 Pauline Mencke
Pauline Mencke Zoé Hanss
Zoé Hanss Ibrahim Boussaad
Ibrahim Boussaad Pierre-Emmanuel Sugier2
Pierre-Emmanuel Sugier2 Rejko Krüger
Rejko Krüger
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 20 August 2020
Sec. Neurogenetics
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00898
This article is part of the Research Topic Celebrating the Diversity of Genetic Research to Dissect the Pathogenesis of Parkinson's Disease View all 22 articles
Cancer and Parkinson's disease (PD) define two disease entities that include opposite concepts. Indeed, the involved mechanisms are at different ends of a spectrum related to cell survival - one due to enhanced cellular proliferation and the other due to premature cell death. There is increasing evidence indicating that patients with neurodegenerative diseases like PD have a reduced incidence for most cancers. In support, epidemiological studies demonstrate an inverse association between PD and cancer. Both conditions apparently can involve the same set of genes, however, in affected tissues the expression was inversely regulated: genes that are down-regulated in PD were found to be up-regulated in cancer and vice versa, for example p53 or PARK7. When comparing glioblastoma multiforme (GBM), a malignant brain tumor with poor overall survival, with PD, astrocytes are dysregulated in both diseases in opposite ways. In addition, common genes, that are involved in both diseases and share common key pathways of cell proliferation and metabolism, were shown to be oppositely deregulated in PD and GBM. Here, we provide an overview of the involvement of PD- and GBM-associated genes in common pathways that are dysregulated in both conditions. Moreover, we illustrate why the simultaneous study of PD and GBM regarding the role of common pathways may lead to a deeper understanding of these still incurable conditions. Eventually, considering the inverse regulation of certain genes in PD and GBM will help to understand their mechanistic basis, and thus to define novel target-based strategies for causative treatments.
There is now accumulating evidence for an inverse association between Parkinson's Disease (PD) and cancer (1–3). Studies suggest that people affected by a neurodegenerative disorder have a reduced incidence for most cancers (4, 5). Molecular studies showed that there is an inverse correlation of the expression of shared genes in PD and cancer: genes down-regulated in PD can be up-regulated in cancer and vice versa (6, 7). These inversely correlated gene expression may affect the same pathways in opposite ways, either involving genetic or environmental factors (5, 8, 9). Shared genetic pathways deregulated in opposite ways are a major focus, particularly those favoring apoptosis and cell proliferation, influencing cell cycle control, DNA repair, and kinase signaling (4). Common mechanisms such as chronic inflammation (10) and immunosenescence, and common risk factors like diabetes and obesity, have been implicated in both conditions (11, 12).
PD is a neurodegenerative disease characterized by three cardinal motor symptoms: tremor, rigidity and bradykinesia resulting from loss of dopaminergic neurons in the substantia nigra pars compacta (13). PD affects 1–2% of the population over 60 years (14). Age of onset before the age of 40 is seen in <5% of the cases in population-based cohorts, which is typical of familial cases of PD with underlying genetic cause like mutations in SNCA, Parkin, PINK1, DJ-1, LRRK2, ATP13A (Table 1). Monogenic forms of PD are rare. In general, genetic factors are claimed to be involved in 5–10% of the cases (14). Histopathological hallmarks of PD are proteolytic inclusions called Lewy bodies (LB) and Lewy neurites containing α-synuclein (47). Cellular hallmarks of PD are an impairment of proper functioning of molecular and organelle degradation pathways like the ubiquitin–proteasome system and autophagy (48). In particular, the process of removing defective mitochondria from the cells is known to be impaired in PD (49). This process is a special form of autophagy, called mitophagy (50), and is regulated by the PD-linked proteins PINK1 and Parkin (51). The impairment of autophagy, lysosomal and mitochondrial function in PD can lead to the accumulation of α-synuclein and defective mitochondria (52) and, ultimately, to neurodegeneration. The diagnostic of PD is mostly a clinical diagnosis as it is based on neurological tests when the PD patients already show motor symptoms. Due to the complexity and heterogeneity of PD, the etiology is not yet fully understood. Therefore, there is no cure for PD and no treatment that will stop the progress of the disease and treatment is only symptomatic, e.g., levodopa therapy. This is why it is important to investigate underlying mechanisms of PD to stratify causative treatments.
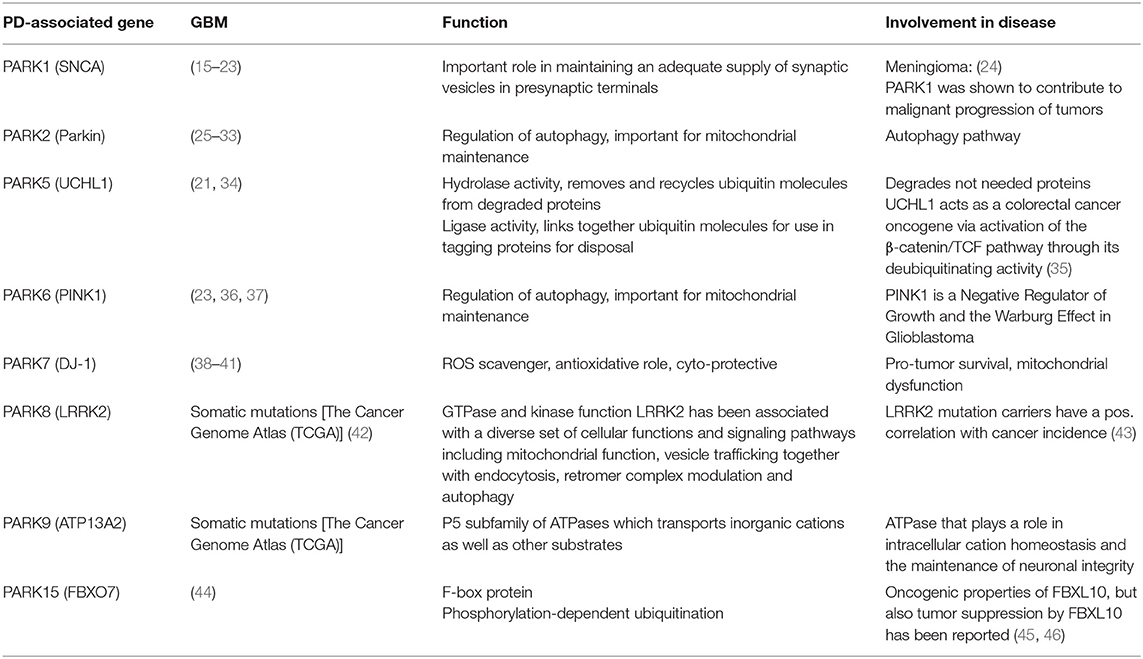
Table 1. Overview PD-genes in GBM.
Glioblastoma multiforme (GBM) is the most malignant tumor of the central nervous system. GBM tumors are most likely developing from astrocytes (53). Based on their histological and clinical features, astrocytomas are classified into four different subtypes according to the WHO classification: Pilocytic astrocytoma, diffuse astrocytoma, anaplastic astrocytoma, and GBM. Pilocytic and diffuse astrocytoma are characterized by a rather low growth rate, while anaplastic astrocytoma and GBM show common uncontrolled proliferation and diffuse tissue penetration (54). GBM is characterized by poor prognosis, low survival rates, and extremely limited opportunities for therapy. Symptoms of GBM are rather unspecific like increased intracranial pressure, including headache and focal or progressive neurologic deficits. Seizures are the presenting symptom in 25% of patients and can occur at a later stage of the disease in 50% of patients (55). Malignant gliomas are the third leading cause of cancer death for people aged between 15 and 34, accounting for 2.5% of the global cancer death toll. GBM has a maximum incidence in patients aged more than 65 years, and is mainly affecting the cerebral hemispheres (54). A cellular hallmark of GBM and all cancers is the so-called Warburg effect which describes the phenomenon that cancer cells use aerobic glycolysis to produce ATP (56). GBM cells are characterized by increased glucose uptake and lactate production (57). GBM cells also use oxidative phosphorylation (OXPHOS) (57). The hypoxic GBM tumor environment allows the constant expression of hypoxia inducible factors 1 alpha and 2 alpha (HIF-1α, HIF-2α). Hypoxia and hypoxia-stabilized HIFs regulate GBM metabolism by stabilizing genes involved in metabolism like the glucose transporters GLUT1 and GLUT3, thereby sustaining an increased glucose uptake of the GBM cells (57). Also, the enzyme catalyzing the first step in glycolysis, hexokinase, is hypoxia/HIF regulated (57). As for PD, the diagnosis of GBM is typically made when first symptoms occur and rely on clinical examination and neuroimaging methods. However, mostly both diseases are diagnosed at an advanced stage of tumor growth or neurodegeneration, respectively. Treatment strategies of GBM are based on a multidisciplinary approach. Current standard therapy is a combination of maximal safe surgical resection of the tumor and subsequent radiation and chemotherapy with temozolomide (Temodar®), an oral alkylating agent. However, even with advances in surgical resection, the prognosis for GBM patients remains poor, with a median survival of 15 months (55).
A common set of genes like the tumor suppressor p53, epidermal growth factor and its receptor EGF(R), the glyoxalase and deglycase DJ-1 and biological processes are deregulated in opposite directions in PD and GBM (6). Particularly, there is evidence that PD-associated genes are involved in GBM pathogenesis (Table 1). A summary of publications examining and exhibiting the involvement of PD-associated genes in GBM is shown in Table 1. Consistent with PD-associated genes being involved in GBM, it is important to note that mutations in the same gene can behave differently if they are germline or somatic mutations. For example, mutations in PARK2 affecting the Parkin protein can cause neuronal cell death in PD if they are present in the germline, or increased cell survival in GBM if they are present in somatic cells like astrocytes (Figure 1). (25). Pathways that are affected in PD and GBM are overlapping but are regulated inversely by alternatively regulated genes. These pathways are regulating cell proliferation and cell metabolism as well as mitochondrial clearance (1). In the following, examples for inversely regulated pathways in PD and GBM are illustrated and the role of commonly involved genes in both diseases in the regulation of these pathways will be outlined.
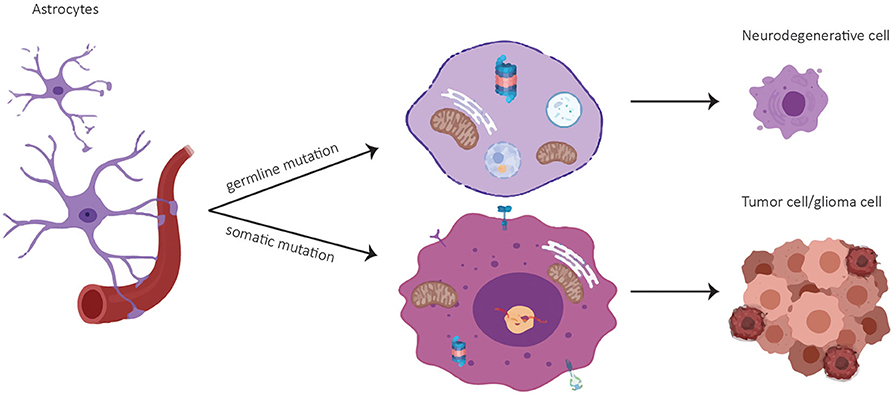
Figure 1. Cell fate of astrocytes depending on mutational status. A germline mutation in a PD-associated gene might result in a neurodegenerative cell whereas a somatic mutation can lead to a tumor cell.
Pro-survival signaling is one of the most important pathways regulating and sustaining cell proliferation. Once dysregulated, uncontrolled cell proliferation can lead to tumorigenesis. This is why cell proliferation and apoptosis need to be in a tight equilibrium, which is well controlled by many mediators.
One key player in the regulation of cell proliferation is the tumor suppressor p53. p53 is upregulated in PD, but downregulated in GBM (Figure 2A) (58–60).
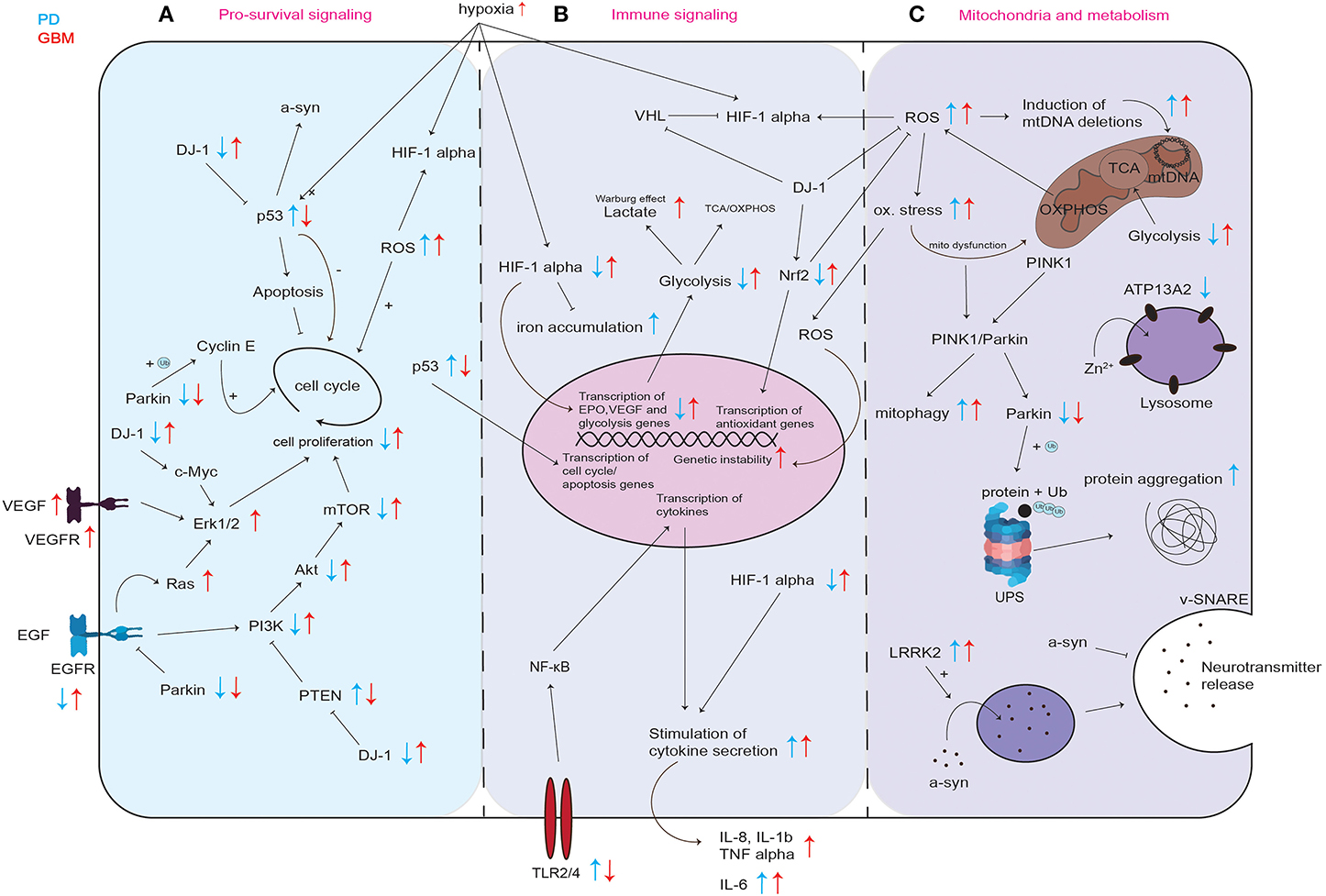
Figure 2. Graphical representation of common cellular pathways described in literature to be dysregulated in PD and GBM. Dysregulation (up- or downregulation) of mediators and proteins of commonly involved mediators and proteins in PD and GBM is illustrated with blue and red arrows, while blue arrows correspond to the situation in PD, red arrows indicate the regulation in GBM. Differential regulation of discussed mediators regarding pro-survival signaling (A) immune signaling (B) and their involvement in mitochondria and metabolism (C). UPS, ubiquitin proteasome system; ox. stress, oxidative stress; mito dysfunction, mitochondrial dysfunction.
p53 inhibits cell proliferation by both blocking cell cycle progression and promoting apoptotic cell death (Figure 2A). This way, p53 provides a clear prevention from stem cell tumor growth and thereby GBM development. p53 itself is also regulated via several stress signals occurring during malignant progression like genotoxic damage, oncogene activation, loss of normal cell contacts, and hypoxia (Figure 2A). This leads to a model where growth inhibitory functions of p53 are normally held dormant, to be unleashed only in nascent cancer cells (61). In PD, the level of p53 and its activity in neurons can increase not only as a result of oxidative stress and DNA damage, but also due to aberrant regulation of its expression for example by mutated or incorrectly cleaved proteins involved in the process of neurodegeneration (58). An increase in p53 expression and its activation results in enhanced expression of genes that are responsible for apoptosis and/or cell cycle arrest and may trigger neuronal cell death (58). In line, Mogi et al. found increased levels of p53 protein in the nigrostriatal dopaminergic region in PD patients compared to controls (62). It was shown that p53 regulates α-synuclein expression since the α-synuclein promoter harbors a p53 responsive element (63). Therefore, an increase in p53 in PD could not only lead to increased apoptosis induction but also to an increase in expression of potentially dysfunctional α-synuclein and to its subsequent aggregation (63). Kato et al. found that DJ-1 inhibits the transcriptional activity of p53 (Figure 2A) (64). Loss of DJ-1 protein in PD could thereby lead to increased expression of p53 target genes leading to cell death. In GBM, p53 is frequently downregulated or inactivated by mutations leading to a reduction in apoptosis induction (Figure 2A) (65) and p53 inactivation positively correlates with GBM tumor invasiveness (66). Zheng et al. showed that central nervous system (CNS)-specific deletion of p53 and Phosphatase And Tensin Homolog (PTEN) in the CNS of mice leads to a high-grade malignant glioma phenotype resembling human GBM (67). These results are in line with the data found at The Cancer Genome Atlas in the exploration mode when looking at the TCGA-GBM data set, which reports PTEN, p53 and EGFR as the most frequently mutated tumor suppressor genes in GBM (https://portal.gdc.cancer.gov).
EGFR is downregulated in PD and upregulated in GBM (Figure 2A). EGFR activates the phosphoinositide 3-kinase (PI3K)-Akt pathway (Figure 2A). The PI3K/Akt signaling pathway is known as one of the most important kinase cascades that mediates crucial cellular functions such as survival, proliferation, migration, and differentiation (68). Activated receptor tyrosine kinases (RTKs) like EGFR activate PI3K through direct binding or through tyrosine phosphorylation of scaffolding adaptors, which can then bind and thereby activate PI3K (Figure 2A). PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3), in a reaction that can be reversed by the PIP3 phosphatase PTEN. AKT can then activate its downstream targets like mTOR, eventually leading to cell proliferation (Figure 2A). It was shown that EGFR endocytosis and degradation are accelerated in Parkin-knockout cells from mouse brain, and EGFR signaling via the PI3K/Akt pathway is reduced (69). Fallon et al. propose that Parkin delays EGFR internalization and degradation, thereby promoting PI3K/Akt signaling (69). Therefore, by decreasing the efficiency of EGFR-mediated Akt signaling in neurons, the loss of Parkin leads to neuronal degeneration (69). In post-mortem brains of idiopathic PD patients, protein levels of EGF and EGFR were shown to be decreased in the prefrontal cortex and the striatum (70). Mutations in EGFR are commonly occurring in GBM (71). These mutations result in EGFR gene amplification and intrinsic alterations of the EGFR structure (71). Brennan et al. showed that gene amplification and mutation of EGFR results in enhanced EGFR activation and is found in about 60% of GBM (72). The most common EGFR mutation in GBM is EGFRvIII, which is caused by the deletion of exon 2–7 leading to constitutively activated EGFR (71, 73, 74). It was shown that EGFR is overexpressed in most of primary GBM and some of the secondary GBM and that EGFR overexpression is associated with more aggressive GBM (75).
In PD, PTEN/PI3K/Akt signaling is down-regulated and therefore causes decreased pro-survival signaling (76). In GBM, PTEN/PI3K/Akt signaling is upregulated (77–79). PTEN negatively regulates PI3K (Figure 2A), thereby inhibiting PI3K/Akt mediated proliferation and cell survival. In PD patient-derived post mortem brains, Sekar et al. found an increase in PTEN levels (80). Absence of PTEN protected dopaminergic neurons in PTEN knockout mice from neuronal death after neurotoxin treatment (81). In another mouse model, depletion of PTEN attenuated the loss of tyrosine hydroxylase-positive (dopaminergic) cells after neurotoxin treatment (82). An increase in PTEN in PD results in decreased pro-survival signaling leading to increased neuronal cell death. In line, it was shown that the ratio of phospho-Akt/total-Akt decreases in dopaminergic neurons indicating a decrease in activation of the pro-survival signaling mediated by Akt upon phosphorylation (83). Overall, an impaired PTEN/PI3K/Akt signaling in PD leading to neuronal cell death can be due to mutations in PD-associated genes regulating Akt signaling [e.g., DJ-1 (84), (Figure 2A)], excessive Akt dephosphorylation, inhibition of Akt activation or oxidative stress (85). In GBM, PTEN/PI3K/Akt signaling is upregulated due to EGFR overexpression or loss of PTEN (78). Mutations or homozygous deletions of PTEN were shown in 36% of the GBM cases that were studied by McLendon et al. and 86% of the GBM harbored at least one genetic event in the receptor tyrosine kinase PI3K pathway (86). High level of phosphorylated Akt was shown to correlate with a poor prognosis for patients with GBM (87). Mutations in the phosphatidylinositol-4,5-bisphosphcxate 3-kinase catalytic subunit alpha (PIK3CA), which is one subunit of PI3K, were shown to induce gliomagenesis (77).
The protein DJ-1 was shown to be inversely regulated in PD and GBM. (Figure 2A). Homozygous mutations in PARK7 (DJ-1) resulting in loss of protein lead to PD (88). DJ-1 expression was shown to be increased in GBM (38, 89, 90). Wang et al. found that high DJ-1 and high β-catenin expression in GBM were significantly associated with high grade and poor prognosis in glioma patients, suggesting DJ-1 levels in GBM as a strong independent prognostic factor (89). DJ-1 also accelerates transformation of tumor cells by c-Myc activating the Erk pathway (91). Hinkle et al. found that GBM tumor tissue expressed DJ-1 protein at significant levels, and typically in a cytoplasmic, non-nuclear manner. They found that immunostaining intensity of DJ-1 varied directly with strong nuclear p53 expression and inversely with EGFR amplification (38). In addition to the fact that DJ-1 negatively regulates pro-apoptotic p53 (Figure 2A) (92), and EGFR signaling is crucial for gliomagenesis (72), these observations suggest that DJ-1 might be involved in tumorigenesis of GBM (38). Toda et al. found that in a serial transplantation study, DJ-1 knockdown resulted in a prolonged survival of mice in secondary transplantation (39). DJ-1 is known to counteract ROS, among others via Nrf2 stabilization leading to the expression of endogenous antioxidant synthesis and ROS-eliminating enzymes like glutathione (Figure 2A) (93, 94). It was shown that a reduction in DJ-1 protein is associated with reduced Nrf2 transcriptional activity and that in PD patients, Nrf2 activation is associated with dysregulated downstream gene expression (93, 95). In contrast, it was found that Nrf2 overexpression accelerates proliferation and oncogenic transformation of glioma cells and that GBM patients have reduced overall survival when Nrf2 levels are upregulated (Figure 2A) (96).
The innate immune system obtains various functions in health and disease. It represents the first line of defense against infection and it is involved in many different processes like tissue repair, wound healing and the clearance of apoptotic cells and cellular debris. An excessive or non-resolving activation of the innate immune system can result in systemic or local inflammatory complications and cause or contribute to the development of neurodegeneration and cancer. In the brain, the innate immune cells are represented by microglia, which regulate brain development, brain maturation, and homeostasis. An impairment of functional microglia through abnormal activation or decreased functionality can occur during aging and during neurodegeneration and the resulting inflammation was shown to be involved in neurodegenerative diseases and cancer (97).
It is well known that hypoxia-inducible factor-1α (HIF-1α) plays an important role in gliomagenesis due to its angiogenesis-promoting effects (98). While HIF-1α is upregulated in GBM, it was shown that HIF-1α is impaired in PD (Figure 2B) (99, 100).
Treatment with MPTP, a prodrug to the neurotoxin MPP+, which causes Parkinsonism symptoms by destroying the dopaminergic neurons, was shown to inhibit HIF-1α accumulation in mice and in dopaminergic cell lines (99). Moreover, Milosevic et al. found that a conditional knock-down of HIF-1α in mice resulted in a 40% decrease in expression of tyrosine hydroxylase, a known marker for dopaminergic neurons, in the substantia nigra of mice (101). In healthy individuals, HIF-1α mediates protection of dopaminergic neurons by regulation of iron homeostasis, improved defense against oxidative stress by upregulation in response to reactive oxygen species (ROS) (Figure 2B) and mitochondrial dysfunction (100). PD is characterized by an accumulation of iron in dopaminergic neurons of the substantia nigra (102). Free cytosolic iron can lead to oxidative stress and trigger α-synuclein aggregation (102). HIF-1α influences iron homeostasis by expression of its target genes ferroportin and heme oxygenase in the substantia nigra which are known to be involved in the attenuation of iron accumulation (100). This way, HIF-1α can counteract iron accumulation (Figure 2B). However, in PD, downregulation of HIF-1α can lead to a dysregulation in iron homeostasis eventually leading to iron accumulation (Figure 2B). In turn, iron accumulation decreases HIF-1α activity, because iron is a necessary cofactor for prolyl hydroxylases that inactivate HIF-1α via subsequent ubiquitinylation through von Hippel-Lindau factor (VHL) (Figure 2B) (102, 103). HIF-1α target genes Erythropoietin (EPO) and vascular endothelial growth factor (VEGF) (Figure 2B) have been shown to contribute to the protection of neurons from PD pathogenesis (100). EPO was shown to be neuroprotective against dopaminergic neurotoxins (104). In rat explants of the ventral mesencephalon, VEGF treatment was shown to be mitogenic for endothelial cells, astrocytes, and could promote growth and survival of neurons and specifically dopaminergic neurons (105). There are accumulating data which suggest that the activation of HIF-1α can exert neuroprotective effects through the induction of intrinsic adaptive mechanisms in neuronal and non-neuronal cells (106). Lee et al. showed that stabilization of HIF-1α leads to the upregulation of several proteins involved in iron efflux and mitochondrial integrity and bioenergetics, cell components that are compromised in PD. This is why Lee's data emphasize the concept that the pharmacological induction of HIF-1α could have neuroprotective effects in PD cells and mice models, with a beneficial impact on dopamine synthesis, iron homeostasis, antioxidant defenses and mitochondrial dysfunction (107).
In contrast to these observations in PD, in GBM, HIF-1α levels are increased (Figure 2B) (108). Liu et al. found that HIF-1α expression was associated with high grade glioma and the overall survival of glioma patients, which indicates that HIF-1α could predict prognosis and provide clinical insights into the therapeutic strategy for GBM patients (109). The lack of oxygen in the GBM microenvironment results from inappropriate neovascularization, irregular blood flow, and excessive consumption of oxygen from the uncontrolled proliferating GBM cells (110). The hypoxia in the GBM tumor induces the expression of genes involved in tumor cell growth and angiogenesis like the signal transducer and activator of transcription 3 (STAT3), which triggers the synthesis of HIF-1α that subsequently induces activation of T-regulatory cells (Tregs) and the production of VEGF (111). Tregs are important modulators of the immune response, and VEGF has known immunosuppressive effects. Moreover, the hypoxic microenvironment causes the transformation of CNS macrophages into tumor-associated macrophages (TAMs), which are capable of adopting immunosuppressive and tumor-supportive phenotypes. Via the STAT3 pathway, this transformation triggers TAMs to enhance angiogenesis and tumor cell invasion (26, 112). Furthermore, HIFs are critical for the upregulation of glycolysis (Figure 2B) (113). Hypoxia is also a known regulator of many other innate immunological functions like cell migration, apoptosis, phagocytosis of pathogens, antigen presentation and production of cytokines, chemokines, and angiogenic and antimicrobial factors (113). In summary, HIF is an important factor in the regulation of the tumor microenvironment due to its central role in promoting proangiogenic and invasive properties. Since HIF activation results in angiogenesis and the emerging vasculature is often abnormal, this leads to a vicious cycle that causes further hypoxia and HIF upregulation in GBM (98).
In PD, increased cytokine levels in response to cellular stress can lead to neuronal cell death whereas in GBM, cytokines like interleukins IL-1β, IL-6, and IL-8 released by the tumor cells, inhibit the immune response and allow the tumor cells to escape the eradication by the immune system (Figure 2B).
IL-6 was found to be increased in the nigrostriatal region and in the cerebrospinal fluid of patients with PD (114). Further, Hofmann et al. found that patients with more severe PD had higher IL-6 levels compared to patients with a milder phenotype (114). In addition, a study from Chen et al. found that patients with PD had elevated levels of transforming growth factor-beta 1 (TGF-β1), IL-6, and IL-1β in cerebrospinal fluid compared to controls (115). In line, it is described that, in autopsy brains of PD, the number of activated microglia, which were among others TNF- α, and IL-6-positive, increased in the substantia nigra and putamen during the progress of PD (116). The activated microglia in PD was observed in various brain regions like the nigro-striatal region, the hippocampus and the cerebral cortex. The levels of IL-6 and TNF- α mRNAs increased in the hippocampus of PD patients (116). It is postulated that cytokines (IL-1β, TNF-α, IL-6) from activated microglia (117) in the substantia nigra and putamen may be initially neuroprotective, but may later turn to be neurotoxic during PD pathogenesis (116).
In contrast to PD, in GBM, the cells can profit from the cytoprotective effects of specific cytokines like IL-1β, IL-6, and IL-8 leading to increased robustness regarding cellular stress (118). As already mentioned, GBM arises from glial cells with surrounding brain parenchyma that contains CNS cells like astrocytes, neurons and microglia, as well as a distinctive extracellular matrix composition. GBM induces a tumor microenvironment characterized by immunosuppressive cytokines secreted by tumor cells, microglia and tumor macrophages. IL-6, IL-10, and TGF-β, and prostaglandin-E collectively inhibit both the innate and adaptive immune systems leading among others to the suppression of natural killer cell activity, T-cell activation and proliferation and induction of T-cell apoptosis (119). IL-1β is a known master pro-inflammatory cytokine that triggers various malignant processes driving oncogenic events such as proliferation and invasiveness (118, 120). Elevated levels of IL-1β were observed in many different GBM cell lines (121) and in human GBM tumor specimens (122). IL-6 was shown to be overexpressed in GBM clinical samples and cell lines and IL-6 gene expression seems to correlate with the aggressiveness of the tumor (123). It was shown that IL-6 is secreted by GBM cells and sustains the cell proliferation by activation of STAT3 pro-survival pathway (124). IL-6 is produced by GBM cells in response to external stimuli or intrinsic factors, for example oncogenic mutations (118). IL-1β and TNF-α induce stabilization of IL-6 mRNA and increase IL-6 biosynthesis (125). Like IL-6, IL-8 is highly expressed and secreted from GBM cell lines, tumor stem cells and human specimens (118). It was shown that the expression of the constitutively active mutant EGFRvIII is associated with significantly higher expression of IL-8 induced by nuclear factor kappa B (NF-κB) (Figure 2B) in human GBM specimens and GBM cell lines (126). In a similar manner as the regulation of IL-6, IL-8 expression can be enhanced by TNF-α, IL-1β or macrophage infiltration (127). Thus, elevated levels of one cytokine like TNF-α for example can lead to an increase in other cytokines. These findings of elevated cytokines and their associated roles in GBM underline the importance of specific cytokines for immune escape mechanisms and tumor proliferation and invasiveness observed in GBM pathogenesis.
Toll-like-receptors (TLRs) are receptors that recognize distinct molecular patterns like lipopolysaccharides, single and double stranded RNAs, hemagglutinin, viral proteins etc. (128), and allow an appropriate immune response to be initiated. The TLR family consists of 10 members (TLR1-10) in humans with different expression profiles and ligands (129). TLR2 is essential for the recognition of peptidoglycans and lipoproteins, whereas TLR4 recognizes bacterial lipopolysaccharide (LPS) (130). TLR2 and TLR4 are both the most important TLRs with regard to innate immune response as they are both implicated in the recognition of endogenous ligands involved in the inflammatory response regardless of the source of infection (131). This is why the implication of TLR2 and TLR4 in PD and GBM will be discussed in the following.
TLR2 and TLR4 are frequently upregulated in PD and downregulated in GBM allowing the tumor cells to escape clearance by the innate immune system. TLR2 and TLR4 were shown to be upregulated in many α-synuclein-overexpressing or toxin-induced animal models (132–135), and accumulating evidence from human studies further implicates these receptors in the pathogenesis of PD (136). Clinical studies revealed that TLR2 expression is increased in PD (137). It was shown that microglial TLR2 is increased in the substantia nigra and the hippocampus in the early stages of PD, but not during the late stages (138), while another study found that TLR2 is increased in the striatum of advanced PD patients (135).
In contrast, GBM cancer stem cells downregulate TLR4 to evade immune suppression (139). Alvarado et al. showed that in GBM, cancer stem cells have low TLR4 expression which enables cell survival by avoiding inhibitory innate immune signaling (e.g., clearance by dendritic cells, cytotoxic T cells, and natural killer cells) that aims to suppress self-renewal of the GBM stem cells (140). This is why TLR agonists that trigger antitumoral immune signaling are being discussed as therapy for GBM (141).
Mitochondria and cellular metabolism are closely linked. Mitochondria host many enzymatic reactions of cellular metabolism like the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) which generate ATP from pyruvate in the presence of oxygen (Figure 2C). In age-related disease, like PD and GBM, damaged mitochondria lead to impaired cellular metabolism (142).
The human brain, even though constituting only 2% of the total body weight, uses ~20% of the body's total oxygen consumption and 60% of our daily glucose intake (143). Furthermore, the brain needs a constant supply of glucose since it lacks fuel stores and cannot store glycogen. This is why cellular changes in glucose metabolism can have high impact on brain cell homeostasis, proliferation and viability.
It was shown that glycolysis and mitochondrial function like respiration are decreased in individuals with PD (Figure 2C) (144–146). In GBM, increased glycolytic activity results from certain oncogenic alterations like c-Myc amplification, PTEN deletion or mutations in p53 (Figure 2C) (147, 148).
While mitochondrial dysfunction in PD can cause increased generation of ROS and subsequent oxidative damage (Figure 2C), it can also result in failing neuronal compensation of their insufficient ATP generation (149). Activation of glycolysis in neurons leads to excessive oxidative stress and apoptosis, suggesting that neurons are predominantly restricted to OXPHOS (150). In line, Hall et al. showed that the majority of ATP used by neurons is produced by OXPHOS (151). Powers et al. found that overexpression of α-synuclein in N27 dopaminergic cells resulted in an impairment in glycolysis, a reduction in glycolytic capacity and mitochondrial respiration (152). This is why an increase in glycolysis as counteract mechanism to neuronal energy failure induced by mitochondrial dysfunction in PD eventually leads to neuronal cell death (153–155). Neurons also metabolize glucose via the pentose phosphate pathway (PPP) to maintain their antioxidant status (156). It was shown that inhibition of the PPP in neuronal cell models causes cell death (157). In rodents, PPP inhibition caused dopaminergic cell death causing motor deficits that resemble Parkinsonism (158). Using postmortem human brain tissue, Dunn et al. characterized glucose metabolism via the PPP in early sporadic PD and controls and observed a down-regulation of PPP enzymes in patients compared to controls (156). This observation suggests that the impairment of the PPP is an early event in sporadic PD (156).
In the absence of oxygen, pyruvate can be metabolized into lactate, a process known as glucose fermentation or anaerobic glycolysis. Rapidly proliferating cells, such as cancer cells, also have the ability to ferment glucose into lactate, even in the presence of abundant oxygen; this process is called aerobic glycolysis. It has been observed already decades ago, that cancer cells, even in aerobic conditions, tend to favor metabolism via glycolysis rather than OXPHOS, which is preferred by most other cells. This phenomenon is called the Warburg effect (56, 159). This is why, in contrast to PD neurons, GBM cells ferment glucose into lactate, even in the presence of abundant oxygen (Figure 2B). Even though ATP production is less efficient in aerobic glycolysis when compared to ATP production via complete oxidative metabolism of glucose, it is being hypothesized that GBM cells use aerobic glycolysis to generate precursors for anabolism to grow and are able to generate enough ATP to sustain their cellular function (160). By modulating glycolysis and altering mitochondrial metabolism, GBM cells generate biomass, namely nucleotides, lipids, proteins, and NADPH by using glycolytic/TCA intermediates (160). Knockdown of glycolytic genes strongly inhibits GBM growth further emphasizing that glycolytic enzymes are essential for GBM growth (148). GBM cells also generate large amounts of lactate for several pro-tumor growth functions (161). Li et al. found that EGFR activation in GBM cells promotes the translocation of phosphoglycerate kinase (PGK1) into mitochondria (162, 163). In the mitochondria, PGK1 phosphorylates and activates pyruvate dehydrogenase kinase that phosphorylates and thereby inhibits pyruvate dehydrogenase and thus mitochondrial pyruvate consumption which eventually leads to enhanced lactate production (162, 163). In addition to the aerobic glycolysis, GBM cells also utilize TCA and OXPHOS (160).
The differential expression of metabolic genes in neurons and astrocytes might explain the differences in glycolysis and OXPHOS rates. For example, neurons lack 6-phosphofructose-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) since it is continuously degraded by the ubiquitin-proteasome pathway. PFKFB3 regulates the biogenesis and degradation of fructose-2,6-bisphosphate, a known glycolytic activator. In contrast, in astrocytes, PFKFB3 is activated by adenosine monophosphate-activated protein kinase (AMPK) and promotes glycolysis (149). In line, it was shown that the expression of PFKFB3 is higher in mouse astrocytes than in murine neurons due to proteasomal degradation in the neurons (164). In neurons, the activation of PFKFB3 results in enhanced glycolysis but eventually leads to cell death since neurons lose their ability to generate glutathione, an essential antioxidant involved in the management of oxidative stress. This means that unlike astrocytes, neurons use glucose to maintain their antioxidant status and not for bioenergetic purposes (164). These findings might help to explain why PD neurons fail to increase their glycolysis rates and why increased glycolysis leads to sustained cell proliferation in astrocyte-originating GBM cells.
Epidemiological evidence suggests that patients with PD have a reduced incidence of primary CNS tumors (165, 166). In contrast, there are a few epidemiological studies that show a positive association of PD with benign and malignant brain tumors, but not specifically with GBM (167–169). However, the problem with these studies is that they do not distinguish between the types of brain cancer, e.g., meningioma or astrocytoma. The described increased risk of all types of brain cancers in PD might be caused by diagnostic misclassification and detection bias. Increased incidence of meningioma in PD patients for example might result from the fact that the symptoms can be wrongly diagnosed as a sign of PD, if the intracranial tumor leads for example to a compression of the basal ganglia resulting in PD symptoms (170–173). Moreover, a positive association of brain tumors and PD can be caused by detection bias as brain tumors can be diagnosed during the clinical work-up for PD (174). Since patients diagnosed with parkinsonism are more likely to have a Magnetic Resonance Imaging at the time of diagnosis, this may explain a higher risk of detecting silent brain tumors (173, 175). The close temporal association between diagnosis of PD and the incidence of brain tumors further leads to the suggestion that brain tumors might be misdiagnosed as PD or vice versa (176). Specifically, for GBM, as it is lethal, it is difficult to study PD in individuals who survived GBM. This is why future studies should focus on evaluating the risk of GBM in PD patients.
Interestingly, there is an increased risk of melanoma in PD patients compared to controls (177–179). In 1985, Dr. Rampen reported a 55-year-old male with PD who developed a local recurrence of a primary melanoma and multiple primary melanomas 4 years after primary excision and 4 months after starting levodopa (180). An increased risk of malignant melanoma in PD patients has been confirmed since in many studies (8, 176, 181, 182). Several hypotheses could account for this association. Since levodopa is a metabolite in the biosynthesis of dopamine and melanin which involves the enzyme tyrosinase, and increased tyrosinase activity is found in melanoma, it was initially hypothesized that levodopa could enhance and stimulate growth on any residual melanoma tissue (183). However, recent studies have refuted a causal association for several reasons (178, 184). In particular, the observation that the risk of melanoma is increased in PD patients before diagnosis argues against an effect of levodopa. Additional explanations may be the existence of shared genetic or environmental factors, or the common embryonic origin of melanocytes and neurons from neural crest cells (178, 185). In addition, mechanistic links caused by common mutations or other alterations in a number of genes or proteins in PD and melanoma could explain the co-occurrence of PD and melanoma (184). Common mechanisms that are dysregulated in PD and melanoma are for example cellular detoxification, melanin biosynthesis or oxidative stress response (184).
Future studies should investigate underlying mechanisms of decreased risk of some cancers and increased risk of other cancers like melanoma in PD patients.
PD and GBM are two highly complex disease entities characterized by multiple cellular changes. Similar mutations within the same gene, for example Parkin (25), can have inverse effects, depending on whether they are germline or somatic mutations and depending on the type of cell in which they occur: a dividing cell in GBM or a post-mitotic neuron in PD. One could hypothesize that neurons are primarily unaffected in GBM due to their postmitotic state. On the contrary, somatic mutations causing tumorigenesis can spread through proliferative astrocytes.
Another inverse association of PD and GBM that requires future causal investigation is the time frame of the pathophysiology of both diseases. While PD is a chronic, generally slowly progressing neurodegenerative disease characterized by gradual neuronal loss, GBM is a rapidly progressing disease with rapid proliferation of glial cells in a much shorter time frame. Possible explanations for these observations are that in PD, the neuronal loss can be compensated for a long time whereas the aggressiveness of GBM due to highly infiltrative growing and metastasizing cells that also display a vast cell heterogeneity leads to a rapid disease progression.
In this review, we showed that there are common pathogenic mechanisms involved in PD and GBM including inversely deregulated pro-survival and immune signaling, mitochondrial dysfunction and metabolic alterations. There is an inverse regulation for p53, EGF(R), PTEN/PI3K/Akt, DJ-1, HIF-1α in PD and GBM. Due to the complexity of both PD and GBM etiology and pathogenesis, future studies need to unveil so far unknown mechanisms of both diseases that will help to better understand and to compare both diseases and to explain why common inverse dysregulated cellular pathways can lead to two such different diseases. Eventually, a deeper understanding of the pathological mechanisms underlying PD and GBM will guide the identification of possibly shared drug targets that need to be modulated inversely for causative treatment of both diseases.
PM wrote the review. ZH, IB, AE, P-ES and RK advised, structured, and reviewed. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Fond National de Recherche within the PEARL programme (FNR/P13/6682797) and the National Centre for Excellence in Research on Parkinson's disease (NCER-PD) programme and by the European Union's Horizon 2020 research and innovation programme under Grant Agreement No 692320 (WIDESPREAD; CENTRE-PD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Devine MJ, Plun-Favreau H, Wood NW. Parkinson's disease and cancer: two wars, one front. Nat Rev Cancer. (2011) 11:812–23. doi: 10.1038/nrc3150
2. Driver JA. Understanding the link between cancer and neurodegeneration. J Geriatr Oncol.. (2012) 3:58–67. doi: 10.1016/j.jgo.2011.11.007
3. Tallaksen CME, Müller U. Cancer and neurodegeneration: time to move beyond Janus? Neurology. (2017) 88:1106–7. doi: 10.1212/WNL.0000000000003727
4. Plun-Favreau H, Lewis PA, Hardy J, Martins LM, Wood NW. Cancer and neurodegeneration: between the devil and the deep blue sea. PLoS Genet. (2010) 6:1–8. doi: 10.1371/journal.pgen.1001257
5. Gao X, Ning Y. Cancer and Parkinson's disease: the odd couple. Drugs Today. (2011) 47:215–22. doi: 10.1358/dot.2011.47.3.1519657
6. Ibáñez K, Boullosa C, Tabarés-Seisdedos R, Baudot A, Valencia A. Molecular evidence for the inverse comorbidity between central nervous system disorders and cancers detected by transcriptomic meta-analyses. PLoS Genet. (2014) 10:1–7. doi: 10.1371/journal.pgen.1004173
7. Klus P, Cirillo D, Botta Orfila T, Gaetano Tartaglia G. Neurodegeneration and cancer: where the disorder prevails. Sci Rep. (2015) 5:1–7. doi: 10.1038/srep15390
8. D'Amelio M, Ragonese P, Sconzo G, Aridon P, Savettieri G. Parkinson's disease and cancer: insights for pathogenesis from epidemiology. Ann N Y Acad Sci. (2009) 1155:324–34. doi: 10.1111/j.1749-6632.2008.03681.x
9. Garcia-Ratés S, Greenfield S. Cancer and neurodegeneration: two sides, same coin? Oncotarget. (2017) 8:22307–8. doi: 10.18632/oncotarget.16190
10. Li Z, Zheng Z, Ruan J, Li Z, Tzeng CM. Chronic inflammation links cancer and Parkinson's disease. Front Aging Neurosci. (2016) 8:1–7. doi: 10.3389/fnagi.2016.00126
11. Ganguli M. Cancer and dementia. Alzheimer Dis Assoc Disord. (2015) 29:177–82. doi: 10.1097/WAD.0000000000000086
12. Noyce AJ, Bandres-Ciga S, Kim J, Heilbron K, Kia D, Hemani G, et al. The Parkinson's Disease Mendelian randomization research portal. Mov Disord. (2019) 34:1864–72. doi: 10.1002/mds.27873
13. Antony PMA, Diederich NJ, Krüger R, Balling R. The hallmarks of Parkinson's disease. FEBS J. (2013) 280:5981–93. doi: 10.1111/febs.12335
14. Tysnes OB, Storstein A. Epidemiology of Parkinson's disease. J Neural Transm. (2017) 124:901–5. doi: 10.1007/s00702-017-1686-y
15. Kawashima M, Suzuki SO, Doh-Ura K, Iwaki T. A-synuclein is expressed in a variety of brain tumors showing neuronal differentiation. Acta Neuropathol. (2000) 99:154–60. doi: 10.1007/PL00007419
16. Tanji K, Imaizumi T, Yoshida H, Mori F, Yoshimoto M, Satoh K, et al. Expression of α-synuclein in a human glioma cell line and its up-regulation by interleukin-1β. Neuroreport. (2001) 12:1909–12. doi: 10.1097/00001756-200107030-00028
17. Stefanova N, Emgård M, Klimaschewski L, Wenning GK, Reindl M. Ultrastructure of α-synuclein-positive aggregations in U373 astrocytoma and rat primary glial cells. Neurosci Lett. (2002) 323:37–40. doi: 10.1016/S0304-3940(02)00117-9
18. Klegeris A, Giasson BI, Zhang H, Maguire J, Pelech S, McGeer PL. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. (2006) 20:2000–8. doi: 10.1096/fj.06-6183com
19. Tousi NS, Buck DJ, Curtis JT, Davis RL. α-Synuclein potentiates interleukin-1β-induced CXCL10 expression in human A172 astrocytoma cells. Neurosci Lett. (2012) 507:133–6. doi: 10.1016/j.neulet.2011.12.001
20. Duan J, Ying Z, Su Y, Lin F, Deng Y. α-Synuclein binds to cytoplasmic vesicles in U251 glioblastoma cells. Neurosci Lett. (2017) 642:148–52. doi: 10.1016/j.neulet.2017.01.067
21. Sánchez-Valle J, Tejero H, Ibáñez K, Portero JL, Krallinger M, Al-Shahrour F, et al. A molecular hypothesis to explain direct and inverse co-morbidities between Alzheimer's Disease, glioblastoma and lung cancer. Sci Rep. (2017) 7:1–12. doi: 10.1038/s41598-017-04400-6
22. Song YC, Lu GX, Zhang HW, Zhong XM, Cong XL, Xue SB, et al. Proteogenomic characterization and integrative analysis of glioblastoma multiforme. Oncotarget. (2017) 8:97304–12. doi: 10.18632/oncotarget.21937
23. Wipfler K, Cornish AS, Guda C. Comparative molecular characterization of typical and exceptional responders in glioblastoma. Oncotarget. (2018) 9:28421–33. doi: 10.18632/oncotarget.25420
24. Ge Y, Xu K. Alpha-synuclein contributes to malignant progression of human meningioma via the Akt/mTOR pathway. Cancer Cell Int. (2016) 16:1–7. doi: 10.1186/s12935-016-0361-y
25. Veeriah S, Taylor BS, Fang F, Yilmaz E, Vivanco I, Janakiraman M, et al. Somatic mutations of the Parkinson's disease–associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet. (2010) 23:1–7. doi: 10.1038/ng.491
26. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. (2006) 66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278
27. Yeo CWS, Ng FSL, Chai C, Tan JMM, Koh GRH, Chong YK, et al. Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res. (2012) 72:2543–53. doi: 10.1158/0008-5472.CAN-11-3060
28. Viotti J, Duplan E, Caillava C, Condat J, Goiran T, Giordano C, et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene. (2014) 33:1764–75. doi: 10.1038/onc.2013.124
29. Feng DD, Cai W, Chen X. The associations between Parkinson's disease and cancer: the plot thickens. Transl Neurodegener. (2015) 4:20. doi: 10.1186/s40035-015-0043-z
30. Maugeri G, D'Amico AG, Magro G, Salvatorelli L, Barbagall O, Saccone S, et al. Expression profile of parkin isoforms in human gliomas. Int J Oncol. (2015) 47:1282–92. doi: 10.3892/ijo.2015.3105
31. Liu K, Li F, Han H, Chen Y, Mao Z, Luo J, et al. Parkin regulates the activity of pyruvate kinase M2. J Biol Chem. (2016) 291:10307–17. doi: 10.1074/jbc.M115.703066
32. Scott TL, Wicker CA, Suganya R, Dhar B, Pittman T, Horbinski C, et al. Polyubiquitination of apurinic/apyrimidinic endonuclease 1 by Parkin. Mol Carcinog. (2017) 56:325–36. doi: 10.1002/mc.22495
33. Yao ZQ, Zhang X, Zhen Y, He XY, Zhao S, Li XF, et al. A novel small-molecule activator of Sirtuin-1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma article. Cell Death Dis. (2018) 9:767. doi: 10.1038/s41419-018-0799-z
34. Sanchez-Diaz PC, Chang JC, Moses ES, Dao T, Chen Y, Hung JY. Ubiquitin carboxyl-Terminal esterase L1 (UCHL1) is associated with stem-like cancer cell functions in pediatric high-grade glioma. PLoS ONE. (2017) 12:1–19. doi: 10.1371/journal.pone.0176879
35. Zhong J, Zhao M, Ma Y, Luo Q, Liu J, Wang J, et al. UCHL1 acts as a colorectal cancer oncogene via activation of the β-catenin/TCF pathway through its deubiquitinating activity. Int J Mol Med. (2012) 30:430–6. doi: 10.3892/ijmm.2012.1012
36. Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, et al. PINK1 is a negative regulator of growth and the warburg effect in glioblastoma. Cancer Res. (2016) 76:4708–19. doi: 10.1158/0008-5472.CAN-15-3079
37. Lee KS, Wu Z, Song Y, Mitra SS, Feroze AH, Cheshier SH, et al. Roles of PINK1, mTORC2, and mitochondria in preserving brain tumor-forming stem cells in a noncanonical Notch signaling pathway. Genes Dev. (2013) 27:2642–7. doi: 10.1101/gad.225169.113
38. Hinkle DA, Mullett SJ, Gabris BE, Hamilton RL. DJ-1 expression in glioblastomas shows positive correlation with p53 expression and negative correlation with epidermal growth factor receptor amplification. Neuropathology. (2011) 31:29–37. doi: 10.1111/j.1440-1789.2010.01124.x
39. Toda Y, Yoshimura R, Itahara M, Imai Y, Yamada K, Uno T, et al. DJ-1 contributes to self-renewal of stem cells in the U87-MG glioblastoma cell line. Anticancer Res. (2019) 39:5983–90. doi: 10.21873/anticanres.13803
40. Jin S, Dai Y, Li C, Fang X, Han H, Wang D. MicroRNA-544 inhibits glioma proliferation, invasion and migration but induces cell apoptosis by targeting PARK7. Am J Transl Res. (2016) 8:1826–37.
41. Haapasalo J, Nordfors K, Granberg KJ, Kivioja T, Nykter M, Haapasalo H, et al. NRF2, DJ1 AND SNRX1 and their prognostic impact in astrocytic gliomas. Histol Histopathol. (2018) 33:791–801. doi: 10.14670/HH-11-973
42. Zhao B, Shen C, Zheng Z, Wang X, Zhao W, Chen X, et al. Peiminine Inhibits glioblastoma in vitro and in vivo through cell cycle arrest and autophagic flux blocking. Cell Physiol Biochem. (2018) 51:1566–83. doi: 10.1159/000495646
43. Agalliu I, San Luciano M, Mirelman A, Giladi N, Waro B, et al. Higher frequency of certain cancers in LRRK2 G2019S mutation carriers with Parkinson disease a pooled analysis. JAMA Neurol. (2015) 72:58–65. doi: 10.1001/jamaneurol.2014.1973
44. Nord H, Hartmann C, Andersson R, Menzel U, Pfeifer S, Piotrowski A, et al. Characterization of novel and complex genomic aberrations in glioblastoma using a 32K BAC array. Neuro Oncol. (2009) 11:803–18. doi: 10.1215/15228517-2009-013
45. Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer NIH Public Access. Nat Rev Cancer. (2014) 14:233–47. doi: 10.1038/nrc3700
46. Teixeira FR, Randle SJ, Patel SP, Mevissen TET, Zenkeviciute G, Koide T, et al. Gsk3β and Tomm20 are substrates of the SCFFbxo7/PARK15 ubiquitin ligase associated with Parkinson's disease. Biochem J. (2016) 473:3563–80. doi: 10.1042/BCJ20160387
47. Inamdar N, Arulmozhi D, Tandon A, Bodhankar S. Parkinsons Disease: genetics and beyond. Curr Neuropharmacol. (2007) 5:99–113. doi: 10.2174/157015907780866893
48. Senkevich K, Gan-Or Z. Autophagy lysosomal pathway dysfunction in Parkinson's disease; evidence from human genetics. Parkinsonism Relat Disord. (2019) 73:60–71. doi: 10.1016/j.parkreldis.2019.11.015
49. Larsen SB, Hanss Z, Krüger R. The genetic architecture of mitochondrial dysfunction in Parkinson's disease. Cell Tissue Res. (2018) 373:21–37. doi: 10.1007/s00441-017-2768-8
50. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. (2012) 441:523–40. doi: 10.1042/BJ20111451
51. Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. (2012) 2:1–8. doi: 10.1038/srep01002
52. Stefanis L. α-Synuclein in Parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:1–23. doi: 10.1101/cshperspect.a009399
53. Jiang Y, Uhrbom L. On the origin of glioma. Ups J Med Sci. (2012) 117:113–21. doi: 10.3109/03009734.2012.658976
54. Silantyev AS, Falzone L, Libra M, Gurina OI, Kardashova KS, Nikolouzakis TK, et al. Current and future trends on diagnosis and prognosis of glioblastoma: from molecular biology to proteomics. Cells. (2019) 8:863. doi: 10.3390/cells8080863
55. Davis ME. Glioblastoma: overview of disease and treatment. Clin J Oncol Nurs. (2016) 20:1–8. doi: 10.1188/16.CJON.S1.2-8
56. Warburg O. Injuring of respiration the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
57. Libby CJ, Tran AN, Scott SE, Griguer C, Anita B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. (2019) 1869:175–88. doi: 10.1016/j.bbcan.2018.01.004
58. Szybinska A, Lesniak W. P53 dysfunction in neurodegenerative diseases - The cause or effect of pathological changes? Aging Dis. (2017) 8:506–18. doi: 10.14336/AD.2016.1120
59. Zhu H, Wang H, Huang Q, Liu Q, Guo Y, Lu J, et al. Transcriptional repression of p53 by PAX3 contributes to gliomagenesis and differentiation of glioma stem cells. Front Mol Neurosci. (2018) 11:187. doi: 10.3389/fnmol.2018.00187
60. Houck AL, Seddighi S, Driver JA. Review of overlapping biology and its implications. Curr Aging Sci. (2019) 11:77–89. doi: 10.2174/1874609811666180223154436
61. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
62. Mogi M, Kondo T, Mizuno Y, Nagatsu T. p53 protein, interferon-γ, and NF-κB levels are elevated in the parkinsonian brain. Neurosci Lett. (2007) 414:94–7. doi: 10.1016/j.neulet.2006.12.003
63. Duplan E, Giordano C, Checler F, Alves Da Costa C. Direct α-synuclein promoter transactivation by the tumor suppressor p53. Mol Neurodegener. (2016) 11:1–9. doi: 10.1186/s13024-016-0079-2
64. Kato I, Maita H, Takahashi-Niki K, Saito Y, Noguchi N, Iguchi-Ariga SMM, et al. Oxidized DJ-1 inhibits p53 by sequestering p53 from promoters in a DNA-binding affinity-dependent manner. Mol Cell Biol. (2013) 33:340–59. doi: 10.1128/MCB.01350-12
65. Zhang Y, Dube C, Gibert M, Cruickshanks N, Wang B, Coughlan M, et al. The p53 pathway in glioblastoma. Cancers. (2018) 10:297. doi: 10.3390/cancers10090297
66. Djuzenova CS, Fiedler V, Memmel S, Katzer A, Hartmann S, Krohne G, et al. Actin cytoskeleton organization, cell surface modification and invasion rate of 5 glioblastoma cell lines differing in PTEN and p53 status. Exp Cell Res. (2015) 330:346–57. doi: 10.1016/j.yexcr.2014.08.013
67. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. P53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. (2008) 455:1129–33. doi: 10.1038/nature07443
68. Marte BM, Downward J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. (1997) 22:355–8. doi: 10.1016/S0968-0004(97)01097-9
69. Fallon L, Bélanger CML, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, et al. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol. (2006) 8:834–42. doi: 10.1038/ncb1441
70. Iwakura Y, Piao YS, Mizuno M, Takei N, Kakita A, Takahashi H, et al. Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson's disease and its model: neurotrophic implication in nigrostriatal neurons. J Neurochem. (2005) 93:974–83. doi: 10.1111/j.1471-4159.2005.03073.x
71. Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets. (2012) 12:197–209. doi: 10.2174/156800912799277557
72. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. (2014) 157:753. doi: 10.1016/j.cell.2014.04.004
73. Chistiakov DA, Chekhonin IV, Chekhonin VP. The EGFR variant III mutant as a target for immunotherapy of glioblastoma multiforme. Eur J Pharmacol. (2017) 810:70–82. doi: 10.1016/j.ejphar.2017.05.064
74. Gao Y, Vallentgoed WR, French PJ. Finding the right way to target EGFR in glioblastomas; Lessons from lung adenocarcinomas. Cancers. (2018) 10:489. doi: 10.3390/cancers10120489
75. Huang PH, Xu AM, White FM. Networks in glioma. Sci Signal. (2009) 2:1–13. doi: 10.1126/scisignal.287re6
76. Timmons S, Coakley MF, Moloney AM, O'Neill C. Akt signal transduction dysfunction in Parkinson's disease. Neurosci Lett. (2009) 467:30–5. doi: 10.1016/j.neulet.2009.09.055
77. Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma – animal models and therapeutic challenges. Bone. (2008) 23:1–7. doi: 10.1111/j.1750-3639.2008.00233.x
78. Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. (2016) 7:33440–50. doi: 10.18632/oncotarget.7961
79. Langhans J, Schneele L, Trenkler N, Von Bandemer H, Nonnenmacher L, Karpel-Massler G, et al. The effects of PI3K-mediated signalling on glioblastoma cell behaviour. Oncogenesis. (2017) 6:1–8. doi: 10.1038/s41389-017-0004-8
80. Sekar S, Taghibiglou C. Elevated nuclear phosphatase and tensin homolog (PTEN) and altered insulin signaling in substantia nigral region of patients with Parkinson's disease. Neurosci Lett. (2018) 666:139–43. doi: 10.1016/j.neulet.2017.12.049
81. Diaz-Ruiz O, Zapata A, Shan L, Zhang Y, Tomac AC, Malik N, et al. Selective deletion of PTEN in dopamine neurons leads to trophic effects and adaptation of striatal medium spiny projecting neurons. Brain Pathol. (2009) 4:e7027. doi: 10.1371/journal.pone.0007027
82. Domanskyi A, Geißler C, Vinnikov IA, Alter H, Schober A, Vogt MA, et al. Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson's disease models. FASEB J. (2011) 25:2898–910. doi: 10.1096/fj.11-181958
83. Malagelada C, Zong HJ, Greene LA. RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci. (2008) 28:14363–71. doi: 10.1523/JNEUROSCI.3928-08.2008
84. Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. (2005) 7:263–73. doi: 10.1016/j.ccr.2005.02.010
85. Greene LA, Levy O, Malagelada C. Akt as a victim, villain and potential hero in Parkinson's disease pathophysiology and treatment. Cell Mol Neurobiol. (2011) 31:969–78. doi: 10.1007/s10571-011-9671-8
86. McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis GM, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. (2008) 455:1061–8. doi: 10.1038/nature07385
87. Majewska E, Szeliga M. AKT/GSK3β signaling in glioblastoma. Neurochem Res. (2017) 42:918–24. doi: 10.1007/s11064-016-2044-4
88. Bonifati V, Rizzu P, Van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science (80-.). (2003) 299:256–9. doi: 10.1126/science.1077209
89. Wang C, Fang M, Zhang M, Li W, Guan H, Sun Y, et al. The positive correlation between DJ-1 and β-catenin expression shows prognostic value for patients with glioma. Neuropathology. (2013) 33:628–36. doi: 10.1111/neup.12041
90. Ariga H. Common mechanisms of onset of cancer and neurodegenerative diseases. Biol Pharm Bull. (2015) 38:795–808. doi: 10.1248/bpb.b15-00125
91. Kim YC, Kitaura H, Iguchi-Ariga SMM, Ariga H. DJ-1, an oncogene and causative gene for familial Parkinson's disease, is essential for SV40 transformation in mouse fibroblasts through up-regulation of c-Myc. FEBS Lett. (2010) 584:3891–5. doi: 10.1016/j.febslet.2010.08.010
92. Fan J, Ren H, Jia N, Fei E, Zhou T, Jiang P, et al. DJ-1 decreases Bax expression through repressing p53 transcriptional activity. J Biol Chem. (2008) 283:4022–30. doi: 10.1074/jbc.M707176200
93. Todorovic M, Wood SA, Mellick GD. Nrf2: a modulator of Parkinson's disease? J Neural Transm. (2016) 123:611–9. doi: 10.1007/s00702-016-1563-0
94. Vomund S, Schäfer A, Parnham MJ, Brüne B, Von Knethen A. Nrf2, the master regulator of anti-oxidative responses. Int J Mol Sci. (2017) 18:1–19. doi: 10.3390/ijms18122772
95. Liu C, Chen Y, Kochevar IE, Jurkunas UV. Decreased DJ-1 leads to impaired Nrf2-regulated antioxidant defense and increased UV-A–induced apoptosis in corneal endothelial cells. Investig Ophthalmol Vis Sci. (2014) 55:5551–60. doi: 10.1167/iovs.14-14580
96. Fan Z, Wirth A-K, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. (2017) 6:e371. doi: 10.1038/oncsis.2017.65
97. Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. (2018) 69:437–49. doi: 10.1146/annurev-med-050715-104343
98. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. (2005) 7:134–53. doi: 10.1215/S1152851704001115
99. Agani FH, Pichiule P, Chavez JC, LaManna JC. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J Biol Chem. (2000) 275:35863–7. doi: 10.1074/jbc.M005643200
100. Zhang Z, Yan J, Chang Y, Yan SS, Shi H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr Med Chem. (2011) 18:4335–43. doi: 10.2174/092986711797200426
101. Milosevic J, Maisel M, Wegner F, Leuchtenberger J, Wenger RH, Gerlach M, et al. Lack of hypoxia-inducible factor-1α impairs midbrain neural precursor cells involving vascular endothelial growth factor signaling. J Neurosci. (2007) 27:412–21. doi: 10.1523/JNEUROSCI.2482-06.2007
102. Zecca L, Youdim MBH, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. (2004) 5:863–73. doi: 10.1038/nrn1537
103. Sofic E, Riederer P, Heinsen H, Beckmann H, Reynolds GP, Hebenstreit G, et al. Increased iron (II1) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm. (1988) 4:132–43.
104. Signore AP, Weng Z, Hastings T, Van Laar AD, Liang Q, Lee YJ, et al. Erythropoietin protects against 6-hydroxydopamine-induced dopaminergic cell death. J Neurochem. (2006) 96:428–43. doi: 10.1111/j.1471-4159.2005.03587.x
105. Silverman WF, Krum JM, Mani N, Rosenstein JM. Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience. (1999) 90:1529–41. doi: 10.1016/S0306-4522(98)00540-5
106. Correia SC, Moreira PI. Hypoxia-inducible factor 1: a new hope to counteract neurodegeneration? J Neurochem. (2010) 112:1–12. doi: 10.1111/j.1471-4159.2009.06443.x
107. Lee DW, Rajagopalan S, Siddiq A, Gwiazda R, Yang L, Beal MF, et al. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. Model for the potential involvement of the hypoxia-unducible factor pathway in Parkinson disease. J Biol Chem. (2009) 284:29065–76. doi: 10.1074/jbc.M109.000638
108. Tanaka H, Sasayama T, Tanaka K, Nakamizo S, Nishihara M, Mizukawa K, et al. MicroRNA-183 upregulates HIF-1α by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J Neurooncol. (2013) 111:273–83. doi: 10.1007/s11060-012-1027-9
109. Liu Q, Cao P. Clinical and prognostic significance of HIF-1α in glioma patients: a meta-analysis. Int J Clin Exp Med. (2015) 8:22073–83.
110. Razavi S-M, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G. Immune evasion strategies of glioblastoma. Front Surg. (2016) 3:1–9. doi: 10.3389/fsurg.2016.00011
111. Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, et al. Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE. (2011) 6:e16195. doi: 10.1371/journal.pone.0016195
112. Wang H, Jiang Z, Na M, Ge H, Tang C, Shen H, et al. PARK2 negatively regulates the metastasis and epithelial-mesenchymal transition of glioblastoma cells via ZEB1. Oncol Lett. (2017) 14:2933–9. doi: 10.3892/ol.2017.6488
113. Harris A, Thompson AAR, Whyte MKB, Walmsley S. HIF-mediated innate immune responses: cell signaling and therapeutic implications. Hypoxia. (2014) 47:47–58. doi: 10.2147/HP.S50269
114. Hofmann KW, Schuh AFS, Saute J, Townsend R, Fricke D, Leke R, et al. Interleukin-6 serum levels in patients with parkinson's disease. Neurochem Res. (2009) 34:1401–4. doi: 10.1007/s11064-009-9921-z
115. Chen X, Hu Y, Cao Z, Liu Q, Cheng Y. Cerebrospinal fluid inflammatory cytokine aberrations in Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis: a systematic review and meta-analysis. Front Immunol. (2018) 9:1–10. doi: 10.3389/fimmu.2018.02122
116. Sawada M, Imamura K, Nagatsu T. Role of cytokines in inflammatory process in Parkinson's disease. J Neural Transm Suppl. (2006) 373–81. doi: 10.1007/978-3-211-45295-0_57
117. Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson's disease. Exp Mol Med. (2006) 38:333–47. doi: 10.1038/emm.2006.40
118. Yeung YT, McDonald KL, Grewal T, Munoz L. Interleukins in glioblastoma pathophysiology: implications for therapy. Br J Pharmacol. (2013) 168:591–606. doi: 10.1111/bph.12008
119. Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the immune system in glioblastoma. Br J Cancer. (2018) 119:1171–81. doi: 10.1038/s41416-018-0258-8
120. Paugh BS, Bryan L, Paugh SW, Wilczynska KM, Alvarez SM, Singh SK, et al. Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J Biol Chem. (2009) 284:3408–17. doi: 10.1074/jbc.M807170200
121. Lu T, Tian L, Han Y, Vogelbaum M, Stark GR. Dose-dependent cross-talk between the transforming growth factor-β and interleukin-1 signaling pathways. Proc Natl Acad Sci USA. (2007) 104:4365–70. doi: 10.1073/pnas.0700118104
122. Sharma V, Dixit D, Koul N, Mehta VS, Sen E. Ras regulates interleukin-1β-induced HIF-1α transcriptional activity in glioblastoma. J Mol Med. (2011) 89:123–36. doi: 10.1007/s00109-010-0683-5
123. Tchirkov A, Rolhion C, Bertrand S, Doré JF, Dubost JJ, Verrelle P. IL-6 gene amplification and expression in human glioblastomas. Br J Cancer. (2001) 85:518–22. doi: 10.1054/bjoc.2001.1942
124. Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. (2002) 21:8404–13. doi: 10.1038/sj.onc.1206047
125. Tanabe K, Matsushima-Nishiwaki R, Yamaguchi S, Iida H, Dohi S, Kozawa O. Mechanisms of tumor necrosis factor-α-induced interleukin-6 synthesis in glioma cells. J Neuroinflammation. (2010) 7:3–10. doi: 10.1186/1742-2094-7-16
126. Bonavia R, Inda M, Vandenberg S, Cheng S, Nagane M, Hadwiger P, et al. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Bone. (2012) 23:1–7. doi: 10.1038/onc.2011.563
127. Hong TM, Teng LJ, Shun CT, Peng MC, Tsai JC. Induced interleukin-8 expression in gliomas by tumor-associated macrophages. J Neurooncol. (2009) 93:289–301. doi: 10.1007/s11060-008-9786-z
128. Bauer S, Müller T, Hamm S. Pattern recognition by toll-like receptors. Adv Exp Med Biol. (2009) 653:15–34. doi: 10.1007/978-1-4419-0901-5_2
129. Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol. (2015) 2015:14.12.1–10. doi: 10.1002/0471142735.im1412s109
130. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:1–8. doi: 10.3389/fimmu.2014.00461
131. Arancibia SA, Beltrán CJ, Aguirre IM, Silva P, Peralta AL, Malinarich F, et al. Toll-like receptors are key participants in innate immune responses. Biol Res. (2007) 40:97–112. doi: 10.4067/S0716-97602007000200001
132. Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. (2013) 4:1–24. doi: 10.1038/ncomms2534
133. Noelker C, Morel L, Lescot T, Osterloh A, Alvarez-Fischer D, Breloer M, et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci Rep. (2013) 3:1–5. doi: 10.1038/srep01393
134. Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E, et al. Regionally-specific microglial activation in young mice overexpressing human wildtype alpha-synuclein. Exp Neurol. (2013) 237:318–34. doi: 10.1016/j.expneurol.2012.06.025
135. Drouin-Ouellet J, St-Amour I, Saint-Pierre M, Lamontagne-Proulx J, Kriz J, Barker RA, et al. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson's disease. Int J Neuropsychopharmacol. (2015) 18:1–11. doi: 10.1093/ijnp/pyu103
136. Kouli A, Horne CB, Williams-Gray CH. Toll-like receptors and their therapeutic potential in Parkinson's disease and α-synucleinopathies. Brain Behav Immun. (2019) 81:41–51. doi: 10.1016/j.bbi.2019.06.042
137. Rietdijk CD, Van Wezel RJA, Garssen J, Kraneveld AD. Neuronal toll-like receptors and neuro-immunity in Parkinson's disease, Alzheimer's disease and stroke. Neuroimmunol Neuroinflammation. (2016) 3:27. doi: 10.20517/2347-8659.2015.28
138. Doorn KJ, Moors T, Drukarch B, van de Berg WDJ, Lucassen PJ, van Dam AM. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun. (2014) 2:1–17. doi: 10.1186/s40478-014-0090-1
139. Finocchiaro G. TLRgeting evasion of immune pathways in glioblastoma. Cell Stem Cell. (2017) 20:422–4. doi: 10.1016/j.stem.2017.03.018
140. Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, et al. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell. (2017) 20:450–61.e4. doi: 10.1016/j.stem.2016.12.001
141. Abarca-Merlin DM, Maldonado-Bernal C, Alvarez-Arellano L, Muthuraju S. Toll-like receptors as therapeutic targets in central nervous system tumors. Biomed Res Int. (2019) 2019:1–9. doi: 10.1155/2019/5286358
142. Barrera G, Gentile F, Pizzimenti S, Canuto RA, Daga M, Arcaro A, et al. Mitochondrial dysfunction in cancer and neurodegenerative diseases: spotlight on fatty acid oxidation and lipoperoxidation products. Antioxidants. (2016) 5:1–25. doi: 10.3390/antiox5010007
143. Siegel GJ, Albers RW, Agranoff BW. (1999). Basic neurochemistry. 6th ed. In: Siegel GJ, Albers RW, Agranoff BW, editors. Molecular, Cellular and Medical Aspects. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK20385/
144. Schapira AHV, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's Disease. J Neurochem. (1990) 54:823–7. doi: 10.1111/j.1471-4159.1990.tb02325.x
145. Requejo-Aguilar R, Bolaños JP. Mitochondrial control of cell bioenergetics in Parkinson's disease. Free Radic Biol Med. (2016) 100:123–37. doi: 10.1016/j.freeradbiomed.2016.04.012
146. Hsu CC, Huang N, Lin PY, Fang SY, Tsai DC, Chen SY, et al. Risk factors for myopia progression in second-grade primary school children in Taipei: a population-based cohort study. Br J Ophthalmol. (2017) 101:1611–7. doi: 10.1136/bjophthalmol-2016-309299
147. Qazi MA, Vora P, Venugopal C, Sidhu SS, Moffat J, Swanton C, et al. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol. (2017) 28:1448–56. doi: 10.1093/annonc/mdx169
148. Zhou W, Wahl DR. Metabolic abnormalities in glioblastoma and metabolic strategies to overcome treatment resistance. Cancers. (2019) 11:1231. doi: 10.3390/cancers11091231
149. Anandhan A, Jacome MS, Lei S, Hernandez-Franco P, Pappa A, Panayiotidis MI, et al. Metabolic dysfunction in Parkinson's Disease: bioenergetics, redox homeostasis and central carbon metabolism. Brain Res Bull. (2017) 133:12–30. doi: 10.1016/j.brainresbull.2017.03.009
150. Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, et al. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. (2016) 5:1–25. doi: 10.7554/eLife.13374
151. Hall CN, Klein-Flügge MC, Howarth C, Attwell D. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci. (2012) 32:8940–51. doi: 10.1523/JNEUROSCI.0026-12.2012
152. Powers R, Lei S, Anandhan A, Marshall DD, Worley B, Cerny RL, et al. Metabolic investigations of the molecular mechanisms associated with Parkinson's disease. Metabolites. (2017) 7:22. doi: 10.3390/metabo7020022
153. Lannuzel A, Michel PP, Höglinger GU, Champy P, Jousset A, Medja F, et al. The mitochondrial complex I inhibitor annonacin is toxic to mesencephalic dopaminergic neurons by impairment of energy metabolism. Neuroscience. (2003) 121:287–96. doi: 10.1016/S0306-4522(03)00441-X
154. Chaudhuri AD, Kabaria S, Choi DC, Mouradian MM, Junn E. MicroRNA-7 promotes glycolysis to protect against 1-methyl-4-phenylpyridinium-induced cell death. J Biol Chem. (2015) 290:12425–34. doi: 10.1074/jbc.M114.625962
155. Hong CT, Chau KY, Schapira AHV. Meclizine-induced enhanced glycolysis is neuroprotective in Parkinson disease cell models. Sci Rep. (2016) 6:6–13. doi: 10.1038/srep25344
156. Dunn L, Allen GFG, Mamais A, Ling H, Li A, Duberley KE, et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol Aging. (2014) 35:1111–5. doi: 10.1016/j.neurobiolaging.2013.11.001
157. Filosa S, Fico A, Paglialunga F, Balestrieri M, Crooke A, Verde P, et al. Failure to increase glucose consumption through the pentose-phosphate pathway results in the death of glucose-6-phosphate dehydrogenase gene-deleted mouse embryonic stem cells subjected to oxidative stress. Biochem J. (2003) 370:935–43. doi: 10.1042/bj20021614
158. Herken H. Neurotoxin-induced impairment of biopterin synthesis and function: initial stage of a Parkinson-like dopamine deficiency syndrome. Neurochem Int. (1990) 17:223–38. doi: 10.1016/0197-0186(90)90145-J
159. Weinhouse S, Warburg O, Burk D, Schade AL. On respiratory impairment in cancer cells. Science. (1956) 124:270–2. doi: 10.1126/science.124.3215.267
160. Agnihotri S, Zadeh G. Metabolic reprogramming in glioblastoma: the influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol. (2016) 18:160–72. doi: 10.1093/neuonc/nov125
161. Kahlon AS, Alexander M, Kahlon A, Wright J. Lactate levels with glioblastoma multiforme. Baylor Univ Med Cent Proc. (2016) 29:313–4. doi: 10.1080/08998280.2016.11929449
162. Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Xia Y, et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol Cell. (2016) 61:705–19. doi: 10.1016/j.molcel.2016.02.009
163. Li X, Zheng Y, Lu Z. PGK1 is a new member of the protein kinome. Cell Cycle. (2016) 15:1803–4. doi: 10.1080/15384101.2016.1179037
164. Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. (2009) 11:747–52. doi: 10.1038/ncb1881
165. Lalonde FM, Myslobodsky M. Are dopamine antagonists a risk factor for breast cancer? An answer from Parkinson's disease. Breast. (2003) 12:280–2. doi: 10.1016/S0960-9776(03)00061-4
166. Diamandis P, Sacher AG, Tyers M, Dirks PB. New drugs for brain tumors? Insights from chemical probing of neural stem cells. Med Hypotheses. (2009) 72:683–7. doi: 10.1016/j.mehy.2008.10.034
167. Wirdefeldt K, Weibull CE, Chen H, Kamel F, Lundholm C, Fang F, et al. Parkinson's disease and cancer: a register-based family study. Am J Epidemiol. (2014) 179:85–94. doi: 10.1093/aje/kwt232
168. Tang CF, Lu MK, Muo CH, Tsai CH, Kao CH. Increased risk of brain tumor in patients with Parkinson's disease: a nationwide cohort study in Taiwan. Acta Neurol Scand. (2016) 134:148–53. doi: 10.1111/ane.12524
169. Ye R, Shen T, Jiang Y, Xu L, Si X, Zhang B. The relationship between Parkinson disease and brain tumor: a meta-analysis. PLoS ONE. (2016) 11:e0164388. doi: 10.1371/journal.pone.0164388
170. Adhiyaman V, Meara J. Meningioma presenting as bilateral parkinsonism. Age Ageing. (2003) 32:456–8. doi: 10.1093/ageing/32.4.456
171. Freeman J, Westerhuis B, Asfora W, Free T, Salem B. Meningioma mimicking Parkinson's disease: a case report and analysis. South Dakota Med. (2013) 66:101–3.
172. Kim J-I, Choi JK, Lee J-W, Hong JY. Intracranial Meningioma-induced Parkinsonism. J Lifestyle Med. (2014) 4:101–3. doi: 10.15280/jlm.2014.4.2.101
173. Fong M, Ghahreman A, Masters L, Huynh W. Large intracranial meningioma masquerading as Parkinson's disease. J Neurol Neurosurg Psychiatry. (2016) 87:1251. doi: 10.1136/jnnp-2015-311531
174. Olsen JH, Friis S, Frederiksen K, McLaughlin JK, Mellemkjaer L, Møller H. Atypical cancer pattern in patients with Parkinson's disease. Br J Cancer. (2005) 92:201–5. doi: 10.1038/sj.bjc.6602279
175. Choi KH, Choi SM, Nam TS, Lee MC. Astrocytoma in the third ventricle and hypothalamus presenting with parkinsonism. J Korean Neurosurg Soc. (2012) 51:144–6. doi: 10.3340/jkns.2012.51.3.144
176. Møller H, Mellemkjaer L, McLaughlin JK, Olsen JH. Occurrence of different cancers in patients with Parkinson's disease. BMJ. (1995) 310:1500. doi: 10.1136/bmj.310.6993.1500
177. Skibba JL, Pinckley J, Gilbert EF, Johnson RO. Multiple primary melanoma following administration of levodopa. Arch Pathol. (1972) 93:556.
178. Liu R, Gao X, Lu Y, Chen H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology. (2011) 76:2002–9. doi: 10.1212/WNL.0b013e31821e554e
179. Catalá-López F, Suárez-Pinilla M, Suárez-Pinilla P, Valderas JM, Gómez-Beneyto M, Martinez S, et al. Inverse and direct cancer comorbidity in people with central nervous system disorders: a meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother Psychosom. (2014) 83:89–105. doi: 10.1159/000356498
180. Rampen FHJ. Levodopa and melanoma: three cases and review of literature. J Neurol Neurosurg Psychiatry. (1985) 48:585–8. doi: 10.1136/jnnp.48.6.585
181. Jansson B, Jankovic J. Low cancer rates among patients with Parkinson's disease. Ann Neurol. (1985) 17:505–9. doi: 10.1002/ana.410170514
182. Tacik P, Currya S, Fujioka S, Strongosky A, Uitti RJ, van Gerpen JA, et al. Cancer in Parkinson's disease. Parkinsonism Relat Disord. (2016) 31:28–33. doi: 10.1016/j.parkreldis.2016.06.014
183. Skinner RB Jr, LeDoux MS. Dermatological disorders in Parkinson's disease. Curr Clin Neurol. (2012) 24:237–42. doi: 10.1007/978-1-60761-429-6_16
184. Bose A, Petsko GA, Eliezer D. Parkinson's disease and melanoma: co-occurrence and mechanisms. J Parkinsons Dis. (2018) 8:385–98. doi: 10.3233/JPD-171263
Keywords: Parkinson's disease, glioblastoma multiforme, pleiotropy, cancer, neurodegeneration
Citation: Mencke P, Hanss Z, Boussaad I, Sugier P-E, Elbaz A and Krüger R (2020) Bidirectional Relation Between Parkinson's Disease and Glioblastoma Multiforme. Front. Neurol. 11:898. doi: 10.3389/fneur.2020.00898
Received: 20 February 2020; Accepted: 13 July 2020;
Published: 20 August 2020.
Edited by:
George Damion Mellick, Griffith University, AustraliaReviewed by:
Linlin Ma, Griffith University, AustraliaCopyright © 2020 Mencke, Hanss, Boussaad, Sugier, Elbaz and Krüger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pauline Mencke, cGF1bGluZS5tZW5ja2VAdW5pLmx1; Rejko Krüger, cmVqa28ua3J1ZWdlckB1bmkubHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.