 Martín Bustelo1,2,3,4†
Martín Bustelo1,2,3,4† Melinda Barkhuizen1†
Melinda Barkhuizen1† Daniel L. A. van den Hove2,5
Daniel L. A. van den Hove2,5 Harry Wilhelm. M. Steinbusch2
Harry Wilhelm. M. Steinbusch2 Martín A. Bruno3C. Fabián Loidl3,4
Martín A. Bruno3C. Fabián Loidl3,4 Antonio W. Danilo Gavilanes1,6*
Antonio W. Danilo Gavilanes1,6*- 1Department of Pediatrics, Maastricht University Medical Center (MUMC), Maastricht, Netherlands
- 2Department of Psychiatry and Neuropsychology, School for Mental Health and Neuroscience (MHeNs), Maastricht University, Maastricht, Netherlands
- 3Instituto de Ciencias Biomédicas, Facultad de Ciencias Médicas, Universidad Católica de Cuyo, San Juan, Argentina
- 4Laboratorio de Neuropatología Experimental, Facultad de Medicina, Instituto de Biología Celular y Neurociencias “Prof. E. De Robertis” (IBCN), Universidad de Buenos Aires, CONICET, Buenos Aires, Argentina
- 5Department of Psychiatry, Psychosomatics and Psychotherapy, University of Würzburg, Würzburg, Germany
- 6Facultad de Ciencias Médicas, Instituto de Investigación e Innovación de Salud Integral, Universidad Católica de Santiago de Guayaquil, Guayaquil, Ecuador
Placental and fetal hypoxia caused by perinatal hypoxic-ischemic events are major causes of stillbirth, neonatal morbidity, and long-term neurological sequelae among surviving neonates. Brain hypoxia and associated pathological processes such as excitotoxicity, apoptosis, necrosis, and inflammation, are associated with lasting disruptions in epigenetic control of gene expression contributing to neurological dysfunction. Recent studies have pointed to DNA (de)methylation, histone modifications, and non-coding RNAs as crucial components of hypoxic-ischemic encephalopathy (HIE). The understanding of epigenetic dysregulation in HIE is essential in the development of new clinical interventions for perinatal HIE. Here, we summarize our current understanding of epigenetic mechanisms underlying the molecular pathology of HI brain damage and its clinical implications in terms of new diagnostic, prognostic, and therapeutic tools.
Introduction
Epigenetics is defined as heritable changes in gene expression that do not result from a change in the DNA sequence (1). Epigenetic regulation plays an essential role during development, and any insult that disrupts physiological developmental epigenetic programming is likely to have long-term consequences. An increasing number of studies link exposure to different adverse factors during early life, including both the gestational and the postnatal period to changes in the epigenome and one's individual suceptibility (2). In this regard, the brain is particularly vulnerable to alterations in the early-life microenvironment, damage induced at this stage may not be evident until the exposure to a new insult triggers it (3).
Perinatal hypoxia represents one of the most common early life insults that ultimately leads to disability or even early death (4, 5). Hypoxia can occur progressively during pregnancy in cases of fetal growth restriction due to placental abnormalities, or can occur acutely during labor and birth, causing peripartum hypoxic-ischemic encephalopathy (HIE). Fetal growth restriction is associated with stillbirths and permanent neurological disability in survivors (6). Management of fetal growth restriction depends on early detection, and timely delivery before stillbirth occurs (7). Advances in this area have focused on detecting the presence of fetal growth restriction, and the degree of hypoxia present in utero.
HIE has a large impact on global child health, with morbidity in 2.5/1,000 live births (4–9 million newborns affected per year worldwide) (8–10). At present, therapeutic hypothermia is the only approved therapy for newborns ≥36 weeks gestational age with moderate-to-severe HIE. Therapeutic hypothermia is thought to work by generally slowing down metabolism and thus counteracting a variety of pathological mechanisms of HIE (11). Several trials have demonstrated that hypothermia is effective in decreasing mortality and decreasing neurocognitive impairments (12, 13), still, this therapy is only partially protective, half of treated newborns still die or develop a lifelong disability, demonstrating the need for the development of other neuroprotective treatment strategies (14).
The features of acute intrapartum hypoxic-ischemia (HI) involve fetal hypoxemia, hypercapnia, and ischemia. In the brain, this leads to metabolic acidosis, cellular necrosis, and activation of apoptotic pathways. After reperfusion/reoxygenation, oxidative metabolism recovers in surviving cells, and most of the neurotoxic cascade is seemingly terminated. This first period immediately following the HI event (0–6 h) is also referred to as the “therapeutic window,” where intervention may prevent secondary damage.
Secondary energy failure (6–48 h following HI) involves potent inflammation as well as oxidative stress induced by reactive oxygen species and free radicals, and failure of mitochondrial oxidative phosphorylation due to permeabilization of the mitochondrial membranes. Ultimately, these processes lead to delayed cell death via necrotic and apoptotic pathways (15). Brain injury continues to evolve even months and years after the initial insult, in the tertiary phase, involving neural scarring and persistent inflammation (16). Traditionally epigenetic changes have been attributed to the tertiary phase, its role in the initial phases of HI induced injury is less well-characterized.
Different in vivo and in vitro models have been used to study the pathological features of HIE. Rodents are the most commonly used animals to model perinatal HI (17), with three major approaches being used. The first model makes use of submersion of the uterine horns containing the term fetal rats in saline. This model mimics a global insult to the fetus at a very low gestational age (18, 19). Considering the fact that brain development in rodents is delayed when compared to that in humans (17), other models use neonatal pups at Postnatal day (P) 3–10 (20) to model insults in the late preterm to term brain. Amongst others, neonatal pups can be subjected to a global hypoxic insult by placing them in a hypoxic chamber, an approach, however, that lacks the ischemic nature of the insult seen in the clinic (20). The most commonly used model, developed by Levine in adult rodents, and adapted by Rice and Vannucci for neonatal rodents (21, 22), induces ischemia by carotid artery occlusion (CAO) followed by exposure to systemic hypoxia (23) inducing an HI insult, here referred as the “HI” model. In vitro models exposing neuronal cultures to oxygen-glucose deprivation (OGD)/reoxygenation have also been used as models of HI induced injury (24).
Epigenetic Dysregulation in Hypoxia-Ischemia
Epigenetic processes regulate both the transcription of the DNA into mRNA and the translation of the mRNA into proteins, acting at multiple levels of control, involving e.g., DNA methylation and hydroxymethylation, chromatin remodeling, and non-coding RNA (ncRNA) regulation (25, 26). As such, DNA is wrapped around histone proteins forming chromatin, where the configuration of chromatin is controlled by post-translational modifications of the associated histone proteins as well as by direct modifications to nucleotides, which collectively form the epigenetic code that controls the transcription of DNA into mRNA (25). This epigenetic code can be modified by “writers,” i.e., enzymes which introduce post-translational modifications on the DNA and histones, “erasers,” which remove the modifications, and “readers,” i.e., specialized proteins which identify and interpret the modifications. The translation of mRNA into proteins is regulated by ncRNAs. HI-induced epigenetic alterations are preserved even in the absence of hypoxia-inducible factors (HIFs) (27, 28) suggesting they may continue to impact upon behavioral phenotypes later in life (Figure 1).
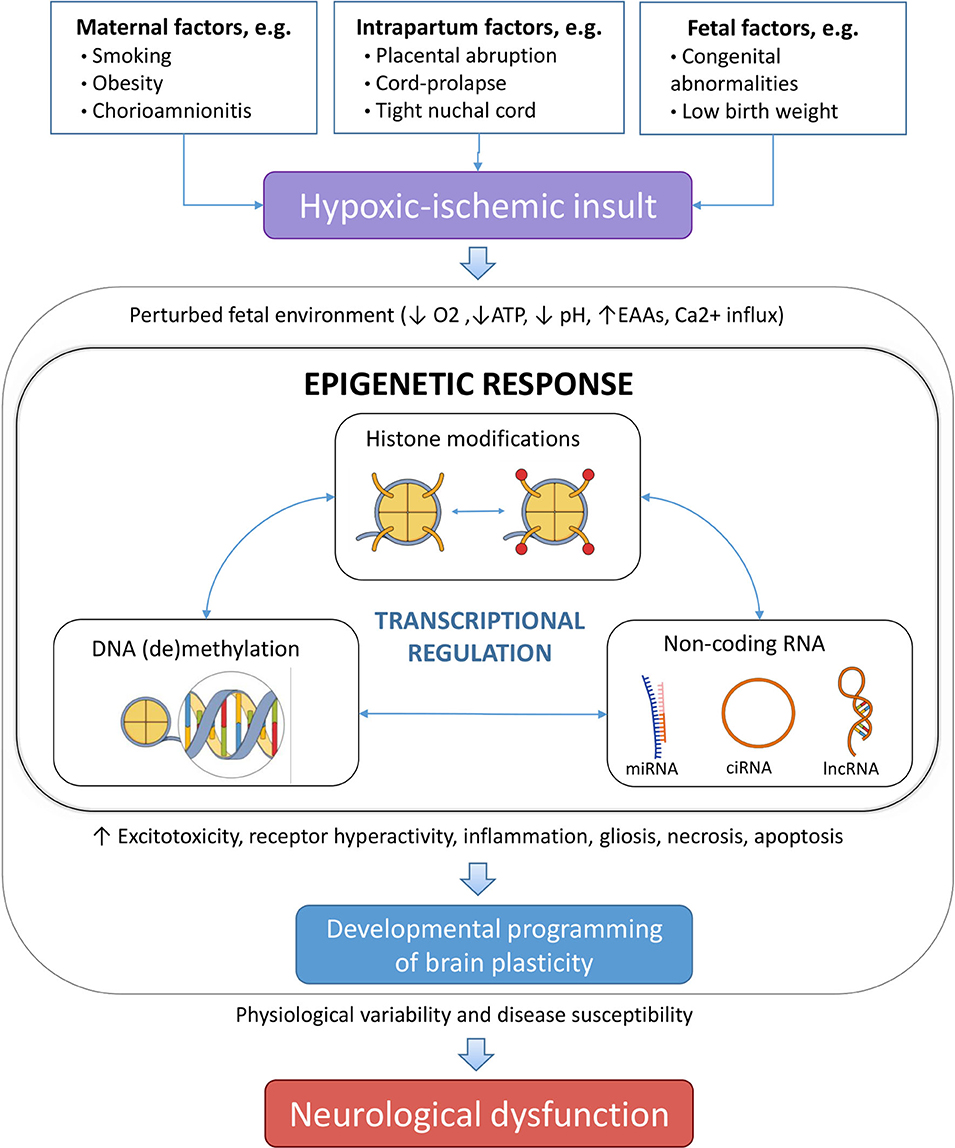
Figure 1. Different risk factors, which can be ante-, peri-, or postnatal can lead to perinatal hypoxia. If prolonged hypoxia results in a perturbed brain environment, leading to modulated epigenetic changes that alter the gene expression profile as an adaption of the fetus to the adverse environment. Different epigenetic mechanisms cooperatively orchestrate this process. As a result, changes in physiology of the neonatal brain can permanently affect the structure and/or functionality, and result in increased disease susceptibility in the offspring. EAAs, excitatory amino acids; miRNA, microRNA; ciRNA, circular RNA; lncRNA, long non-coding RNA.
Hypoxia-Inducible Factor-1
Hypoxia-inducible factor-1 (HIF-1) is the main effector of cellular hypoxia, being an oxygen-sensitive transcription factor. HIF-1 comprises two subunits, i.e., HIF-1α and HIF-1β (29). The stability and activity of the oxygen-regulated subunit HIF-1α are regulated by post-translational modifications that control its proteasomal degradation. In hypoxia, activation of HIFs is mediated via the inhibition of dioxygenases, such as the Jumonji-C (JmjC) domain-containing histone demethylases. As a result, HIF-1α accumulates, binds to a hypoxic response element (HRE), and leads to the transcriptional induction of several genes involved in adaptation to hypoxia, including genes involved in angiogenesis, iron metabolism, and glucose metabolism.
HIF-1 expression levels, protein stabilization, and its association with HRE are under tight epigenetic regulation, including promoter methylation and microRNA (miRNAs) control (30–32). Conversely, HIF-1 controls the expression of several epigenetic regulators. Studies have shown that DNA methylation regulates HIF-1α transcription regulating the promoter activity at HRE-152 (33) and SP1.
DNA Methylation
DNA methylation comprises the addition of a methyl group at the carbon 5 position on the pyrimidine ring of cytosines, creating 5-methylcytosine (5-mC) (34). Cytosine methylation provides a stable epigenetic mark with long-term transcriptional effects. DNA methylation is catalyzed by DNA methyltransferases (DNMTs). These enzymes can work in different ways depending on the local chromatin microenvironment, acting as DNA methyltransferase or DNA dehydroxymethylase (35). The methylation status of the DNA is “read” by methyl CpG binding proteins (MBPs), like the methyl-CpG-binding protein 2 (MECP2) which recruits HDACs to repress transcription (36). Recent evidence indicates that during early postnatal development neuronal genomes accumulate uniquely high levels of two alternative forms of methylation, i.e., non-CpG methylation and DNA hydroxymethylation (37). DNA methylation marks can be “erased” either passively through inhibition of the DNMTs or actively by Ten-eleven translocation (TET) enzymes. TETs are 2-oxoglutarate-dependent dioxygenases that mediate DNA hydroxymethylation and their activity is oxygen-dependent (38).
Non-coding RNAs
In humans, only 1–2% of the genome encodes proteins, while non-coding RNAs (ncRNAs) that do not produce proteins are the great majority of human transcripts (39). As such, ncRNAs, including microRNAs (miRNAs), long non-coding RNAs (lncRNAs), small nucleolar RNAs, and circular RNAs (circRNAs), have important roles in regulating gene expression playing diverse regulatory roles (40). The limited availability of human fetal and neonatal tissue has resulted in studies using animal models and in vitro studies as a major alternative for studying the role of ncRNAs in newborns under physiological and pathophysiological conditions.
miRNAs
MiRNAs are a large class of short regulatory RNAs, about 19–22 nucleotides in length, which silence gene expression by binding to the 3′-untranslated region of target genes. The majority of miRNAs are enriched in the developing brain, which signifies their role in neural development (41). Consequently, disruption of miRNAs in the perinatal period is likely to have long-term consequences (42, 43). Several miRNAs termed hypoxamirs have been shown to be induced by hypoxia, and control the cellular response to hypoxia via HIFs. These include miRNAs that regulate HIF1-α signaling (31, 44–50) and miRNAs that contain hypoxia-responsive elements that are transcribed in response to HIF1-α activation and act downstream of the HIFs (31, 47, 49, 51–53). miRNA expression is highly tissue- and disease-specific, which, together with their remarkable stability in circulation, makes them potential diagnostic biomarker candidates, still none has made it to a clinical setting (Table 1).

Table 1. Clinical implications of non-coding RNAs in perinatal hypoxia ischemia studies: SYMBOLS: ↑, Upregulation; ↓, Downregulation; –, Not specified.
LncRNAs
Long-non coding RNAs (LncRNAs) are ncRNAs larger than 200 nucleotides that act as a competing endogenous RNAs controlling gene expression by sponging miRNAs, and binding and inactivating chromosomes (78). LncRNAs fold into complex secondary and tertiary structures defining their interactions and function, providing a scaffold for proteins to form regulatory complexes (79, 80). Studies have demonstrated that LncRNAs play an important role in the regulation of gene expression, particularly during CNS development (81), with nearly 40% of LncRNAs reported to be specifically expressed in the CNS and involved in brain development and related disorders (82, 83).
Histone Modifications
DNA is wrapped around octamers of histone (H) proteins containing two copies of H2A, H2B, H3, and H4 proteins forming nucleosomes that are bounded by H1 linker histones. H3 and H4 have N-terminal tails that extend beyond the nucleosome and are permissive to modifications, such as acetylation, methylation, glycosylation, ubiquitination, farnesylation, citrullination, and ADP-ribosylation. These modifications alter the charge of the amino acid residues of histones, resulting in relaxed DNA (euchromatin), accessible for the transcriptional machinery, or condensed DNA (heterochromatin) where the transcriptional machinery cannot access (84).
Altogether, these modifications encode the histone code, that, depending on the type and locus of the modifications, the relative location of histones within or toward a gene, and the combination of histone modifications, can lead to enormously diverse readouts (85). Histone modifications can act in different ways depending on the context, increasing the complexity of the histone code. The best-described histone marks in HI brain damage are histone acetylation and methylation.
Histone Methylation
Many enzymes participate in the control of histone methylation, including histone methyltransferases (writers), histone demethylases (erasers), and proteins that recognize the methylated state (readers) (86).
Histone demethylation is carried out by histone lysine demethylases (KDM2–7), which can remove both activating and repressing methyl groups from the chromatin (87, 88). Studies exposing cell lines to hypoxia have shown increased expression of the KDM3 gene, which removes methyl groups from repressive H3K9 sites and activates gene expression (89, 90). Methyl modifications take place mostly on lysine (K) residues of H3 and H4, being the main histone methylation site subject to epigenetic variation H4K20 (91). Interestingly, in vitro experiments suggest that JmjC enzymes can act as molecular oxygen sensors in the cell (92).
Histone Acetylation
Histone acetylation by histone acetyltransferases (HATs) relaxes chromatin formation, promoting transcription, whilst the deacetylation of histone tails by histone deacetylases (HDACs) causes the chromatin to condense, thereby inhibiting transcription (93). HATs exert their effects in collaboration with proteins like p300/CBP, PCAF, SRC that can associate and regulate the transcription of HIF-1α (94). HDACs are divided into 4 classes based on their domain organization. Class I, II, and IV HDACs depend on zinc as a co-factor, whilst class III HDACs known as sirtuins (SIRTs), depending on the co-factor NAD+ (95). Class I and class IIa HDACs enhances HIF-1α stability by directly binding to the oxygen-dependent degradation domain of HIF-1α and class IIb HDACs promote HIF-1α transcriptional activity (96, 97).
Potential Biomarkers
miRNAs
MIR210 is the master hypoxamir and its expression is induced under hypoxia in many cell types. MIR210 acts downstream of HIF-1α, repressing several key processes to lower cellular energy requirements during hypoxia (31, 47, 98, 99). Human studies have consistently shown MIR210 upregulation in placenta (57, 100–103) and plasma (104) from preeclampsia pregnancies, and also in intrauterine growth restriction (56) (Table 1). Studies using maternal miRNAs derived from the placenta circulate in the maternal blood during pregnancy and may serve as non-invasive biomarkers. Studies using maternal plasma (57), and blood (55) have shown an elevation in MIR210 in both chronic and acute fetal hypoxia. miRNAs produced in the placenta circulate in the maternal blood during pregnancy and can be used as non-invasive biomarkers for hypoxia in-utero and HIE, allowing early interventions.
When perinatal HIE is diagnosed, it is critical to grade the injury to decide whether or not to subject the patient to hypothermic treatment. The ideal biomarker for HIE will quickly define the grade of HIE, and it should discriminate newborns with mild HIE, for whom hypothermia therapy is not indicated, from newborns with moderate HIE, who are eligible for this treatment. Levels of MIR210 in neonatal patient blood in combination with MIR374a, S100B protein, and Neuron-specific enolase (NSE) have shown high accuracy in distinguishing HIE patients from healthy newborns, but also between mild, moderate, and severe HIE (54). These biomarkers also showed prognostic value as their levels correlated with neonatal behavioral neurological assessment scores. Increased levels of MiR210 were corroborated in plasma using piglet newborn model of HI (58).
Notably, MIR374a is downregulated in the umbilical cord blood after global HIE in humans (59). The extent of down-regulation of MIR374a corresponded to the severity of the insult and, as such, combining the levels of MIR374a with MIR210 may have prognostic value. Other studies in human neonates have shown a correlation between the downregulation of MIR181b, MIR199a, and MIR376c in the umbilical cord blood after HI, and the severity of the hypoxic insult (60, 61). Combining the levels of MIR181b and its target ubiquitin C-terminal hydrolase-L1 (UCH-L1) has diagnostic value, possibly enabling discrimination between moderate and severe HIE.
A principal component of HI brain damage is the inflammatory response and associated excitotoxicity (105). HI activates astrocytes and microglia that participate in the inflammatory response leading to increased levels of pro-inflammatory cytokines (106, 107). In this context, a second key hypoxamir is MIR21, a miRNA that could modulate inflammation. MIR-21 also controls several other key processes after HI, including cellular proliferation and migration, mitochondrial function, apoptosis, and HIF-1α stabilization and signaling (47, 49). MIR21 has been shown to be increased in cases of severe preterm fetal growth restriction compared to controls, and combined expression profiles of MIR21 and MIR20b in maternal blood were shown to be associated with the grade of fetal hypoxia at birth (55).
For the use of miRNAs as biomarkers, optimization of protocols for extraction and analysis of circulating miRNAs still needs further improvement toward selectivity and specificity (108). Remarkably, a study in human neonates demonstrated that it is feasible to extract sufficient miRNA from a single dried peripheral blood spot (109), with expression patterns that correlate well with those from EDTA-blood. This method eliminates potential sources of error, associated with blood collection and centrifugation. It is relatively cheap, technically easier to obtain, and easier for transportation and storing. Optimization and standardization of protocols for miRNA analysis will help the implementation into a clinical setting.
One consideration when examining miRNAs profiles after HI is that people of discrepant ethnicities might respond to the injury in different ways, therefore study results obtained in one ethnic group might not be appropriate for another cohort (54). Large-scale studies covering diverse ethnicities should be conducted to address this question.
LncRNAs
In addition to ncRNAs, elevated levels of circulating cell-free fetal DNA (cffDNA) and cell-free fetal RNA (cffRNA) have been proposed as an early indicator of damages caused by perinatal hypoxia, that could be used as early biomarkers for preeclampsia or HIE. A study using newborn piglets exposed to hypoxia-reoxygenation revealed tendencies to higher concentrations of cffDNA in the cerebrospinal fluid in comparison to controls (110). This indicates that CffDNA and cffRNA levels in maternal blood, and also cell-free RNA of placental origin, could have potential applications as biomarkers for the screening and diagnosis of preeclampsia (111).
Potential Treatments
HIF-1α
In the brains of both fetuses and posnatal (P) 12 rat pups, fetal hypoxia resulted in global DNA hypomethylation and a continuous increase in HIF-1α mRNA and protein, and increased brain injury in response to hypoxia and ischemia (112). In the early stages of neonatal cerebral ischemia, inhibition of HIF-1α has been shown to be neuroprotective. In P7 HI rats inhibition of HIF-1α by 2-methoxyestradiol (2ME2) immediately after HI protected neuronal cells, attenuated blood-brain-barrier (BBB) disruption, and reduced brain edema (113). In contrast, the stabilization of HIF-1α with dimethyloxalylglycine (DMOG), increased BBB permeability and brain edema.
The c-glycosylated flavonoid, vitexin (5, 7, 4-trihydroxyflavone-8-glucoside) is a natural compound found in many medicinal plants, that has HIF-1α inhibitor activity (114). Intraperitoneal administration of vitexin immediately (5 min) after the HI insult in perinatal rats attenuated the increase in HIF-1α and vascular endothelial growth factor (VEGF), reduced infarct size, improved brain edema, BBB disruption, and neuronal cell death, and improved the neurobehavioral outcomes (115). Pretreatment with vitexin before HI showed the same results (116), diminishing the pro-apoptotic signaling pathway by inhibiting the phosphorylation of Ca2+/Calmodulin-dependent protein kinase II, and increasing the BCL-2/BAX protein ratio 24 h after injury. Animals pretreated with vitexin showed reduced brain infarct volume, brain atrophy, and improved neurobehavioral outcomes. Vitexin has also been proposed as a treatment for HI induced epilepsy (117). Treatment with vitexin suppress brain HI induced electrical activity in neonatal rats, by inhibiting the Na-K-Cl cotransporter (NKCC1), and preventing HI induced BBB leakage, and inflammatory cytokine and neutrophil infiltration. These results support further scientific exploration of vitexin as therapy for perinatal for HIE.
DNA (de)Methylation
Defining the direct vs. indirect effects of hypoxia on DNA methylation in HIE populations is challenging. DNA methylation is time, tissue, and cell-type-specific, which poses a challenge in human studies that usually analyze peripheral blood (118). Studies in animal models have shown a causal effect of gestational and perinatal acute hypoxia on the regulation of gene-specific DNA methylation in mediating the neonatal programming of hypoxic sensitivity and the resulting consequences on the developing fetus and offspring. The first report associating DNA methylation to HI brain damage came from a model of ischemia/reperfusion using adult rats, showing that HI generated a 3- to 4-fold increase in methyl group incorporation in the brain (119). Transgenic animals expressing reduced DNA methyltransferase levels did not show this increase in DNA methylation and were resistant to HI brain injury.
In addition, evidence from studies following stroke indicated that the generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) directly modify cytosine residues chemically, by promoting DNA hydroxymethylation (120, 121). Moreover, peroxides involved in stroke have been shown to induce nucleobase modifications like 5-chlorocytosine, which mimics 5-mC and induces improper Dnmt1 methylation within CpG sequences, resulting in gene silencing (122). These findings associated oxidative stress and epigenetic changes via chemical DNA modifications and altering DNA-protein interactions.
In the perinatal period, the majority of studies involving DNA methylation in HI have investigated the effect of a preconditioning stimulus on methylation and subsequent vulnerability to the HI insults. In ischemic tolerance, exposure to a sublethal ischemic (preconditioning) event protects the brain against a subsequent severe ischemic challenge, producing tolerance.
In rat studies, mild fetal asphyxia during the last week of gestation caused global changes in gene transcription at birth, with down-regulation of most mRNA transcripts in the brain, and upregulation of DNMT1 and DMT3L, various HDACs, the Polycomb group ring finger 2 (PCGF2) and the methyl-CpG-binding protein-2 (MeCP2) (123). Fetal asphyxia preconditioning protected against subsequent severe perinatal global HI, reducing postnatal mortality and behavioral deficits after perinatal HI. Concomitantly, fetal asphyctic preconditioning lowered acute cytokine infiltration and modulated the transcriptional response to perinatal asphyxia. This effect was mediated by epigenetic changes, particularly involving histone deacetylation (124–126).
Other evidence suggest that prenatal insults generally tend to increase vulnerability to neonatal HI. In studies using rat models, HI induced gene-specific DNA hypermethylation. In the developing rat fetus, fetal hypoxia (GD 15 to 21) increased methylation of the glucocorticoid receptor (GR) gene (Nr3c1) promoter and repression of the GR in the brain, leading to an increased brain vulnerability to hypoxic-ischemic injury (127). In this way, methylation controls the expression patterns of GR, being key in the stress-mediated programming of GR expression (127, 128).
As mentioned, HIF-1α and DNA hypomethylation participate in fetal stress-mediated programming of HI sensitive phenotypes (112). Maternal hypoxia in rats, during the last week of gestation, reduced global methylation levels in the fetal brain and affected methylation of the HIF-1α gene. More specifically, hypomethylation induced by either maternal hypoxia, or pharmacological treatment with the DNMT inhibitor 5-aza-dC increased the vulnerability of the fetus to subsequent neonatal HI and worsened neurobehavioral outcomes in the rat pups. Interestingly, inhibiting HIF-1α with 2ME could counteract some of the damaging effects of hypomethylation.
Prenatal nicotine exposure has shown to increase brain infarct size after subsequent neonatal HI in male rats (129, 130). This increased susceptibility was linked to a down-regulation of angiotensin II receptor expression in the brain and hypermethylation of the angiotensin II type 2-receptor (At2r) promoter.
miRNAs
miRNAs and its regulated genes have also been proposed as therapeutic targets. Induction of miRNAs inhibiting pro-apoptotic pathways and inhibition of miRNAs implicated in HI-induced inflammation and apoptosis has shown therapeutic potential in animal models. Further characterization of these findings will help them advance into clinical trials.
MiR139-5p is down-regulated in P10 rat brains after HI treatment, and in cultured neurons exposed to OGD (65). The expression of MiR139-5p correlates inversely with the expression of one of its targets, the pro-apoptotic protein human growth transformation dependent protein (HGTD-P) (131). Interestingly, administration of MiR139-5p agomir attenuates HI brain damage, which is concurrent with the downregulation of HGTD-P expression. This effect was shown even at 12 h after the insult, indicating that targeting epigenetic pathways could extend the therapeutic window in HI brain damage.
The selective α2-adrenoreceptor agonist dexmedetomidine has been shown to provide neuroprotection in HI by inhibiting apoptosis, oxidative activity, Notch/NF-κB activation (132), and inflammation (133). MiR129-5p targets the type III procollagen gene (COL3A1) and has shown therapeutic potential in P7 HI rats mimicking and enhancing the neuroprotective effect of dexmedetomidine (66).
Hypoxia-induced brain injury appears to downregulate the expression of the MiR23-27 cluster leading to increased apoptosis. Overexpression of MiR23a/b and MiR27a/b was shown to exert neuroprotective effects in the late stage of mouse gestation (GD20) after global maternal hypoxia, by reducing apoptotic pathways including the expression of Apoptotic protease factor-1 (Apaf-1) (74). Indirectly increasing MiR23a expression by reducing the expression of the lncRNA Growth arrest-specific 5 (GAS5), that binds to MiR23a, reduced infarct size after HI in P10 rats (67).
One pathological mechanism of HI damage is the induction of endoplasmic reticulum stress, which activates the unfolded protein response (UPR). The UPR induces activation of stress sensor signaling pathways, like the RNase inositol requiring enzyme-1 alpha (IRE1α) pathway, leading to inflammation and neuronal cell death. In this context, MiR17-5p, a substrate of IRE1α, is a target that has shown therapeutic potential (68). Animal studies have shown that MiR17-5p mimic before HI, prevented inflammasome activation reducing brain infarct volume. Moreover, intranasal administration of the IRE1α inhibitor STF-083010 1h post-HI attenuated MiR17-5p downregulation and brain injury, and improved neurological behavior outcomes. It has also been reported that activation of the nuclear receptor peroxisome proliferator-activated receptor beta/delta (PPAR-β/δ) in P10 HI rat, by intranasal delivery of the agonist GW0742, could induce MiR17-5p levels (69), diminishing apoptosis, brain infarct area, brain atrophy, and improving neurological function post HI.
In vitro findings showed that primary neonatal rat microglial cells exposed to hypoxia experience downregulation of MiR21 expression and upregulation of one of its targets, the apoptosis-inducing factor Fas ligand (FasL) (77). Overexpression of FasL post hypoxic microglial activation increased neuronal apoptosis, which can be partially reversed by ectopic expression of MiR21. MiR592-5p targets inflammation by targeting the prostaglandin D2 receptor. HI in P7 rats reduces the expression of MiR592-5p and increases prostaglandin D2 expression in the hippocampus, which is detrimental (75), induction of this miRNA could also be beneficial in HI.
MiR124 is the most abundant brain-specific miRNA and plays key roles in neuronal development. Multiple studies have shown a neuroprotective effect of MiR124 in adult HI models (134–137), which has not been validated for the perinatal period. Animal studies showed that MiR124 was down-regulated in the hippocampus after global neonatal hypoxia in rats (72). MiR124 warrants further investigation in neonatal HI models as it may have therapeutic value.
A potential strategy to use miRNAs as therapeutic targets is comprised of inhibiting miRNAs by using antagomirs, or naturally occurring compounds with similar effects such as curcumin or resveratrol. MiR210 expression steadily increases over the first 24 h after HI in P10 rats, disrupting glucocorticoid receptor-mediated neuroprotection and increasing leakiness of the BBB (62–64). Inhibition of MiR210 using an intracerebroventricular injection of complementary locked nucleic acid oligonucleotides increased BDNF signaling, reduced neuronal death and infarct size, and improved functional recovery of the animals. In vitro studies also support these findings (138). Although this suggests that MiR210 inhibitors could be used as a potential therapy in HI brain injury, further characterization of its spatiotemporal expression, regulation, targets, and physiological and pathogenic effects in HIE is still required.
NF-κB triggers MiR155, inducing pro-inflammatory effects compromising the BBB integrity. Inhibition of MiR155 is neuroprotective in adult HI models by reducing brain cytokines (139), but its role in neonatal models is underexplored to date. The neuroprotective actions of resveratrol and curcumin in neonatal HI (140–142) may in part be due to the downregulation of MiR155 and MiR21 by resveratrol, and the downregulation of MiR21 by curcumin, combined with epigenetic and anti-oxidant mechanisms (143, 144). MiR153 is reported to be a neuron-related miRNA (50). Inhibition of MiR153 protects neurons against OGD/R-induced injury by increasing the expression of Nuclear factor erythroid 2-related factor 2 (Nrf2) and heme oxygenase-1 (HO-1) signaling.
Sleep problems associated with circadian rhythm disruptions are common in children after mild-moderate HIE (145), and disruptions in miRNAs likely contribute to these abnormalities. HI in P7 rats leads to an increased expression of MiR325-3p in the pineal gland (73). This disrupts melatonin signaling by targeting Aralkylamine N-acetyltransferase (AANAT), a key protein that controls melatonin synthesis, and this leads to circadian rhythm disturbances. Reducing MiR325-3p has been shown to prevent this impairment after OGD in vitro.
Long-Non Coding and Circular RNAs
Studies in human neonates diagnosed with HIE and animal models have shown that lncRNAs are aberrantly expressed under hypoxic conditions and might be implicated in regulating the expression of protein-coding genes involved in pathological processes associated to HI brain damage. A microarray study in human neonates showed that neonatal HI dramatically changed the expression patterns of numerous ncRNAs in peripheral whole blood (146), including 376 lncRNAs and 126 mRNAs involved in the immune system and nervous system. A study on the P7 HI rat cortex identified 7,157 differentially expressed mRNA transcripts, and 328 differentially expressed lncRNAs targeting mRNAs involved in inflammatory and immune responses, wounding, and neurological system processes (147). Specifically, JAK-STAT, NF-κB, and TLR signaling pathways were altered. Targeting genes in these pathways is proposed as a therapeutic strategy.
LncRNAs have shown potential therapeutic applications in animal models. Various studies have pointed to the inhibition of HI-induced lncRNAs as a therapeutic approach to induce neuroprotection. A second microarray profiling study in P10 rats exposed to HI pointed in the same direction, finding expression differences in 322 lncRNAs and 375 coding genes in hippocampal and cortex tissue 24 h after the insult (71). Upregulated protein-coding genes were also involved in inflammation and wounding, while repair and neurogenesis pathways were downregulated. BC088414, which plays a role in apoptosis by regulating caspase 6 (Casp6) and in adrenergic signaling by regulating the beta-2 adrenergic receptor (Adrb2), was the most significantly upregulated lncRNA. Inhibition of BC088414 with a siRNA in PC12 cells exposed to OGD resulted in the downregulation of Adrb2 and Casp6, increased cell proliferation, and decreased apoptosis.
The lncRNA maternally expressed gene 3 (Meg3) induces cell death in ischemia by binding to the p53 DNA binding domain (148). A study in P7 HI mice hippocampus showed that the Meg3 sponges MiR129-5p, a miRNA with neuroprotective activity, and abolishes the effect of dexmedetomidine therapy (76). The silencing of Meg3 and upregulation of MiR129-5p enhanced the therapeutic effect of dexmedetomidine. Finally, the lncRNA Growth arrest-specific 5 (Gas5) was also shown upregulated in HI models (67). Gas5 sponges MiR23a thereby preventing its neuroprotective action. Inhibition of Gas5 by intracerebroventricular delivery of Gas5 small hairpin RNA has been shown to reduce brain infarct size and diminish functional sequelae in rats, suggesting its use as therapy for the treatment of HI brain injury.
CircRNAs are a subtype of lncRNAs that form a closed loop. CircRNAs play crucial roles as miRNA sponges, and recent studies highlighted the role of these molecules in hypoxic regulation. An exploratory study in P3 HI rats found 98 dysregulated circRNAs in the brain (70). One of the top hits, termed chr6:48820833|48857932, targets HIF-1α signaling by sponging the HIF-1α-targeting-miRNAs MiR433-3p and MiR206-3p. Another study in P10 HI rats discovered a total of 66 circRNAs differentially expressed in HI brain damage rats compared to controls (149). Numerous mRNAs transcribed from the host genes of altered circRNAs were associated with brain damage and neural regeneration processes. These results indicate a novel focus for future studies investigating the molecular mechanism underlying HIE and potentials new biomarkers and treatments through modulating circRNAs.
Histone Methylation
Enhancer of zeste homolog 2 (EZH2), a catalytic subunit of Polycomb repressive complex 2 (PRC2), plays an important role in mammalian CNS development (150). EZH2 participates in hippocampal learning, memory, and neurogenesis through trimethylation at H3K27 (H3K27me3), which silences downstream genes, including, among others, BDNF and PTEN (151). In HIE, autophagy is increased in the hippocampus, and inhibition of autophagy provides neuroprotection. In this regard, volatile anesthetics, i.e., sevoflurane and isoflurane, have shown to be neuroprotective against HI brain damage in neonatal rats (152, 153) (Table 2). Diverse studies have shown sevoflurane inhibits excessive hippocampal autophagy increasing the expression of EZH2, H3K27me3, and decreased expression of PTEN induced by HI, improving the behavioral outcome (154).
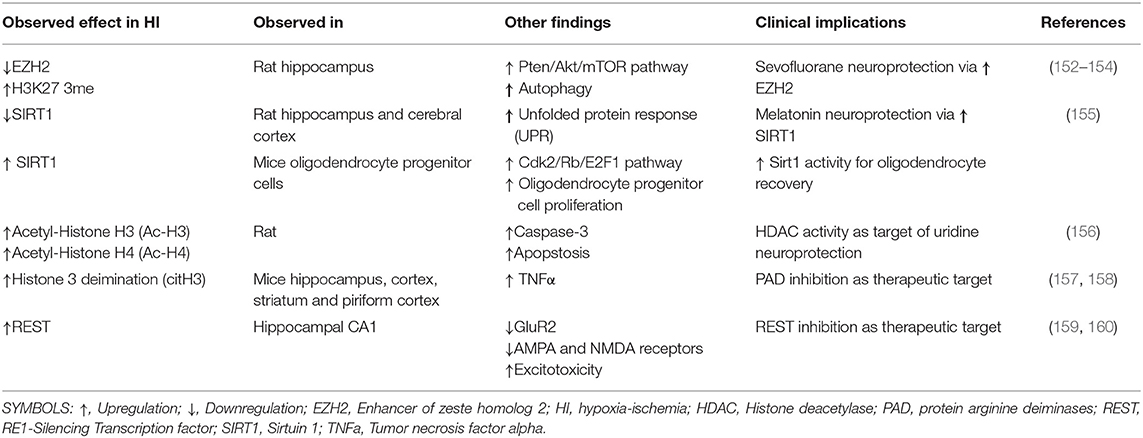
Table 2. Clinical implications of histone modifications in perinatal hypoxia ischemia studies.
Histone Acetylation
Many studies have shown the implications of HATs in perinatal HI damage. Fetal asphyxia in E17 rats leads to an upregulation of class I HDACs HDAC1, HDAC2, HDAC3, and the HAT MYST3 (123). The same stimulus upregulated HDAC1 and class IIb HDAC10 and HDAC11, after severe perinatal asphyxia (126). The HDAC SIRT1 plays an important role in the regulation of oligodendrocyte progenitor cell proliferation and oligodendrocyte regeneration after neonatal brain injury. Neonatal hypoxia in P3 mice has been shown to enhance SIRT1 and SIRT1/Cdk2 complex formation through HIF1α activation, leading to an enhanced oligodendrocyte progenitor cell proliferation (161). Enhancing SIRT1 activity may promote oligodendrocyte recovery after diffuse white matter injury. SIRT1 expression was significantly reduced after neonatal HI in P7 rat pups (155) in cells that activate the unfolded protein response (UPR). In that same study, melatonin neuroprotection involved the prevention of SIRT1 downregulation and UPR activation.
From a therapeutic perspective, HDAC inhibitors (HDACi's) represent the most widely studied epigenetic drugs as several existing marketed drugs and natural dietary metabolites inhibit HDACs. Regarding HI, HDAC inhibition reduces HIF-1α signaling, which is generally protective against HI (96, 112). Numerous HDAC inhibitors have shown beneficial effects in either focal or global neonatal HI. For example, the marketed anticonvulsant sodium valproate inhibits class I HDAC activity and has shown potential in treating neonatal HI in rats (162). Several naturally occurring bioactive molecules, such as trichostatin A (163), sodium butyrate (164), curcumin (142), quercetin (165), resveratrol (72, 140, 141), and uridine (156) have also proven protective effects in focal neonatal HI, the effects of which were shown to be at least partially dependent on HDAC inhibition. The mechanism(s) by which HDACi's provide neuroprotection include prevention of oxidation, suppression of inflammation, and reduction of apoptosis. Importantly, histone acetylation/methylation precedes DNMT or TET methylcytosine dioxygenase binding and promoter methylation/demethylation, and these mechanisms function cooperatively (166). Still, many potential problems remain to be addressed before clinical use of selective HDACi's for the treatment of HIE.
Other Histone Modifications and Epigenetic Complexes
Protein deimination (citrullination) is a post-translational modification that converts the amino acid arginine into citrulline, is caused by Ca+2-regulated peptidylarginine deiminases (PADs), and can act on histone tails.
H3 citrullination (citH3) by PAD4 is associated with gene regulation and the formation of neutrophil extracellular traps in response to infection. PAD activity is induced under HI with or without lipopolysaccharide stimulation. Selective and targeted pharmacological PAD inhibition following HI can be a therapeutic target to enhance neuroprotection (157). Hypothermia following the insult inhibits this pathway and affect the Ca2+-regulated PAD activation (158).
A large degree of epigenetic variation is controlled by epigenetic regulatory complexes. Several epigenetic regulatory complexes may play a role in neonatal HI injuries. The repressor element-1 (RE1) silencing transcription factor (REST) is the main regulator of neurogenesis and neuronal fate. Neuronal genes controlled by REST contain an RE1 motif. As such, REST assembles to this site with its co-repressor CoREST and recruits a number of epigenetic regulators including HDACs and histone methyltransferases to the promoters of target genes to achieve epigenetic changes (167, 168). It has been demonstrated that REST is a HI-responsive gene, regulating around 20% of the hypoxia-repressed genes. As an example, HI has been shown to upregulate REST expression in the hippocampal CA1, suppressing GluR2 gene expression (159). Interestingly, the downregulation of REST expression with antisense oligodeoxynucleotides has been demonstrated to be neuroprotective 72 h post OGD. REST was also shown to orchestrate epigenetic changes and silencing of miR-132 in insulted CA1 neurons (169) and has been shown to bind directly to the HIF-1α promoter in order to repress HIF-1α transcription after prolonged hypoxia (159, 170, 171). Moreover, REST has been demonstrated to repress the transcription of AMPA and NMDA glutamate receptor subunits (168), which are involved in mediating excitotoxicity after HIE (18). Recently, it has been shown that adult rats overexpress REST after CAO (160), and knocking down REST with an intracerebral siRNA injection increased the expression of its target genes, attenuated apoptosis, and infarct volume, and improved post-ischemic functional recovery. Altogether, although the actual therapeutic potential of REST in the perinatal period has not yet been explored, these data suggest REST reflects a promising new therapeutic target to treat acute hypoxic brain damage.
Discussion
There is an urgent need for new therapies for neonatal HI, as currently, therapeutic hypothermia for term infants is not fully protective against HIE, and the disability burden after neonatal HI remains high. Evidently, epigenetic mechanisms play a key role in the pathological cascade after neonatal HI. The cellular hypoxia response, mediated by HIF-1α and other hypoxia-inducible factors, is controlled by an intricate multi-leveled epigenetic network centered on HIF-1a. A better understanding of this intricate regulatory circuit will provide us with new diagnostic tools and therapeutic approaches for HIE. As an example, HIF-1α signaling controls the expression of various miRNAs, whileHIF-1α by itself is intricately controlled by different miRNAs. Apart from HIF-1α signaling, numerous miRNAs regulate a myriad of other processes relevant for HI, ranging from well-studied processes like inflammation and apoptosis to circadian rhythm disturbances after HIE. Some hypoxia-regulated miRNAs in the maternal blood are promising in terms of identifying pregnancies at risk of fetal hypoxia, permitting early intervention, whereas preclinical interventions using miRNAs are promising in combination with therapeutic hypothermia. Together with miRNAs, HDACs hold promise as biomarkers and therapeutic targets. HDAC inhibitors represent the most advanced agents in this respect as several existing marketed drugs and natural dietary metabolites have been shown to directly inhibit HDACs.
Still, no single epigenetic mark has demonstrated enough reliability and reproducibility to be used as a biomarker or as a therapeutic target in a clinical setting. For this purpose, replication and/or validation studies in larger cohorts are needed. In conclusion, even though numerous advancements have been made in understanding the pathophysiology of perinatal HI brain damage, still our knowledge on the role of epigenetics in HI is very limited. As such, new discoveries on epigenetics may mark the beginning of an etiopathogenic research revolution in neurodevelopmental disorders, and continued exploration of this area is of great promise.
Author Contributions
MBu, MBa, MAB, AG, CL, and DH conceived the presented idea. MBu and MBa wrote the first draft. DH and HS edited the draft. AG, MAB, and CL provided funding for the manuscript. All authors read the final version of the manuscript.
Funding
This research was partially supported by the Sistema de Investigación y Desarrollo (SINDE) and the Vicerrectorado de Investigación y Posgrado of the Universidad Católica de Santiago de Guayaquil, Guayaquil, Ecuador. MBu was funded by Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) of Argentina and the Foundation of Pediatrics, Maastricht University Medical Center+ (MUMC+). CL was supported by Universidad de Buenos Aires (UBACyT - 20020160100150BA). All views expressed in this article are those of the authors and do not represent the views of the funding agencies.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
BBB, blood-brain barrier; DNMTs, DNA methyltransferases; E, embryonic day; HDACs, histone deacetylases; HI, hypoxia-ischemia; HIE, hypoxic-ischemic encephalopathy; HIF, hypoxia inducible factor; HIF-1α, Hypoxia-inducible factor 1α; HRE, hypoxia response element; JmjC, Jumonji-C histone demethylase; KDMs, histone lysine demethylases; P, postnatal day; REST, repressor element-1 (RE1) silencing transcription factor; TETs, ten-eleven translocation enzymes; 5-aza-dC, 5-aza-2′-deoxycytidine.
References
1. Choudhuri S. From Waddington's epigenetic landscape to small noncoding RNA: some important milestones in the history of epigenetics research. Toxicol Mech Methods. (2011) 21:252–74. doi: 10.3109/15376516.2011.559695
2. Bolton JL, Molet J, Ivy A, Baram TZ. New insights into early-life stress and behavioral outcomes. Curr Opin Behav Sci. (2017) 14:133–9. doi: 10.1016/j.cobeha.2016.12.012
3. Lahiri DK, Maloney B, Zawia NH. The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases. Mol Psychiatry. (2009) 14:992. doi: 10.1038/mp.2009.82
4. Nelson KB, Leviton A. How much of neonatal encephalopathy is due to birth asphyxia? Am J Dis Child. (1991) 145:1325–31. doi: 10.1001/archpedi.1991.02160110117034
5. Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev. (2010) 86:329–38. doi: 10.1016/j.earlhumdev.2010.05.010
6. Frøen JF, Gardosi JO, Thurmann A, Francis A, Stray-Pedersen B. Restricted fetal growth in sudden intrauterine unexplained death. Acta Obstet Gynecol Scand. (2004) 83:801–7. doi: 10.1111/j.0001-6349.2004.00602.x
7. Maršál K. Obstetric management of intrauterine growth restriction. Best Pract Res Clin Obstetr Gynaecol. (2009) 23:857–70. doi: 10.1016/j.bpobgyn.2009.08.011
8. Graham EM, Ruis KA, Hartman AL, Northington FJ, Fox HE. A systematic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. Am J Obstet Gynecol. (2008) 199:587–95. doi: 10.1016/j.ajog.2008.06.094
9. Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, et al. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet. (2010) 375:1969–87. doi: 10.1016/S0140-6736(10)60549-1
10. Wachtel EV, Hendricks-Muñoz KD. Current management of the infant who presents with neonatal encephalopathy. Curr Probl Pediatr Adolesc Health Care. (2011) 41:132–53. doi: 10.1016/j.cppeds.2010.12.002
11. Lemyre B, Chau V. Hypothermia for newborns with hypoxic-ischemic encephalopathy. Paediatr Child Health. (2018) 23:285–91. doi: 10.1093/pch/pxy028
12. Natarajan G, Pappas A, Shankaran S. Outcomes in childhood following therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy (HIE). Semin Perinatol. (2016) 40:549–55. doi: 10.1053/j.semperi.2016.09.007
13. Rivero-Arias O, Eddama O, Azzopardi D, Edwards AD, Strohm B, Campbell H. Hypothermia for perinatal asphyxia: trial-based resource use and costs at 6–7 years. Arch Dis Childh Fetal Neonatal Ed. (2019) 104:F285–92. doi: 10.1136/archdischild-2017-314685
14. Sarkar S, Barks JD. Systemic complications and hypothermia. Semin Fetal Neonatal Med. (2010) 15:270–5. doi: 10.1016/j.siny.2010.02.001
15. Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol. (2011) 69:743–58. doi: 10.1002/ana.22419
16. Robertson NJ, Cowan FM, Cox IJ, Edwards AD. Brain alkaline intracellular pH after neonatal encephalopathy. Ann Neurol. (2002) 52:732–42. doi: 10.1002/ana.10365
17. Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. (2013) 106:1–16. doi: 10.1016/j.pneurobio.2013.04.001
18. Barkhuizen M, Van den Hove D, Vles J, Steinbusch H, Kramer B, Gavilanes A. 25 years of research on global asphyxia in the immature rat brain. Neurosci Biobehav Rev. (2017) 75:166–82. doi: 10.1016/j.neubiorev.2017.01.042
19. Vazquez-Borsetti P, Pena E, Rojo Y, Acuna A, Loidl FC. Deep hypothermia reverses behavioral and histological alterations in a rat model of perinatal asphyxia. J Comp Neurol. (2019) 527:362–71. doi: 10.1002/cne.24539
20. Takada S, Sampaio C, Allemandi W, Ito P, Takase L, Nogueira M. A modified rat model of neonatal anoxia: development and evaluation by pulseoximetry, arterial gasometry and Fos immunoreactivity. J Neurosci Methods. (2011) 198:62–9. doi: 10.1016/j.jneumeth.2011.03.009
21. Rice JE, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. (1981) 9:131–41. doi: 10.1002/ana.410090206
22. Vannucci RC, Vannucci SJ. A model of perinatal hypoxic-ischemic brain damage A. Ann N Y Acad Sci. (1997) 835:234–49. doi: 10.1111/j.1749-6632.1997.tb48634.x
23. Edwards AB, Feindel KW, Cross JL, Anderton RS, Clark VW, Knuckey NW, et al. Modification to the Rice-Vannucci perinatal hypoxic-ischaemic encephalopathy model in the P7 rat improves the reliability of cerebral infarct development after 48 hours. J Neurosci Methods. (2017) 288:62–71. doi: 10.1016/j.jneumeth.2017.06.016
24. Loetscher PD, Rossaint J, Rossaint R, Weis J, Fries M, Fahlenkamp A, et al. Argon: neuroprotection in in vitro models of cerebral ischemia and traumatic brain injury. Crit Care. (2009) 13:R206. doi: 10.1186/cc8214
25. Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. (2010) 28:1069–78. doi: 10.1038/nbt.1678
26. Biswas S, Rao CM. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol. (2018) 837:8–24. doi: 10.1016/j.ejphar.2018.08.021
27. Watson JA, Watson CJ, McCann A, Baugh J. Epigenetics: the epicenter of the hypoxic response. Epigenetics. (2010) 5:293–6. doi: 10.4161/epi.5.4.11684
28. Tsai YP, Wu KJ. Epigenetic regulation of hypoxia-responsive gene expression: focusing on chromatin and DNA modifications. Int J Cancer. (2014) 134:249–56. doi: 10.1002/ijc.28190
29. Ke Q, Costa M. Hypoxia-inducible factor-1. (HIF-1). Mol Pharmacol. (2006) 70:1469–80. doi: 10.1124/mol.106.027029
30. Nguyen MP, Lee S, Lee YM. Epigenetic regulation of hypoxia inducible factor in diseases and therapeutics. Arch Pharm Res. (2013) 36:252–63. doi: 10.1007/s12272-013-0058-x
31. Ma Q, Xiong F, Zhang L. Gestational hypoxia and epigenetic programming of brain development disorders. Drug Discov Today. (2014) 19:1883–96. doi: 10.1016/j.drudis.2014.09.010
32. Hancock RL, Dunne K, Walport LJ, Flashman E, Kawamura AJE. Epigenetic regulation by histone demethylases in hypoxia. Epigenomics. (2015) 7:791–811. doi: 10.2217/epi.15.24
33. Koslowski M, Luxemburger U, Türeci Ö, Sahin U. Tumor-associated CpG demethylation augments hypoxia-induced effects by positive autoregulation of HIF-1α. Oncogene. (2011) 30:876. doi: 10.1038/onc.2010.481
34. Razin A, Riggs AD. DNA methylation and gene function. Science. (1980) 210:604–10. doi: 10.1126/science.6254144
35. van der Wijst MG, Venkiteswaran M, Chen H, Xu G-L, Plösch T, Rots MG. Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics. (2015) 10:671–6. doi: 10.1080/15592294.2015.1062204
36. Monteggia LM, Kavalali ET. Rett syndrome and the impact of MeCP2 associated transcriptional mechanisms on neurotransmission. Biol Psychiatry. (2009) 65:204–10. doi: 10.1016/j.biopsych.2008.10.036
37. Kinde B, Gabel HW, Gilbert CS, Griffith EC, Greenberg ME. Reading the unique DNA methylation landscape of the brain: non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci USA. (2015) 112:6800–6. doi: 10.1073/pnas.1411269112
38. Ye D, Xiong Y. Cancer: suffocation of gene expression. Nature. (2016) 537:42. doi: 10.1038/nature19426
39. Fu X-D. Non-coding RNA: a new frontier in regulatory biology. Nat Sci Rev. (2014) 1:190–204. doi: 10.1093/nsr/nwu008
40. Qu Z, Adelson DL. Evolutionary conservation and functional roles of ncRNA. Front Genet. (2012) 3:205–205. doi: 10.3389/fgene.2012.00205
41. Coolen M, Bally-Cuif L. Chapter 18 - MicroRNAs in brain development. In: Sen CK, editor. MicroRNA in Regenerative Medicine. Oxford: Academic Press. (2015). p. 447–88.
42. Cui H, Yang L. Analysis of microRNA expression detected by microarray of the cerebral cortex after hypoxic-ischemic brain injury. J Craniofac Surg. (2013) 24:2147–52. doi: 10.1097/SCS.0b013e3182a243f3
43. Ponnusamy V, Yip PK. The role of microRNAs in newborn brain development and hypoxic ischaemic encephalopathy. Neuropharmacology. (2019) 148:55–65. doi: 10.1016/j.neuropharm.2018.11.041
44. Taguchi A, Yanagisawa K, Tanaka M, Cao K, Matsuyama Y, Goto H, et al. Identification of hypoxia-inducible factor-1α as a novel target for miR-17-92 microRNA cluster. Cancer Res. (2008) 68:5540–5. doi: 10.1158/0008-5472.CAN-07-6460
45. Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1α and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Cicul Res. (2009) 104:879–86. doi: 10.1161/CIRCRESAHA.108.193102
46. Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J Clin Invest. (2010) 120:4141–54. doi: 10.1172/JCI42980
47. Nallamshetty S, Chan SY, Loscalzo J. Hypoxia: a master regulator of microRNA biogenesis and activity. Free Radic Biol Med. (2013) 64:20–30. doi: 10.1016/j.freeradbiomed.2013.05.022
48. Bartoszewska S, Kochan K, Piotrowski A, Kamysz W, Ochocka RJ, Collawn JF, et al. The hypoxia-inducible miR-429 regulates hypoxia-inducible factor-1 α expression in human endothelial cells through a negative feedback loop. FASEB J. (2014) 29:1467–79. doi: 10.1096/fj.14-267054
49. Xu X, Kriegel AJ, Jiao X, Liu H, Bai X, Olson J, et al. miR-21 in ischemia/reperfusion injury: a double-edged sword? Physiol Genomics. (2014) 46:789–97. doi: 10.1152/physiolgenomics.00020.2014
50. Ji Q, Gao J, Zheng Y, Liu X, Zhou Q, Shi C, et al. Inhibition of microRNA-153 protects neurons against ischemia/reperfusion injury in an oxygen–glucose deprivation and reoxygenation cellular model by regulating Nrf2/HO-1 signaling. J Biochem Mol Toxicol. (2017) 31:e21905. doi: 10.1002/jbt.21905
51. Crosby ME, Kulshreshtha R, Ivan M, Glazer PM. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. (2009) 69:1221–9. doi: 10.1158/0008-5472.CAN-08-2516
52. Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, et al. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell. (2009) 35:856–67. doi: 10.1016/j.molcel.2009.09.006
53. Agrawal R, Pandey P, Jha P, Dwivedi V, Sarkar C, Kulshreshtha R. Hypoxic signature of microRNAs in glioblastoma: insights from small RNA deep sequencing. BMC Genomics. (2014) 15:686. doi: 10.1186/1471-2164-15-686
54. Wang Z, Liu Y, Shao M, Wang D, Zhang YJB. Combined prediction of miR-210 and miR-374a for severity and prognosis of hypoxic–ischemic encephalopathy. Brain Behav. (2018) 8:e00835. doi: 10.1002/brb3.835
55. Whitehead CL, Teh WT, Walker SP, Leung C, Larmour L, Tong SJ. Circulating microRNAs in maternal blood as potential biomarkers for fetal hypoxia in-utero. PLoS ONE. (2013) 8:e78487. doi: 10.1371/journal.pone.0078487
56. Awamleh Z, Gloor GB, Han VKM. Placental microRNAs in pregnancies with early onset intrauterine growth restriction and preeclampsia: potential impact on gene expression and pathophysiology. BMC Med Genomics. (2019) 12:91. doi: 10.1186/s12920-019-0548-x
57. Ishibashi O, Ohkuchi A, Ali MM, Kurashina R, Luo S-S, Ishikawa T, et al. Hydroxysteroid. (17-β) dehydrogenase 1 is dysregulated by miR-210 and miR-518c that are aberrantly expressed in preeclamptic placentas: a novel marker for predicting preeclampsia. Hypertension. (2012) 59:265–73. doi: 10.1161/HYPERTENSIONAHA.111.180232
58. Garberg HT, Huun MU, Baumbusch LO, Åsegg-Atneosen M, Solberg R, Saugstad OD. Temporal profile of circulating microRNAs after global hypoxia-ischemia in newborn piglets. Neonatology. (2017) 111:133–9. doi: 10.1159/000449032
59. Looney AM, Walsh BH, Moloney G, Grenham S, Fagan A, O'Keeffe GW, et al. Downregulation of umbilical cord blood levels of miR-374a in neonatal hypoxic ischemic encephalopathy. J Pediatr. (2015) 167:269–73.e262. doi: 10.1016/j.jpeds.2015.04.060
60. Looney A, O'Sullivan M, Ahearne C, Finder M, Felderhoff-Mueser U, Boylan G, et al. Altered expression of umbilical cord blood levels of miR-181b and its downstream target mUCH-L1 in infants with moderate and severe neonatal hypoxic-ischaemic encephalopathy. Mol Neurobiol. 56:3657–63. doi: 10.1007/s12035-018-1321-4
61. O'Sullivan MP, Looney AM, Moloney GM, Finder M, Hallberg B, Clarke G, et al. Validation of altered umbilical cord blood microRNA expression in neonatal hypoxic-ischemic encephalopathy. JAMA Neurol. (2018) 76:333–41. doi: 10.1001/jamaneurol.2018.4182
62. Ma Q, Dasgupta C, Li Y, Bajwa NM, Xiong F, Harding B, et al. Inhibition of microRNA-210 provides neuroprotection in hypoxic–ischemic brain injury in neonatal rats. Neurobiol Dis. (2016) 89:202–12. doi: 10.1016/j.nbd.2016.02.011
63. Ma Q, Dasgupta C, Li Y, Huang L, Zhang L. MicroRNA-210 suppresses junction proteins and disrupts blood-brain barrier integrity in neonatal rat hypoxic-ischemic brain injury. Int J Mol Sci. (2017) 18:1356. doi: 10.3390/ijms18071356
64. Wang L, Ke J, Li Y, Ma Q, Dasgupta C, Huang X, et al. Inhibition of miRNA-210 reverses nicotine-induced brain hypoxic-ischemic injury in neonatal rats. Int J Biol Sci. (2017) 13:76. doi: 10.7150/ijbs.17278
65. Qu Y, Wu J, Chen D, Zhao F, Liu J, Yang C, et al. MiR-139-5p inhibits HGTD-P and regulates neuronal apoptosis induced by hypoxia–ischemia in neonatal rats. Neurobiol Dis. (2014) 63:184–93. doi: 10.1016/j.nbd.2013.11.023
66. Zhou XM, Liu J, Wang Y, Zhang SL, Zhao X, Zhang MH. microRNA-129-5p involved in the neuroprotective effect of dexmedetomidine on hypoxic-ischemic brain injury by targeting COL3A1 through the Wnt/β-catenin signaling pathway in neonatal rats. J Cell Biochem. (2018) 120:6908–19. doi: 10.1002/jcb.26704
67. Zhao R-B, Zhu L-H, Shu J-P, Qiao L-X, Xia Z-K. GAS5 silencing protects against hypoxia/ischemia-induced neonatal brain injury. Biochem Biophys Res Commun. (2018) 497:285–91. doi: 10.1016/j.bbrc.2018.02.070
68. Chen D, Dixon BJ, Doycheva DM, Li B, Zhang Y, Hu Q, et al. IRE1alpha inhibition decreased TXNIP/NLRP3 inflammasome activation through miR-17-5p after neonatal hypoxic-ischemic brain injury in rats. J Neuroinflamm. (2018) 15:32. doi: 10.1186/s12974-018-1077-9
69. Gamdzyk M, Doycheva DM, Malaguit J, Enkhjargal B, Tang J, Zhang JH. Role of PPAR-beta/delta/miR-17/TXNIP pathway in neuronal apoptosis after neonatal hypoxic-ischemic injury in rats. Neuropharmacology. (2018) 140:150–61. doi: 10.1016/j.neuropharm.2018.08.003
70. Zhu L, Zhao R, Huang L, Mo S, Yu Z, Jiang L, et al. Circular RNA expression in the brain of a neonatal rat model of periventricular white matter damage. J Cell Biochem. (2018) 49:2264–76. doi: 10.1159/000493829
71. Zhao F, Qu Y, Liu J, Liu H, Zhang L, Feng Y, et al. Microarray profiling and co-expression network analysis of LncRNAs and mRNAs in neonatal rats following hypoxic-ischemic brain damage. Sci Rep. (2015) 5:13850. doi: 10.1038/srep13850
72. Isac S, Panaitescu AM, Spataru A, Iesanu M, Totan A, Udriste A, et al. Trans-resveratrol enriched maternal diet protects the immature hippocampus from perinatal asphyxia in rats. Neurosci Lett. (2017) 653:308–13. doi: 10.1016/j.neulet.2017.06.003
73. Yang Y, Sun B, Huang J, Xu L, Pan J, Fang C, et al. Up-regulation of miR-325-3p suppresses pineal aralkylamine N-acetyltransferase. (Aanat) after neonatal hypoxia–ischemia brain injury in rats. Brain Res. (2017) 1668:28–35. doi: 10.1016/j.brainres.2017.05.001
74. Chen Q, Xu J, Li L, Li H, Mao S, Zhang F, et al. MicroRNA-23a/b and microRNA-27a/b suppress Apaf-1 protein and alleviate hypoxia-induced neuronal apoptosis. Cell Death Dis. (2014) 5:e1132. doi: 10.1038/cddis.2014.92
75. Sun L-Q, Guo G-L, Zhang S, Yang L-L. Effects of microRNA-592-5p on hippocampal neuron injury following hypoxic-ischemic brain damage in neonatal mice-involvement of PGD2/DP and PTGDR. Cell Physiol Biochem. (2018) 45:458–73. doi: 10.1159/000486923
76. Zhou XM, Liu J, Wang Y, Zhang MH. Silencing of long noncoding RNA MEG3 enhances cerebral protection of dexmedetomidine against hypoxic-ischemic brain damage in neonatal mice by binding to miR-129-5p. J Cell Biochem. (2018) 120:7978–88. doi: 10.1002/jcb.28075
77. Zhang L, Dong LY, Li YJ, Hong Z, Wei WS. miR-21 represses FasL in microglia and protects against microglia-mediated neuronal cell death following hypoxia/ischemia. Glia. (2012) 60:1888–95. doi: 10.1002/glia.22404
78. Bao M-H, Szeto V, Yang BB, Zhu S-Z, Sun H-S, Feng Z-P. Long non-coding RNAs in ischemic stroke. Cell Death Dis. (2018) 9:281. doi: 10.1038/s41419-018-0282-x
79. Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. (2016) 73:2491–509. doi: 10.1007/s00018-016-2174-5
80. Cuevas-Diaz Duran R, Wei H, Kim DH, Wu JQ. Long non-coding RNA s: important regulators in the development, function and disorders of the central nervous system. Neuropathol Appl Neurobiol. (2019) 45:538–56. doi: 10.1111/nan.12541
81. An S, Song J-J. The coded functions of noncoding RNAs for gene regulation. Mol Cells. (2011) 31:491–6. doi: 10.1007/s10059-011-1004-8
82. Bond AM, VanGompel MJ, Sametsky EA, Clark MF, Savage JC, Disterhoft JF, et al. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat Neurosci. (2009) 12:1020. doi: 10.1038/nn.2371
83. Briggs JA, Wolvetang EJ, Mattick JS, Rinn JL, Barry G. Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron. (2015) 88:861–77. doi: 10.1016/j.neuron.2015.09.045
84. Lawrence M, Daujat S, Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. (2016) 32:42–56. doi: 10.1016/j.tig.2015.10.007
85. Jenuwein T, Allis CD. Translating the histone code. Science. (2001) 293:1074–80. doi: 10.1126/science.1063127
86. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. (2017) 49:e324. doi: 10.1038/emm.2017.11
87. Johansson C, Tumber A, Che K, Cain P, Nowak R, Gileadi C, et al. The roles of Jumonji-type oxygenases in human disease. Epigenomics. (2014) 6:89–120. doi: 10.2217/epi.13.79
88. Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal. (2014) 26:1598–603. doi: 10.1016/j.cellsig.2014.03.030
89. Pollard PJ, Loenarz C, Mole DR, McDonough MA, Gleadle JM, Schofield CJ, et al. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor. (HIF)-1α. Biochem J. (2008) 416:387–94. doi: 10.1042/BJ20081238
90. Sar A, Ponjevic D, Nguyen M, Box AH, Demetrick DJ. Identification and characterization of demethylase JMJD1A as a gene upregulated in the human cellular response to hypoxia. Cell Tissue Res. (2009) 337:223–34. doi: 10.1007/s00441-009-0805-y
91. Balakrishnan L, Milavetz B. Decoding the histone H4 lysine 20 methylation mark. Crit Rev Biochem Mol Biol. (2010) 45:440–52. doi: 10.3109/10409238.2010.504700
92. Batie M, Rocha S. JmjC histone demethylases act as chromatin oxygen sensors. Mol Cell Oncol. (2019) 6:1608501. doi: 10.1080/23723556.2019.1608501
93. Brandl A, Heinzel T, Krämer, OH. Histone deacetylases: salesmen and customers in the post-translational modification market. Biol Cell. (2009) 101:193–205. doi: 10.1042/BC20080158
94. Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA. (1996) 93:12969–73. doi: 10.1073/pnas.93.23.12969
95. Cho Y, Cavalli V. HDAC signaling in neuronal development and axon regeneration. Curr Opin Neurobiol. (2014) 27:118–26. doi: 10.1016/j.conb.2014.03.008
96. Kim S-H, Jeong J-W, Park J, Lee J-W, Seo JH, Jung B-K, et al. Regulation of the HIF-1α stability by histone deacetylases. Oncol Rep. (2007) 17:647–51. doi: 10.3892/or.17.3.647
97. Schoepflin ZR, Shapiro IM, Risbud MV. Class I and IIa HDACs mediate HIF-1α stability through PHD2-dependent mechanism, while HDAC6, a class IIb member, promotes HIF-1α transcriptional activity in nucleus pulposus cells of the intervertebral disc. J Bone Miner Res. (2016) 31:1287–99. doi: 10.1002/jbmr.2787
98. Chan YC, Banerjee J, Choi SY, Sen CK. miR-210: The master hypoxamir. Microcirculation. (2012) 19:215–23. doi: 10.1111/j.1549-8719.2011.00154.x
99. Li JY, Yong TY, Michael MZ, Gleadle JM. MicroRNAs: are they the missing link between hypoxia and pre-eclampsia? Hypertens Pregn. (2014) 33:102–14. doi: 10.3109/10641955.2013.832772
100. Pineles BL, Romero R, Montenegro D, Tarca AL, Han YM, Kim YM, et al. Distinct subsets of microRNAs are expressed differentially in the human placentas of patients with preeclampsia. Am J Obstet Gynecol. (2007) 196:261.e26-6. doi: 10.1016/j.ajog.2007.01.008
101. Zhu X-M, Han T, Sargent IL, Yin G-W, Yao Y-Q. Differential expression profile of microRNAs in human placentas from preeclamptic pregnancies vs normal pregnancies. Am J Obstetr Gynecol. (2009) 200:661–7. doi: 10.1016/j.ajog.2008.12.045
102. Mayor-Lynn K, Toloubeydokhti T, Cruz AC, Chegini N. Expression profile of microRNAs and mRNAs in human placentas from pregnancies complicated by preeclampsia and preterm labor. Reprod Sci. (2011) 18:46–56. doi: 10.1177/1933719110374115
103. Fu G, Brkic J, Hayder H, Peng C. MicroRNAs in human placental development and pregnancy complications. Int J Mol Sci. (2013) 14:5519–44. doi: 10.3390/ijms14035519
104. Zhang Y, Fei M, Xue G, Zhou Q, Jia Y, Li L, et al. Elevated levels of hypoxia-inducible microRNA-210 in pre-eclampsia: new insights into molecular mechanisms for the disease. J Cell Mol Med. (2012) 16:249–59. doi: 10.1111/j.1582-4934.2011.01291.x
105. Fernández-López D, Natarajan N, Ashwal S, Vexler ZS. Mechanisms of perinatal arterial ischemic stroke. J Cerebral Blood Flow Metab. (2014) 34:921–32. doi: 10.1038/jcbfm.2014.41
106. Badiola N, Malagelada C, Llecha N, Hidalgo J, Comella JX, Sabriá J, et al. Activation of caspase-8 by tumour necrosis factor receptor 1 is necessary for caspase-3 activation and apoptosis in oxygen–glucose deprived cultured cortical cells. Neurobiol Dis. (2009) 35:438–47. doi: 10.1016/j.nbd.2009.06.005
107. Algra SO, Groeneveld KM, Schadenberg AW, Haas F, Evens FC, Meerding J, et al. Cerebral ischemia initiates an immediate innate immune response in neonates during cardiac surgery. J Neuroinflamm. (2013) 10:796. doi: 10.1186/1742-2094-10-24
108. Keller A, Meese E. Can circulating miRNAs live up to the promise of being minimal invasive biomarkers in clinical settings? Wiley Interdisc Rev. (2016) 7:148–56. doi: 10.1002/wrna.1320
109. Ponnusamy V, Kapellou O, Yip E, Evanson J, Wong LF, Michael-Titus A, et al. A study of microRNAs from dried blood spots in newborns after perinatal asphyxia: a simple and feasible biosampling method. Pediatr Res. (2015) 79:799–805. doi: 10.1038/pr.2015.276
110. Manueldas S, Benterud T, Rueegg CS, Garberg HT, Huun MU, Pankratov L, et al. Temporal patterns of circulating cell-free DNA (cfDNA) in a newborn piglet model of perinatal asphyxia. PLoS ONE. (2018) 13:e0206601. doi: 10.1371/journal.pone.0206601
111. McCarthy FP, Ryan RM, Chappell LC. Prospective biomarkers in preterm preeclampsia: a review. Pregn Hypertens. (2018) 14:72–8. doi: 10.1016/j.preghy.2018.03.010
112. Li Y, Ma Q, Halavi S, Concepcion K, Hartman RE, Obenaus A, et al. Fetal stress-mediated hypomethylation increases the brain susceptibility to hypoxic–ischemic injury in neonatal rats. Exp Neurol. (2016) 275:1–10. doi: 10.1016/j.expneurol.2015.10.007
113. Chen W, Jadhav V, Tang J, Zhang JH. HIF-1 alpha inhibition ameliorates neonatal brain damage after hypoxic-ischemic injury. Acta Neurochir Suppl. (2008) 102:395–9. doi: 10.1007/978-3-211-85578-2_77
114. Choi HJ, Eun JS, Kim BG, Kim SY, Jeon H, Soh Y. Vitexin, an HIF-1α inhibitor, has anti-metastatic potential in PC12 cells. Mol Cells. (2006) 22:291–9.
115. Min JW, Hu JJ, He M, Sanchez RM, Huang WX, Liu YQ, et al. Vitexin reduces hypoxia-ischemia neonatal brain injury by the inhibition of HIF-1alpha in a rat pup model. Neuropharmacology. (2015) 99:38–50. doi: 10.1016/j.neuropharm.2015.07.007
116. Min JW, Kong WL, Han S, Bsoul N, Liu WH, He XH, et al. Vitexin protects against hypoxic-ischemic injury via inhibiting Ca2+/Calmodulin-dependent protein kinase II and apoptosis signaling in the neonatal mouse brain. Oncotarget. (2017) 8:25513–24. doi: 10.18632/oncotarget.16065
117. Luo WD, Min JW, Huang WX, Wang X, Peng YY, Han S, et al. Vitexin reduces epilepsy after hypoxic ischemia in the neonatal brain via inhibition of NKCC1. J Neuroinflamm. (2018) 15:186. doi: 10.1186/s12974-018-1221-6
118. Nanduri J, Semenza GL, Prabhakar NR. Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L1096–100. doi: 10.1152/ajplung.00325.2017
119. Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, et al. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. (2000) 20:3175–81. doi: 10.1523/JNEUROSCI.20-09-03175.2000
120. Valinluck V, Tsai H-H, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain. (MBD) of methyl-CpG binding protein 2. (MeCP2). Nucleic Acids Res. (2004) 32:4100–8. doi: 10.1093/nar/gkh739
121. Zhao H, Han Z, Ji X, Luo Y. Epigenetic regulation of oxidative stress in ischemic stroke. Aging Dis. (2016) 7:295. doi: 10.14336/AD.2015.1009
122. Lao VV, Herring JL, Kim CH, Darwanto A, Soto U, Sowers LC. Incorporation of 5-chlorocytosine into mammalian DNA results in heritable gene silencing and altered cytosine methylation patterns. Carcinogenesis. (2009) 30:886–93. doi: 10.1093/carcin/bgp060
123. Cox-Limpens KE, Vles JS, Schlechter J, Zimmermann LJ, Strackx E, Gavilanes AW. Fetal brain genomic reprogramming following asphyctic preconditioning. BMC Neurosci. (2013) 14:61. doi: 10.1186/1471-2202-14-61
124. Strackx E, Van den Hove DL, Prickaerts J, Zimmermann L, Steinbusch HW, Blanco CE, et al. Fetal asphyctic preconditioning protects against perinatal asphyxia-induced behavioral consequences in adulthood. Behav Brain Res. (2010) 208:343–51. doi: 10.1016/j.bbr.2009.11.040
125. Vlassaks E, Strackx E, Vles J, Nikiforou M, Martinez-Martinez P, Kramer BW, et al. Fetal asphyctic preconditioning modulates the acute cytokine response thereby protecting against perinatal asphyxia in neonatal rats. J Neuroinflamm. (2013) 10:14. doi: 10.1186/1742-2094-10-14
126. Cox-Limpens KE, Vles JS, van den Hove DL, Zimmermann LJ, Gavilanes AW. Fetal asphyctic preconditioning alters the transcriptional response to perinatal asphyxia. BMC Neurosci. (2014) 15:67. doi: 10.1186/1471-2202-15-67
127. Gonzalez-Rodriguez PJ, Xiong F, Li Y, Zhou J, Zhang L. Fetal hypoxia increases vulnerability of hypoxic–ischemic brain injury in neonatal rats: role of glucocorticoid receptors. Neurobiol Dis. (2014) 65:172–9. doi: 10.1016/j.nbd.2014.01.020
128. McGowan PO, Sasaki A, D'alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. (2009) 12:342. doi: 10.1038/nn.2270
129. Li Y, Xiao D, Dasgupta C, Xiong F, Tong W, Yang S, et al. Perinatal nicotine exposure increases vulnerability of hypoxic–ischemic brain injury in neonatal rats: role of angiotensin II receptors. Stroke. (2012) 43:2483–90. doi: 10.1161/STROKEAHA.112.664698
130. Li Y, Xiao D, Yang S, Zhang L. Promoter methylation represses AT2R gene and increases brain hypoxic–ischemic injury in neonatal rats. Neurobiol Dis. (2013) 60:32–8. doi: 10.1016/j.nbd.2013.08.011
131. Qu Y, Mao M, Zhao F, Zhang L, Mu D. Proapoptotic role of human growth and transformation-dependent protein in the developing rat brain after hypoxia-ischemia. Stroke. (2009) 40:2843–8. doi: 10.1161/STROKEAHA.109.553644
132. Ren X, Ma H, Zuo Z. Dexmedetomidine postconditioning reduces brain injury after brain hypoxia-ischemia in neonatal rats. J Neuroimmune Pharmacol. (2016) 11:238–47. doi: 10.1007/s11481-016-9658-9
133. Liu YJ, Wang DY, Yang YJ, Lei WF. Effects and mechanism of dexmedetomidine on neuronal cell injury induced by hypoxia-ischemia. BMC Anesthesiol. (2017) 17:117. doi: 10.1186/s12871-017-0413-4
134. Doeppner TR, Doehring M, Bretschneider E, Zechariah A, Kaltwasser B, Müller B, et al. MicroRNA-124 protects against focal cerebral ischemia via mechanisms involving Usp14-dependent REST degradation. Acta Neuropathol. (2013) 126:251–65. doi: 10.1007/s00401-013-1142-5
135. Sun Y, Luo ZM, Guo XM, Su DF, Liu X. An updated role of microRNA-124 in central nervous system disorders: a review. Front Cell Neurosci. (2015) 9:193. doi: 10.3389/fncel.2015.00193
136. Taj SH, Kho W, Riou A, Wiedermann D, Hoehn MJB. MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials. (2016) 91:151–65. doi: 10.1016/j.biomaterials.2016.03.025
137. Doeppner TR, Kaltwasser B, Sanchez-Mendoza EH, Caglayan AB, Bähr M, Hermann DM, et al. Lithium-induced neuroprotection in stroke involves increased miR-124 expression, reduced RE1-silencing transcription factor abundance and decreased protein deubiquitination by GSK3β inhibition-independent pathways. J Cereb Blood Flow Metab. (2017) 37:914–26. doi: 10.1177/0271678X16647738
138. Voloboueva LA, Sun X, Xu L, Ouyang Y-B, Giffard RG. Distinct effects of miR-210 reduction on neurogenesis: increased neuronal survival of inflammation but reduced proliferation associated with mitochondrial enhancement. J Neurosci. (2017) 37:3072–84. doi: 10.1523/JNEUROSCI.1777-16.2017
139. Pena-Philippides JC, Caballero-Garrido E, Lordkipanidze T, Roitbak T. In vivo inhibition of miR-155 significantly alters post-stroke inflammatory response. J Neuroinflammation. (2016) 13:287. doi: 10.1186/s12974-016-0753-x
140. West T, Atzeva M, Holtzman DM. Pomegranate polyphenols and resveratrol protect the neonatal brain against hypoxic-ischemic injury. Dev Neurosci. (2007) 29:363–72. doi: 10.1159/000105477
141. Karalis F, Soubasi V, Georgiou T, Nakas CT, Simeonidou C, Guiba-Tziampiri O, et al. Resveratrol ameliorates hypoxia/ischemia-induced behavioral deficits and brain injury in the neonatal rat brain. Brain Res. (2011) 1425:98–110. doi: 10.1016/j.brainres.2011.09.044
142. Cui X, Song H, Su J. Curcumin attenuates hypoxic-ischemic brain injury in neonatal rats through induction of nuclear factor erythroid-2-related factor 2 and heme oxygenase-1. Exp Ther Med. (2017) 14:1512–8. doi: 10.3892/etm.2017.4683
143. Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr. (2011) 6:93. doi: 10.1007/s12263-011-0222-1
144. Vahid F, Zand H, Nosrat–Mirshekarlou E, Najafi R, Hekmatdoost AJG. The role dietary of bioactive compounds on the regulation of histone acetylases and deacetylases: a review. Gene. (2015) 562:8–15. doi: 10.1016/j.gene.2015.02.045
145. Ding X, Cheng Z, Sun B, Huang J, Wang L, Han X, et al. Distinctive sleep problems in children with perinatal moderate or mild hypoxic-ischemia. Neurosci Lett. (2016) 614:60–4. doi: 10.1016/j.neulet.2015.12.061
146. Dong X, Zhao Y, Huang Y, Yu L, Yang X, Gao FJJ, et al. Analysis of long noncoding RNA expression profiles in the whole blood of neonates with hypoxic-ischemic encephalopathy. J Cell Biochem. (2018) 120:8499–509. doi: 10.1002/jcb.28138
147. Zhou H, Wang X, Cheng R, Hou X, Chen Y, Feng Y, et al. Analysis of long non-coding RNA expression profiles in neonatal rats with hypoxic-ischemic brain damage. J Neurochem. (2019) 149–346–61. doi: 10.1111/jnc.14689
148. Yan H, Yuan J, Gao L, Rao J, Hu J. Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke. Neuroscience. (2016) 337:191–9. doi: 10.1016/j.neuroscience.2016.09.017
149. Jiang L, Li H, Fan Z, Zhao R, Xia Z. Circular RNA expression profiles in neonatal rats following hypoxic-ischemic brain damage. Int J Mol Med. (2019) 43:1699–708. doi: 10.3892/ijmm.2019.4111
150. Henriquez B, Bustos FJ, Aguilar R, Becerra A, Simon F, Montecino M, et al. Ezh1 and Ezh2 differentially regulate PSD-95 gene transcription in developing hippocampal neurons. Mol Cell Neurosci. (2013) 57:130–43. doi: 10.1016/j.mcn.2013.07.012
151. Zhang J, Ji F, Liu Y, Lei X, Li H, Ji G, et al. Ezh2 regulates adult hippocampal neurogenesis and memory. J Neurosci. (2014) 34:5184–99. doi: 10.1523/JNEUROSCI.4129-13.2014
152. Ren X, Wang Z, Ma H, Zuo Z. Sevoflurane postconditioning provides neuroprotection against brain hypoxia–ischemia in neonatal rats. Neurol Sci. (2014) 35:1401–4. doi: 10.1007/s10072-014-1726-4
153. Xu Y, Xue H, Zhao P, Yang Y, Ji G, Yu W, et al. Isoflurane postconditioning induces concentration-and timing-dependent neuroprotection partly mediated by the GluR2 AMPA receptor in neonatal rats after brain hypoxia–ischemia. J Anesth. (2016) 30:427–36. doi: 10.1007/s00540-015-2132-7
154. Xue H, Xu Y, Wang S, Wu Z-Y, Li X-Y, Zhang Y-H, et al. Sevoflurane post-conditioning alleviates neonatal rat hypoxic-ischemic cerebral injury via Ezh2-regulated autophagy. Drug Des DevTher. (2019) 13:1691–706. doi: 10.2147/DDDT.S197325
155. Carloni S, Albertini MC, Galluzzi L, Buonocore G, Proietti F, Balduini W. Melatonin reduces endoplasmic reticulum stress and preserves sirtuin 1 expression in neuronal cells of newborn rats after hypoxia–ischemia. J Pineal Res. (2014) 57:192–9. doi: 10.1111/jpi.12156
156. Koyuncuoglu T, Turkyilmaz M, Goren B, Cetinkaya M, Cansev M, Alkan T. Uridine protects against hypoxic-ischemic brain injury by reducing histone deacetylase activity in neonatal rats. Restor Neurol Neurosci. (2015) 33:777–84. doi: 10.3233/RNN-150549
157. Lange S, Rocha-Ferreira E, Thei L, Mawjee P, Bennett K, Thompson PR, et al. Peptidylarginine deiminases: novel drug targets for prevention of neuronal damage following hypoxic ischemic insult. (HI) in neonates. J Neurochem. (2014) 130:555–62. doi: 10.1111/jnc.12744
158. Lange S. Peptidylarginine deiminases as drug targets in neonatal hypoxic-ischemic encephalopathy. Front Neurol. (2016) 7:22. doi: 10.3389/fneur.2016.00022
159. Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, et al. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci. (2003) 23:2112–21. doi: 10.1523/JNEUROSCI.23-06-02112.2003
160. Morris-Blanco KC, Kim T, Bertogliat MJ, Mehta SL, Chokkalla AK, Vemuganti R. Inhibition of the epigenetic regulator REST ameliorates ischemic brain injury. Mol Neurobiol. (2019) 56:2542–50. doi: 10.1007/s12035-018-1254-y
161. Jablonska B, Gierdalski M, Chew L-J, Hawley T, Catron M, Lichauco A, et al. Sirt1 regulates glial progenitor proliferation and regeneration in white matter after neonatal brain injury. Nat Commun. (2016) 7:13866. doi: 10.1038/ncomms13866
162. Kabakus N, Ay I, Aysun S, Söylemezoglu F, Özcan A, Celasun B. Protective effects of valproic acid against hypoxic-ischemic brain injury in neonatal rats. J Child Neurol. (2005) 20:582–7. doi: 10.1177/08830738050200070801
163. Fleiss B, Nilsson MKL, Blomgren K, Mallard C. Neuroprotection by the histone deacetylase inhibitor trichostatin A in a model of lipopolysaccharide-sensitised neonatal hypoxic-ischaemic brain injury. J Neuroinflamm. (2012) 9:70. doi: 10.1186/1742-2094-9-70
164. Jaworska J, Zalewska T, Sypecka J, Ziemka-Nalecz M. Effect of the HDAC inhibitor, sodium butyrate, on neurogenesis in a rat model of neonatal hypoxia–ischemia: potential mechanism of action. Mol Neurobiol. (2019) 56:6341–70. doi: 10.1007/s12035-019-1518-1
165. Qu X, Qi D, Dong F, Wang B, Guo R, Luo M, et al. Quercetin improves hypoxia-ischemia induced cognitive deficits via promoting remyelination in neonatal rat. Brain Res. (2014) 1553:31–40. doi: 10.1016/j.brainres.2014.01.035
166. Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. (2001) 15:2343–60. doi: 10.1101/gad.927301
167. Qureshi IA, Gokhan S, Mehler MF. REST and CoREST are transcriptional and epigenetic regulators of seminal neural fate decisions. Cell Cycle. (2010) 9:4477–86. doi: 10.4161/cc.9.22.13973
168. Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, et al. Repressor element-1 silencing transcription factor. (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci USA. (2012) 109:E962–71. doi: 10.1073/pnas.1121568109
169. Hwang J-Y, Kaneko N, Noh K-M, Pontarelli F, Zukin RS. The gene silencing transcription factor REST represses miR-132 expression in hippocampal neurons destined to die. J Mol Biol. (2014) 426:3454–66. doi: 10.1016/j.jmb.2014.07.032
170. Cavadas MA, Mesnieres M, Crifo B, Manresa MC, Selfridge AC, Scholz CC, et al. REST mediates resolution of HIF-dependent gene expression in prolonged hypoxia. Sci Rep. (2015) 5:17851. doi: 10.1038/srep17851
Keywords: hypoxic-ischemic encephalopathy, biomarker, hypoxia, ischemia, microRNAs, histone modifications, DNA methylation
Citation: Bustelo M, Barkhuizen M, van den Hove DLA, Steinbusch HWM, Bruno MA, Loidl CF and Gavilanes AWD (2020) Clinical Implications of Epigenetic Dysregulation in Perinatal Hypoxic-Ischemic Brain Damage. Front. Neurol. 11:483. doi: 10.3389/fneur.2020.00483
Received: 22 November 2019; Accepted: 04 May 2020;
Published: 09 June 2020.
Edited by:
Claire Thornton, Royal Veterinary College (RVC), United KingdomReviewed by:
Wang-Tso Lee, National Taiwan University Hospital, TaiwanErnest Marshall Graham, Johns Hopkins University, United States
Copyright © 2020 Bustelo, Barkhuizen, van den Hove, Steinbusch, Bruno, Loidl and Gavilanes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio W. D. Gavilanes, ZGFuaWxvLmdhdmlsYW5lc0BtdW1jLm5s
†These authors have contributed equally to this work