 Kathrin Janitzky
Kathrin Janitzky- Department of Neurology, Carl von Ossietzky University Oldenburg, Oldenburg, Germany
The locus coeruleus (LC) is a small brainstem nucleus with widely distributed noradrenergic projections to the whole brain, and loss of LC neurons is a prominent feature of age-related neurodegenerative diseases, such as Alzheimer's disease (AD) and Parkinson's disease (PD). This article discusses the hypothesis that in early stages of neurodegenerative diseases, the discharge mode of LC neurons could be changed to a persistent high tonic discharge, which in turn might impair phasic discharge. Since phasic discharge of LC neurons is required for the release of high amounts of norepinephrine (NE) in the brain to promote anti-inflammatory and neuroprotective effects, persistent high tonic discharge of LC neurons could be a key factor in the progression of neurodegenerative diseases. Transcutaneous vagal stimulation (t-VNS), a non-invasive technique that potentially increases phasic discharge of LC neurons, could therefore provide a non-pharmacological treatment approach in specific disease stages. This article focuses on LC vulnerability in neurodegenerative diseases, discusses the hypothesis that a persistent high tonic discharge of LC neurons might affect neurodegenerative processes, and finally reflects on t-VNS as a potentially useful clinical tool in specific stages of AD and PD.
Introduction
The locus coeruleus (LC) is a small nucleus located in the brainstem near the fourth ventricle and is composed of noradrenergic (NAergic) cells. Despite comprising only roughly 30,000–50,000 neurons in the adult human brain (1–4), the LC consists of extensively branched efferent axons that project throughout the brain and spinal cord (1, 5–14). LC neurons project to all layers of the cortex (15) and have dense projections to the hippocampus (16) as well as the frontal cortex (17).
As part of the ascending reticular activating system, the LC affects consciousness, wakefulness, and attentiveness (14) by projecting axons into the whole brain and thereby activating neural networks across many brain regions synchronously. There is evidence for a functional and topographic order within the LC. LC neurons in the dorso-rostral part project to the neocortex and the hippocampus, whereas more caudo-ventrally located neurons project to the cerebellum and the spinal cord (11, 18–21). Furthermore, tracing studies revealed that individual LC neurons receive input from 9 to 15 different brain regions indicating a largely integrative input to a single LC neuron. The release of NE locally from the soma of LC neurons activates somatodendritic α2-autoreceptors that inhibit neuronal activity via an auto-inhibitory mechanism (22, 23).
Since the loss of LC neurons is a shared feature of neurodegenerative diseases, especially Parkinson's disease (PD) and Alzheimers's disease (AD) (24–26), a better understanding of the role of the LC in AD and PD may provide important insights into the underlying mechanism of these neurodegenerative diseases.
The first part of this article focuses on the neuroprotective effects of NE. Next, it discusses potential mechanisms underlying the selective vulnerability of LC neurons to neurofibrillary tangles (NFTs)- and β-amyloid (Aβ) pathology in AD as well as α-synuclein (αSyn)-pathology in PD, respectively. Finally, a hypothesis discussing the potential relevance of changes in the discharge mode of LC neurons for neurodegeneration is presented together with t-VNS as a non-invasive technique to modulate LC activity with potentially neuroprotective effects.
LC and Neuroprotection
Besides its role as a conventional neurotransmitter in the synapse, extrasynaptically released NE has a paracrine-type of anti-inflammatory and neuroprotective effect on surrounding neurons, glia cells and microvessels (1, 27, 28). Therefore, extrasynaptically released NE decreases toxin-induced inflammatory processes [for review, see (29, 30)], endotoxin-mediated inflammation (31) and Aβ induced neuroinflammation in the brain (24, 25, 30, 32–39). Furthermore, LC neurons innervate the cerebral vasculature throughout the brain via extensive varicosities for non-synaptic release of NE (38, 40), hence playing an important role in maintaining the blood-brain barrier (3, 38, 41, 42).
Moreover, LC neurons exhibit neuroprotective properties through the secretion of brain-derived neurotrophic factor (BDNF) and nerve growth factor (24, 32, 43). BDNF is synthesized in LC neurons (44, 45) and anterogradely transported and released from axon terminals in the projection areas in an activity-dependent manner (45–47). BDNF induces neurotrophic activity, promotes the survival of NAergic neurons and increases axonal sprouting of LC neurons at the terminal sites (45, 48, 49).
N-2-Chloroethyl-N-ethyl-2-bromobenzylamine hydrochloride (DSP-4), a selective neurotoxin for the LC-NAergic system in the rodent brain, is accumulated in NAergic nerve terminals, and damages them, thus resulting in rapid and long lasting loss of NE (50). DSP-4 increases the expression of proinflammatory factors, like inducible nitric oxide synthase (iNOS) (24, 30), and nitric oxide production, which in turn enhance the processing of amyloid precursor protein (APP) to Aβ (24, 51, 52). After treatment with the selective NAergic neurotoxin DSP-4, surviving LC neurons exhibit a regenerative axon sprouting in the target regions as a compensatory mechanism (45, 53), which seems to be important for the temporary maintenance of extracellular NE levels.
Thus, understanding the role of the LC in the process of neurodegeneration may start with the question: What makes LC neurons vulnerable to aging-related neuropathology?
Potential Factors for the Vulnerability of LC Neurons
Why LC neurons are vulnerable to aging and aging-related neurodegenerative diseases, such as PD and AD, is not completely understood. One potential reason for the vulnerability of LC neurons might be intense mitochondrial demand caused by sustained cellular excitability attributable to autonomous pacemaking activity in these neurons, which results in mitochondrial dysfunction and cumulative oxidant stress (54–56).
A common feature of LC neurons is autonomous pacemaking. Small-conductance Ca2+-activated K+ (SK) channels are essential regulators of the intrinsic pacemaking of LC neurons and the activation of SK channels is primarily coupled to Ca2+ influx via the opening of L- and T-type calcium (Ca2+) channels (55, 57, 58). Activity-dependent Ca2+ entry through L-type Ca2+ channels enables feed-forward stimulation of mitochondrial oxidative phosphorylation, and thereby helps to prevent bioenergetic shortage when activity needs to be sustained, but in turn leads to basal mitochondrial oxidant stress (59). Hence, autonomous pacemaking caused by Ca2+ signaling in LC neurons requires elevated mitochondrial activity, leading to oxidative stress under basal conditions (55, 59–62), and thus resulting in elevated susceptibility to mitochondrial impairment.
Another potential factor for the vulnerability of LC neurons are their highly branched, long and thinly myelinated or unmyelinated axons that cause high energy demand, because ATP requirements for propagation of axon potentials grow exponentially with the level of branching (63, 64).
Oxidative stress together with required mitochondrial oxidative phosphorylation to sustain neurotransmitter release and cellular excitability, could interfere with key cellular functions, such as degradation of damaged and misfolded proteins (59), promoting protein aggregation and finally resulting in cell death. Subsequently, the brain is deprived of its NAergic innervation which may be a key step in the early stages of neurodegenerative diseases, such as AD and PD (64, 65).
Physiology of LC Neurons
LC neurons show two different discharge modes, tonic and phasic. In the tonic mode, LC neurons show irregular but continuous firing patterns at 1–6 Hz whereas during the phasic mode, LC neurons fire in short (<300 ms) bursts of higher frequencies (10–15 Hz) that can occur spontaneously or associated with salient stimuli (1, 66, 67). Tonic discharge is high during stress and agitation, moderate during active wakefulness, low during drowsiness and completely absent during REM sleep (1, 66, 68, 69). Complete silencing of LC neurons during REM sleep may be due to elevated inhibitory GABAergic input from the ventral medulla (70–72). There is an inverted U-shaped correlation between tonic and phasic discharge, in such a way that phasic discharge to salient stimuli in the environment is optimal at a moderate tonic discharge level (1).
LC neurons are electrotonically coupled through gap junctions between dendrites outside of the nucleus, in the peri-coerulear region (73, 74). The strength of coupling changes between both discharges modes with increased coupling during phasic activation and decreased coupling in the tonic mode. The shift between the two discharge modes is thought to be modulated by the anterior cingulate (ACC) and the orbitofrontal cortices (OFC) of the prefrontal cortex (PFC) (1).
The PFC is important for a number of cognitive and executive functions (75), and strongly innervated and modulated by NAergic ascending projections from the LC. Aston-Jones and Cohen proposed that glutamatergic projections from the OFC and the ACC back to the LC are important in generating the patterns of LC activity (1). Besides, corticotrophin-releasing hormone (CRF)-containing afferences from the paraventricular nucleus of the hypothalamus and the central nucleus of the amygdala, increase tonic firing (74). Furthermore, LC neuronal activity is inhibited by local GABAergic interneurons, located dorsomedial to the LC nucleus, which hyperpolarize LC cells and reset their spontaneous activity (76).
Stress-Induced Changes in LC Activity
Sustained tonic activity during waking is metabolically demanding and may render LC neurons a vulnerable target to stress (77). Particularly, stress-induced high tonic activity, mediated in part by the stress-related neuropeptide CRF, causes vulnerability to damage induced by high energy demands (33, 78) and makes LC neurons stress-sensitive. Stressful stimuli activate the hypothalamic-pituitary-adrenal axis and cause a release of CRF. CRF-immunoreactive fibers densely innervate the pericoerulear region that contains the dendrites of LC neurons (79) and CRF increases the tonic discharge of LC neurons (80–84). CRF peptide promotes the tonic discharge mode of LC neurons with a decreased maximum magnitude and slower onset, but a much longer duration of activity (74). Furthermore, stress seems to cause long-lasting changes in the LC that directly impact LC function and induce morphological alterations in LC neurons, such as proliferation of dendrites and axons (80, 85–94).
As a protection against these changes, stress-induced increase of NE release triggers auto-inhibitory mechanisms via α2-autoreceptors on LC neurons (78, 95, 96), which induce hyperpolarization and decrease the sensitivity of LC neurons to stimulation. This negative feedback mechanism protects LC neurons against stress-induced changes and damage of these autoregulatory mechanisms may contribute to neurodegenerative diseases like AD and PD (74, 78).
In conclusion, LC neurons are metabolically demanding and highly vulnerable to stress. Given that death of LC neurons is a shared feature of PD and AD (24–26), the damage of autoregulatory mechanisms that protect LC neurons from stress-induced changes might be involved in neurodegenerative processes of both diseases.
LC and AD Pathology
Neurofibrillary tangles (NFTs) are aggregates of the microtubule-associated protein tau and increasing levels of tau pathology characterize the advancing stages in the development of AD. The LC is the first brainstem structure that displays pretangle material [for details see (97–100)], and thus axonal projections from the LC to the transentorhinal region could be important for the anterograde induction of tau pathology (97). Since aggregation of tau in the LC is one of the first pathological hallmarks of AD and precedes cortical tau pathology, it may act as a seed for subsequent spreading of tau pathology throughout the brain (38, 97, 98, 100–103). As hyperphosphorylated tau levels in the LC increase, the volume of the LC decreases in early stages of AD (102, 104). While total numbers of LC neurons are relatively stable until Braak stage II, they are significantly reduced in Braak stages III-VI, and analyses in the human brain revealed that as the Braak stage increased by one unit, the average LC volume decreased by 8.4% (38, 104). A loss of 30% characterizes the transition to MCI and a 55% reduction represents AD (105).
Since NE released from LC neurons is needed to maintain Aβ clearance, the progression of LC degeneration contributes to Aβ pathology in AD (36, 106), and further degeneration of LC neurons might be triggered by an Aβ-mediated failure in the anterograde and retrograde transport of neurotrophic factors like BDNF in LC axons (99, 107, 108). The bidirectional relationship between Aβ pathology and LC degeneration may lead to an exponential progression of the disease, because increased Aβ-levels exaggerate LC degeneration, which in turn reduces NE levels in the terminal fields that diminish the internalization of Aβ by microglia, adding to increased Aβ pathology (35). Progressive loss of LC neurons and the concomitant decrease of NE levels in the brain diminish anti-inflammatory and neuroprotective mechanisms and finally result in an exacerbation of Aβ-induced pro-inflammatory processes and neurotoxicity (31, 109), as well as tau pathology (101, 110).
NE deficiency in the cortex impairs the activation of microglia, the induction of their migration toward amyloid plaques and the stimulation of the internalization and clearance of Aβ (35). Furthermore, NE deficiency results in an increased tau phosphorylation and compromises neurogenesis in the dentate gyrus, dendritic arborization of new neurons and synaptic plasticity (111).
Smaller fusiform cells located in the dorsal part of the LC (23, 112, 113) that project to forebrain regions such as the PFC (23), are characterized by a high density of voltage-gated Ca2+ channels, which enables higher spontaneous firing frequencies (57). Furthermore, this subpopulation of PFC-projecting LC neurons is more excitable and responsive to glutamate than LC neurons projecting to other cortical circuitries (114). This subpopulation of LC neurons that can be distinguished from other LC neurons on the basis of their anatomical projections, molecular phenotypes, and electrophysiological properties, may be more vulnerable to activity-dependent cellular dysfunctions and the neurodegenerative processes in AD because of their higher basal discharge rates (114).
In summary, it can be concluded that NFT pathology causes dysfunction of LC neurons resulting in decreased NE levels in target regions, which contributes to Aβ pathology. This, in turn, accelerates LC degeneration and diminishes anti-inflammatory and neuroprotective effects of NE, resulting in increased Aβ plaque load. Therefore, LC degeneration and Aβ pathology synergistically interact to generate neurodegeneration in AD.
LC and PD Pathology
In PD, α-synuclein (αSyn)-positive deposits, called Lewy bodies, can be found in the LC in Braak stage II, and thus earlier than in the substantia nigra pars compacta (SNc) (115, 116). It is believed that αSyn burden may lead to neuronal dysfunction and impaired neurotransmitter release, but αSyn pathology does not correlate well with cell death (2, 117, 118). Accordingly, αSyn may contribute to neurodegeneration in PD, but is not likely to be the sole reason (119). Furthermore, there is evidence that αSyn can be transferred across synapses and spread within postsynaptic cells in a prion-like fashion (3, 120).
A loss of LC neurons can be found throughout the rostral-caudal extent of the nucleus (20), earlier and in a greater magnitude as compared to the SNc (20, 121), with the onset of LC pathology occurring more than 10 years before the clinical diagnose of PD (122, 123).
Neuromelanin (NM) is an autophagic product synthesized via oxidation of catecholamines and subsequent reactions, and it is the main iron storage mechanism in neurons that protects them from iron-mediated neurotoxicity caused by superoxide free radicals (124–127). Although intraneuronal NM is neuroprotective, NM released by dying neurons can trigger neuroinflammation via activation of microglia (128). Postmortem studies have shown NM accumulation in LC neurons with increasing age (129). Several studies found an inverted U-shaped correlation between NM accumulation in LC cells and age with peak levels around 60 years, followed by an age-related decline related to loss of LC neurons (130, 131). However, some studies reported a linear age-related increase of NM in LC neurons without age-related decline (132). Nevertheless, under pathological conditions involving LC degeneration, the pigment is diminished (132, 133).
NM-sensitive Magnetic resonance imaging (NM-MRI) allows for in vivo visualization of the LC by exploiting the presence of NM (125, 134–138). Studies indicate that NM-MRI can detect structural alterations in the LC in early disease stages (139), even in patients with rapid eye movement (REM) sleep behavioral disorder (RBD) (140, 141), thus indicating NM-MRI as a potential biomarker in prodromal stages of neurodegenerative diseases [(138, 142) for a review see (143, 144)].
RBD is a prodrome of α-synucleinopathies, like PD, that appears 10 or more years before the first motor symptoms occur (145–147). Patients with RBD show fully developed αSyn pathology in the LC, equivalent to the pathology found in patients diagnosed with PD, while exhibiting normal nigrostriatal dopaminergic innervation (148). This begs the question if there are special characteristics of LC neurons, which are relevant to early αSyn pathology and Lewy body burden (106, 149–151).
One feature of LC neurons that may affect their sensitivity for PD-pathology is the presence of elevated Ca2+ concentrations in the cytosol (119). Autonomous pacemaking of LC neurons caused by voltage-sensitive Ca2+ channels (57) requires extensive Ca2+ entry to stimulate oxidative phosphorylation, which promotes high levels of reactive oxygen species (ROS) production, thus elevating oxidative stress (55, 152). Thus, sustained neurotransmitter release and neuronal excitability of LC neurons require high energetic demands that could impair other key cellular functions, such as degradation of misfolded proteins and promote their accumulation in intracellular inclusions (59). αSyn is widely expressed in the nervous system and located in presynaptic terminals, where it is involved in the regulation of synaptic vesicle exocytosis (153–157). Furthermore, αSyn interferes with Ca2+ homeostasis (59, 158–160), for instance by increasing ion permeability of the lysosomal and plasma membrane (161–163). Extracellularly accumulated αSyn increases the permeability of Ca2+ channels resulting in increased cytoplasmatic Ca2+ (119). Hence, αSyn oligomers are able to increase internal Ca2+ concentrations and Ca2+, in turn, increases αSyn oligomerization with cytotoxic effects (164–166). This positive feedback cycle of αSyn oligomerization and increased internal Ca2+ concentration could make LC neurons more vulnerable to PD pathology, because of Ca2+ channel-mediated pacemaking and the cellular burden associated with it (57). NE stabilizes αSyn in a soluble, monomeric form, thereby preventing the formation of toxic oligomers and enabling the disaggregation of existing fibrils (167). Consequently, decreased NE levels increase αSyn oligomerization, and therefore, LC degeneration and αSyn pathology synergistically interact to induce neurodegeneration in PD.
Discussion
Persistent High Tonic Discharge of LC Neurons and Potential Implications for Neurodegenerative Diseases
The findings mentioned above suggest that AD and PD are both characterized by a significant degeneration of LC neurons, despite having distinct pathologies (22, 106, 121). Postmortem studies reveal disease-specific patterns of LC cell loss, affecting the whole LC in PD. In AD, particularly the rostral and dorsal parts of the LC are affected, along with cortical-projecting LC neurons, while the caudal and ventral parts that contain non-cortical-projecting neurons are spared (20, 104). Slight differences in LC pathology in AD and PD could be a result of different underlying neuropathological mechanisms that may depend on the internal organization of the LC nucleus, the modulation of neuronal activity and the complexity of axonal projections of LC cells. The LC is not a single functional entity of neurons that function as a whole in order to control global arousal. Instead, LC neurons appear to be a collection of NAergic cells with sub-specializations according to their anatomical projections (114, 168–170), electrical properties (75, 168) and co-transmitter content, with distinctive roles in regulating brain function (168, 171).
The loss of LC neurons occurs earlier and in a greater magnitude than the atrophy of the hippocampus in AD and the loss of dopaminergic cells in the SNc in PD (25, 26, 104, 106, 121, 150, 172–175). However, LC pathology starts much earlier than evidence of cell loss. Grinberg and colleagues showed tau pathology in the LC in Braak stage 0, but no significantly decreased number of LC neurons until Braak stage III (104), indicating that LC neurons may survive substantial tau burden even with impaired NAergic neurotransmission (176). Moreover, αSyn pathology in the LC is evident early in the premotor phase of PD (Braak stage II), prior to the involvement of the dopaminergic SNc (Braak stage III) (177), but LC neurons can survive with αSyn pathology for years before significant cell loss is evident (20, 26, 121). Thus, tau- and αSyn pathology is verifiable in LC neurons many years before a significant loss of LC neurons is detectable in AD and PD, respectively (45, 106, 150, 174, 178–180).
Previous studies suggest that early damage to the LC in preclinical or prodromal AD may result in a persistent state of high tonic activity (181), which might be detrimental to brain functions that require phasic responses. Wang et al. (182) have shown that the mean firing rate of LC neurons increases significantly 2 and 4 weeks after unilateral lesion of the nigrostriatal pathway in the rat by local injection of 6-hydroxydopamine (6-OHDA) into the right substantia nigra pars compacta (SNc). Furthermore, the percentage of neurons with irregular firing patterns increased significantly. The authors postulated that 6-OHDA lesions of the SNc caused loss of LC neurons and decreased NE concentration in the LC of 6-OHDA-lesioned rats, which in turn resulted in overactivity of residual LC neurons. Moreover, patch-clamp data from Parkin-knockout mice also showed increased spontaneous firing of aged LC neurons caused by alterations in calcium-dependent excitability (183). In contrast, however, Miguelez et al. (184) found no effects of 6-OHDA infusion into the right medial forebrain bundle on the number of spontaneously active LC neurons, but in turn reported a significantly lower basal firing rate after 6-OHDA infusion and postulated that changes in firing rate may be attributed to dopaminergic degeneration. Another study showed an increase in α2-adrenoceptor mRNA in the LC in 6-OHDA-lesiond rats (185), and α2-adrenoceptors modulate the firing rate of the LC by inhibiting neuronal activity (186). With respect to these apparently opposing results, new mouse models, which overexpress αSyn in the LC seem to be a promising approach, since these models capture some cardinal morphological changes in human PD more closely (187, 188). Unfortunately, no electrophysiological LC data based on these models have been reported to date. The fundamental problem, however, off all the reported electrophysiological findings is that in all studies, recordings have been conducted under anesthesia, which impacts spontaneous LC activity (66, 189–192), and thus limits any conclusions about tonic LC discharge in awake animals, let alone patients.
Since recording from LC neurons is a challenging and invasive task, human data are lacking to date. Therefore, to make inferences about LC activity in humans, one has to rely on more indirect measures, like event-related potentials (ERPs), for instance the P300. The P300 is an event-related potential (ERP) that can be recorded in humans from the scalp in an auditory oddball paradigm. In contrast to former reviews that discuss glutamate as the most important neurotransmitter for the generation of the P300 as well as the cholinergic system and GABAergic influences as important modulators (193), more recent reviews suggest that dopamine and NE are the most important modulators for the generation of the P300 (194–196). The P300 can be devided into two subcomponents, the P3a and the P3b, respectively (196). P3a seems to depend on dopaminergic (DAergic) activity and is thought to reflect a novelty-driven orienting response to distractors that can be recorded more frontally on the scalp (194). On the other hand, P3b seems to be related to memory and decision making driven by phasic NAergic LC activity and can be recorded from more temporal-parietal areas (195, 196). Hence, a dual-transmitter P300 hypothesis was assumed that associated DAergic neurotransmission with P3a and LC-NAergic neurotransmission with P3b (194). Furthermore, Nieuwenhuis et al. (195) suggest that the P300 reflects phasic activity of the LC-NAergic system, and recent studies suggest that phasic LC activity depends on background tonic discharge of LC neurons in an inverted U-shaped manner, with highest phasic discharge rates at moderate levels of tonic LC activity, which would in turn create the largest P3b amplitudes (1, 197).
Also, animal studies suggest that the P300 can be interpreted as a cortical correlate of the phasic LC response (195, 197–199), and the hypothesis that phasic LC activity contributes to P300 generation during a target detection task is consistent with the fact that both phasic LC activity and the P300 depend on the motivational significance of the eliciting stimulus as well as underlying attentional mechanisms and show congruent latencies in response to target stimuli [for review see (195)]. Hence, it was suggested that the P300 reflects NE mediated enhancement of gain in the cerebral cortex induced by phasic LC activity and thus enhances cortical encoding of salient stimuli (1, 67, 195, 198).
In addition, studies in PD patients have shown a reduced P300 amplitude (200–206) or an increase in P300 latency (207, 208), whereas in AD patients an increase in P300 latency was found (209–213). If the hypothesis given above is correct and an optimal rate of phasic LC activity is contingent upon a moderate level of tonic activity (1), then a reduction in P300 amplitude and/or increase in P300 latency might reflect impaired phasic activity caused by persistent high tonic LC activity.
Furthermore, persistent high tonic discharge of LC neurons during REM sleep could explain RBD, an early feature of neurodegenerative disorders, including PD (214). RBD is characterized by REM sleep without atonia (RSWA), leading an individual to “act out” dreams, and lesions of the LC and subcoeruleus (SubC) nucleus complex have been theorized to be one possible cause of RBD (215, 216). Glutamatergic neurons in the ventral part of the SubC project to the ventromedial medulla and the spinal cord, who's GABA and glycine neurons inhibit motoneurons and initiate REM sleep atonia (214). These glutamatergic neurons in the SubC get inhibitory afferents from NAergic LC neurons (217, 218). Because normally, tonic discharge of LC neurons is completely absent during REM sleep (66), the SubC is not inhibited by the LC during that state and can thus promote REM sleep muscle paralysis. Therefore, a constantly high tonic discharge of LC neurons during REM sleep could result in an over-inhibition of the SubC and thus explain RSWA.
Moreover, persistent high tonic LC activity during sleep could contribute to the accumulation of protein aggregation and promote neurodegeneration by an inhibition of the glymphatic system (219). The glymphatic system is a macroscopic waste clearance system formed by astroglial cells (220). It is turned on during sleep and enables the elimination of potentially neurotoxic waste products, including Aβ [for details see (219)]. Hence, dysfunction of the glymphatic system could thus contribute to the accumulation of misfolded or hyperphosphorylated proteins and could thereby render the brain more vulnerable to developing a neurodegenerative pathology, because all neurodegenerative diseases are characterized by accumulation of aggregated proteins (221), e.g., misfolded Aβ and NFT in AD or misfolded αSyn in PD, respectively.
A distinct subpopulation of LC neurons in the dorsal part of the nucleus that innervates the PFC (168, 222) and the hippocampus (170), seems to be particularly vulnerable to the pathophysiological processes in AD. Since these LC cells are characterized by greater synaptic excitability, higher spontaneous firing frequencies, and higher susceptibility to glutamate (75), they could be more vulnerable to stress and hyperactivity-dependent cell death. If LC neurons in the dorsal part of the nucleus are indeed the first set of neurons affected by the pathophysiological processes, then this might result in persistent high tonic activity and impaired task-related phasic activity in those cells (181), which in turn may result in compromised PFC and hippocampal functions (64, 176).
Transient silence of LC neurons during REM sleep and prior to each non-REM sleep spindle seems to be important for synaptic plasticity and essential for hippocampus-dependent memory consolidation (223). Swift and colleagues could show that increased LC activity during sleep has no effect on the stability and duration of sleep states, but impairs learning related signatures of non-REM and REM sleep (223). Since glutamatergic projections from the SubC to the medial septum innervate the hippocampus and promote the generation of the theta electroencephalographic activity characterizing REM sleep (218), a persistent high tonic discharge of LC neurons could impair theta oscillation in the hippocampus during REM sleep, which has been shown to be important for consolidation of hippocampal-dependent memories (224, 225).
Since LC neurons in the dorsal part of the nucleus show minimally overlapping projections to the OFC, the medial PFC, and anterior cingulate (ACC) cortex (75), respectively, neuronal activity in individual prefrontal subregions could also be impaired, resulting in compromised cognitive and executive functions, e.g. shifting of the attentional focus and behavioral adaptation in a changing environment. NAergic projections of LC neurons to the PFC are critical for the ability to rapidly switch attention between stimuli and stimulus categories (226–229), and therefore tests of cognitive flexibility could possibly be used to determine LC dysfunction in early stages of AD (230) and PD (231).
Consequences of Persistent and Abnormally High Tonic Discharge of LC Neurons
Neuronal plasticity is essential to adapt to a changing environment through strengthening, weakening or adding of synaptic connections or by promoting neurogenesis (232). Alteration in synaptic plasticity is an early feature in AD with abnormally suppressed efficacy of neuronal plasticity linked to cognitive decline (232). Since deprivation of cortical NAergic innervation is associated with reduced expression of genes important for synaptic plasticity in the cerebral cortex, the NAergic system seems to have a gating function for neuronal plasticity (233). Oberman and Pascual-Leone (232) hypothesize that cortical “hyperplasticity” in autism spectrum disorder (ASD) may provide protection for this population against the development of age-related cognitive decline and AD, and it was assumed that in autism, tonic LC discharge is affected, which in turn has a protective effect against later development of AD (234).
Assuming a persistent and abnormally high tonic discharge of LC neurons in the dorsal part of the nucleus in early stages of AD (20, 64, 104, 170, 181), phasic discharge of these cells—which requires a moderate tonic discharge level—could be impaired as a result. Because the release of high amounts of NE requires phasic spiking of LC neurons (32, 65, 235–238), NE levels in the cortex might not be high enough in this scenario to activate low-affinity β-adrenoceptors that facilitate the anti-inflammatory and neuroprotective effects of NE (32, 35, 65). It can be assumed that sustained high tonic activity would induce compensatory mechanisms that could initially help to maintain homeostasis (85, 89, 92–94), such as axon sprouting in the terminal fields, augmented synthesis of NE by increased activity of tyrosine hydroxylase and dopamine beta-hydroxylase, and decreased NE transporter activity (174, 180, 239–242). All these compensatory mechanisms, however, would further serve to increase the energy demand and oxidative stress of LC neurons. As a protection against these changes, NE could trigger auto-inhibitory mechanisms via α2-autoreceptors on LC neurons (78, 95, 96), which induce hyperpolarization and decrease the sensitivity of LC neurons to dendritic stimulation. These autoregulatory mechanisms may protect LC neurons, but could on the other hand interfere with LC functions, because they further impair the ability of LC neurons to create selective phasic responses, as described above. Subsequently, sustained increase of basal mitochondrial oxidant stress in tonically discharging LC neurons could contribute to an impaired capability to maintain other key cellular functions, such as the degradation of damaged or misfolded proteins (59), giving rise to axon terminal degeneration as an adaptation to excessively high metabolic demand. The consequences of decreased NE levels in the PFC and hippocampus are increased neuro-inflammation and neurodegeneration, and finally, increased amounts of damaged axonal proteins, such as αSyn and tau-protein may promote aggregation and accelerate cell death of LC neurons. If a dysbalance between tonic and phasic discharge of LC neurons is part of the problem—due to the fact that sustained high tonic discharge impairs phasic discharge to salient stimuli—then modulation of afferent stimulation may be an option to normalize LC functions.
t-VNS, a Potential Non-invasive Technique to Increase Phasic Discharge of LC Neurons
Transcutaneous vagal stimulation (t-VNS) is a novel non-invasive brain stimulation technique that increases activation of the LC and NE release (243), via activation of the auricular branch of the vagal nerve at the external ear (244), and fMRI studies in humans show that t-VNS and invasive vagal nerve stimulation (i-VNS) activate the same afferent vagal projection sites (245). The vagal nerve innervates the nucleus of the solitary tract (NTS), which directly modulates the activity of LC neurons via monosynaptic excitatory projections (246, 247) and indirectly excites the LC via the nucleus paragigantocellularis, providing pathways by which VNS could directly drive short latency spiking in the LC (248, 249). Stronger activation of LC dendrites in the peri-coerulear area via t-VNS may significantly influence the activity of LC neurons, for instance via changes in their electrotonic coupling (250) due to gap junctions between dendrites in the peri-coerulear region (73, 74). Previous studies show that when LC neurons are isolated from the peri-coerulear dendritic region, synchronous activity is reduced or abolished (73). This suggests that the modulation of electrical interactions between dendrites in the peri-coerulear area can stimulate synchronous activity within the LC. Hence, t-VNS may increase electrotonic coupling, and in that way may promote phasic activation and decrease tonic activation of LC neurons, which in turn could normalize the dysbalance in LC activity and facilitate phasic spiking of LC neurons required for the release of NE levels high enough to activate low-affinity β-adrenoceptors to promote anti-inflammatory and neuroprotective mechanisms (32, 35, 65).
Electrophysiological studies in rats show that i-VNS increases the firing rate of LC neurons above their baseline activity (246, 251–253). Moreover, VNS causes a significant increase in the percentage of LC neurons firing in bursts (251–253), which in turn leads to a greater NE release in terminal fields as compared to single pulses (254). Short trains of VNS drive rapid, phasic neural activity in the LC (248), and 30 second trains of VNS increase firing rates of LC neurons and NE concentrations in the cortex and hippocampus on the order of minutes to hours (246, 248, 251, 253, 255).
Since direct measurements would require invasive procedures there is a lack of human data on LC activity. However, a recent review of Burger and colleagues discusses the P300 as a potential biomarker for t-VNS effects (256) that seems to represent the phasic activity of the LC-NAergic system. Studies investigating t-VNS effects on P3b in an oddball paradigm in healthy subjects found increased P300/P3b amplitudes compared to sham stimulation (257, 258). Furthermore, consistent with the hypothesis that P3b reflects the activation of the LC-NA system, no effects of t-VNS stimulation on P3a were found (257, 259). However, there are other studies that could not confirm those data (260, 261).
If phasic LC activity in humans could be assessed by P3b measurements, a reduction in P3b amplitude and/or increase in latency in PD or AD patients could possibly indicate t-VNS as a valid non-invasive treatment option. Indeed, we have shown that t-VNS in healthy subjects leads to an increase in P3b amplitude and a reduction in P3b latency (258). Based on the literature cited above, these findings might be interpreted as t-VNS having a positive influence on the imbalance between tonic and phasic discharge by shifting LC activity toward increased phasic activation. If this presumption can stand the test of further experimental scrutiny, then t-VNS may have potential as a clinical tool used to normalize the imbalance between tonic and phasic LC activity in patients in certain stages of neurodegenerative diseases, and possibly have additional benefits by promoting anti-inflammatory and neuroprotective effects which require sufficiently large NE levels related to phasic LC discharge (32, 35, 65, 262). Recently, auricular t-VNS has been shown to have neuroprotective effects on dopaminergic neurons in 6-OHDA-treated rats and the authors suggested that these effects might be related to the inhibition of neuroinflammation (262). Thus, these results indicate t-VNS as a prospectively useful tool with potential anti-inflammatory and neuroprotective effects in early stages of neurodegenerative diseases, like AD and PD (Figure 1).
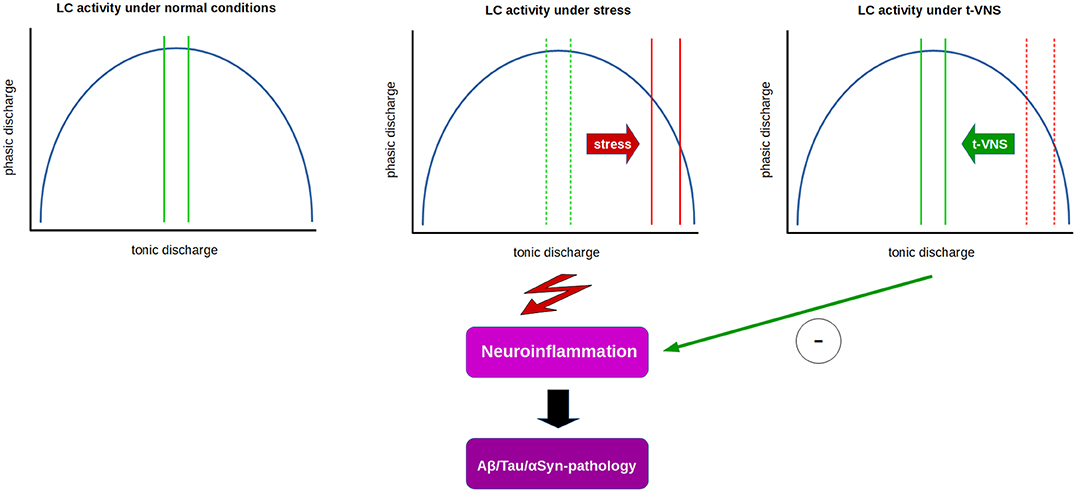
Figure 1. Relation between tonic and phasic LC discharge (blue curve) and proposed effects of t-VNS treatment. Under normal conditions (left), a moderate range of tonic activity (indicated by green lines) allows for an optimal level of phasic activity in response to salient stimuli. Under constant stress (middle), LC tonic activity is thought to shift from moderate to high tonic discharge (solid red lines) which in turn impairs phasic discharge, presumably facilitating neuroinflammatory processes and contributing to neurodegeneration. t-VNS treatment (right) is proposed to restore tonic discharge to a moderate range, thus enabling an optimal level of phasic discharge, and presumably inhibiting neuroinflammatory/neurodegenerative processes by neuroprotective effects of NE.
Concluding Remarks
Studies suggest that early damage to the LC in preclinical or prodromal phases of neurodegenerative diseases, such as AD and PD, may result in an abnormally persistent state of high tonic activity of the LC (181) that impairs phasic discharge, which requires a moderate tonic activity level. Since phasic LC discharge is essential for optimization of cognitive and neural network function (67), as well as anti-inflammatory and neuroprotective effects, a potential facilitation of phasic LC activity by t-VNS might be a useful clinical tool in early stages of neurodegenerative diseases, like AD and PD (Figure 1).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
KJ developed the presented hypotheses and wrote the manuscript.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Dr. Tim Wanger, Dr. Fabian Fincke, Dr. Michael T. Lippert, and Isabella Ampomah for their help in editing the manuscript.
Abbreviations
AD, Alzheimers's disease; Aβ, amyloid-beta; ACC, anterior cingulate cortex; αSyn, α-synuclein; BDNF, brain derived neurotrophic factor; CRF, corticotrophin-releasing hormone; LC, locus coeruleus; NA, noradrenaline; NE, norepinephrine; NFT, neurofibrillary tangles; NTS, nucleus of the solitary tract; OFC, orbitofrontal cortex; PD, Parkinson's disease; PFC, prefrontal cortex; RBD, rapid eye movement sleep behavioral disorder; SNc, substantia nigra pars compacta; SubC, subcoeruleus; t-VNS, transcutaneous vagal stimulation.
References
1. Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. (2005) 28:403–50. doi: 10.1146/annurev.neuro.28.061604.135709
2. Espay AJ, LeWitt PA, Kaufmann H. Norepinephrine deficiency in Parkinson's disease: the case for noradrenergic enhancement. Mov Disord. (2014) 29:1710–9. doi: 10.1002/mds.26048
3. Vermeiren Y, De Deyn PP. Targeting the norepinephrinergic system in Parkinson's disease and related disorders: the locus coeruleus story. Neurochem Int. (2017) 102:22–32. doi: 10.1016/j.neuint.2016.11.009
4. Zorec R, Parpura V, Verkhratsky A. Preventing neurodegeneration by adrenergic astroglial excitation. FEBS J. (2018) 285:3645–56. doi: 10.1111/febs.14456
5. Aston-Jones G. Locus coeruleus, A5 and A7 noradrenergic cell groups. In: Paxinos G, editor. The Rat Nervous System. San Diego, CA: Elsevier Academic (2004). p. 259–94.
6. Aston-Jones G, Foote SL, Bloom FE. Anatomy and physiology of locus coeruleus neurons: functional implications. In: Ziegler M, Lake CR, editors. Norepinephrine: Frontiers of Clinical Neuroscience. Vol. 2. Baltimore, MD: Williams and Wilkins (1984). p. 92–116.
7. Jones BE, Halaris AE, McIlhany M, Moore RY. Ascending projections of the locus coeruleus in the rat. I axonal transport in central noradrenaline neurons. Brain Res. (1977) 127:1–21. doi: 10.1016/0006-8993(77)90377-8
8. Jones BE, Yang T-Z. The efferent projections from the reticular formation and the locus coeruleus studied by anterograde and retrograde axonal transport in the rat. J Compar Neurol. (1985) 242:56–92. doi: 10.1002/cne.902420105
9. Loughlin SE, Foote SL, Bloom FE. Efferent projections of nucleus locus coeruleus: topographic organization of cells of origin demonstrated by three-dimensional reconstruction. Neuroscience. (1986) 18:291–306. doi: 10.1016/0306-4522(86)90155-7
10. Moore RY, Bloom FE. Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci. (1979) 2:113–68. doi: 10.1146/annurev.ne.02.030179.000553
11. Rho H-J, Kim J-H, Lee S-H. Function of selective neuromodulatory projections in the mammalian cerebral cortex: comparison between cholinergic and noradrenergic systems. Front Neural Circuits. (2018) 12:47. doi: 10.3389/fncir.2018.00047
12. Samuels E, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr Neuropharmacol. (2008) 6:235–53. doi: 10.2174/157015908785777229
13. Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part II: physiological and pharmacological manipulations and pathological alterations of locus coeruleus activity in humans. Curr Neuropharmacol. (2008) 6:254–85. doi: 10.2174/157015908785777193
14. Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci. (2009) 10:211–23. doi: 10.1038/nrn2573
15. Oleskevich S, Descarries L, Lacaille J. Quantified distribution of the noradrenaline innervation in the hippocampus of adult rat. J Neurosci. (1989) 9:3803–15. doi: 10.1523/JNEUROSCI.09-11-03803.1989
16. Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-B-hydroxylase as a marker. J Compar Neurol. (1975) 163:467–505. doi: 10.1002/cne.901630406
17. Morrison J, Molliver M, Grzanna R. Noradrenergic innervation of cerebral cortex: widespread effects of local cortical lesions. Science. (1979) 205:313–6. doi: 10.1126/science.451605
18. Agster KL, Mejias-Aponte CA, Clark BD, Waterhouse BD. Evidence for a regional specificity in the density and distribution of noradrenergic varicosities in rat cortex. J Compar Neurol. (2013) 521:2195–207. doi: 10.1002/cne.23270
19. Benarroch EE. The locus ceruleus norepinephrine system: functional organization and potential clinical significance. Neurology. (2009) 73:1699–704. doi: 10.1212/WNL.0b013e3181c2937c
20. German DC, Manaye KF, White CL, Woodward DJ, McIntire DD, Smith WK, et al. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. (1992) 32:667–76. doi: 10.1002/ana.410320510
21. Marcyniuk B, Man DMA, Yates PO. Loss of nerve cells from locus coeruleus in Alzheimer's disease is topographically arranged. Neurosci Lett. (1986) 64:247-252. doi: 10.1016/0304-3940(86)90336-8
22. Chamba G, Weissmann D, Rousset C, Renaud B, Pujol JF. Distribution of alpha-1 and alpha-2 binding sites in the rat locus coeruleus. Brain Res Bull. (1991) 26:185–93. doi: 10.1016/0361-9230(91)90225-9
23. Schwarz LA, Luo L. Organization of the locus coeruleus-norepinephrine system. Curr Biol. (2015) 25:R1051–6. doi: 10.1016/j.cub.2015.09.039
24. Choudhary P, Pacholko AG, Palaschuk J, Bekar LK. The locus coeruleus neurotoxin, DSP4, and/or a high sugar diet induce behavioral and biochemical alterations in wild-type mice consistent with Alzheimers related pathology. Metab Brain Dis. (2018) 33:1563–71. doi: 10.1007/s11011-018-0263-x
25. Marien MR, Colpaert FC, Rosenquist AC. Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res Rev. (2004) 45:38–78. doi: 10.1016/j.brainresrev.2004.02.002
26. Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia Nigra in Alzheimer and Parkinson diseases. Arch Neurol. (2003) 60:337. doi: 10.1001/archneur.60.3.337
27. Beaudet A, Descarries L. The monoamine innervation of rat cerebral cortex: synaptic and nonsynaptic axon terminals. Neuroscience. (1978) 3:851–60. doi: 10.1016/0306-4522(78)90115-X
28. Séguéla P, Watkins KC, Geffard M, Descarries L. Noradrenaline axon terminals in adult rat neocortex: an immunocytochemical analysis in serial thin sections. Neuroscience. (1990) 35:249–64. doi: 10.1016/0306-4522(90)90079-J
29. Feinstein DL, Heneka MT, Gavrilyuk V, Russo CD, Weinberg G, Galea E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int. (2002) 41:357–65. doi: 10.1016/S0197-0186(02)00049-9
30. Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, et al. Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: implications for Alzheimer's disease. J Neurosci. (2002) 22:2434–42. doi: 10.1523/JNEUROSCI.22-07-02434.2002
31. Feinstein DL, Galea E, Reis DJ. Norepinephrine suppresses inducible nitric oxide synthase activity in rat astroglial cultures. J Neurochem. (1993) 60:1945–8. doi: 10.1111/j.1471-4159.1993.tb13425.x
32. Counts SE, Mufson EJ. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J Neurochem. (2010) 113:649–60. doi: 10.1111/j.1471-4159.2010.06622.x
33. Feinstein DL, Kalinin S, Braun D. Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: noradrenergic signaling system. J Neurochem. (2016) 139:154–78. doi: 10.1111/jnc.13447
34. Gavrilyuk V, Dello Russo C, Heneka MT, Pelligrino D, Weinberg G, Feinstein DL. Norepinephrine increases IκBα expression in astrocytes. J Biol Chem. (2002) 277:29662–8. doi: 10.1074/jbc.M203256200
35. Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, et al. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proce Natl Acad Sci USA. (2010) 107:6058–63. doi: 10.1073/pnas.0909586107
36. Kalinin S, Gavrilyuk V, Polak PE, Vasser R, Zhao J, Heneka MT, et al. Noradrenaline deficiency in brain increases β-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. (2007) 28:1206–14. doi: 10.1016/j.neurobiolaging.2006.06.003
37. Madrigal JLM, Kalinin S, Richardson JC, Feinstein DL. Neuroprotective actions of noradrenaline: effects on glutathione synthesis and activation of peroxisome proliferator activated receptor delta. J Neurochem. (2007) 103:2092–101. doi: 10.1111/j.1471-4159.2007.04888.x
38. Satoh A, Iijima KM. Roles of tau pathology in the locus coeruleus (LC) in age-associated pathophysiology and Alzheimer's disease pathogenesis: potential strategies to protect the LC against aging. Brain Res. (2019) 1702:17–28. doi: 10.1016/j.brainres.2017.12.027
39. Weinshenker D. Functional consequences of locus coeruleus degeneration in Alzheimers disease. Curr Alzheimer Res. (2008) 5:342–5. doi: 10.2174/156720508784533286
40. Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev. (2003) 42:33–84. doi: 10.1016/S0165-0173(03)00143-7
41. Kalinin S, Feinstein DL, Xu H-L, Huesa G, Pelligrino DA, Galea E. Degeneration of noradrenergic fibres from the locus coeruleus causes tight-junction disorganisation in the rat brain. Eur J Neurosci. (2006) 24:3393–400. doi: 10.1111/j.1460-9568.2006.05223.x
42. Pamphlett R. Uptake of environmental toxicants by the locus ceruleus: a potential trigger for neurodegenerative, demyelinating and psychiatric disorders. Med Hypotheses. (2014) 82:97–104. doi: 10.1016/j.mehy.2013.11.016
43. Debeir T, Marien M, Ferrario J, Rizk P, Prigent A, Colpaert F, et al. In vivo upregulation of endogenous NGF in the rat brain by the alpha2-adrenoreceptor antagonist dexefaroxan: potential role in the protection of the basalocortical cholinergic system during neurodegeneration. Exp Neurol. (2004) 190:384–95. doi: 10.1016/j.expneurol.2004.08.023
44. Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, et al. Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci. (1998) 18:2808–21. doi: 10.1523/JNEUROSCI.18-08-02808.1998
45. Ishida Y, Shirokawa T, Miyaishi O, Komatsu Y, Isobe K. Age-dependent changes in projections from locus coeruleus to hippocampus dentate gyrus and frontal cortex. Eur J Neurosci. (2000) 12:1263–70. doi: 10.1046/j.1460-9568.2000.00017.x
46. Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, et al. Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. (1997) 272:8837–40. doi: 10.1074/jbc.272.14.8837
47. Thoenen H. Neurotrophins and neuronal plasticity. Science. (1995) 270:593–8. doi: 10.1126/science.270.5236.593
48. Matsunaga W, Shirokawa T, Isobe K. BDNF is necessary for maintenance of noradrenergic innervations in the aged rat brain. Neurobiol. Aging. (2004) 25:341–8. doi: 10.1016/S0197-4580(03)00093-9
49. Matsunaga W, Isobe K, Shirokawa T. Involvement of neurotrophic factors in aging of noradrenergic innervations in hippocampus and frontal cortex. Neurosci Res. (2006) 54:313–8. doi: 10.1016/j.neures.2005.12.013
50. Ross SB, Stenfors C. DSP4, a selective neurotoxin for the locus coeruleus noradrenergic system. A review of its mode of action. Neurotox Res. (2014) 27:15–30. doi: 10.1007/s12640-014-9482-z
51. Guix FX, Wahle T, Vennekens K, Snellinx A, Chávez-Gutiérrez L, Ill-Raga G, et al. Modification of γ-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer's disease. EMBO Mol Med. (2012) 4:660–73. doi: 10.1002/emmm.201200243
52. Kwak Y-D, Wang R, Li J, Zhang Y-W, Xu H, Liao F-F. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener. (2011) 6:17. doi: 10.1186/1750-1326-6-17
53. Fritschy J-M, Grzanna R. Restoration of ascending noradrenergic projections by residual locus coeruleus neurons: compensatory response to neurotoxin-induced cell death in the adult rat brain. J Compar Neurol. (1992) 321:421–41. doi: 10.1002/cne.903210309
54. Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Ann NY Acad Sci. (2008) 1147:53–60. doi: 10.1196/annals.1427.002
55. Sanchez-Padilla J, Guzman JN, Ilijic E, Kondapalli J, Galtieri DJ, Yang B, et al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci. (2014) 17:832–40. doi: 10.1038/nn.3717
56. Surmeier DJ, Guzman JN, Sanchez-Padilla J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson's disease. Cell Calcium. (2010) 47:175–82. doi: 10.1016/j.ceca.2009.12.003
57. Matschke LA, Bertoune M, Roeper J, Snutch TP, Oertel WH, Rinné S, et al. A concerted action of L- and T-type Ca2+ channels regulates locus coeruleus pacemaking. Mol Cell Neurosci. (2015) 68:293–302. doi: 10.1016/j.mcn.2015.08.012
58. Matschke LA, Rinné S, Snutch TP, Oertel WH, Dolga AM, Decher N. Calcium-activated SK potassium channels are key modulators of the pacemaker frequency in locus coeruleus neurons. Mol Cell Neurosci. (2018) 88:330–41. doi: 10.1016/j.mcn.2018.03.002
59. Surmeier DJ, Schumacker PT, Guzman JD, Ilijic E, Yang B, Zampese E. Calcium and Parkinson's disease. Biochem Biophys Res Commun. (2017) 483:1013–9. doi: 10.1016/j.bbrc.2016.08.168
60. Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, et al. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci. (2012) 15:1414–21. doi: 10.1038/nn.3209
61. Liddell JR, White AR. Nexus between mitochondrial function, iron, copper and glutathione in Parkinson's disease. Neurochem Int. (2018) 117:126–38. doi: 10.1016/j.neuint.2017.05.016
62. Williams JT, North RA, Shefner SA, Nishi S, Egan TM. Membrane properties of rat locus coeruleus neurones. Neuroscience. (1984) 13:137–56. doi: 10.1016/0306-4522(84)90265-3
63. Pissadaki EK, Bolam JP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Front Comput Neurosci. (2013) 7:13. doi: 10.3389/fncom.2013.00013
64. Weinshenker D. Long road to ruin: noradrenergic dysfunction in neurodegenerative disease. Trends Neurosci. (2018) 41:211–23. doi: 10.1016/j.tins.2018.01.010
65. Mather M, Harley CW. The locus coeruleus: essential for maintaining cognitive function and the aging brain. Trends Cogn Sci. (2016) 20:214–26. doi: 10.1016/j.tics.2016.01.001
66. Aston-Jones G, Bloom F. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. (1981) 1:876–86. doi: 10.1523/JNEUROSCI.01-08-00876.1981
67. Vazey EM, Moorman DE, Aston-Jones G. Phasic locus coeruleus activity regulates cortical encoding of salience information. Proc Natl Acad Sci USA. (2018) 115:E9439–48. doi: 10.1073/pnas.1803716115
68. Hobson J, McCarley R, Wyzinski P. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. (1975) 189:55–8. doi: 10.1126/science.1094539
69. Rasmussen K, Morilak DA, Jacobs BL. Single unit activity of locus coeruleus neurons in the freely moving cat. Brain Res. (1986) 371:324–34. doi: 10.1016/0006-8993(86)90370-7
70. Gervasoni D, Darracq L, Fort P, Soulière F, Chouvet G, Luppi P-H. Electrophysiological evidence that noradrenergic neurons of the rat locus coeruleus are tonically inhibited by GABA during sleep. Eur J Neurosci. (1998) 10:964–70. doi: 10.1046/j.1460-9568.1998.00106.x
71. Neves RM, van Keulen S, Yang M, Logothetis NK, Eschenko O. Locus coeruleus phasic discharge is essential for stimulus-induced gamma oscillations in the prefrontal cortex. J Neurophysiol. (2018) 119:904–20. doi: 10.1152/jn.00552.2017
72. Weber F, Chung S, Beier KT, Xu M, Luo L, Dan Y. Control of REM sleep by ventral medulla GABAergic neurons. Nature. (2015) 526:435–8. doi: 10.1038/nature14979
73. Ishimatsu M, Williams JT. Synchronous activity in locus coeruleus results from dendritic interactions in pericoerulear regions. J Neurosci. (1996) 16:5196–204. doi: 10.1523/JNEUROSCI.16-16-05196.1996
74. Valentino RJ, Van Bockstaele E. Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur J Pharmacol. (2008) 583:194–203. doi: 10.1016/j.ejphar.2007.11.062
75. Chandler DJ, Waterhouse BD, Gao W-J. New perspectives on catecholaminergic regulation of executive circuits: evidence for independent modulation of prefrontal functions by midbrain dopaminergic and noradrenergic neurons. Front Neural Circuits. (2014) 8:53. doi: 10.3389/fncir.2014.00053
76. Jin X, Li S, Bondy B, Zhong W, Oginsky MF, Wu Y, et al. Identification of a group of GABAergic neurons in the dorsomedial area of the locus coeruleus. PLoS ONE. (2016) 11:e0146470. doi: 10.1371/journal.pone.0146470
77. Sharma Y, Xu T, Graf WM, Fobbs A, Sherwood CC, Hof PR, et al. Comparative anatomy of the locus coeruleus in humans and nonhuman primates. J Compar Neurol. (2010) 518:963–71. doi: 10.1002/cne.22249
78. Ross JA, Reyes BAS, Van Bockstaele EJ. Amyloid beta peptides, locus coeruleus-norepinephrine system and dense core vesicles. Brain Res. (2019) 1702:46–53. doi: 10.1016/j.brainres.2018.03.009
79. Valentino RJ, Page M, Van Bockstaele E, Aston-Jones G. Corticotropin-releasing factor innervation of the locus coeruleus region: distribution of fibers and sources of input. Neuroscience. (1992) 48:689–705. doi: 10.1016/0306-4522(92)90412-U
80. Borodovitsyna O, Joshi N, Chandler D. Persistent stress-induced neuroplastic changes in the locus coeruleus/norepinephrine system. Neural Plasticity. (2018) 2018:1–14. doi: 10.1155/2018/1892570
81. Curtis AL, Leiser SC, Snyder K, Valentino RJ. Predator stress engages corticotropin-releasing factor and opioid systems to alter the operating mode of locus coeruleus norepinephrine neurons. Neuropharmacology. (2012) 62:1737–45. doi: 10.1016/j.neuropharm.2011.11.020
82. Snyder K, Wang W-W, Han R, McFadden K, Valentino RJ. Corticotropin-releasing factor in the norepinephrine nucleus, locus coeruleus, facilitates behavioral flexibility. Neuropsychopharmacology. (2011) 37:520–30. doi: 10.1038/npp.2011.218
83. Valentino RJ, Foote SL, Aston-Jones G. Corticotropin-releasing factor activates noradrenergic neurons of the locus coeruleus. Brain Res. (1983) 270:363–7. doi: 10.1016/0006-8993(83)90615-7
84. Zitnik GA. Control of arousal through neuropeptide afferents of the locus coeruleus. Brain Res. (2016) 1641:338–50. doi: 10.1016/j.brainres.2015.12.010
85. Cibelli G, Corsi P, Diana G, Vitiello F, Thiel G. Corticotropin-releasing factor triggers neurite outgrowth of a catecholaminergic immortalized neuron via cAMP and MAP kinase signalling pathways. Eur J Neurosci. (2001) 13:1339–48. doi: 10.1046/j.0953-816x.2001.01510.x
86. Fan Y, Chen P, Li Y, Zhu M-Y. Effects of chronic social defeat on expression of dopamine β-hydroxylase in rat brains. Synapse. (2013) 67:300–12. doi: 10.1002/syn.21641
87. George SA, Knox D, Curtis AL, Aldridge JW, Valentino RJ, Liberzon I. Altered locus coeruleus-norepinephrine function following single prolonged stress. Eur J Neurosci. (2012) 37:901–9. doi: 10.1111/ejn.12095
88. Mamalaki E, Kvetnansky R, Brady LS, Gold PW, Herkenham M. Repeated immobilization stress alters tyrosine hydroxylase, corticotropin-releasing hormone and corticosteroid receptor messenger ribonucleic acid levels in rat brain. J Neuroendocrinol. (1992) 4:689–99. doi: 10.1111/j.1365-2826.1992.tb00220.x
89. Nakamura S, Sakaguchi T, Aoki F. Electrophysiological evidence for terminal sprouting of locus coeruleus neurons following repeated mild stress. Neurosci Lett. (1989) 100:147–52. doi: 10.1016/0304-3940(89)90675-7
90. Sabban EL, Serova LI, Newman E, Aisenberg N, Akirav I. Changes in gene expression in the locus coeruleus-amygdala circuitry in inhibitory avoidance PTSD model. Cell Mol Neurobiol. (2017) 38:273–80. doi: 10.1007/s10571-017-0548-3
91. Salim S, Hite B, Eikenburg DC. Activation of the CRF1receptor causes ERK1/2 mediated increase in GRK3 expression in CATHa cells. FEBS Lett. (2007) 581:3204–10. doi: 10.1016/j.febslet.2007.06.006
92. Swinny JD, Valentino RJ. Corticotropin-releasing factor promotes growth of brain norepinephrine neuronal processes through Rho GTPase regulators of the actin cytoskeleton in rat. Eur J Neurosci. (2006) 24:2481–90. doi: 10.1111/j.1460-9568.2006.05129.x
93. Swinny JD, O'Farrell E, Bingham BC, Piel DA, Valentino RJ, Beck SG. Neonatal rearing conditions distinctly shape locus coeruleus neuronal activity, dendritic arborization, and sensitivity to corticotrophin-releasing factor. In J Neuropsychopharmacol. (2009) 13:515. doi: 10.1017/S146114570999037X
94. Van Bockstaele EJ, Reyes BAS, Valentino RJ. The locus coeruleus: a key nucleus where stress and opioids intersect to mediate vulnerability to opiate abuse. Brain Res. (2010) 1314:162–74. doi: 10.1016/j.brainres.2009.09.036
95. Huang H-P, Zhu F-P, Chen X-W, Xu Z-QD, Zhang CX, Zhou Z. Physiology of quantal norepinephrine release from somatodendritic sites of neurons in locus coeruleus. Front Mol Neurosci. (2012) 5:29. doi: 10.3389/fnmol.2012.00029
96. Trueta C, De-Miguel FF. Extrasynaptic exocytosis and its mechanisms: a source of molecules mediating volume transmission in the nervous system. Front Physiol. (2012) 3:319. doi: 10.3389/fphys.2012.00319
97. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. (2011) 70:960–9. doi: 10.1097/NEN.0b013e318232a379
98. Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. (2010) 121:171–81. doi: 10.1007/s00401-010-0789-4
99. Ponnusamy R, McNerney MW, Moghadam S, Salehi A. Assessing disease-modifying effects of norepinephrine in Down syndrome and Alzheimer's disease. Brain Res. (2019) 1702:3–11. doi: 10.1016/j.brainres.2017.09.035
100. Stratmann K, Heinsen H, Korf H-W, Del Turco D, Ghebremedhin E, Seidel K, et al. Precortical phase of Alzheimer's disease (AD)-related tau cytoskeletal pathology. Brain Pathol. (2015) 26:371–86. doi: 10.1111/bpa.12289
101. Chalermpalanupap T, Weinshenker D, Rorabaugh JM. Down but not out: the consequences of pretangle tau in the locus coeruleus. Neural Plasticity. (2017) 2017:1–9. doi: 10.1155/2017/7829507
102. Ehrenberg AJ, Nguy AK, Theofilas P, Dunlop S, Suemoto CK, Di Lorenzo Alho AT, et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer's disease. Neuropathol Appl Neurobiol. (2017) 43:393–408. doi: 10.1111/nan.12387
103. Theofilas P, Dunlop S, Heinsen H, Grinberg LT. Turning on the light within: subcortical nuclei of the isodentritic core and their role in Alzheimer's disease pathogenesis. J Alzheimer's Dis. (2015) 46:17–34. doi: 10.3233/JAD-142682
104. Theofilas P, Ehrenberg AJ, Dunlop S, Di Lorenzo Alho AT, Nguy A, Leite REP, et al. Locus coeruleus volume and cell population changes during Alzheimer's disease progression: a stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimer's Dement. (2017) 13:236–46. doi: 10.1016/j.jalz.2016.06.2362
105. Eser RA, Ehrenberg AJ, Petersen C, Dunlop S, Mejia MB, Suemoto CK, et al. Selective vulnerability of brainstem nuclei in distinct tauopathies: a postmortem study. J Neuropathol Exp Neurol. (2018) 77:149–61. doi: 10.1093/jnen/nlx113
106. Peterson AC, Li CR. Noradrenergic dysfunction in Alzheimer's and Parkinson's diseases-an overview of imaging studies. Front Aging Neurosci. (2018) 10:127. doi: 10.3389/fnagi.2018.00127
107. Seifert B, Eckenstaler R, Rönicke R, Leschik J, Lutz B, Reymann K, et al. Amyloid-beta induced changes in vesicular transport of BDNF in hippocampal neurons. Neural Plasticity. (2016) 2016:1–15. doi: 10.1155/2016/4145708
108. Yenkoyan K, Fereshetyan K, Matinyan S, Chavushyan V, Aghajanov M. The role of monoamines in the development of Alzheimer's disease and neuroprotective effect of a proline rich polypeptide. Progress Neuro Psychopharmacol Biol Psychiatry. (2018) 86:76–82. doi: 10.1016/j.pnpbp.2018.05.013
109. Madrigal JLM, Russo CD, Gavrilyuk V, Feinstein DL. Effects of noradrenaline on neuronal NOS2 expression and viability. Antioxidants Redox Signal. (2006) 8:885–92. doi: 10.1089/ars.2006.8.885
110. Betts MJ, Ehrenberg AJ, Hämmerer D, Düzel E. Commentary: locus coeruleus ablation exacerbates cognitive deficits, neuropathology, and lethality in P301S tau transgenic mice. Front Neurosci. (2018) 12:401. doi: 10.3389/fnins.2018.00401
111. Dang V, Medina B, Das D, Moghadam S, Martin KJ, Lin B, et al. Formoterol, a long-acting β2 adrenergic agonist, improves cognitive function and promotes dendritic complexity in a mouse model of down syndrome. Biol Psychiatry. (2014) 75:179–88. doi: 10.1016/j.biopsych.2013.05.024
112. Grzanna R, Molliver ME. The locus coeruleus in the rat: an immunohistochemical delineation. Neuroscience. (1980) 5:21–40. doi: 10.1016/0306-4522(80)90068-8
113. Swanson LW. The locus coeruleus: a cytoarchitectonic, golgi and immunohistochemical study in the albino rat. Brain Res. (1976) 110:39–56. doi: 10.1016/0006-8993(76)90207-9
114. Chandler DJ, Gao W-J, Waterhouse BD. Heterogeneous organization of the locus coeruleus projections to prefrontal and motor cortices. Proc Natl Acad Sci USA. (2014) 111:6816–21. doi: 10.1073/pnas.1320827111
115. Braak E, Sandmann-Keil D, Rub U, Gai WP, de Vos RA, Steur EN, et al. alpha-synuclein immunopositive Parkinson's disease-related inclusion bodies in lower brain stem nuclei. Acta Neuropathol. (2001) 101:195–201. doi: 10.1007/s004010000247
116. Sommerauer M, Fedorova TD, Hansen AK, Knudsen K, Otto M, Jeppesen J, et al. Evaluation of the noradrenergic system in Parkinson's disease: an 11C-MeNER PET and neuromelanin MRI study. Brain. (2017) 141:496–504. doi: 10.1093/brain/awx348
117. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. (2012) 9:13–24. doi: 10.1038/nrneurol.2012.242
118. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. (2017) 18:101–13. doi: 10.1038/nrn.2016.178
119. Post MR, Lieberman OJ, Mosharov EV. Can Interactions between α-synuclein, dopamine and calcium explain selective neurodegeneration in Parkinson's disease? Front Neurosci. (2018) 12:161. doi: 10.3389/fnins.2018.00161
120. Brundin P, Li J-Y, Holton JL, Lindvall O, Revesz T. Research in motion: the enigma of Parkinson's disease pathology spread. Nat Rev Neurosci. (2008) 9:741–5. doi: 10.1038/nrn2477
121. Rommelfanger KS, Weinshenker D. Norepinephrine: the redheaded stepchild of Parkinson's disease. Biochem Pharmacol. (2007) 74:177–90. doi: 10.1016/j.bcp.2007.01.036
122. Baloyannis SJ, Costa V, Baloyannis IS. Morphological alterations of the synapses in the locus coeruleus in Parkinson's disease. J Neurol Sci. (2006) 248:35–41. doi: 10.1016/j.jns.2006.05.006
123. Hawkes CH, Del Tredici K, Braak H. A timeline for Parkinson's disease. Parkinsonism Relat Disord. (2010) 16:79–84. doi: 10.1016/j.parkreldis.2009.08.007
124. Double KL, Gerlach M, Schünemann V, Trautwein AX, Zecca L, Gallorini M, et al. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem Pharmacol. (2003) 66:489–94. doi: 10.1016/S0006-2952(03)00293-4
125. Sulzer D, Cassidy C, Horga G, Kang UJ, Fahn S, Casella L, et al. Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson's disease. NPJ Parkinson's Dis. (2018) 4:11. doi: 10.1038/s41531-018-0047-3
126. Wang J, Li Y, Huang Z, Wan W, Zhang Y, Wang C, et al. Neuromelanin-sensitive magnetic resonance imaging features of the substantia nigra and locus coeruleus in de novo Parkinson's disease and its phenotypes. Eur J Neurol. (2018) 25:949–e73. doi: 10.1111/ene.13628
127. Zecca L, Casella L, Albertini A, Bellei C, Zucca FA, Engelen M, et al. Neuromelanin can protect against mediated oxidative damage in system modeling iron overload of brain aging and Parkinson's disease. J Neurochem. (2008) 106:1866–75. doi: 10.1111/j.1471-4159.2008.05541.x
128. Zucca FA, Basso E, Cupaioli FA, Ferrari E, Sulzer D, Casella L, et al. Neuromelanin of the human substantia Nigra: an update. Neurotox Res. (2013) 25:13–23. doi: 10.1007/s12640-013-9435-y
129. Liu KY, Acosta-Cabronero J, Cardenas-Blanco A, Loane C, Berry AJ, Betts MJ, et al. In vivo visualization of age-related differences in the locus coeruleus. Neurobiol Aging. (2019) 74:101–11. doi: 10.1016/j.neurobiolaging.2018.10.014
130. Manaye KF, McIntire DD, Mann DMA, German DC. Locus coeruleus cell loss in the aging human brain: a non-random process. J Compar Neurol. (1995) 358:79–87. doi: 10.1002/cne.903580105
131. Mann DMA, Yates PO. Lipoprotein pigments-their relationship to aging in the human nervous system. Brain. (1974) 97:489–98. doi: 10.1093/brain/97.1.489
132. Zucca FA, Bellei C, Giannelli S, Terreni MR, Gallorini M, Rizzio E, et al. Neuromelanin and iron in human locus coeruleus and substantia nigra during aging: consequences for neuronal vulnerability. J Neural Trans. (2006) 113:757–67. doi: 10.1007/s00702-006-0453-2
133. Zecca L, Zucca F, Albertini A, Rizzio E, Fariello RG. A proposed dual role of neuromelanin in the pathogenesis of Parkinson' s disease. Neurology. (2006) 67:S8–S11. doi: 10.1212/WNL.67.7_suppl_2.S8
134. Betts MJ, Cardenas-Blanco A, Kanowski M, Jessen F, Düzel E. In vivo MRI assessment of the human locus coeruleus along its rostrocaudal extent in young and older adults. NeuroImage. (2017) 163:150–9. doi: 10.1016/j.neuroimage.2017.09.042
135. Hämmerer D, Callaghan MF, Hopkins A, Kosciessa J, Betts M, Cardenas-Blanco A, et al. Locus coeruleus integrity in old age is selectively related to memories linked with salient negative events. Proc Natl Acad Sci USA. (2018) 115:2228–33. doi: 10.1073/pnas.1712268115
136. Priovoulos N, Jacobs HIL, Ivanov D, Uludag K, Verhey FRJ, Poser BA. High-resolution in vivo imaging of human locus coeruleus by magnetization transfer MRI at 3T and 7T. NeuroImage. (2018) 168:427–36. doi: 10.1016/j.neuroimage.2017.07.045
137. Sasaki M, Shibata E, Tohyama K, Takahashi J, Otsuka K, Tsuchiya K, et al. Neuromelanin magnetic resonance imaging of locus ceruleus and substantia nigra in Parkinson's disease. NeuroReport. (2006) 17:1215–8. doi: 10.1097/01.wnr.0000227984.84927.a7
138. Trujillo P, Petersen KJ, Cronin MJ, Lin Y-C, Kang H, Donahue MJ, et al. Quantitative magnetization transfer imaging of the human locus coeruleus. NeuroImage. (2019) 200:191–98. doi: 10.1016/j.neuroimage.2019.06.049
139. Miyoshi F, Ogawa T, Kitao S-I, Kitayama M, Shinohara Y, Takasugi M, et al. Evaluation of Parkinson disease and Alzheimer disease with the use of neuromelanin MR imaging and123I-metaiodobenzylguanidine scintigraphy. Am J Neuroradiol. (2013) 34:2113–8. doi: 10.3174/ajnr.A3567
140. Ehrminger M, Latimier A, Pyatigorskaya N, Garcia-Lorenzo D, Leu-Semenescu S, Vidailhet M, et al. The coeruleus/subcoeruleus complex in idiopathic rapid eye movement sleep behaviour disorder. Brain. (2016) 139:1180–8. doi: 10.1093/brain/aww006
141. García-Lorenzo D, Longo-Dos Santos C, Ewenczyk C, Leu-Semenescu S, Gallea C, Quattrocchi G, et al. The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson's disease. Brain. (2013) 136:2120–9. doi: 10.1093/brain/awt152
142. Chen X, Huddleston DE, Langley J, Ahn S, Barnum CJ, Factor SA, et al. Simultaneous imaging of locus coeruleus and substantia nigra with a quantitative neuromelanin MRI approach. Magnetic Resonance Imaging. (2014) 32:1301–6. doi: 10.1016/j.mri.2014.07.003
143. Betts MJ, Kirilina E, Otaduy MCG, Ivanov D, Acosta-Cabronero J, Callaghan MF, et al. Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain. (2019) 142:2558–71. doi: 10.1093/brain/awz193
144. Liu KY, Marijatta F, Hämmerer D, Acosta-Cabronero J, Düzel E, Howard RJ. Magnetic resonance imaging of the human locus coeruleus: a systematic review. Neurosci Biobehav Rev. (2017) 83:325–55. doi: 10.1016/j.neubiorev.2017.10.023
145. Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol. (2009) 8:464–74. doi: 10.1016/S1474-4422(09)70068-7
146. Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. (2013) 14:744–8. doi: 10.1016/j.sleep.2012.10.009
147. Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson's disease in primary care: a case-control study. Lancet Neurol. (2015) 14:57–64. doi: 10.1016/S1474-4422(14)70287-X
148. Knudsen K, Fedorova TD, Hansen AK, Sommerauer M, Otto M, Svendsen KB, et al. In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol. (2018) 17:618–28. doi: 10.1016/S1474-4422(18)30162-5
149. Braak H, Rüb U, Del Tredici K. Cognitive decline correlates with neuropathological stage in Parkinson's disease. J Neurol Sci. (2006) 248:255–8. doi: 10.1016/j.jns.2006.05.011
150. Chan-Palay V, Asan E. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J Compar Neurol. (1989) 287:373–92. doi: 10.1002/cne.902870308
151. Szot P. Common factors among Alzheimer's disease, Parkinson's disease, and epilepsy: possible role of the noradrenergic nervous system. Epilepsia. (2012) 53:61–6. doi: 10.1111/j.1528-1167.2012.03476.x
152. De Oliveira RB, Petiz LL, Lim R, Lipski J, Gravina FS, Brichta AM, et al. Crosstalk between mitochondria, calcium channels and actin cytoskeleton modulates noradrenergic activity of locus coeruleus neurons. J Neurochem. (2019) 149:471–87. doi: 10.1111/jnc.14692
153. Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron. (2013) 79:1044–66. doi: 10.1016/j.neuron.2013.09.004
154. Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. -Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. (2010) 329:1663–7. doi: 10.1126/science.1195227
155. Imaizumi T, Yamashita K, Taima K, Ishikawa A, Yoshida H, Satoh K. Effect of peroxisome proliferator-activated receptor-γ ligands on the expression of retinoic acid-inducible gene-I in endothelial cells stimulated with lipopolysaccharide. Prostaglandins Other Lipid Mediat. (2005) 78:46–54. doi: 10.1016/j.prostaglandins.2005.02.006
156. Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, et al. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. (2006) 26:11915–22. doi: 10.1523/JNEUROSCI.3821-06.2006
157. Mosharov EV. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. (2006) 26:9304–11. doi: 10.1523/JNEUROSCI.0519-06.2006
158. Duda J, Pötschke C, Liss B. Converging roles of ion channels, calcium, metabolic stress, and activity pattern of substantia nigradopaminergic neurons in health and Parkinson's disease. J Neurochem. (2016) 139:156–78. doi: 10.1111/jnc.13572
159. Michel PP, Hirsch EC, Hunot S. Understanding dopaminergic cell death pathways in parkinson disease. Neuron. (2016) 90:675–91. doi: 10.1016/j.neuron.2016.03.038
160. Ottolini D, Calí T, Szabò I, Brini M. Alpha-synuclein at the intracellular and the extracellular side: functional and dysfunctional implications. Biol. Chem. (2017) 398. doi: 10.1515/hsz-2016-0201
161. Angelova PR, Ludtmann MHR, Horrocks MH, Negoda A, Cremades N, Klenerman D, et al. Ca2+is a key factor in α-synuclein-induced neurotoxicity. J Cell Sci. (2016) 129:1792–801. doi: 10.1242/jcs.180737
162. Bourdenx M, Bezard E, Dehay B. Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front Neuroanatomy. (2014) 8:83. doi: 10.3389/fnana.2014.00083
163. Subramaniam M, Althof D, Gispert S, Schwenk J, Auburger G, Kulik A, et al. Mutant α-synuclein enhances firing frequencies in dopamine substantia Nigra neurons by oxidative impairment of A-type potassium channels. J Neurosci. (2014) 34:13586–99. doi: 10.1523/JNEUROSCI.5069-13.2014
164. Ding TT, Lee S-J, Rochet J-C, Lansbury PT. Annular α-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes†. Biochemistry. (2002) 41:10209–17. doi: 10.1021/bi020139h
165. Gosavi N, Lee H-J, Lee JS, Patel S, Lee S-J. Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem. (2002) 277:48984–92. doi: 10.1074/jbc.M208194200
166. Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, et al. α-synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. (2002) 322:1089–102. doi: 10.1016/S0022-2836(02)00735-0
167. Fischer AF, Matera KM. Stabilization of alpha-synuclein oligomers in vitro by the neurotransmitters, dopamine and norepinephrine: the effect of oxidized catecholamines. Neurochem Res. (2015) 40:1341–9. doi: 10.1007/s11064-015-1597-y
168. Hirschberg S, Li Y, Randall A, Kremer EJ, Pickering AE. Functional dichotomy in spinal- vs prefrontal-projecting locus coeruleus modules splits descending noradrenergic analgesia from ascending aversion and anxiety in rats. eLife. (2017) 6:e29808. doi: 10.7554/eLife.29808.027
169. Kebschull JM, Garcia da Silva P, Reid AP, Peikon ID, Albeanu DF, Zador AM. High-throughput mapping of single-neuron projections by sequencing of barcoded RNA. Neuron. (2016) 91:975–87. doi: 10.1016/j.neuron.2016.07.036
170. Loughlin SE, Foote SL, Grzanna R. Efferent projections of nucleus locus coeruleus: morphologic subpopulations have different efferent targets. Neuroscience. (1986) 18:307–19. doi: 10.1016/0306-4522(86)90156-9
171. Simpson K, Lin R. Brain Norepinephrine: Neurobiology and Therapeutics. Cambridge: Cambridge University Press (2007).
172. Kelly SC, He B, Perez SE, Ginsberg SD, Mufson EJ, Counts SE. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer's disease. Acta Neuropathol Commun. (2017) 5:8. doi: 10.1186/s40478-017-0411-2
173. McMillan PJ, White SS, Franklin A, Greenup JL, Leverenz JB, Raskind MA, et al. Differential response of the central noradrenergic nervous system to the loss of locus coeruleus neurons in Parkinson's disease and Alzheimer's disease. Brain Res. (2011) 1373:240–52. doi: 10.1016/j.brainres.2010.12.015
174. Szot P. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer's disease and dementia with lewy bodies. J Neurosci. (2006) 26:467–78. doi: 10.1523/JNEUROSCI.4265-05.2006
175. Theofilas P, Ehrenberg AJ, Nguy A, Thackrey JM, Dunlop S, Mejia MB, et al. Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer's disease Braak stages: a quantitative study in humans. Neurobiol Aging. (2018) 61:1–12. doi: 10.1016/j.neurobiolaging.2017.09.007
176. Braak H, Del Tredici K. Where, when, and in what form does sporadic Alzheimer's disease begin? Curr Opin Neurol. (2012) 25:708–14. doi: 10.1097/WCO.0b013e32835a3432
177. Braak H, Tredici KD, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
178. Delaville C, Deurwaerdère PD, Benazzouz A. Noradrenaline and Parkinson's Disease. Front Syst Neurosci. (2011) 5:31. doi: 10.3389/fnsys.2011.00031
179. Herrero MT, Hirsch EC, Kastner A, Ruberg M, Luquin MR, Laguna J, et al. Does neuromelanin contribute to the vulnerability of catecholaminergic neurons in monkeys intoxicated with MPTP? Neuroscience. (1993) 56:499–511. doi: 10.1016/0306-4522(93)90349-K
180. Iversen LL, Rossor MN, Reynolds GP, Hills R, Roth M, Mountjoy CQ, et al. Loss of pigmented dopamine-β-hydroxylase positive cells from locus coeruleus in senile dementia of alzheimer's type. Neurosci Lett. (1983) 39:95–100. doi: 10.1016/0304-3940(83)90171-4
181. Elman JA, Panizzon MS, Hagler DJ, Eyler LT, Granholm EL, Fennema-Notestine C, et al. Task-evoked pupil dilation and BOLD variance as indicators of locus coeruleus dysfunction. Cortex. (2017) 97:60–9. doi: 10.1016/j.cortex.2017.09.025
182. Wang T, Zhang Q-J, Liu J, Wu Z-H, Wang S. Firing activity of locus coeruleus noradrenergic neurons increases in a rodent model of Parkinsonism. Neurosci Bull. (2009) 25:15–20. doi: 10.1007/s12264-009-1023-z
183. Key J, Mueller AK, Gispert S, Matschke L, Wittig I, Corti O, et al. Ubiquitylome profiling of Parkin-null brain reveals dysregulation of calcium homeostasis factors ATP1A2, Hippocalcin and GNA11, reflected by altered firing of noradrenergic neurons. Neurobiol Dis. (2019) 127:114–30. doi: 10.1016/j.nbd.2019.02.008
184. Miguelez C, Grandoso L, Ugedo L. Locus coeruleus and dorsal raphe neuron activity and response to acute antidepressant administration in a rat model of Parkinson's disease. Int J Neuropsychopharmacol. (2010) 14:187–200. doi: 10.1017/S146114571000043X
185. Alachkar A, Brotchie JM, Jones OT. Changes in the mRNA levels of α2A and α2C adrenergic receptors in rat models of Parkinson's Disease and l-DOPA-induced dyskinesia. J Mol Neurosci. (2011) 46:145–52. doi: 10.1007/s12031-011-9539-x
186. Miguelez C, Aristieta A, Cenci MA, Ugedo L. The locus coeruleus is directly implicated in L-DOPA-induced dyskinesia in parkinsonian rats: an electrophysiological and behavioural study. PLoS ONE. (2011) 6:e24679. doi: 10.1371/journal.pone.0024679
187. Henrich MT, Geibl FF, Lee B, Chiu W-H, Koprich JB, Brotchie JM, et al. A53T-α-synuclein overexpression in murine locus coeruleus induces Parkinson's disease-like pathology in neurons and glia. Acta Neuropathol Commun. (2018) 6:39. doi: 10.1186/s40478-018-0541-1
188. Sotiriou E, Vassilatis DK, Vila M, Stefanis L. Selective noradrenergic vulnerability in alpha-synuclein transgenic mice. Neurobiol Aging. (2010) 31:2103–14. doi: 10.1016/j.neurobiolaging.2008.11.010
189. Eschenko O, Magri C, Panzeri S, Sara SJ. Noradrenergic neurons of the locus coeruleus are phase locked to cortical up-down states during sleep. Cerebral Cortex. (2011) 22:426–35. doi: 10.1093/cercor/bhr121
190. Lestienne R, Hervé-Minvielle A, Robinson D, Briois L, Sara S. Slow oscillations as a probe of the dynamics of the locus coeruleus-frontal cortex interaction in anesthetized rats. J PhysiolParis. (1997) 91:273–84. doi: 10.1016/S0928-4257(97)82407-2
191. Safaai H, Neves R, Eschenko O, Logothetis NK, Panzeri S. Modeling the effect of locus coeruleus firing on cortical state dynamics and single-trial sensory processing. Proc Natl Acad Sci USA. (2015) 112:12834–9. doi: 10.1073/pnas.1516539112
192. Totah NK, Neves RM, Panzeri S, Logothetis NK, Eschenko O. The locus coeruleus is a complex and differentiated neuromodulatory system. Neuron. (2018) 99:1055–68.e6. doi: 10.1016/j.neuron.2018.07.037
193. Frodl-Bauch T, Bottlender R, Hegerl U. Neurochemical substrates and neuroanatomical generators of the event-related P300. Neuropsychobiology. (1999) 40:86–94. doi: 10.1159/000026603
194. Huang W-J, Chen W-W, Zhang X. The neurophysiology of P 300 - an integrated review. Eur Rev Med Pharmacol Sci. (2015) 19:1480–8.
195. Nieuwenhuis S, Aston-Jones G, Cohen JD. Decision making, the P3, and the locus coeruleus–norepinephrine system. Psychol Bull. (2005) 131:510–32. doi: 10.1037/0033-2909.131.4.510
196. Polich J. Updating P300: an integrative theory of P3a and P3b. Clin Neurophysiol. (2007) 118:2128–48. doi: 10.1016/j.clinph.2007.04.019
197. Murphy PR, Robertson IH, Balsters JH, O'connell RG. Pupillometry and P3 index the locus coeruleus-noradrenergic arousal function in humans. Psychophysiology. (2011) 48:1532–43. doi: 10.1111/j.1469-8986.2011.01226.x
198. De Taeye L, Vonck K, van Bochove M, Boon P, Van Roost D, Mollet L, et al. The P3 event-related potential is a biomarker for the efficacy of vagus nerve stimulation in patients with epilepsy. Neurotherapeutics. (2014) 11:612–22. doi: 10.1007/s13311-014-0272-3
199. Nieuwenhuis S, De Geus EJ, Aston-Jones G. The anatomical and functional relationship between the P3 and autonomic components of the orienting response. Psychophysiology. (2011) 48:162–75. doi: 10.1111/j.1469-8986.2010.01057.x
200. Antal A, Pfeiffer R, Bodis-Wollner I. Simultaneously evoked primary and cognitive visual evoked potentials distinguish younger and older patients with Parkinson's disease. J Neural Transm. (1996) 103:1053–67. doi: 10.1007/BF01291790
201. Hansch EC, Syndulko K, Cohen SN, Goldberg ZI, Potvin AR, Tourtellotte WW. Cognition in Parkinson disease: an event-related potential perspective. Ann Neurol. (1982) 11:599–607. doi: 10.1002/ana.410110608
202. Li M, Kuroiwa Y, Wang L, Kamitani T, Takahashi T, Suzuki Y, et al. Early sensory information processes are enhanced on visual oddball and S1-S2 tasks in Parkinson's disease: a visual event-related potentials study. Parkinsonism Relat Disord. (2003) 9:329–40. doi: 10.1016/S1353-8020(02)00094-9
203. Özmüş G, Yerlikaya D, Gökçeoglu A, Emek Savaş DD, Çakmur R, Dönmez Çolakoglu B, et al. Demonstration of early cognitive impairment in Parkinson's disease with visual P300 responses. Noro Psikiyatr Ars. (2017) 54:21–7. doi: 10.5152/npa.2016.12455
204. Philipova D, Gatchev G, Vladova T, Georgiev D. Event-related potentials in Parkinsonian patients under auditory discrimination tasks. Int J Psychophysiol. (1997) 27:69–78. doi: 10.1016/S0167-8760(97)00783-6
205. Wang L, Kuroiwa Y, Kamitani T, Takahashi T, Suzuki Y, Hasegawa O. Effect of interstimulus interval on visual P300 in Parkinson's disease. J Neurol Neurosurg Psychiatry. (1999) 67:497–503. doi: 10.1136/jnnp.67.4.497
206. Wang H, Wang Y, Wang D, Cui L, Tian S, Zhang Y. Cognitive impairment in Parkinson's disease revealed by event-related potential N270. J Neurol Sci. (2002) 194:49–53. doi: 10.1016/S0022-510X(01)00674-8
207. Tachibana H, Aragane K, Kawabata K, Sugita M. P3 latency change in aging and Parkinson disease. Arch Neurol. (1997) 54:296–302. doi: 10.1001/archneur.1997.00550150054016
208. Toda K, Tachibana H, Sugita M, Konishi K. P300 and reaction time in Parkinson's disease. J Geriatric Psychiatry Neurol. (1993) 6:131–6. doi: 10.1177/089198879300600301
209. Dauwels J, Vialatte F, Cichocki A. Diagnosis of Alzheimers disease from EEG signals: where are we standing? Curr Alzheimer Res. (2010) 7:487–505. doi: 10.2174/156720510792231720
210. Drago V, Babiloni C, Bartrés-Faz D, Caroli A, Bosch B, Hensch T, et al. Disease tracking markers for Alzheimer's disease at the prodromal (MCI) stage. J Alzheimer's Dis. (2011) 26:159–99. doi: 10.3233/JAD-2011-0043
211. Fruehwirt W, Dorffner G, Roberts S, Gerstgrasser M, Grossegger D, Schmidt R, et al. Associations of event-related brain potentials and Alzheimer's disease severity: a longitudinal study. Progress Neuro Psychopharmacol Biol Psychiatry. (2019) 92:31–8. doi: 10.1016/j.pnpbp.2018.12.013
212. Howe AS, Bani-Fatemi A, De Luca V. The clinical utility of the auditory P300 latency subcomponent event-related potential in preclinical diagnosis of patients with mild cognitive impairment and Alzheimer's disease. Brain Cogn. (2014) 86:64–74. doi: 10.1016/j.bandc.2014.01.015
213. Olichney JM, Yang J-C, Taylor J, Kutas M. Cognitive event-related potentials: biomarkers of synaptic dysfunction across the stages of Alzheimer's disease. J Alzheimer's Dis. (2011) 26:215–28. doi: 10.3233/JAD-2011-0047
214. Bassetti CL, Bargiotas P. REM sleep behavior disorder. Front Neurol Neurosci. (2017) 41:104–16. doi: 10.1159/000478914
215. Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. (2007) 130:2770–88. doi: 10.1093/brain/awm056
216. Hendricks JC, Morrison AR, Mann GL. Different behaviors during paradoxical sleep without atonia depend on pontine lesion site. Brain Res. (1982) 239:81–105. doi: 10.1016/0006-8993(82)90835-6
217. Boissard R, Fort P, Gervasoni D, Barbagli B, Luppi P-H. Localization of the GABAergic and non-GABAergic neurons projecting to the sublaterodorsal nucleus and potentially gating paradoxical sleep onset. Eur J Neurosci. (2003) 18:1627–39. doi: 10.1046/j.1460-9568.2003.02861.x
218. Iranzo A. The REM sleep circuit and how its impairment leads to REM sleep behavior disorder. Cell Tissue Res. (2018) 373:245–66. doi: 10.1007/s00441-018-2852-8
219. Jessen NA, Munk ASF, Lundgaard I, Nedergaard M. The glymphatic system: a beginner's guide. Neurochem Res. (2015) 40:2583–99. doi: 10.1007/s11064-015-1581-6
220. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. (2013) 342:373–7. doi: 10.1126/science.1241224
221. Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. (2004) 10:S10–7. doi: 10.1038/nm1066
222. Li Y, Hickey L, Perrins R, Werlen E, Patel AA, Hirschberg S, et al. Retrograde optogenetic characterization of the pontospinal module of the locus coeruleus with a canine adenoviral vector. Brain Res. (2016) 1641:274–90. doi: 10.1016/j.brainres.2016.02.023
223. Swift KM, Gross BA, Frazer MA, Bauer DS, Clark KJD, Vazey EM, et al. Abnormal locus coeruleus sleep activity alters sleep signatures of memory consolidation and impairs place cell stability and spatial memory. Curr Biol. (2018) 28:3599–609.e4. doi: 10.1016/j.cub.2018.09.054
224. Boyce R, Glasgow SD, Williams S, Adamantidis A. Causal evidence for the role of REM sleep theta rhythm in contextual memory consolidation. Science. (2016) 352:812–6. doi: 10.1126/science.aad5252
226. Lapiz MDS, Bondi CO, Morilak DA. Chronic treatment with desipramine improves cognitive performance of rats in an attentional set-shifting test. Neuropsychopharmacology. (2006) 32:1000–10. doi: 10.1038/sj.npp.1301235
227. Tait DS, Brown VJ, Farovik A, Theobald DE, Dalley JW, Robbins TW. Lesions of the dorsal noradrenergic bundle impair attentional set-shifting in the rat. Eur J Neurosci. (2007) 25:3719–24. doi: 10.1111/j.1460-9568.2007.05612.x
228. McGaughy J, Ross RS, Eichenbaum H. Noradrenergic, but not cholinergic, deafferentation of prefrontal cortex impairs attentional set-shifting. Neuroscience. (2008) 153:63–71. doi: 10.1016/j.neuroscience.2008.01.064
229. Janitzky K, Lippert MT, Engelhorn A, Tegtmeier J, Goldschmidt J, Heinze H-J, et al. (2015). Optogenetic silencing of locus coeruleus activity in mice impairs cognitive flexibility in an attentional set-shifting task. Front Behav Neurosci. 9:286. doi: 10.3389/fnbeh.2015.00286
230. Chandler D, Waterhouse BD. Evidence for broad versus segregated projections from cholinergic and noradrenergic nuclei to functionally and anatomically discrete subregions of prefrontal cortex. Front Behav Neurosci. (2012) 6:20. doi: 10.3389/fnbeh.2012.00020
231. Farina E, Gattellaro G, Pomati S, Magni E, Perretti A, Cannata AP, et al. Researching a differential impairment of frontal functions and explicit memory in early Parkinson's disease. Eur J Neurol. (2000) 7:259–67. doi: 10.1046/j.1468-1331.2000.00066.x
232. Oberman LM, Pascual-Leone A. Hyperplasticity in Autism Spectrum Disorder confers protection from Alzheimer's disease. Med Hypotheses. (2014) 83:337–42. doi: 10.1016/j.mehy.2014.06.008
233. Cirelli C, Tononi G. Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J Neurosci. (2000) 20:9187–94. doi: 10.1523/JNEUROSCI.20-24-09187.2000
234. Fluegge K. Does MeCP2 deficiency in autism confer protection against later development of Alzheimer's disease? A reply to Oberman and Pascual-Leone. Med Hypotheses. (2016) 92:18–20. doi: 10.1016/j.mehy.2016.04.020
235. Braun D, Madrigal JL, Feinstein DL. Noradrenergic regulation of glial activation: molecular mechanisms and therapeutic implications. Curr Neuropharmacol. (2014) 12:342–52. doi: 10.2174/1570159X12666140828220938
236. Robertson IH. A noradrenergic theory of cognitive reserve: implications for Alzheimer's disease. Neurobiol Aging. (2013) 34:298–308. doi: 10.1016/j.neurobiolaging.2012.05.019
237. Yang JH, Lee EO, Kim SE, Suh Y-H, Chong YH. Norepinephrine differentially modulates the innate inflammatory response provoked by amyloid-β peptide via action at β-adrenoceptors and activation of cAMP/PKA pathway in human THP-1 macrophages. Exp Neurol. (2012) 236:199–206. doi: 10.1016/j.expneurol.2012.05.008
238. Mather M, Clewett D, Sakaki M, Harley CW. Norepinephrine ignites local hotspots of neuronal excitation: how arousal amplifies selectivity in perception and memory. Behav Brain Sci. (2016) 39:e200. doi: 10.1017/S0140525X15000667
239. Gannon M, Wang Q. Complex noradrenergic dysfunction in Alzheimer's disease: low norepinephrine input is not always to blame. Brain Res. (2019) 1702:12–6. doi: 10.1016/j.brainres.2018.01.001
240. Giubilei F, Calderaro C, Antonini G, Sepe-Monti M, Tisei P, Brunetti E, et al. Increased lymphocyte dopamine β-hydroxylase immunoreactivity in Alzheimer's disease: compensatory response to cholinergic deficit? Dement Geriatric Cogn Disord. (2004) 18:338–41. doi: 10.1159/000080128
241. Miyata S, Nagata H, Yamao S, Nakamura S, Kameyama M. Dopamine-β-hydroxylase activities in serum and cerebrospinal fluid of aged and demented patients. J Neurol Sci. (1984) 63:403–9. doi: 10.1016/0022-510X(84)90163-1
242. Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA. Changes in adrenoreceptors in the prefrontal cortex of subjects with dementia: evidence of compensatory changes. Neuroscience. (2007) 146:471–80. doi: 10.1016/j.neuroscience.2007.01.031
243. Colzato LS, Wolters G, Peifer C. Transcutaneous vagus nerve stimulation (tVNS) modulates flow experience. Exp Brain Res. (2017) 236:253–7. doi: 10.1007/s00221-017-5123-0
244. Peuker ET, Filler TJ. The nerve supply of the human auricle. Clin Anatomy. (2002) 15:35–7. doi: 10.1002/ca.1089
245. Butt MF, Albusoda A, Farmer AD, Aziz Q. The anatomical basis for transcutaneous auricular vagus nerve stimulation. J Anatomy. (2020) 236:588–611. doi: 10.1111/joa.13122
246. Groves DA, Bowman EM, Brown VJ. Recordings from the rat locus coeruleus during acute vagal nerve stimulation in the anaesthetised rat. Neurosci Lett. (2005) 379:174–9. doi: 10.1016/j.neulet.2004.12.055
247. Van Bockstaele EJ, Peoples J, Telegan P. Efferent projections of the nucleus of the solitary tract to peri-locus coeruleus dendrites in rat brain: evidence for a monosynaptic pathway. J Comp Neurol. (1999) 412:410–28. doi: 10.1002/(SICI)1096-9861(19990927)412:3<410::AID-CNE3>3.0.CO;2-F
248. Hulsey DR, Riley JR, Loerwald KW, Rennaker RL, Kilgard MP, Hays SA. Parametric characterization of neural activity in the locus coeruleus in response to vagus nerve stimulation. Exp Neurol. (2017) 289:21–30. doi: 10.1016/j.expneurol.2016.12.005
249. Ruffoli R, Giorgi FS, Pizzanelli C, Murri L, Paparelli A, Fornai F. The chemical neuroanatomy of vagus nerve stimulation. J Chem Neuroanatomy. (2011) 42:288–96. doi: 10.1016/j.jchemneu.2010.12.002
250. Williams JT, Bobker DH, Harris GC. Synaptic potentials in locus coeruleus neurons in brain slices. Neurobiol Locus Coeruleus. (1991) 167–72. doi: 10.1016/S0079-6123(08)63806-6
251. Dorr AE. Effect of vagus nerve stimulation on serotonergic and noradrenergic transmission. J Pharmacol Exp Therapeutics. (2006) 318:890–8. doi: 10.1124/jpet.106.104166
252. Manta S, Dong J, Debonnel G, Blier P. Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J Psychiatry Neurosci. (2009) 34:272–80.
253. Manta S, El Mansari M, Debonnel G, Blier P. Electrophysiological and neurochemical effects of long-term vagus nerve stimulation on the rat monoaminergic systems. Int J Neuropsychopharmacol. (2012) 16:459–70. doi: 10.1017/S1461145712000387
254. Florin-Lechner SM, Druhan JP, Aston-Jones G, Valentino RJ. Enhanced norepinephrine release in prefrontal cortex with burst stimulation of the locus coeruleus. Brain Res. (1996) 742:89–97. doi: 10.1016/S0006-8993(96)00967-5
255. Roosevelt RW, Smith DC, Clough RW, Jensen RA, Browning RA. Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain Res. (2006) 1119:124–32. doi: 10.1016/j.brainres.2006.08.048
256. Burger AM, D'Agostini M, Verkuil B, Van Diest I. Moving beyond belief: a narrative review of potential biomarkers for transcutaneous vagus nerve stimulation. Psychophysiology. (2020) e13571. doi: 10.1111/psyp.13571
257. Lewine JD, Paulson K, Bangera N, Simon BJ. Exploration of the impact of brief noninvasive vagal nerve stimulation on EEG and event-related potentials. Neuromodulation. (2019) 22:564–72. doi: 10.1111/ner.12864
258. Rufener KS, Geyer U, Janitzky K, Heinze H-J, Zaehle T. Modulating auditory selective attention by non-invasive brain stimulation: differential effects of transcutaneous vagal nerve stimulation and transcranial random noise stimulation. Eur J Neurosci. (2018) 48:2301–9. doi: 10.1111/ejn.14128
259. Ventura-Bort C, Wirkner J, Genheimer H, Wendt J, Hamm AO, Weymar M. Effects of transcutaneous vagus nerve stimulation (tVNS) on the P300 and alpha-amylase level: a pilot study. Front Hum Neurosci. (2018) 12:202. doi: 10.3389/fnhum.2018.00202
260. Warren C, Tona K, Ouwerkerk L, van Paridon J, Poletiek F, van Steenbergen H, et al. The neuromodulatory and hormonal effects of transcutaneous vagus nerve stimulation as evidenced by salivary alpha amylase, salivary cortisol, pupil diameter, and the P3 event-related potential. Brain Stimul. (2019) 12:635–42. doi: 10.1016/j.brs.2018.12.224
261. Fischer R, Ventura-Bort C, Hamm A, Weymar M. Transcutaneous vagus nerve stimulation (tVNS) enhances conflict-triggered adjustment of cognitive control. Cogn Affect Behav Neurosci. (2018) 18:1–14. doi: 10.3758/s13415-018-0596-2
Keywords: locus coeruleus, phasic and tonic discharge, Alzheimers's disease, Parkinson's disease, norepinephrine, transcutaneous vagal stimulation, neurodegeneration, neuroprotection
Citation: Janitzky K (2020) Impaired Phasic Discharge of Locus Coeruleus Neurons Based on Persistent High Tonic Discharge—A New Hypothesis With Potential Implications for Neurodegenerative Diseases. Front. Neurol. 11:371. doi: 10.3389/fneur.2020.00371
Received: 20 December 2019; Accepted: 14 April 2020;
Published: 12 May 2020.
Edited by:
Sabine Liebscher, Ludwig-Maximilians-Universität München, GermanyReviewed by:
Cristina Miguelez, University of the Basque Country, SpainMatthew Betts, German Center for Neurodegenerative Diseases (DZNE), Germany
Copyright © 2020 Janitzky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathrin Janitzky, ay5qYW5pdHpreUBnbXguZGU=