
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci. , 05 September 2018
Sec. Neuropharmacology
Volume 12 - 2018 | https://doi.org/10.3389/fnins.2018.00556
 Shekher Mohan1*
Shekher Mohan1* Emily J. Koller2
Emily J. Koller2 Jawad A. Fazal2
Jawad A. Fazal2 Gabriela De Oliveria2
Gabriela De Oliveria2 Anna I. Pawlowicz2
Anna I. Pawlowicz2 Sylvain Doré2,3*
Sylvain Doré2,3*Background: The release of inflammatory molecules such as prostaglandins (e.g., PGF2α) is associated with brain damage following an intracerebral hemorrhagic (ICH) stroke; however, the role of PGF2α and its cognate FP receptor in ICH remains unclear. This study focused on investigating the role of the FP receptor as a target for novel neuroprotective drugs in a preclinical model of ICH, aiming to investigate the contribution of the PGF2α-FP axis in modulating functional recovery and anatomical outcomes following ICH.
Results: Neurological deficit scores in FP−/− mice were significantly higher compared to WT mice 72 h after ICH (6.1 ± 0.7 vs. 3.1 ± 0.8; P < 0.05). Assessing motor skills, the total time mice stayed on the rotating rod was significantly less in FP−/−mice compared to WT mice 24 h after ICH (27.0 ± 7.5 vs. 52.4 ± 11.2 s; P < 0.05). Using grip strength to quantify forepaw strength, results showed that the FP−/− mice had significantly less strength compared to WT mice 72 h after ICH (96.4 ± 17.0 vs. 129.6 ± 5.9 g; P < 0.01). In addition to the behavioral outcomes, histopathological measurements were made. In Cresyl violet stained brain sections, the FP−/− mice showed a significantly larger lesion volume compared to the WT (15.0 ± 2.2 vs. 3.2 ± 1.7 mm3; P < 0.05 mice.) To estimate the presence of ferric iron in the peri-hematoma area, Perls' staining was performed, which revealed that FP−/− mice had significantly greater staining than the WT mice (186.3 ± 34.4% vs. 86.9 ± 13.0% total positive pixel counts, P < 0.05). Immunoreactivity experiments on brain sections from FP−/− and WT mice post-ICH were performed to monitor changes in microgliosis and astrogliosis using antibodies against Iba1 and GFAP respectively. These experiments showed that FP−/− mice had a trend toward greater astrogliosis than WT mice post-ICH.
Conclusion: We showed that deletion of the PGF2α FP receptor exacerbates behavioral impairments and increases lesion volumes following ICH compared to WT-matched controls.Detailed mechanisms responsible for these novel results are actively being pursued.
Each year, approximately 795,000 Americans suffer a stroke, of which approximately 13% are attributed to intracerebral hemorrhagic (ICH) stroke (Go et al., 2014). Other than clinical management of the patient with surgical and supportive methods, there are no effective therapies for the treatment of ICH. New methods of treatment for post-stroke patients are essential to improving outcomes following a hemorrhagic brain injury. An ICH stroke is caused by the rupture of a blood vessel within the brain. While the primary injury is caused by the mass effect, a secondary injury is caused by the components of blood that make up the hematoma. For example, edema, and inflammation are major clinical concerns that are mediated by erythrocyte lysis and the release of hemoglobin and heme. Blood components released from the breakdown of the hematoma (e.g., hemoglobin, heme, and iron) result in irreversible neuronal cell death and neurological deficits (Xi et al., 2006). Following ICH, neuronal survival can be affected by changes in phenotype and function of microglia following trauma and the release of blood breakdown products. Understanding the physiopathology involved during the secondary injury of an ICH may provide further insight into inflammatory pathways and therefore potential novel targets for therapeutics.
Microglia, or infiltrating neutrophils, travel to the site of the stroke and engulf blood components and cell debris (Zhao et al., 2007). Part of this repair response involves the secretion of prostaglandins. Prostaglandins are signaling molecules that are generated and released upon cell damage and are involved in the inflammatory cascade (Minghetti et al., 1997). In the aftermath of a stroke, some prostaglandins and their receptors can be neuroprotective, while others can contribute to the injury. For example, using the mouse model of ICH stroke, our lab has shown that activation of the PGE2 EP1 receptor can protect against neurotoxicity and when deleted, the same receptor can exacerbate neurological outcomes following ICH (Singh et al., 2013). Also, using the same mouse model of ICH, our lab has shown that EP2−/− and EP3−/− mice have less ICH-induced injury compared to WT control mice (Leclerc et al., 2015b,c).
Despite the abundance of arachidonic acid in the brain, the function of its metabolite, PGF2α, is poorly understood. However, PGF2α is known to play a significant role in the initiation of parturition, renal function, control of cerebral blood flow, and intraocular pressure principally by an increase in uveoscleral outflow of aqueous humor, autoregulation in newborn piglets, contraction of arteries, and myocardial dysfunction. Pathological conditions in humans influence PGF2α levels in cerebrospinal fluid, where elevated levels of PGF2α were measured following epilepsy, meningtitis, brain injury, and stroke. The FP receptor is another G-protein coupled receptor that binds selectively to PGF2α, which is originally synthesized from arachidonic acid (Sugimoto et al., 1994, 1997). Cyclooxygenase enzymes control the rate of transformation of arachidonic acid into the prostaglandin PGH2, which can then be converted by prostaglandin synthases into PGF2α and other prostanoids such as PGE2, PGD2, PGI2, and TxA2 (Doré, 2006). Activation of the FP receptor triggers Gαq protein-coupled mechanisms involving Ca2+ signaling, IP3 turnover and activation of protein kinase C (Toh et al., 1995). This FP receptor-mediated increase in levels of Ca2+ may have accounted for the increased levels of brain injury and excitotoxicity measured by our group using in a mouse model of ischemic stroke (Saleem et al., 2009; Kim et al., 2012). However, until recently, our group found that deleting, the FP receptor may also attenuate brain injury as found in a mouse model of traumatic brain injury (Glushakov et al., 2013). Currently, the role of the FP receptor in hemorrhagic stroke is undetermined and thus our goal is to elucidate the role of the FP receptor in a preclinical model of intracerebral hemorrhagic stroke.
Studies were performed on 2–4-month-old male adult WT (24–29 g) and FP receptor knockout (FP−/−) (15-21 g) C57BL/6 mice. The FP−/− mice developed normally, gained weight at a rate equal to that of the WT mice and had no gross anatomical or behavioral abnormalities when compared to the WT littermates (Glushakov et al., 2013). Prior to all experiment's, PCR-genotyping was performed on all littermates and the WT mice were separated from the FP−/− mice. All animal protocols were approved by the Institutional Animal Care and Use Committee of University of Florida and conducted in accordance with guidelines established by the National Institutes of Health. All mice were bred, maintained, and housed in the university's vivarium under controlled conditions (23 ± 2°C; 12 h reverse light/dark cycle), with access to food and water ad libitum.
ICH was induced in age matched male WT and FP−/− mice using collagenase VII-S (0.04 units in 0.4 μL saline) (Cat. No. 9001-12-1, Sigma-Aldrich, St. Louis, MO). All mice were anesthetized with isoflurane (4% initial and 2% maintenance) and immobilized on a stereotaxic frame. A single unilateral intrastriatal injection of collagenase was given at the following coordinates relative to the bregma: 0.4 mm anterior, 2.4 mm lateral, and 3.4 mm from the dura in both WT and FP−/− mice (Wang et al., 2006). Collagenase was infused at 0.2 μL/min using a stereotaxic automated injector (Stoelting, Wood Dale, IL). The needle was left in place for 10 min and then slowly removed over a 15-min period. Rectal temperature was monitored and maintained at 37.0 ± 0.5°C using a homeothermic blanket system to prevent hypothermia throughout the surgery. After the surgical procedure, the incision was closed with low toxic tissue adhesive, 3M™ Vetbond (Fischer Scientific, Pittsburgh, PA) and each mouse received a 1 mL intraperitoneal injection of warm saline to prevent dehydration. All mice were then transferred to incubators with a temperature maintained at 37.0 ± 0.5°C for recovery and monitored for 2–4 h and survived for 72 h post-ICH injury.
Neurological functions were assessed daily post ICH (24–72 h) in the following order: neurological deficit scores (NDS), grip strength test, and accelerating rotarod test. All assessments were performed during the dark cycle (awake phase) by investigators blinded to the genotype, and for consistency, tests were performed in the same morning period of each day post-ICH. NDS scoring was measured using the 24-point scale (Clark et al., 1998). Briefly, this NDS assessment includes six individual tests (body symmetry, gait, climbing, circling behavior, front limb symmetry, and compulsory circling) scored from 0, indicating normal performance, up to 4 points on a basis of increasing severity. The sum of the scores from individual tests was reported as the NDS. The accelerating rotarod test was used to assess motor deficits using the Rota Rod Rotamex 5 machine and software (Columbus Instruments International, Columbus, OH) following ICH injury (Jones and Roberts, 1968). The rotarod tests motor deficits and coordination and is comprised of a rotating barrel, which accelerates from 4 to 30 revolutions/min over the course of 5 min. The time in seconds at which each animal fell from the barrel was recorded from a single trial using the provided software. Prior to surgery, mice were trained once daily over the course of 3 days, the average of these training periods then serving as the baseline. The grip strength test was used to assess forelimb strength by measurement using the Animal Grip Strength System (San Diego Instruments, San Diego, CA). Each mouse was placed over a steel grid by the tail so that its forelimbs were allowed to grip a single steel bar before being gently pulled backwards (away) from the bar by the tail until the grip was released. Each mouse had five consecutive trials with a 1-min rest period between trials. The data was reported as the average value of maximal force recorded before the mouse released the bar.
The hemoglobin content of each brain subjected to ICH was quantified with Drabkin's reagent (Sigma-Aldrich), as described previously (Choudhri et al., 1997; Wang and Doré, 2007a). Briefly, mice were anesthetized 5 h after ICH, and transcardially perfused with 30 mL of normal saline. The brain was dissected into ipsilateral and contralateral sides, treated individually as follows. Each sample was homogenized for 2 min in 1 mL of distilled water and then centrifuged at 13,000 × g for 30 min. Eighty microliters of Drabkin's reagent was added to a 20 μL aliquot of supernatant (which contained hemoglobin) and allowed to stand for 15 min at room temperature. The concentration of cyanomethemoglobin produced was measured at 540 nm. A standard curve, reflecting the amount of hemoglobin present was generated by adding incremental volumes of blood (0, 0.5, 1.0, 2.0, 4.0, and 8.0 μL), obtained by cardiac puncture of anesthetized control mice, to 100 μL of lysate from the tissue of normal caudate putamen. Results from at least three samples per mouse were averaged.
All mice were euthanized 72 h post ICH and transcardially perfused with 4% paraformaldehyde in phosphate-buffered saline (PBS). Brains were harvested, post-fixed in perfusion solution for 48 h., and then cryopreserved in 30% sucrose/PBS solution for up to 3 days. All brains were sectioned using the Leica CM 1850 cryostat and mounted onto slides to make 10 sets of 16 sections, each 30 μM thick, equally distributed through the entire brain. Slides were then stored at −80°C until processed for histological analysis. Cresyl violet staining was used to measure corticostriatal lesion volumes. Perls' staining was used to assess the perihematoma amounts of ferric iron (Hill and Switzer, 1984). A Perls' reaction is produced through the addition of an 2% potassium ferrocyanide, which combines with Fe3+ to form ferric ferrocyanide, producing a bright blue color. Slides were then counterstained with nuclear fast red. Microglia and astrocyte involvement in ICH injury were studied by immunostaining using polyclonal rabbit anti-Iba1 (1:1000; Wako Bioproducts, Richmond, VA) and anti-GFAP (1:1000; DAKO, Carpinteria, CA) to measure microgliosis and astrogliosis respectively. Following overnight incubation with primary antibodies, sections were incubated with avidin-peroxidase-labeled biotin complex secondary antibodies (1:1000; BA-500, Vector Laboratories, Burlingame, CA) for 1 h Vectastain ABC and DAB SK-4100 kits (Vector Laboratories) were used per the manufacturer's protocol.
All slides were scanned using the ScanScope CS (Aperio Technologies, Inc., Vista, CA) and ImageScope software (Aperio Technologies, Inc.) was used to perform quantitative analysis. Quantitative analysis of lesion volume was performed on 8 sections taken from 5 slides per mouse, allowing for an analysis that represented of the whole brain. Quantification of Perls' positive reaction was performed similarly to the method used to calculate lesion volume; however, only the number of positive counts around the lesion was measured. The ImageScope Positive Pixel Count algorithm was used for quantification after the appropriate brain regions were outlined. To perform quantitative analysis of cortical Iba1 and GFAP immunoreactivity, four to five sections from five slides per mouse were selected. Microgliosis and astrogliosis were analyzed by placing identically sized boxes of 1000 × 1000 pixels in both the ipsilateral and contralateral motor cortex. Data is presented as the relative ipsilateral to contralateral signal for signal normalization per area quantified. For each section within the cortex, an area immediately lateral to the lesion was selected for quantification and the intensity of Iba1 and GFAP immunoreactivity was evaluated by means of relative positive pixel counts. An analyst blinded to the experimental groups performed the entire quantifying procedure.
All data is expressed as mean ± standard error of the mean and statistical difference between the two groups was analyzed using an unpaired two-tailed Student's t-test. For the neurological deficit scores (NDS), non-parametric data was calculated using the Mann–Whitney U test. When appropriate, statistical comparisons between multiple groups were done using one-way ANOVA followed by Turkey's multiple comparison tests. Statistical differences were considered significant if P < 0.05. All data was analyzed through GraphPad Prism 6.0 software (GraphPad Software Inc., La Jolla, CA).
Neurobehavioral functional testing was performed at 24, 48, and 72 h post ICH by investigators blinded to the genotype. When compared to WT controls, the NDS of FP−/− mice was significantly higher than WT mice after ICH (6.1 ± 0.7 vs. 3.1 ± 0.8; P < 0.05) (Figure 1A). In addition to neurological deficit analysis, rotarod and grip strength tests were performed after ICH. At 24, 48, and 72 h post ICH the FP−/− mice showed reduced rotarod performances (seconds) when compared to baseline function (Figure 1B). However, only FP−/− mice showed significantly reduced performance at 24 h post ICH compared to the baseline (30.3 ± 7.8 vs. 74.4 ± 15.3 s; P < 0.05) (Figure 1B). Additionally, when compared at 24 h post-ICH, the FP−/− mice had significantly lower rotarod performance compared to WT mice (30.3 ± 7.8 vs. 58.7 ± 8.0 s; P < 0.05) (Figure 1C).
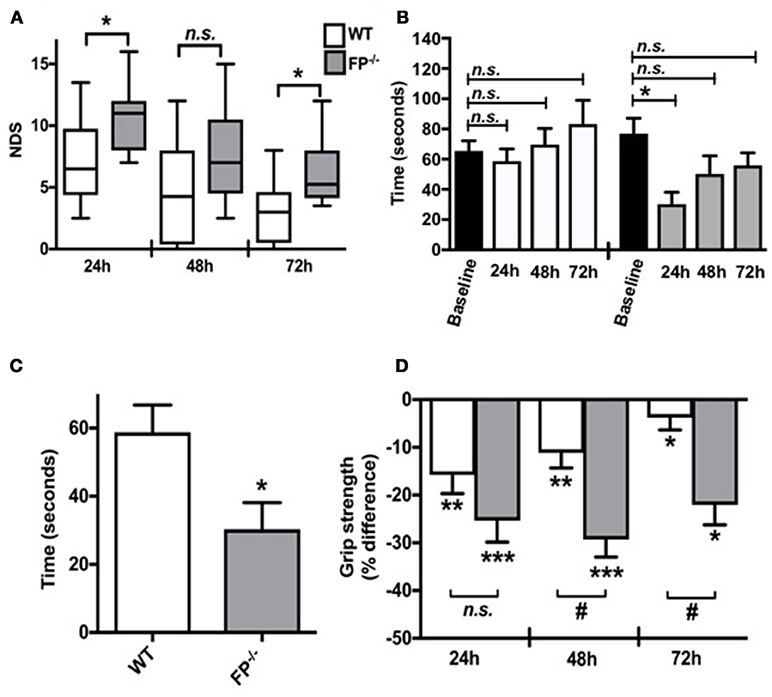
Figure 1. Effect of PGF2α-FP receptor deletion on functional neurological outcomes after ICH. Neurobehavioral testing of WT and FP−/− mice was performed by investigators blinded to genotype at 24, 48, and 72 h post-ICH. (A) At 24 and 72 h post-ICH, FP−/− mice had significantly more neurological deficit scoring compared to WT mice, and recovery was seen in both FP−/− and WT mice. (B,C) WT and FP−/− mice had similar baseline rotarod performances. Only the FP−/− mice had significantly reduced latency to fall when compared to baseline function 24 h post-ICH (P < 0.05). The FP−/− also had a greater recovery time in returning to baseline; whereas, this comparison was not obvious for the WT mice. (D) Changes in grip strength in FP−/− and WT mice 24, 48, and 72 h post-ICH was depicted on the bar graph. Both groups showed significant changes in grip strength at all times points; however, at 48 and 72 h post-ICH FP−/− mice showed significantly greater deficits in grip strength compared to the WT mice (P < 0.001). Both groups recovered, although the FP−/− mice recovery was slower than the WT mice. All comparisons included n = 7–10 WT and n = 11 FP−/− mice, and statistics were calculated using a two-way repeated measures analysis of variance with Newman–Keuls multiple comparisons test (NDS and rotarod) and a paired Student's t-test versus baseline (grip strength). ns = not significant, *P < 0.05. **P < 0.01. *** and #P < 0.001. FP = F prostanoid receptor subtype; WT = wildtype.
The grip strength test was used as an additional test to assess neuromuscular function following ICH by measuring maximal muscle strength of forelimbs. Forelimb strength was measured in five consecutive trials, with a 1-min rest between trials. Grip strength was recorded as the maximal force (in grams) and the changes at 24, 48, and 72 h post ICH were reported as the percentage (%) difference as compared to baseline (before ICH). Both FP−/− and WT mice showed significant deficit with improvements in grip strength only measured in the WT mice 24, 48 and 72 h post ICH (FP−/− mice: 24 h: −25.1 ± 4.6%; 48 h:−27.8 ± 4.4%; 72 h: −17.7 ± 5.0% and WT mice: 24 h:−24.3 ± 2.6%; 48 h: −16.9 ± 3.9%; 72 h: −6.8 ± 3.5%). At 48 h and 72 h post ICH, FP−/− mice had greater deficits in grip strength than compared to WT mice (48 h:−27.8 ± 4.4 % vs. −16.9 ± 3.9% and 72 h: −17.7 ± 5.0 % vs. −6.8 ± 3.5 % respectively) (P < 0.001) (Figure 1D).
Collagenase-induced ICH in mice consistently produces intrastriatal hematoma as evident from Cresyl violet staining (Figure 2A). Following analysis of the quantification, the FP−/− mice had a greater lesion volume than the WT mice 72 h post ICH (15.0 ± 2.3 vs. 3.2 ± 1.7 mm3 respectively; P < 0.01).
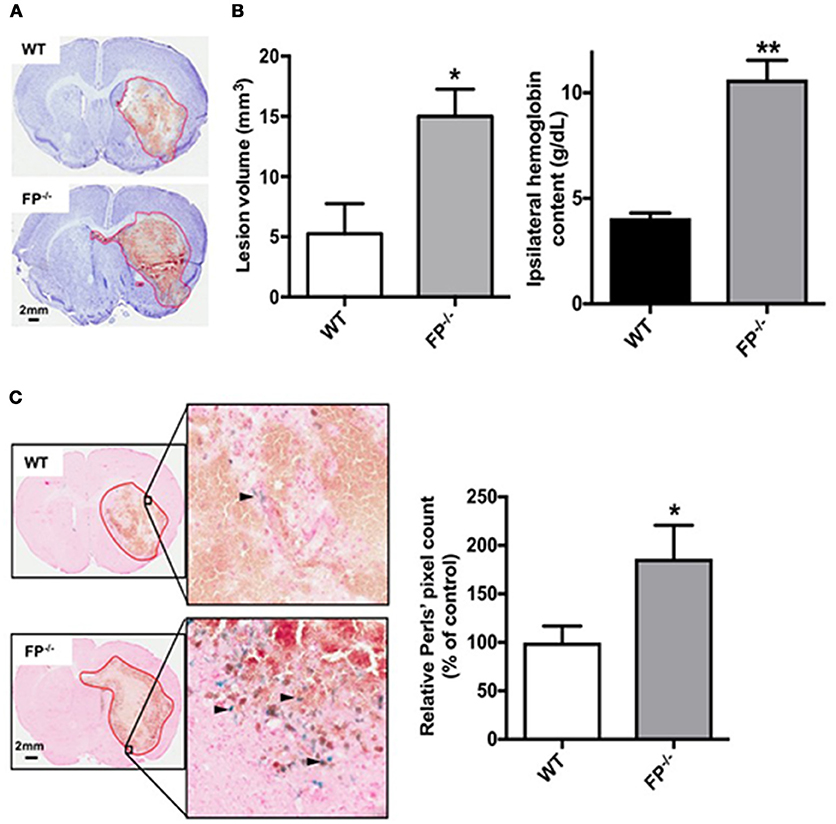
Figure 2. Genetic deletion of the PGF2α-FP receptor increases brain injury after ICH. WT and FP−/− mice underwent ICH and were euthanized at 72 h for determination of lesion volume by Cresyl violet staining of brain sections. (A) Representative images of coronal brain sections from WT (upper panel) and FP−/− mice (lower panel) show FP−/− mice as having a greater lesion volume. Images were obtained from a single animal and demonstrate the characteristic hematoma profile for WT and FP−/− mice, captured adjacent to the needle insertion site and representing maximal hematoma size. Quantification of lesion volumes showed that FP−/− mice had significantly greater ICH-induced brain injury. (WT: n = 7, FP−/−: n = 10, *P < 0.05). (B) In a separate cohort, quantification of hemoglobin content volumes at 72 h showed that FP−/− mice had significantly greater ICH-induced hemoglobin content compared to WT mice (WT: n = 3, FP−/−: n = 3, **P < 0.01). (C) Genetic deletion of the FP receptor increased brain ferric iron content, as represented by Perls staining post-ICH. Representative high magnification images of coronal brain sections show Perls staining (blue) in peri-hematomal regions in WT (upper panel) and FP−/− mice (lower panel). Square selections in the inserts denote magnified regions. Quantification of blue positive pixel count in the ipsilateral hemisphere showed that FP−/− mice had significantly greater ferric iron deposition (WT: n = 6, FP−/−: n = 6, *P < 0.05).
To better understand the potential cellular mechanisms of action in FP−/− mice, two additional independent measurements were taken. First, hemoglobin content was measured in WT and FP−/− mice 5 h post ICH. Both the WT and FP−/− mice had greater hemoglobin levels on the site-of collagenase injection (ipsilateral hemisphere). However, the FP−/− mice (10.63 g/dL) had significantly more hemoglobin compared to WT mice (4.06 g/dL) (P < 0.01) (Figure 2B). No hemoglobin was measured on the contralateral tissue and therefore this region served as an internal control. Secondly, brain sections were assessed for the deposition of ferric iron as estimated using the Perls' staining (blue), and the levels of ferric iron were noted primarily in the perihematomal regions. Quantification of blue positive pixel count showed that FP−/− mice had more ferric iron in the ipsilateral hemisphere than WT mice (186.3 ± 34.4% vs. 100.0 ± 16.8%; P < 0.05) (Figure 2C). Perls' positive staining was present only in or around the perihematomal region, while none was present in the contralateral hemisphere.
Microglia and astrocyte immunoreactivities were estimated in the cortical region of FP−/− and WT mice brain sections using anti-Iba1 and anti-GFAP immunohistochemistry (Figure 3). ICH injury caused apparent microglia activation as detected via increased Iba1 immunoreactivity in the cortical region surrounding the lesion (area marked in red). The FP−/− mice had a trend toward greater Iba1 immunoreactivity of microglia than WT mice (4.1 ± 1.8% vs. 2.8 ± 0.4%) (P = 0.09) (Figure 3A).
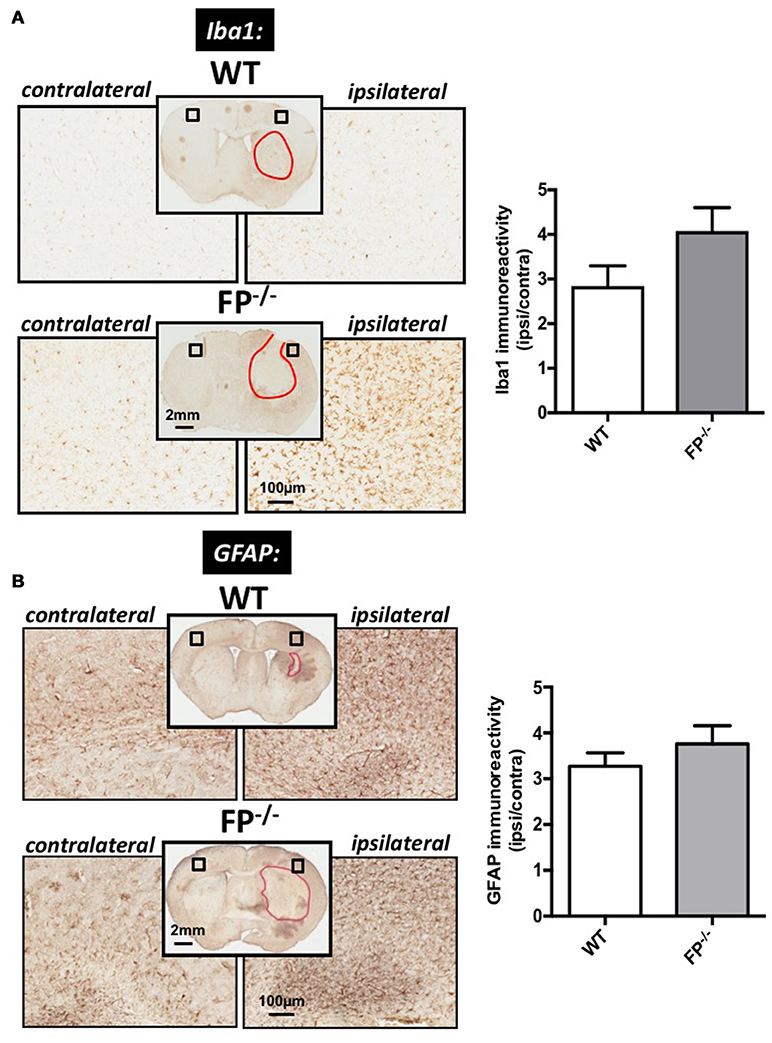
Figure 3. Effect of PGF2α-FP receptor deletion on microgliosis and astrogliosis post-ICH. At 72 h after ICH, WT, and FP−/− mice were euthanized and brains processed for Iba1 and GFAP immunohistochemistry in order to evaluate cortical gliosis. (A) Representative high magnification images of coronal brain sections show the ipsilateral and contralateral cortex for WT (upper panels) and FP−/− mice (lower panels). Square selections in the inserts denote magnified regions. Quantification of brown positive pixel count demonstrated that FP−/− mice had a trend toward greater cortical Iba1 immunoreactivity then WT mice (WT: n = 10, FP−/−: n = 12). This trend in microglial activation was accompanied by morphological changes. (B) Quantification of brown positive pixel count demonstrated that FP−/− mice had relatively the same level of cortical GFAP immunoreactivity as WT mice (WT: n = 7, FP−/−: n = 8). All data was normalized to the corresponding contralateral equivalent areas. Iba1 = Ionized calcium-binding adapter protein; GFAP = glial fibrillary acidic protein.
To study astrogliosis, GFAP immunoreactivity was used. Similar to the microglia marker, the GFAP levels were greater in the ipsilateral cortical region compared with the corresponding contralateral area in the ICH-treated animals. The changes in GFAP levels in the FP−/− mice (3.7 ± 0.4%) were not significantly different when compared to WT mice (3.2 ± 0.3%) (Figure 3B).
This study investigated the role of the toxic molecule prostaglandin F2α (PGF2α) in a mouse model of ICH stroke. The main finding of this study is that the FP−/− mice have an increased susceptibility to ICH-induced injury compared to WT mice. Here, we have documented for the first time the unique and significant role of the FP receptor in exacerbating ICH injury, potentially by interfering in the functions of microglia against iron and/or heme-induced neuronal death. Our data shows that FP−/− mice had significantly greater neurological deficits compared to WT controls. For example, the NDS of FP−/− mice was significantly higher than WT mice after ICH (P < 0.05, Figure 1A). In addition to neurological deficit analysis, rotarod, and grip strength tests were performed after ICH. For the rotarod, FP−/− mice showed significantly reduced performance at 24 h post-ICH compared to the baseline (P < 0.05, Figure 1B). The grip strength test was used as an additional test to assess neuromuscular function following ICH by measuring maximal muscle strength of forelimbs. Both FP−/− and WT mice showed significant deficit in terms of improvements in grip strength 24, 48, and 72 h post ICH with the FP−/− mice showing a greater deficit in grip strength compared to WT mice at the 48 and 72 h post ICH (Figure 1C).
Previous data have revealed that there is no significant difference in the morphology of cerebral vasculature and anastomoses in FP−/− and WT mice brains (Glushakov et al., 2013). Therefore, the hemoglobin content 5 h post ICH between FP−/− and WT mice may not be attributed to structural differences in micro-vasculature of the brain. However, greater amounts of hemoglobin following ICH might be attributed to the functions of the microvasculature. For example, the FP−/− mice may have weaker blood vessel walls which may therefore be more likely to rupture under stress, such as that induced by collagenase. Also, the greater neurological deficits measured in the FP−/− mice may also have been due to the greater hemoglobin content. Hemoglobin levels in the brain following hemorrhagic stroke can not only disrupt the blood brain barrier but can also up-regulate nitric oxide synthase and peroxynite formation, which would lead to further neuronal death (Ding et al., 2014). Additionally, greater expression of hemoglobin proteins (α- and β-globin) have been measured and found to be localized in neurons and microglial cells following ICH in rats (He et al., 2011). The same group also found that levels of heme and iron may cause an increase in the expression of endogenous hemoglobin after ICH.
The mouse PGF2α-FP receptor is reported to have the highest homology to the PGE2-EP1 receptor, and when activated, this receptor can increase levels of IP3 and intracellular levels of Ca2+ (Sugimoto et al., 1994; Mohan et al., 2012). This may explain why our findings with the FP−/− mice are consistent with our previous findings with the EP1−/− mice using the same ICH model; the EP1−/− mice showed greater deteriorated outcomes compared to WT control mice (Singh et al., 2013). Our groups also found recently that use of EP1 receptor agonists improved anatomical outcomes and functional recovery (Leclerc et al., 2015a). In contrast to ICH, the genetic deletion and/or pharmacological blockade of either the EP1 or FP receptor types attenuated brain injury and improved neurological outcomes in excitotoxicity and mouse ischemic stroke models (Ahmad et al., 2006, 2008; Saleem et al., 2007, 2009). The difference in the role of these receptors between ischemic and hemorrhagic strokes demonstrates a uniqueness and dynamism in functionality that is determined by the type of brain injury. Differences in the vasculature might be a possible mechanism that could lead to greater lesion volumes in FP−/− mice post ICH. We have previously shown that FP−/− mice do not present with any significantly altered gross vascular anatomy of the brain; although we cannot rule-out changes in proteins as being responsible for the regulation of neovascularization. For example, stromal cell-derived factor 1 (SDF-1), which is controlled by endothelial cells, could alter ICH outcomes (Seo et al., 2009; Glushakov et al., 2013). These findings therefore direct our attention toward the mechanisms and cells involved in the clearance of blood in examining the etiology of increased cerebral injury in FP−/− mice post ICH.
The role of the FP receptor in ischemic versus hemorrhagic stroke may be determined by the state and level of humoral neuroinflammation in intracerebral hemorrhage as compared to ischemic stroke. For example, the expression levels of pro-inflammatory cytokines IL-1β and TNFα increased early (3 h post ICH) following collagenase-induced ICH (Liesz et al., 2011). Microglia have been identified as the main producers of the early increased levels of intracerebral IL-1β and TNFα (Wang and Dore, 2007b; Wang et al., 2007). Furthermore, previous studies have investigated the activation of microglia/macrophage and leukocyte invasion after experimental ICH (Xue and Del Bigio, 2000, 2003; Loftspring et al., 2009). Nevertheless, the role of the FP receptor in microglia mediated inflammation remains largely unknown and therefore further in vitro studies are warranted. This study suggests that a mild activation state of microglia could be different in FP−/− mice compared to WT mice post ICH. Whether changes in microglial activation significantly translate into changes in functions of the microglia remains to be explored. This study demonstrated that the FP−/− mice had greater perihematomal Perls' pixel count and may support that the microglia had a reduced ability to remove ferric iron released from lysed blood cells post ICH. Previous studies have shown that microglia and astrocytes express the FP receptor and, more recently, it has been demonstrated that PGF2α may enhance the clearance of β amyloid by its agonism of liver X receptors (LXR)/ retinoid X receptors (RXR) expressed on microglia (Zhuang et al., 2013). Previous evidence has shown that LXRs are expressed at high levels in the brain and when stimulated, can cause changes in the expression of inflammatory genes in microglia and macrophages (Wang et al., 2002; Joseph et al., 2003; Zelcer et al., 2007; Cui et al., 2012). To conclude, if the LXR is involved in ICH further studies would be necessary, as evidence suggests that PGF2α can regulate the LXR. However, using an ischemic stroke model, activation of the LXR promoted neuroprotection and reduced inflammation via the inhibition of nuclear factor κB (Morales et al., 2008; Cheng et al., 2010). Thus, it is possible that in this study FP−/− mice showed greater ICH-induced lesions with greater blood and ferric iron accumulation because of diminished phagocytic capability, resulting in decreased clearance of red blood cells and therefore increased brain injury.
In response to injury, astrocytes have a diverse role and it is well known that reactive astrocytes (gliosis) form a glial scar that contains the damaged area as shown following our ICH protocol. However, following injury and/or neurodegeneration, reactive gliosis, which involves alterations in functioning and phenotype of different glial cells, may augment brain damage. Studies have revealed that the astrocyte response to gross brain damage leads to anisomorphic (disorganized) astrogliosis that reinforces a cascade of events that eventually increases brain injury. Anisomorphic astrogliosis can inhibit neurite outgrowth and increase levels of the inducible form of nitric oxide synthase (iNOS) and nitric oxide (NO) which can possess cytotoxic properties and contribute to neuronal death (Gibbons and Dragunow, 2006). The elevated levels of NO released from activated astrocytes might be the most relevant in ICH-induced injury as NO is a vasodilator (Chi et al., 2015; Crobeddu et al., 2015; Munoz et al., 2015). Therefore, increased vasodilation could lead to greater ICH-induced secondary injury. In this study, we saw increased ipsilateral astrogliosis in both FP−/− and WT mice post-ICH; however, no significance was found between the two genotype groups.
Microglia and astrocytes become activated following brain injury and release factors that contribute to the functional state of the blood-brain barrier. Here, we show that microglia may be more “reactive” than astrocytes in FP−/− mice compared to WT mice. Furthermore, compared to previously published work by our group using EP1−/−, EP2−/−, and EP3−/− mice post ICH, the levels of micro- and astrogliosis are contradictory compared to the FP−/− mice used in this study (Singh et al., 2013; Leclerc et al., 2015b,c). The decreased level of micro- and astrogliosis in EP1−/−, EP2−/−, and EP3−/− mice was used to account for the changes in functional and anatomical outcomes post ICH. We found that the FP−/− mice had a trend toward greater microgliosis, suggesting that this state of activation in the FP−/− mice could potentially be responsible for the changes in functional and anatomical outcomes presented here. Further in vivo and in vitro studies are necessary to elucidate the role of the FP receptor in glial cells are necessary. Proposed future experiments include performing in vivo experiments using the FP receptor selective antagonist and agonist to measure any FP receptor mediated-neuroprotection in this and other ICH models. Neuropharmacological experiments designed to specifically study the FP receptor will help clarify the respective role of the glial-neuronal axis after ICH.
In this study, we have provided evidence that suggests a neuroprotective role for the FP receptor following ICH. Our results show that deletion of the FP receptor increases brain injury, functional deficits and increases the deposition of ferric iron post-ICH. However, without following this work with further experiments that utilize a FP receptor selective antagonist and agonist, caution should be used when interpreting the potential for the FP receptor as a therapeutic target for the treatment of ICH. Our findings are similar to those found with EP1−/− mice post-ICH, and recently our group has shown that following activation of the EP1 receptor, astrogliosis, neutrophil infiltration, blood-brain barrie breakdown, and functional recovery all improved (Leclerc et al., 2015a). Based on these findings, we hypothesize that the activation of the FP receptor will result in measurable changes in the improvement of functional and anatomical outcomes following ICH. Until then, we remain hopeful that the FP receptor, similar to the related EP1 receptor, is a viable therapeutic target for the treatment of ICH.
SM and SD designed the study, analyzed, and interpreted the results and wrote the manuscript. SM performed the surgical procedures, performed the blinded behavioral testing, and harvest brains for analysis with the assistance of the other lab members. EK, JF, GD, and AP coordinated and performed tissue sections and contributed to histological staining, quantification and data analysis. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by grants from the National Institutes of Health (RO1NS046400 to SD) and (F32NS078933 to SM) and special thank you to all members of the Doré lab for helpful discussions and technical support.
Ahmad, A. S., Kim, Y. T., Ahmad, M., Maruyama, T., and Doré, S. (2008). Selective blockade of PGE2 EP1 receptor protects brain against experimental ischemia and excitotoxicity, and hippocampal slice cultures against oxygen-glucose deprivation. Neurotox. Res. 14, 343–351. doi: 10.1007/BF03033858
Ahmad, A. S., Saleem, S., Ahmad, M., and Doré, S. (2006). Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol. Sci. 89, 265–270. doi: 10.1093/toxsci/kfj022
Cheng, O., Ostrowski, R. P., Liu, W., and Zhang, J. H. (2010). Activation of liver X receptor reduces global ischemic brain injury by reduction of nuclear factor-kappaB. Neuroscience 166, 1101–1109. doi: 10.1016/j.neuroscience.2010.01.024
Chi, O. Z., Rah, K. H., Barsoum, S., Liu, X., and Weiss, H. R. (2015). Inhibition of neuronal nitric oxide synthase improves microregional O balance in cerebral ischemia-reperfusion. Neurol. Res. doi: 10.1179/1743132815y.0000000062. [Epub ahead of print].
Choudhri, T. F., Hoh, B. L., Solomon, R. A., Connolly, E. S., and Pinsky, D. J. (1997). Use of a spectrophotometric hemoglobin assay to objectively quantify intracerebral hemorrhage in mice. Stroke 28, 2296–2302. doi: 10.1161/01.STR.28.11.2296
Clark, W., Gunion-Rinker, L., Lessov, N., and Hazel, K. (1998). Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke 29, 2136–2140. doi: 10.1161/01.STR.29.10.2136
Crobeddu, E., Pilloni, G., Tardivo, V., Fontanella, M. M., Panciani, P. P., Spena, G., et al. (2015). Role of nitric oxide and mechanisms involved into cerebral injury after subarachnoid hemorrhage: is nitric oxide a possible answer to cerebral vasospasm? J. Neurosurg. Sci. 60, 385–391.
Cui, W., Sun, Y., Wang, Z., Xu, C., Peng, Y., and Li, R. (2012). Liver X receptor activation attenuates inflammatory response and protects cholinergic neurons in APP/PS1 transgenic mice. Neuroscience 210, 200–210. doi: 10.1016/j.neuroscience.2012.02.047
Ding, R., Chen, Y., Yang, S., Deng, X., Fu, Z., Feng, L., et al. (2014). Blood-brain barrier disruption induced by hemoglobin in vivo: involvement of up-regulation of nitric oxide synthase and peroxynitrite formation. Brain Res. 1571, 25–38. doi: 10.1016/j.brainres.2014.04.042
Doré, S. (2006). GPCR antagonists as an alternative to COX-2 inhibitors: a case for the PGE2 EP1 receptor. Trends Pharmacol. Sci. 27, 458–460. doi: 10.1016/j.tips.2006.07.001
Gibbons, H. M., and Dragunow, M. (2006). Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res. 1084, 1–15. doi: 10.1016/j.brainres.2006.02.032
Glushakov, A. V., Robbins, S. W., Bracy, C. L., Narumiya, S., and Doré, S. (2013). Prostaglandin F2alpha FP receptor antagonist improves outcomes after experimental traumatic brain injury. J. Neuroinflammation 10:132. doi: 10.1186/1742-2094-10-132
Go, A. S., Mozaffarian, D., Roger, V. L., Benjamin, E. J., Berry, J. D., Blaha, M. J., et al. (2014). Executive summary: heart disease and stroke statistics−2014 update: a report from the American Heart Association. Circulation 129, 399–410. doi: 10.1161/01.cir.0000442015.53336.12
He, Y., Hua, Y., Keep, R. F., Liu, W., Wang, M. M., and Xi, G. (2011). Hemoglobin expression in neurons and glia after intracerebral hemorrhage. Acta Neurochir. Suppl. 111, 133–137. doi: 10.1007/978-3-7091-0693-8_22
Hill, J. M., and Switzer, R. C. (1984). The regional distribution and cellular localization of iron in the rat brain. Neuroscience 11, 595–603. doi: 10.1016/0306-4522(84)90046-0
Jones, B. J., and Roberts, D. J. (1968). The quantiative measurement of motor inco-ordination in naive mice using an acelerating rotarod. J. Pharm. Pharmacol. 20, 302–304. doi: 10.1111/j.2042-7158.1968.tb09743.x
Joseph, S. B., Castrillo, A., Laffitte, B. A., Mangelsdorf, D. J., and Tontonoz, P. (2003). Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 9, 213–219. doi: 10.1038/nm820
Kim, Y. T., Moon, S. K., Maruyama, T., Narumiya, S., and Doré, S. (2012). Prostaglandin FP receptor inhibitor reduces ischemic brain damage and neurotoxicity. Neurobiol. Dis. 48, 58–65. doi: 10.1016/j.nbd.2012.06.003
Leclerc, J. L., Ahmad, A. S., Singh, N., Soshnik-Schierling, L., Greene, E., Dang, A., et al. (2015a). Intracerebral hemorrhage outcomes following selective blockade or stimulation of the PGE EP1 receptor. BMC Neurosci. 16:48. doi: 10.1186/s12868-015-0182-2
Leclerc, J. L., Lampert, A. S., Diller, M. A., and Doré, S. (2015b). Genetic deletion of the prostaglandin E2 E prostanoid receptor subtype 3 improves anatomical and functional outcomes after intracerebral hemorrhage. Eur. J. Neurosci. 41, 1381–1391. doi: 10.1111/ejn.12909
Leclerc, J. L., Lampert, A. S., Diller, M. A., Immergluck, J. B., and Doré, S. (2015c). Prostaglandin E2 EP2 receptor deletion attenuates intracerebral hemorrhage-induced brain injury and improves functional recovery. ASN Neuro. 7:1759091415578713. doi: 10.1177/1759091415578713
Liesz, A., Middelhoff, M., Zhou, W., Karcher, S., Illanes, S., and Veltkamp, R. (2011). Comparison of humoral neuroinflammation and adhesion molecule expression in two models of experimental intracerebral hemorrhage. Exp. Transl. Stroke Med. 3:11. doi: 10.1186/2040-7378-3-11
Loftspring, M. C., McDole, J., Lu, A., Clark, J. F., and Johnson, A. J. (2009). Intracerebral hemorrhage leads to infiltration of several leukocyte populations with concomitant pathophysiological changes. J. Cereb. Blood Flow Metab. 29, 137–143. doi: 10.1038/jcbfm.2008.114
Minghetti, L., Nicolini, A., Polazzi, E., Créminon, C., Maclouf, J., and Levi, G. (1997). Inducible nitric oxide synthase expression in activated rat microglial cultures is downregulated by exogenous prostaglandin E2 and by cyclooxygenase inhibitors. Glia 19, 152–160. doi: 10.1002/(SICI)1098-1136(199702)19:2<152::AID-GLIA6>3.0.CO;2-2
Mohan, S., Ahmad, A. S., Glushakov, A. V., Chambers, C., and Doré, S. (2012). Putative role of prostaglandin receptor in intracerebral hemorrhage. Front. Neurol. 3:145. doi: 10.3389/fneur.2012.00145
Morales, J. R., Ballesteros, I., Deniz, J. M., Hurtado, O., Vivancos, J., Nombela, F., et al. (2008). Activation of liver X receptors promotes neuroprotection and reduces brain inflammation in experimental stroke. Circulation 118, 1450–1459. doi: 10.1161/CIRCULATIONAHA.108.782300
Muñoz, M. F., Puebla, M., and Figueroa, X. F. (2015). Control of the neurovascular coupling by nitric oxide-dependent regulation of astrocytic Ca(2+) signaling. Front. Cell. Neurosci. 9:59. doi: 10.3389/fncel.2015.00059
Saleem, S., Ahmad, A. S., Maruyama, T., Narumiya, S., and Doré, S. (2009). PGF(2alpha) FP receptor contributes to brain damage following transient focal brain ischemia. Neurotox. Res. 15, 62–70. doi: 10.1007/s12640-009-9007-3
Saleem, S., Li, R. C., Wei, G., and Doré, S. (2007). Effects of EP1 receptor on cerebral blood flow in the middle cerebral artery occlusion model of stroke in mice. J. Neurosci. Res. 85, 2433–2440. doi: 10.1002/jnr.21399
Seo, J., Kim, Y. O., and Jo, I. (2009). Differential expression of stromal cell-derived factor 1 in human brain microvascular endothelial cells and pericytes involves histone modifications. Biochem. Biophys. Res. Commun. 382, 519–524. doi: 10.1016/j.bbrc.2009.03.049
Singh, N., Ma, B., Leonardo, C. C., Ahmad, A. S., Narumiya, S., and Dore, S. (2013). Role of PGE(2) EP1 receptor in intracerebral hemorrhage-induced brain injury. Neurotox. Res. 24, 549–559. doi: 10.1007/s12640-013-9410-7
Sugimoto, Y., Hasumoto, K., Namba, T., Irie, A., Katsuyama, M., Negishi, M., et al. (1994). Cloning and expression of a cDNA for mouse prostaglandin F receptor. J. Biol. Chem. 269, 1356–1360.
Sugimoto, Y., Yamasaki, A., Segi, E., Tsuboi, K., Aze, Y., Nishimura, T., et al. (1997). Failure of parturition in mice lacking the prostaglandin F receptor. Science 277, 681–683. doi: 10.1126/science.277.5326.681
Toh, H., Ichikawa, A., and Narumiya, S. (1995). Molecular evolution of receptors for eicosanoids. FEBS Lett. 361, 17–21. doi: 10.1016/0014-5793(95)00129-W
Wang, J., and Doré, S. (2007a). Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 130(Pt. 6), 1643–1652. doi: 10.1093/brain/awm095
Wang, J., and Doré, S. (2007b). Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 27, 894–908. doi: 10.1038/sj.jcbfm.9600403
Wang, J., Zhuang, H., and Doré, S. (2006). Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobio. Dis. 22, 473–476. doi: 10.1016/j.nbd.2005.12.009
Wang, L., Schuster, G. U., Hultenby, K., Zhang, Q., Andersson, S., and Gustafsson, J. A. (2002). Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 99, 13878–13883. doi: 10.1073/pnas.172510899
Wang, Q., Tang, X. N., and Yenari, M. A. (2007). The inflammatory response in stroke. J. Neuroimmunol. 184, 53–68. doi: 10.1016/j.jneuroim.2006.11.014
Xi, G., Keep, R. F., and Hoff, J. T. (2006). Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 5, 53–63. doi: 10.1016/S1474-4422(05)70283-0
Xue, M., and Del Bigio, M. R. (2000). Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci. Lett. 283, 230–232. doi: 10.1016/S0304-3940(00)00971-X
Xue, M., and Del Bigio, M. R. (2003). Comparison of brain cell death and inflammatory reaction in three models of intracerebral hemorrhage in adult rats. J. Stroke Cerebrovasc. Dis. 12, 152–159. doi: 10.1016/S1052-3057(03)00036-3
Zelcer, N., Khanlou, N., Clare, R., Jiang, Q., Reed-Geaghan, E. G., Landreth, G. E., et al. (2007). Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 10601–10606. doi: 10.1073/pnas.0701096104
Zhao, X., Sun, G., Zhang, J., Strong, R., Song, W., Gonzales, N., et al. (2007). Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann. Neurol. 61, 352–362. doi: 10.1002/ana.21097
Zhuang, J., Zhang, H., Zhou, R., Chen, L., Chen, J., and Shen, X. (2013). Regulation of prostaglandin F2alpha against beta amyloid clearance and its inflammation induction through LXR/RXR heterodimer antagonism in microglia. Prostaglandins Other Lipid Mediat. 106, 45–52. doi: 10.1016/j.prostaglandins.2013.09.002
Keywords: astrogliosis, hemorrhagic stroke, microgliosis, neuroinflammation, neuroprotection, prostaglandins, prostanoids
Citation: Mohan S, Koller EJ, Fazal JA, De Oliveria G, Pawlowicz AI and Doré S (2018) Genetic Deletion of PGF2α-FP Receptor Exacerbates Brain Injury Following Experimental Intracerebral Hemorrhage. Front. Neurosci. 12:556. doi: 10.3389/fnins.2018.00556
Received: 13 December 2017; Accepted: 23 July 2018;
Published: 05 September 2018.
Edited by:
Jacob Raber, Oregon Health Science University, United StatesReviewed by:
Lance Johnson, University of Kentucky, United StatesCopyright © 2018 Mohan, Koller, Fazal, De Oliveria, Pawlowicz and Doré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shekher Mohan, c21vaGFuQG1hbmNoZXN0ZXIuZWR1
Sylvain Doré, c2RvcmVAdWZsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.