 Matthew J. Reid
Matthew J. Reid Paula Beltran-Lobo
Paula Beltran-Lobo Louisa Johnson
Louisa Johnson Beatriz Gomez Perez-Nievas
Beatriz Gomez Perez-Nievas Wendy Noble
Wendy Noble- Department of Basic and Clinical Neuroscience, Institute of Psychiatry, Psychology and Neuroscience, King's College London, London, United Kingdom
Tauopathies are a group of neurodegenerative diseases characterized by the progressive accumulation across the brain of hyperphosphorylated aggregates of the microtubule-associated protein tau that vary in isoform composition, structural conformation and localization. Tau aggregates are most commonly deposited within neurons but can show differential association with astrocytes, depending on the disease. Astrocytes, the most abundant neural cells in the brain, play a major role in synapse and neuronal function, and are a key component of the glymphatic system and blood brain barrier. However, their contribution to tauopathy progression is not fully understood. Here we present a brief overview of the association of tau with astrocytes in tauopathies. We discuss findings that support a role for astrocytes in the uptake and spread of pathological tau, and we describe how alterations to astrocyte phenotype in tauopathies may cause functional alterations that impedes their ability to support neurons and/or cause neurotoxicity. The research reviewed here further highlights the importance of considering non-neuronal cells in neurodegeneration and suggests that astrocyte-directed targets that may have utility for therapeutic intervention in tauopathies.
Introduction
Tauopathies are a heterogeneous group of neurodegenerative diseases in which the deposition of hyperphosphorylated tau aggregates in affected brain regions accompanies synapse and neuron loss (1, 2). Primary tauopathies exhibit tau aggregates as the predominant pathological hallmark and include a diverse family of frontal-temporal lobar dementia (FTLD) subtypes referred to as FTLD-tau, and progressive supranuclear palsy (PSP) and Pick's disease (PiD). Alzheimer's disease (AD) is considered a secondary tauopathy owing to the presence of extracellular amyloid-beta (Aß) plaques, and is the most common cause of dementia (1).
Tau proteins undergo several post-translational and other modifications in disease (2). Modified forms of tau spreads from the original site of deposition to anatomically connected regions by a “prion-like” mechanism, whereby tau proteopathic seeds passively recruit tau monomers (3). The mechanisms underlying tau release, uptake and spread are not fully understood. It has long been acknowledged that in some tauopathies astrocytes accumulate tau leading to characteristic disease neuropathology. Accumulating evidence now suggests that astrocytes may actively participate in tau spread and/or clearance mechanisms by actively internalizing tau. This review summarizes the association of tau with astrocytes in tauopathies, and discusses the evidence implicating astrocytes in tau spread, as well as the impact of tauopathy brain environments on physiological astrocytic functions.
Tau Protein
Human tau is encoded by the MAPT gene on chromosome 17 which comprises 16 exons. Exons 2, 3, and 10 undergo alternative splicing to produce the six main tau isoforms present in the adult human central nervous system (CNS) (4). Alternative splicing of exon 10 gives rise to tau isoforms containing either three or four microtubule binding repeats (referred to as 3R or 4R tau) in the C-terminal region, and alternative splicing of exons 2 and 3 produces tau proteins with zero, one or two inserts in the N-terminal tail (0N, 1N, or 2N tau, respectively). A conserved proline-rich domain is found between these two spliced regions and is known to be important for tau interactions with other proteins, including actin (5). Tau isoforms are developmentally regulated; the shortest 0N3R isoform is expressed in the fetal brain whereas in the adult human brain 3R and 4R isoforms are equally represented (6). Tau has a number of key functions, the most recognized of which is stabilizing microtubules in the axons of neurons, however tau roles in other important physiological functions such as axonal transport, DNA protection, cell signaling at the membrane, and synaptic vesicle release, have been described (2, 7). Tau is primarily expressed in neurons (8), but is known to be expressed to a lesser extent in glial cells (9–12).
Monomeric tau is water soluble and resists aggregation (7). In tauopathies, tau undergoes extensive post-translational and other modifications including, but not limited to, phosphorylation, acetylation, nitration, SUMOylation, glycosylation, ubiquitination, cleavage, and aggregation (2). The best studied of these is phosphorylation. There are 85 potential phosphorylation sites in 2N4R tau (13) and increased phosphorylation of tau, alongside other tau modifications, can reduce tau affinity for microtubules, increase cytoplasmic tau concentrations and promote tau oligomerisation and aggregation (2). Differential extents of tau modifications lead to the accumulation of heterogeneous pools of modified tau between, and within, different tauopathies. Recently, Dujardin et al. (14) found variations in the relative abundance of soluble, oligomeric and seed-competent species of hyperphosphorylated tau in tauopathy brain. Specific post-translational modifications were found to influence tau seeding capacity, and tau seeding potential strongly correlated with the rate of clinical symptoms/disease progression.
The isoform composition of tau aggregates, as well as the structure of tau filaments, also differs between tauopathies. In AD, both paired helical and straight filaments contain identical protofilament cores comprising residues 306–378 that define the aggregatory seed/core (15). This structure differs from the folds of tau filaments observed in Pick's disease (16) and tau filaments of chronic traumatic encephalopathy (CTE) have a unique hydrophobic core (17). A novel fold in corticobasal degeneration (CBD) tau has now also been discovered (18). These features may be important for the tau lesions that arise in different tauopathies (Table 1).
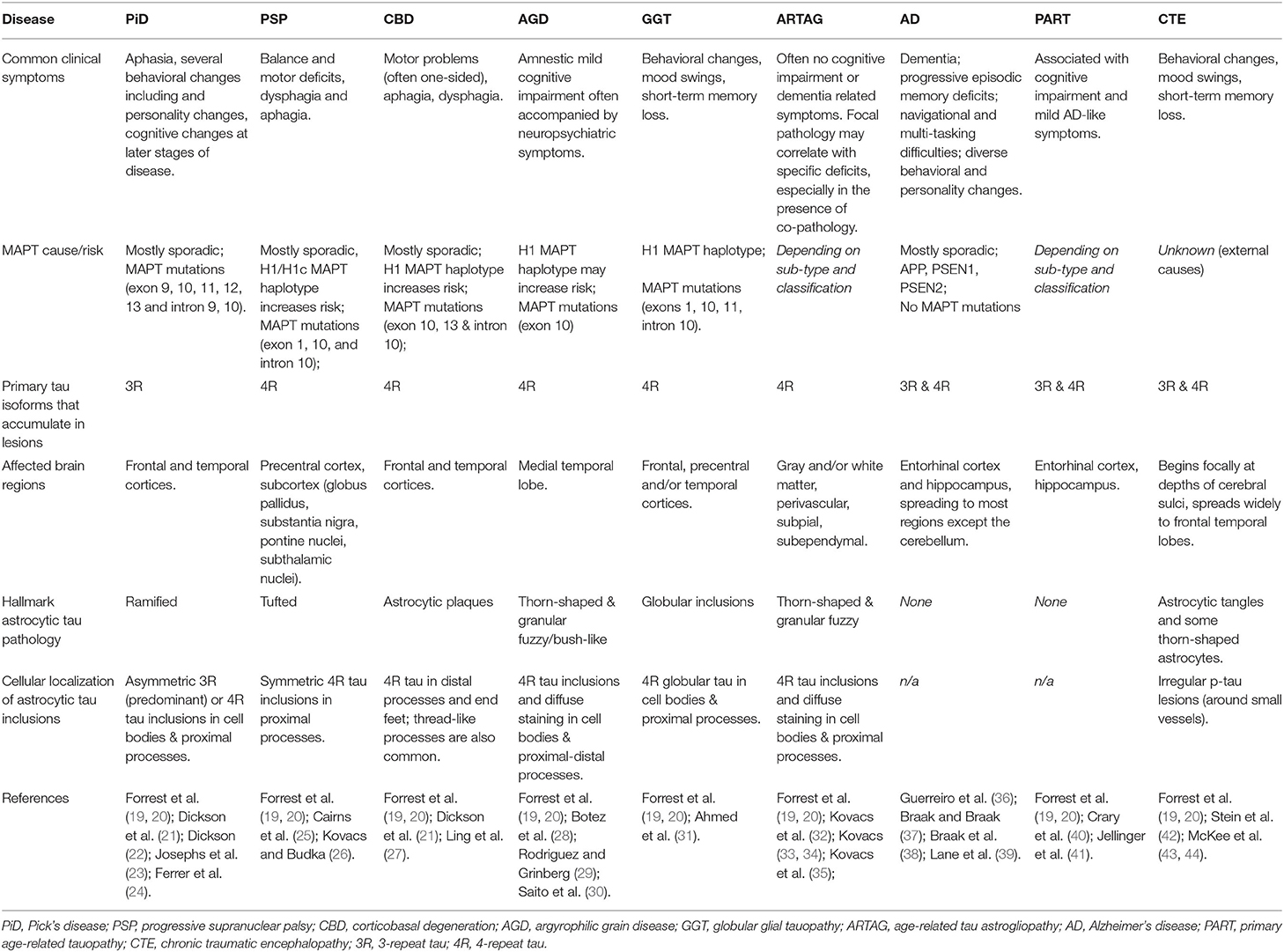
Table 1. Overview of the main clinical, genetic, molecular, and pathological features of tauopathies, including description of astrocyte abnormalities.
Astrocytes in Health and Disease
Astrocytes are organized into distinct domains, and each astrocyte can connect with thousands of neurons, allowing them to coordinate synaptic activity in the CNS (45, 46). Astrocytes were long considered as supporting cells in the brain, providing metabolic and nutritional support for neurons. However, astrocytes are critical for neuronal function due to their ability to sense changes in neuronal activity through their complement of cell surface receptors, and to modulate neuronal activity by releasing gliotransmitters and gliomodulators, as well as controlling the availability of glutamate, GABA, and energy substrates (45, 47, 48). Hence, astrocytes are now known to be actively involved in synaptic transmission (49), neural circuit maintenance (50) and long-term potentiation (51). In addition, astrocytic end-feet are a structural component of the blood-brain barrier (BBB), and together with endothelial cells and pericytes have a central role in the regulation of blood flow (52). Furthermore, astrocyte end-feet are crucial for the glymphatic system of the brain, a perivascular network that allows for exchange of interstitial and cerebrospinal fluid (CSF), providing a route for clearance of molecules and proteins including Aβ (53, 54).
In neurodegenerative disease brain, astrocytes undergo pathological changes in responses to changes in the local brain environment that precede neuronal loss (55). These morphologically and functionally modified astrocytes are often termed “reactive.” Reactive astrocytes show considerable heterogeneity related to their localization in the brain and the severity and length of injury/insult to their local environment (56). Reactive astrocytes are traditionally characterized by increased levels of glial fibrillary acidic protein (GFAP), which allows cytoskeletal and morphological arrangements as astrocytes alter their function (57, 58). The accumulation of GFAP-immunopositive astrocytes is common in neurodegenerative diseases. For example, reactive astrocytes are often found surrounding plaques in AD (59, 60). Indeed, levels of GFAP-reactive astrocytes are closely associated with dementia in AD (61). While increased GFAP is also found in aged brain (62), new evidence suggests that there are subgroups of astrocytes, with varying levels of GFAP expression, that distinguish aging from AD, at least in mice (63). Alterations in GFAP expression have also been noted in primary tauopathies including PSP, PiD and corticobasal degeneration (CBD) (24).
Functional changes in reactive astrocytes are well-documented and include impaired gliotransmitter release (64), alterations in calcium signaling (65), deficient ability to regulate glutamate levels at neuronal synapses and aberrant GABA release (58). In addition, astrocytes are now recognized to contribute to neuroinflammatory responses that accelerate the progression of neurodegenerative diseases (59, 66, 67). For example, reactive astrocytes increase their production and release of pro-inflammatory cytokines, complement components, and reactive oxygen species, alongside downregulating anti-inflammatory, and repair proteins to induce neurotoxicity in diseased environments (59, 68–70). Recent seminal findings proposed that astrocytes respond to their local environment by adopting “A1-neurotoxic” or “A2-neuroprotective” phenotypes (71). Secretion of Il-1α, TNFα, and C1q by microglia in response to damage, induces astrocytes to upregulate their expression of a specific cluster of “A1” genes, lose their trophic and synaptic support for neurons, and induce neuron death (71). Markers of A1 astrocytes are upregulated in AD and other neurodegenerative diseases (71), strongly implicating microglia-astrocyte communications in neurodegeneration. However, it is likely that there is a spectrum of reactive astrocyte states in different brain regions, throughout aging and disease progression (63, 72), similar to dynamic microglial responses in disease (25).
The Association of Astrocytes With Tauopathy
Tau aggregates accumulate in both neurons and astrocytes in different tauopathies. In AD, tau aggregates containing both 3R and 4R tau deposit as intraneuronal neurofibrillary tangles and there is scant evidence of astrocytic tau inclusions (73). In contrast, astrocytic tau pathology is the defining feature of several FTLD-tau subtypes (Table 1). In PSP, a neuropathological diagnosis criterion is “tufted” astrocytes that show 4R tau aggregates in their proximal processes (26, 74). CBD has extensive clinical overlap with PSP. In CBD, astrocytic plaques containing 4R tau deposits that mark distal and end processes are an exclusive feature in most (19), but not all cases (75). Thread-like tau-positive astrocytic processes are also common in CBD (21, 27). Argyrophilic grain disease (AGD) is a rare tauopathy that is characterized by 4R tau-immunopositive astrocytes, described as thorn-shaped and fuzzy/bush-like, in the medial temporal lobe (19, 28, 30). In contrast, PiD is typically characterized by neuronal 3R tau inclusions, predominantly in granular neurons in the hippocampus, frontal and temporal cortices (22, 23). “Ramified” astrocytes immunopositive for tau have also been reported in PiD, but they are not considered a major pathological hallmark of the disease (21, 24). Several rarer tauopathy subtypes that show 4R tau-immunopositive globular inclusions, predominantly in oligodendrocytes, and more rarely in the cytoplasm, and proximal processes of astrocytes, are collectively termed globular glial tauopathy (GGT) (31).
A spectrum of FTLD-tau subtypes that accumulate both 3R and 3R tau in neurofibrillary tangles (NFTs), typically occurring in cognitively normal aged individuals, is referred as primary age-related tauopathy (PART) (40, 41). Depending on the co-occurrence of Aß pathology, PART can be histologically classified as “definite PART” in the absence of Aß deposits, or “possible PART” when a limited number of Aß deposits are present (40). Although the neuropathological characteristics of PART can overlap with other tauopathies, particularly AD, PART shows a lower threshold of amyloid load, and appears to have a more limited impact on cognition (40, 76). Tau pathology in PART is predominantly neuronal and found in the CA2 hippocampal subfield, with little evidence of astrocytic tau deposits (40, 77). In contrast, age-related tau astrogliopathy (ARTAG) describes a spectrum of abnormal tau pathology, predominantly in the aged brain, that is characterized by thorn-shaped and granular or fuzzy astrocytes containing phosphorylated tau (32, 33). ARTAG can present alongside more typical tau pathology in tauopathies such as CBD (33, 34), but is not always linked with dementia (78). In a recent detailed review, Kovacs (34) describe two distinct distribution patterns of ARTAG. They describe ARTAG as a consequence of repeated mechanical damage (related to CTE), or chronic damage such as blood-brain barrier dysfunction. Furthermore, they propose that the location and type (white vs. gray matter) of ARTAG pathology may result in decompensation of cognitive functions, the rate of which may be influenced by co-existing pathologies (34). It is important to note that the presence of astrocytic tau accumulations in the absence of dementia may suggest that tau-containing astrocytes are not damaging in tau-associated neurodegeneration, or at least in ARTAG, and may internalize tau aggregates as a means of clearing damaging protein species.
Finally, chronic traumatic encephalopathy (CTE) is caused by mild repetitive head injuries. 3R and 4R tau-positive aggregates are common in CTE, however the tau aggregates that accumulate in astrocytes are predominantly 4R and localize in astrocytes near small vessels in the cerebral sulci of the frontal and temporal cortices (42, 43, 79). Thorn-shaped astrocytes are also observed subpial and periventricular regions, an interesting link to ARTAG (34, 44).
Do Astrocytes Contribute to Tau Pathology Spread?
Neurofibrillary tangles have long been acknowledged to follow a stereotypical temporospatial pattern of spread from the entorhinal cortex as AD progresses (38). Recent evidence indicates that differences in the tau species that deposit in characteristic tau lesions may confer specific neuronal vulnerabilities and/or prion-like spread of tau (14, 80). Mouse models that express wild-type 3R and 4R human tau isoforms in appropriate ratios recapitulate the same cell type vulnerabilities that typify human tauopathies when injected with human tau extracts, including the development of tufted astrocytes in PSP tau-injected mice, and astroglial plaques in CBD tau-injected mice (81). These data raise the possibility that astrocytes actively contribute to the spread of pathological forms of tau, particularly in PSP and CBD. That tau spreads in a prion-like manner trans-synaptically along anatomical connections was elegantly shown in transgenic mice in which mutant human (P301L) FTLD-causing tau expression was restricted to layer II neurons in the entorhinal cortex. Following local tau aggregation, tau “seeds” were found to spread to the hippocampus and onwards as mice aged (82, 83). Notably, PHF1-positive tau was detected in GFAP-positive astrocytes in the hippocampus of older mice, suggesting that astrocytes internalize and may contribute to tau spread (82) (Figure 1).
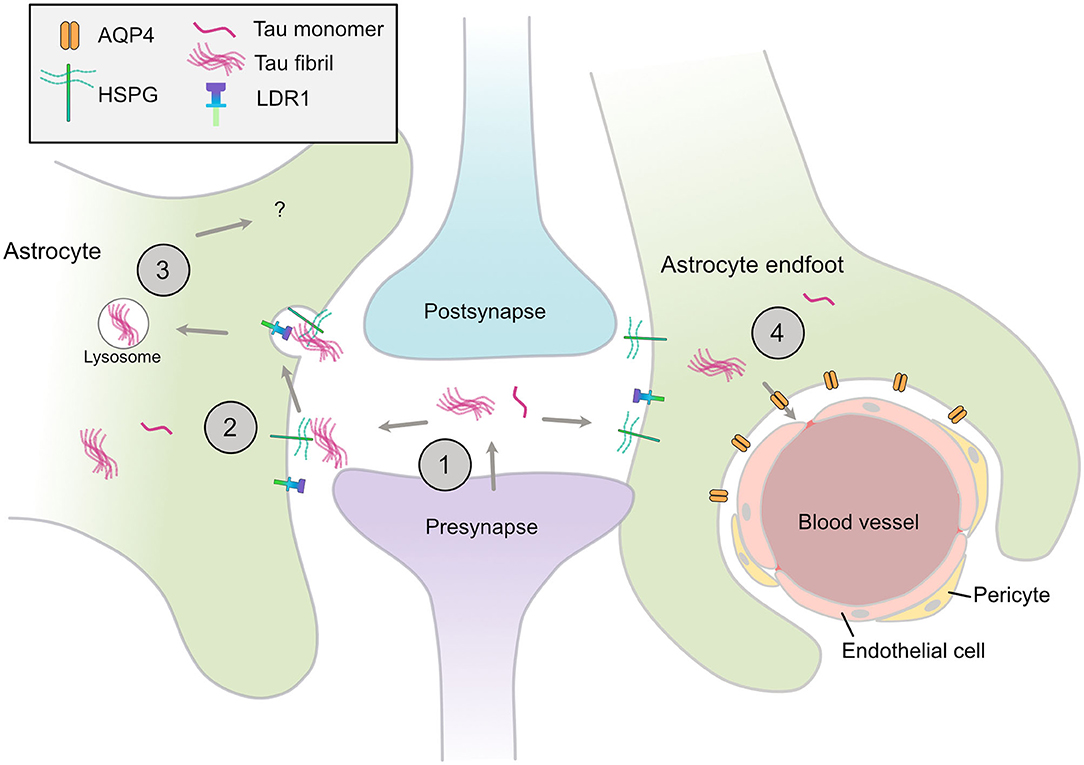
Figure 1. Astrocytic mechanisms that may contribute to spread of tau pathology. (1) Tau monomers and aggregates are released from neurons via various mechanisms, including from the pre-synapse, (2) Astrocytes have specific HSPGs and receptors such as LDR1 that may mediate the uptake of tau aggregates, (3) These aggregates may be internalized and processed by various mechanisms, include lysosomal degradation, (4) Disruption of AQP4 in perivascular astrocytic end-feet may contribute to the disrupted tau clearance and the accumulation of tau aggregates in the CNS. HSPG, heparin sulfate proteoglycan; LDR1, low density lipoprotein receptor-related protein 1; AQP4, aquaporin-4.
Heparan sulfate proteoglycans (HSPGs) are a well-conserved group of proteoglycans expressed on the cell surface of astrocytes and neurons (84, 85) that mediate targeted endocytosis (84), including that of purified prion proteins in vitro (86, 87). HSPGs were recently shown to interact with protein aggregates including α-synuclein, Aβ and tau (88–90). HSPGs regulate the uptake of synthetic tau fibrils (89) and human brain-derived tau (91) in human immortalized cell lines and mouse primary neuronal cultures. HSPGs vary in the length of their glycosaminoglycan chains and sulfation patterns, properties that are important for tau uptake in human embryonic kidney cells (92) and human iPSC derived neurons (93). Interestingly, tau fibrils are efficiently internalized in a HSPG-dependent manner by primary astrocytes exogenously expressing transcription factor EB (TFEB), a master regulator of lysosomal biogenesis (94). In contrast, monomeric tau appears to be taken up by astrocytes using an HSPG-independent mechanism (95). Together this suggests that multiple mechanisms are involved in tau uptake by astrocytes, that may be specific to tau aggregation state or conformation, as well as the HSPG profile of the cell type (96).
HSPGs can also partner with cell surface receptors to mediate the intake of protein aggregates. For example, HSPGs interact with members of the low-density lipoprotein receptor (LDLR) such as LRP1, to facilitate Aβ uptake and degradation by astrocytes (97, 98). Knockdown of LRP1 was recently shown to block the uptake of monomeric and oligomeric tau in a human neuroglioma cell line, and partially inhibit uptake of sonicated tau fibrils (99), warranting further investigation into how astrocytic LRP1 may mediate tau uptake and spread in tauopathies.
Astrocytes are an integral part of the glymphatic system of the brain, a clearance system of soluble proteins and solutes. The astrocytic water channel aquaporin-4 (AQP4), expressed at the astrocyte end feet, facilitates this process and is important for Aβ clearance (53, 100). Disruption to AQP4 may also contribute to tauopathy progression. In a mouse model of CTE, knockout of AQP4 exacerbated neurofibrillary tau pathology and neurodegeneration (101). Distinct phosphorylation marks in AQP4 have been reported in human post-mortem ARTAG samples relative to controls (102) that are suggested to increase water permeability of AQP4. However, the functional implications of these modifications in ARTAG remain to be explored (103, 104). A recent transcriptional analysis of cognitively-impaired subjects and controls showed that components of the dystrophin-associated complex, which anchors AQP4 at the perivascular astrocytic end foot, are associated with phosphorylated tau levels in the temporal cortex (54). This analysis also revealed other astrocyte endfoot candidate genes that significantly correlate with temporal cortex tau pathology. The authors speculate that endfoot functions of astrocytes may play a role in the accumulation of tau aggregates throughout the brain. Although AQP4 might contribute to the clearance of aberrant proteins early in the disease process, this function could become impaired at later stages, hindering the clearance of pathogenic tau.
Tau Effects on Astrocyte Function
In addition to potential roles in tau spread, internalization of pathological forms of tau has been shown to disrupt a myriad of astrocytic functions, central for the maintenance and support of neurons. Oligomeric tau uptake alters calcium signaling and gliotransmitter release (e.g. ATP) via Ca2+-dependant mechanisms, to disrupt post-synaptic currents and downregulate pre- and post-synaptic markers in neuronal-astrocyte co-cultures (64), together suggesting that tau-induced changes to astrocyte function are toxic to neighboring neurons, at least in vitro. Astrocytes isolated from a transgenic tauopathy model (P301S) expressing a 4R mutant tau isoform also acquired early functional deficiencies that impaired their ability to support neurons in culture (105). Astrocytes from mouse models of tauopathies also show altered expression of neuronally regulated genes (106), indicating that the accumulation of abnormal tau species is sufficient to drive transcriptional and likely functional changes in astrocytes, via altered neuron-astrocyte interactions. In addition, human astrocytes differentiated from iPSCs harboring FTD-causing MAPT mutations display an increased vulnerability to oxidative stress and elevated protein ubiquitination, alongside disease-associated transcriptomic alterations (107).
The immune-related functions of astrocytes are a major contributor to neuroinflammatory response that directly alter neuronal integrity in neurodegenerative diseases (52). In particular, the complement cascade, which also involves microglia, has an important role in the accumulation of beta-amyloid pathology (108, 109). C3 is a major component of the complement cascade and is highly expressed in reactive astrocytes (71). C3, as well as its downstream receptor C3aR1, that is mainly expressed by microglia, (9), is upregulated in postmortem tauopathy brain and correlates with cognitive decline during disease progression (110). Levels of C3 also correlate with tau amounts in AD CSF (111). Ablation of C3aR or C3 in mouse models of tauopathy reversed neuronal loss and neurodegeneration (110, 111), alongside reduced numbers of GFAP-reactive hypertrophied astrocytes being apparent upon C3aR knockout (110). These data indicate that complement activation downstream of astrocyte reactivity may be an important driver of tauopathy.
Astrocytes, together with microglia, are also hypothesized to induce synaptic loss and neurotoxicity in tauopathies, as they do during development (112), through dysregulated synaptic pruning (113). Sleep deprivation is common in AD (114), where it is believed to be both a cause and consequence of neurodegenerative changes (114). Sleep deprivation leads to enhanced tau release and spread (115), alongside astrocyte-mediated synapse elimination (116). It is therefore possible that astrocyte engulfment of tau-containing synapses may be one route by which astrocytes contribute to tau spread in AD.
Ultimately, cross-talk between astrocytes and microglia forms part of a complex innate immune response that may be exacerbated during tauopathies in response to protein aggregates. Deeper investigation of these pathways may reveal novel targets that can be exploited to slow or halt disease progression.
Discussion
Recent evidence has highlighted that altered astrocyte functions have detrimental consequences for neurons and may be a driver of neurodegenerative diseases. Astrocytes are closely associated with the accumulation of pathological forms of tau in tauopathies. There is some evidence that astrocytes internalize tau aggregates, via mechanisms that are not yet fully understood, and contribute to tau pathology spread across the brain and tau aggregate clearance via the glymphatic system. However, astrocytes show significant regional heterogeneity and more work is needed to better understand the contribution of different astrocyte subtypes in affected brain regions at different disease stages. Such understanding may aid in the development of astrocyte-targeted therapies for tauopathies. Astrocyte-targeted therapeutic approaches have been well-described elsewhere including by Sadik and Liddelow (70), and could include antagonists that prevent tau uptake by astrocytes to reduce tau spread, agents that prevent the release of neurotoxic astrocyte secretions or their uptake by neurons, or therapies that restore physiological astrocyte functions including their trophic support for neurons and synapses, maintenance of the blood brain barrier, and roles in the glymphatic clearance of protein aggregates.
Author Contributions
MR, PB-L, LJ, BP-N, and WN wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Work in our laboratories is supported by funding from Alzheimer's Research UK; ARUK-PHD2018-002 to WN, BP-N, and PB-L and ARUK-RF2014-2 to BP-N Medical Research Council, British Biotechnology and Biological Sciences Research Council, and the van Geest Charitable Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Prince M, Albanese E, Guerchert M, Prina M, Ferri C, Mazzotti DR, et al. World Alzheimer Report 2014: Dementia and Risk Reduction, an Analysis of Protective and Modifiable Factors. Alzheimer's Disease International. (2014). Available at: https://www.alz.co.uk/research/WorldAlzheimerReport2014.pdf.
2. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. (2017) 133:665–704. doi: 10.1007/s00401-017-1707-9
3. Jucker M, Walker LC. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci. (2018) 21:1341–9. doi: 10.1038/s41593-018-0238-6
4. Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta Mol Basis Dis. (2005) 1739:91–103. doi: 10.1016/j.bbadis.2004.08.010
5. He HJ, Wang XS, Pan R, Wang DL, Liu MN, He RQ. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. (2009) 10:81. doi: 10.1186/1471-2121-10-81
6. Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. (1990) 9:4225–30. doi: 10.1002/j.1460-2075.1990.tb07870.x
7. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. (2016) 17:5–21. doi: 10.1038/nrn.2015.1
8. Binder LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. (1985) 101:1371–8. doi: 10.1083/jcb.101.4.1371
9. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
10. Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA. (2015) 112:7285–90. doi: 10.1073/pnas.1507125112
11. Seiberlich V, Bauer NG, Schwarz L, Ffrench-Constant C, Goldbaum O, Richter-Landsberg C. Downregulation of the microtubule associated protein Tau impairs process outgrowth and myelin basic protein mRNA transport in oligodendrocytes. Glia. (2015) 63:1621–35. doi: 10.1002/glia.22832
12. McKenzie AT, Wang M, Hauberg ME, Fullard JF, Kozlenkov A, Keenan A, et al. Brain cell type specific gene expression and co-expression network architectures. Sci Rep. (2018) 8:1–19. doi: 10.1038/s41598-018-27293-5
13. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. (2009) 15:112–9. doi: 10.1016/j.molmed.2009.01.003
14. Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes A. R, Kamath T. V, et al. (2020). Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med. (2020) 26:1256–63. doi: 10.1038/s41591-020-0938-9
15. Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature. (2017) 547:185–90. doi: 10.1038/nature23002
16. Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature. (2018) 561:137–40. doi: 10.1038/s41586-018-0454-y
17. Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. (2019) 568:420–3. doi: 10.1038/s41586-019-1026-5
18. Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, et al. Novel tau filament fold in corticobasal degeneration. Nature. (2020) 580:283–7. doi: 10.1038/s41586-020-2043-0
19. Forrest SL, Kril JJ, Halliday GM. Cellular and regional vulnerability in frontotemporal tauopathies. Acta Neuropathol. (2019) 138:705–27. doi: 10.1007/s00401-019-02035-7
20. Forrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain. (2018) 141:521–34. doi: 10.1093/brain/awx328
21. Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-Tau (FTLD-Tau). J Mol Neurosci. (2011) 45:384–9. doi: 10.1007/s12031-011-9589-0
22. Dickson DW. Neuropathology of Pick's disease. Neurology. (2001) 56:S16–S20. doi: 10.1212/WNL.56.suppl_4.S16
23. Josephs KA, Hodges JR, Snowden JS, MacKenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. (2011) 122:137–53. doi: 10.1007/s00401-011-0839-6
24. Ferrer I, López-González I, Carmona M, Arregui L, Dalfó E, Torrejón-Escribano B, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol. (2014) 73:81–97. doi: 10.1097/NEN.0000000000000030
25. Cairns NJ, Bigio EH, Mackenzie IRA, Neumann M, Lee VMY, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol. (2007) 114:5–22. doi: 10.1007/s00401-007-0237-2
26. Kovacs GG, Budka H. Current concepts of neuropathological diagnostics in practice: neurodegenerative diseases. Clin Neuropathol. (2010) 29:271–88. doi: 10.5414/NPP29271
27. Ling H, Kovacs GG, Vonsattel JPG, Davey K, Mok KY, Hardy J, et al. Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain. (2016) 139:3237–52. doi: 10.1093/brain/aww256
28. Botez G, Probst A, Ipsen S, Tolnay M. Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol. (1999) 98:251–6. doi: 10.1007/s004010051077
29. Rodriguez RD, Grinberg LT. Argyrophilic grain disease: an underestimated tauopathy. Dement Neuropsychol. (2015) 9:2–8. doi: 10.1590/S1980-57642015DN91000002
30. Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y, et al. Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol. (2004) 63:911–8. doi: 10.1093/jnen/63.9.911
31. Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol. (2013) 126:537–44. doi: 10.1007/s00401-013-1171-0
32. Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. (2016) 131:87–102. doi: 10.1007/s00401-015-1509-x
33. Kovacs G. Understanding the Relevance of Aging-Related Tau Astrogliopathy (ARTAG). Neuroglia. (2018) 1:339–50. doi: 10.3390/neuroglia1020023
34. Kovacs GG. Astroglia and tau: new perspectives. Front Aging Neurosci. (2020) 12:1–14. doi: 10.3389/fnagi.2020.00096
35. Kovacs GG, Robinson GL, Xie SX, Lee EB, Grossman M, Wolk DA, et al. Evaluating the patterns of aging-related tau astrogliopathy unravels novel insights into brain aging and neurodegenerative diseases. J Neuropathol Exp Neurol. (2017) 76:270–88. doi: 10.1093/jnen/nlx007
36. Guerreiro RJ, Gustafson DR, Hardy J. The genetic architecture of Alzheimer's disease: beyond APP, PSENS and APOE. Neurobiol Aging. (2012) 33:437–56. doi: 10.1016/j.neurobiolaging.2010.03.025
37. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
38. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. (2011) 70:960–9. doi: 10.1097/NEN.0b013e318232a379
39. Lane CA, Hardy J, Schott JM. Alzheimer's disease. Eur J Neurol. (2018). 25:59–70. doi: 10.1111/ene.13439
40. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. (2014) 128:755–66. doi: 10.1007/s00401-014-1349-0
41. Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. (2015) 129:757–62. doi: 10.1007/s00401-015-1407-2
42. Stein TD, Alvarez VE, McKee AC. Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther. (2014) 6:4. doi: 10.1186/alzrt234
43. McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. (2015) 25:350–64. doi: 10.1111/bpa.12248
44. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Dirk Keene C, Litvan I, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. (2016) 131:75–86. doi: 10.1007/s00401-015-1515-z
45. Parpura V, Basarsky TA, Liu F, Jeftinijatt K. Glutamate-mediated astrocyte-neuron signalling. Nature. (1994) 369:744–7. doi: 10.1038/369744a0
46. Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. (2006) 29:547–53. doi: 10.1016/j.tins.2006.08.004
47. Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. (2005) 6:626–40. doi: 10.1038/nrn1722
48. Choi SS, Lee HJ, Lim I, Satoh JI, Kim SU. Human astrocytes: secretome profiles of cytokines and chemokines. PLoS ONE. (2014) 9:e92325. doi: 10.1371/journal.pone.0092325
49. Santello M, Toni N, Volterra A. Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci. (2019) 22:154–66. doi: 10.1038/s41593-018-0325-8
50. Mederos S, González-Arias C, Perea G. Astrocyte–neuron networks: a multilane highway of signaling for homeostatic brain function. Front Synaptic Neurosci. (2018) 10:45. doi: 10.3389/fnsyn.2018.00045
51. Lushnikova I, Skibo G, Muller D, Nikonenko I. Synaptic potentiation induces increased glial coverage of excitatory synapses in CA1 Hippocampus. (2009) 762:753–62. doi: 10.1002/hipo.20551
52. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
53. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. (2012) 4:147ra111. doi: 10.1126/scitranslmed.3003748
54. Simon MJ, Wang MX, Murchison CF, Roese NE, Boespflug EL, Woltjer RL, et al. Transcriptional network analysis of human astrocytic endfoot genes reveals region-specific associations with dementia status and tau pathology. Sci Rep. (2018) 8:1–16. doi: 10.1038/s41598-018-30779-x
55. Kersaitis C, Halliday GM, Kril JJ. Regional and cellular pathology in frontotemporal dementia: relationship to stage of disease in cases with and without Pick bodies. Acta Neuropathol. (2004) 108:515–23. doi: 10.1007/s00401-004-0917-0
56. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. (2012) 32:6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012
57. Kamphuis W, Kooijman L, Orre M, Stassen O, Pekny M, Hol EM. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer's disease. Glia. (2015) 63:1036–56. doi: 10.1002/glia.22800
58. Acosta C, Anderson HD, Anderson CM. Astrocyte dysfunction in Alzheimer disease. J Neurosci Res. (2017) 95:2430–47. doi: 10.1002/jnr.24075
59. Bouvier DS, Jones EV, Quesseveur G, Davoli MA, Ferreira TA, Quirion R, et al. High resolution dissection of reactive glial nets in Alzheimer's Disease. Sci Rep. (2016) 6:1–15. doi: 10.1038/srep24544
60. Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Astrogliosis: an integral player in the pathogenesis of Alzheimer's disease. Prog Neurobiol. (2016) 144:121–41. doi: 10.1016/j.pneurobio.2016.01.001
61. Perez-Nievas BG, Stein TD, Tai HC, Dols-Icardo O, Scotton TC, Barroeta-Espar I, et al. Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain. (2013) 136:2510–26. doi: 10.1093/brain/awt171
62. Wruck W, Adjaye J. Meta-analysis of human prefrontal cortex reveals activation of GFAP and decline of synaptic transmission in the aging brain. Acta Neuropathol Commun. (2020) 8:1–18. doi: 10.1186/s40478-020-00907-8
63. Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, et al. Disease-associated astrocytes in Alzheimer's disease and aging. Nat Neurosci. (2020) 23:701–6. doi: 10.1038/s41593-020-0624-8
64. Piacentini R, Li Puma DD, Mainardi M, Lazzarino G, Tavazzi B, Arancio O, et al. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia. (2017) 65:1302–16. doi: 10.1002/glia.23163
65. Shigetomi E, Saito K, Sano F, Koizumi SC. Aberrant calcium signals in reactive astrocytes: a key process in neurological disorders. Int J Mol Sci. (2019) 20:996. doi: 10.3390/ijms20040996
66. Phillips EC, Croft CL, Kurbatskaya K, O'Neill MJ, Hutton ML, Hanger DP, et al. (2013). Astrocytes and neuroinflammation in Alzheimer's disease. Biochem Soc Trans. 42:1321–5. doi: 10.1042/BST20140155
67. Bright F, Ittner LM, Halliday GM. Neuroinflammation in frontotemporal dementia. Nat Rev Neurol. (2019) 15:540–55. doi: 10.1038/s41582-019-0231-z
68. Lian H, Yang L, Cole A, Sun L, Chiang ACA, Fowler SW, et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's Disease. Neuron. (2015) 85:101–15. doi: 10.1016/j.neuron.2014.11.018
69. Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. (2017) 12:1–16. doi: 10.1186/s13024-017-0192-x
70. Sadick JS, Liddelow SA. Don't forget astrocytes when targeting Alzheimer's disease. Br J Pharmacol. (2019) 176:3585–98. doi: 10.1111/bph.14568
71. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
72. Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. (2018) 22:269–85. doi: 10.1016/j.celrep.2017.12.039
73. Garwood CJ, Ratcliffe LE, Simpson JE, Heath PR, Ince PG, Wharton SB. Review: astrocytes in Alzheimer's disease and other age-associated dementias: a supporting player with a central role. Neuropathol Appl Neurobiol. (2017) 43:281–98. doi: 10.1111/nan.12338
74. Vainchtein ID, Molofsky AV. Astrocytes and microglia: in sickness and in health. Trends Neurosci. (2020) 43:144–54. doi: 10.1016/j.tins.2020.01.003
75. Ling H, Gelpi E, Davey K, Jaunmuktane Z, Mok KY, Jabbari E, et al. Fulminant corticobasal degeneration: a distinct variant with predominant neuronal tau aggregates. Acta Neuropathol. (2020) 139:717–34. doi: 10.1007/s00401-019-02119-4
76. Besser LM, Crary JF, Mock C, Kukull WA. Comparison of symptomatic and asymptomatic persons with primary age-related tauopathy. Neurology. (2017) 89:1707–15. doi: 10.1212/WNL.0000000000004521
77. Jellinger KA. Different patterns of hippocampal tau pathology in Alzheimer's disease and PART. Acta Neuropathol. (2018) 136:811–3. doi: 10.1007/s00401-018-1894-z
78. Lace G, Ince PG, Brayne C, Savva GM, Matthews FE, de Silva R, et al. Mesial temporal astrocyte tau pathology in the MRC-CFAS ageing brain cohort. Dement Geriatr Cogn Disord. (2012) 34:15–24. doi: 10.1159/000341581
79. McKee AC, Stein TD, Nowinski CJ, Stern RA, Daneshvar DH, Alvarez VE, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. (2013) 136:43–64. doi: 10.1093/brain/aws307
80. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA. (2013) 110:9535–40. doi: 10.1073/pnas.1301175110
81. He Z, McBride JD, Xu H, Changolkar L, Kim SJ, Zhang B, et al. Transmission of tauopathy strains is independent of their isoform composition. Nat Commun. (2020) 11:7. doi: 10.1038/s41467-019-13787-x
82. De Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. (2012) 73:685–97. doi: 10.1016/j.neuron.2011.11.033
83. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE. (2012) 7:1–9. doi: 10.1371/journal.pone.0031302
84. Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. (2001) 11:75–82. doi: 10.1016/S0962-8924(00)01897-3
85. Sarrazin S, Lamanna WC, Esko JD. Heparan Sulfate Proteoglycans. Cold Spring Harb Perspect Biol. (2011) 3:a004952. doi: 10.1101/cshperspect.a004952
86. Schonberger O, Horonchik L, Gabizon R, Papy-Garcia D, Barritault D, Taraboulos A. Novel heparan mimetics potently inhibit the scrapie prion protein and its endocytosis. Biochem Biophys Res Commun. (2003) 312:473–9. doi: 10.1016/j.bbrc.2003.10.150
87. Horonchik L, Tzaban S, Ben-Zaken O, Yedidia Y, Rouvinski A, Papy-Garcia D, et al. Heparan sulfate is a cellular receptor for purified infectious prions. J Biol Chem. (2005) 280:17062–7. doi: 10.1074/jbc.M500122200
88. Kanekiyo T, Zhang J, Liu Q, Liu CC, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-β uptake. J Neurosci. (2011) 31:1644–51. doi: 10.1523/JNEUROSCI.5491-10.2011
89. Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA. (2013) 110:E3138–E3147. doi: 10.1073/pnas.1301440110
90. Ihse E, Yamakado H, Wijk XM, Van Lawrence R, Esko JD. Cellular internalization of alpha- synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Rep. (2017) 7:9008. doi: 10.1038/s41598-017-08720-5
91. Puangmalai N, Bhatt N, Montalbano M, Sengupta U, Gaikwad S, Mcallen S, et al. Internalization mechanisms of brain-derived tau oligomers from patients with Alzheimer ' s disease, progressive supranuclear palsy and dementia with Lewy bodies. Cell Death Dis. (2020) 11:314. doi: 10.1038/s41419-020-2503-3
92. Stopschinski BE, Holmes BB, Miller GM, Manon VA, Vaquer-Alicea J, Prueitt WL, et al. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus -synuclein and -amyloid aggregates. J Biol Chem. (2018) 293:10826–40. doi: 10.1074/jbc.RA117.000378
93. Rauch JN, Chen JJ, Sorum AW, Miller GM, Sharf T, See SK, et al. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci Rep. (2018) 8:1–10. doi: 10.1038/s41598-018-24904-z
94. Martini-Stoica H, Cole AL, Swartzlander DB, Chen F, Wan YW, Bajaj L, et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J Exp Med. (2018) 215:2355–77. doi: 10.1084/jem.20172158
95. Perea JR, López E, Díez-Ballesteros JC, Ávila J, Hernández F, Bolós M. Extracellular monomeric tau is internalized by astrocytes. Front Neurosci. (2019) 13:442. doi: 10.3389/fnins.2019.00442
96. Tselnicker IF, Boisvert MM, Allen NJ. The role of neuronal versus astrocyte-derived heparan sulfate proteoglycans in brain development and injury. Biochem Soc Trans. (2014) 42:1263–9. doi: 10.1042/BST20140166
97. Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer's disease. Front Aging Neurosci. (2014) 6:93. doi: 10.3389/fnagi.2014.00093
98. Liu C, Hu J, Zhao N, Wang J, Wang N, Cirrito JR, et al. Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J Neurosci. (2017) 37:4023–31. doi: 10.1523/JNEUROSCI.3442-16.2017
99. Rauch JN, Luna G, Guzman E, Audouard M, Challis C, Sibih YE, et al. LRP1 is a master regulator of tau uptake and spread. Nature. (2020) 1:1–5. doi: 10.1038/s41586-020-2156-5
100. Benarroch EE. Aquaporin-4, homeostasis, and neurologic disease. Neurology. (2007) 69:2266–2268. doi: 10.1212/01.wnl.0000286385.59836.e2
101. Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. (2014) 34:16180–93. doi: 10.1523/JNEUROSCI.3020-14.2014
102. Ferrer I, García MA, González IL, Lucena DD, Villalonga AR, Tech MC, et al. Aging-related tau astrogliopathy (ARTAG): not only tau phosphorylation in astrocytes. Brain Pathol. (2018) 28:965–85. doi: 10.1111/bpa.12593
103. Han Z, Wax MB, Patil RV. Regulation of aquaporin-4 water channels by phorbol ester-dependent protein phosphorylation. J Biol Chem. (1998) 273:6001–4. doi: 10.1074/jbc.273.11.6001
104. Kitchen P, Day RE, Taylor LHJ, Salman MM, Bill RM, Conner MT, et al. Identification and molecular mechanisms of the rapid tonicity-induced relocalization of the aquaporin 4 channel. J Biol Chem. (2015) 290:16873–81. doi: 10.1074/jbc.M115.646034
105. Sidoryk-Wegrzynowicz M, Gerber YN, Ries M, Sastre M, Tolkovsky AM, Spillantini MG. Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol Commun. (2017) 5:89. doi: 10.1186/s40478-017-0478-9
106. Hasel P, Dando O, Jiwaji Z, Baxter P, Todd AC, Heron S, et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat Commun. (2017)8:15132. doi: 10.1038/ncomms15132
107. Hallmann AL, Araúzo-Bravo MJ, Mavrommatis L, Ehrlich M, Röpke A, Brockhaus J, et al. Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci Rep. (2017) 7:1–10. doi: 10.1038/srep42991
108. Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. (2011) 48:1592–603. doi: 10.1016/j.molimm.2011.04.003
109. Lian H, Litvinchuk A, Chiang ACA, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of alzheimer's disease. J Neurosci. (2016) 36:577–89. doi: 10.1523/JNEUROSCI.2117-15.2016
110. Litvinchuk A, Wan YW, Swartzlander DB, Chen F, Cole A, Propson NE, et al. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer's disease. Neuron. (2018) 100:1337–53.e5. doi: 10.1016/j.neuron.2018.10.031
111. Wu T, Dejanovic B, Gandham VD, Carano RAD, Sheng M, Hanson JE, et al. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy article. Cell Rep. (2019) 28:2111–23.e6. doi: 10.1016/j.celrep.2019.07.060
112. Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. (2013) 504:394–400. doi: 10.1038/nature12776
113. Henstridge CM, Tzioras M, Paolicelli RC. Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front Cell Neurosci. (2019) 13:63. doi: 10.3389/fncel.2019.00063
114. Noble W, Spires-Jones TL. Sleep well to slow Alzheimer's progression? Science. (2019) 363:813–4. doi: 10.1126/science.aaw5583
115. Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. (2019) 363:880–4. doi: 10.1126/science.aav2546
Keywords: tau, astrocyte, tauopathy, prion-like propagation, Alzheimer's disease, glia
Citation: Reid MJ, Beltran-Lobo P, Johnson L, Perez-Nievas BG and Noble W (2020) Astrocytes in Tauopathies. Front. Neurol. 11:572850. doi: 10.3389/fneur.2020.572850
Received: 15 June 2020; Accepted: 24 August 2020;
Published: 24 September 2020.
Edited by:
Sonia Do Carmo, McGill University, CanadaReviewed by:
Margaret Ellen Flanagan, Northwestern Medicine, United StatesAnthoula Charalampos Tsolaki, Aristotle University of Thessaloniki, Greece
Todd Cohen, University of North Carolina at Chapel Hill, United States
Copyright © 2020 Reid, Beltran-Lobo, Johnson, Perez-Nievas and Noble. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wendy Noble, d2VuZHkubm9ibGVAa2NsLmFjLnVr