 Yuanyuan Xiang
Yuanyuan Xiang Cuicui Liu
Cuicui Liu Shan Li
Shan Li Qinjian Sun
Qinjian Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 11 September 2020
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00901
This article is part of the Research Topic Immunogenetics Advances in Non-glial CNS Tumors View all 8 articles
Lymphomatoid granulomatosis (LYG) is an infrequent lymphoproliferative disease that typically involves the lungs, but may also affect the central nervous system (CNS). Isolated CNS involvement is very rare, and its clinicopathological features have not been fully elucidated. Here, we systematically reviewed the English literature through PubMed to collect all relevant case reports and small case series with pathologically confirmed primary CNS-LYG. A total of 29 relevant articles with 40 cases were included in this systemic review. In cases where T cells and B cells were compared, T cells were predominant in 19 (79.2%), and B cells were predominant in 5 (20.8%). The overall infection rate of EBV was 48.1% (13/27), among which the infection rate was 40.9% (9/22) in immunocompetent patients and 80% (4/5) in immunodeficient (HIV-infected) patients. Among the patients who underwent pathological grading, 35.7% (5/14) were at grade I, 42.9% (6/14) were at grade II, and 21.4% were at grade III. In conclusion, primary CNS-LYG is closely related to EBV infection and some cases may be predominantly T-cell phenotype. Surgical resection may be effective for mass-like lesions, although there is still a lack of standard therapeutic regimen. Accurate grading of lesions is essential for treatment selection and prognosis evaluation.
Lymphomatoid granulomatosis (LYG) is a rare Epstein–Barr virus (EBV)-associated lymphoproliferative disease, characterized by B-cell proliferation of uncertain malignant potential accompanied by extensive reactive T-cell infiltrate (1–3). This disorder was first described by Liebow et al. (4). To date, its prevalence has not been accurately measured and the information is mainly from case reports and small case series. The most frequently involved site of LYG is the lung (90%), but it may also affect multiple extrapulmonary sites, including the skin (25–50%), the kidney (30–40%), the liver (29%), and the central nervous system (CNS) (26%) (4–7). In rare cases, patients with LYG initially present with neurological symptoms and lack of symptoms in the lungs and other regions, which may result in significant delays in diagnosis (8, 9). Since the isolated CNS involvement is very rare, its clinicopathological features have not been fully elucidated yet. We herein conduct a systematic literature review of primary CNS-LYG, focusing on summarizing its neuroimaging, histopathological features and the relationship with EBV infection.
We used the following terms to search the PubMed database: “lymphomatoid granulomatosis,” “central nervous system,” “brain,” and “cerebral.” Among the patients reported, we selected the adults with pathological evidence of CNS involvement and excluded those with extra CNS involvement. We only evaluated the English literature, and the references of each pertinent study were also reviewed to search possible pertinent articles.
The last PubMed search was performed on January 1, 2019. A total of 29 relevant articles with 40 cases were included in this systemic review (Table 1). There were 26 males and 13 females, with a male/female ratio of 2.0, while in one case, gender was not reported. The mean age was 47.75 years (ranging from 19 to 74). Except for 3 patients who did not describe the follow-up, the average follow-up time of the remaining 37 patients was 27.3 months (ranging from 3 weeks to 221 months).
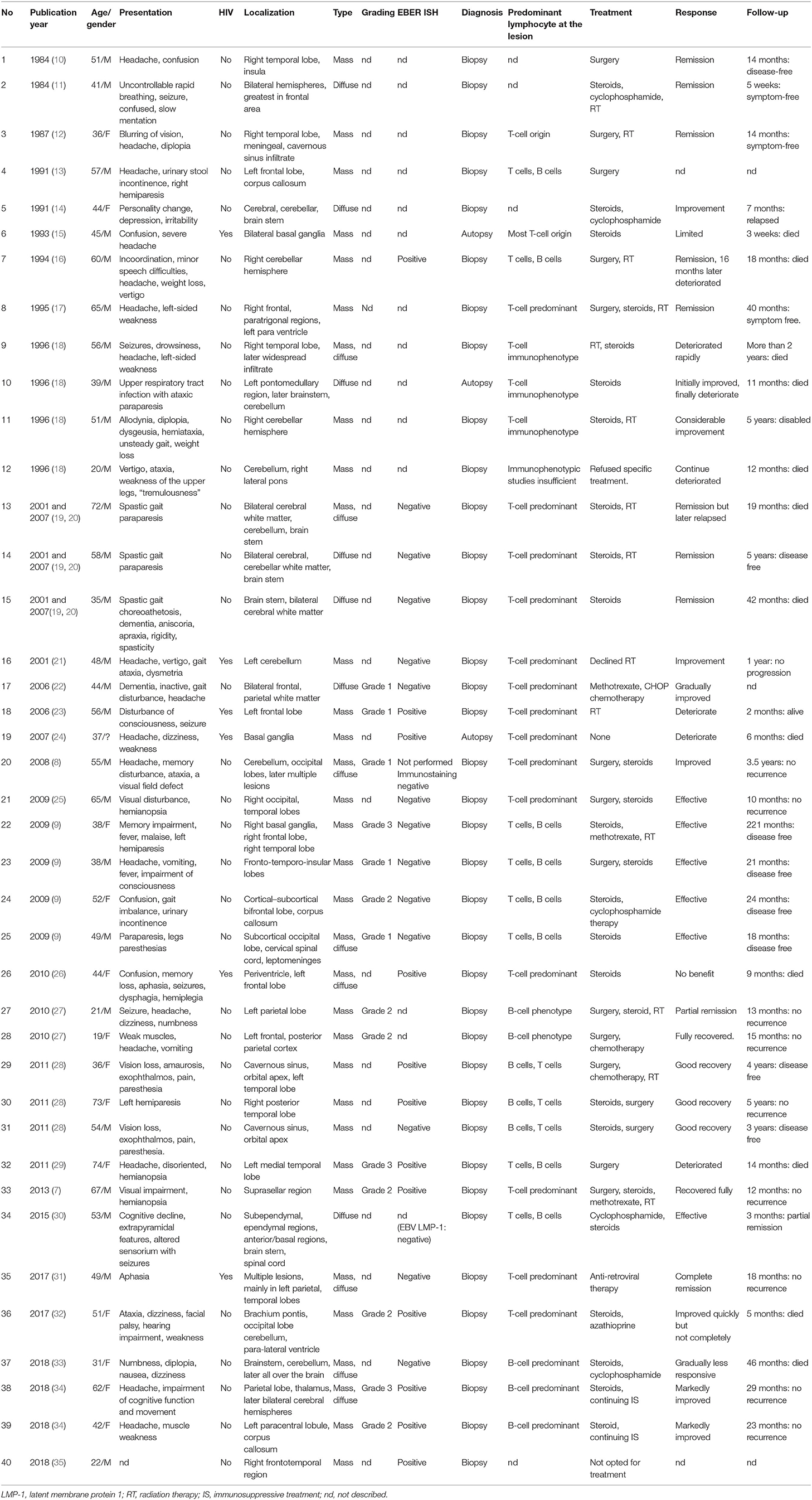
Table 1. Literature review of 40 cases of primary CNS-LYG.
In our review, except for 1 case that did not describe the main symptoms, 17 patients had headaches (43.6%) in the remaining 39 patients. Of 40 patients, 25 had supratentorial lesions, 5 patients had infratentorial lesions, while the remaining 10 patients had lesions both above and below the tentorium or in the spinal cord. Twenty-five patients (62.5%) manifested as mass-like lesions, 7 patients (17.5%) presented as diffuse infiltrating changes, while in 8 cases (20%), there were both space-occupying and infiltrating lesions.
Except for 5 cases that did not describe T cells and B cells in the brain lesions, T cells were predominant in 19 cases, B cells were predominant in 5 cases, and 11 cases described both T-cell and B-cell infiltrates, but did not compare them in the remaining 35 cases.
In our review of the 27 patients who described the details of EBV infection, 24 were detected by in situ hybridization of EBV-encoded RNA (EBER ISH), 1 was detected by EBV latent membrane protein 1 (LMP-1), 1 was detected by both EBER ISH and EBV-PCR, and 1 was detected by immunostaining. Of these 27 patients, 12 were positive for EBER ISH in brain biopsy or autopsy, 1 was positive for CSF EBV-PCR but negative for EBER ISH in brain biopsy a month later, and 14 were negative. Therefore, the positive rate of EBV infection was 48.1% in our review of CNS-LYG.
Six patients were found to combine with HIV infection, 3 of whom were positive for EBER ISH, 2 were negative, and 1 did not describe the details of EBV infection. One of the EBER ISH negative patients was positive for EBV-PCR in CSF a month before and the other EBER ISH negative patient was infected with HIV-2.
Fourteen patients underwent pathological grading, including 5 patients at grade I, 6 patients at grade II, and 3 patients at grade III.
Fifteen patients underwent surgical resection and 14 of them had mass-like lesions, while 1 patient manifested as both mass-like and diffuse infiltrating changes. After surgical resection (grade III), the symptoms of one patient continued to deteriorate, and later died after giving up treatment, and in another patient, complete remission lasting 16 months after treating with surgery and radiotherapy before the lesion transformed into a lymphoma, while in a third patient, the effect was not described. Of the remaining 12 patients who underwent surgery, 1 patient was relieved only by surgical resection, and 11 patients underwent surgery combined with steroids, chemotherapy, or radiotherapy, and achieved partial or complete remission during follow-up without recurrence.
Twelve patients died during follow-up, and among the three patients who underwent autopsy, two patients completed biopsy while alive. Among the 25 surviving patients, 1 patient experienced symptom deterioration, 1 patient relapsed, and 1 patient was severely disabled during follow-up; the remaining 22 patients achieved partial or complete remission after treatment, with good quality of life and no recurrence. Of the six HIV-infected patients, one deteriorated and three died during follow-up, while the other two patients had a better prognosis, including one with HIV-2 and the other with complete remission after first-line anti-retroviral therapy.
LYG is a rare EBV-associated, T-cell rich, B-cell lymphoproliferative systemic disease (systemic-LYG) (3), which is usually a male preponderance and generally peaks between the fifth to sixth decades (36). CNS involvement occurs in approximately 30% of patients (19), and it is a serious prognostic factor, with a mortality rate of 86% compared with 66% for patients without CNS after 14 months (5).
Isolated CNS involvement has been rarely reported and the diagnosis of primary CNS-LYG relies mainly upon biopsy and lacks other specific tests. So far, we have only found 40 cases of primary CNS-LYG in the English literature (7–35) (Table 1).
Various clinical symptoms, such as headache, seizure, diplopia, hemiplegia, altered consciousness, Parkinsonism, dementia, etc. (6, 37), may occur in primary CNS-LYG depending on the site involved, and primary CNS-LYG may involve any part of the CNS, such as the cerebrum, cerebellum, basal ganglia, brainstem, etc., without specificity. In our review, headaches were the most common symptoms and supratentorial involvement was more common.
The brain lesions of LYG mainly manifest as mass-like or diffuse infiltrating changes (38), which may be single or multiple. Primary CNS-LYG might more frequently manifest as intracranial mass-like lesions than CNS involvement of systemic LYG (9). Mass-like lesions were more common than diffuse infiltrating changes in the present systematic review of primary CNS-LYG.
Although not specific, MRI examination is indispensable for detecting intracranial lesions and monitoring changes during follow-up (39). The multiple punctate, linear, and ringlike enhancement can be seen on Gadolinium-enhanced T1-weighted imaging of primary CNS-LYG patients, which is likely to represent the pathological features of involving the vessel wall and perivascular tissue (19, 30).
Nishihara and coworkers pointed out in their case report that in the same lesion site, FDG-PET scan showed a low uptake indicating necrotic lesions, while methionine (MET)-PET scan showed a high uptake indicating hypercellular proliferative lesions (25). This mismatch accumulation was considered to be characteristic of LYG (25). In addition to PET-CT, other advanced imaging techniques, such as MRS and T2*-weighted MRI have also been applied to the study of LYG. However, the diagnostic value of these techniques for primary CNS-LYG still needs further study.
LYG is characterized by an angiocentric and angiodestructive infiltrate of lymphocytes, histiocytes and occasional plasma cells. Inflammatory cells, such as neutrophils and eosinophils, usually do not exist. Necrosis is variable and more pronounced in higher-grade lesions. Well-formed granulomas do not exist (6). “Lymphomatoid granulomatosis” is named because its clinical manifestations are similar to granulomatous polyangiitis and its histological features are similar to lymphoma. The term “granulomatosis” is not the actual granulomatous inflammation (40).
Whereas, a predominant B-cell phenotype of systemic LYG is generally accepted, the results of this review support the idea that some cases of primary CNS-LYG may be predominantly T-cell phenotype. All the HIV-infected patients reviewed showed T-cell dominant angiocentric infiltrate in brain tissue histopathological examination (15, 21, 23, 24, 26, 31). Nishihara et al. analyzed T-cell receptor (TcR) and immunoglobulin heavy chain (IgH) gene rearrangements in their cases of CNS-LYG and proposed that a large part of CNS-LYG could be classified as a low-grade malignant lymphoproliferative disease with essential T-cell phenotype (20). However, primary CNS-LYG is still a controversial disease, and its exact immunophenotype needs further study.
According to the WHO classification, LYG grading is based on the proportion of large lymphoid cells, the number of EBV-positive B cells per high-power field and the amount of necrosis (41). About 50% of grade II and 70% of grade III LYG present as clonal immunoglobulin gene rearrangements, but grade I lesion rearrangements are not common (42).
Due to the presence of many mimickers, primary CNS-LYG is diagnostically challenging (43–46), and should be differentiated from both space-occupying and diffuse cerebral lesions (30), such as primary CNS lymphoma, vasculitis, Wegener's granulomatosis, malignant glioma, metastatic tumors, chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroid (CLIPPERS), multiple sclerosis, etc. Primary CNS lymphoma is an important differential diagnosis of LYG grade III; however, it lacks the pattern of vascular invasion/destruction seen in primary CNS-LYG, although with a unique histopathological feature of angiocentric growth. In addition, some patients of LYG grade III may still have spontaneous remission with the change of immune status (47). Both vasculitis and primary CNS-LYG manifest as vascular damage caused by polymorphic infiltrate of the vessel wall, resulting in infarct-like tissue necrosis. The significant angiocentric lymphoid infiltrate, evidence of EBV-positive cells and lack of eosinophils or giant cells suggest a diagnosis of primary CNS-LYG, whereas the presence of granulocytes, eosinophils, and microabscess suggests vasculitis.
EBV is associated with a series of lymphoproliferative disorders. LYG is a rare EBV-driven B-cell lymphoproliferative process known for its angiocentricity and angioinvasiveness (48), and it is speculated that these patients have EBV immune surveillance disorders. In LYG, organ damage may be partly mediated by the host's immune response to EBV, while the pathological damage in most other EBV-driven B-cell lymphoproliferative diseases is caused by the direct expansion of EBV-infected B cells (49). Post-transplantation lymphoproliferative disease (PTLD) is an EBV-related B-cell lymphoproliferative process presenting as polymorphous or monomorphous, and shares many clinical features with LYG. In most cases, PTLD can be differentiated from LYG by the lack of a background of angiocentric T-cell infiltrate, the presence of prominent plasmacytic differentiation, and elevated median viral loads (42). Currently, the number of EBV-positive B cells per high-power field is used as part of the LYG grading criteria in the 2008 WHO classification (41), and grading is essential for guiding treatment and assessing prognosis.
Lucantoni et al. (9) reported that unlike systemic LYG, primary CNS-LYG appears not to be associated with EBV, while our review has shown that the positive rate of EBV infection was 40.9% (9/22) for immunocompetent patients, 80% (4/5) for immunodeficient (HIV-infected) patients, with a ratio of ~1:2. Thus, we speculate that the association of EBV with primary CNS-LYG is not restricted to patients with severe immunodeficiency and EBV may also be implicated in immunocompetent patients. However, it is still unclear whether EBV actually causes the disease or it is only a reflection of secondary activation, so large series of cases are needed to clarify this issue. AIDS-related primary CNS-LYG may be triggered by reactivation of EBV in the CNS under immunosuppressive conditions, followed by active T-cell infiltrates (31). However, it remains unknown whether these immunocompetent patients have other occult immunodeficiency. In addition, since treatment options and follow-up times were varied, we were unable to analyze the effect of EBV on survival time.
No standard therapeutic regimen has been established for primary CNS-LYG, and alternative treatment options include surgical resection, steroids, chemotherapy, radiation therapy, and anti-retroviral therapy for HIV patients. In our review, surgical resection may be effective for mass-like lesions.
Regarding the prognosis of primary CNS-LYG, it seems to be correlated with a better life expectancy than systemic-LYG with CNS involvement (9) and is closely associated with grading. In the present review, 1 patient developed lymphoma 16 months after complete remission and then died 2 months later (16). The biopsy of another patient showed LYG and focal conversion to lymphoma (11). However, neither case was graded, because pathological grading was not achieved in the 1980s and 1990s. We speculate that the most likely pathological grade for the two patients was grade III, for LYG grade III lesion is considered a subtype of diffuse large B-cell lymphoma (47).
Primary CNS-LYG is rarely reported, and its diagnosis relies mainly upon biopsy and differential diagnosis should consider diffuse and mass-like cerebral lesions. MRI examination is crucial for detecting intracranial lesions and monitoring changes during follow-up, although not specific. Unlike systemic LYG, some cases of primary CNS-LYG may be predominantly T-cell phenotype. Primary CNS-LYG is also closely related to EBV infection, which is not only in patients with severe immunodeficiency but also in immunocompetent patients. So far, there is no standard therapeutic regimen established for primary CNS-LYG and surgical resection may be effective for mass-like lesions. Accurate lesion grading is crucial for selecting the appropriate treatment and assessing prognosis.
XL and YXi conceived the study, performed literature research, and wrote the paper. CL, YXu, SL, and YS helped to perform literature research. XL, JL, and QS revised the paper. All authors contributed to the article and approved the submitted version.
This study was supported by the Key Technology Research and Development Program of Shandong (Project numbers: 2011GGH21838, 2015GGH318020, and 2016GSF201068).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Jancar J. Progress in the classification of myeloid and lymphoid neoplasms. From REAL to WHO concept. Adv Clin Path. (2000) 4:59–76.
2. Heslop HE. Biology and treatment of Epstein-Barr virus-associated non-Hodgkin lymphomas. Hematol Am Soc Hematol Educ Prog. (2005) 260–6. doi: 10.1182/asheducation-2005.1.260
3. Colby TV. Current histological diagnosis of lymphomatoid granulomatosis. Mod Pathol. (2012) 25(Suppl. 1):S39–42. doi: 10.1038/modpathol.2011.149
4. Liebow AA, Carrington CR, Friedman PJ. Lymphomatoid granulomatosis. Hum Pathol. (1972) 3:457–558. doi: 10.1016/S0046-8177(72)80005-4
5. Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer. (1979) 43:360–73.
6. Katzenstein AL, Doxtader E, Narendra S. Lymphomatoid granulomatosis: insights gained over 4 decades. Am J Surg Pathol. (2010) 34:e35–48. doi: 10.1097/PAS.0b013e3181fd8781
7. Cetin B, Benekli M, Akyurek N, Senturk S, Koklu H, Karakus E, et al. Isolated primary lymphomatoid granulomatosis of central nervous system. Indian J Hematol Blood Transfus. (2013) 29:39–42. doi: 10.1007/s12288-011-0123-x
8. Okuda T, Akai F, Kataoka K, Taneda M. A case of lymphomatoid granulomatosis followed for 14 months on the basis of clinical and histological findings. Brain Tumor Pathol. (2008) 25:33–8. doi: 10.1007/s10014-007-0227-z
9. Lucantoni C, De Bonis P, Doglietto F, Esposito G, Larocca LM, Mangiola A, et al. Primary cerebral lymphomatoid granulomatosis: report of four cases and literature review. J Neurooncol. (2009) 94:235–42. doi: 10.1007/s11060-009-9834-3
10. Schmidt BJ, Meagher-Villemure K, Del Carpio J. Lymphomatoid granulomatosis with isolated involvement of the brain. Ann Neurol. (1984) 15:478–81. doi: 10.1002/ana.410150513
11. Sunderrajan EV, Passamonte PM. Lymphomatoid granulomatosis presenting as central neurogenic hyperventilation. Chest. (1984) 86:634–6. doi: 10.1378/chest.86.4.634
12. Kerr RS, Hughes JT, Blamires T, Teddy PJ. Lymphomatoid granulomatosis apparently confined to one temporal lobe. Case Rep J Neurosurg. (1987) 67:612–5. doi: 10.3171/jns.1987.67.4.0612
13. Bae WK, Lee KS, Kim PN, Kim IY, Lee BH, Lee KS, et al. Lymphomatoid granulomatosis with isolated involvement of the brain–case report. J Korean Med Sci. (1991) 6:255–9. doi: 10.3346/jkms.1991.6.3.255
14. Kerslake R, Rowe D, Worthington BS. CT and MR imaging of CNS lymphomatoid granulomatosis. Neuroradiology. (1991) 33:269–71. doi: 10.1007/BF00588234
15. George JC, Caldemeyer KS, Smith RR, Czaja JT. CNS lymphomatoid granulomatosis in AIDS: CT and MR appearances. AJR Am J Roentgenol. (1993) 161:381–3. doi: 10.2214/ajr.161.2.8333381
16. Hamilton MG, Demetrick DJ, Tranmer BI, Curry B. Isolated cerebellar lymphomatoid granulomatosis progressing to malignant lymphoma. Case report J Neurosurg. (1994) 80:314–20. doi: 10.3171/jns.1994.80.2.0314
17. Sekhon LH, Morgan MK, Salisbury EL, Grace J, Lamont PJ. Recurrent lymphomatoid granulomatosis and isolated CNS involvement. J Clin Neurosci. (1995) 2:163–6. doi: 10.1016/0967-5868(95)90011-X
18. Kermode AG, Robbins PD, Carroll WM. Cerebral lymphomatoid granulomatosis. J Clin Neurosci. (1996) 3:346–53. doi: 10.1016/S0967-5868(96)90031-4
19. Tateishi U, Terae S, Ogata A, Sawamura Y, Suzuki Y, Abe S, et al. MR imaging of the brain in lymphomatoid granulomatosis. Am J Neuroradiol. (2001) 22:1283–90.
20. Nishihara H, Tateishi U, Itoh T, Nagashima K, Tanaka S. Immunohistochemical and gene rearrangement studies of central nervous system lymphomatoid granulomatosis. Neuropathology. (2007) 27:413–8. doi: 10.1111/j.1440-1789.2007.00804.x
21. Issakhanian M, Chang L, Cornford M, Witt M, Speck O, Goldberg M, et al. HIV-2 infection with cerebral toxoplasmosis and lymphomatoid granulomatosis. J Neuroimaging. (2001) 11:212–6. doi: 10.1111/j.1552-6569.2001.tb00037.x
22. Kawai N, Miyake K, Nishiyama Y, Yamamoto Y, Sasakawa Y, Haba R, et al. FDG-PET findings of the brain in lymphomatoid granulomatosis. Ann Nucl Med. (2006) 20:683–7. doi: 10.1007/BF02984680
23. Kiryu S, Okubo T, Takeuchi K, Inoue Y, Endo T, Odawara T, et al. Magnetic resonance imaging and diffusion tensor analysis of lymphomatoid granulomatosis of the brain. Acta Radiol. (2006) 47:509–13. doi: 10.1080/02841850600644790
24. Wyen C, Stenzel W, Hoffmann C, Lehmann C, Deckert M, Fatkenheuer G. Fatal cerebral lymphomatoid granulomatosis in an HIV-1-infected patient. J Infect. (2007) 54:e175–8. doi: 10.1016/j.jinf.2006.11.003
25. Nishihara H, Nakasato M, Sawa H, Murakami H, Yamamoto D, Moriyama K, et al. A case of central nervous system lymphomatoid granulomatosis; characteristics of PET imaging and pathological findings. J Neurooncol. (2009) 93:275–8. doi: 10.1007/s11060-008-9771-6
26. Gonzalez-Valcarcel J, Corral I, Quereda C, Alonso-Canovas A, Aparicio Hernandez M, de Felipe Mimbrera A, et al. Primary cerebral lymphomatoid granulomatosis as an immune reconstitution inflammatory syndrome in AIDS. J Neurol. (2010) 257:2106–8. doi: 10.1007/s00415-010-5669-2
27. Hu YH, Shao ED, Wu JL, Meng XB. Isolated neurological involvement of lymphomatoid granulomatosis. Chin Med J. (2010) 123:3163–6. doi: 10.3760/cma.j.issn.0366-6999.2010.21.041
28. Gonzalez-Darder JM, Vera-Roman JM, Pesudo-Martinez JV, Cerda-Nicolas M, Ochoa E. Tumoral presentation of primary central nervous system lymphomatoid granulomatosis. Acta Neurochir. (2011) 153:1963–70. doi: 10.1007/s00701-011-1088-0
29. Takeishi G, Moroki K, Kawasoe T, Fukushima T, Yokogami K, Nabeshima K, et al. Spontaneous regression and regrowth of central nervous system lymphomatoid granulomatosis: case report. Neurol Med Chir. (2011) 51:801–4. doi: 10.2176/nmc.51.801
30. Patil AK, Alexander M, Nair B, Chacko G, Mani S, Sudhakar S. Clinical, imaging and histopathological features of isolated CNS lymphomatoid granulomatosis. Indian J Radiol Imaging. (2015) 25:56–9. doi: 10.4103/0971-3026.150149
31. Kano Y, Kodaira M, Ushiki A, Kosaka M, Yamada M, Shingu K, et al. The complete remission of acquired immunodeficiency syndrome-associated isolated central nervous system lymphomatoid granulomatosis: a case report and review of the literature. Intern Med. (2017) 56:2497–501. doi: 10.2169/internalmedicine.8776-16
32. Wang X, Huang D, Huang X, Zhang J, Ran Y, Lou X, et al. Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS): A lymphocytic reactive response of the central nervous system? A case report. J Neuroimmunol. (2017) 305:68–71. doi: 10.1016/j.jneuroim.2017.01.014
33. Tian D, Zhu X, Xue R, Zhao P, Yao Y. Case 259: primary central nervous system lymphomatoid granulomatosis mimicking chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS). Radiology. (2018) 289:572–7. doi: 10.1148/radiol.2018161475
34. Ahle G, Uro-Coste E, Taieb G, de Broucker T. Primary lymphomatoid granulomatosis in the central nervous system: a report of three cases. Neuropathology. (2019) 39:168–9. doi: 10.1111/neup.12546
35. Sigamani E, Chandramohan J, Nair S, Chacko G, Thomas M, Mathew LG, et al. Lymphomatoid granulomatosis: a case series from South India. Indian J Pathol Microbiol. (2018) 61:228–32. doi: 10.4103/IJPM.IJPM_471_17
36. Sykorova A, Campr V, Kasparova P, Kocova E, Belada D, Trneny M, et al. [Lymphomatoid granulomatosis - the past and present]. Vnitr Lek. (2014) 60:225–38.
37. Gitelson E, Al-Saleem T, Smith MR. Review: lymphomatoid granulomatosis: challenges in diagnosis and treatment. Clin Adv Hematol Oncol. (2009) 7:68–70.
38. Mizuno T, Takanashi Y, Onodera H, Shigeta M, Tanaka N, Yuya H, et al. A case of lymphomatoid granulomatosis/angiocentric immunoproliferative lesion with long clinical course and diffuse brain involvement. J Neurol Sci. (2003) 213:67–76. doi: 10.1016/S0022-510X(03)00127-8
39. Patsalides AD, Atac G, Hedge U, Janik J, Grant N, Jaffe ES, et al. Lymphomatoid granulomatosis: abnormalities of the brain at MR imaging. Radiology. (2005) 237:265–73. doi: 10.1148/radiol.2371041087
40. Sukswai N, Lyapichev K, Khoury JD, Medeiros LJ. Diffuse large B-cell lymphoma variants: an update. Pathology. (2020) 52:53–67. doi: 10.1016/j.pathol.2019.08.013
41. Sabattini E, Bacci F, Sagramoso C, Pileri SA. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica. (2010) 102:83–7.
42. Song JY, Pittaluga S, Dunleavy K, Grant N, White T, Jiang L, et al. Lymphomatoid granulomatosis–a single institute experience: pathologic findings and clinical correlations. Am J Surg Pathol. (2015) 39:141–56. doi: 10.1097/PAS.0000000000000328
43. Braham E, Ayadi-Kaddour A, Smati B, Ben Mrad S, Besbes M, El Mezni F. Lymphomatoid granulomatosis mimicking interstitial lung disease. Respirology. (2008) 13:1085–7. doi: 10.1111/j.1440-1843.2008.01351.x
44. Bartosik W, Raza A, Kalimuthu S, Fabre A. Pulmonary lymphomatoid granulomatosis mimicking lung cancer. Interact Cardiovasc Thorac Surg. (2012) 14:662–4. doi: 10.1093/icvts/ivr083
45. Ammannagari N, Srivali N, Price C, Ungprasert P, Leonardo J. Lymphomatoid granulomatosis masquerading as pneumonia. Ann Hematol. (2013) 92:981–3. doi: 10.1007/s00277-012-1643-7
46. Srivali N, Thongprayoon C, Cheungpasitporn W, Ungprasert P. Lymphomatoid granulomatosis mimicking vasculitis. Ann Hematol. (2016) 95:345–6. doi: 10.1007/s00277-015-2507-8
47. Sugita Y, Muta H, Ohshima K, Morioka M, Tsukamoto Y, Takahashi H, et al. Primary central nervous system lymphomas and related diseases: pathological characteristics and discussion of the differential diagnosis. Neuropathology. (2016) 36:313–24. doi: 10.1111/neup.12276
48. Dunleavy K, Roschewski M, Wilson WH. Lymphomatoid granulomatosis and other Epstein-Barr virus associated lymphoproliferative processes. Curr Hematol Malig Rep. (2012) 7:208–15. doi: 10.1007/s11899-012-0132-3
49. Teruya-Feldstein J, Jaffe ES, Burd PR, Kanegane H, Kingma DW, Wilson WH, et al. The role of Mig, the monokine induced by interferon-gamma, and IP-10, the interferon-gamma-inducible protein-10, in tissue necrosis and vascular damage associated with Epstein-Barr virus-positive lymphoproliferative disease. Blood. (1997) 90:4099–105. doi: 10.1182/blood.V90.10.4099
Keywords: lymphomatoid granulomatosis, central nervous system, primary, imaging, neuropathology, Epstein-Barr virus
Citation: Xiang Y, Liu C, Xue Y, Li S, Sui Y, Li J, Sun Q and Liu X (2020) Primary Central Nervous System Lymphomatoid Granulomatosis: Systemic Review. Front. Neurol. 11:901. doi: 10.3389/fneur.2020.00901
Received: 12 October 2019; Accepted: 13 July 2020;
Published: 11 September 2020.
Edited by:
Adriana Olar, Medical University of South Carolina, United StatesReviewed by:
Jan Styczynski, University of Bydgoszcz, PolandCopyright © 2020 Xiang, Liu, Xue, Li, Sui, Li, Sun and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaohui Liu, c2x5eWx4aEAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.