Abstract
Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative disease that is defined by loss of upper and lower motor neurons, associated with accumulation of protein aggregates in cells. There is also pathology in extra-motor areas of the brain, Possible causes of cell death include failure to deal with the aggregated proteins, glutamate toxicity and mitochondrial failure. ALS also involves abnormalities of metabolism and the immune system, including neuroinflammation in the brain and spinal cord. Strikingly, there are also abnormalities of the peripheral immune system, with alterations of T lymphocytes, monocytes, complement and cytokines in the peripheral blood of patients with ALS. The precise contribution of the peripheral immune system in ALS pathogenesis is an active area of research. Although some trials of immunomodulatory agents have been negative, there is strong preclinical evidence of benefit from immune modulation and further trials are currently underway. Here, we review the emerging evidence implicating peripheral immune alterations contributing to ALS, and their potential as future therapeutic targets for clinical intervention.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease, defined by the presence of muscle weakness and the progressive death of upper and lower motor neurons (1). ALS leads to respiratory failure with the length of survival being predicted by respiratory muscle weakness (2). However, ALS is more than just a motor neurone disease. ALS also has extra-motor features, including cognitive and behavioral disturbance (3–5). ALS is markedly heterogeneous in clinical features, such as site of onset of weakness and rate of progression (6, 7), and is more common in men than in women (8).
ALS can be sporadic (SALS) or familial (FALS), although the distinction can be difficult to assign (9). Genetic susceptibility (10, 11) and environmental exposure (12) contribute to the pathogenesis of ALS, possibly through a multi-stage process (13, 14). Causative genes exist in patients with FALS, and mutations in these genes occur in some patients with SALS (15). Calculations suggest that 61% of the variance in risk of developing ALS is due to genetic factors (16), which means that ~40% of the variance in risk is due to non-genetic factors, which could include environmental exposures. The pathological features of ALS include aggregation of insoluble protein within cells (17), but the type of protein aggregate varies among patients. It has been thought that the majority of patients have accumulation of tar DNA binding protein 43 (TDP-43), (as well as others), with a small group of patients having accumulations of superoxide dismutase 1 (SOD1) (18–20). However, recent evidence suggests that SOD1 may aggregate in the spinal cord in a majority of ALS patients (21, 22). The genes that cause ALS usually encode for proteins or polypeptides that accumulate within cells or are involved in the metabolism of protein aggregates (19, 23). There is evidence that some of the aggregated proteins can transfer from cell to cell in a prion-like fashion (24, 25) which could explain the characteristic spread of weakness from the site of onset to other regions.
A number of possible pathways of disease have been described, including mitochondrial dysfunction, glutamate excitotoxicity (26, 27), problems with autophagy (28) and altered RNA metabolism (29). Furthermore, the death of motor neurons can be “non-cell autonomous,” meaning that other types of cells such as astrocytes, microglia and possibly oligodendrocytes can drive motor neuron death (30, 31). There has been considerable research on the type of cell death that occurs in ALS. It has been previously thought that neuronal cell death in ALS is due to apoptosis (32–35) which is mediated through caspases. Evidence for apoptosis in ALS has been found with TUNEL staining of human tissues (36) and with measurements of bcl-2 (37). Others found increased p53 in ALS (38). In ALS there is also evidence of caspase activation (35). However, more recently there has been a suggestion that necroptosis, an inflammatory form of cell death which is caspase independent and involves RIP kinase activation, is a common form of cell death in neurodegenerative disease (39). Necroptosis is the mechanism of cell death from glutamate toxicity (40), which is one of the most important mechanisms proposed for the pathogenesis of ALS. There is evidence that necroptosis occurs in a cell culture model of ALS (41). Mutations in optineurin, a rare genetic cause of ALS, allow the activation of RIP kinases to promote necroptosis (42). More recently still, ferroptosis, an oxidative form of cell death (43), has been reported to occur in ALS (44).
The death of motor neurons, possibly stimulated by the pathways described above, and occurring through one of the types of cell death described above, is the cardinal feature of ALS. However, the pathology of ALS in the brain and spinal cord also involves more than death of motor neurons, with evidence of involvement of the immune system (45). There is neuroinflammation with microglial activation and a modest level of T lymphocyte infiltration (46–48). In ALS patients, microglial activation is visible with PET imaging suggestive of an ongoing neuroinflammatory process (49). Such inflammatory pathology could be a reaction to cellular damage. Once established, such inflammation could aggravate disease. However, it must be also noted that the immune system can also be protective, particularly after injury (50, 51). Thus, the role of the immune system in pathogenesis could be either harmful or helpful, and work is required to delineate the precise role of each immune pathway to ALS pathology.
There is also evidence of abnormality of the peripheral immune system in ALS, and this is the topic of the present review. As with inflammation in the CNS, peripheral immune activation could be a reaction to tissue damage, but once established, could exacerbate disease. This review will focus on describing the abnormalities of circulating blood cells, different immune system proteins, and their key inflammatory mediators, cytokines. These are summarized in Tables 1, 2. To consider whether the immune abnormalities contribute to disease pathogenesis, we list some evidence that these abnormalities are correlated with human disease or are pathogenic in animal models of ALS. If immune abnormalities contribute to pathogenesis, then modification of the immune response could be beneficial to patients, so we also highlight the results of forthcoming and completed clinical trials of immune interventions in ALS.
Table 1
| Cell | Change in ALS | Evidence of disease association | Reference(s) |
|---|---|---|---|
| Total leukocytes | Increased | Level correlates with rapidity of progression | (52) |
| Granulocytes/neutrophils | Increased granulocytes, increased neutrophils, increased ratio of neutrophils to monocytes | Treatment with mastinib reduces axonal degeneration in animal model | (53–56) |
| CD4+T cells | Conflicting reports, most suggest an increase | CD4+ T cells are protective in animal model | (54, 57–60) |
| CD8+ T cells | Conflicting reports | Cytotoxic cell cause death of motor neurones in animal model | (54, 57, 59, 61, 62) |
| NK T cells | Increased | Reduction in numbers led to prolonged survival in animal model | (61, 63) |
| Treg cells | Reduced and dysfunctional | Inverse correlation with rate of progression | (57, 61, 64–66) |
| CD14+ monocytes | Variable reports of numbers, but evidence of activation, increased ratio of classical to non-classical monocytes | Monocyte activation correlates with disease progression | (52, 57, 58, 67–69) |
Changes in peripheral blood cells in ALS.
Table 2
| Protein | Changes in ALS | Evidence for role in pathogenesis | References |
|---|---|---|---|
| IgG | Increased | Passive transfer leads to motor neurone degeneration | (70–72) |
| Complement | Increased complement in ALS | Lack of C5a is protective in animal model | (59, 73–75) |
| Tumor necrosis factor | Increased | Mixed effects on motor neuron survival, depending on receptor | (76–82) |
| Interleukin 1β | Increased | Blocking IL-1 led to prolonged survival in animal model of ALS | (77, 79, 83) |
| Interleukin 33 | Reduced | Treatment with IL33 reduced disease in animal model of ALS | (84, 85) |
| Interleukin 6 | Increased | Genetic variation of IL-6 receptor influences the severity of ALS. However, IL6 deficiency has no effect of animal model | (79, 86–90) |
| Interleukin 17 | Increased | Unknown- but usually pro-inflammatory | (91) |
| C reactive protein | Increased | Unlikely—this is evidence of inflammation | (89, 92–94) |
Changes in peripheral blood proteins in ALS.
Abnormalities of Peripheral Blood Cells
Total Leukocyte Count/Granulocytes
Several studies have provided evidence of immune activation in the peripheral blood in ALS. The total leukocyte count is elevated in patients with ALS, and correlates with progression of disease (52). The ratio of neutrophils to monocytes was also shown to be increased (53), as was the total number of granulocytes (54). A micro-array study further confirmed evidence for mild neutrophilia in ALS patients (55). In the SOD1G93A transgenic mouse ALS model, circulating neutrophils are increased (73), and neutrophils and mast cells are present along peripheral motor axons, with masitinib treatment leading to reduction of axonal damage (56). This suggests these cells are harmful and contribute to disease progression.
Lymphocytes
CD4+ T Cells
Some studies demonstrate increased levels of CD4+ helper T lymphocytes in patients with ALS (54, 57, 58), but others have found reduced numbers of these cells (59). It is possible that this variation is related to the variation in immune responsiveness of individuals. CD4+ T cells in the CNS are thought to be neuroprotective in an animal model of ALS (60), and a lack of CD4+ T cell mediated neuroprotection could be detrimental, in patients with reduced numbers. This protection is mediated through Treg cells that are discussed below.
CD8+ T Cells
There are reports of reduced levels of CD8+ cytotoxic T lymphocytes in ALS (57), reports of increased levels of CD8+ cytotoxic T lymphocytes (54, 61), and reports of no alterations in these cells (59). Once again this could be related in part to individual variability. In the SOD1G93A mouse ALS model, cytotoxic lymphocytes cause death of motor neurons (62), so increased numbers could be detrimental.
NKT Cells
NK T cells recognize lipid antigens through CD1,and secrete an array of cytokines (95). A study in people with ALS found increased levels of natural killer T (NKT) cells (61). In the SOD1G93A mouse model, there are also increased NKT cells, especially in the liver (63); furthermore treatment that reduced the numbers of peripheral NKT cells led to prolongation of life-span, suggesting that these cells are harmful in ALS.
Th 17 Cells
The co-stimulatory pathway activated through CD40 ligand is upregulated in some human subjects with ALS (96), and there is thought to be a particular activation of Th-17 T lymphocytes (97). Th17 lymphocytes are pro-inflammatory and thought to be harmful, but can exhibit plasticity and change to other less harmful functions (98).
Treg Cells
Much work has focused on regulatory T cells (Tregs) in ALS (99). There are reduced levels of Tregs in ALS patients (57, 61, 64), and these cells are also found to be dysfunctional (65). The level of Tregs correlates inversely with progression of disease (64). Another study also found that there was an inverse correlation between Treg numbers and the rate of disease progression (66). In a human trial, three patients were given autologous expanded Tregs (100), which showed a possible reduction in the rates of disease progression during infusion periods. A trial has been commenced to determine whether rapamycin, which increases levels of Tregs through the mToR pathway, can lead to increased levels of Tregs in ALS (101).
In SOD1G93A mice, there is evidence of dysfunction of Tregs and transfer of wild-type Tregs delays onset of disease (102). Another study in SOD1 transgenic mice showed that transfer of Tregs slowed disease progression (66). These studies are a promising area of research because of the suggestion that Tregs are able to control or reduce disease activity, but clearly requires larger, controlled and blinded human studies to validate their therapeutic potential.
NK Cells
NK cells are cells of the innate immune system, that mediate cytotoxicity. There is an increase in NK cells in patients with ALS compared to controls (52, 54). NK cells are found in the CNS of SOD1 G93A mutant mice where they are thought to be harmful. Thus, NK cells could possibly be pathogenic and a trial of anti-NK therapy has been proposed in ALS (http://grantome.com/grant/NIH/R21-NS102960-01A1).
Monocytes
Monocyte Classification
With measurements of expression of CD14 (the lipopolysaccharide receptor) and CD16 (the FcγIII receptor), monocytes can be separated into three groups; these are classical (CD14++ CD16−), intermediate (CD14++CD16+) and non-classical (CD14+CD16++) (103, 104). HLA DR is expressed in CD16+ monocytes, while CD14+ monocytes reduce HLADR expression when activated (105). Other markers can also be used to distinguish monocyte subsets (106).
Monocyte Numbers and Proportions
There is a report of a mild increase in CD14+ monocyte numbers in ALS (52). However, another study reported reduced levels of CD14+ cells in the early stage of disease (57). There is also a report that there is no difference on the numbers of CD14+ monocytes between patients and controls (58). In addition, it has been reported there is a reduction in CD16− monocytes in ALS (53). These variations could be explained by differences in methodology and the lack of clear demarcation between these monocyte populations in flow cytometry gating strategies. There are also reports of alterations in the proportions of monocytes in ALS, with an increase in the ratio of classical to non-classical monocytes (67, 107).
In addition to population shifts, there have been reports of alterations in monocyte activation in ALS. CD14+CD16− classical monocytes in ALS show an inflammatory microRNA profile (68). Another study reported increased production of neurotoxic cytokines by monocytes from twins with ALS compared to the unaffected twin (108). Increased peripheral monocyte expression of inflammatory genes correlates with disease progression (69). Another study reported expression of activation markers on monocytes but reduced expression of HLA-DR (57). In another study, patients with ALS could be separated into groups, with one group showing increased HLA-DR expression on monocytes (54). Another study found that there was increased expression of HLA-DR on CD14+ monocytes in ALS and this correlated with the rate of disease progression (58). Another study used exosomes to activate monocytes, and found that monocytes from ALS patients were less responsive than those from healthy individuals (109). ALS monocytes are less responsive to purinergic stimulation than those from controls (110).
As outlined above, the pathology of ALS is characterized by the accumulation of aggregates of proteins in neurons. There is now evidence of abnormal accumulation/location of these proteins in monocytes. For example, altered location of TDP43 in monocytes of patients with genetic mutations in TARDBP, the gene encoding TDP-43 has been demonstrated (111). There is also a report that C9orf72 is expressed in myeloid cells and that expression in monocytes increases after activation (112). Ablation of the mouse homologue of C9orf72 led to macrophage dysfunction and microglial activation (113).
There is evidence that peripheral monocytes enter the CNS in ALS (107), although this is controversial. In SOD1G93A mice, numbers of inflammatory monocytes correlated with disease progression (114). Activated macrophages are found around degenerating nerve (115) and at the neuro-muscular junction in mouse models of ALS (116). These experimental studies suggest that a shift toward activated monocytes in ALS could contribute to ALS progression through secretion of inflammatory and potentially neurotoxic mediators. Further research is needed to precisely define the role of the monocyte in ALS.
Abnormalities of Immune Proteins in Peripheral Blood
Immunoglobulin Levels
Some of the first studies of the role of the immune system in ALS were concerned with the presence of antibodies in the blood of subject with ALS, particularly reports of antibodies to voltage gated calcium channels (117, 118). In addition, there have been studies of non-specific changes in antibodies, as a recent study has shown an increase in IgG levels in subjects with ALS compared to controls (70). In mice, an experimental study showed that prolonged intra-peritoneal injection of immunoglobulin from human subjects with ALS led to loss of spinal motor neurons and loss of muscle strength (71). An earlier study showed that passive transfer of purified immunoglobulin from ALS patients led to motor neuron degeneration and accumulation of calcium containing organelles (72).
Complement System
There is clear evidence of activation of innate immune complement system in human subjects with ALS, with raised C5a levels and increased expression of C5a on human leukocytes (74). A two dimensional gel electrophoresis was used to study serum proteins in ALS subjects and found that components of complement C3 were increased compared to controls (75) and another study using nephelometry showed increased levels of complement C3 in the blood of ALS patients (59). Animal studies also indicate a role for terminal complement activation in motor neuron degeneration. In SOD1 and TDP43 animal models of ALS there is evidence of complement activation (119, 120), and genetic deficiency or pharmacological inhibition of the C5a receptor, C5aR1, is protective in rodent SOD1G93A models (73, 121–123). A comprehensive review of the involvement of complement in ALS has recently been written (124).
Cytokines
Tumor Necrosis Factor (TNF)
There are increased levels of TNF and soluble TNF receptor in the blood of patients with ALS (76–78). A meta-analysis found that TNF levels were significantly increased in ALS (79). RNA-seq analysis has identified TNF as a contributor to inflammation in the spinal cord of ALS patients (125). It is unknown whether this inflammation is harmful or beneficial. It has been suggested that TNF is harmful and that reduction would be beneficial (80). On the other hand, TNF stimulates a survival pathway in motor neurons and could be beneficial (81, 82).
In SOD1 mutant mice, signaling through the TNF receptor 2 lead to motor neurone death (126), whereas signaling through TNF receptor 1 was harmful (127). The recent suggestions that TNF inhibitors could be a risk factor for ALS (128) could indicate that TNF is beneficial in some way.
Interleukin 1 (IL-1)
Interleukin 1 exists as a family of proteins (129). One study found that interleukin 1 β (IL-1β) was undetectable in ALS patients (130) but other studies have found increased levels (77). A meta-analysis found that IL-1β was significantly increased in ALS (79). Pathways involving IL-1 are thought to be involved in ALS pathogenesis as shown in SOD1 and TDP-43 animal models (83, 131, 132). A proteomic study of plasma from ALS patients showed activation of pathways associated with inflammation and activation of two networks centered on NF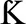 B and IL-1 (133). In animal studies, blocking IL-1 led to prolonged survival (83). In humans, there has been a pilot study that showed that blocking IL-1 with Anakinra was safe in ALS, although there was no prolongation of survival (134).
B and IL-1 (133). In animal studies, blocking IL-1 led to prolonged survival (83). In humans, there has been a pilot study that showed that blocking IL-1 with Anakinra was safe in ALS, although there was no prolongation of survival (134).
Interleukin 33 (IL-33)
IL-33, a cytokine related to IL-1, has a role both in inflammation, and in metabolism (135, 136). IL-33 binds to receptor, ST2. Levels of IL-33 are reduced in ALS, and levels of soluble ST2 are increased in ALS (84). In a study in SOD1G93A transgenic mice, IL-33 treatment ameliorated disease (85), suggesting this is a key downstream mediator of ALS progression.
Interleukin 6 (IL-6)
IL-6 is considered to be a pro-inflammatory cytokine, and is part of an acute phase response, however, it also has some documented anti-inflammatory effects. Plasma levels of IL-6 are increased in ALS (79, 86, 87), and this was supported by a meta-analysis (79). One study suggested that this was a response to hypoxia rather than to the disease itself (137) (see below). IL-6 has been suggested to have a role in endothelial damage in ALS (138). Genetic variation in the IL-6 receptor has been shown to modify the severity of ALS (88). Treatment with the IL-6 blocking antibody toclizumab reduced levels of IL-6 and other cytokines in cells from some ALS patients (89); this study did not look for effects on clinical signs. In SOD1 mutant mice, IL-6 deficiency did not affect the severity of disease (90).
Interleukin 17 (IL-17)
IL-17 is a pro-inflammatory cytokine that also responds to stress (139, 140). Increased levels of IL-17 are reported in the serum of subjects with ALS (91, 141), but to date IL-17 has not been explored clinically as a therapeutic target.
Interleukin 13 (IL-13)
IL-13 regulates T lymphocytes and has been implicated in autoimmune disease (142). IL-13 levels are elevated in the blood of patients with ALS (77). IL-13 producing T lymphocytes have been found in the blood of subjects with ALS and correlate with the rate of disease progression (91, 143).
Interleukin 18 (IL-18)
IL-18 is another member of the IL-1 family of cytokines and stimulates many lymphoid cells (144). Although levels of IL-18 are increased in patients with ALS (130), there is no information about relation of IL-18 to disease activity to date.
Chemokines
Chemokines are small proteins that are involved in chemotaxis and activation of granulocytes and lymphocytes. In the CNS, chemokines also have a role in signaling between cells (145). The expression of MCP-1 receptor (CCR2) is reduced on circulating monocytes in ALS (146). Another study showed significantly increased expression of CXCR3, CXCR4, CCL2, and CCL5 on T lymphocytes in ALS patients compared to healthy controls (147). There are higher levels of the chemokine MCP-1 in patients with a shorter diagnostic delay, which is a marker of more severe rapidly progressing disease (148).
Other Evidence of Systemic Inflammation
There is also evidence of increased levels of C reactive protein and erythrocyte sedimentation rate (ESR) in subjects with ALS compared to controls, and evidence that levels correlate with the levels of disability as measured by the ALS functional rating scale (89, 92–94). Levels of lipopolysaccharide are elevated in patients with ALS, (149), as have levels of nitric oxide, suggesting systemic inflammation (78).
Evidence of Hypoxia
In ALS, there is evidence of hypoxia in neurons, and this is thought to contribute to pathogenesis. This can be seen as increased levels of hypoxia inducible factor−1α (150). There is also thought to be dysregulation of the pathways that protect from hypoxia (151, 152). In the peripheral blood monocytes of ALS patients there is also dysregulation of hypoxia pathways (153). A gene expression study found evidence of hypoxia related genes in peripheral blood of ALS patients (55). In an animal model of ALS, hypoxia aggravates the loss of motor neurons (154). The significance of these findings is presently unclear, but this is further evidence of peripheral immune changes in ALS.
NF-κ B Pathways
Nuclear factor κB (NF-κ B) is a protein complex that regulates the transcription of DNA. Evidence that NF-κ B is important in ALS comes from studies showing genetic abnormalities in optineurin (155). Analysis of cell transfection showed that the nonsense and missense mutations of OPTN abolished the inhibition of activation of NF-κ B. The authors proposed that NF-κ B is the final common pathway in ALS pathogenesis, and that inhibitors of NF-κ B could be used to treat ALS. Further, in animal studies it has also been found that the NF-κ B p65 subunit is a binding partner for TDP-43 and that dysregulation of TDP-43 leads to activation of NF-κ B (156). NF-κ B is expressed in astrocytes (157), and in activated microglia in ALS spinal cord (158).
Other evidence of a possible role of NF-κ B in ALS comes from a role for hypoxia in ALS. (153, 159) (see above). NF-κ B is activated during acute hypoxia and acts to up-regulate inflammatory factors such as IL-6, cyclo-oxygenase (COX 2), TNF-α, and prostaglandin E-2 (PGE-2) (159). Reactive oxygen species lead to induction of NF-κ B. This is mainly in lymphoid cells but also in neurones. It has been suggested that NF- B is a transcription factor controlled by hypoxia and may contribute to neurological disorders (160).
In neurodegenerative disease it is thought that NF-κ B can augment cell death (161). There is some evidence about the role of NF-κ B from animal models of ALS. In SOD1G93A mutant mice, treatment with a PPAR inhibitor led to clinical improvement and reduced expression of iNOS and NF-κ B reactivity (162). Phenylbutyrate induced NF-κ B translocation to the nucleus in ALS mice, and this led to reduced motor neuron death (163). Intrathecal injection of an adenovirus containing insulin like growth factor led to slowing of disease through inhibition of NF-κ B in an animal model of ALS (164). However, inhibition of NF-κ B in astrocytes did not reduce disease in ALS mice (165).
The above-mentioned studies focus predominantly on the role of NF-κ B within the CNS in ALS. Little is known about NF-κ B in the peripheral immune system in ALS, but given the central role of NF-κ B in the biology of the immune system (166), this warrants further study.
Immunometabolic Changes
There is considerable interaction between the immune system and metabolic pathways, which is a rapidly growing research field being known as “immunometabolism” (167, 168). For survival, metabolism and the immune system need to be linked, because there needs to be a mechanism for balancing the energy needed for basal and defensive processes (169). In ALS, there is evidence of alterations in metabolism (170). There are reports of alteration in the levels of metabolic proteins such as adipokines. This includes IL-6 but also other proteins such as leptin and adiponectin (86, 171). A proteomic study found dysregulation of pathways involved in lipid metabolism (133). In particular, there was dysregulation of the Liver X receptor/Retinoid X receptor (LXR/RXR) and the Farnesoid X receptor/Retinoid X receptor (FXR/RXR) pathways that are at the intersection of immunology and metabolism.
Evidence From Completed Clinical Trials
It is attractive to consider that modulation of the immune response will be a useful therapy in ALS. If neuroinflammation enhances disease activity, then control of neuroinflammation should be helpful (172), possibly by enhancing the protective immunity (173).
Overall, clinical trials of new disease-modifying therapies in ALS have been disappointing (174). Several of these trials have used medications that act on the peripheral immune system. Total body irradiation and stem cell therapy were of no benefit in ALS (175). Earlier attempts at immune therapy included treatment with intravenous immunoglobulin, (176), with cyclophosphamide, (177) and with azathioprine and prednisone which were also of no benefit (178). Glatiramer acetate, a synthetic polypeptide with immune effects that is used in multiple sclerosis, further demonstrated no benefit in ALS (179).
Minocycline, an anti-inflammatory agent, also failed in a trial in ALS, and in fact, patients on this treatment had a worse outcome (180). Celecoxib, another anti-inflammatory agent, also failed its clinical end-point (181). Sodium chlorite (NP001) which was proposed to deactivate macrophages, was also more recently shown to be unsuccessful (182, 183).
Masitinib, a tyrosine kinase inhibitor that targets mast cells, microglia, and macrophages despite showing positive results in SOD1 transgenic mice (184), failed in its phase II study in humans (185). Finally, a trial of granulocyte colony stimulating factor led to a decrease in levels of MCP-1 and IL-17 in subjects with ALS (186).
Is There an inflammatory Subgroup?
One of the challenging features of ALS is its heterogeneity- of clinical features, of rate of progression and also in the underlying pathological aggregation of proteins. This heterogeneity could indicate that the pathogenesis of disease varies among patients, and there could be sub- groups of patients in whom immune processes are more or less important.
A study of gene expression indicated that patients can be grouped into patients with higher expression of IL-6R and myeloid lineage-specific genes, and patients with higher expression of IL-23A and lymphoid-specific genes (55). The results from a clinical trial of Toclizumab also led the authors to note that Toclizumab reduced IL-6 and other cytokines in cells from some ALS patients (i.e., an “inflammatory group”) but not others (89).
Interaction Among the Nervous System, the Immune System and the Gut Microbiota
The gut microbiota has been increasingly recognized as playing an important role in human health, and has been implicated in neurodegenerative disease including ALS, as we have recently reviewed (187). One of the many functions of the gut microbiota is to regulate the immune system. It is therefore possible that some of the immune abnormalities in ALS are linked to the gut microbiota. However, this field is complex and analysis requires large numbers of subjects so more remains to be discovered regarding this possible interaction.
Immunogenetics of ALS
If immune genes played a role in the susceptibility to ALS or modified the course of ALS, this would be evidence of involvement of the immune system in disease. In autoimmune diseases, there is an association of disease with HLA loci (188). This is not the case in ALS, except for a possible association with HLA class I antigens (189, 190).
The association with HLA class I antigens could be due to linkage with the haemochromatosis locus (HFE), which is found in the HLA region. Some years ago, an association with the H63D polymorphism was reported (191). More recently, a meta-analysis has discounted this association but instead suggested an association with the C282Y polymorphism (192).
There are numerous polymorphisms that affect the immune system. These have been linked to autoimmune diseases such as multiple sclerosis and type I diabetes (193) but not to ALS (194). However, it would seem likely that genetic variation in immune genes could influence the immune abnormalities described above, For example, polymorphisms in cytokine genes can influence the levels of cytokines such as IL-6, (195) and TNFα (196) and the IL33/ST2 pathway (197).
The field of immunogenetics of ALS would appear to be a fruitful topic for further exploration, and possibly could explain why some patients have a stronger immune response than others, and why some patients show an “inflammatory” phenotype. Immunogenetics, and variation in the immune response to disease could therefore contribute to the known heterogeneity of ALS.
Conclusion
There is clear evidence of immune activation in some patients with ALS and in animal models of disease. It is possible that there is a subgroup of patients in whom inflammatory pathways are important in pathogenesis. In some cases, the immune abnormalities are correlated with disease severity, but it is not clear whether this is cause or effect. The immune system has both harmful and beneficial effects and there is a need to focus research efforts on enhancing the beneficial effects of protective immunity. Clinical trials so far have been disappointing, but there is still scope for further attempts at immune intervention to ameliorate this disease.
Statements
Author contributions
PM conceived the idea and wrote the first draft. JL, TW, and RH read and revised the manuscript.
Funding
This study was supported by funding from the National Health and Medical Research Council (NHMRC; Project grant 1082271 to PM and TW). TW was supported by a NHMRC Career Development Fellowship (1105420).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
BrownRHAl-ChalabiA. Amyotrophic lateral sclerosis. New Engl J Med. (2017) 377:162−72. 10.1056/NEJMra1603471
2.
BaumannFHendersonRDMorrisonSCBrownMHutchinsonNDouglasJAet al. Use of respiratory function tests to predict survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2009) 11:194–202. 10.1080/17482960902991773
3.
McCombePAWrayNRHendersonRD. Extra-motor abnormalities in amyotrophic lateral sclerosis: another layer of heterogeneity. Expert Rev Neurother. (2017) 17:561–77. 10.1080/14737175.2017.1273772
4.
XuZAlruwailiARSHendersonRDMcCombePA. Screening for cognitive and behavioural impairment in amyotrophic lateral sclerosis: frequency of abnormality and effect on survival. J Neurol Sci. (2017) 376:16–23. 10.1016/j.jns.2017.02.061
5.
AlruwailiARPannekKCoulthardAHendersonRKurniawanNDMcCombeP. A combined tract-based spatial statistics and voxel-based morphometry study of the first MRI scan after diagnosis of amyotrophic lateral sclerosis with subgroup analysis. J Neuroradiol. (2018) 45:41–8. 10.1016/j.neurad.2017.03.007
6.
BeghiEChioACouratierPEstebanJHardimanOLogroscinoGet al. The epidemiology and treatment of ALS: Focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph Lateral Scler. (2011) 12:1–10. 10.3109/17482968.2010.502940
7.
BeghiEMenniniTBendottiCBiginiPLogroscinoGChioAet al. The heterogeneity of amyotrophic lateral sclerosis: a possible explanation of treatment failure. Curr Med Chem. (2007) 14:3185–200. 10.2174/092986707782793862
8.
McCombePAHendersonRD. Effects of gender in amyotrophic lateral sclerosis. Gend Med. (2010) 7:557–70. 10.1016/j.genm.2010.11.010
9.
ByrneSElaminMBedePHardimanO. Absence of consensus in diagnostic criteria for familial neurodegenerative diseases. J Neurol Neurosurg Psychiatr. (2012) 83:365–7. 10.1136/jnnp-2011-301530
10.
MarangiGTraynorBJ. Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res. (2015) 14:75–93. 10.1016/j.brainres.2014.10.009
11.
RentonAEChioATraynorBJ. State of play in amyotrophic lateral sclerosis genetics. Nature Neurosci. (2014) 17:17–23. 10.1038/nn.3584
12.
FangFKamelFLichtensteinPBelloccoRSparenPSandlerDPet al. Familial aggregation of amyotrophic lateral sclerosis. Ann Neurol. (2009) 66:94–9. 10.1002/ana.21580
13.
Al-ChalabiACalvoAChioAColvilleSEllisCMHardimanOet al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. (2014) 13:1108–13. 10.1016/S1474-4422(14)70219-4
14.
ChioAMazziniLD'AlfonsoSCorradoLCanosaAMogliaCet al. The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology. (2018) 91:e635–42. 10.1212/WNL.0000000000005996
15.
LattanteSConteAZollinoMLuigettiMDel GrandeAMarangiGet al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology. (2012) 79:66–72. 10.1212/WNL.0b013e31825dceca
16.
Al-ChalabiAFangFHanbyMFLeighPNShawCEYeWet al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatr. (2010) 81:1324–6. 10.1136/jnnp.2010.207464
17.
BlokhuisAMGroenEJKoppersMvan den BergLHPasterkampRJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. (2013) 125:777–94. 10.1007/s00401-013-1125-6
18.
HardimanOAl-ChalabiAChioACorrEMLogroscinoGRobberechtWet al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. (2017) 3:17071. 10.1038/nrdp.2017.85
19.
SaberiSStaufferJESchulteDJRavitsJ. Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol Clin. (2015) 33:855–76. 10.1016/j.ncl.2015.07.012
20.
Da CruzSBuiASaberiSLeeSKStaufferJMcAlonis-DownesMet al. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol. (2017) 134:97–111. 10.1007/s00401-017-1688-8
21.
ForsbergKGraffmoKPakkenbergBWeberMNielsenMMarklundSet al. Misfolded SOD1 inclusions in patients with mutations in C9orf72 and other ALS/FTD-associated genes. J Neurol Neurosurg Psychiatr. (2019) 90:861–9. 10.1136/jnnp-2018-319386
22.
PareBLehmannMBeaudinMNordstromUSaikaliSJulienJPet al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci Rep. (2018) 8:14223. 10.1038/s41598-018-31773-z
23.
LeeYBBaskaranPGomez-DezaJChenHJNishimuraALSmithBNet al. C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum Mol Genet. (2017) 26:4765–77. 10.1093/hmg/ddx350
24.
AyersJICashmanNR. Prion-like mechanisms in amyotrophic lateral sclerosis. Handb Clin Neurol. (2018) 153:337–54. 10.1016/B978-0-444-63945-5.00018-0
25.
ManieckaZPolymenidouM. From nucleation to widespread propagation: a prion-like concept for ALS. Virus Res. (2015) 207:94–105. 10.1016/j.virusres.2014.12.032
26.
RothsteinJD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. (2009) 65 (Suppl. 1):S3–9. 10.1002/ana.21543
27.
Al-ChalabiAJonesATroakesCKingAAl-SarrajSvan den BergLH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. (2012) 124:339–52. 10.1007/s00401-012-1022-4
28.
ChenSZhangXSongLLeW. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. (2012) 22:110–6. 10.1111/j.1750-3639.2011.00546.x
29.
DroppelmannCACampos-MeloDIshtiaqMVolkeningKStrongMJ. RNA metabolism in ALS: when normal processes become pathological. Amyotrop Lateral Scler Frontotemp Degener. (2014) 15:321–6. 10.3109/21678421.2014.881377
30.
SerioAPataniR. Concise review: the cellular conspiracy of amyotrophic lateral sclerosis. Stem Cells. (2018) 36:293–303. 10.1002/stem.2758
31.
YamanakaKKomineO. The multi-dimensional roles of astrocytes in ALS. Neurosci Res. (2018) 126:31–8. 10.1016/j.neures.2017.09.011
32.
MartinLJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. (1999) 58:459–71. 10.1097/00005072-199905000-00005
33.
GueganCPrzedborskiS. Programmed cell death in amyotrophic lateral sclerosis. J Clin Invest. (2003) 111:153–61. 10.1172/JCI200317610
34.
SathasivamSIncePGShawPJ. Apoptosis in amyotrophic lateral sclerosis: a review of the evidence. Neuropathol Appl Neurobiol. (2001) 27:257–74. 10.1046/j.0305-1846.2001.00332.x
35.
IlzeckaJ. Serum caspase-9 levels are increased in patients with amyotrophic lateral sclerosis. Neurol Sci. (2011) 33:825–9. 10.1007/s10072-011-0837-4
36.
TomikBAdamekDPierzchalskiPBanaresSDudaAPartykaDet al. Does apoptosis occur in amyotrophic lateral sclerosis? TUNEL experience from human amyotrophic lateral sclerosis (ALS) tissues. Folia Neuropathol. (2005) 43:75–80.
37.
TroostDAtenJMorsinkFde JongJM. Apoptosis in amyotrophic lateral sclerosis is not restricted to motor neurons. Bcl-2 expression is increased in unaffected post-central gyrus. Neuropathol Appl Neurobiol. (1995) 21:498–504. 10.1111/j.1365-2990.1995.tb01096.x
38.
MartinLJ. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis. (2000) 7:613–22. 10.1006/nbdi.2000.0314
39.
FayazSMSuvanish KumarVSRajanikantGK. Necroptosis: who knew there were so many interesting ways to die?CNS Neurol Disord Drug Targets. (2014) 13:42–51. 10.2174/18715273113126660189
40.
XuXChuaCCZhangMGengDLiuCFHamdyRCet al. The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res. (2010) 1343:206–12. 10.1016/j.brainres.2010.04.080
41.
ReDBLe VercheVYuCAmorosoMWPolitiKAPhaniSet al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. (2014) 81:1001–8. 10.1016/j.neuron.2014.01.011
42.
ItoYOfengeimDNajafovADasSSaberiSLiYet al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. (2016) 353:603–8. 10.1126/science.aaf6803
43.
ConradMPrattDA. The chemical basis of ferroptosis. Nat Chem Biol. (2019) 15:1137–147. 10.1038/s41589-019-0408-1
44.
DevosDMoreauCKyhengMGarconGRollandASBlascoHet al. A ferroptosis-based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci Rep. (2019) 9:2918. 10.1038/s41598-019-39739-5
45.
McCombePAHendersonRD. The role of immune and inflammatory mechanisms in ALS. Curr Mol Med. (2011) 11:246–54. 10.2174/156652411795243450
46.
TroostDvan den OordJJde JongJMSwaabDF. Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin Neuropathol. (1989) 8:289–94.
47.
TroostDvan den OordJJVianney de JongJM. Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. (1990) 16:401–10. 10.1111/j.1365-2990.1990.tb01276.x
48.
HolmoyT. T cells in amyotrophic lateral sclerosis. Eur J Neurol. (2008) 15:360–6. 10.1111/j.1468-1331.2008.02065.x
49.
TurnerMRCagninATurkheimerFEMillerCCShawCEBrooksDJet al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. (2004) 15:601–9. 10.1016/j.nbd.2003.12.012
50.
SchwartzMKipnisJ. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med. (2001) 7:252–8. 10.1016/S1471-4914(01)01993-1
51.
SchwartzMCohenILazarov-SpieglerOMoalemGYolesE. The remedy may lie in ourselves: prospects for immune cell therapy in central nervous system protection and repair. J Mol Med. (1999) 77:713–7. 10.1007/s001099900047
52.
MurdockBJZhouTKashlanSRLittleRJGoutmanSAFeldmanEL. Correlation of peripheral immunity with rapid amyotrophic lateral sclerosis progression. JAMA Neurol. (2017) 74:1446–54. 10.1001/jamaneurol.2017.2255
53.
MurdockBJBenderDEKashlanSRFigueroa-RomeroCBackusCCallaghanBCet al. Increased ratio of circulating neutrophils to monocytes in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e242. 10.1212/NXI.0000000000000242
54.
GustafsonMPStaffNPBornschleglSButlerGWMaasMLKazamelMet al. Comprehensive immune profiling reveals substantial immune system alterations in a subset of patients with amyotrophic lateral sclerosis. PLoS ONE. (2017) 12:e0182002. 10.1371/journal.pone.0182002
55.
SwindellWRKruseCPSListEOBerrymanDEKopchickJJ. ALS blood expression profiling identifies new biomarkers, patient subgroups, and evidence for neutrophilia and hypoxia. J Transl Med. (2019) 17:170. 10.1186/s12967-019-1909-0
56.
TriasEKingPHSiYKwonYVarelaVIbarburuSet al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight. (2018) 3:123249. 10.1172/jci.insight.123249
57.
MantovaniSGarbelliSPasiniAAlimontiDPerottiCMelazziniMet al. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J Neuroimmunol. (2009) 210:73–9. 10.1016/j.jneuroim.2009.02.012
58.
ZhangRGasconRMillerRGGelinasDFMassJHadlockKet al. Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol. (2005) 159:215–24. 10.1016/j.jneuroim.2004.10.009
59.
ChenXFengWHuangRGuoXChenYZhengZet al. Evidence for peripheral immune activation in amyotrophic lateral sclerosis. J Neurol Sci. (2014) 347:90–5. 10.1016/j.jns.2014.09.025
60.
ChiuIMChenAZhengYKosarasBTsiftsoglouSAVartanianTKet al. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci USA. (2008) 105:17913–18. 10.1073/pnas.0804610105
61.
RentzosMEvangelopoulosESeretiEZouvelouVMarmaraSAlexakisTet al. Alterations of T cell subsets in ALS: a systemic immune activation?Acta Neurol Scand. (2012) 125:260–4. 10.1111/j.1600-0404.2011.01528.x
62.
CoqueESalsacCEspinosa-CarrascoGVargaBDegauqueNCadouxMet al. Cytotoxic CD8+ T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc Natl Acad Sci USA. (2019) 116:2312–7. 10.1073/pnas.1815961116
63.
FinkelsteinAKunisGSeksenyanARonenABerkutzkiTAzoulayDet al. Abnormal changes in NKT cells, the IGF-1 axis, and liver pathology in an animal model of ALS. PLoS ONE. (2011) 6:e22374. 10.1371/journal.pone.0022374
64.
HenkelJSBeersDRWenSRiveraALToennisKMAppelJEet al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. (2013) 5:64–79. 10.1002/emmm.201201544
65.
BeersDRZhaoWWangJZhangXWenSNealDet al. ALS patients' regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight. (2017) 2:e89530. 10.1172/jci.insight.89530
66.
SheeanRKMcKayFCCretneyEByeCRPereraNDTomasDet al. Association of regulatory T-cell expansion with progression of amyotrophic lateral sclerosis: a study of humans and a transgenic mouse model. JAMA Neurol. (2018) 75:681–9. 10.1001/jamaneurol.2018.0035
67.
McGillRSFNgoSThorpeKHeggieSRuitenbergMHendersonRDet al. Monocytes and neurtophils are associated with clinical features in amyotrophic lateral sclerosis. Brain Commun. (2020) fcaa013. 10.1093/braincomms/fcaa013
68.
ButovskyOSiddiquiSGabrielyGLanserAJDakeBMurugaiyanGet al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. (2012) 122:3063–87. 10.1172/JCI62636
69.
ZhaoWBeersDRHootenKGSieglaffDHZhangAKalyana-SundaramSet al. Characterization of gene expression phenotype in amyotrophic lateral sclerosis monocytes. JAMA Neurol. (2017) 74:677–85. 10.1001/jamaneurol.2017.0357
70.
SalehIAZesiewiczTXieYSullivanKLMillerAMKuzmin-NicholsNet al. Evaluation of humoral immune response in adaptive immunity in ALS patients during disease progression. J Neuroimmunol. (2009) 215:96–101. 10.1016/j.jneuroim.2009.07.011
71.
ObalINogradiBMeszlenyiVPataiRRickenGKovacsGGet al. Experimental motor neuron disease induced in mice with long-term repeated intraperitoneal injections of serum from ALS patients. Int J Mol Sci. (2019) 20. 10.3390/ijms20102573
72.
PullenAHDemestreMHowardRSOrrellRW. Passive transfer of purified IgG from patients with amyotrophic lateral sclerosis to mice results in degeneration of motor neurons accompanied by Ca2+ enhancement. Acta Neuropathol. (2004) 107:35–46. 10.1007/s00401-003-0777-z
73.
LeeJDKumarVFungJNRuitenbergMJNoakesPGWoodruffTM. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Br J Pharmacol. (2017) 174:689–99. 10.1111/bph.13730
74.
MantovaniSGordonRMacmawJKPflugerCMHendersonRDNoakesPGet al. Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood. J Neuroimmunol. (2014) 276:213–8. 10.1016/j.jneuroim.2014.09.005
75.
GoldknopfILShetaEABrysonJFolsomBWilsonCDutyJet al. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson's disease. Biochem Biophys Res Commun. (2006) 342:1034–39. 10.1016/j.bbrc.2006.02.051
76.
CeredaCBaiocchiCBongioanniPCovaEGuareschiSMetelliMRet al. TNF and sTNFR1/2 plasma levels in ALS patients. J Neuroimmunol. (2008) 194:123–31. 10.1016/j.jneuroim.2007.10.028
77.
LuCHAllenKOeiFLeoniEKuhleJTreeTet al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e244. 10.1212/NXI.0000000000000244
78.
BabuGNKumarAChandraRPuriSKKalitaJMisraUK. Elevated inflammatory markers in a group of amyotrophic lateral sclerosis patients from northern India. Neurochem Res. (2008) 33:1145–9. 10.1007/s11064-007-9564-x
79.
HuYCaoCQinXYYuYYuanJZhaoYet al. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: a meta-analysis study. Sci Rep. (2017) 7:9094. 10.1038/s41598-017-09097-1
80.
BeersDRAppelSH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. (2019) 18:211–20. 10.1016/S1474-4422(18)30394-6
81.
YinXRenMJiangHCuiSWangSJiangHet al. Downregulated AEG-1 together with inhibited PI3K/Akt pathway is associated with reduced viability of motor neurons in an ALS model. Mol Cell Neurosci. (2015) 68:303–13. 10.1016/j.mcn.2015.08.009
82.
OsawaYBannoYNagakiMBrennerDANaikiTNozawaYet al. TNF-alpha-induced sphingosine 1-phosphate inhibits apoptosis through a phosphatidylinositol 3-kinase/Akt pathway in human hepatocytes. J Immunol. (2001) 167:173–80. 10.4049/jimmunol.167.1.173
83.
MeissnerFMolawiKZychlinskyA. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc Natl Acad Sci USA. (2010) 107:13046–50. 10.1073/pnas.1002396107
84.
LinCYPflugerCMHendersonRDMcCombePA. Reduced levels of interleukin 33 and increased levels of soluble ST2 in subjects with amyotrophic lateral sclerosis. J Neuroimmunol. (2012) 249:93–5. 10.1016/j.jneuroim.2012.05.001
85.
KorhonenPPollariEKanninenKMSavchenkoELehtonenSWojciechowskiSet al. Long-term interleukin-33 treatment delays disease onset and alleviates astrocytic activation in a transgenic mouse model of amyotrophic lateral sclerosis. IBRO Rep. (2019) 6:74–86. 10.1016/j.ibror.2019.01.005
86.
NgoSTSteynFJHuangLMantovaniSPflugerCMWoodruffTMet al. Altered expression of metabolic proteins and adipokines in patients with amyotrophic lateral sclerosis. J Neurol Sci. (2015) 357:22–7. 10.1016/j.jns.2015.06.053
87.
Pronto-LaborinhoAPintoSGromichoMPereiraMSwashMde CarvalhoM. Interleukin-6 and amyotrophic lateral sclerosis. J Neurol Sci. (2019) 398:50–3. 10.1016/j.jns.2019.01.026
88.
Wosiski-KuhnMRMArounleutPMartinMCaressJCartwrightMBowserRet al. IL6 receptor358Ala variant and trans-signaling are disease modifiers in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. (2019) 6::e631. 10.1212/NXI.0000000000000631
89.
MizwickiMTFialaMMagpantayLAzizNSayreJLiuGet al. Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am J Neurodegener Dis. (2012) 1:305–15.
90.
HanYRipleyBSeradaSNakaTFujimotoM. Interleukin-6 deficiency does not affect motor neuron disease caused by superoxide dismutase 1 mutation. PLoS ONE. (2016) 11:e0153399. 10.1371/journal.pone.0153399
91.
FialaMChattopadhayMLaCATseELiuGLourencoEet al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J. Neuroinflammation. (2010) 7:76. 10.1186/1742-2094-7-76
92.
KeizmanDRogowskiOBerlinerSIsh-ShalomMMaimonNNefussyBet al. Low-grade systemic inflammation in patients with amyotrophic lateral sclerosis. Acta Neurol Scand. (2009) 119:383–9. 10.1111/j.1600-0404.2008.01112.x
93.
CorciaPBlascoHBeltranSAndresCVourc'hPCouratierP. C-reactive protein: a promising biomarker in ALS?Rev Neurol. (2018) 174:104–5. 10.1016/j.neurol.2017.07.001
94.
LunettaCLizioAMaestriESansoneVAMoraGMillerRGet al. Serum C-reactive protein as a prognostic biomarker in amyotrophic lateral sclerosis. JAMA Neurol. (2017) 74:660–7. 10.1001/jamaneurol.2016.6179
95.
TeytonL. New directions for natural killer T cells in the immunotherapy of cancer. Front Immunol. (2017) 8:1480. 10.3389/fimmu.2017.01480
96.
LincecumJMVieiraFGWangMZThompsonKDe ZutterGSKiddJet al. From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat Genet. (2010) 42:392–9. 10.1038/ng.557
97.
SaresellaMPianconeFTortorellaPMarventanoIGattiACaputoDet al. T helper-17 activation dominates the immunologic milieu of both amyotrophic lateral sclerosis and progressive multiple sclerosis. Clin Immunol. (2013) 148:79–88. 10.1016/j.clim.2013.04.010
98.
SandquistIKollsJ. Update on regulation and effector functions of Th17 cells. F1000Research. (2018) 7:205. 10.12688/f1000research.13020.1
99.
ThonhoffJRSimpsonEPAppelSH. Neuroinflammatory mechanisms in amyotrophic lateral sclerosis pathogenesis. Curr Opin Neurol. (2018) 31:635–9. 10.1097/WCO.0000000000000599
100.
ThonhoffJRBeersDRZhaoWPleitezMSimpsonEPBerryJDet al. Expanded autologous regulatory T-lymphocyte infusions in ALS: a phase I, first-in-human study. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e465. 10.1212/NXI.0000000000000465
101.
MandrioliJD'AmicoRZucchiEGessaniAFiniNFasanoAet al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine. (2018) 97:e11119. 10.1097/MD.0000000000011119
102.
BanerjeeRMosleyRLReynoldsADDharAJackson-LewisVGordonPHet al. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS ONE. (2008) 3:e2740. 10.1371/journal.pone.0002740
103.
Ziegler-HeitbrockL. Blood monocytes and their subsets: established features and open questions. Front Immunol. (2015) 6:423. 10.3389/fimmu.2015.00423
104.
VlacilAKSchuettJSchiefferBGroteK. Variety matters: Diverse functions of monocyte subtypes in vascular inflammation and atherogenesis. Vasc Pharmacol. (2019) 113:9–19. 10.1016/j.vph.2018.12.002
105.
AbelesRDMcPhailMJSowterDAntoniadesCGVergisNVijayGKet al. Vergani CD14 D, CD16 and HLA-DR reliably identifies human monocytes and their subsets in the context of pathologically reduced HLA-DR expression by CD14(hi)/CD16(neg) monocytes: expansion of CD14(hi)/CD16(pos) and contraction of CD14(lo)/CD16(pos) monocytes in acute liver failure. Cytometry Part A J Int Soc Anal Cytol. (2012) 81:823–34. 10.1002/cyto.a.22104
106.
OngSMTengKNewellEChenHChenJLoyTet al. A novel, five-marker alternative to CD16-CD14 gating to identify the three human monocyte subsets. Front Immunol. (2019) 10:1761. 10.3389/fimmu.2019.01761
107.
ZondlerLMullerKKhalajiSBliederhauserCRufWPGrozdanovVet al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol. (2016) 132:391–411. 10.1007/s00401-016-1548-y
108.
LamLChinLHalderRCSagongBFameniniSSayreJet al. Epigenetic changes in T-cell and monocyte signatures and production of neurotoxic cytokines in ALS patients. FASEB J. (2016) 30:3461–73. 10.1096/fj.201600259RR
109.
ZondlerLFeilerMSFreischmidtARufWPLudolphACDanzerKMet al. Impaired activation of ALS monocytes by exosomes. Immunol Cell Biol. (2017) 95:207–14. 10.1038/icb.2016.89
110.
LiuJPrellTStubendorffBKeinerSRingerTGunkelAet al. Down-regulation of purinergic P2X7 receptor expression and intracellular calcium dysregulation in peripheral blood mononuclear cells of patients with amyotrophic lateral sclerosis. Neurosci Lett. (2016) 630:77–83. 10.1016/j.neulet.2016.07.039
111.
De MarcoGLomartireACalvoARissoADe LucaEMostertMet al. Monocytes of patients with amyotrophic lateral sclerosis linked to gene mutations display altered TDP-43 subcellular distribution. Neuropathol Appl Neurobiol. (2017) 43:133–53. 10.1111/nan.12328
112.
RizzuPBlauwendraatCHeetveldSLynesEMCastillo-LizardoMDhingraAet al. C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol Commun. (2016) 4:37. 10.1186/s40478-016-0306-7
113.
O'RourkeJGBogdanikLYanezALallDWolfAJMuhammadAKet al. C9orf72 is required for proper macrophage and microglial function in mice. Science. (2016) 351:1324–9. 10.1126/science.aaf1064
114.
GascoSZaragozaPGarcia-RedondoACalvoACOstaR. Inflammatory and non-inflammatory monocytes as novel prognostic biomarkers of survival in SOD1G93A mouse model of Amyotrophic Lateral Sclerosis. PLoS ONE. (2017) 12:e0184626. 10.1371/journal.pone.0184626
115.
ChiuIMPhatnaniHKuligowskiMTapiaJCCarrascoMAZhangMet al. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc Natl Acad Sci USA. (2009) 106:20960–5. 10.1073/pnas.0911405106
116.
Van DykeJMSmit-OistadIMMacranderCKrakoraDMeyerMGSuzukiM. Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS). Exp Neurol. (2016) 277:275–82. 10.1016/j.expneurol.2016.01.008
117.
EngelhardtJISiklosLKomuvesLSmithRGAppelSH. Antibodies to calcium channels from ALS patients passively transferred to mice selectively increase intracellular calcium and induce ultrastructural changes in motoneurons. Synapse. (1995) 20:185–99. 10.1002/syn.890200302
118.
KimuraFSmithRGDelbonoONyormoiOSchneiderTNastainczykWet al. Amyotrophic lateral sclerosis patient antibodies label Ca2+ channel alpha 1 subunit. Ann Neurol. (1994) 35:164–71. 10.1002/ana.410350207
119.
LeeJDKamaruzamanNAFungJNTaylorSMTurnerBJAtkinJDet al. Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflamm. (2013) 10:119. 10.1186/1742-2094-10-119
120.
LeeJDLevinSCWillisEFLiRWoodruffTMNoakesPG. Complement components are upregulated and correlate with disease progression in the TDP-43(Q331K) mouse model of amyotrophic lateral sclerosis. J Neuroinflamm. (2018) 15:171. 10.1186/s12974-018-1217-2
121.
WoodruffTMCostantiniKJCraneJWAtkinJDMonkPNTaylorSMet al. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol. (2008) 181:8727–34. 10.4049/jimmunol.181.12.8727
122.
WangHALeeJDLeeKMWoodruffTMNoakesPG. Complement C5a-C5aR1 signalling drives skeletal muscle macrophage recruitment in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Skelet Musc. (2017) 7:10. 10.1186/s13395-017-0128-8
123.
WoodruffTMLeeJDNoakesPG. Role for terminal complement activation in amyotrophic lateral sclerosis disease progression. Proc Natl Acad Sci USA. (2014) 111:E3–4. 10.1073/pnas.1321248111
124.
ParkerSEHantonAMStefanouSNNoakesPGWoodruffTMLeeJD. Revisiting the role of the innate immune complement system in ALS. Neurobiol Dis. (2019) 127:223–32. 10.1016/j.nbd.2019.03.003
125.
BrohawnDGO'BrienLCBennettJPJr. RNAseq analyses identify tumor necrosis factor-mediated inflammation as a major abnormality in ALS spinal cord. PLoS ONE. (2016) 11:e0160520. 10.1371/journal.pone.0160520
126.
TortaroloMVallarolaALidonniciDBattagliaEGensanoFSpaltroGet al. Lack of TNF-alpha receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J Neurochem. (2015) 135:109–24. 10.1111/jnc.13154
127.
BrambillaLGuidottiGMartoranaFIyerAMAronicaEValoriCFet al. Disruption of the astrocytic TNFR1-GDNF axis accelerates motor neuron degeneration and disease progression in amyotrophic lateral sclerosis. Hum Mol Genet. (2016) 25:3080–95. 10.1093/hmg/ddw161
128.
PetitpainNDevosDBagheriHRocherFGouraudAMasmoudiKet al. Is TNF inhibitor exposure a risk factor for amyotrophic lateral sclerosis?Fund Clin Pharmacol. (2019) 33:689–94. 10.1111/fcp.12480
129.
PalomoJDietrichDMartinPPalmerGGabayC. The interleukin (IL)-1 cytokine family–Balance between agonists and antagonists in inflammatory diseases. Cytokine. (2015) 76:25–37. 10.1016/j.cyto.2015.06.017
130.
ItalianiPCarlesiCGiungatoPPuxedduIBorroniBBossuPet al. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J Neuroinflamm. (2014) 11:94. 10.1186/1742-2094-11-94
131.
PasinelliPHouseweartMKBrownRHJrClevelandDW. Caspase-1 and−3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. (2000) 97:13901–6. 10.1073/pnas.240305897
132.
DeoraVLeeJDAlbornozEAMcAlaryLJagarajCJRobertsonAABet al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia. (2020) 68:407–21. 10.1002/glia.23728
133.
XuZLeeANouwensADavid HendersonRAnn MccombeP. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotrop Lateral Scler Frontotemporal Degener. (2018) 19:362–76. 10.1080/21678421.2018.1433689
134.
MaierADeigendeschNMullerKWeishauptJHKrannichARohleRet al. Interleukin-1 antagonist anakinra in amyotrophic lateral sclerosis–a pilot study. PLoS ONE. (2015) 10:e0139684. 10.1371/journal.pone.0139684
135.
TuLYangL. IL-33 at the crossroads of metabolic disorders and immunity. Front Endocrinol. (2019) 10:26. 10.3389/fendo.2019.00026
136.
MilovanovicMVolarevicVRadosavljevicGJovanovicIPejnovicNArsenijevicNet al. IL-33/ST2 axis in inflammation and immunopathology. Immunol Res. (2012) 52:89–99. 10.1007/s12026-012-8283-9
137.
MoreauCDevosDBrunaud-DanelVDefebvreLPerezTDesteeAet al. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia?Neurology. (2005) 65:1958–60. 10.1212/01.wnl.0000188907.97339.76
138.
Garbuzova-DavisSEhrhartJSanbergPRBorlonganCV. Potential role of humoral IL-6 cytokine in mediating pro-inflammatory endothelial cell response in amyotrophic lateral sclerosis. Int J Mol Sci. (2018) 19. 10.3390/ijms19020423
139.
McGeachyMJCuaDJGaffenSL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. 10.1016/j.immuni.2019.03.021
140.
MatsuzakiGUmemuraM. Interleukin-17 family cytokines in protective immunity against infections: role of hematopoietic cell-derived and non-hematopoietic cell-derived interleukin-17s. Microbiol Immunol. (2018) 62:1–13. 10.1111/1348-0421.12560
141.
RentzosMRombosANikolaouCZogaMZouvelouVDimitrakopoulosAet al. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation?Acta Neurol Scand. (2010) 122:425–9. 10.1111/j.1600-0404.2010.01333.x
142.
MaoYMZhaoCNLengJLengRXYeDQZhengSGet al. Interleukin-13: A promising therapeutic target for autoimmune disease. Cytok Growth Factor Rev. (2019) 45:9–23. 10.1016/j.cytogfr.2018.12.001
143.
ShiNKawanoYTateishiTKikuchiHOsoegawaMOhyagiYet al. Increased IL-13-producing T cells in ALS: positive correlations with disease severity and progression rate. J Neuroimmunol. (2007) 182:232–5. 10.1016/j.jneuroim.2006.10.001
144.
YasudaKNakanishiKTsutsuiH. Interleukin-18 in health and disease. Int J Mol Sci. (2019) 20. 10.3390/ijms20030649
145.
TrettelFDi CastroMALimatolaC. Chemokines: key molecules that orchestrate communication among neurons, microglia and astrocytes to preserve brain function. Neuroscience. (2019). 10.1016/j.neuroscience.2019.07.035. [Epub ahead of print].
146.
ZhangRGasconRMillerRGGelinasDFMassJLanceroMet al. MCP-1 chemokine receptor CCR2 is decreased on circulating monocytes in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol. (2006) 179:87–93. 10.1016/j.jneuroim.2006.06.008
147.
PernerCPernerFStubendorffBForsterMWitteOWHeidelFHet al. Dysregulation of chemokine receptor expression and function in leukocytes from ALS patients. J Neuroinflamm. (2018) 15:99. 10.1186/s12974-018-1135-3
148.
KuhleJLindbergRLRegeniterAMehlingMSteckAJKapposLet al. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur J Neurol. (2009) 16:771–4. 10.1111/j.1468-1331.2009.02560.x
149.
ZhangRMillerRGGasconRChampionSKatzJLanceroMet al. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol. (2009) 206:121–4. 10.1016/j.jneuroim.2008.09.017
150.
NomuraEOhtaYTadokoroKShangJFengTLiuXet al. Imaging hypoxic stress and the treatment of amyotrophic lateral sclerosis with dimethyloxalylglycine in a mice model. Neuroscience. (2019) 415:31–43. 10.1016/j.neuroscience.2019.06.025
151.
SatoKMorimotoNKurataTMimotoTMiyazakiKIkedaYet al. Impaired response of hypoxic sensor protein HIF-1alpha and its downstream proteins in the spinal motor neurons of ALS model mice. Brain Res. (2012) 1473:55–62. 10.1016/j.brainres.2012.07.040
152.
NagaraYTateishiTYamasakiRHayashiSKawamuraMKikuchiHet al. Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1alpha in amyotrophic lateral sclerosis. Brain Pathol. (2013) 23:534–46. 10.1111/bpa.12040
153.
MoreauCGossetPKluzaJBrunaud-DanelVLassallePMarchettiPet al. Deregulation of the hypoxia inducible factor-1alpha pathway in monocytes from sporadic amyotrophic lateral sclerosis patients. Neuroscience. (2011) 172:110–7. 10.1016/j.neuroscience.2010.10.040
154.
KimSMKimHLeeJSParkKSJeonGSShonJet al. Intermittent hypoxia can aggravate motor neuronal loss and cognitive dysfunction in ALS mice. PLoS ONE. (2013) 8:e81808. 10.1371/journal.pone.0081808
155.
MaruyamaHMorinoHItoHIzumiYKatoHWatanabeYet al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. (2010) 465:223–6. 10.1038/nature08971
156.
SwarupVPhaneufDDupreNPetriSStrongMKrizJet al. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med. (2011) 208:2429–47. 10.1084/jem.20111313
157.
MigheliAPivaRAtzoriCTroostDSchifferD. c-Jun D JNK/SAPK kinases transcription factor NF-kappa B are selectively activated in astrocytes. J Neuropathol Exp Neurol. (1997) 56:1314–22. 10.1097/00005072-199712000-00006
158.
FrakesAEFerraiuoloLHaidet-PhillipsAMSchmelzerLBraunLMirandaCJet al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. (2014) 81:1009–23. 10.1016/j.neuron.2014.01.013
159.
MoreauCDevosDGossetPBrunaud-DanelVTonnelABLassallePet al. [Mechanisms of deregulated response to hypoxia in sporadic amyotrophic lateral sclerosis: a clinical study]. Rev Neurol. (2010) 166:279–83. 10.1016/j.neurol.2009.05.018
160.
KaltschmidtBBaeuerlePAKaltschmidtC. Potential involvement of the transcription factor NF-kappa B in neurological disorders. Mol Aspects Med. (1993) 14:171–90. 10.1016/0098-2997(93)90004-W
161.
CamandolaSMattsonMP. NF-kappa B as a therapeutic target in neurodegenerative diseases. Expert Opin Ther Targets. (2007) 11:123–32. 10.1517/14728222.11.2.123
162.
KiaeiMKipianiKChenJCalingasanNYBealMF. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. (2005) 191:331–6. 10.1016/j.expneurol.2004.10.007
163.
RyuHSmithKCameloSICarrerasILeeJIglesiasAHet al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. (2005) 93:1087–98. 10.1111/j.1471-4159.2005.03077.x
164.
HuHLinHDuanWCuiCLiZLiuYet al. Intrathecal injection of scAAV9-hIGF1 prolongs the survival of ALS model mice by inhibiting the NF-kB pathway. Neuroscience. (2018) 381:1–10. 10.1016/j.neuroscience.2018.02.004
165.
CrosioCValleCCasciatiAIaccarinoCCarriMT. Astroglial inhibition of NF-kappaB does not ameliorate disease onset and progression in a mouse model for amyotrophic lateral sclerosis (ALS). PLoS ONE. (2011) 6:e17187. 10.1371/journal.pone.0017187
166.
ZhangQLenardoMJBaltimoreD. 30 years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. 10.1016/j.cell.2016.12.012
167.
HotamisligilGS. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity. (2017) 47:406–20. 10.1016/j.immuni.2017.08.009
168.
HotamisligilGS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. 10.1038/nature21363
169.
WangALuanHHMedzhitovR. An evolutionary perspective on immunometabolism. Science. (2019) 363:eaar3932. 10.1126/science.aar3932
170.
IoannidesZANgoSTHendersonRDMcCombePASteynFJ. Altered metabolic homeostasis in amyotrophic lateral sclerosis: mechanisms of energy imbalance and contribution to disease progression. Neuro Degener Dis. (2016) 16:382–97. 10.1159/000446502
171.
NagelGPeterRSRosenbohmAKoenigWDupuisLRothenbacherDet al. Adipokines, C-reactive protein and Amyotrophic Lateral Sclerosis - results from a population- based ALS registry in Germany. Sci Rep. (2017) 7:4374. 10.1038/s41598-017-04706-5
172.
MosleyRLGendelmanHE. Control of neuroinflammation as a therapeutic strategy for amyotrophic lateral sclerosis and other neurodegenerative disorders. Exp Neurol. (2010) 222:1–5. 10.1016/j.expneurol.2009.12.018
173.
KosloskiLMHaDMHutterJAStoneDKPichlerMRReynoldsADet al. Adaptive immune regulation of glial homeostasis as an immunization strategy for neurodegenerative diseases. J Neurochem. (2010) 114:1261–76. 10.1111/j.1471-4159.2010.06834.x
174.
PetrovDMansfieldCMoussyAHermineO. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment?Front Aging Neurosci. (2017) 9:68. 10.3389/fnagi.2017.00068
175.
AppelSHEngelhardtJIHenkelJSSiklosLBeersDRYenAAet al. Hematopoietic stem cell transplantation in patients with sporadic amyotrophic lateral sclerosis. Neurology. (2008) 71:1326–34. 10.1212/01.wnl.0000327668.43541.22
176.
MeucciNNobile-OrazioEScarlatoG. Intravenous immunoglobulin therapy in amyotrophic lateral sclerosis. J Neurol. (1996) 243:117–20. 10.1007/BF02444000
177.
SmithSAMillerRGMurphyJRRingelSP. Treatment of ALS with high dose pulse cyclophosphamide. J Neurol Sci. (1994) 124 (Suppl.):84–7. 10.1016/0022-510X(94)90188-0
178.
WerdelinLBoysenGJensenTSMogensenP. Immunosuppressive treatment of patients with amyotrophic lateral sclerosis. Acta Neurol Scand. (1990) 82:132–4. 10.1111/j.1600-0404.1990.tb01602.x
179.
MeiningerVDroryVELeighPNLudolphARobberechtWSilaniV. Glatiramer acetate has no impact on disease progression in ALS at 40 mg/day: a double- blind, randomized, multicentre, placebo-controlled trial. Amyotrop Lateral Scler. (2009) 10:378–83. 10.3109/17482960902803432
180.
GordonPHMooreDHMillerRGFlorenceJMVerheijdeJLDoorishCet al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. (2007) 6:1045–53. 10.1016/S1474-4422(07)70270-3
181.
CudkowiczMEShefnerJMSchoenfeldDAZhangHAndreassonKIRothsteinJDet al. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol. (2006) 60:22–31. 10.1002/ana.20903
182.
MillerRGZhangRBlockGKatzJBarohnRKasarskisEet al. NP001 regulation of macrophage activation markers in ALS: a phase I clinical and biomarker study. Amyotrop Lateral Scler Frontotemp Degener. (2014) 15:601–9. 10.3109/21678421.2014.951940
183.
MillerRGBlockGKatzJSBarohnRJGopalakrishnanVCudkowiczMet al. Randomized phase 2 trial of NP001-a novel immune regulator: Safety and early efficacy in ALS. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e100. 10.1212/NXI.0000000000000100
184.
TriasEIbarburuSBarreto-NunezRBabdorJMacielTTGuilloMet al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflamm. (2016) 13:177. 10.1186/s12974-016-0620-9
185.
MoraJSGengeAChioAEstolCJChaverriDHernandezMet al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotrop Lateral Scler Frontotemp Degener. (2019) 21:5–14. 10.1080/21678421.2019.1632346
186.
ChioAMoraGBellaVLCaponnettoCMancardiGSabatelliMet al. Repeated courses of granulocyte colony-stimulating factor in amyotrophic lateral sclerosis: clinical and biological results from a prospective multicenter study. Muscle Nerve. (2011) 43:189–95. 10.1002/mus.21851
187.
McCombePAHendersonRDLeeALeeJDWoodruffTMRestuadiRet al. Gut microbiota in ALS: possible role in pathogenesis?Expert Rev Neurother. (2019) 1-21. 10.1080/14737175.2019.1623026
188.
RoseNRBonaC. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol Today. (1993) 14:426–30. 10.1016/0167-5699(93)90244-F
189.
ChiarottoGBNardoGTroleseMCFrancaMCJrBendottiCRodrigues de OliveiraAL. The emerging role of the major histocompatibility complex class I in amyotrophic lateral sclerosis. Int J Mol Sci. (2017) 18:2298. 10.3390/ijms18112298
190.
AntelJPRichmanDPArnasonBG. Immunogenetics and amyotrophic lateral sclerosis. UCLA Forum Med Sci. (1976) 71:151–71.
191.
GoodallEFGreenwayMJvan MarionICarrollCBHardimanOMorrisonKE. Association of the H63D polymorphism in the hemochromatosis gene with sporadic ALS. Neurology. (2005) 65:934–7. 10.1212/01.wnl.0000176032.94434.d4
192.
LiMWangLWangWQiXLTangZY. Mutations in the HFE gene and sporadic amyotrophic lateral sclerosis risk: a meta-analysis of observational studies. Braz J Med Biol Res. (2014) 47:215–22. 10.1590/1414-431X20133296
193.
YeJGillespieKMRodriguezS. Unravelling the roles of susceptibility loci for autoimmune diseases in the post-GWAS Era. Genes. (2018) 9. 10.3390/genes9080377
194.
WeiLTianYChenYWeiQChenFCaoBet al. Identification of TYW3/CRYZ and FGD4 as susceptibility genes for amyotrophic lateral sclerosis. Neurol Genet. (2019) 5:e375. 10.1212/NXG.0000000000000375
195.
WooPHumphriesSE. IL-6 polymorphisms: a useful genetic tool for inflammation research?J Clin Invest. (2013) 123:1413–4. 10.1172/JCI67221
196.
CajadoCCerqueiraBACoutoFDMoura-NetoJPVilas-BoasWDoreaMJet al. TNF-alpha and IL-8: serum levels and gene polymorphisms (−308G>A and−251A>T) are associated with classical biomarkers and medical history in children with sickle cell anemia. Cytokine. (2011) 56:312–7. 10.1016/j.cyto.2011.07.002
197.
WeiZHLiYYHuangSQTanZQ. Genetic variants in IL-33/ST2 pathway with the susceptibility to hepatocellular carcinoma in a Chinese population. Cytokine. (2018) 118:124–9. 10.1016/j.cyto.2018.03.036
Summary
Keywords
amyotrophic lateral sclerosis (ALS), T lymphocytes, monocyte, cytokine, inflammation, immunity
Citation
McCombe PA, Lee JD, Woodruff TM and Henderson RD (2020) The Peripheral Immune System and Amyotrophic Lateral Sclerosis. Front. Neurol. 11:279. doi: 10.3389/fneur.2020.00279
Received
21 September 2019
Accepted
25 March 2020
Published
21 April 2020
Volume
11 - 2020
Edited by
Sandra Amor, VU University Medical Center, Netherlands
Reviewed by
Maria F. Cano-Abad, Autonomous University of Madrid, Spain; Stanley Hersh Appel, Houston Methodist Research Institute, United States
Updates
Copyright
© 2020 McCombe, Lee, Woodruff and Henderson.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela A. McCombe Pamela.McCombe@uq.edu.au
This article was submitted to Multiple Sclerosis and Neuroimmunology, a section of the journal Frontiers in Neurology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.