Asano, T., et al. (2013). BSTA promotes mTORC2-mediated phosphorylation of Akt1 to suppress expression of FoxC2 and stimulate adipocyte differentiation. Sci. Signal. 6:ra2. doi: 10.1126/scisignal.2003295
Yiu, G., and He, Z. (2006). Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 7, 617–627. doi: 10.1038/nrn1956
Yuan, M., Pino, E., Wu, L., Kacergis, M., and Soukas, A. A. (2012). Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J. Biol. Chem. 287, 29579–29588. doi: 10.1074/jbc.M112.386854
Zinzalla, V., Stracka, D., Oppliger, W., and Hall, M. N. (2011). Activation of mTORC2 by association with the ribosome. Cell 144, 757–768. doi: 10.1016/j.cell.2011.02.014
Keywords: axon regeneration, PTEN/PI3K/Akt, optic nerve, mTOR, GSK3β
Citation: Zhang J, Yang D, Huang H, Sun Y and Hu Y (2018) Coordination of Necessary and Permissive Signals by PTEN Inhibition for CNS Axon Regeneration. Front. Neurosci. 12:558. doi: 10.3389/fnins.2018.00558
Received: 31 May 2018; Accepted: 24 July 2018;
Published: 13 August 2018.
Edited by:
Wenbo Zhang, The University of Texas Medical Branch at Galveston, United StatesReviewed by:
Bo Chen, Icahn School of Medicine at Mount Sinai, United StatesFengquan Zhou, Johns Hopkins University, United States
Copyright © 2018 Zhang, Yang, Huang, Sun and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Hu, aHV5YW5nQHN0YW5mb3JkLmVkdQ==