
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 30 June 2020
Sec. Neurogenetics
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00527
This article is part of the Research TopicCelebrating the Diversity of Genetic Research to Dissect the Pathogenesis of Parkinson's DiseaseView all 22 articles
 Diana A. Olszewska1,2,3*
Diana A. Olszewska1,2,3* Allan McCarthy1,4
Allan McCarthy1,4 Alexandra I. Soto-Beasley2Ronald L. Walton2Brian Magennis1Russell L. McLaughlin5Orla Hardiman6,7
Alexandra I. Soto-Beasley2Ronald L. Walton2Brian Magennis1Russell L. McLaughlin5Orla Hardiman6,7 Owen A. Ross2,3,8†
Owen A. Ross2,3,8† Tim Lynch1,3†
Tim Lynch1,3†Multiple studies implicate heterozygous GBA mutations as a major genetic risk factor for Parkinson's disease (PD); however, the frequency of mutations has never been examined in PD patients from the Irish population. We prospectively recruited 314 unrelated Irish PD patients (UK Brain Bank Criteria) and 96 Irish healthy controls (without any signs or family history of parkinsonism) attending. The Dublin Neurological Institute (DNI). Complete exon GBA Sanger sequencing analysis with flanking intronic regions was performed. The GBA carrier frequency was 8.3% in PD and 3.1% in controls. We identified a number of potentially pathogenic mutations including a p.G195E substitution and a p.G377C variant, previously described in a case study of Gaucher's disease in Ireland. On genotype–phenotype assessment hallucinations, dyskinesia, and dystonia were more prevalent in GBA-PD. The genetic etiology of PD in Ireland differs from the continental Europe as seen with the lower LRRK2 and higher than in most European countries GBA mutation frequency. Determining genetic risk factors in different ethnicities will be critical for future personalized therapeutic approach.
Glucocerebrosidase gene (GBA) encodes B-glucocerebrosidase enzyme hydrolyzing glucocerebroside to glucose and ceramide. While homozygous or compound heterozygous GBA mutations alter glucocerebrosidase activity and result in a recessive lysosomal-storage disorder, Gaucher's disease, heterozygous variants have been implicated in Parkinson's disease (PD) pathogenesis (1–3). In fact, GBA mutations have been identified as one of the strongest known genetic risk factors for PD (5–20% of PD patients are reported to harbor GBA mutations depending on ethnicity) (2). Initially, GBA-related PD was thought to reflect the clinical phenotype of idiopathic PD (3); however, current evidence suggests unique motor (e.g., frequent falls, freezing of gait, dyskinesia, faster progression), non-motor [autonomic symptoms, younger age at onset (2.6–0.9 year earlier age at onset)] (4), increased prevalence of rapid eye movement (REM) sleep behavior disorder (RBD) and daytime sleepiness (5) features and cognitive impairment (frequent cognitive decline and hallucinations) (5–7).
The GBA gene is located on chromosome 1q22 and consists of 11 coding exons. The existence of a highly homologous pseudogene (GBAP1) (sharing 96% of exonic sequence) located 16 kb downstream of the functional GBA gene makes GBA sequencing studies challenging (8). Sequencing of all exons and using long-range PCR primers specific for the functional GBA gene is the most reliable method for genetic screening. However, many centers continue to restrict the analysis to the most common variants or exons where most of the mutations are reported (exons 9 and 10) (9). GBA p.L444P and p.N370S are the two most common pathogenic substitutions among 335 reported, responsible for 50–70% of cases (1). Based on homozygous and compound heterozygous mutations resulting in specific subtypes of Gaucher's disease, GBA mutations can be classified as “mild” (p.N370S and p.R496H) and “severe” [p.L444P, p.D380A, p.R131C, p.D409H, p.R463C, p.R257Q, p.V394L, c.1263-1317del, and RecNciI-a recombinant allele (p.L444P-A456P-V460V)] (10). Carriers of mild mutations are reported to have 2.2-fold higher risk of PD and mean age at onset of 58.1 (±10.6), while carriers of severe mutations have 9.92- to 21.29-fold increased risk of PD and mean age at onset of 52.1 (±11.2) (10, 11). GBA p.E326K homozygous and compound heterozygous mutations do not cause Gaucher's disease; thus, there may be a distinct mechanism predisposing to PD in carriers of p.E326K. Until recently, believed to be a benign polymorphism, now p.E326K is an established risk factor (large meta-analyses) causing rapid motor progression of PD (β = 3.42; 95% CI, 0.66–6.17; p = 0.02) (12, 13), cognitive decline, and the development of RBD among those who did not have the disorder at baseline (5, 6, 11). Similarly, there is an emerging trend in the literature to classify p.T369M as a risk variant (with the reported effect size similar to that of p.E326K, baseline RBD, associated cognitive decline, and higher hazard ratio of reaching H&Y3) (4, 6, 14, 15).
The prevalence of GBA mutations can vary according to ethnicity (10) (e.g., mutations are common in Ashkenazi Jewish populations). Locally derived data are important for further genetic characterization, development of local guidelines, enrolment in clinical trials, and search for the disease-modifying treatments. Large Irish families, small population size of the country (Republic of Ireland population, 4.79 million in 2017, as per the most recent census) (16), and relative isolation from the continental Europe make the Irish population ideal for genetic studies. We and others reported that genetics of PD in the Irish population differs from that in the continental Europe, e.g., LRRK2 mutations are rare (17). We have shown in a recent epidemiological study that the population structure in Ireland differs from the rest of the Europe, and this may be related to the Celtic ancestry (18). We have also demonstrated that the prevalence of another neurological condition, amyotrophic lateral sclerosis (ALS) in Ireland, differs from other European countries (19). The prevalence of GBA in PD and genotype-phenotype correlation has never been studied in Ireland, and we hypothesized that similarly to LRRK2, it may differ from that in other populations.
The ethical approval (1/378/1,300) was granted by the Mater Misericordiae University Hospital (MMUH), Dublin, Ireland. Informed written consent was obtained.
This is an observational cross-sectional study.
Three hundred fourteen unrelated Irish patients over age 18 diagnosed with PD (UK Brain Bank Criteria) were recruited from a tertiary referral center, Dublin Neurological Institute (DNI), at the MMUH, Dublin, Ireland. Patient's attending the DNI reside in the Dublin city or rural areas of the country. To expand the diversity of participants, an additional notice about the study recruitment was posted on the Irish Parkinson's Association website. Secondary and atypical forms of parkinsonism were excluded. Ninety-six healthy controls (friends or spouses of patients) without any signs or family history of parkinsonism, over age 18 were recruited from the DNI.
Genomic DNA was isolated from peripheral blood lymphocytes using QIAmp Blood Midi Kit. Genetic analysis was performed at the Department of Neuroscience, Mayo Clinic, Jacksonville, FL, USA. All PD patients were prescreened for the most common genetic causes of autosomal dominant disease (LRRK2, SNCA, VPS35) and patients with young onset PD (YOPD) (age, <50) also for autosomal recessive PD (PARKIN, PINK1, DJ1). PD patients (n = 314) were tested for specific point mutations; LRRK2 p.G2019S, SNCA p. A53T, and p.A30P and VPS35 p.D620N (TaqMan Allelic Discrimination Assay, on Quant Studio System Real-Time PCR System). Multiplex ligation-dependent probe amplification (MLPA) analysis was employed to determine the dosage alterations in SNCA. PD patients with YOPD (n = 81) were investigated by Sanger sequencing of all coding exons plus 25 base pairs of exon–intron boundary of PARKIN (12 exons), PINK1 (8 exons), and DJ1 (6 exons) genes. MLPA analysis was also performed.
GBA sequencing of all 11 coding exons plus 25 base pairs of exon–intron boundary was performed on all patients and 96 controls and confirmed bidirectionally. PCR and primer sequences are available in Supplementary Tables 1, 2. The pathogenicity of the variants was determined based on the current literature and in silico tools [Polyphen v2 (20), SIFT (21), Mutation Taster (22)] (Table 1). Results are presented in Table 2. Both newer and well-established historical nomenclature (GBA protein −39 amino acids) for GBA variants annotation was used.
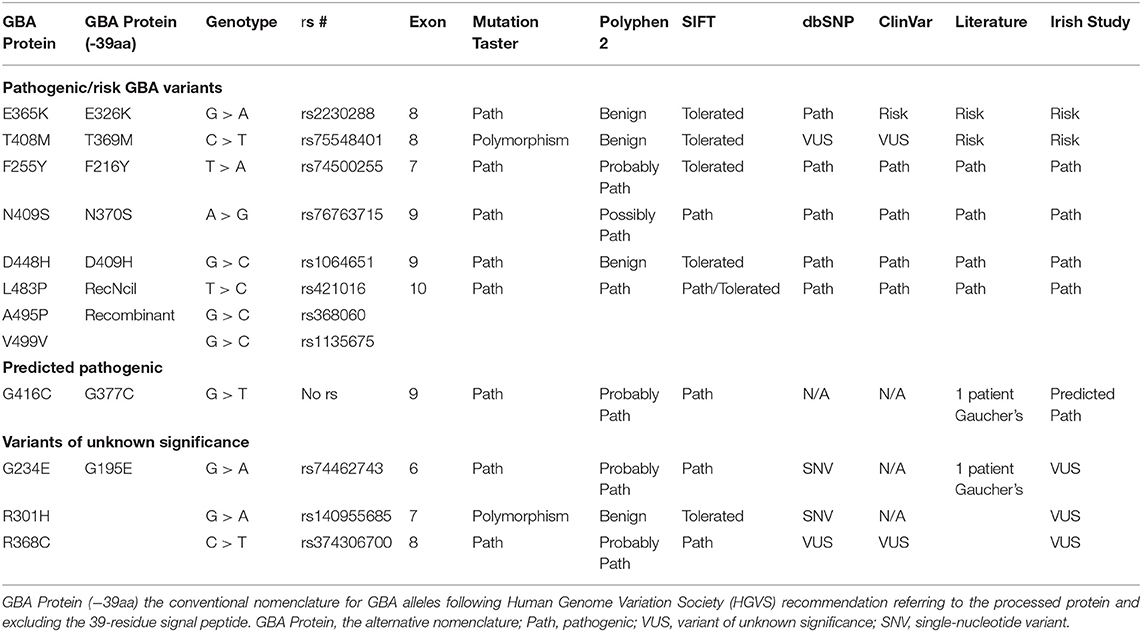
Table 1. Pathogenicity prediction results for GBA genotypes found in Irish Parkinson's disease (PD) patients and controls: pathogenic/risk variants, predicted pathogenic, and VUS genotypes.
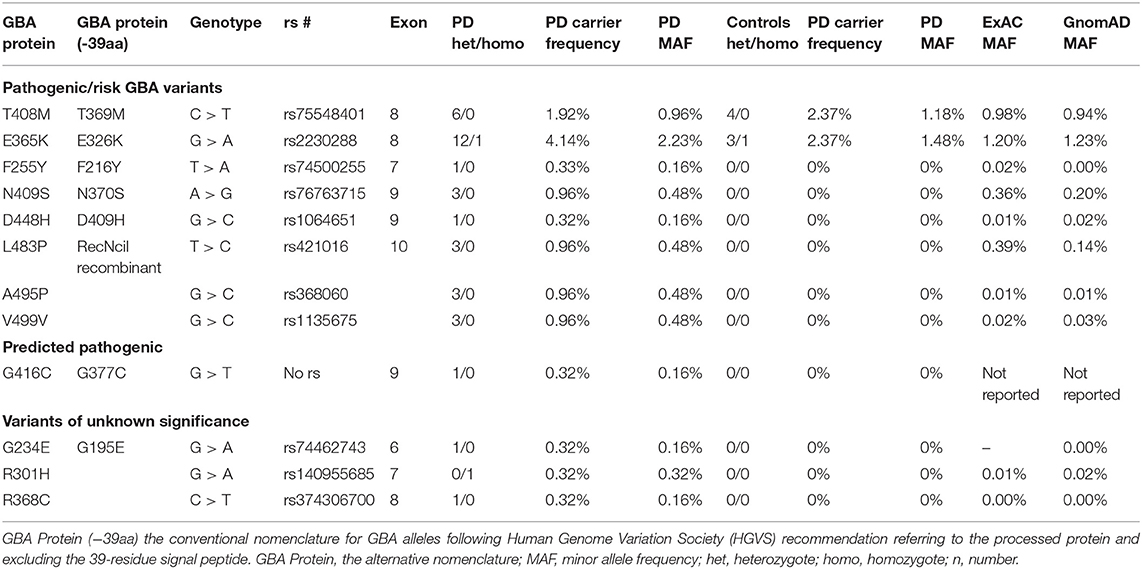
Table 2. Pathogenic/risk variants and variant of unknown significance (VUS) genotypes, carrier frequency, and minor allele frequency in Irish Parkinson's disease (PD) patients and controls and comparison to ExAC and GnomAD databases.
Samples from two patients were tested for β-glucosidase enzyme level and chitoriosidase level by the use of a validated functional assay (fluorimetric assay method) (23) at the Guy's Hospital, London, United Kingdom.
Statistical analysis was performed using IBM SPSS Statistics, Version 22.0. Cohort characteristics were assessed using descriptive statistics. Bivariate associations between categorical variables were calculated using Pearson chi-square tests (X2) or Fisher's exact tests (when expected cell counts ≤ 5 observations, e.g., genotype frequencies comparison). Where continuous variables were normally distributed, independent sample t-tests were used, and where not, Mann–Whitney U-tests. The results were deemed statistically significant where p < 0.05. Two hypotheses were tested. The first was that the presence of a GBA variant increased the risk of PD in the Irish population. Logistic regression model was used to control for age and gender. The second hypothesis was that the GBA variant carriers have earlier age-at-PD onset (14). A linear regression model was fitted with age at onset as the dependent variable and the presence or absence of a GBA variant as the independent variable (gender as a covariate). Linear and logistic regression models were used to test whether the frequency of the motor complications, wearing off, dyskinesia, dystonia, freezing of gait, hallucinations, dementia, and Unified Parkinson's Disease Rating Score part III (UPDRS-III) differed between carriers and non-carriers. Gender, age, and disease duration were considered as covariates. In the sensitivity analysis, each of the motor complications was also controlled for medications, UPDRS-III score, and disease subtype.
We screened 314 Irish patients with PD of which 62.4% (n = 196) were male and 37.6% (n = 118) were female, with mean age at inclusion of 64.94 ± 10.69 years, mean age at onset of 56.23 ± 12.04 years, and mean disease duration of 8.62 ± 6.97 years. The majority had tremor-predominant disease (55.7%), followed by postural instability gait disorder (PIGD) (34.1%) and mixed subtypes (10.2%). Motor complications were seen in 43.3%. Family history of PD was present in 173 patients (55.1%). A group of 96 Irish controls [51% men (n = 49), 49% women (n = 47), mean age at inclusion (61.15 ± 14.5)] was studied to assess the mutation frequency in the ethnically matched population.
We examined 314 patients for genes associated with autosomal dominant PD. Among 314 patients, we identified one PD patient with LRRK2 G2019S mutation positive for GBA benign intronic variant (exon 7−18 bp). We did not identify any carriers of p.A53T or p.A30P in SNCA or p.D620N in VPS35. There were no patients with SNCA dosage alterations.
On examination of the 81 patients with YOPD, we identified one homozygote carrier (p.G430D/p.G430D), three compound heterozygote (p.Leu112fsX163/p.R275W; p.G430D/Ex 4&5del; p.R275W/Ex3 del), and three heterozygote carriers [p.R275W/wt. (n = 1); p.P437L/wt (n = 2)] in PARKIN gene. None of the PARKIN gene carriers had variants in GBA detected. No homozygous or compound heterozygous carriers of PINK1 or DJ1 were detected.
We detected 26 carriers of GBA pathogenic/risk variants in PD and 3 in controls [p.E326K, P.T369M, p.N370S, p.F216Y, p.D409H, and RecNcil (p.L444P-A456P-V460V)] (Tables 2, 3). These variants were found in 8.3% of PD patients and 3.1% of controls (p = 0.08, X2). The result remained non-significant after controlling for age and gender [odd's ratio (OR), 3.2 (p = 0.06; 95%CI, 0.94–10.97). RecNcil and p.N370S carrier frequency in PD was 1.9% [RecNcil, n = 3, minor allele frequency (MAF) = 0.48%; p.N370S, n = 3, MAF = 0.478%); these variants represented 18.2% (6/33) of the pathogenic/risk variants and were not present in controls. The frequency of GBA mutations in familial PD was 8.1% (n = 14) and 8.6% (n = 12) in sporadic PD. The most common variants detected in PD were the risk variants: p.E326K (12 heterozygotes including p.E326K/G377C and p.E326K/T369M and 1 homozygote, p.E326K/E326K) and p.T369M (n = 6, including mentioned above p.E326K/T369M), followed by the pathogenic variants: RecNcil (n = 3) and p.N370S (n = 3) (Tables 2–4). Additionally, we detected three variants of unknown significance (VUS), p.G195E, p.R301H, and p.R368C, in three PD patients and none in controls (Tables 1, 4). We also identified 11 carriers of benign, known intronic variants (rs140335079, T > A: three heterozygotes, five homozygotes; exon 7–17 bp, G > C: one homozygote; rs377143075, T > C: two heterozygotes).
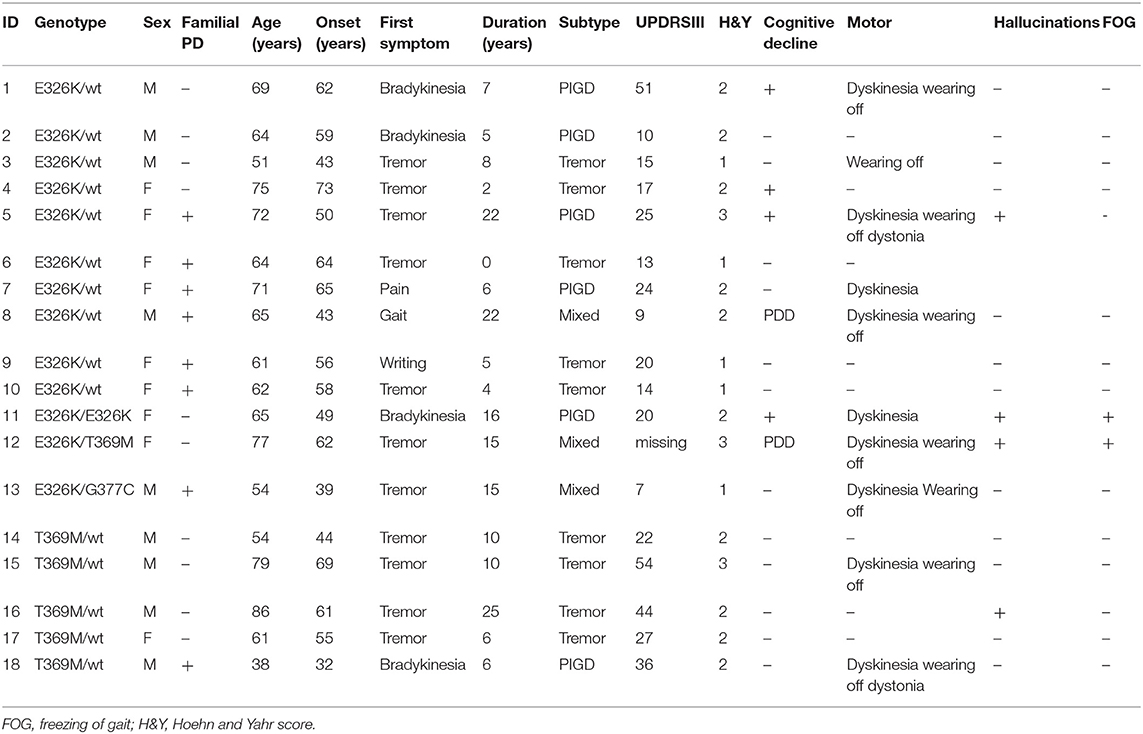
Table 3. Genotype–phenotype correlation in risk variants of GBA carriers with Parkinson's disease (PD).
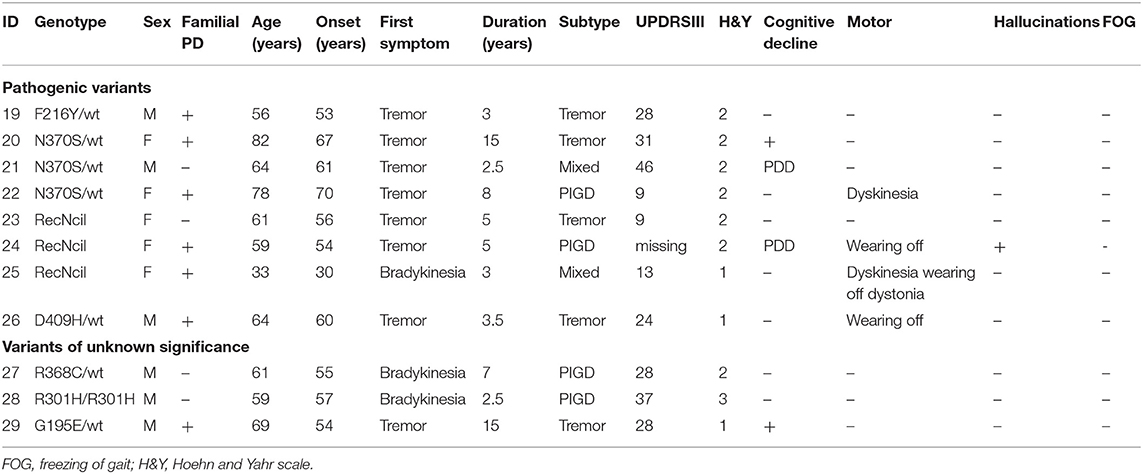
Table 4. Genotype–phenotype correlation in GBA pathogenic variants and variants of unknown significance carriers with Parkinson's disease (PD).
The mean age at onset for the mild mutation carriers (p.N370S n = 3) was 66 (±4.58) years, while for the severe mutations (p.D409H, n = 1; RecNcil, n = 3), it was 50 (±13.56) years (p = 0.11, independent t-test). The mean age at onset for the risk variants carriers (p.E326K, n = 12; p.T369M, n = 6 including p.E326K/p.T369M) was 54.66 (±11.15).
We found a VUS p.R301H variant, which has not been previously reported either in ExAC (24) or gnomAD (24) databases in a homozygous state. The affected was a man with PIGD from age 57 years, normal cognition, and Hoehn and Yahr (H&Y) stage 3 who was asymptomatic for Gaucher's disease. We detected two very interesting variants. p.G195E reported in Gaucher's disease, but not in PD, predicted to be pathogenic by in silico tools (25) in a 69-year-old man. β-Glucosidase enzyme level was measured, and it was at 10.3 nmol/h/mg protein (normal range, 8.4–32.8), and plasma chitoriosidase level was normal. The patient had unilateral, tremor-predominant, levodopa-responsive PD and normal cognition from age 54 years (Figure 13-1). Later on, his cognition declined (MoCA score was 25/30: visuospatial/executive abilities and delayed recall were affected), but hallucinations were not present. He had mild drooling of saliva during the nighttime and REM sleep behavior disorder (RBD). His medications included carbidopa/levodopa 25/100 mg four times daily, mirabegron 50 mg for urinary frequency, and citalopram 15 mg for well-controlled depression. He was also on a continuous positive airway pressure (CPAP) machine for an obstructive sleep apnea. His MRI brain was normal, and dopamine transporter single photon emission computerized tomography (DaT scan) showed a decreased dopamine tracer uptake more pronounced on the left side of the brain. On examination he was hypophonic, had slight rigidity in all limbs, and was bradykinetic more on the right than on the left side. He had bilateral postural and kinetic hand tremor without rest tremor. He did not have any other motor complications.
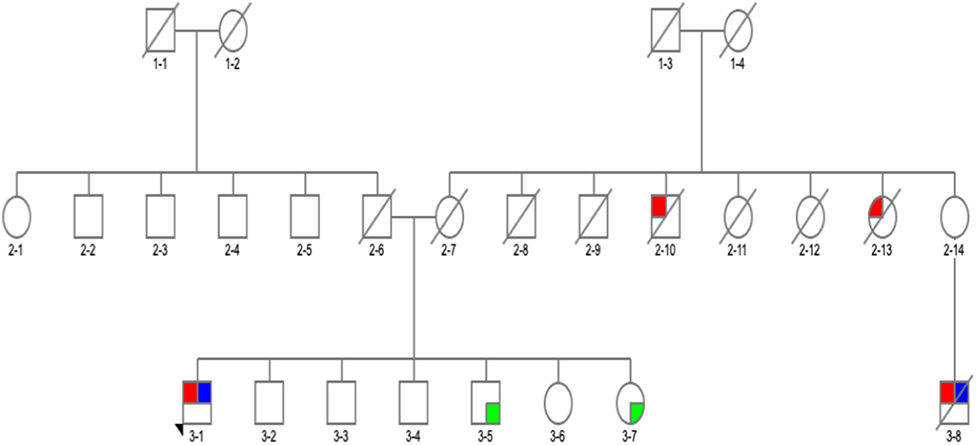
Figure 1. Family pedigree of the patient with GBA p.G195E variant in exon 6, rs74462743: red, affected with Parkinson's disease (PD); blue, tested and positive for p.G195E variant; green, tested and negative for p.G195E variant; arrow, proband; diagonal line, deceased.
There was a PD history in his maternal aunt (deceased) (Figure 12-13), maternal uncle (deceased) (Figure 12-10) and in his 67-year-old cousin (deceased) (Figure 13-8). He had tremor-predominant PD since age 51 years and was positive for the same variant. Initially, he was started on mirapexin for 2 years. He developed hallucinations at age 56 (benign, mainly animals) treated with quetiapine 25 mg in the morning and 50 mg at night. He became forgetful, developed dementia (MoCA score, 7/30), and was commenced on donepezil 10 mg once a day and memantine 10 mg twice a day. He then was switched to carbidopa/levodopa preparation 25/100 mg two tablets three times daily. His other non-motor symptoms included depression—stable on venlafaxine (effexor XL) 150 and 75 mg, and RBD. He was unable to turn in bed and get out of a chair without help and had freezing of gait episodes. There was no wearing off or dyskinesia present. He needed assistance with cutting food, dressing up, and hygiene. His swallow became affected with occasional choking episodes; therefore, he was commenced on thickened fluid diet. He had a urinary catheter in situ due to the difficulties arising from the urinary urgency. The biggest problem was agitation in the evening. At the time of recruitment, he was wheelchair bound, and his UPDRS-III score was 28. On examination, there was slight, however ongoing, resting tremor and bilateral, severe bradykinesia, and rigidity. The dopamine uptake on the DaT scan at age 65 was reported as profoundly decreased. The variant was absent in two asymptomatic siblings of the proband age 57 and 67 (Table 3). Neurological examination was normal in both cases, and MoCA test was 30/30 in the sister (Figure 13-7) and 29/30 in the brother (Figure 13-5). There were no subtle signs of Gaucher's disease with normal hematology, biochemistry, and liver tests.
The second interesting variant found was p.G377C (c.1246 G > T) variant (not reported in databases), predicted to be pathogenic by in silico tools (Tables 1, 2) in a 54-year-old man (Figure 23-1). On further analysis, the β-glucosidase enzyme level was 10.8 nmol/h/mg protein (normal range, 8.4–32.8). The enzyme level in simultaneous controls was 13.6, 14.2, and 14.4. The level of plasma chitotriosidase was normal (as expected in a heterozygous asymptomatic for Gaucher's disease patient). The patient had tremor-predominant PD and normal cognition since age 39 years. There were no hallucinations. At 42 years old, he developed dyskinesia, which was treated by a deep brain stimulator (DBS) at age 49. He also had dystonia and micrographia (the majority of the words was not legible when written). At the time of recruitment, he was on amantadine 100 mg twice a day, selegiline 5 mg, slow release levodopa preparation at night (half-sinemet CR), and carbidopa/levodopa 50/12.5 mg five times a day. On examination, there was hypophonia, moderately stooped posture, and slow gait. PD was present in his mother diagnosed at age 45 (Figure 22-6), who died at 73, maternal aunt diagnosed in her 70s (deceased at 78) (Figure 22-9), and maternal grandfather (Figure 21-3).
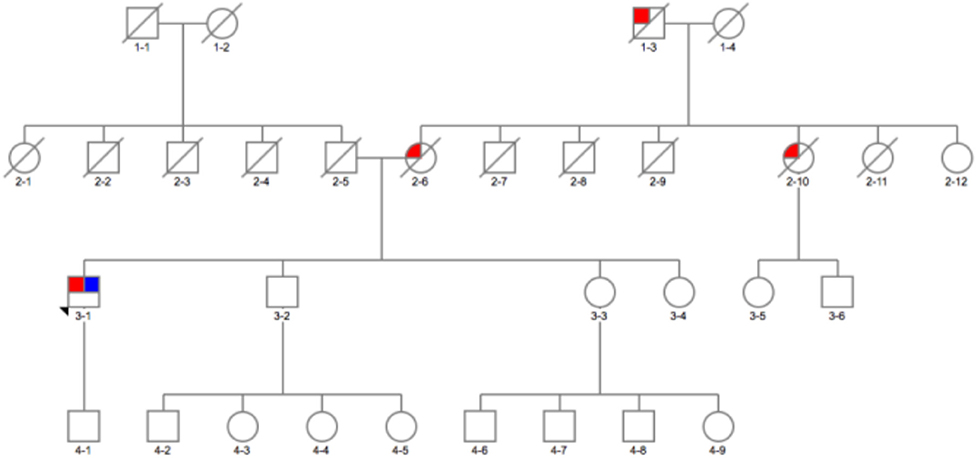
Figure 2. Family pedigree of the patient with GBA p.E326K variant in exon 8 and p.G377C variant in exon 9: red, affected with Parkinson's disease (PD); blue, tested and positive for p.E326K/G377C variant; arrow, proband; diagonal line, deceased.
On genotype-phenotype assessment between 26 mutation/risk variant carriers and 285 non-carriers, the median age at onset in both groups was the same (57 years), carriers were more likely to be a female (53.8% carrier women vs. 46.2% carrier men, p = 0.08, X2), and the majority (65.4%) of carriers had late onset PD (n = 17, p = 0.31, X2) and reported having a positive PD family history (53.8%, n = 14, p = 0.87, X2) (Table 5). However, these results were statistically non-significant (Table 1). Cognitive problems were present in 34.6% of carriers in comparison to 29.8% in non-carriers; however, the p-value was non-significant. Hallucinations were four times more prevalent in carriers than non-carriers [p = 0.01, OR 3.97 (95%CI 1.434–11.015), Fisher's exact test]. This remained true after adjusting for age, gender, disease duration, dementia, cognitive impairment, and medications. While additionally controlled for the presence of Parkin homozygous/compound heterozygous or LRRK2 mutations, the prevalence of hallucinations in GBA mutation carriers remained significantly higher than in GBA non-carriers [p = 0.04, OR 4 (95%CI 1.1–15.3)]. In terms of motor complications dyskinesia [p = 0.003, OR 3.36 (95%CI 1.46–7.75, X2 test)] and dystonia [p = 0.009, OR 12.261 (95% CI 2.34–64.212, Fisher's exact test)] were more prevalent in carriers vs. non-carriers and were independently associated with the carrier status after controlling for age, gender, disease duration, medications, UPDRS III score, and subtypes. When the presence of dyskinesia and dystonia was controlled for the presence of PARKIN homozygous/compound heterozygous or LRRK2 mutations, the result for dyskinesia remained significant [dyskinesia: p = 0.007, OR 4.37 (95%CI 1.49–12.85)], but it became non-significant for dystonia (p = 0.088). There was no association found between other variables and GBA status (Table 5).
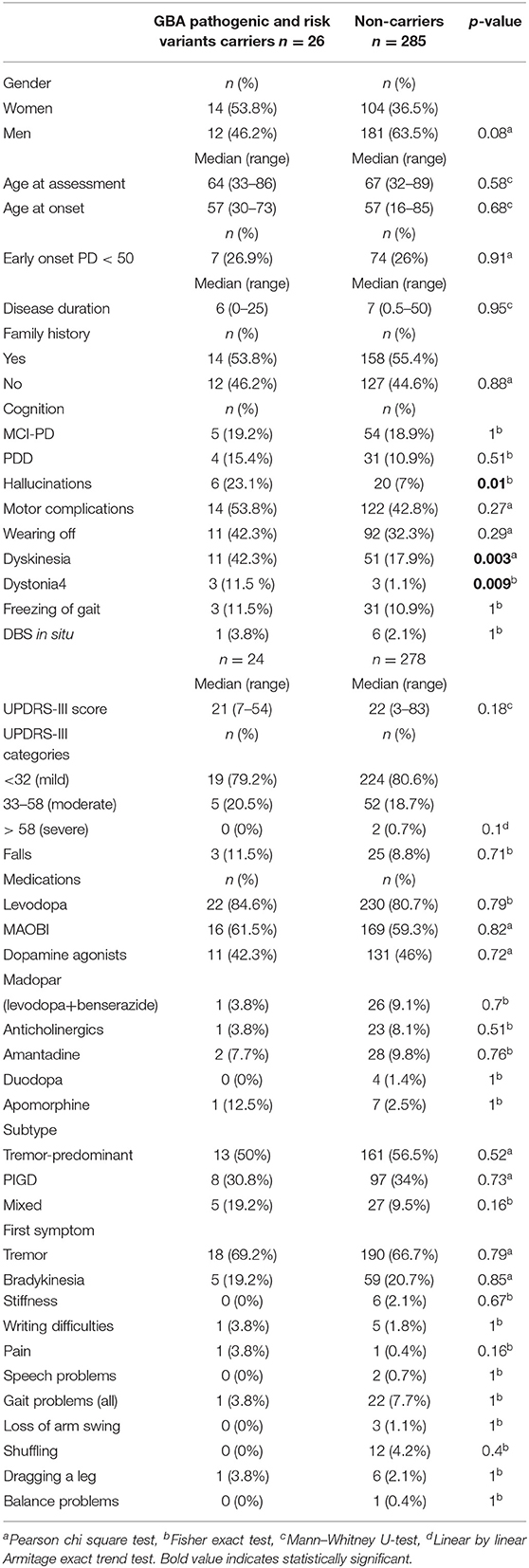
Table 5. Genotype–phenotype assessment in GBA pathogenic/risk variants carriers with Parkinson's disease.
GBA mutations carried in the heterozygous state are a strong risk factor for developing α-synucleinopathy including PD. GBA variants also appear to act as phenotypic modifiers affecting cognition and motor progression of PD. The frequency of GBA variants differs across populations (9, 26, 27), and herein, we show that patients with PD in Ireland have a relatively high frequency of GBA mutations (PD, 8.3%; controls, 3.1%). The frequency is higher than that of the Portuguese (6.1%, 14/230) (28), Greek (4.7%, 8/172) (29), Flanders–Belgian population (4.5%, 12/266) (30), and British (4.2%, 33/790) (10), and slightly above the average European carrier frequency (6.7%, 76/1,130) (16). The carrier frequency both in Irish patients and controls was high when compared to that reported in most European studies with the exception of Spain (9.8%, 22/225) (31). The higher GBA frequency in Spain could be related to the higher rate of Jewish ancestry in the Iberian Peninsula (32).
While the number of p.N370S and RecNcil carriers was equal in Irish PD (with no carriers of p.L444P found), it has been reported that p.N370S is more prevalent in the Europeans and Ashkenazi Jews (70%), and p.L444P is the most common mutation in Chinese (62%) (27, 33). It could be argued that p.T369M should not be included in our pathogenic/risk group, as the number of p.T369M carriers was greater in the control group p.T369M (n = 6) than in the PD group (n = 5 heterozygotes and n = 1 compound heterozygote with p.E326K); however, we did include p.T369M variant in the pathogenic/risk group in agreement with the most recent literature (4, 6, 14, 15).
We detected a p.G377C (p.G416C, c.1246G > T) variant. The G > T nucleotide change in position 416 has not been found in the available databases; however, a change G > A (p.G416S, rs121908311) in the same position is classified as pathogenic. The β-glucosidase enzyme level in a heterozygote carrier within the normal range found in our patient is consistent with the report by Alcalay et al. (26) (p.E326K does not cause Gaucher's disease even in a homozygote state, and the enzymatic level is also within the normal range) (26). The p.G377C variant was described in one case study of Gaucher's disease from Northern Ireland (34) as a compound heterozygote, but not in PD. Only homozygous or compound heterozygous variants cause Gaucher's disease; therefore, p.G377C was necessary to contribute to the disease in the case report by Illingworth (34). Moreover, the level of plasma chitotriosidase was 8,000 in the Illingworth (34) case, further supporting symptomatic Gaucher's disease (the level of chitotriosidase is only elevated in symptomatic patients with Gaucher's disease). Taken together, these evidence support the likely pathogenicity of this variant.
We also identified a p.G195E variant implicated in Gaucher's disease (25) that cosegregated with PD, which merits further investigation (the at-risk siblings are now older than the affected, but we cannot exclude the development of PD at a later age due to the intrafamilial PD heterogeneity). This finding may suggest a more significant role of p.G195E in PD; however, this variant needs to be further explored.
There was no association of gender with pathogenic/risk variants in PD in keeping with findings from Lesage et al. where there was no difference detected (9). Male/female ratio was also reported 5:1–3:2 in other studies (35). Combined MCI-PD (n = 5/26) and PDD (n = 4/26) occurred in 34.6%. This is in keeping with other studies (24–48%) (36). The more common cognitive decline (six-fold increased dementia risk) has been reported in the literature (5, 6, 31), but the prevalence of cognitive impairment and dementia analyzed either separately or as one group in our study did not differ between carriers and non-carriers (however, the sample was small). The GBA mutation/risk variant presence was independently associated with hallucinations in our study in keeping with other studies (31). The three-fold higher occurrence of dyskinesia in the GBA carrier group in comparison to non-carriers in our study is in keeping with other reports. The higher occurrence of dyskinesia suggests that GBA carriers may be more sensitive to medications and genetic assessment in the appropriate patient should be taken into consideration (5, 9, 36) (similarly to the levodopa sensitivity and more frequent dyskinesia resulting in small levodopa doses being used for parkin mutations carriers). While the number of patients with YOPD in the study (n = 81) may seem high, and this group is interesting in itself from the genetic perspective, these were neither specifically preselected for the recruitment nor were they related in any way (only probands were reported in this study).
There are several strengths of our study including the examination of the GBA prevalence in Irish PD for the first time, the full sequencing of coding regions (and exon-intron boundaries) of the GBA gene on the 3.14% of the Irish PD population (314 PD patients recruited/10,000 number of PD patients in Ireland) (limited screening for p.N370S and p.L444P would result in 81.8% of the mutations being missed in our study), comparison to the ethnically matched control group with a high genotyping success rate, and comprehensive investigation of other causes of autosomal dominant and recessive PD (making this study the most comprehensive and up-to-date report of genetics of PD in the Irish population).
Our study also has a few limitations: it was an observational cross-sectional study; therefore, the progression of the disease could not be assessed, and a longitudinal study should be performed in the future. It should be noted that we were unable to reliably determine phase for the two individuals with a presumed compound heterozygous carrier state, both carried the risk variant E326K (E326K/T369M, n = 1; E326K/G377C, n = 1); however, these subjects would fall into the risk/pathogenic group regardless of being in “cis” or in “trans” status. While we acknowledge the small sample size of the control group in comparison to the PD group most likely leading to the non-significance of the GBA prevalence data, in our opinion, having a control group is an important part of any GBA study.
With the advent of new screening technologies, GBA carriers will be encountered more frequently in our clinics, and it will be essential to prepare a mutation-specific approach to the management of PD. Efforts to find a disease-modifying treatment for GBA carriers with PD are ongoing. There are currently two clinical trials underway, one trial examining ambroxol, an over-the-counter medication used to reduce mucus production in respiratory tract disorders, and its influence on motor and cognitive progression in GBA carriers, and a placebo controlled phase 2 double-blind study (MOVES-PD) of GZ/SAR4027671—a molecule capable of crossing brain-blood barrier (37, 38). Another recently completed trial showed that the target engagement and CSF penetration were accomplished in PD patients treated with oral ambroxol. While the CSF glucocerebrosidase activity decreased, the glucocerebrosidase protein levels and alpha-synuclein levels increased and UPDRS-III score improved by a mean of 6.8 points. Notably, these changes occurred both in patients with and without GBA mutations (39). There are no recommendations of how to proceed, where the risk of PD is disclosed, but preventative approaches may be rapidly approaching. Patients should be informed about the increased possibility of cognitive decline, depression, falls, autonomic vulnerability, family (35) planning, and disease progression [carriers are reported to die earlier at 75.7 years (SD 5.5) than non-carriers 80.9 (6.6)] (40). Treatment options for GBA mutation carriers should be a little bit distinct and focus on the avoidance of medications increasing the risk of falls (lowering blood pressure), worsening cognitive status, and deferral of levodopa therapy (36). Further research is required, but personalized therapeutic approach for PD may be closer than we might think.
We present the most comprehensive and up-to-date overview of genetics of PD in the Irish population. We, for the first time, showed the link between GBA and PD in Ireland. In agreement with our hypothesis, the findings of our study suggest that the GBA prevalence in PD is higher than in most European countries, and genetic background of Irish PD patients warrants further studies.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Mater Misericordiae University Hospital, Dublin, Ireland 1/378/1300. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
DO: study idea and design, study recruitment and phenotypic assessment, DNA extraction, GBA sequencing, and laboratory work, interpretation of the results and statistical analysis, preparation of the first draft of manuscript, correction, and approval of the final draft. AM: study recruitment and phenotypic assessment, DNA extraction, critique, and approval of the final draft. AS-B and RW: laboratory work, critique, and approval of the final draft. BM: study recruitment, critique, and approval of the final draft. RM and OH: critique and approval of the final draft. OR: study idea, lead of the laboratory work and expertise, overlooking the laboratory work at the Mayo Clinic, expertise in the interpretation of the results, critique, and approval of the final draft. TL: study idea and design, lead of the neurological expertise, overlooking the study in the Dublin Neurological Institute, critique, and approval of the final draft. All authors: read and approved the final version on the manuscript.
RM was supported by the MND Association of England, Wales and Northern Ireland (957-799), and OH was supported by Science Foundation Ireland (15/SPP/3244). Mayo Clinic is an American Parkinson Disease Association (APDA) Mayo Clinic Information and Referral Center, an APDA Center for Advanced Research and the Mayo Clinic Lewy Body Dementia Association (LBDA) Research Center of Excellence. OR was supported by the National Institutes of Health (NIH; R01 NS78086, U54 NS100693, U54 NS110435), the US Department of Defense (W81XWH-17-1-0249), The Little Family Foundation, the Mayo Clinic Center for Individualized Medicine, and the Michael J. Fox Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to this resource. A full list of contributing groups can be found at http://gnomad.broadinstitute.org/about. The authors would like to acknowledge the statistical advice obtained from Dr. Ricardo Segurado, UCD CSTAR.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.00527/full#supplementary-material
1. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. (2012) 11:986–98. doi: 10.1016/S1474-4422(12)70190-4
2. O'Regan G, deSouza RM, Balestrino R, Schapira AH. Glucocerebrosidase mutations in Parkinson Disease. J Parkinsons Dis. (2017) 7:411–22. doi: 10.3233/JPD-171092
3. Schapira AHV. Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci. (2015) 66:37–42. doi: 10.1016/j.mcn.2015.03.013
4. Blauwendraat C, Heilbron K, Vallerga CL, Bandres-Ciga S, von Coelln R, Pihlstrøm L, et al. Parkinson's disease age at onset genome-wide association study: defining heritability, genetic loci, and α-synuclein mechanisms. Mov Dis. (2019) 34:866–75. doi: 10.1002/mds.27659
5. Iwaki H, Blauwendraat C, Leonard HL, Liu G, Maple-Grødem J, Corvol JC, et al. Genetic risk of Parkinson disease and progression: an analysis of 13 longitudinal cohorts. Neurol Genet. (2019) 5:e348. doi: 10.1212/NXG.0000000000000354
6. Iwaki H, Blauwendraat C, Leonard HL, Kim JJ, Liu G, Maple-Grødem J, et al. Genomewide association study of Parkinson's disease clinical biomarkers in 12 longitudinal patients' cohorts. Mov Disord. (2019) 34:1839–50. doi: 10.1002/mds.27845
7. Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AH. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. (2015) 72:201–8. doi: 10.1001/jamaneurol.2014.2950
8. Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. (1989) 4:87–96. doi: 10.1016/0888-7543(89)90319-4
9. Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, et al. Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet. (2011) 20:202–10. doi: 10.1093/hmg/ddq454
10. Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. (2015) 84:880–7. doi: 10.1212/WNL.0000000000001315
11. Blauwendraat C, Reed X, Krohn L, Heilbron K, Bandres-Ciga S, Tan M, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson's disease and Lewy body dementia. Brain. (2020) 143:234–48. doi: 10.1093/brain/awz350
12. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. (2014) 46:989–93. doi: 10.1038/ng.3043
13. Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. (2016) 73:1217–24. doi: 10.1001/jamaneurol.2016.2245
14. Gan Or Z, Roy N, Alcalay RN, Makarious MB, Scholz SW, Blauwendraat C. Classification of GBA variants and their effects in synucleinopathies. Mov Dis. (2019) 34:1581–2. doi: 10.1002/mds.27803
15. Mallett V, Ross JP, Alcalay RN, Ambalavanan A, Sidransky E, Dion PA, et al. GBA p.T369M substitution in Parkinson disease: polymorphism or association? A meta-analysis. Neurol Genet. (2016) 2:e104. doi: 10.1212/NXG.0000000000000104
16. CSO Statistical Release. Central Statistics Office. (2017) Available Online at: http://www.cso.ie/en/releasesandpublications/er/pme/populationandmigrationestimatesapril2017/ (accessed May, 2020).
17. Gosal D, Ross OA, Wiley J, Irvine GB, Johnston JA, Toft M, et al. Clinical traits of LRRK2-associated Parkinson's disease in Ireland: a link between familial and idiopathic PD. Parkinsonism Relat. Disord. (2005) 11:349–52. doi: 10.1016/j.parkreldis.2005.05.004
18. Byrne RP, Martiniano R, Cassidy LM, Carrigan M, Hellenthal G, Hardiman O, et al. Insular Celtic population structure and genomic footprints of migration. PLoS Genet. (2018) 14:e1007152. doi: 10.1371/journal.pgen.1007152
19. Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al., Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet. (2013) 50:776–83. doi: 10.1136/jmedgenet-2013-101795
20. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
21. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
22. Schwarz JM, Cooper DN, Schuelke M, Seelow D. Mutation Taster 2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
23. Motabar O, Goldin E, Leister W, Liu K, Southall N, Huang W, et al. A high throughput glucocerebrosidase assay using the natural substrate glucosylceramide. Anal Bioanal Chem. (2012) 402:731–9. doi: 10.1007/s00216-011-5496-z
24. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
25. Grace ME, Desnick RJ, Pastores GM. Identification and expression of acid beta-glucosidase mutations causing severe type 1 and neurologic type 2 Gaucher disease in non-Jewish patients. J Clin Invest. (1997) 99:2530–7. doi: 10.1172/JCI119437
26. Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. (2015) 138:2648–58. doi: 10.1093/brain/awv179
27. Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology. (2008) 70:2277–83. doi: 10.1212/01.wnl.0000304039.11891.29
28. Bras J, Paisan-Ruiz C, Guerreiro R, Ribeiro MH, Morgadinho A, Januario C, et al. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Portugal. Neurobiol Aging. (2009) 30:1515–7. doi: 10.1016/j.neurobiolaging.2007.11.016
29. Kalinderi K, Bostantjopoulou S, Paisan-Ruiz C, Katsarou Z, Hardy J, Fidani L. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Greece. Neurosci Lett. (2009) 452:87–9. doi: 10.1016/j.neulet.2009.01.029
30. Crosiers D, Verstraeten A, Wauters E, Engelborghs S, Peeters K, Mattheijssens M, et al., Mutations in glucocerebrosidase are a major genetic risk factor for Parkinson's disease and increase susceptibility to dementia in a flanders-belgian cohort. Neurosci Lett. (2016) 629:160–4. doi: 10.1016/j.neulet.2016.07.008
31. Setó-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson's disease course. Mov Disord. (2012) 27:393–9. doi: 10.1002/mds.24045
32. Bandrés-Ciga S, Price TR, Barrero FJ, Escamilla-Sevilla F, Pelegrina J, Arepalli S, et al. Genome-wide assessment of Parkinson's disease in a Southern Spanish population. Neurobiol Aging. (2016) 45:213.e3–9. doi: 10.1016/j.neurobiolaging.2016.06.001
33. Chen J, Li W, Zhang T, Wang YI, Jiang XJ, Xu ZQ. Glucocerebrosidase gene mutations associated with Parkinson's disease: a met-analysis in a Chinese population. PLoS ONE. (2014) 9:e115747. doi: 10.1371/journal.pone.0115747
34. Illingworth MA, O'Connor B, Stewart F, Vellodi A, Jones S, Hughes J, Peake D. Poster 035: a novel mutation in rapidly progressive late onset intermediate neuronopathic Gaucher disease. Dev Med Child Neurol. (2011) 53.
35. Zampieri S, Cattarossi S, Bembi B, Dardis A. GBA analysis in next-generation era: pitfalls, challenges, and possible solutions. J Mol Diagn. (2017) 19:733–41. doi: 10.1016/j.jmoldx.2017.05.005
36. Giladi N, Mirelman A, Thaler A, Orr-Urtreger A. A personalized approach to Parkinson's disease patients based on founder mutation analysis. Front Neurol. (2016) 7:71. doi: 10.3389/fneur.2016.00071
37. Clinical Trials Finder, Search Words GBA and Parkinson's Disease. (2018) Available Online at: https://clinicaltrials.gov/ct2/show/NCT02914366?term=gba+and+parkinson&draw=2&rank=6 (accessed May, 2020).
38. Silveira CRA, MacKinley J, Coleman K, Li Z, Finger E, Bartha R, et al. Ambroxol as a novel disease-modifying treatment for Parkinson's disease dementia: protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. (2019) 19:20. doi: 10.1186/s12883-019-1252-3
39. Mullin S, Smith L, Lee K, D'Souza G, Woodgate P, Elfein J, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. (2020) 77:427–34. doi: 10.1001/jamaneurol.2019.4611
Keywords: GBA, glucocerebrosidase, Ireland, sequencing, Parkinson's disease
Citation: Olszewska DA, McCarthy A, Soto-Beasley AI, Walton RL, Magennis B, McLaughlin RL, Hardiman O, Ross OA and Lynch T (2020) Association Between Glucocerebrosidase Mutations and Parkinson's Disease in Ireland. Front. Neurol. 11:527. doi: 10.3389/fneur.2020.00527
Received: 01 February 2020; Accepted: 13 May 2020;
Published: 30 June 2020.
Edited by:
Christos Proukakis, University College London, United KingdomReviewed by:
Stephen Mullin, University of Plymouth, United KingdomCopyright © 2020 Olszewska, McCarthy, Soto-Beasley, Walton, Magennis, McLaughlin, Hardiman, Ross and Lynch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana A. Olszewska, ZGlhbmEuYW5nZWxpa2Eub2xzemV3c2thQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.