
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Neurol. , 28 August 2020
Sec. Multiple Sclerosis and Neuroimmunology
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00784
 Christopher M. Dwyer1,2*
Christopher M. Dwyer1,2* Linda Thien-Trang Nguyen1
Linda Thien-Trang Nguyen1 Luke M. Healy3
Luke M. Healy3 Ranjan Dutta4Samuel Ludwin3Jack Antel3
Ranjan Dutta4Samuel Ludwin3Jack Antel3 Michele D. Binder5,6Trevor J. Kilpatrick1,2
Michele D. Binder5,6Trevor J. Kilpatrick1,2We propose that multiple sclerosis (MS) is best characterized as a syndrome rather than a single disease because different pathogenetic mechanisms can result in the constellation of symptoms and signs by which MS is clinically characterized. We describe several cellular mechanisms that could generate inflammatory demyelination through disruption of homeostatic interactions between immune and neural cells. We illustrate that genomics is important in identifying phenocopies, in particular for primary progressive MS. We posit that molecular profiling, rather than traditional clinical phenotyping, will facilitate meaningful patient stratification, as illustrated by interactions between HLA and a regulator of homeostatic phagocytosis, MERTK. We envisage a personalized approach to MS management where genetic, molecular, and cellular information guides management.
Multiple sclerosis is a complex autoimmune disease of the central nervous system (CNS), characterized pathologically by inflammatory demyelination (1). The diagnosis of MS is standardized using an array of clinical, imaging, and laboratory measures, most recently described in the 2017 revised McDonald criteria (2). These require evidence of inflammatory activity within the CNS with dissemination in time and space. Temporal dissemination is not required if isolated intrathecal synthesis of oligoclonal proteins is identified. These criteria reflect a consensus based on the synthesis of clinical and paraclinical data, agnostic to the pathogenetic mechanism(s) driving neuroinflammation. We propose that this clinical phenotype of MS will be increasingly viewed as a syndrome—rather than a single disease state—because these diagnostic elements can be generated by varying pathogenetic mechanisms.
There is precedent to support this contention. Neuromyelitis optica (NMO) presents with inflammatory demyelination of the optic nerves and spinal cord and was previously considered an MS variant. We now recognize NMO is distinct, driven by unique autoantibodies (3, 4). Compellingly, NMO is now more appropriately referred to as neuromyelitis optica spectrum disorder (NMOSD), reflecting that the condition can be triggered in multiple ways, for example, by antibodies targeting aquaporin-4 expressed on astrocytes or, alternatively, by antibodies directed against the oligodendrocyte-specific myelin oligodendrocyte glycoprotein (5, 6). This demonstrates that common clinical presentations sometimes unwittingly group phenocopies, reflecting more than one pathogenic mechanism.
The concept that MS is heterogeneous was previously championed by Lucchinetti and Lassmann who described four distinct histological patterns of acute MS (7). They posited that MS was a disease of varying etiology and pathogenesis and suggested that therapy might be tailored to the individual on this basis. Unfortunately, no convincing biomarkers have emerged to enable reliable and valid clinical stratification according to these parameters and the idea was not uniformly accepted (8). However, recent advances in identifying genomic, immuno-biological, and environmental contributors to MS pathogenesis call for reexamination of this issue. For example, Trapp et al. recently described MS histopathology that demonstrated neurodegeneration with cortical and spinal myelin loss but without cerebral white-matter demyelination, so-called myelocortical MS (MCMS). While clinical and MRI findings in MCMS and typical MS are indistinguishable, it is possible that the pathophysiologies driving these presentations are markedly different, requiring different therapeutic approaches (9).
Genetic factors and environmental exposures contribute to MS susceptibility, although, consistent with a syndrome-based hypothesis, no algorithm that meaningfully quantifies individual risk has been identified (10). Caucasians heterozygous for HLA-DRB1*1501 within the major histocompatibility complex (MHC) have an odds ratio for developing MS of ~3.0, representing the strongest susceptibility allele for MS (11). Over 200 other susceptibility loci have been identified via genome-wide association studies (GWAS), but each conveys an odds ratio of 1.2 or less (12). Prior infection by the Epstein–Barr virus (EBV) appears necessary, but not sufficient, to cause MS (13). Other environmental factors that contribute to risk include smoking, hypovitaminosis D, and obesity (14, 15). How these various factors contribute at an individual level to pathogenesis and clinical phenotype remains unknown.
How can we interrogate the pathogenesis of MS more effectively? The neurological signs and symptoms of MS are non-parametric traits and attempts to use them to meaningfully sub-stratify the disease have not been helpful (16). The alternative, as proven successful in other contexts, is to classify using cellular and molecular profiling (17). If successful, such an approach would have major implications for both prognostication and therapeutic targeting in MS.
Next-generation sequencing of MS pedigrees, although hampered by incomplete penetrance and limited due to pedigree scarcity, could also offer new insights (18). Enticingly, mutations in genes implicated in cholesterol metabolism and oxysterol synthesis have been recently reported in MS pedigrees (19). It will be important to determine whether these mutations are also more prevalent in sporadic MS, similar to the genetic links identified between pedigree-based and sporadic Parkinson's disease (20).
A subset of progressive MS patients demonstrate mutations pathogenic for other neurological conditions (21). Certain of the identified genes could be implicated in MS pathophysiology through, for example, influences upon myelin composition or microglial development (22–24). It will be important to understand what factors, in addition to these mutations, are necessary to induce the inflammation and neurodegeneration implicated in MS progression.
If MS is a syndrome, are there ways that the condition can be classified holistically beyond clinical definitions? The traditional view that MS reflects T-effector activity induced by one or more autoantigens provides such a framework. This perspective has been very recently advanced by Martin et al. who identified auto-proliferative T-effector clones in MS patients during remission rather than relapse, as might have been presumed a priori (25). T-effector cell reactivity was induced in a HLA-DRB1*1501-positive MS patient by presentation of a peptide derivative of Ras guanine nucleotide-releasing protein 2 (RASGRP2), a calcium sensor expressed within the CNS. Whether this pathogenic mechanism applies to the broader MS population and whether RASGRP2 is an initiator of disease or a reflection of epitope spreading awaits clarification.
The influence that specific molecules exert upon adaptive immunity and disease pathogenesis can be contextual. For example, we discovered that the co-stimulatory molecule CD40 is a risk gene for MS (26). We found the responsible single-nucleotide polymorphism (SNP) is in the Kozak consensus sequence, which drives expression of the CD40 protein and that, paradoxically, this genetic variation leads to reduced CD40 in B lymphocytes and other mononuclear cells (27). Intriguingly, this SNP also conveys susceptibility to Graves' disease and rheumatoid arthritis but susceptibility in these diseases rests with the high-expressing allele. Clearly, there are subtleties in molecular signaling that influence autoimmune disease phenotype in ways we are yet to understand. This perspective, although challenging, is not iconoclastic; single antigens can induce either tolerance or pathogenic T-effector cell-induced autoimmunity depending on the route of autoantigen administration (28), and the same myelin basic protein-derived peptide can induce anti- and pro-inflammatory responses in T cells derived from MS patients (29).
The innate immune system also plays a central role in MS pathogenesis and potentially in MS initiation as recently reported by the International Multiple Sclerosis Genetics Consortium (30). This builds on earlier work, demonstrating that in a subset of pathological specimens oligodendrocyte injury and microglial activation appear to precede T-lymphocyte infiltration (31). The innate immune system is composite; it includes professional antigen-presenting cells (APCs) such as dendritic cells and mononuclear cells such as microglia and macrophages that can present antigen and, in a context-dependent manner, either promote or inhibit inflammation via cytokine production (32). Mononuclear cells can also phagocytose pathogens, cellular debris, and even living cells (33). Intimate associations between macrophages and neural cells occur in MS, suggesting innate immune cells could disrupt axons and myelin (34). Conversely, innate immune activation and phagocytosis of myelin debris appear necessary for oligodendrocyte differentiation and remyelination (35). Recent work has also identified key differences between peripheral macrophages and central microglia with respect to expression profiling and functional activity, with microglia being more likely to assume an anti-inflammatory, reparative phenotype (36). In a context-dependent manner, microglia can also assume either a pro- or anti-inflammatory function (37).
How can we assimilate these various perspectives concerning disease pathogenesis? An overarching hypothesis of chronic inflammatory demyelination posits a breakdown in homeostatic interactions between a target cell (presumably the oligodendrocyte) and the innate and adaptive immune systems, recognizing some trafficking of lymphocytes occurs through the healthy CNS (38). This breakdown could be orchestrated by several mechanisms.
A favored pathology (Figure 1, Scenario 1) embraces the classical perspective of an adaptive immune system inappropriately induced by antigenic stimuli to target neural cells, potentially via molecular mimicry. Exogenous or CNS antigen is first presented in regional lymphoid tissue adjacent to the entry site, typically in deep cervical lymph nodes where either tolerance or auto-reactivity can be induced, depending on the nature of the antigen and its immune processing prior to presentation (39). Where auto-reactivity is induced, full activation of the lymphocyte population requires the expression of appropriately configured self-antigen by perivascular mononuclear cells within the CNS (40).
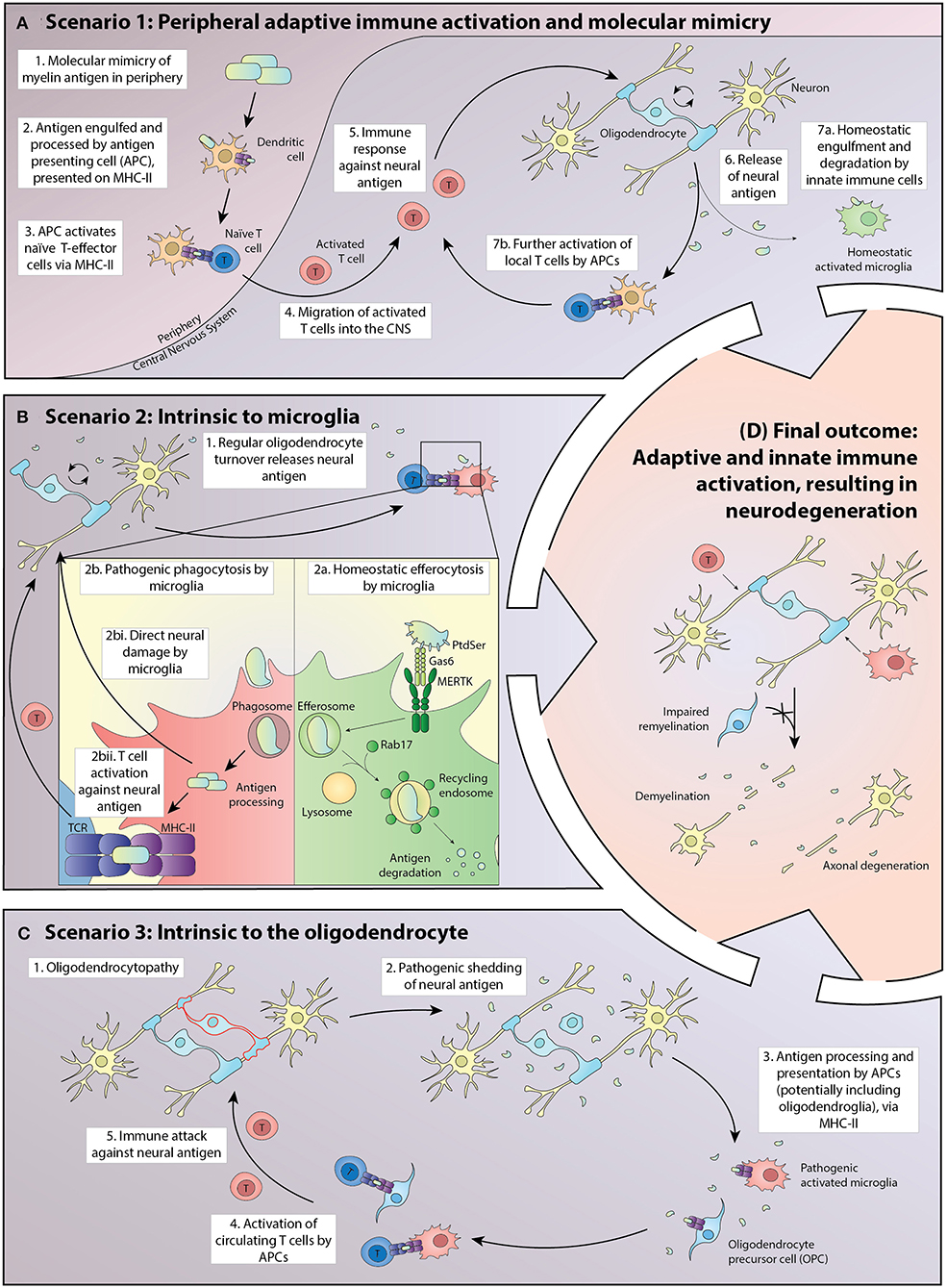
Figure 1. Potential scenarios driving the pathogenesis of MS in which homeostatic interactions between oligodendroglia and the adaptive and innate immune systems break down, leading to inflammatory demyelination, and eventually to neurodegeneration. (A) In Scenario 1, exogenous antigen is processed in peripheral lymphoid tissue and presented to naïve T cells via MHC Class II. A small proportion of the activated T-effector cells traffic through the CNS where some turnover of neural cell macromolecules, including myelin proteins, occurs. Those molecules are either degraded by the innate immune system or are presented at a basal level by antigen-presenting cells (APCs), principally microglia but potentially also oligodendrocytes and their progenitors. Where the exogenous antigen exhibits molecular mimicry to a neural antigen, there is the potential for further activation of inflammatory T cells within the CNS. This leads to a positive feedback loop in which the T-effector cells initiate CNS damage, resulting in increased presentation of neural antigen by APCs and accelerated proliferation and activation of T-effectors locally. (B) In Scenario 2, the processing of neural antigens within APCs becomes corrupted, with the balance shifting from efferocytosis in which molecules are digested, to one in which peptides are presented via MHC class II. Homeostatic mechanisms potentially corrupted include the presentation of phospholipids to the APC by a limited number of cell surface receptors, including MERTK and its ligands proteins and Gas6. MERTK signaling primes the cell to efferocytosis as does the intracellular protein Rab7, which activates lysosomal induced molecular degradation rather than antigen presentation. In this scenario, microglia both have an initial pathogenic role and activate naïve T cells that normally transit through the CNS and to draining cervical lymph nodes in an immune surveillance role, but with potential for reactivity against neural antigens once homeostasis is disrupted. (C) In Scenario 3, pathology intrinsic to oligodendrocytes results in the presentation of neuronal antigens by APCs, including microglia and potentially oligodendroglia. The initial pathology is an oligodendrocytopathy with acceleration of disease driven via activation of microglia and eventually of circulating T cells. (D) Independent of the initiating event, breakdown in homeostatic interactions within the CNS ultimately leads to relatively stereotyped pathology characterized by oligodendrocyte loss, demyelination, and axonal degeneration.
Disturbance in the processing of oligodendrocyte antigens normally cleared by the innate immune system (Figure 1, Scenario 2) could represent another mechanism by which homeostasis is disrupted. Sculpting of the oligodendrocyte membrane is an active, normal developmental process and potentially extends throughout life (41). Insults either directly or indirectly targeting microglia could catalyze a maladaptive response leading to spurious Toll-like receptor (TLR) activation, as previously demonstrated in systemic lupus erythematosus (SLE) (42). Aberrant TLR activation could corrupt the processing and presentation of oligodendrocytic antigens. Cytokine and reactive oxygen species produced by activated microglia also could be an important early pathogenic event, independent of adaptive immune system activation (43). This pathophysiology would accordingly bypass the presentation of phospholipids to the APC by pro-homeostatic mechanisms, including MERTK and its ligands Proteins and Gas6. MERTK signaling primes the cell to efferocytosis as does the intracellular protein Rab7, which activates lysosomal induced molecular degradation rather than antigen presentation.
A third mechanism (Figure 1, Scenario 3) could involve pathology intrinsic to the oligodendrocyte, possibly established developmentally but most commonly revealed in adult life. A long prodromal period of this nature is typical of other neurodegenerative diseases and is sometimes identified in MS following detection of asymptomatic neuroinflammatory lesions on magnetic resonance imaging undertaken for unrelated reasons (44). The oligodendrocyte is a metabolically active cell with ongoing turnover of myelin throughout life (45). Corruption of the nature or kinetics of oligodendrocyte metabolism, leading to pathogenic shedding of antigen, could represent the first insult predisposing to inflammatory demyelination (46). The concept that oligodendroglia are active immunomodulators in MS has also been recently further championed by Castelo-Branco et al., having identified disease-specific oligodendroglia that express the major histocompatibility complex (MHC) in the MS brain (47). Oligodendroglia that participate in antigen presentation were also recently described (48).
Irrespective of the disease initiator, in established MS oligodendrocytes are targeted and the adaptive and innate immune systems are activated. Corruption of oligodendrocyte viability leads to antigen processing within the CNS, with macrophages and microglia expressing MHC class II and costimulatory molecules (49–51). In this context, a subset of the repertoire of lymphocytes that normally traffic through the CNS can become activated, with some of the cells exposed to self-antigen trafficking back to the cervical lymph nodes (52) to accelerate an adaptive immune response systemically, a process which plays out in all our described scenarios (Figure 1) (53–55). Regional stratification of adaptive immune targeting has been reported in animal models, with myelin-reactive T cells observed to infiltrate white matter while T-cells recognizing the neuronal protein synuclein target gray matter, a phenomenon that could contribute to the duality of MS immunopathology, with cortical destruction by synuclein-reactive T cells representing a mediator of neurodegeneration (56).
Homeostasis can be reestablished by innate immune cells when they engulf and digest apoptotic cells and necrotic debris without presenting self-antigens via the MHC (57). In contrast, the innate immune system can present antigen, induce inflammation, and recruit the adaptive immune system, which is an appropriate response to pathogens but, when corrupted, promotes autoimmunity (58). In MS, the disruption of homeostasis enables antigens released from the oligodendrocyte or molecules expressed at paranodes to be presented via innate immune cells to pathogenic T-effector cells.
Whether homeostasis is restored, or autoimmunity promoted, will be dependent upon the molecular environment. Homeostatic phagocytosis by innate immune cells is facilitated by “eat me” signals including phosphatidylserine (PtdSer) and other phospholipids actively “flipped” onto the external surface of the cell membrane of apoptotic cells (59). These lipids are also found in myelin debris. Several secreted and cell surface receptors expressed by phagocytes recognize these lipids and respond to the “eat me” signal. One of these recognition molecules is the receptor tyrosine kinase, MERTK, which is encoded by a susceptibility gene for MS (60, 61). Ligands for MERTK, namely, Protein S (ProS) and growth arrest-specific 6 (Gas6), act as opsonins or molecular bridges between PtdSer presented by apoptotic cells and MERTK expressing phagocytes. The resultant signaling within the phagocytic cell influences how the engulfed proteins are processed. Under the influence of MERTK, these proteins potentially bypass the phagosomes that would otherwise facilitate antigen presentation by MHC and predispose to autoimmunity (Figure 1, Scenario 2) (62). Interestingly, in SLE, increased soluble MERTK (which negates the anti-inflammatory effect of membrane-bound MERTK signaling by APCs) is associated with increased autoantibody production, a marker of disease activity (63).
MERTK is a member of the TAM (TYRO3, AXL, and MERTK) family of receptor tyrosine kinases. The TAMs are part of a dynamic, compensatory system that responds to inflammatory insults. TAM receptor expression on innate immune cells is upregulated in response to phagocytosis, via activation of the retinoic acid pathway (64).
We identified MERTK as both a susceptibility gene for MS and an expression quantitative trait locus (eQTL) within innate immune cells in the peripheral blood (60). The predominant susceptibility alleles of MERTK for a majority of the MS population drive high expression of MERTK in innate immune cells. However, for people homozygous for HLA-DRB1*1501, itself a major susceptibility gene for MS, there is enhanced susceptibility and severity of the disease among those homozygous for low expressing MERTK alleles. This stratification of risk and severity based on HLA-DRB1*1501 status suggests the contextual influence of MERTK is via antigen presentation. This provides further support for the concept of disease heterogeneity and the need for targeted molecular phenotyping.
In the majority of MS cases, the direct contribution of adaptive immunity from the peripheral blood diminishes with time (65). Concurrently, immune therapies that target adaptive immune responses lose efficacy (66). Whether disease pathogenesis in progressive disease reflects a shift to intrinsic neurodegeneration, persistent activity of pathogenic adaptive immune cells behind an intact blood–brain barrier or corruption of innate immune system activity that subsequently leads to neurodegeneration, remains unknown.
MERTK expressed by microglia and by infiltrating macrophages could promote the efficient removal of debris, once MS is initiated. There are precedents for this function. For example, MERTK is important in preventing retinitis pigmentosa by abrogating the accumulation of debris from photoreceptor cells (67). The level of expression of MERTK on human microglia strongly correlates with the capacity of these cells to engulf myelin debris, of importance as the ability to clear myelin from the damaged CNS is essential for myelin repair (68, 69). Our preliminary analysis of pathological material indicates that for patients with secondary progressive MS, there is robust expression of MERTK in periplaque microglia, although the level of expression varies between specimens (Ranjan Dutta, personal communication), indicating that this phagocytic role is likely to be relevant to MS but of variable importance between individuals.
Demyelinating diseases can be acquired or inherited. Among inherited leukodystrophies, cerebral adrenoleukodystrophy (X-ALD) is exceptional for associated T cell-mediated inflammation. CSF examination of patients with X-ALD reveals oligoclonal T-cell expansion, as seen in MS (70). X-ALD is caused by mutations in the X-linked ABCD1 gene that encodes a peroxisomal transporter. The maintenance of myelinated axons requires continual turnover of myelin membranes and oligodendroglial peroxisomes play an intimate role in lipid metabolism in that context. Perturbations in the removal of lipid debris by oligodendrocytes could facilitate an inflammatory cascade in which local microglia/macrophages become autoreactive. Specifically, a failure to metabolize eicosanoids, which are potent mediators of inflammation and are increased in a range of neurodegenerative conditions, could result from intrinsic peroxisome dysfunction, leading to the phenotype seen in X-ALD but also in a subset of MS patients (31, 46, 71).
We believe it is essential to adopt a precision medicine, biomarker-based approach that looks beyond a singular focus on adaptive immune responses to further benefit patients with MS, in particular those at risk of progressive disease. Understanding what drives MS progression, and recognizing that this may be heterogeneous, is essential for the efficient deployment of novel therapeutic interventions.
Our preliminary findings suggest that at least 10% of people currently diagnosed with primary progressive MS could harbor mutations for other genetic neurodegenerative diseases (21). This is particularly germane now that anti-CD20 immunotherapy is reported to benefit a subset of patients with primary progressive disease (72). Clearly, the expense and risks of this therapy should be restricted to people who demonstrably have MS rather than phenocopies. Moreover, once patients begin treatment with therapies specific to progressive MS their subsequent disease course must be monitored with valid, reliable, and responsive biomarkers to establish—at the individual level—that benefit is being achieved in order to justify incumbent risks. Serial assessment of phosphorylated neurofilament light chain levels in either blood or CSF is one biomarker with potential in this regard (73).
In addition, the MS clinic of the future will need to embrace genomics to detect phenocopies and substratify the disease in meaningful ways. For example, our data already indicate that a key modulator of innate immune function, MERTK, influences not only susceptibility but also potentially disease severity but in an HLA-dependent manner (60, 61). If next-generation therapies targeting MERTK and other innate immune modulators become available, it will be important to determine, a priori, which patients will benefit; for example, the potentiation of MERTK signaling for therapeutic benefit could be predicated on HLA-DR15 homozygosity to enhance a tolerogenic phenotype (60). The variable expression of MERTK by microglia in autopsy specimens indicates the need to understand whether variance in microglial activity, as can be determined by PET-based imaging (74), correlates with clinical phenotype. Such information could inform future translational research, by identifying patients who would be most likely to benefit from therapies targeting microglial activity.
Finally, although the promise of regenerative therapy has garnered significant attention, we believe that most impactful therapeutic advances in MS will involve the prevention of CNS damage (66). Only by understanding the diversity of pathogenetic mechanisms driving neuroinflammation and degeneration in MS-like presentations will we achieve this goal.
CD and TK wrote the manuscript. LN produced the figure. LN, LH, RD, SL, JA, and MB provided conceptual input and edited the manuscript. All authors read and approved the final manuscript.
This work was supported by MS Research Australia Post-Graduate Scholarship (to CD), Australian Government Research Training Program Scholarship (to LN), and National Health and Medical Research Council Investigator Grant (to TK). The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Compston. McAlpine's Multiple Sclerosis. Philadelphia, PA: Churchill Livingstone Elsevier (2005).
2. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
3. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International Panel for: international consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
4. Borisow N, Mori M, Kuwabara S, Scheel M, Paul F. Diagnosis and treatment of NMO spectrum disorder and MOG-Encephalomyelitis. Front Neurol. (2018) 9:888. doi: 10.3389/fneur.2018.00888
5. Paul F, Jarius S, Aktas O, Bluthner M, Bauer O, Appelhans H, et al. Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS Med. (2007) 4:e133. doi: 10.1371/journal.pmed.0040133
6. Probstel AK, Rudolf G, Dornmair K, Collongues N, Chanson JB, Sanderson NS, et al. Anti-MOG antibodies are present in a subgroup of patients with a neuromyelitis optica phenotype. J Neuroinflamm. (2015) 12:46. doi: 10.1186/s12974-015-0256-1
7. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
9. Trapp BD, Vignos M, Dudman J, Chang A, Fisher E, Staugaitis SM, et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: a retrospective study. Lancet Neurol. (2018) 17:870–84. doi: 10.1016/S1474-4422(18)30245-X
10. De Jager PL, Chibnik LB, Cui J, Reischl J, Lehr S, Simon KC, et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. Lancet Neurol. (2009) 8:1111–9. doi: 10.1016/S1474-4422(09)70275-3
11. Barcellos LF, Oksenberg JR, Begovich AB, Martin ER, Schmidt S, Vittinghoff E, et al. Multiple sclerosis genetics: HLA-DR2 dose effect on susceptibility to multiple sclerosis and influence on disease course. Am J Hum Genet. (2003) 72:710–6. doi: 10.1086/367781
12. International Multiple Sclerosis Genetics Consortium (IMSGC), Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. (2013) 45:1353–60. doi: 10.1038/ng.2770
13. Lucas RM, Hughes AM, Lay ML, Ponsonby AL, Dwyer DE, Taylor BV, et al. Epstein-Barr virus and multiple sclerosis. J Neurol Neurosurg Psychiatry. (2011) 82:1142–8. doi: 10.1136/jnnp-2011-300174
14. Koch-Henriksen N, Sorensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol. (2010) 9:520–32. doi: 10.1016/S1474-4422(10)70064-8
15. Hedstrom AK, Olsson T, Alfredsson L. The role of environment and lifestyle in determining the risk of multiple sclerosis. Curr Top Behav Neurosci. (2015) 26:87–104. doi: 10.1007/7854_2015_372
16. Uitdehaag M. Clinical outcome measures in multiple sclerosis. Handb Clin Neurol. (2014) 122:393–404. doi: 10.1016/B978-0-444-52001-2.00016-9
17. Wiestner A, Staudt LM. Towards molecular diagnosis and targeted therapy of lymphoid malignancies. Semin Hematol. (2003) 40:296–307. doi: 10.1016/S0037-1963(03)00194-X
18. Aivo J, Hanninen A, Ilonen J, Soilu-Hanninen M. Vitamin D3 administration to MS patients leads to increased serum levels of latency activated peptide (LAP) of TGF-beta. J Neuroimmunol. (2015) 280:12–5. doi: 10.1016/j.jneuroim.2015.01.005
19. Traboulsee AL, Sadovnick AD, Encarnacion M, Bernales CQ, Yee IM, Criscuoli MG, et al. Common genetic etiology between “multiple sclerosis-like” single-gene disorders and familial multiple sclerosis. Hum Genet. (2017) 136:705–14. doi: 10.1007/s00439-017-1784-9
20. Klein C, Westenberger A. Genetics of parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:a008888. doi: 10.1101/cshperspect.a008888
21. Jia X, Madireddy L, Caillier S, Santaniello A, Esposito F, Comi G, et al. Genome sequencing uncovers phenocopies in primary progressive multiple sclerosis. Ann Neurol. (2018) 84:51–63. doi: 10.1002/ana.25263
22. Cloake NC, Yan J, Aminian A, Pender MP, Greer JM. PLP1 mutations in patients with multiple sclerosis: identification of a new mutation and potential pathogenicity of the mutations. J Clin Med. (2018) 7:342. doi: 10.3390/jcm7100342
23. Saitoh BY, Yamasaki R, Hayashi S, Yoshimura S, Tateishi T, Ohyagi Y, et al. A case of hereditary diffuse leukoencephalopathy with axonal spheroids caused by a de novo mutation in CSF1R masquerading as primary progressive multiple sclerosis. Mult Scler. (2013) 19:1367–70. doi: 10.1177/1352458513489854
24. Nicholson AM, Baker MC, Finch NA, Rutherford NJ, Wider C, Graff-Radford NR, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology. (2013) 80:1033–40. doi: 10.1212/WNL.0b013e31828726a7
25. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell. (2018) 175:85–100.e23. doi: 10.1016/j.cell.2018.08.011
26. Australia C. New Zealand multiple sclerosis genetics: genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet. (2009) 41:824–8. doi: 10.1038/ng.396
27. Field J, Shahijanian F, Schibeci S, Australia and New Zealand MS Genetics Consortium (ANZgene) Johnson L Laverick L Parnell G . The MS risk allele of CD40 is associated with reduced cell-membrane bound expression in antigen presenting cells: implications for gene function. PLoS ONE. (2015) 10:e0127080. doi: 10.1371/journal.pone.0127080
28. Weiner HL, Friedman A, Miller A, Khoury SJ, al-Sabbagh A, Santos L, et al. Oral tolerance: immunologic mechanisms and treatment of animal and human organ-specific autoimmune diseases by oral administration of autoantigens. Annu Rev Immunol. (1994) 12:809–37. doi: 10.1146/annurev.iy.12.040194.004113
29. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. the altered peptide ligand in relapsing MS study group. Nat Med. (2000) 6:1176–82. doi: 10.1038/80525
30. International Multiple Sclerosis Genetics: Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. (2019) 365:6460. doi: 10.1126/science.aav7188
31. Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. (2004) 55:458–68. doi: 10.1002/ana.20016
32. Parnell GP, Booth DR. The multiple sclerosis (MS) genetic risk factors indicate both acquired and innate immune cell subsets contribute to ms pathogenesis and identify novel therapeutic opportunities. Front Immunol. (2017) 8:425. doi: 10.3389/fimmu.2017.00425
33. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. (2014) 15:209–16. doi: 10.1038/nrn3710
34. Chiang CS, Powell HC, Gold LH, Samimi A, Campbell IL. Macrophage/microglial-mediated primary demyelination and motor disease induced by the central nervous system production of interleukin-3 in transgenic mice. J Clin Invest. (1996) 97:1512–24. doi: 10.1172/JCI118574
35. Hagemeyer N, Hanft KM, Akriditou MA, Unger N, Park ES, Stanley ER, et al. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. (2017). 134:441–58. doi: 10.1007/s00401-017-1747-1
36. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. (2014) 17:131–43. doi: 10.1038/nn.3599
37. Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao CC, Ardura-Fabregat A, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. (2018) 557:724–8. doi: 10.1038/s41586-018-0119-x
38. Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. (2010) 120:1368–79. doi: 10.1172/JCI41911
39. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of cns immune privilege. Trends Immunol. (2015) 36:569–77. doi: 10.1016/j.it.2015.08.006
40. Martin R, Sospedra M, Rosito M, Engelhardt B. Current multiple sclerosis treatments have improved our understanding of MS autoimmune pathogenesis. Eur J Immunol. (2016) 46:2078–90. doi: 10.1002/eji.201646485
41. Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci. (2016) 19:995–8. doi: 10.1038/nn.4325
42. Schell SL, Soni C, Fasnacht MJ, Domeier PP, Cooper TK, Rahman ZSM. Mer receptor tyrosine kinase signaling prevents self-ligand sensing and aberrant selection in germinal centers. J Immunol. (2017) 199:4001–15. doi: 10.4049/jimmunol.1700611
43. Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. (2013) 339:156–61. doi: 10.1126/science.1227901
44. Giovannoni G. The neurodegenerative prodrome in multiple sclerosis. Lancet Neurol. (2017) 16:413–4. doi: 10.1016/S1474-4422(17)30127-8
45. Jungalwala FB, Dawson RM. The turnover of myelin phospholipids in the adult and developing rat brain. Biochem J. (1971) 123:683–93. doi: 10.1042/bj1230683
46. Kassmann CM, Nave KA. Oligodendroglial impact on axonal function and survival - a hypothesis. Curr Opin Neurol. (2008) 21:235–41. doi: 10.1097/WCO.0b013e328300c71f
47. Falcao AM, van Bruggen D, Marques S, Meijer M, Jakel S, Agirre E, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. (2018) 24:1837–44. doi: 10.1038/s41591-018-0236-y
48. Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J, et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun. (2019) 10:3887. doi: 10.1038/s41467-019-11638-3
49. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci USA. (2014) 111:16029–34. doi: 10.1073/pnas.1406508111
50. Italiani P, Mazza EM, Lucchesi D, Cifola I, Gemelli C, Grande A, et al. Transcriptomic profiling of the development of the inflammatory response in human monocytes in vitro. PLoS ONE. (2014) 9:e87680. doi: 10.1371/journal.pone.0087680
51. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. (2006) 177:7303–11. doi: 10.4049/jimmunol.177.10.7303
52. van Zwam M, Huizinga R, Melief MJ, Wierenga-Wolf AF, van Meurs M, Voerman JS, Biber KP, et al. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med. (2009) 87:273–86. doi: 10.1007/s00109-008-0421-4
53. McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. (2005) 11:335–9. doi: 10.1038/nm1202
54. Negi N, Das BK. CNS: not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol. (2018) 37:57–68. doi: 10.1080/08830185.2017.1357719
55. Kawakami N, Lassmann S, Li Z, Odoardi F, Ritter T, Ziemssen T, et al. The activation status of neuroantigen-specific T cells in the target organ determines the clinical outcome of autoimmune encephalomyelitis. J Exp Med. (2004) 199:185–97. doi: 10.1084/jem.20031064
56. Lodygin D, Hermann M, Schweingruber N, Flugel-Koch C, Watanabe T, Schlosser C, et al. beta-Synuclein-reactive T cells induce autoimmune CNS grey matter degeneration. Nature. (2019) 566:503–8. doi: 10.1038/s41586-019-0964-2
57. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. (2001) 411:207–11. doi: 10.1038/35075603
58. Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. (2002) 196:135–40. doi: 10.1084/jem.20012094
59. Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. (2007) 7:964–74. doi: 10.1038/nri2214
60. Binder MD, Fox AD, Merlo D, Johnson LJ, Giuffrida L, Calvert SE, et al. Common and low frequency variants in MERTK are independently associated with multiple sclerosis susceptibility with discordant association dependent upon HLA-DRB1*15:01 status. PLoS Genet. (2016) 12:e1005853. doi: 10.1371/journal.pgen.1005853
61. Ma GZ, Stankovich J, Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene) Kilpatrick TJ Binder MD Field J. Polymorphisms in the receptor tyrosine kinase MERTK gene are associated with multiple sclerosis susceptibility. PLoS ONE. (2011) 6:e16964. doi: 10.1371/journal.pone.0016964
62. Yin C, Kim Y, Argintaru D, Heit B. Rab17 mediates differential antigen sorting following efferocytosis and phagocytosis. Cell Death Dis. (2016) 7:e2529. doi: 10.1038/cddis.2016.431
63. Zizzo G, Guerrieri J, Dittman LM, Merrill JT, Cohen PL. Circulating levels of soluble MER in lupus reflect M2c activation of monocytes/macrophages, autoantibody specificities and disease activity. Arthritis Res Ther. (2013) 15:R212. doi: 10.1186/ar4407
64. A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. (2009) 31:245–58. doi: 10.1016/j.immuni.2009.06.018
65. Bruck W, Stadelmann C. Inflammation and degeneration in multiple sclerosis. Neurol Sci. (2003) 24(Suppl. 5):S265–7. doi: 10.1007/s10072-003-0170-7
66. Stangel M, Kuhlmann T, Matthews PM, Kilpatrick TJ. Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat Rev Neurol. (2017) 13:742–54. doi: 10.1038/nrneurol.2017.139
67. Prasad D, Rothlin CV, Burrola P, Burstyn-Cohen T, Lu Q, Garcia de Frutos P, et al. TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci. (2006) 33:96–108. doi: 10.1016/j.mcn.2006.06.011
68. Healy LM, Perron G, Won SY, Michell-Robinson MA, Rezk A, Ludwin SK, et al. MerTK is a functional regulator of myelin phagocytosis by human myeloid cells. J Immunol. (2016) 196:3375–84. doi: 10.4049/jimmunol.1502562
69. Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJ. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia. (2001) 35:204–12. doi: 10.1002/glia.1085
70. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. (2000) 192:393–404. doi: 10.1084/jem.192.3.393
71. Barnett MH, Henderson AP, Prineas JW. The macrophage in MS: just a scavenger after all? Pathology and pathogenesis of the acute MS lesion. Mult Scler. (2006) 12:121–32. doi: 10.1191/135248506ms1304rr
72. Gelfand JM, Cree BAC, Hauser SL. Ocrelizumab and other CD20+ B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics. (2017) 14:835–41. doi: 10.1007/s13311-017-0557-4
73. Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. (2019) 90:870–81. doi: 10.1136/jnnp-2018-320106
Keywords: multiple sclerosis, syndrome, demyelination, innate immunity, pathophysiology
Citation: Dwyer CM, Nguyen LT, Healy LM, Dutta R, Ludwin S, Antel J, Binder MD and Kilpatrick TJ (2020) Multiple Sclerosis as a Syndrome—Implications for Future Management. Front. Neurol. 11:784. doi: 10.3389/fneur.2020.00784
Received: 20 April 2020; Accepted: 25 June 2020;
Published: 28 August 2020.
Edited by:
Robert Weissert, University of Regensburg, GermanyReviewed by:
Jorge Correale, Fundación para la Lucha contra las Enfermedades Neurológicas de la Infancia (FLENI), ArgentinaCopyright © 2020 Dwyer, Nguyen, Healy, Dutta, Ludwin, Antel, Binder and Kilpatrick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher M. Dwyer, Y2hyaXMuZHd5ZXJAdW5pbWVsYi5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.