 Carol L. Thompson1 Jonathan P. Wisor2 Chang-Kyu Lee1 Sayan D. Pathak1 Dmitry Gerashchenko2 Kimberly A. Smith1 Shanna R. Fischer1 Chihchau L. Kuan1 Susan M. Sunkin1 Lydia L. Ng1 Christopher Lau1 Michael Hawrylycz1 Allan R. Jones1 Thomas S. Kilduff2 Edward S. Lein1*
Carol L. Thompson1 Jonathan P. Wisor2 Chang-Kyu Lee1 Sayan D. Pathak1 Dmitry Gerashchenko2 Kimberly A. Smith1 Shanna R. Fischer1 Chihchau L. Kuan1 Susan M. Sunkin1 Lydia L. Ng1 Christopher Lau1 Michael Hawrylycz1 Allan R. Jones1 Thomas S. Kilduff2 Edward S. Lein1*
- 1 Allen Institute for Brain Science, Seattle, WA, USA
- 2 Biosciences Division, SRI International, Menlo Park, CA, USA
Sleep deprivation (SD) leads to a suite of cognitive and behavioral impairments, and yet the molecular consequences of SD in the brain are poorly understood. Using a systematic immediate-early gene (IEG) mapping to detect neuronal activation, the consequences of SD were mapped primarily to forebrain regions. SD was found to both induce and suppress IEG expression (and thus neuronal activity) in subregions of neocortex, striatum, and other brain regions. Laser microdissection and cDNA microarrays were used to identify the molecular consequences of SD in seven brain regions. In situ hybridization (ISH) for 222 genes selected from the microarray data and other sources confirmed that robust molecular changes were largely restricted to the forebrain. Analysis of the ISH data for 222 genes (publicly accessible at http://sleep.alleninstitute.org) provided a molecular and anatomic signature of the effects of SD on the brain. The suprachiasmatic nucleus (SCN) and the neocortex exhibited differential regulation of the same genes, such that in the SCN genes exhibited time-of-day effects while in the neocortex, genes exhibited only SD and waking (W) effects. In the neocortex, SD activated gene expression in areal-, layer-, and cell type-specific manner. In the forebrain, SD preferentially activated excitatory neurons, as demonstrated by double-labeling, except for striatum which consists primarily of inhibitory neurons. These data provide a characterization of the anatomical and cell type-specific signatures of SD on neuronal activity and gene expression that may account for the associated cognitive and behavioral effects.
Introduction
Sleep is necessary for normal neural function, including synaptic plasticity and homeostatic synaptic downscaling (reviewed in Tononi and Cirelli, 2006; Vyazovskiy et al., 2008). Sleep deprivation (SD) affects cognition, attention, memory, and emotional behaviors controlled by higher brain regions such as the neocortex, hippocampus, and amygdala (Yoo et al., 2007). There is evidence that specific anatomic areas are differentially activated by SD. Imaging studies have shown differential enhancement or suppression of neural activity in specific cortical areas occurs during sleeping, waking, and SD (Nofzinger, 2005; Chee and Chuah, 2008). Specific anatomic areas also control the regulation of sleep. The electrical activity of the cerebral cortex, as measured by the electroencephalogram (EEG), provides the primary electrophysiological characteristics that are used to define different stages of sleep as well as to distinguish sleep from wakefulness. EEG activity is the product of intrinsic electrical rhythms generated within the cortex and a dynamic interplay between the thalamus and the cortex (Steriade, 2006). In contrast, the transitions between, and duration of, different sleep and behavioral states are regulated by subcortical waking- and sleep-active brain regions. These structures include the hypocretin-containing (Hcrt) neurons in the tuberal hypothalamus, histaminergic tuberomammillary nuclei (TMN), noradrenergic locus coeruleus (LC), serotonergic raphe nuclei, cholinergic basal forebrain (BF), and GABAergic ventrolateral preoptic nucleus (Saper et al., 2005), as well as the circadian pacemaker in the suprachiasmatic nucleus (SCN).
Immediate-early gene (IEG)-based “activity-mapping” has been used by several laboratories to identify neuronal activation under different states of sleep and wakefulness (Sherin et al., 1996; Cirelli and Tononi, 2000b; Terao et al., 2003a; Modirrousta et al., 2005; Gerashchenko et al., 2008), a technique which has helped identify regions important in sleep, such as the ventrolateral preoptic nucleus (Sherin et al., 1996). More recently, investigators have applied microarray-based profiling methods to brain regions such as the cerebral cortex, cerebellum and hypothalamus to determine gene expression changes associated with spontaneous sleep and wake, SD, and recovery sleep (RS) after SD (Cirelli and Tononi, 1998, 2000a; Cirelli et al., 2004; Cirelli, 2006; Terao et al., 2006; Mackiewicz et al., 2007). Such transcriptomic approaches have estimated that 5–10% of cortical transcripts and 10% of total transcripts are regulated in response to time-of-day or sleep/wake state (Panda et al., 2002; Cirelli et al., 2004). These studies have generally concluded that the major effect of SD is upregulation of gene expression, and IEG expression is increased overall in cortex. There are two major limitations of these studies. First is the use of gross anatomical regions (e.g., whole cerebral cortex) consisting of numerous heterogenous cell types and regions which may both reduce the detection of region- or cell type-specific gene expression as well as provide an overarching conclusion about the cortex based upon only the most prevalent changes. Second is the lack of precise validation to examine the cellular specificity of the gene expression changes. Two more recent studies have further examined the dynamics of the transcriptome in response to sleep and wake in discrete brain nuclei, and these studies have concluded that there are many regionally specific genes responding to sleep/wake state that may go undetected in studies of gross brain regions (Conti et al., 2007; Winrow et al., 2009).
The identification of the genes and anatomical regions activated during sleep and responsive to SD could be important to defining the function of sleep in biochemical and molecular terms, as well as in understanding the mechanisms underlying sleep homeostasis. The goals of the current study were threefold: (1) to map brain regions activated by sleeping, waking, and SD using IEG expression, (2) to profile the molecular changes occurring in these brain regions, and (3) to characterize the responses of these genes to behavioral conditions with cellular resolution. To achieve these goals, we combined genome-wide microarray analysis with high-throughput in situ hybridization (ISH) (Lein et al., 2007). These data provide a comprehensive neuroanatomical, cellular, and molecular map of gene induction across behavioral conditions including sleeping, waking, and SD.
Materials and Methods
Experimental Animals, Electroencephalographic/Electromyographic State Determination, Sleep Deprivation
All procedures involving animals adhered to the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee at SRI International. Six-week-old male C57BL/6J mice (Jackson Labs West, Sacramento, CA, USA) were entrained to a 12 h light/12 h dark cycle (LD 12:12) with food and water ad libitum for at least 3 weeks before use (9–11 weeks). Animals were assigned to five treatment groups (n = 74 per group): 6 h SD, 4 h RS after 6 h SD, time-matched controls for SD and RS groups (SDC and RSC, respectively), or 6 h into the dark phase of the LD cycle, representative of a predominantly waking condition (W). SD commenced at the beginning of the light period [Zeitgeber time (ZT) 0–6]. Mice were kept awake by a combination of cage tapping, introduction of foreign objects (e.g., balled paper towels), cage rotation, and stroking of vibrissae and fur with an artist’s paintbrush (Terao et al., 2003b). “Gentle handling” procedures have been used extensively to induce SD (Borbely et al., 1984; Tobler and Jaggi, 1987; Deboer et al., 1994; Huber et al., 2000; Wisor et al., 2008). Mice were euthanized by cervical dislocation and decapitated at ZT6 (SD and SDC groups), ZT10 (RS and RSC groups), or ZT18 (W group). The W group mice were euthanized in darkness and the eyes enucleated in dim red light prior to dissection in white light. The brains were frozen in Tissue-Tek OCT mounting medium (Sakura Finetek, Torrance, CA, USA).
Of 370 mice used (74 per group), the first 50 mice (10 per group) were subjected to EEG/EMG assessment of sleep states for electrophysiological validation of SD protocols described previously (Terao et al., 2000, 2003b; Wisor et al., 2008). Time-of-day (SDC and RSC) control mice spent the majority of time asleep (63.4% and 62.8%, respectively) during the first 6 h of the light phase. In contrast, SD and RS groups spent 1.0% and 3.5% of time asleep during this interval as a consequence of SD, resulting in a 98.4% sleep loss in the SD group and 94.5% in the RS group. The W group spent 17.2% of time asleep during the 6 h preceding termination of recording, corresponding to the first 6 h of the dark phase (when C57BL/6 mice are most active), and thus time spent asleep was 72.8% lower in this group sacrificed at ZT18 than in the SDC group sacrificed at ZT6.
High-Throughput in Situ Hybridization
Non-isotopic colorimetric ISH was performed as described previously (Lein et al., 2007). Entrez gene IDs, probe sequences and sources are provided in Table S1 in Supplementary Material. Riboprobes were hybridized to 25 μm thick post-fixed sections sampled uniformly across a brain hemisphere, such that each gene was analyzed at 200 μm sampling density in the sagittal plane. Double fluorescent ISH was performed using a variation of this protocol, as described previously (Thompson et al., 2008). Riboprobes were labeled during in vitro transcription with either digoxigenin-UTP (DIG) or dinitrophenyl-11-UTP (DNP; Perkin Elmer, Waltham, MA, USA). DIG and DNP probes were hybridized simultaneously, with separate probe-specific tyramide amplification steps using either anti-DIG-HRP with tyramide-biotin or anti-DNP-HRP with tyramide-DNP. Visualization was achieved through labeling with either streptavidin-Alexa-Fluor 488 or anti-DNP-Alexa-Fluor 555 (Invitrogen/Molecular Probes, Carlsbad, CA, USA). Colorimetric and fluorescent ISH slides were scanned using an automated microscopy platform (Lein et al., 2007).
Laser Microdissection and RNA Isolation
Frozen brains were cryosectioned at 10 μm onto PEN-foil slides (Leica Microsystems, Inc., Bannockburn, IL, USA). After drying for 30 min at room temperature, slides were used immediately or frozen at −80°C. Slides were lightly Nissl stained to allow cytoarchitectural visualization. Slides were fixed in ice cold 70% ethanol for 30 s, washed 15 s in nuclease-free water, stained in 0.7% cresyl violet in 0.05% NaOAc, pH 3.1 (Harleco) for 2 min, nuclease-free water for 15 s, followed by 15–20 s each in 50%, 75%, and 95% ethanol, and finally twice in 100% ethanol for 20–25 s. Slides were air-dried for 2 min, and dessicated in a vacuum for 1 h. Laser microdissection was performed on a Leica LMD6000 (Leica Microsystems, Inc., Bannockburn, IL, USA), using the Nissl stain as a guide to identify target brain regions. Tissue was collected for orbital cortex (ORB), SCN, hypocretin neurons (Hcrt), TMN, posteromedial cortical amygdala (PMCo), entorhinal cortex (ENT), and LC. Collection of the Hcrt area/tuberal hypothalamus was further guided by labeling for preprohypocretin mRNA using ISH on adjacent tissue sections. Microdissected tissue was collected directly into RLT buffer with β-mercaptoethanol from the RNeasy kit (Qiagen Inc., Valencia, CA, USA). Samples were volume-adjusted, vortexed, centrifuged, and frozen. RNA was isolated independently for each animal and brain region following the manufacturer’s directions.
RNA Amplification and Microarray Hybridizations
All procedures beginning with RNA amplification through microarray processing were performed by GenUS Biosystems, Inc. (Northbrook, IL, USA). RNA integrity and concentration were evaluated with Agilent RNA6000 Pico Lab Chip on the Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). For each sample, 5 ng of total RNA was amplified using MessageAmpII (Applied Biosystems, Foster City, CA, USA). For the majority of samples, RNA was amplified from an individual biological replicate, and each amplification product was applied to one array. However, due to consistently low yields of total RNA isolated from the TMN brain region, three TMN RNA amplifications and microarrays were generated from the pooling of two individual biological replicates each (one SD, one SDC, and one W microarray); however, an individual RNA sample was never used for more than one array. Following the second round of reverse transcription, second-strand cDNA synthesis, and purification of double-stranded cDNA, in vitro transcription was performed using T7 RNA polymerase in the presence of Biotin-11-UTP. The concentration and purity of the amplified cRNA was measured by OD 260/280 and the quality was assessed using an Agilent RNA6000 Nano Lab Chip on the Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Purified cRNA (10 μg) was fragmented to uniform size (∼0.1 kb) and applied to CodeLink Mouse Whole Genome Bioarrays (formerly supplied by GE Healthcare, Piscataway, NJ; now supplied by Applied Microarrays, Inc, Tempe, AZ, USA) in hybridization buffer. CodeLink Mouse Whole Genome arrays are comprised of 36,000 30-mer probes designed to conserved exons across the transcripts of targeted genes. Arrays were hybridized (37°C for 18 h) in a shaking incubator, washed in 0.75X Tris-buffered saline (pH 7.6) at 46°C for 1 h, and stained with Cy5-Streptavidin dye conjugate for 30 min. Rinsed and dried arrays were scanned with a GenePix™ 4000B scanner [Molecular Devices, (formerly Axon Instruments), Sunnyvale, CA] at 5 μm resolution. Codelink Expression Analysis software (Applied Microarrays, Inc, Tempe, AZ, USA) was used for gridding and feature intensity extraction from the scanned array images.
Microarray Analysis
The preprocessing of the microarray data set was done using Rosetta Resolver (Rosetta Biosoftware, Seattle, WA, USA) where its error model (Weng et al., 2006) is applied as part of the Amersham/CodeLink-Intensity Experiment Builder pipeline. The preprocessed intensity data were exported to R (www.r-project.org) to undo batch bias by COMBAT normalization (Johnson et al., 2007) and to do the following analyses using Bioconductor library functions (Gentleman et al., 2004). Genes differentially expressed across the five groups in each of the seven brain regions were identified by ANOVA analysis with p < 0.05 and the empirical estimated FDR = 4.83 ± 2.5% which is estimated by 100 random permutations of group assignments (Xie et al., 2005). For each of these identified genes, the normalized expression values of four replicate samples were averaged, z-transformed, and arranged to form a trend vector, a 5-tuple of expression values at the treatment groups, SD, SDC, RS, RSC, and W. An hierarchical clustering was applied with (1-Pearson correlation) as its distance metric to detect trends in each brain region, and to detect common trends across the seven brain regions. All microarray data are available at GEO (accession no. GSE23628).
Image Registration and Automated Quantification
For a given gene and condition, a set of 20 ISH images spanning a sagittal hemisphere with 200 μm sampling density comprised an “image series.” Each image series was quantified and registered in 3D to a common anatomical reference framework (Lein et al., 2007), such that individual reference atlas regions were assigned values for the metric expression energy. For each tissue section, an adaptive threshold algorithm (Ng et al., 2007) classified individual pixels as belonging to expressing cells. Expression energy is the average product of the classification mask with ISH intensity computed over each atlas region (Ng et al., 2009).
Difference Grid Heat MAP Generation
Following image registration and automated quantification, a 3D representation of ISH expression energy was generated for a given image series with a (200 μm)3 voxel size. In order to calculate spatial differences in expression for a given gene across two different experimental conditions for three replicate sets, the 3D expression file for each image series was first normalized to the mean expression energy of its experimental set (set of image series for all five conditions assayed on the same day) and averaged across three biological replicates for a given gene and condition. Pairwise computations were then performed to create the difference heat maps. The “difference grids” were computed by subtracting the average expression energy between one experimental condition (e.g., SD) and its relevant comparison condition (e.g., SDC) for a given gene, and were viewable in a 2D format. 3D difference grids were calculated from a fold difference between two conditions.
Manually-Assisted Optical Densitometry
In situ hybridization and Nissl images from the same brain were first aligned as a stack. An area of interest (AOI) was drawn on an adjacent Nissl-stained section using ImagePro v. 6.0 (MediaCybernetics, Bethesda, MD, USA). The AOI was then transferred to an adjacent aligned ISH image, and adjusted as necessary. Subsequently, expressing cells (expression objects) were segmented and optical density was computed for each AOI, using custom algorithm software based upon quantification performed for the Allen Brain Atlas (Ng et al., 2007). Expression objects were detected within each AOI (a) and the integrated object optical density (IOD) was calculated as IOD(a) = ΣOD(p), such that the optical density of individual expression object pixels (p) are summed over the AOI (a). IOD measurements were tallied for each AOI, summed over different sections containing the same brain region, and normalized to total AOI area.
Results
IEG Mapping Identified Numerous Brain Regions Differentially Affected by Behavioral State
In order to identify brain regions specifically activated by SD and RS, IEG expression was analyzed using a standardized high-throughput ISH platform (Lein et al., 2007). IEG expression provides a cellular resolution “readout” of recent neuronal activity (Sagar et al., 1988; Morgan and Curran, 1991). Altogether, nine IEGs were analyzed by colorimetric and/or fluorescent ISH over the five experimental conditions with three replicates each (Arc, Egr1, Egr2, Egr3, Fos, Fosb, Fosl2, Nr4a1, Nr4a3; all colorimetric data are available at http://sleep.alleninstitute.org). These IEG were selected because there were multiple reports that each gene changed in response to SD (Table S2 in Supplementary Material).
Arc and Nr4a1 were particularly robust examples of condition-specific expression patterns, shown in Figure 1. Arc mRNA expression was highest in SD in the neocortex, hippocampus, piriform cortex and amygdala (Figure 1A). Mice allowed to sleep for 4 h following SD (RS mice) showed a similar pattern to SD, although with reduced signal intensity, suggesting a “recovery” from the sleep-deprived state. Sleeping controls (SDC, RSC) showed dramatically lower Arc expression overall. Finally, W showed robust IEG induction relative to sleeping controls, although the W pattern was quite different from SD.
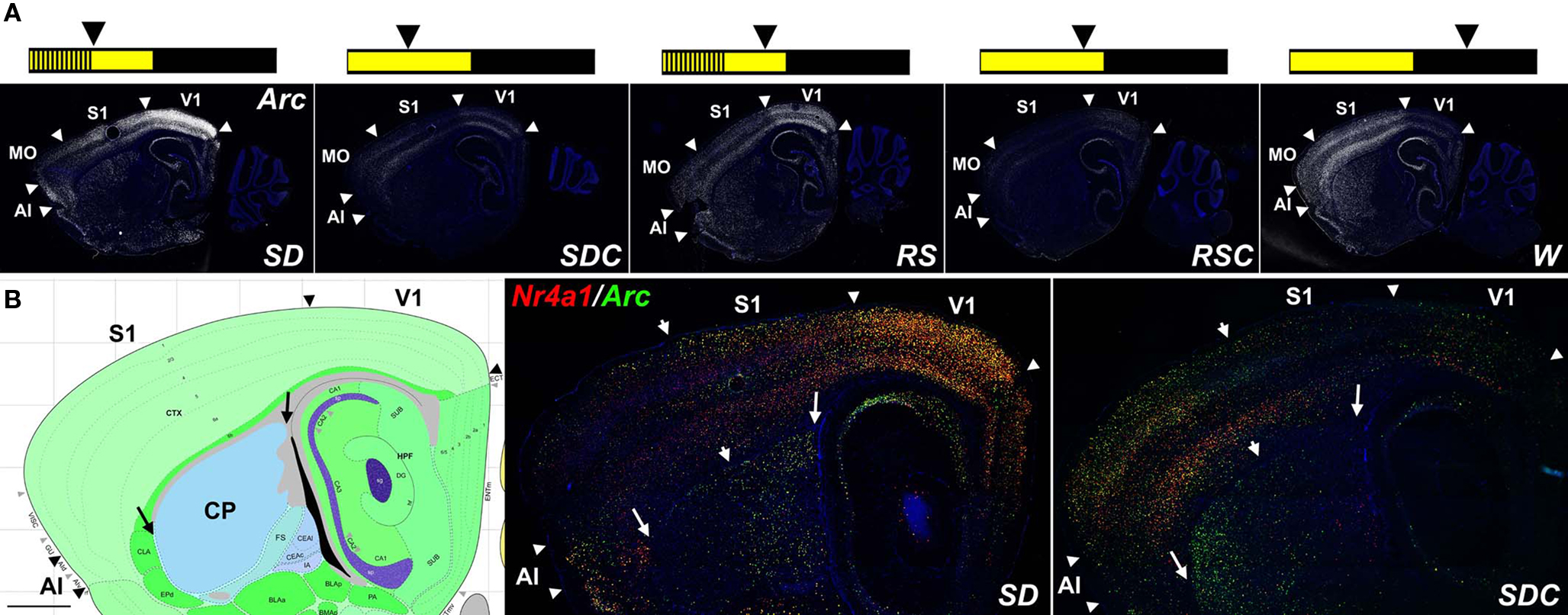
Figure 1. IEG mapping of neuronal activation in response to behavioral state. (A) Fluorescent Arc ISH (white) shows robust induction in SD, RS, and W relative to sleeping controls SDC and RSC, most notably in forebrain structures including discrete neocortical regions and the striatum. Summary schematics of the treatment conditions are shown across the top, in which the lights on portion of the LD12:12 light cycle is in yellow and the lights off portion in black; vertical lines represent sleep deprivation, and arrowheads indicate the time of sacrifice for each of the five experimental groups. (B) Double fluorescent ISH for Nr4a1 (red) and Arc (green) for SD and SDC, with extensive cellular colocalization of these two mRNAs (yellow cells). Arrowheads in (A) and (B) delineate major cortical areas, large arrows in (B) delineate the boundaries of the caudate putamen (CP)and arrowheads in (B) divide zones of higher rostral (left in SD) or caudal (right in SDC) IEG expression in the CP and neocortex. Left panel in (B) is an annotated reference atlas panel (Dong, 2008), plane-matched to the ISH images (right panels). All sections were counterstained with DAPI (blue). AI, agranular insular cortex; MO, motor cortex somatomotor cortex; S1, primary somatosensory cortex; V1, primary visual cortex. Scale bars: 1 mm.
Strikingly, in the neocortex and caudate putamen, SD had the effect of both inducing and repressing IEG expression in distinct complementary anatomic subcompartments as compared to SDC, with virtually no part of these brain regions remaining static (Figure 1B). Arc and Nr4a1 expression was elevated in SD in primary visual cortex and, to a lesser extent, in the neighboring caudal somatosensory cortex, agranular insular cortex and orbitofrontal cortex (not shown). Conversely, Arc and Nr4a1 in SDC mice were typically highest in motor and rostral somatosensory cortices. Similarly, IEG induction was seen selectively in the caudal dorsolateral caudate putamen of SD mice, while the converse pattern (highest in rostral and ventral caudate putamen) was present in SDC mice. Arc and Nr4a1 showed highly similar patterns, at both an anatomical and cellular level, as shown by extensive co-labeling using double fluorescent ISH (yellow cells in Figure 1B).
In order to perform quantification and statistical analyses of the ISH image data, colorimetric ISH data for each gene were algorithmically quantified and mapped onto a common 3D anatomical coordinate framework using methods developed as part of the Allen Brain Atlas project (Lein et al., 2007). This methodology enabled automated quantification of gene expression in each ISH experiment for several hundred brain regions, as delineated in the Allen Reference Atlas (Dong, 2008), and is demonstrated in Figure 2 using Arc as an example. Signal detection algorithms were used to detect expressing cells (represented as an “expression mask” in Figures 2C,F) and register expression information to the Allen Reference Atlas (Dong, 2008), shown in Figures 2D,G as a projection of the reference atlas volume back on to the tissue sections. These algorithms allowed statistical analysis of expression summarized by classically annotated brain structures, or analyzed in a grid volume with (200 μm)3 voxels overlaid on the 3D reference atlas volume. Using the grid volume, differential gene expression could be computed at a voxel level between pairs of conditions (e.g., SD versus SDC), with presentation of the results in both 2D and 3D formats. Briefly, triplicate data for each gene within a behavioral condition were averaged at the voxel level in 3D and used to generate a smoothed “difference grid” representing subtractive or fold change comparisons between conditions. As shown in Figures 2H–L, averaged Arc expression in SD and SDC mice was used to calculate an SD/SDC ratio in 3D coordinate space (Figure 2J). Difference grids could also be calculated as a subtraction (SD-SDC) and projected in 2D (Figure 2L). For example, the SD-induced Arc expression pattern illustrated in Figure 1, with induction in specific neocortical areas, caudate putamen and piriform cortex, is clearly visible on a smoothed 2D projection of the 3D difference grid (Figure 2L).
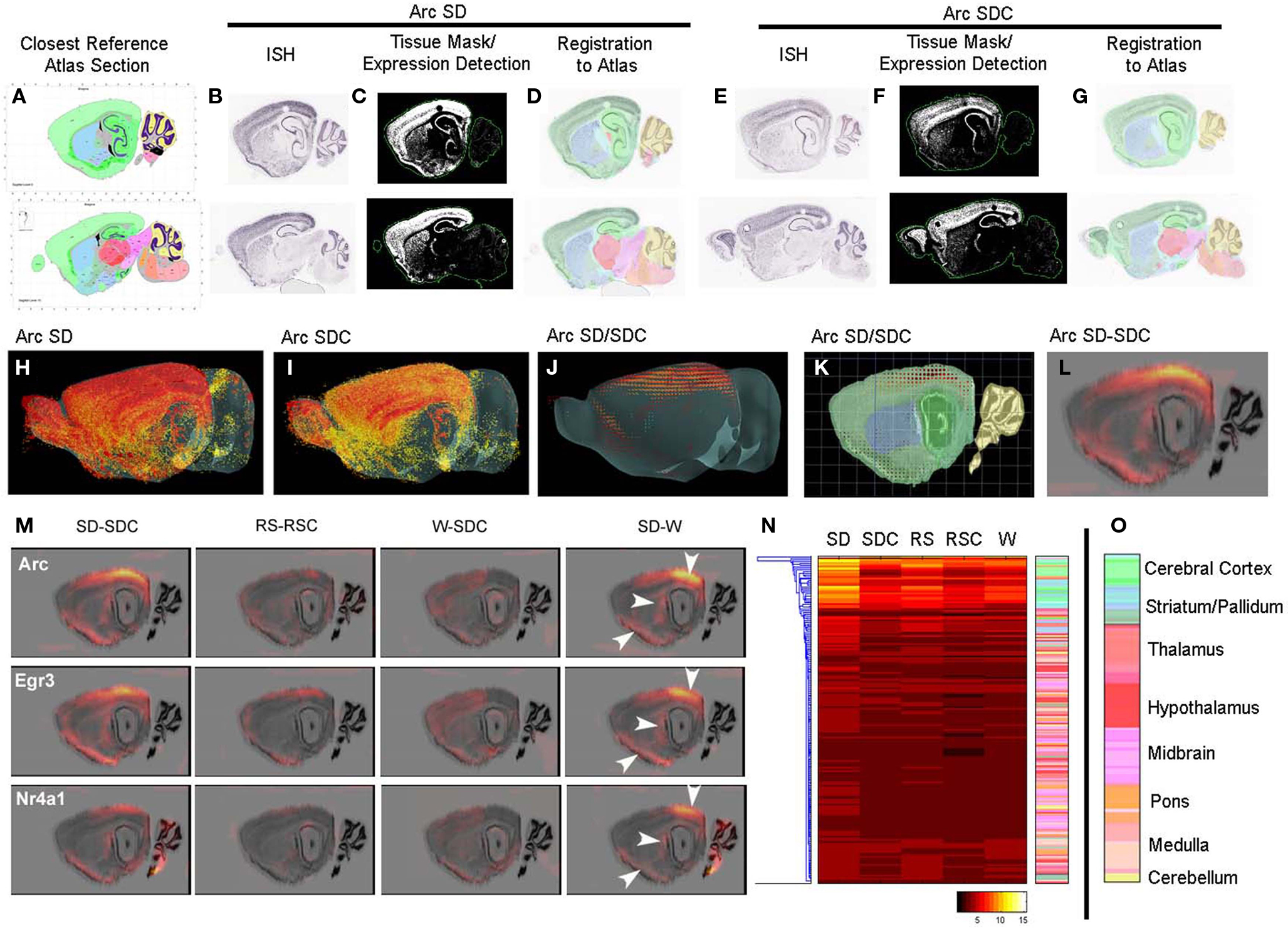
Figure 2. Automated quantification and mapping of ISH data. (A–L) Quantification and registration of ISH data to an anatomical coordinate framework allowing statistical analysis of differential image-based gene expression across conditions, illustrated for Arc expression in SD (B–D) and SDC (E–G). Primary ISH data (B,E) is algorithmically segmented to create a quantified expression mask (C,F), and registered in 3D to a common anatomical coordinate framework based on an annotated reference atlas (A,D,G). ISH data are quantified in voxels of (200 μm)3, which can be visualized in 3D (H,I) and used to compute differential expression between conditions at this resolution (J,K). This 3D “difference grid” can be projected in 2D (superimposed on a Nissl section in (L)) and used to analyze differential expression at the level of brain region or voxel. (M) Difference grids of Arc, Egr3, and Nr4a1 show similar activation patterns by SD for these IEGs in specific anatomical structures including the neocortex, caudate putamen and piriform cortex visible at this resolution (arrows). SD leads to dramatic quantitative changes relative to SDC (left panel) and waking (right panel), while less robust differences are seen between W and SDC and between RS and RSC (middle panels). (N) Hierarchical clustering of Arc quantification across 209 brain regions over five behavioral conditions. Individual rows represent specific anatomical regions, color-coded by their position in the hierarchical structural ontology of the Allen Reference Atlas (right panel). The heatmap is colored by relative expression, based upon the metric expression energy (see Materials and Methods), with white/yellow representing high expression and black representing low expression, as denoted in the color key at the bottom. (O) Color key for anatomical regions as shown in (N).
Overall, different IEGs had highly concordant condition-specific expression patterns. For example, Arc, Egr1, and Nr4a1 (Figure 2M) as well as Fos, Egr3, and Fosl2 (not shown), showed robust induction in visual and caudal somatosensory cortex, piriform cortex, cortical amygdala, and dorsolateral portions of the caudate putamen in SD mice (Figure 2M). The effect of SD is particularly robust, and is evident in both the SD-SDC and SD-W comparisons. W-SDC showed consistent induction of IEG mRNAs across many forebrain regions. Gene-specific patterns were also observed, including elevated Fos and Nr4a1 expression in the cerebellum (Figure 2M) and Arc in the pons following SD, as well as decreased Fos expression in part of the ventral posteromedial nucleus of the thalamus (VPM) following SD (Figure 3). Lastly, the algorithmic quantification enabled a complete characterization of gene expression across annotated anatomic regions, shown in Figure 2N as a heat map for the gene Arc, which plots the expression level of Arc across 209 brain regions (y-axis, with the key to structure colors in Figure 2O) for each of the five conditions. The highest expression level (yellow on heatmap) demonstrates that SD induced widespread expression of Arc across much of the forebrain and was most prominent in cortical, hippocampal and olfactory (green), and striatal (blue) regions (Figure 2N).
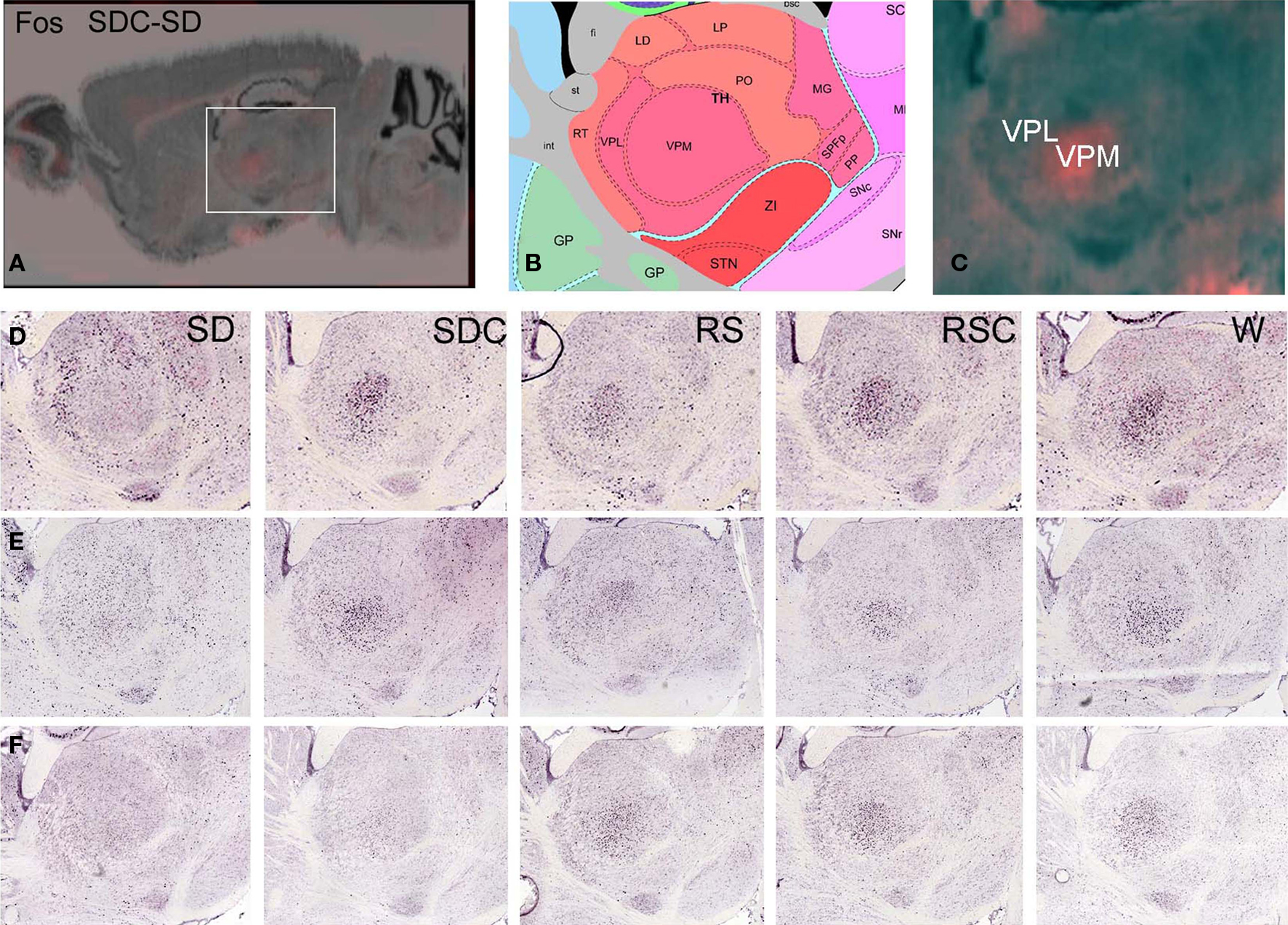
Figure 3. Difference grid analysis identified decreased Fos expression in the ventral posteromedial nucleus of the thalamus during SD. Difference grid representation of upregulated Fos expression in SDC versus SD, showing higher expression (orange) in SDC than SD in VPM at low magnification (A) and high magnification (C). Corresponding reference atlas panel is shown in (B). (D–F) Colorimetric Fos ISH demonstrating differential Fos mRNA localization in SD, with lowest levels in VPM in SD relative to all other conditions, with higher SD expression in surrounding thalamic areas, for three replicate experiments.
Molecular Signatures of Sleep/Wake and Sleep Deprivation Identified by Genetic Profiling
To identify additional genes regulated by behavioral state or time-of-day, DNA microarrays were used to profile seven brain regions. IEG mapping was used to select three subregions of the neocortex, amygdala and hippocampus, structures linked to known detrimental effects of SD on cognitive, emotional and mnemonic abilities. Orbitofrontal cortex (ORB) showed robust SD-induced IEG expression as measured by both standard manual densitometry and the automated quantification (e.g., Arc in Figure 4A). Although relatively modest changes in IEG expression were seen in the hippocampus proper (Ammon’s horn and dentate gyrus), robust SD induction was observed in the entorhinal cortex (ENT) that provides the major input to the dentate gyrus (e.g., Egr3 in Figure 4B). In the amygdala, the deep nuclei (lateral, basal, and accessory basal) provide the majority of the input to the hippocampus for the emotional association with memory (Pikkarainen et al., 1999; Pikkarainen and Pitkanen, 2001); however, the most robust changes were in the neighboring posteromedial cortical amygdala (PMCO; e.g., Egr3 in Figure 4C). These three regions also exhibited statistically significant widespread induction of other IEGs in response to SD as measured by densitometry (data not shown). Although the visual cortex was a site of strong Arc induction after SD, the activation of this area was likely a reflection of the recent activation of visual pathways by light rather than a phenomenon related to the known detrimental effects of SD on cognition, emotion, or memory, and thus was not selected for further study.
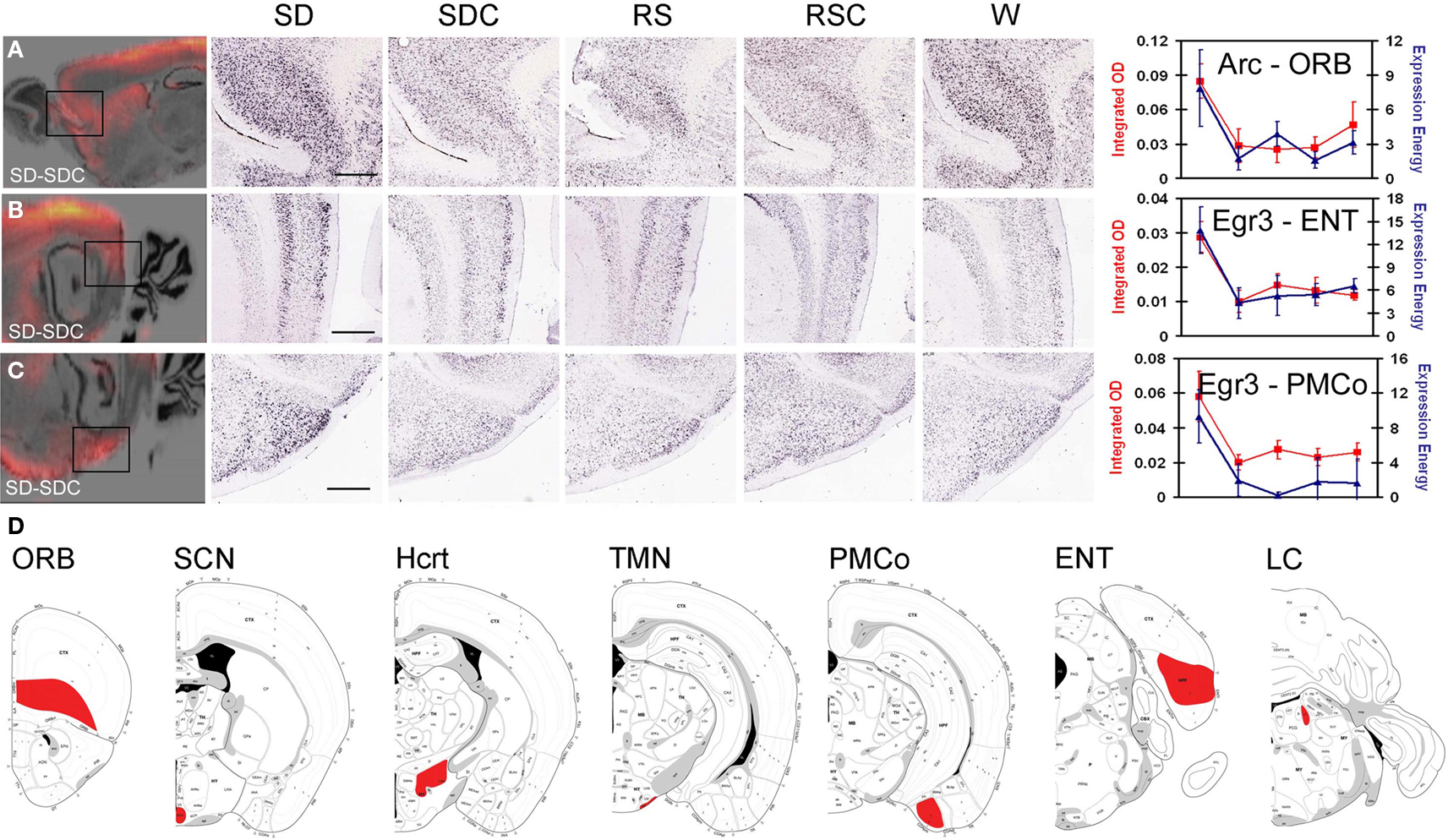
Figure 4. Regions assayed by laser microdissection and microarray. IEG mapping identified three forebrain regions affected by SD, including orbitofrontal cortex [ORB, (A)], entorhinal cortex [ENT, (B)] and posteromedial cortical amygdala [PMCo, (C)]. Left panels show difference grids for SD-SDC for Arc (A) and Egr3 (B,C). Middle panels show colorimetric ISH data for all five behavioral conditions at high magnification corresponding to the boxed areas in the difference grids. Right panels show quantification of SD induction using automated informatics quantification (blue) and manual integrated optical density measurements (red). Scale bars: 500 μm. (D) Coronal schematics indicating brain regions collected for laser microdissection (red) shown on atlas images from rostral (left) to caudal (right).
Based on these observations, ORB, PMCO, and ENT were isolated by laser microdissection across all five conditions for microarray analysis. Four regions known to be involved in the neural control of behavioral states and circadian rhythms were also analyzed in parallel: the LC, TMN, tuberal hypothalamus (location of the Hcrt neurons) and SCN (Figure 4D). High quality RNA was isolated from four biological replicates for each brain region and condition, and a small sample RNA amplification technique was used for DNA microarray analyses on the GE Codelink platform.
After normalization of the microarray data, differentially expressed genes were identified for each brain region (one-way ANOVA, p < 0.05). In order to visualize the dynamics of these genes across the five experimental conditions, microarray expression values were plotted as “trends.” The averaged replicate microarray values of each gene were z-transformed to create an individual gene trend across the five conditions, and then the genes for each brain region were clustered into 25 distinct trend clusters. To identify similar expression dynamics occurring across different brain regions, the trend clusters from all brain regions were then hierarchically clustered. Figures 5A–I shows nine of the trend types (rows) observed across the seven brain regions (columns), where each trend is plotted as five points corresponding to SD, SDC, RS, RSC, and W, respectively.
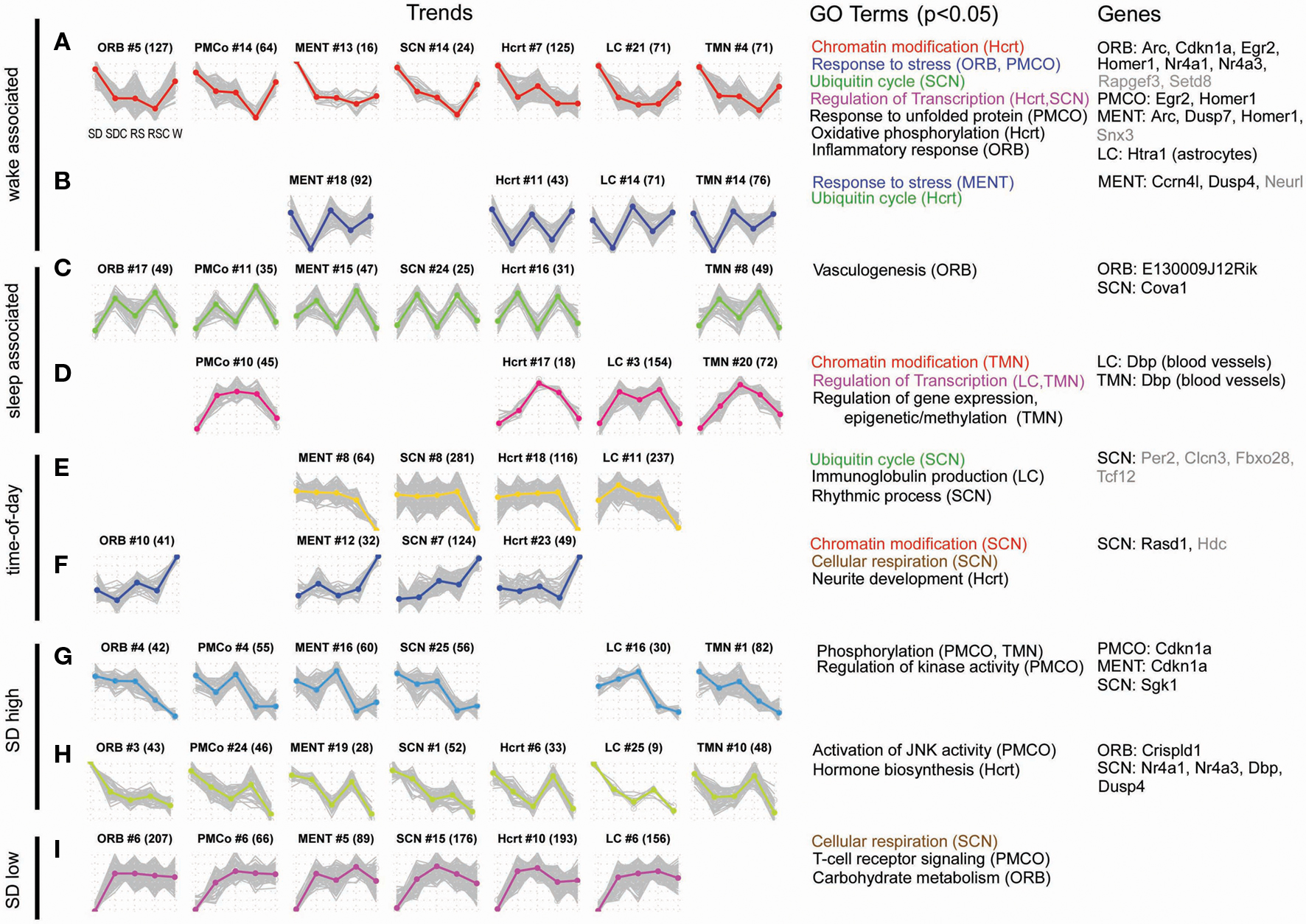
Figure 5. Gene expression showed a diversity of responses to time-of-day and treatment condition. Differentially expressed genes were identified by one-way ANOVA with a p-value cutoff of 0.05. For each identified gene, the expression values of four replicates were averaged and z-transformed (plotted on y-axis) and arranged to form a trend vector, with the expression values of the treatment groups plotted in the following order: SD, SDC, RS, RSC, and W [x-axis of each trend; labels shown in the first plot in (A)]. Hierarchical clustering was used to cluster differentially expressed genes into 25 trends per each of 7 regions, resulting in a total of 175 trends. For each cluster, the average expression trend of the cluster [plotted as colored, bolded line in (A–I)] was extracted by averaging the individual gene trend vectors (plotted as gray lines in (A–I)]. In order to compare similar trends across brain regions, the 175 expression trends were hierarchically clustered (not shown), and 48 of 175 trends are shown (A–I). At the top of each individual trend plot, the structure name and cluster number are shown with the number of genes within each cluster in parentheses. Gene ontology (GO) terms which were enriched in a gene cluster (DAVID, p < 0.05) are shown (structures exhibiting those terms are listed in parentheses). Color-coding of GO terms shows terms that are shared across multiple trend types. The right-most column lists examples of genes which have been validated by ISH for each structure either in the original ISH screen (gray font), or with triplicate data (black font).
Two trends could be generally characterized as “wake-associated” or possibly activity-associated, with upregulation of gene expression in SD and W, either with or without a “recovery” during RS (Figures 5A,B). Numerous IEG, markers of neuronal activity, exhibited wake-associated expression in the microarray data (Arc, Egr2, Homer1, Nr4a1). Consistent with previous reports (Cirelli et al., 2004; Mackiewicz et al., 2007), gene ontology (GO) analysis of these trends identifies “response to stress” (Figure 5A ORB and PMCO trends and Figure 5B MENT trend; Table S3 in Supplementary Material), “response to unfolded protein,” and “inflammatory response” (Figure 5A) as enriched functional categories. These trends are exemplified by Cdkn1a, also known as p21WAF1/CIP1, a cell cycle gene regulated in response to DNA damage that has also been shown to be a circadian clock-controlled gene in some tissues (Grechez-Cassiau et al., 2008). Many genes from this trend type were later validated by ISH. Two other trends could be characterized as “sleep-associated” (Figures 5C,D), with or without a recovery during RS. GO analysis suggested that genes involved in vasculogenesis and epigenetic regulation of gene expression were enriched in sleep-associated gene clusters. Genes involved in the “regulation of transcription” were enriched in both wake- and sleep-associated trends (Figures 5A,D).
The trends shown in Figures 5E,F are likely driven by time-of-day or light-dark cycle, with the defining feature being either an up- (Figure 5F) or down- (Figure 5E) regulation during ZT18/W (dark phase). The SCN trend illustrated in Figure 5E includes genes associated with the GO term “rhythmic process” (p = 0.016), appropriate for a time-of-day trend occurring in the master circadian clock, and includes the well-known clock gene Per2. Two other GO terms were associated with both time-of-day and sleep/wake regulation. Ubiquitin-mediated proteolysis is important for tightly regulating the levels of some of the key circadian clock proteins (such as Per2). Genes involved in the “ubiquitin cycle” were down-regulated in the SCN in wake, consistent with a previous report that these genes exhibit circadian oscillation in the SCN (Panda et al., 2002). However, these genes also exhibited wake-associated expression in the SCN and Hcrt (Figures 5A,B). Similarly, “chromatin modification” has previously only been implicated in the control of circadian transcription (Etchegaray et al., 2003), but in the present study, genes associated with chromatin modification were affected by both time-of-day in the SCN (Figure 5F) as well as sleep/wake in the Hcrt and TMN, respectively (Figures 5A,D).
Only 48 of 175 trends are shown in Figure 5. The majority of the remaining trends show patterns of gene expression that reflect the combined influence of time-of-day, sleep/wake, SD, and RS-specific patterns. The last three trend types demonstrate some of the diversity of expression patterns. In Figures 5G,H, the only feature in common is upregulation in SD that is distinct from expression in W, while the trends in Figure 5I all exhibit downregulation of expression in SD. Genes differentially expressed between SD and SDC were identified (t-test, fold change > 1.5, p < 0.05). Out of 34,967 unique probes available on the microarray, approximately 4–6% of genes were significantly changed by SD for each brain region. Altogether across the seven brain regions, ∼22.6% of the probes were affected by SD in at least one brain region.
Anatomic Signatures of Sleep Deprivation Using High-Throughput in Situ Hybridization
In order to place the molecular characterization of SD into a cellular/anatomical context, high-throughput colorimetric ISH was used to screen 222 candidate genes (including the IEGs described above; gene list and primers in Table S1 in Supplementary Material) chosen to provide a broad survey of state-, condition-, and region-specific gene expression. Candidate selection was based on a combination of previously published studies documenting dynamic gene regulation across behavioral states or in response to SD (Table S2 in Supplementary Material) as well as the microarray experiments in the present study. ANOVA (across five conditions) p-values and t-test (SD versus SDC) p-values and fold change are provided for ISH candidates in Table S4 in Supplementary Material. The full ISH data set is available at http://sleep.alleninstitute.org. The screen was used to select 53 genes (including IEGs) showing the most robust qualitative changes across behavioral conditions, and these genes were processed on an additional two sets of animals to obtain triplicate data. Generally, the effects of SD elicited the most robust changes in gene expression, and thus the triplicate dataset heavily represents activation by SD. These dynamic genes included the following gene ontologies: circadian rhythm and rhythmic process (Per1, Dbp, Prok2, Prokr, Egr2, Ccrn4l), response to stress (Vwf, Prok2, Scg2, Crh, Bdnf, Cdkn1a, Cfp, Hspb1, Sgk), and regulation of apoptosis and progression through cell cycle (Prok2, Scg2, Pdia3, Bdnf, Cdkn1a, Nr4a1, Fosb, Snf1lk, Junb, Fos).
A brain-wide (Figure 6) and gene-specific (Figure 7) signature of the effects of SD on gene expression was generated using an algorithmic process. The set of 53 genes with triplicate ISH data were mapped to the reference atlas framework (Figure 2). The effects of SD for this gene set were widespread across the brain, with 65 of 209 brain regions showing statistically significant (p < 0.05) differential expression between SD and SDC for at least 6 and up to 24 genes in each region (Figure 6). Forebrain regions typically exhibited the most robust SD-associated changes across the most genes (Figure 6A; blue histogram bars represent striatum/pallidum; green histogram bars represent cerebral cortex), including olfactory structures (anterior olfactory nucleus, piriform cortex and main olfactory bulb), cerebral cortex, striatum, nucleus accumbens and hippocampus. In contrast, the hypothalamus and brainstem were notably unresponsive at a regional level to the effects of SD.
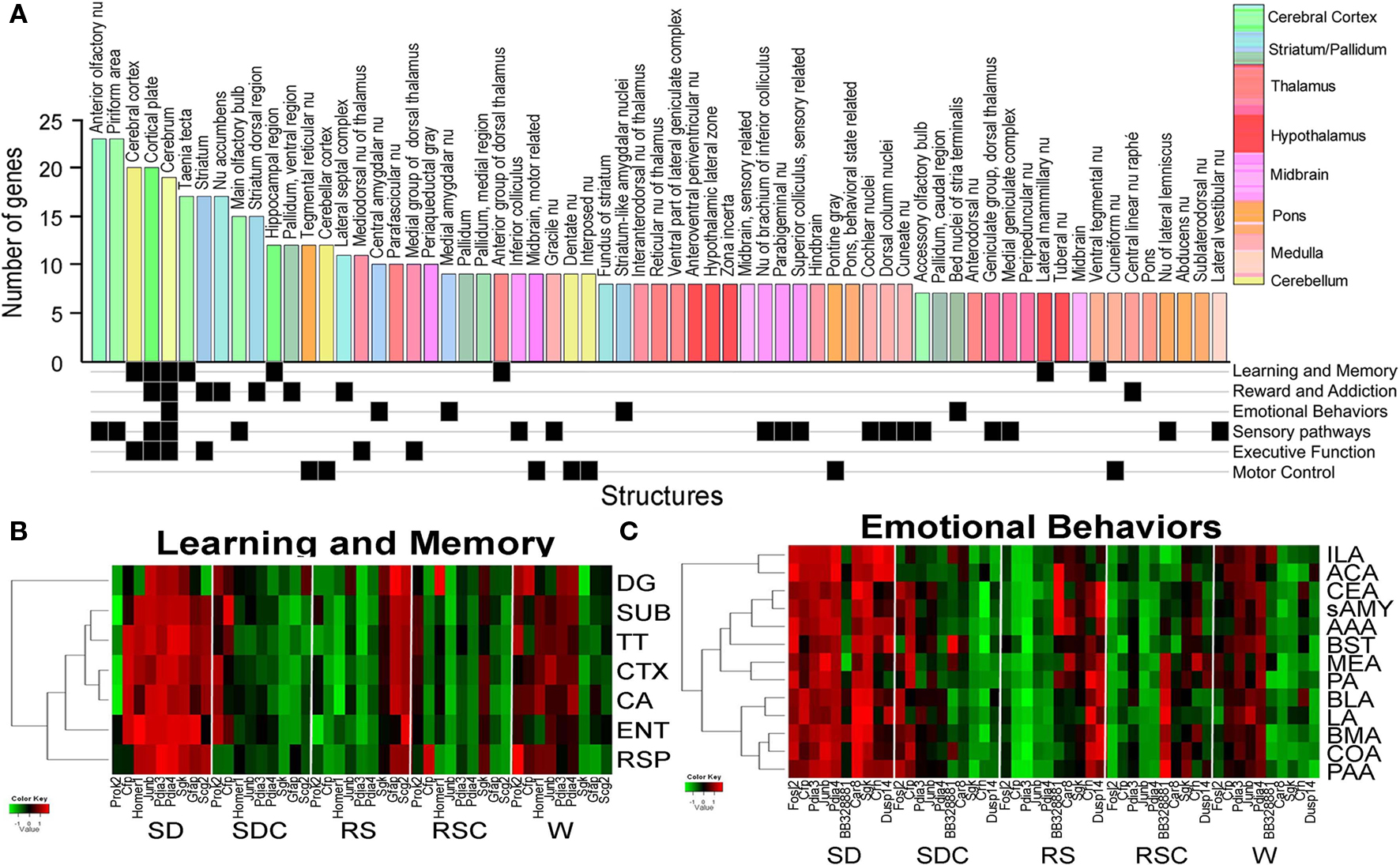
Figure 6. Signatures of sleep deprivation by anatomic region. (A) The histogram shows the number of genes exhibiting differential expression between SD and SDC (ANOVA p < 0.05) per anatomic region based upon automated quantification of triplicate ISH data from 53 genes using expression energy. Histogram bars are color-coded by the anatomic ontology based upon the Allen Reference Atlas for adult mouse brain, and the key to the colors is shown to the far right. Below the histogram, anatomic regions are annotated for involvement in different functional pathways based on the published literature. (B,C) Heatmaps showing the response of genes to five behavioral conditions for subsets of brain regions (y-axis) involved in either learning and memory (B) or emotional behaviors (C). Along the x-axis, genes which exhibited differential gene expression across five conditions for any of the given brain regions as determined by triplicate ISH (ANOVA, p < 0.05) are shown. Abbreviations: DG, dentate gyrus; SUB, subiculum; TT, tania tecta; CTX, cerebral cortex; CA, Ammon’s Horn CA region; ENT, entorhinal cortex; RSP, retrosplenial cortex; ILA, infralimbic area; ACA, anterior cingulate area; CEA, central nucleus of amygdala; sAMY, striatum-like amygdalar nuclei; AAA, anterior amygdalar area; BST, bed nuclei of stria terminalis; MEA, medial amygdalar nucleus; PA, posterior amygdalar nucleus; BLA, basolateral amygdala; LA, lateral amygdala; BMA, basomedial amygdala; COA, cortical amygdala; PAA, piriform-amygdalar area.
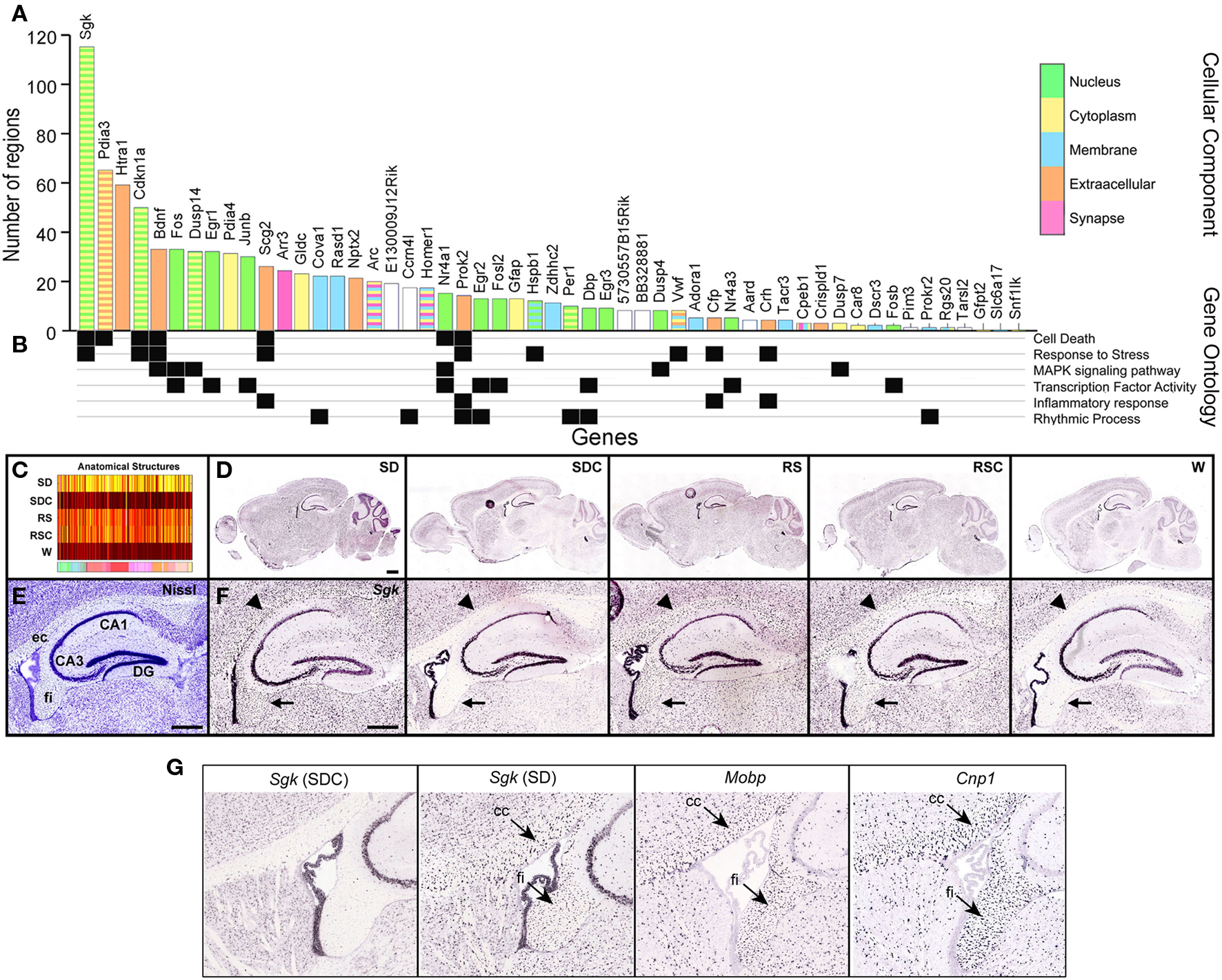
Figure 7. Signatures of sleep deprivation by gene. (A) The histogram shows the number of the 209 brain regions examined that showed statistically different expression in SD versus SDC (ANOVA p < 0.05) for the same ISH data set used in Figure 6. Histogram bars are color-coded by GO terminology for cellular component (key to far right). Note that some genes belong to multiple cellular component categories, as indicated by striped bars. (B) Annotation of genes for selected molecular function, biological process, or KEGG pathway terms. (C) Automated quantification across all five conditions showed that Sgk1, the gene showing the broadest anatomical response to SD in (A), exhibited widespread upregulation by both SD and by time-of-day (RS, RSC). (D,F) ISH data for Sgk1 across five behavioral conditions on a whole sagittal brain section (D) and in the hippocampus at high magnification (F). Sgk1 is upregulated selectively in oligodendrocyte–rich white matter tracts including the external capsule (ec, arrowhead) and fimbria (fi, arrow), delineated in a plane-matched Nissl-stained section (E). (G) Sgk expression in SDC strongly resembles the expression patterns of two well-known oligodendrocyte markers, Mobp and Cnp1. DG, dentate gyrus; CA1, CA3, hippocampal subfields. Scale bars: (D): 1 mm; (E,F): 500 μm.
The neuroanatomical distribution of changes in gene expression could indicate the associated behavioral functions affected by SD. Therefore, the brain regions shown in Figure 6A were manually annotated for their involvement in different functional pathways (Figure 6A, bottom). SD-affected gene expression in many brain regions involved in sensory pathways (visual, auditory, and somatosensory), as previously observed with IEG mapping. Brain regions associated with reward and addiction as well as learning and memory also exhibited numerous SD-associated changes. To further determine how these genes and brain regions were responding across all five behavioral conditions, we plotted gene expression for the associated brain regions for two functional pathways (learning and memory and emotional behaviors), including additional brain regions not part of the original 209 structures assessed in the algorithmic quantification pipeline (Figures 6B,C).
Most of the brain regions involved in learning and memory exhibited gene activation in SD, with some genes also being activated in either RS or W (but not both; Figure 6B). Interestingly, the three genes which exhibited activation in SD and RS but not W included two genes from the “response to stress” GO category (Sgk, Scg2), suggesting that SD specifically (but not W) may function as a stressor that induces gene expression which does not completely recover following 4 h of RS. Genes activated in SD and W, and thus presumably associated more generally with wakefulness and consequently neuronal activity, include two IEGs, Homer1, and Junb, which are commonly interpreted as markers of neuronal activity. While most of the brain regions in learning and memory behaved similarly, the dentate gyrus showed the greatest deviation from the group, exhibiting less SD- or W-induced gene expression.
Thirteen brain regions associated with emotional behaviors, including amygdalar regions and bed nucleus of stria terminalis, were also assessed for changes in gene expression (Figure 6C). Most of the genes shown in Figure 6C exhibit broad activation by SD across all of the brain regions associated with emotional behaviors. Similar to the learning and memory group, most genes exhibited activation either in SD and W or SD and RS across the majority of brain regions. Again, the genes which exhibited induction in SD and RS (but not W) included genes from the GO category “response to stress” (Sgk, Crh, Cfp), as well as Dusp14, which is in the stress-activated MAPK signaling pathway (note that Sgk was also upregulated in RSC). And once again, the genes represented in the awake and active groups SD and W (not RS), include two classical IEGs: Fosl2 and Junb.
Overall, there was a significant effect of SD on 50 of the 53 genes analyzed by ISH (Figure 7A). Some genes exhibited SD-associated changes in gene expression in very restricted brain regions (minimum of one brain region) and ranged to SD-associated changes in gene expression broadly across the brain (up to 115 brain structures for Sgk1). Within this 50 gene set, the median was 11 brain structures affected/gene. Genes that were significantly affected by SD in at least 20 brain regions were heavily selective for nuclear (green bars) and extracellular or secreted (orange bars) genes such as Bdnf or Scg2, suggesting that SD or its associated wakefulness resulted in changes in transcriptional regulatory events and or cell-cell signaling. Correspondingly, GO terms for “transcription factor activity” (Figure 7B) were well-represented in the set of 53 genes. The genes exhibiting the broadest range of changes across the brain also tended to belong to related gene ontology categories cell death, stress response and MAPK signaling. Serum/glucocorticoid-regulated kinase 1 (Sgk1) exhibited the broadest response to SD, with significant differential expression across nearly twice as many reference atlas structures as the most responsive IEGs. The expression heatmap for Sgk1 (Figure 7C) showed a generalized upregulation by SD as well as regulation by time-of-day with peak expression at the two ZT10 timepoints (RS and RSC). Examination of the primary ISH data explained the anatomical breadth of this gene regulation, in that Sgk1 appeared to be induced in oligodendrocytes across the entire brain (Figures 7D–F), based upon the distribution and density of labeled cells in white matter tracts. Expression in other cell types, including hippocampal CA3 pyramidal cells and the choroid plexus, appeared relatively unchanged across all conditions.
Independent Modes of Gene Regulation in SCN and Neocortex
The molecular components of the circadian clock are expressed nearly ubiquitously, with the existence of an autonomous circadian clock in each cell that is then synchronized via various mechanisms to the overall rhythm of the tissue or organism (Antle and Silver, 2005). Thus, while the SCN is considered the site of the master circadian clock that synchronizes the other tissues, circadian rhythms are observed in most other tissues and cell types, although the phase of these rhythms may lag behind the SCN (Yoo et al., 2004). The SCN was a target of analysis by both high-throughput ISH and microarray. Based upon our ISH analysis, differential gene expression in the SCN was overwhelmingly dependent upon time-of-day, and only a single gene, Gfap, was determined to be robustly induced by SD in the SCN as shown by ISH (Figure 8D). A number of genes were identified with differential gene expression in both SCN and ORB but, surprisingly, circadian regulation in the SCN was not predictive of gene regulation in other regions such as the neocortex. For example, in Figure 8, the microarray trends and ISH data of four genes (Snf1lk, Dbp, Rasd1, Nr4a1) are shown for ORB and SCN. Each of these genes were predominantly regulated by time-of-day in the SCN, with either peak expression during the day (Snf1lk, Dbp, Nr4a1) or peak expression at night (Rasd1). In contrast, the expression of Snf1lk, Rasd1, and Nr4a1 was elevated in the ORB during SD in both the microarray data and in the ISH. Furthermore, Dbp displayed an inverse time-of-day rhythm in the ORB compared to SCN, with peak expression in ORB at ZT10 and ZT18 as opposed to the trough expression at ZT18 in the SCN. Dbp expression in the ORB was reduced by SD.
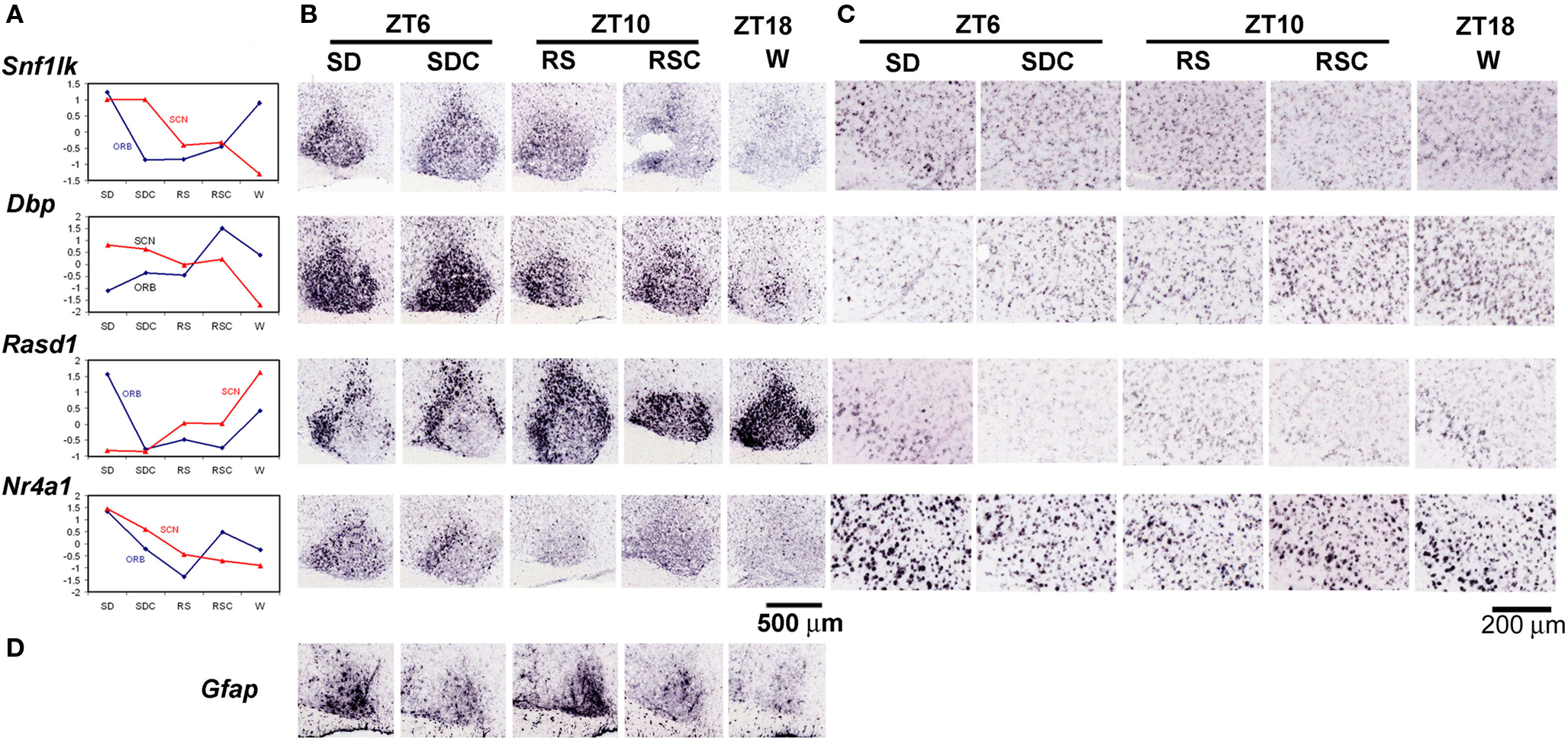
Figure 8. Differential effects of behavioral condition on the SCN and orbital cortex. (A) Microarray values shown as z scores for four genes, Snf1lk, Dbp, Rasd1, and Nr4a1, for both orbital cortex (blue) and SCN (red), demonstrating the different trends shown for the same gene across different brain regions. (B,C) Colorimetric ISH on sagittal sections through the SCN (B) and orbital cortex (C) across five behavioral conditions for the four genes shown in (A). (D) Induction of Gfap in the SCN by SD. Labels across the top indicate the circadian time (ZT6, 10, or 18) and whether there was an experimental manipulation (SD or RS). Scale bar in (B): 500 μm. Scale bar in (C): 200 μm.
Region and Cell Type-Specificity of SD-Induced Gene Expression in Neocortex and Caudate Putamen
The neocortex can be divided into functional areas (visual, somatosensory, motor, agranular insular, and orbitofrontal) as well as into classically described layers easily separated by histological characteristics (layers 1–6). Furthermore, within each of the layers exist numerous cell types including subsets of both inhibitory and excitatory neurons. Colorimetric ISH can be used to detect both areal- and laminar-specificity of gene expression. In the neocortex, mRNA induction by SD was observed in visual cortex, somatosensory cortex, agranular insular cortex and orbitofrontal cortex (Figure 9). Some genes were induced in all of these cortical areas (e.g., Arc, Scg2, Nptx2, Rgs20, Fos, Rasd1; Figures 9A–E,G), while induction of other genes exhibited areal specificity and were limited to visual and somatosensory cortex (e.g., Bdnf, Ccrn4l; Figures 9F,H), or just to visual cortex (Crh, Figure 9J). Crispld1 showed induction in somatosensory and agranular insular cortex but not visual cortex (Figure 9I). Laminar distribution of SD-induced gene expression in the neocortex also ranged broadly, from widespread induction across all layers (e.g., Arc, Scg2; Figures 9A,B) to highly layer-specific (layer 2/3 for Rasd1, Crispld1, Crh; layer 2 for Ccrn4l; Figures 9G–J). Genes with the highest specificity after SD were observed in superficial cortical layers and induction was limited or (most pronounced in some cases) to an extremely sparse and specific population of neurons in layer 2 (e.g., Nptx2, Ccrn4l; Figures 9C,H).
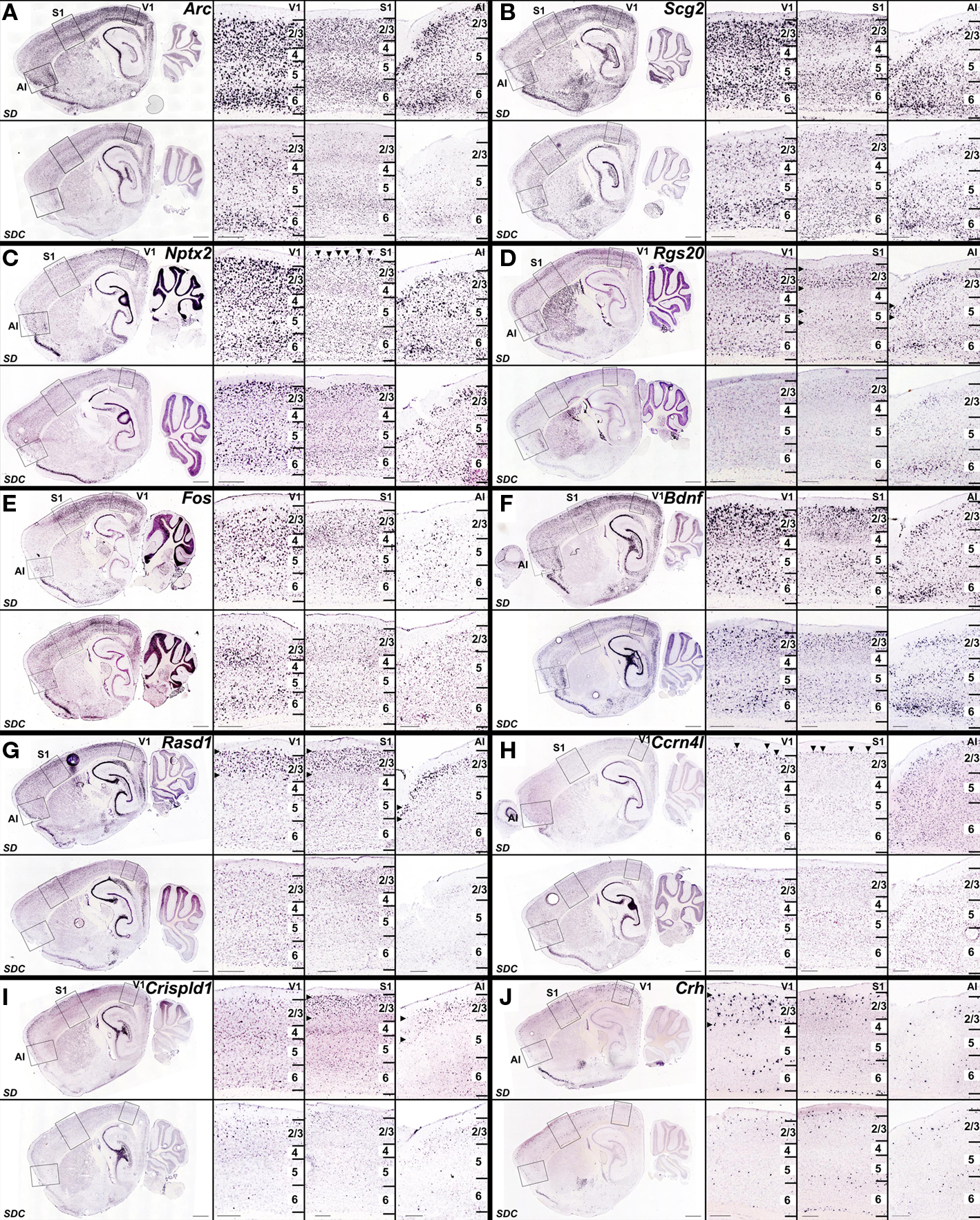
Figure 9. Heterogeneous cellular patterns of cortical gene upregulation by SD. (A–J) Colorimetric ISH for genes that were upregulated in SD (upper panels) relative to SDC (lower panels), shown at low magnification on a sagittal section through the lateral hippocampus (left), and at high magnification (right panels) in visual cortex (V1), somatosensory cortex (S1) and agranular insular cortex (AI) corresponding to boxed regions in low magnification images. Specific laminar and regional patterns of gene upregulation by SD varied from gene to gene, from broad upregulation in all three regions in all layers [e.g., Arc (A), Scg2 (B)], to increasingly layer- and region-specific upregulation for Nptx2 (C), Rgs20 (D), Fos (E), Bdnf (F), Rasd1 (G), Ccrn4l (H), Crispld1 (I), and Crh (J). Horizontal arrowheads in (D,G,I,J) indicate layer-specific SD induction, and arrowheads in (C) and (H) delineate selective gene induction in neurons at the upper boundary of layer 2. Scale bars: low magnification images, 1 mm; high magnification images, 250 μm.
Double-labeling with marker genes for inhibitory (Gad1) and excitatory (Slc17a7) neurons demonstrated that the vast majority of gene upregulation was restricted to excitatory neurons. For example, there was no co-labeling of Arc (Figures 10A–C), Nr4a1 or Nptx2 (Figures 10H–K) with Gad1, the major synthetic enzyme for GABA that marks inhibitory neurons. Unlike Arc, Nr4a1 and Nptx2, Fos co-labeled a very small percentage of Gad1-positive neurons (Figures 10D,E), although the vast majority of Fos-positive neurons co-labeled with the excitatory neuron-specific vesicular glutamate transporter 1 (Slc17a7; Figures 10F,G). Expression of Rasd1 in layer 2/3 partially overlapped with Gad1 (Figures 10L,M), although there was also expression of Rasd1 in SDC (not shown), so it is unclear whether the specific neurons showing SD induction of Rasd1 were inhibitory neurons.
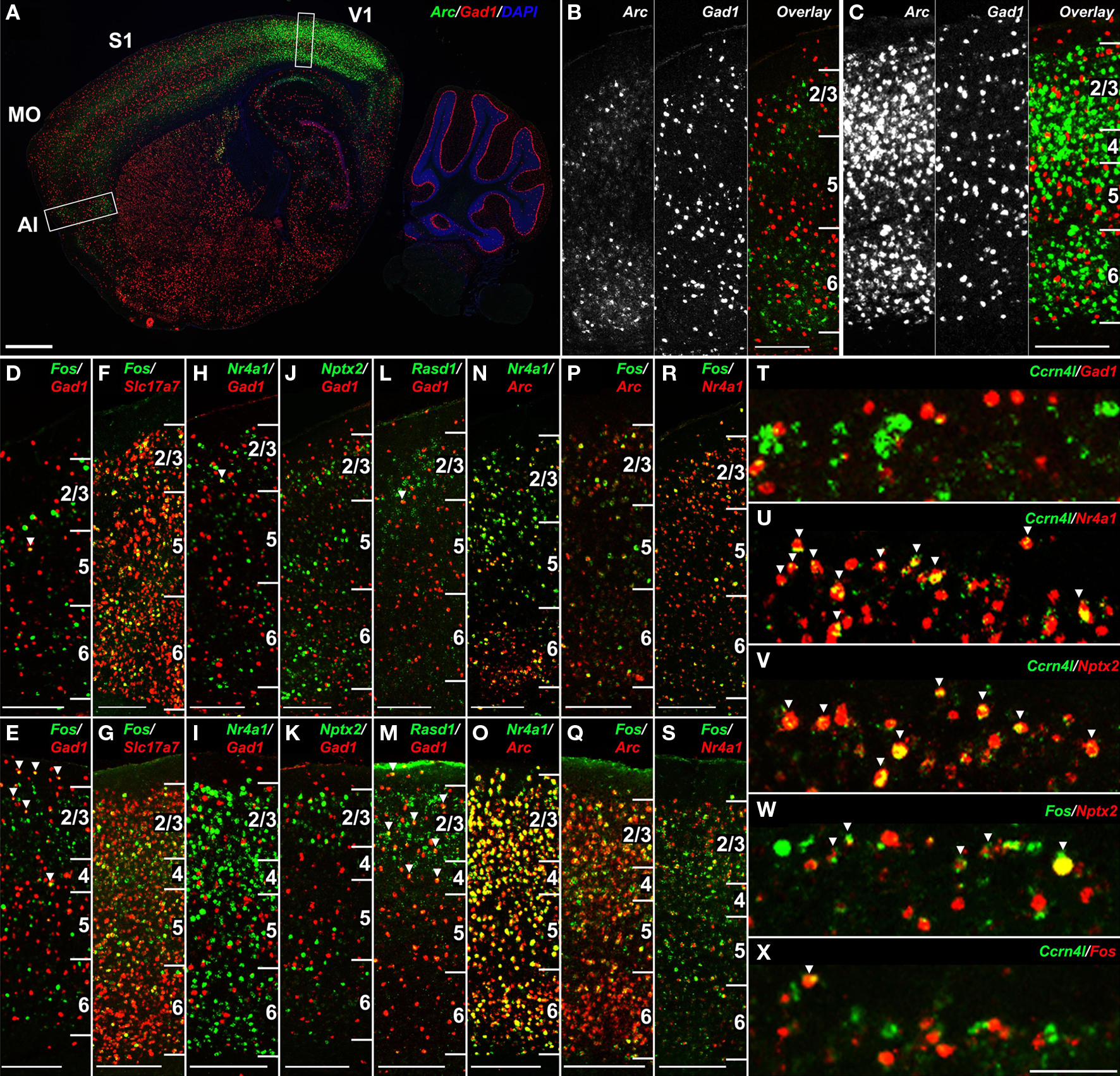
Figure 10. SD-induced transcripts in the neocortex label overlapping populations of excitatory neurons. (A–C) Double fluorescent ISH for Arc (green) and Gad1 (red) at low magnification (A) and high magnification in agranular insular cortex (B) and visual cortex (C) corresponding to boxed regions in (A), demonstrating lack of co-labeling in the neocortex. (D–S) Cellular colocalization of genes showing cortical gene upregulation following SD with canonical inhibitory (Gad1) and excitatory (Slc17a7) neuronal markers and with one another in agranular insular cortex (D,F,H,J,L,N,P,R) and primary visual cortex (E,G,I,K,M,O,Q,S). Fos is largely restricted to excitatory neurons (D–G), neither Nr4a1 nor Nptx2 colocalize with Gad1 (H–K), and Rasd1 partially overlaps with Gad1 (L,M). Arc, Nr4a1 and Fos label partially overlapping neuronal populations (N–S). (T–X) Gene-specific cellular specificity in layer 2 of somatosensory cortex following SD. Ccrn4l is expressed in a subset of Nr4a1 and Nptx2-positive excitatory neurons (T–V), Fos partially overlaps with Nptx2 (W), yet Ccrn4l and Fos label nearly non-overlapping populations (X). Numbers in (B–S) delineate cortical layers. Scale bars: (A), 1 mm; (B–S), 250 μm; (T–X), 100 μm.
Double-labeling was also used to determine whether genes were induced by SD in the same cell populations. Arc, Nr4a1 and Fos labeled largely overlapping cellular populations, with some single-gene labeled neurons in each comparison in visual and agranular insular cortices (Figures 10N–S). On the other hand, the robust induction of Ccrn4l in a sparse population of neurons in layer 2 of somatosensory cortex, which was almost entirely excitatory based on limited co-labeling with Gad1 (Figure 10T), was entirely a subset of Nr4a1-expressing neurons (Figure 10U). Ccrn4l-expressing neurons was also entirely a subset of Nptx2, another gene displaying robust layer 2 induction (Figure 10V). Although Nptx2 showed partial overlap with Fos in layer 2 (Figure 10W), Ccrn4l and Fos expression were surprisingly mutually exclusive (Figure 10X), indicating that Nptx2 induction by SD in layer 2 labeled two separate cell populations.
The use of ISH to confirm differential gene expression identified from the microarray dataset provided a characterization of these genes across the brain, far more comprehensive than the seven brain regions specifically targeted for microarray. For instance, ISH revealed that the caudate putamen was a frequent site of robust SD-induced gene expression, although this region was not included in the microarray experiments. Robust gene induction by SD was observed across many genes in the caudal dorsolateral caudate putamen and, in several cases (e.g., Nts, Arr3), the effect of SD was nearly restricted to this region (Figures 11C,E). The numbers of labeled cells in the caudate putamen varied widely from gene to gene. For example, Arc and Nr4a1 labeled large numbers of striatal neurons (Figures 11A,B), Nts and Ccrn4l labeled smaller numbers (Figures 11C,D) and Arr3 labeled only a handful of neurons per brain section (Figure 11E). In general, the different SD-induced genes labeled partially overlapping subsets of neurons in SD, with the sparser populations generally labeling subsets of the more broadly expressing genes. For example, Nts is entirely a subset of Nr4a1-positive cells (Figure 11F), while Ccrn4l and Nts labeled partially overlapping cell populations (Figure 11I). To identify the striatal neuron subtypes labeled by SD-induced genes, co-labeling was performed against two neuropeptides which are non-overlapping markers for the two subcategories of medium spiny neurons in the caudate putamen, accounting for 95% of striatal neurons in rodents (Tepper and Bolam, 2004). Proenkephalin 1 (Penk1) is expressed in neurons in the indirect striatopallidal pathway, and prodynorphin (Pdyn) is expressed in neurons in the direct striatonigral pathway (Kawaguchi, 1997). Expression of Nr4a1 in SD partially overlapped with Pdyn (Figure 11G) and Ccrn4l partially overlapped with Penk1 (Figure 11J), suggesting that these genes are expressed in both subsets of medium spiny neurons. However, Nts expression was completely exclusive of Pdyn expression (Figure 11H), indicating that Nts induction by SD is exclusive of the direct striatonigral pathway, and is mostly likely occurring in neurons of the indirect striatopallidal pathway. Overall, these data show that SD affects gene expression reproducibly across several gross brain regions (e.g., neocortex and caudate putamen), within each of these regions there are complicated cell type-specific responses that likely reflect some combination of differential timecourses of induction and gene-specific induction in specific neuronal subtypes.
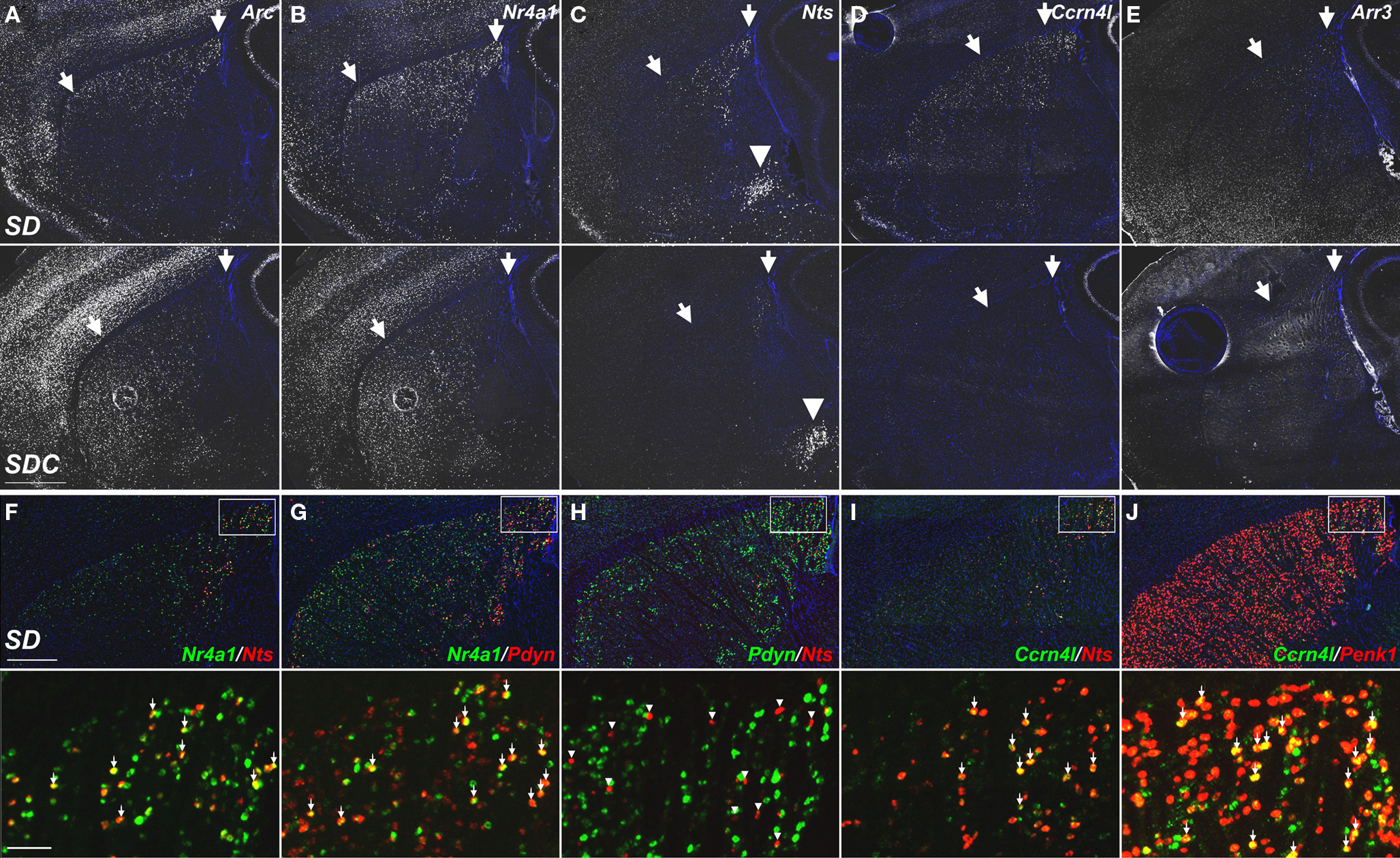
Figure 11. Molecular phenotyping neuronal populations exhibiting SD-induced gene expression in the dorsolateral caudate putamen. (A–E) Fluorescent ISH images showing selective induction of Arc, Nr4a1, Nts, Ccrn4l, and Arr3 in the caudal dorsolateral caudate putamen of SD (upper panels) and SDC (lower panels) groups. Striatal expression of Nts, Ccrn4l, and Arr3 was specific to SD, with no expression in SDC (C–E). Arrows indicate the approximate region showing SD gene upregulation. SD induction of Nts was specific to the caudate putamen, while expression in the amygdala [arrowheads in (C)] was unaffected by SD. (F–J) Cellular colocalization of SD-induced mRNAs using double fluorescent ISH. Upper panels show co-labeling across the entire dorsal caudate putamen, and lower panels show a magnified view corresponding to the boxed areas. Nts extensively colocalizes with Nr4a1 (F) and Ccrn4l (I), but does not overlap with Pdyn (H). Arrows in (F,G,I,J) indicate cells expressing both respective gene products. Arrowheads in H indicate Nts-expressing neurons that do not co-label with Pdyn. All sections were counterstained with DAPI (blue). Scale bars: (A–E), 1 mm; (F–J) upper panels, 500 μm; lower panels, 50 μm.
Discussion
Mapping the Effects of Sleep Deprivation
Previous studies have documented gene regulation by sleep/wake and SD (Cirelli and Tononi, 1998; Cirelli et al., 2004; Terao et al., 2006; Mackiewicz et al., 2007), visual input (Schoups et al., 1995), and circadian time (Panda et al., 2002). The current study utilized both a genome-scale survey of gene expression for specific brain regions (microarray) and a cellular resolution approach (ISH) that provided anatomic localization of gene activation. The behavioral paradigm was designed to identify gene regulation associated with arousal state, SD, and RS after SD. Environmental condition is an important driver of gene expression, as some of the gene induction occurred in visual structures in mice awake during the light phase (e.g., SD). In general, gene activation patterns were related to recent sleep history and environmental conditions and largely reflected presumed neuronal activity. Other examples of gene regulation included global time-of-day- and SD-associated induction of Sgk1 in oligodendrocytes, and SD-induced cortical expression of Crispld1 in all cortical regions except visual cortex. The observed changes in gene expression can reflect changes either in the level of transcription or in mRNA stability.
Despite a lengthy history of the investigation of the impact of behavioral paradigms on IEG expression, the present study demonstrates the value of understanding the nuances of the effects of SD on neuronal activity. The highest expression of IEGs and many other genes occurred in SD and W conditions as previously described (Cirelli and Tononi, 2000a; Terao et al., 2003a). Although the similarity between IEG expression in cortex or brain in SD and W has previously been interpreted as a correlate of waking, the cellular resolution ISH data demonstrated that the precise anatomical distributions of IEG mRNAs were distinct between these two conditions, and thus may be reflective of distinct molecular pathways or physiological functions. In addition, while IEGs were generally higher in SD and to a lesser extent in W, specific subcompartments of the neocortex, caudate putamen, and thalamus could be found where IEG expression was higher in sleep as compared to SD or W, indicating that IEG expression was not simply a correlate of waking. Finally, RS mice often showed a pattern similar to SD for many genes and an expression level only slightly lower than SD, suggesting that 4 h post-SD was sufficient to allow a partial recovery from SD to occur. Consistent with these findings, previous reports have shown similar molecular induction profiles in SD and RS conditions (Terao et al., 2003a).
Genes Affected by Behavioral State
Although most previous studies have interpreted gene regulation by sleep or wakefulness as a binary division of states, or as a longitudinal investigation of the cumulative effects of increasing SD, one study evaluated the effect of 6 h SD by microarray of whole brain mRNA at four different timepoints in the day (Maret et al., 2007), and showed that SD largely disrupted rhythmic expression of genes by time-of-day. In the current study, the responses of genes to SD, RS, and time-of-day have been assessed by microarray as a spectrum of brain region-specific “trends,” identifying trends that appear to be time-of-day-specific, SD-specific, sleep/wake-specific, as well as a mixture of time-of-day, sleep/wake, and SD- or RS-dependent effects on gene expression. Furthermore, by identifying clusters of genes sharing the same trend across experimental conditions (suggesting possible co-regulation), we identified unique GO categories that were associated with the specific trends.
Like other studies that have identified stress response genes and unfolded protein response genes that increase in the cerebral cortex with prolonged waking (Terao et al., 2003b; Cirelli et al., 2004; Mackiewicz et al., 2007; Zhu et al., 2008), we observed these GO categories associated with SD- and W-induction in orbital and entorhinal cortex, and cortical amygdala (Figures 5A,B). ISH also provided confirmation of behavioral condition-specific changes in gene expression for several stress response genes, including Sgk, Cdkn1a, Bdnf, Scg2, Prok2, Hspb1, and Crh. Sgk has been previously identified as an SD-responsive gene in the cerebral cortex by differential display (Cirelli and Tononi, 2000a), and in the LC and dorsal raphe by microarray (Conti et al., 2007). In the current study Sgk, known for its rapid regulation by stress-induced corticosterone levels, was also broadly upregulated across the brain in response to SD and time-of-day, but the upregulation appeared to occur predominantly in oligodendrocytes, not in neurons. The robust induction of the glial marker Gfap in the SCN by SD may also be related to stress. Diurnal Gfap immunoreactivity in the SCN was found to be dependent on corticosterone in adrenalectomized rats (Maurel et al., 2000). If SD-induced Gfap expression is also dependent on corticosterone, it could suggest a glial mechanism to provide stress feedback to circadian rhythm. These data suggest that SD induces a stress hormone-based response predominantly in glia rather than in neurons. A recent study evaluated the contribution of stress to molecular markers of SD using adrenalectomized mice and, using microarrays of forebrain mRNA, identified large numbers of genes whose expression in response to SD was attributable to stress, including Sgk (Mongrain et al., 2010). However, many genes were reportedly induced by SD despite adrenalectomy, including many of the genes characterized in the present study, such as the majority of the IEGs, Bdnf, Homer1, Dbp, Per2, Hspb1, and Dusp4.
There have been previous reports that circadian clock genes may be involved in sleep homeostasis in the cerebral cortex, particularly Per1, Per2, and Dbp (Franken et al., 2007). In the present study, ISH demonstrated that the expression of Per1 (data not shown), Per2 (n = 1; data not shown), Dbp, as well as non-circadian clock genes including Snf1lk, Rasd1, and Nr4a1, was primarily regulated by time-of-day in the SCN and by SD in the neocortex. It is not clear whether the occurrence of SCN-specific circadian regulation combined with cortex-specific sleep-regulation are general phenomenon for many genes, or whether the observance of this type regulation for Snf1lk, for instance, implies that it should be considered as a putative circadian clock gene. Salt-inducible kinase, Snf1lk, could be involved in circadian “gating,” whereby light-induced phase-shifting of the circadian rhythm is gated to restrict the effect of light by time-of-day. Induction of this kinase in the SCN during the light phase could allow Snf1lk to prevent CREB-mediated phase-shifting (Antle and Silver, 2005) during the light-insensitive phase through phosphorylation and repression of CREB (Katoh et al., 2004).
Another gene, Arr3, a presumed retina-specific gene, is not expressed in the unmanipulated state at ZT6, but is instead only detected in the brain following SD, restricted to sparse cells in striatum, cortical amygdala, and superior colliculus. Arrestins desensitize and recycle G protein-coupled receptors (Luttrell and Lefkowitz, 2002). Thus, arrestin upregulation could act as a protective mechanism to desensitize signaling pathways following sustained neuronal activity.
Anatomical Signatures of Sleep Deprivation
These data delineate a discrete set of structures that likely mediate the major and immediate consequences of SD. Informatics-based mapping of differential gene expression in ISH experiments revealed the greatest number of genes induced by SD in the neocortex, hippocampus, olfactory structures, amygdala and striatum (Figure 6). Surprisingly, despite the identification of numerous gene candidates by microarray profiling of the sleep regulatory tuberal hypothalamus (Hcrt neurons), TMN and LC, no robust qualitative changes in gene expression were seen in these regions by ISH. The failure to detect changes in these nuclei reflects the general observation noted above that the hypothalamus and brainstem were notably unresponsive to the effects of SD (Figure 6).
Expression patterns of genes induced by SD were typically seen in topographically restricted, subfield- and/or cell type-specific patterns within each major forebrain region. Cortical and striatal IEG activation may be directly linked, as topographically-specific IEG induction in the striatum has been shown to result from activation of cortical areas projecting to those striatal regions (Sgambato et al., 1997; Miyachi et al., 2005), and the dorsolateral striatum is known to be involved in sensorimotor integration. Furthermore, the striatum sends a massive inhibitory projection to the thalamus, where the VPM nucleus, relaying somatosensory information to the cortex, showed a specific reduction in Fos labeling in SD (Figure 3). These results suggest differential activation of thalamocorticostriatal circuits in sleeping versus SD. In this regard, there is increasing interest in the role of dopamine in sleep and the response to SD (Monti and Jantos, 2008; Volkow et al., 2009).
The anatomic localization of SD-induced changes in the neocortex may be relevant to the synaptic homeostasis hypothesis of sleep function (Tononi and Cirelli, 2006). In this hypothesis, wakefulness results in increased synaptic potentiation in cortical circuits, while sleep is responsible for synaptic downscaling. A homeostatic relationship between potentiation and sleep is proposed whereby the amount of wake-associated synaptic potentiation correlates with an increase in slow wave activity in the subsequent sleep phase, and this slow wave activity is implicated in synaptic downscaling during sleep. Evidence for the role of wakefulness in synaptic potentiation comes in part by the upregulation in spontaneous waking of genes associated with long-term potentiation, such as Arc, Bdnf, Egr1, Homer1, and Nptx2 (Tononi and Cirelli, 2006). In the present study Arc, Bdnf, and Nptx2 (Figure 9) were all shown to be induced by SD broadly across different layers of the cortex, suggesting that they may have a widespread role in the cortex during wakefulness towards the regulation of synaptic potentiation. However, several other genes (Rasd1, Ccrn4l, Crispld1) exhibited SD-associated expression specifically in superficial layers 2/3. There is evidence that layers 2/3 are involved in propagation of slow waves across the neocortex during sleep (Sanchez-Vives and McCormick, 2000). Thus it is possible that broad upregulation of synapse-related genes may be associated with wake-associated potentiation, while the specific upregulation of genes in L2/3 by SD could alter the propagation and homeostatic regulation of slow waves, which in turn impacts synaptic downscaling. Future studies would be needed to evaluate the role of the SD-affected cell types in the superficial neocortex in slow wave propagation.
The brain regions most affected by SD are broadly consistent with cognitive, mnemonic and emotional deficits elicited by SD. Frontal cortical regions are associated with executive function, while the hippocampal formation is critical for memory encoding and retrieval. Cerebral blood flow imaging studies in humans have shown that SD alters responses of the striatum to spatial memory tasks (Orban et al., 2006). A particularly intriguing finding is the upregulation of neurotensin (Nts) in the dorsolateral caudate putamen by SD. Nts is implicated in schizophrenia, and has been suggested to mediate actions of antipsychotic drugs and psychostimulants (Dobner et al., 2003). Typical antipsychotic drugs increase Fos and Nts expression in the dorsolateral striatum (Merchant et al., 1992), while intrastriatal Nts administration increases extracellular levels of dopamine, GABA and glutamate (Ferraro et al., 1995, 1997, 1998, 2001). The induction of Nts by SD in the dorsolateral striatum may also indicate a hyperactivation of dopaminergic circuitry in sensorimotor gating functions. Consistent with such a role, human studies measuring reaction times to a visual task have shown changes in thalamocortical gating affecting attentional and decision-making functions as a consequence of total SD (Corsi-Cabrera et al., 1999).
Molecular Phenotyping of Neurons Affected by SD Indicates Cellular Heterogeneity
Double-labeling experiments demonstrated the cellular specificity of gene induction by SD. For instance, gene induction by SD in the cortex varied from widespread expression to layer- or areal-specific, and some genes showed further cellular specificity to scattered cell types or a thin band of neurons in layer 2. Systematic co-labeling of SD-induced genes with one another and with canonical cell type markers yielded an exceptionally complex data set, resulting in the following organizing principles. First, the vast majority of SD-induced gene expression occurred in excitatory neurons, except for the caudate putamen which is populated almost entirely by inhibitory projection neurons. Second, the majority of activity-regulated genes label largely the same anatomical regions, but with only partially overlapping excitatory neuron populations. This could be interpreted as the result of sampling IEGs that have differential activation timecourses and cell type-specificity of induction. There was one case of complete non-overlap in somatosensory cortex as well. Both nocturnin (Ccrn4l) and Fos labeled subsets of Nr4a1 and Nptx2-positive neurons in superficial layer 2, yet Fos and Ccrn4l did not co-label the same cells. Ccrn4l, an mRNA deadenylase that was initially identified as a circadian-controlled gene in Xenopus (Green and Besharse, 1996), is expressed rhythmically in multiple mammalian tissues (Wang et al., 2001), and is acutely inducible in cell culture by serum shock or TPA (Garbarino-Pico et al., 2007). A reasonable hypothesis is that Ccrn4l, which speeds mRNA decay by removing polyA tails, is a late response gene induced after Fos levels have returned to baseline and may even be causative for Fos mRNA decay. Why Ccrn4l localization would be specific to cortical layer 2 and part of the striatum is unclear. Lastly, among affected GABAergic neurons, cellular specificity of gene induction was observed in the striatum. Nts expression was exclusive from Pdyn expression in striasomal compartments, indicating that Nts was not induced in the direct striatonigral pathway.
Summary and Perspective
The present study represents a “discovery science” approach to understanding the anatomical and molecular correlates of sleeping, waking, and SD. The use of IEG mapping to identify regions affected by SD allowed an empirical selection of targets for microarray analysis. The use of high-throughput ISH (>200 genes) to validate and characterize changes in gene expression in response to SD provided a large dataset amenable to informatics-based analyses to statistically identify brain regions and genes affected by behavioral state. These data provide a wealth of knowledge for future studies. We conclude that SD leads to the induction of numerous genes in forebrain regions relevant to brain functions affected by SD, including cognition, memory and emotional control, and within specific cellular populations that have not previously been studied in detail in the context of SD, such as the striatum and superficial layers of the neocortex. Understanding how these cell populations may be connected to the circuitry involved in higher cognitive functions is an essential next step to understanding the significance of the anatomical localization of the effects of SD. Second, a number of the genes identified have not previously been described in the context of SD, particularly Arr3, Ccrn4l, Rasd1, Nptx2, Crispld1, Rgs20, and Snf1lk, in addition to other genes listed in Figure 7 which were not discussed specifically including Htra1, Tacr3, Dusp7, and Pim3. Some of these genes may provide potential targets for pharmacological intervention to alleviate the consequences of SD or may help to understand protective mechanisms against prolonged activity or waking. Lastly, a major goal of the project was to create a resource for presenting the data set to the sleep research community to fuel discovery across the field. Consequently, the complete ISH data set is publicly available at http://sleep.alleninstitute.org.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by the Department of Defense USAMRAA award # W81XWH-06-1-0131 and NIH RO1 HL59658. The authors wish to thank the Allen Institute founders, Paul G. Allen and Jody Patton, for their vision, encouragement, and support. The authors also wish to acknowledge Tracy Lemon, Rebecca Morgan, and Theresa Zwingman for assistance with laser microdissection, Sabrina Gates for RNA purification, Sheana Parry and Randy Ng for assistance with quantification, Jolene Kidney for advice on dissection/embedding, Suvro Datta for help with densitometry, Lia Noges for assessment of gene expression, Conor Kelly for assistance with figure preparation, Paul Wohnoutka and Chinh Dang for supporting production and presentation of the data online, and Maheen Hassan and Kristy Silveira for assistance with surgery, EEG recording and behavioral state classification.
References
Antle, M. C., and Silver, R. (2005). Orchestrating time: arrangements of the brain circadian clock. Trends Neurosci. 28, 145–151.
Borbely, A. A., Tobler, I., and Hanagasioglu, M. (1984). Effect of sleep deprivation on sleep and EEG power spectra in the rat. Behav. Brain Res. 14, 171–182.
Chee, M. W., and Chuah, L. Y. (2008). Functional neuroimaging insights into how sleep and sleep deprivation affect memory and cognition. Curr. Opin. Neurol. 21, 417–423.
Cirelli, C. (2006). Cellular consequences of sleep deprivation in the brain. Sleep Med. Rev. 10, 307–321.
Cirelli, C., Gutierrez, C. M., and Tononi, G. (2004). Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron 41, 35–43.
Cirelli, C., and Tononi, G. (1998). Differences in gene expression between sleep and waking as revealed by mRNA differential display. Brain Res. Mol. Brain Res. 56, 293–305.
Cirelli, C., and Tononi, G. (2000a). Gene expression in the brain across the sleep-waking cycle. Brain Res. 885, 303–321.
Cirelli, C., and Tononi, G. (2000b). On the functional significance of c-fos induction during the sleep-waking cycle. Sleep 23, 453–469.
Conti, B., Maier, R., Barr, A. M., Morale, M. C., Lu, X., Sanna, P. P., Bilbe, G., Hoyer, D., and Bartfai, T. (2007). Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol. Psychiatry 12, 167–189.
Corsi-Cabrera, M., Arce, C., Del Rio-Portilla, I. Y., Perez-Garci, E., and Guevara, M. A. (1999). Amplitude reduction in visual event-related potentials as a function of sleep deprivation. Sleep 22, 181–189.
Deboer, T., Franken, P. and Tobler, I. (1994). Sleep and cortical temperature in the Djungarian hamster under baseline conditions and after sleep deprivation. J. Comp. Physiol. 174, 145–155.
Dobner, P. R., Deutch, A. Y., and Fadel, J. (2003). Neurotensin: dual roles in psychostimulant and antipsychotic drug responses. Life Sci. 73, 801–811.
Dong, H. W. (2008). The Allen Reference Atlas: A Digital Color Brain Atlas of C57BL/6J Male Mouse. Hoboken: John Wiley and Sons.
Etchegaray, J. P., Lee, C., Wade, P. A., and Reppert, S. M. (2003). Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 421, 177–182.
Ferraro, L., Antonelli, T., O’Connor, W. T., Fuxe, K., Soubrie, P., and Tanganelli, S. (1998). The striatal neurotensin receptor modulates striatal and pallidal glutamate and GABA release: functional evidence for a pallidal glutamate-GABA interaction via the pallidal-subthalamic nucleus loop. J. Neurosci. 18, 6977–6989.
Ferraro, L., O’Connor, W. T., Antonelli, T., Fuxe, K., and Tanganelli, S. (1997). Differential effects of intrastriatal neurotensin(1-13) and neurotensin(8-13) on striatal dopamine and pallidal GABA release. A dual-probe microdialysis study in the awake rat. Eur. J. Neurosci. 9, 1838–1846.
Ferraro, L., Tanganelli, S., O’Connor, W. T., Bianchi, C., Ungerstedt, U., and Fuxe, K. (1995). Neurotensin increases endogenous glutamate release in the neostriatum of the awake rat. Synapse 20, 362–364.
Ferraro, L., Tomasini, M. C., Fernandez, M., Bebe, B. W., O’Connor, W. T., Fuxe, K., Glennon, J. C., Tanganelli, S., and Antonelli, T. (2001). Nigral neurotensin receptor regulation of nigral glutamate and nigroventral thalamic GABA transmission: a dual-probe microdialysis study in intact conscious rat brain. Neuroscience 102, 113–120.
Franken, P., Thomason, R., Heller, H. C., and O’Hara, B. F. (2007). A non-circadian role for clock-genes in sleep homeostasis: a strain comparison. BMC Neurosci. 8, 87. doi: 10.1186/1471-2202-8-87.
Garbarino-Pico, E., Niu, S., Rollag, M. D., Strayer, C. A., Besharse, J. C., and Green, C. B. (2007). Immediate early response of the circadian polyA ribonuclease nocturnin to two extracellular stimuli. RNA 13, 745–755.
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., Ellis, B., Gautier, L., Ge, Y., Gentry, J., Hornik, K., Hothorn, T., Huber, W., Iacus, S., Irizarry, R., Leisch, F., Li, C., Maechler, M., Rossini, A. J., Sawitzki, G., Smith, C., Smyth, G., Tierney, L., Yang, J. Y., and Zhang, J. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80.
Gerashchenko, D., Wisor, J. P., Burns, D., Reh, R. K., Shiromani, P. J., Sakurai, T., de la Iglesia, H. O., and Kilduff, T. S. (2008). Identification of a population of sleep-active cerebral cortex neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 10227–10232.
Grechez-Cassiau, A., Rayet, B., Guillaumond, F., Teboul, M., and Delaunay, F. (2008). The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J. Biol. Chem. 283, 4535–4542.
Green, C. B., and Besharse, J. C. (1996). Identification of a novel vertebrate circadian clock-regulated gene encoding the protein nocturnin. Proc. Natl. Acad. Sci. U.S.A. 93, 14884–14888.
Huber, R., Deboer, T. and Tobler, I. (2000). Effects of sleep deprivation on sleep and sleep EEG in three mouse strains: empirical data and simulations. Brain Res. 857, 8–19.
Johnson, W. E., Li, C., and Rabinovic, A. (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127.
Katoh, Y., Takemori, H., Min, L., Muraoka, M., Doi, J., Horike, N., and Okamoto, M. (2004). Salt-inducible kinase-1 represses cAMP response element-binding protein activity both in the nucleus and in the cytoplasm. Eur. J. Biochem. 271, 4307–4319.
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., Boe, A. F., Boguski, M. S., Brockway, K. S., Byrnes, E. J., Chen, L., Chen, L., Chen, T. M., Chin, M. C., Chong, J., Crook, B. E., Czaplinska, A., Dang, C. N., Datta, S., Dee, N. R., Desaki, A. L., Desta, T., Diep, E., Dolbeare, T. A., Donelan, M. J., Dong, H. W., Dougherty, J. G., Duncan, B. J., Ebbert, A. J., Eichele, G., Estin, L. K., Faber, C., Facer, B. A., Fields, R., Fischer, S. R., Fliss, T. P., Frensley, C., Gates, S. N., Glattfelder, K. J., Halverson, K. R., Hart, M. R., Hohmann, J. G., Howell, M. P., Jeung, D. P., Johnson, R. A., Karr, P. T., Kawal, R., Kidney, J. M., Knapik, R. H., Kuan, C. L., Lake, J. H., Laramee, A. R., Larsen, K. D., Lau, C., Lemon, T. A., Liang, A. J., Liu, Y., Luong, L. T., Michaels, J., Morgan, J. J., Morgan, R. J., Mortrud, M. T., Mosqueda, N. F., Ng, L. L., Ng, R., Orta, G. J., Overly, C. C., Pak, T. H., Parry, S. E., Pathak, S. D., Pearson, O. C., Puchalski, R. B., Riley, Z. L., Rockett, H. R., Rowland, S. A., Royall, J. J., Ruiz, M. J., Sarno, N. R., Schaffnit, K., Shapovalova, N. V., Sivisay, T., Slaughterbeck, C. R., Smith, S. C., Smith, K. A., Smith, B. I., Sodt, A. J., Stewart, N. N., Stumpf, K. R., Sunkin, S. M., Sutram, M., Tam, A., Teemer, C. D., Thaller, C., Thompson, C. L., Varnam, L. R., Visel, A., Whitlock, R. M., Wohnoutka, P. E., Wolkey, C. K., Wong, V. Y., Wood, M., Yaylaoglu, M. B., Young, R. C., Youngstrom, B. L., Yuan, X. F., Zhang, B., Zwingman, T. A., and Jones, A. R. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176.
Luttrell, L. M., and Lefkowitz, R. J. (2002). The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell. Sci. 115, 455–465.
Mackiewicz, M., Shockley, K. R., Romer, M. A., Galante, R. J., Zimmerman, J. E., Naidoo, N., Baldwin, D. A., Jensen, S. T., Churchill, G. A., and Pack, A. I. (2007). Macromolecule biosynthesis: a key function of sleep. Physiol. Genomics 31, 441–457.
Maret, S., Dorsaz, S., Gurcel, L., Pradervand, S., Petit, B., Pfister, C., Hagenbuchle, O., O’Hara, B. F., Franken, P., and Tafti, M. (2007). Homer1a is a core brain molecular correlate of sleep loss. Proc. Natl. Acad. Sci. U.S.A. 104, 20090–20095.
Maurel, D., Sage, D., Mekaouche, M., and Bosler, O. (2000). Glucocorticoids up-regulate the expression of glial fibrillary acidic protein in the rat suprachiasmatic nucleus. Glia 29, 212–221.
Merchant, K. M., Dobner, P. R., and Dorsa, D. M. (1992). Differential effects of haloperidol and clozapine on neurotensin gene transcription in rat neostriatum. J. Neurosci. 12, 652–663.
Miyachi, S., Hasegawa, Y. T., and Gerfen, C. R. (2005). Coincident stimulation of convergent cortical inputs enhances immediate early gene induction in the striatum. Neuroscience 134, 1013–1022.
Modirrousta, M., Mainville, L., and Jones, B. E. (2005). Orexin and MCH neurons express c-Fos differently after sleep deprivation vs. recovery and bear different adrenergic receptors. Eur. J. Neurosci. 21, 2807–2816.
Mongrain, V., Hernandez, S. A., Pradervand, S., Dorsaz, S., Curie, T., Hagiwara, G., Gip, P., Heller, H. C., and Franken, P. (2010). Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep 33, 1147–1157.
Monti, J. M., and Jantos, H. (2008). The roles of dopamine and serotonin, and of their receptors, in regulating sleep and waking. Prog. Brain Res. 172, 625–646.
Morgan, J. I., and Curran, T. (1991). Stimulus-transcription coupling in the nervous system: involvement of the inducible proto-oncogenes fos and jun. Annu. Rev. Neurosci. 14, 421–451.
Ng, L., Bernard, A., Lau, C., Overly, C. C., Dong, H. W., Kuan, C., Pathak, S., Sunkin, S. M., Dang, C., Bohland, J. W., Bokil, H., Mitra, P. P., Puelles, L., Hohmann, J., Anderson, D. J., Lein, E. S., Jones, A. R., and Hawrylycz, M. (2009). An anatomic gene expression atlas of the adult mouse brain. Nat. Neurosci. 12, 356–362.
Ng, L., Pathak, S. D., Kuan, C., Lau, C., Dong, H., Sodt, A., Dang, C., Avants, B., Yushkevich, P., Gee, J. C., Haynor, D., Lein, E., Jones, A., and Hawrylycz, M. (2007). Neuroinformatics for genome-wide 3D gene expression mapping in the mouse brain. IEEE/ACM Trans. Comput. Biol. Bioinform. 4, 382–393.
Orban, P., Rauchs, G., Balteau, E., Degueldre, C., Luxen, A., Maquet, P., and Peigneux, P. (2006). Sleep after spatial learning promotes covert reorganization of brain activity. Proc. Natl. Acad. Sci. U.S.A. 103, 7124–7129.
Panda, S., Antoch, M. P., Miller, B. H., Su, A. I., Schook, A. B., Straume, M., Schultz, P. G., Kay, S. A., Takahashi, J. S., and Hogenesch, J. B. (2002). Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109, 307–320.
Pikkarainen, M., and Pitkanen, A. (2001). Projections from the lateral, basal and accessory basal nuclei of the amygdala to the perirhinal and postrhinal cortices in rat. Cereb. Cortex 11, 1064–1082.
Pikkarainen, M., Ronkko, S., Savander, V., Insausti, R., and Pitkanen, A. (1999). Projections from the lateral, basal, and accessory basal nuclei of the amygdala to the hippocampal formation in rat. J. Comp. Neurol. 403, 229–260.
Sagar, S. M., Sharp, F. R., and Curran, T. (1988). Expression of c-fos protein in brain: metabolic mapping at the cellular level. Science 240, 1328–1331.
Sanchez-Vives, M. V., and McCormick, D. A. (2000). Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 3, 1027–1034.
Saper, C. B., Scammell, T. E., and Lu, J. (2005). Hypothalamic regulation of sleep and circadian rhythms. Nature 437, 1257–1263.
Schoups, A. A., Elliott, R. C., Friedman, W. J., and Black, I. B. (1995). NGF and BDNF are differentially modulated by visual experience in the developing geniculocortical pathway. Brain Res. Dev. Brain Res. 86, 326–334.
Sgambato, V., Abo, V., Rogard, M., Besson, M. J., and Deniau, J. M. (1997). Effect of electrical stimulation of the cerebral cortex on the expression of the Fos protein in the basal ganglia. Neuroscience 81, 93–112.
Sherin, J. E., Shiromani, P. J., McCarley, R. W., and Saper, C. B. (1996). Activation of ventrolateral preoptic neurons during sleep. Science 271, 216–219.
Steriade, M. (2006). Grouping of brain rhythms in corticothalamic systems. Neuroscience 137, 1087–1106.
Tepper, J. M., and Bolam, J. P. (2004). Functional diversity and specificity of neostriatal interneurons. Curr. Opin. Neurobiol. 14, 685–692.
Terao, A., Greco, M. A., Davis, R. W., Heller, H. C., and Kilduff, T. S. (2003a). Region-specific changes in immediate early gene expression in response to sleep deprivation and recovery sleep in the mouse brain. Neuroscience 120, 1115–1124.
Terao, A., Peyron, C., Ding, J., Wurts, S. W., Edgar, D. M., Heller, H. C. and Kilduff, T. S. (2000). Prepro-hypocretin (prepro-orexin) expression is unaffected by short-term sleep deprivation in rats and mice. Sleep 23, 867–874.
Terao, A., Steininger, T. L., Hyder, K., Apte-Deshpande, A., Ding, J., Rishipathak, D., Davis, R. W., Heller, H. C., and Kilduff, T. S. (2003b). Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience 116, 187–200.
Terao, A., Wisor, J. P., Peyron, C., Apte-Deshpande, A., Wurts, S. W., Edgar, D. M., and Kilduff, T. S. (2006). Gene expression in the rat brain during sleep deprivation and recovery sleep: an Affymetrix GeneChip study. Neuroscience 137, 593–605.
Thompson, C. L., Pathak, S. D., Jeromin, A., Ng, L. L., MacPherson, C. R., Mortrud, M. T., Cusick, A., Riley, Z. L., Sunkin, S. M., Bernard, A., Puchalski, R. B., Gage, F. H., Jones, A. R., Bajic, V. B., Hawrylycz, M. J., and Lein, E. S. (2008). Genomic anatomy of the hippocampus. Neuron 60, 1010–1021.
Tobler, I. and Jaggi, K. (1987). Sleep and EEG spectra in the Syrian hamster (Mesocricetus auratus) under baseline conditions and following sleep deprivation. J. Comp. Physiol. 161, 449–459.
Tononi, G., and Cirelli, C. (2006). Sleep function and synaptic homeostasis. Sleep Med. Rev. 10, 49–62.
Volkow, N. D., Tomasi, D., Wang, G. J., Telang, F., Fowler, J. S., Wang, R. L., Logan, J., Wong, C., Jayne, M., and Swanson, J. M. (2009). Hyperstimulation of striatal D2 receptors with sleep deprivation: Implications for cognitive impairment. Neuroimage 45, 1232–1240.
Vyazovskiy, V. V., Cirelli, C., Pfister-Genskow, M., Faraguna, U., and Tononi, G. (2008). Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat. Neurosci. 11, 200–208.
Wang, Y., Osterbur, D. L., Megaw, P. L., Tosini, G., Fukuhara, C., Green, C. B., and Besharse, J. C. (2001). Rhythmic expression of Nocturnin mRNA in multiple tissues of the mouse. BMC Dev. Biol. 1, 9. doi: 10.1186/1471-213X-1-9.
Weng, L., Dai, H., Zhan, Y., He, Y., Stepaniants, S. B., and Bassett, D. E. (2006). Rosetta error model for gene expression analysis. Bioinformatics 22, 1111–1121.
Winrow, C. J., Tanis, K. Q., Rigby, A. M., Taylor, R. R., Serikawa, K., McWhorter, M., Tokiwa, G. Y., Marton, M. J., Stone, D. J., Koblan, K. S., and Renger, J. J. (2009). Refined anatomical isolation of functional sleep circuits exhibits distinctive regional and circadian gene transcriptional profiles. Brain Res. 1271, 1–17.
Wisor, J. P., Pasumarthi, R. K., Gerashchenko, D., Thompson, C. L., Pathak, S., Sancar, A., Franken, P., Lein, E. S. and Kilduff, T. S. (2008). Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. J. Neurosci. 28, 7193–7201.
Xie, Y., Pan, W., and Khodursky, A. B. (2005). A note on using permutation-based false discovery rate estimates to compare different analysis methods for microarray data. Bioinformatics 21, 4280–4288.
Yoo, S. H., Yamazaki, S., Lowrey, P. L., Shimomura, K., Ko, C. H., Buhr, E. D., Siepka, S. M., Hong, H. K., Oh, W. J., Yoo, O. J., Menaker, M., and Takahashi, J. S. (2004). PERIOD2:LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. U.S.A. 101, 5339–5346.
Yoo, S. S., Gujar, N., Hu, P., Jolesz, F. A., and Walker, M. P. (2007). The human emotional brain without sleep – a prefrontal amygdala disconnect. Curr. Biol. 17, R877–R878.
Keywords: in situ hybridization, sleep deprivation, gene expression, microarray
Citation: Thompson CL, Wisor JP, Lee C-K, Pathak SD, Gerashchenko D, Smith KA, Fischer SR, Kuan CL, Sunkin SM, Ng LL, Lau C, Hawrylycz M, Jones AR, Kilduff TS and Lein ES (2010) Molecular and anatomical signatures of sleep deprivation in the mouse brain. Front. Neurosci. 4:165. doi: 10.3389/fnins.2010.00165
Received: 15 May 2010;
Paper pending published: 16 June 2010;
Accepted: 23 August 2010;
Published online: 21 October 2010
Edited by:
Paul Franken, Université de Lausanne, SwitzerlandReviewed by:
Valérie Mongrain, Université de Lausanne, SwitzerlandBruce F. O’Hara, University of Kentucky, USA
Copyright: © 2010 Thompson, Wisor, Lee, Pathak, Gerashchenko, Smith, Fischer, Kuan, Sunkin, Ng, Lau, Hawrylycz, Jones, Kilduff and Lein. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Edward S. Lein, Allen Institute for Brain Science, 551 N. 34th Street, Ste. 200, Seattle, WA 98103, USA. e-mail: edl@alleninstitute.org