 Qin Chen
Qin Chen Kejal Kantarci
Kejal Kantarci- 1Department of Neurology, West China Hospital of Sichuan University, Chengdu, China
- 2Department of Radiology, Mayo Clinic, Rochester, MN, United States
Frontotemporal lobar degeneration (FTLD) is a neurodegenerative disorder characterized by behavioral changes, language abnormality, as well as executive function deficits and motor impairment. In about 30–50% of FTLD patients, an autosomal dominant pattern of inheritance was found with major mutations in the MAPT, GRN, and the C9orf72 repeat expansion. These mutations could lead to neurodegenerative pathology years before clinical symptoms onset. With potential disease-modifying treatments that are under development, non-invasive biomarkers that help determine the early brain changes in presymptomatic FTLD patients will be critical for tracking disease progression and enrolling the right participants into the clinical trials at the right time during the disease course. In recent years, there is increasing evidence that a number of imaging biomarkers show the abnormalities during the presymptomatic stage. Imaging biomarkers of presymptomatic familial FTLD may provide insight into the underlying neurodegenerative process years before symptom onset. Structural magnetic resonance imaging (MRI) has demonstrated cortical degeneration with a mutation-specific neurodegeneration pattern years before onset of clinical symptoms in presymptomatic familial FTLD mutation carriers. In addition, diffusion tensor imaging (DTI) has shown the loss of white matter microstructural integrity in the presymptomatic stage of familial FTLD. Furthermore, proton magnetic resonance spectroscopy (1H MRS), which provides a non-invasive measurement of brain biochemistry, has identified early neurochemical abnormalities in presymptomatic MAPT mutation carriers. Positron emission tomography (PET) imaging with [18F]-fluorodeoxyglucose (FDG) has demonstrated the glucose hypometabolism in the presymptomatic stage of familial FTLD. Also, a novel PET ligand, 18F-AV-1451, has been used in this group to evaluate tau deposition in the brain. Promising imaging biomarkers for presymptomatic familial FTLD have been identified and assessed for specificity and sensitivity for accurate prediction of symptom onset and tracking disease progression during the presymptomatic stage when clinical measures are not useful. Furthermore, identifying imaging biomarkers for the presymptomatic stage is important for the design of disease-modifying trials. We review the recent progress in imaging biomarkers of the presymptomatic phase of familial FTLD and discuss the imaging techniques and analysis methods, with a focus on the potential implication of these imaging techniques and their utility in specific mutation types.
Introduction
Frontotemporal lobar degeneration (FTLD) is a progressive neurodegenerative disorder characterized by behavioral abnormalities, language impairment, impaired social cognition, and executive function, as well as progressive supranuclear palsy (PSP), corticobasal syndrome (CBS), or motor neuron disease (1). FTLD has a strong genetic background with an autosomal dominant pattern of inheritance in about 30–50% FTLD patients (2) with major mutations in the microtubule-associated protein tau gene (MAPT) (3), progranulin (GRN), and the repeat expansions in the chromosome 9 open reading frame 72 gene (C9orf72).
Mutations in the gene encoding the microtubule-associated protein tau (MAPT) on chromosome 17 was first reported in 1998 (4) and have been found in many kindreds with familial FTLD. The vast majority of known mutations occurring in the coding region are in the repeats, causing the reduced ability of the mutant tau proteins to interact with microtubules, leading to hyperphosphorylated tau accumulation in glia and neurons causing neurodegeneration, white-matter integrity alterations, and brain atrophy years before clinical symptom onset (3, 5–8). Subtypes of MAPT mutations were associated with different types of tauopathies. These are mutations inside exon 10 (i.e., N279K, S305N, and P301L) and outside exon 10 (i.e., R406W and V337M). Mutations inside exon 10 tend to form four tandem microtubule-binding domain repeat (4R-tau) pathology rather than 3 repeat (3R-tau) pathology (9), while mutations outside exon 10 tend to form mixed 3R/4R tau pathology.
In 2006, loss-of-function mutations in the progranulin gene (GRN) were first reported to cause familial FTLD (10, 11). There are more than 70 different pathogenetic GRN mutations that have been identified until this date (http://www.molgen.ua.ac.be/FTDmutations). Mutations in the GRN gene leads to a loss of progranulin levels through haploinsufficiency, and the intraneuronal aggregation of TAR DNA-binding protein (TDP)-43 protein (12). The most frequent clinical phenotypes of GRN mutation carriers were behavioral variant frontotemporal dementia (bvFTD), CBS, and primary progressive aphasia (PPA) (13, 14). GRN mutation carriers have FTLD with TDP-43 inclusions and present with a diverse clinical phenotype and a highly heterogeneous age of disease onset.
In 2011, the hexanucleotide GGGGCC (G4C2) repeat expansions of the chromosome 9 open reading frame 72 gene (C9orf72) C9orf72 gene were found as a common cause of both FTD and amyotrophic lateral sclerosis (ALS) (15, 16). The most common clinical phenotypes associated with C9orf72 expansions are bvFTD (17), ALS, or the combination of both in one person (18). Cases with the C9orf72 repeat expansion with histopathological correlation had TDP-43 depositions (18).
These mutations associated with familial FTLD could lead to brain neurodegeneration years before symptom onset. Investigation of families with the presence of MAPT, GRN, or C9orf72 provide a unique opportunity to shed light on early neurodegenerative changes and also identify biomarkers for tracking disease progression in future disease-modifying trials. In recent years, there is growing evidence that a number of imaging biomarkers show abnormalities during the presymptomatic stage. Imaging biomarkers of presymptomatic familial FTLD may provide insight into the underlying neurodegenerative process years before symptom onset.
Discussion
Structural MRI
Structural magnetic resonance imaging (sMRI) has captured cortical degeneration with a mutation-specific neurodegeneration pattern years before onset of clinical symptoms in presymptomatic familial FTLD mutation carriers (6, 19–32). Studies of sMRI using different analysis methods, such as region of interest (ROI) in specific brain regions, cortical thickness analysis and voxel-based morphometry (VBM), could capture the gray matter and white matter volumes, cortical thickness, and the subcortical gray matter volume.
MAPT_sMRI
Previous cross-sectional MRI studies demonstrated atrophy in the anteromedial temporal lobe and orbitofrontal cortex in asymptomatic MAPT mutation carriers (6, 19, 20), while others found no difference between asymptomatic MAPT mutation carriers and controls (6, 7, 33). Recently, two longitudinal studies from a cohort of asymptomatic MAPT mutation carriers have reported that hippocampal volumes decline during a 2-year follow-up, but no cortical atrophy was found in longitudinal analysis with 4 years of follow-up (21, 22). During a 10-year follow-up, the rates of temporal lobe atrophy were accelerated in asymptomatic MAPT mutation carriers who were asymptomatic during follow-up, while accelerated atrophy rates in the temporal, parietal and frontal lobes were reported in MAPT mutation carriers who became symptomatic compared to non-carriers (23). These data altogether suggest the consistent finding of early involvement of the anterior–medial temporal lobe in asymptomatic MAPT mutation carriers. Taken together, the cross-sectional and longitudinal studies of lobar cortical atrophy in MAPT mutation carriers suggest a sequential pattern throughout the disease course. The cortical volume appears to decline in the temporal lobe early in the asymptomatic stage, with an acceleration of atrophy rates along with development of symptoms, followed by the frontal and parietal lobe atrophy with sparing of the occipital lobe (23) (Table 1).
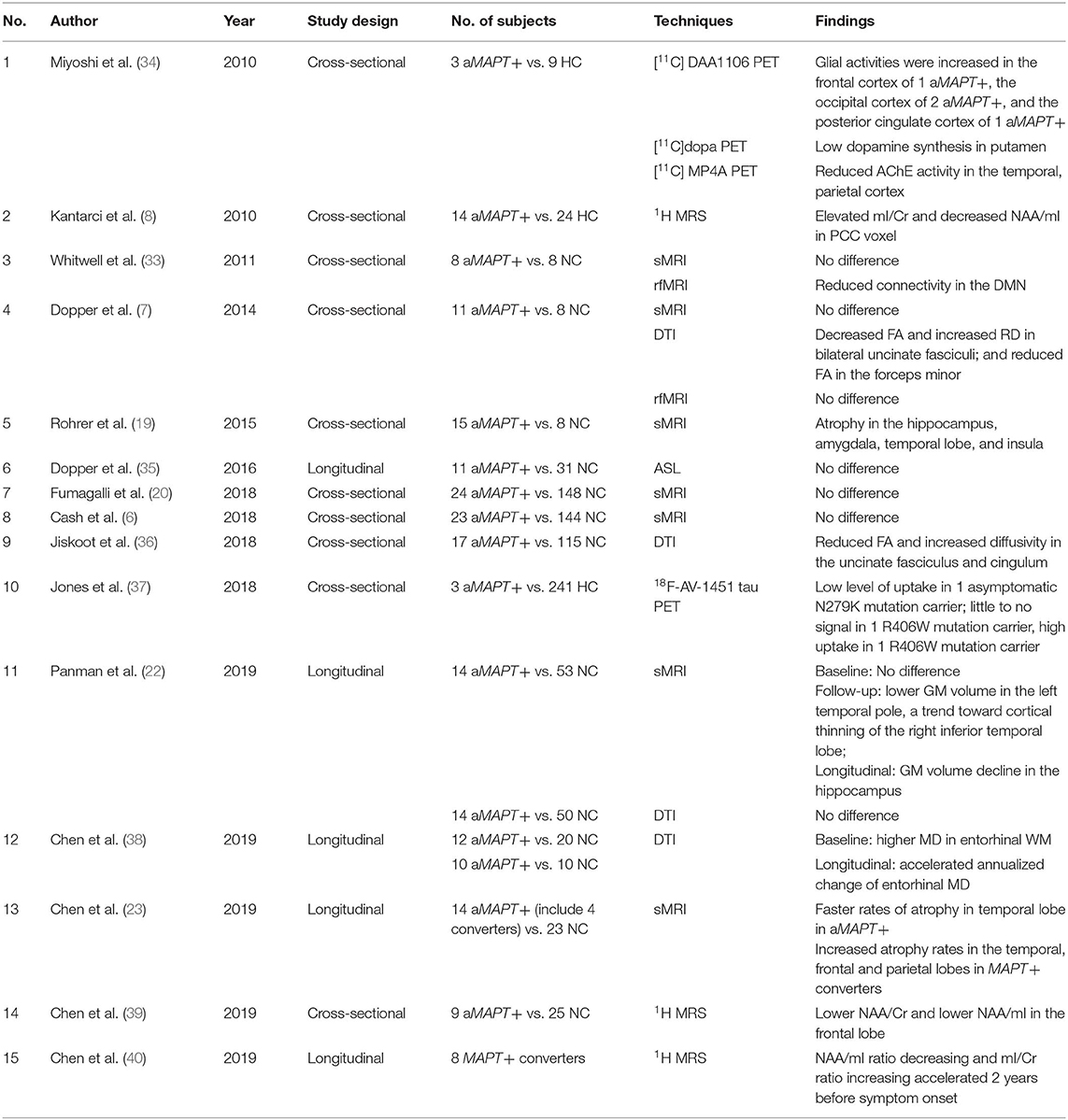
Table 1. Studies investigating asymptomatic MAPT mutation vs. controls.
Across different subtypes of MAPT mutations (IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M), similar patterns of atrophy were reported in the later symptomatic phase (41). However, the atrophy pattern in the early disease phase may be varied across the different subtypes of MAPT mutations. For example, patients with MAPT N279K mutations have prominent motor symptoms early in the disease process (42–44) (Figure 1A), and therefore involvement of the primary and supplementary motor cortices may be expected in this sub-mutation group (45–49). The N279K MAPT mutation carriers' trajectories of lobar atrophy, such as the supplemental motor cortex involvement, may be specific to the suspected 4R-tau associated neurodegeneration in N279K kindred. Further studies are needed to characterize the trajectories of rates of cortical atrophy across the different subtypes of MAPT mutations.
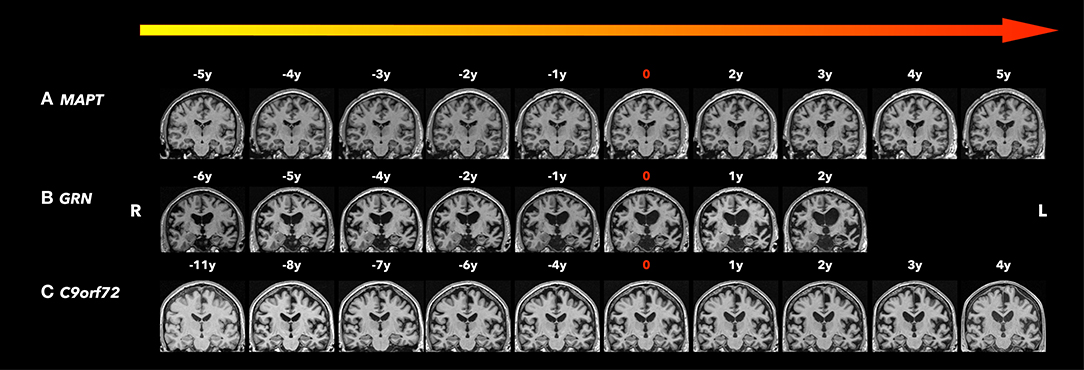
Figure 1. Coronal T1-weighted magnetic resonance imaging (MRI). Brain MRIs (coronal T1-weighted images) of the three familiar FTLD mutation carriers who progressed from the asymptomatic to the symptomatic stage are shown in the timeline with years before and after conversion. 0 indicates the actual symptom onset time point. (A) A male patient with MAPT N279K mutation whose first clinical manifestation was difficulties in remembering street names and people's names with Kokmen short test of mental status (36/38) at age 43. Two years after symptom onset, his forgetfulness progressed, with resting tremor in the left lower limbs, as well as some impulsivity and disinhibition, and was diagnosed with bvFTD and Parkinsonism. Five years after symptom onset, his cognitive function, parkinsonism, and behavior changes significantly worsened and mostly by motor impairment. Focal adjacent mesial temporal lobe atrophy mainly in the right side was observed 5 years before symptom onset and became bilateral hippocampal atrophy with generalized cerebral atrophy most marked in the bilateral parietotemporal regions. The structural brain change seems to be slower than his clinic symptom progression. (B) A male patient GRN mutation carrier whose first clinical manifestation was word-finding difficulties at age 68. Two years after symptom onset, his word-finding difficulties progressed, with additional executive dysfunction, forgetfulness, and tendency to repeat questions. He also had apathy, irritability, and dysphoria, and was diagnosed with mixed PPA/bvFTD syndrome. Asymmetrical (left greater than right) atrophy was observed 6 years before symptom onset, and became progressively asymmetric affecting the frontal, temporal, and parietal lobes as he aged. (C) A male patient C9orf72 mutation carrier whose first clinical manifestation was a change in motivation, spontaneity, and empathy at age 73 and was diagnosed as behavior MCI. After 4 years from symptom onset, his behavior change slightly progressed with subtleties in empathy, emotional blunting, irritability, and subtle apathy, still diagnosed as behavior MCI. While his clinic symptoms seem to be stable, the moderate parenchymal volume loss was observed 8 years before symptom onset most involved in the frontal and temporal areas, and progressed affecting the whole brain with greatest atrophy in the fronto-temporal regions as he aged.
GRN_sMRI
In FTLD patients with GRN mutations, the pattern of brain atrophy was reported to be asymmetric and widespread, predominantly involving the frontal, inferior parietal, and posterior temporal cortices (6, 13, 50–54). In asymptomatic GRN mutation carriers, the structural MRI studies were inconsistent (24–27).
Although most cross-sectional (6, 7, 20, 32, 55–57) and longitudinal structural MRI studies (22) reported no gray matter volume differences between asymptomatic GRN mutation carriers and controls, a few recent studies found a gray matter atrophy pattern mainly involving the frontal (25), parietal (58), and temporal lobes (27). Brain atrophy was reported in the insula early, 15 years before the expected symptom onset, followed by the temporal and parietal lobes at 10 years before the expected symptom onset, then in the striatum at 5 years before the expected symptom onset (19). In a longitudinal study of asymptomatic GRN mutation carriers with a 20-month follow-up, no gray matter volume loss was found at baseline, but left inferior and middle temporal gyri atrophy was reported 20 months later (26). Reduced gray matter density was also reported in bilateral orbitofrontal, anterior temporal, and insular cortices at baseline with greater annualized GM density changes in right orbitofrontal and left occipital cortices during follow-up (27). A recent study reported a sequential pattern of regional cortical atrophy rates with up to 10 years of follow-up, involving the frontal and parietal lobe cortices early during the asymptomatic stage, followed by the temporal lobe cortex after symptom onset in GRN mutation carriers (28) (Table 2).
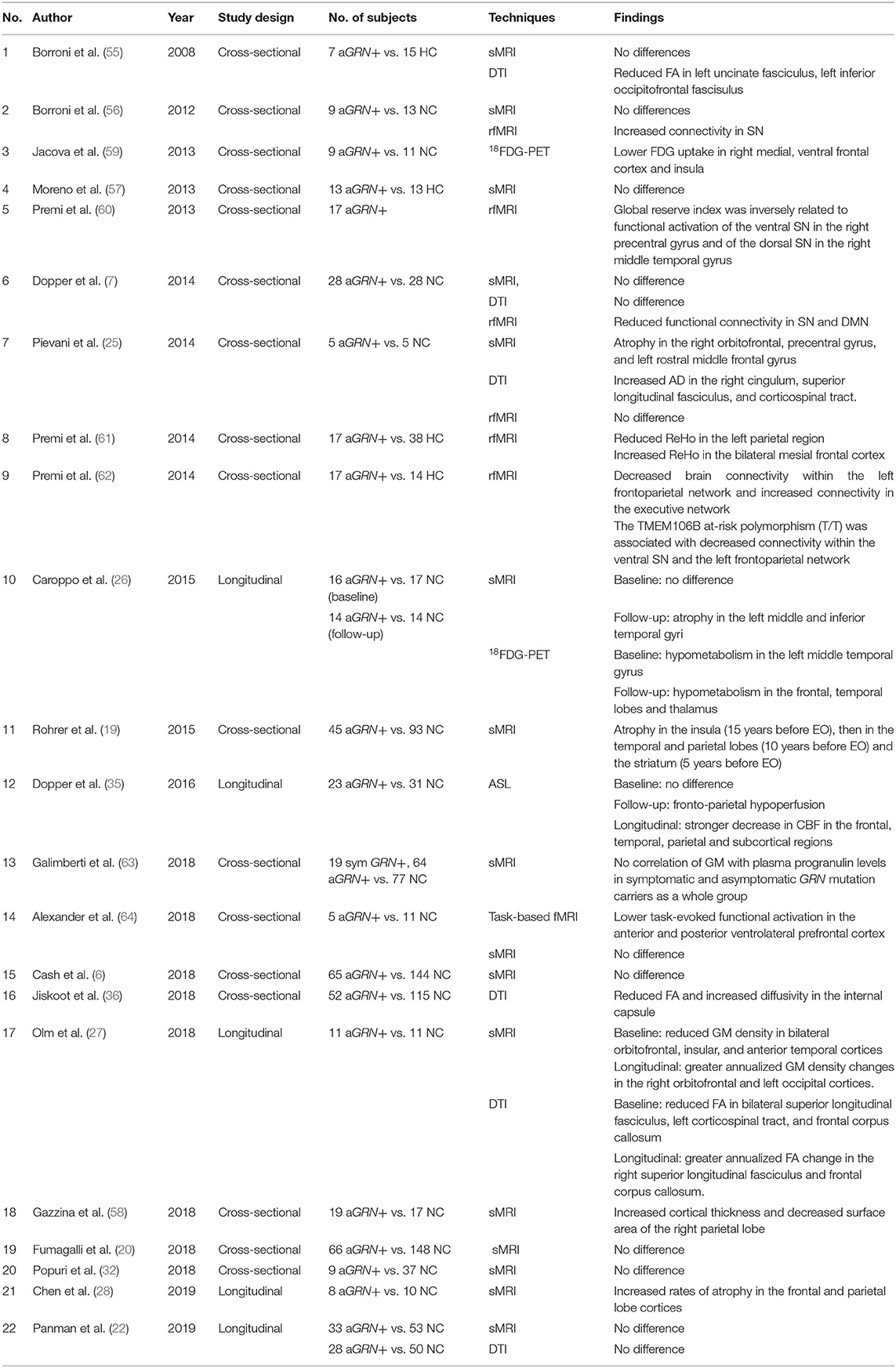
Table 2. Studies investigating asymptomatic GRN mutation vs. controls.
These inconsistencies might be due to the age at examination, the proximity to onset of clinical symptoms, associated lobar cortical atrophy changes, the sample size of GRN mutation carriers, and the statistical analysis methodology with different sensitivity and specificity. For example, voxel-based analysis needs larger effect sizes and corrections for multiple comparisons to detect differences in cortical volumes between groups than the targeted ROI analysis. Furthermore, many studies have demonstrated the heterogeneity of clinical presentation during the early phase and variability in age of onset in GRN mutations carriers (65, 66). In addition, investigations in different time windows during the disease course may capture the variable findings in the laterality of lobar cortical atrophy across individuals with GRN mutations. It may partly explain the inconsistent results of cortical atrophy among the prior cross-sectional studies of asymptomatic GRN mutation carriers.
In GRN mutation carriers, asymmetric atrophy is a common finding with left or right asymmetry reported even within the same family (51, 54). Asymmetric cortical atrophy was found early, 5 years before expected symptom onset in asymptomatic GRN mutation carriers (19), while the underlying pathological cause for the asymmetry remains unclear. In contrast, symmetric rates of brain atrophy were also found in symptomatic GRN mutation carriers in a longitudinal study (67). This discrepancy suggests that asymmetry in cortical atrophy may be more predominant during the early stages, becoming more symmetric during the later stages of the disease (67). Also, as noted above, the specific phenotype and associated lobar cortical atrophy changes and the differences in analysis methods may be underlying these discrepancies. It is important to note that laterality occurs at various stages of neurodegenerative disease progression, which may increase the variability in atrophy rates in individual GRN mutation carriers (Figure 1B).
C9orf72_sMRI
In asymptomatic C9orf72 mutation carriers, cross-sectional structural MRI studies consistently found a diffuse pattern of atrophy including frontal, temporal, parietal, insular, and posterior cortical regions, as well as subcortical volumes of thalamus, hippocampus, and the cerebellum (6, 22, 29–32), while very few studies reported no difference between asymptomatic C9orf72 mutation carriers and healthy controls (20, 68). The atrophy of subcortical regions is estimated to occur as early as 25 years before the expected symptom onset in C9orf72 mutation carriers, and later involves temporal and frontal lobes at around 20 years before the expected symptom onset, and finally involving the cerebellum at around 10 years before the expected symptom onset (19). The early involvement of subcortical regions is a specific atrophy pattern in asymptomatic C9orf72 mutation carriers that distinguishes them from other FTLD mutation carriers.
To date, two longitudinal investigations have reported that there is no difference in the trajectory of cortical atrophy rates between asymptomatic C9orf72 mutation carriers and controls (22, 68), suggesting that longer time follow-up duration is needed to track the trajectory in asymptomatic C9orf72 mutation carriers (Table 3). Figure 1C shows serial structural MRIs from a C9orf72 mutation carriers who converted from asymptomatic phase to symptomatic phase during follow up.
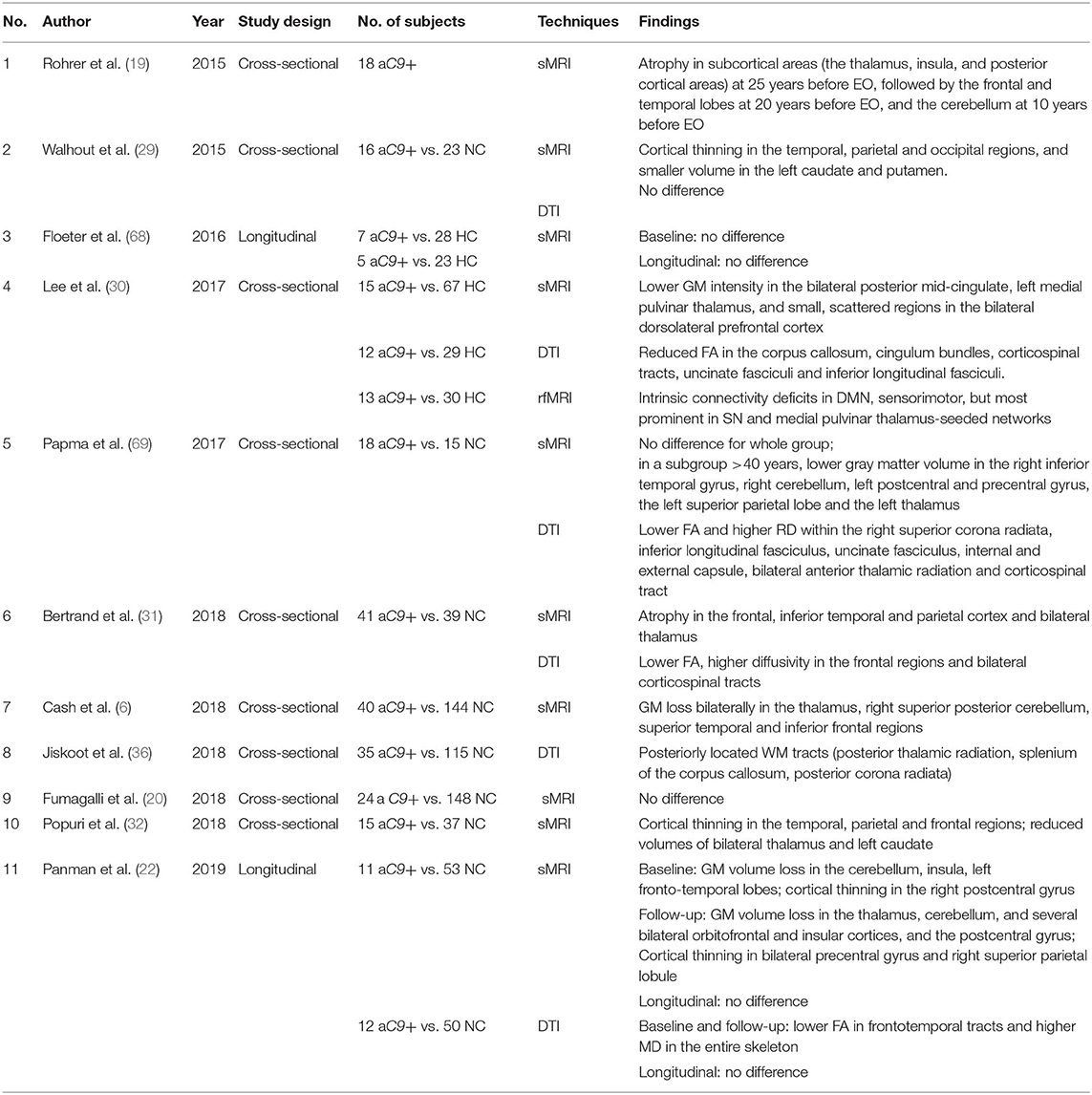
Table 3. Studies investigating asymptomatic C9orf72 mutation vs. controls.
A few studies on familial FTLD have combined different mutation carriers as one group and compared them with non-carriers (Table 4). Combined asymptomatic GRN and C9orf72 mutation carriers demonstrated overlapping atrophy in the insula (70), which has also been reported in a study combining asymptomatic MAPT, GRN, and C9orf72 mutation carriers (6). No difference was observed in both cross-sectional (7) and longitudinal investigations (21) in the combined cohort of asymptomatic MAPT and GRN mutation carriers. However, in a small cohort of 5 MAPT mutation and 3 GRN mutation carriers who converted from asymptomatic to symptomatic phase, extensive temporal and frontal cortical atrophy were observed, while no gray matter volume loss was observed longitudinally in asymptomatic MAPT and GRN mutation carriers who remained asymptomatic compared to non-carriers (21).
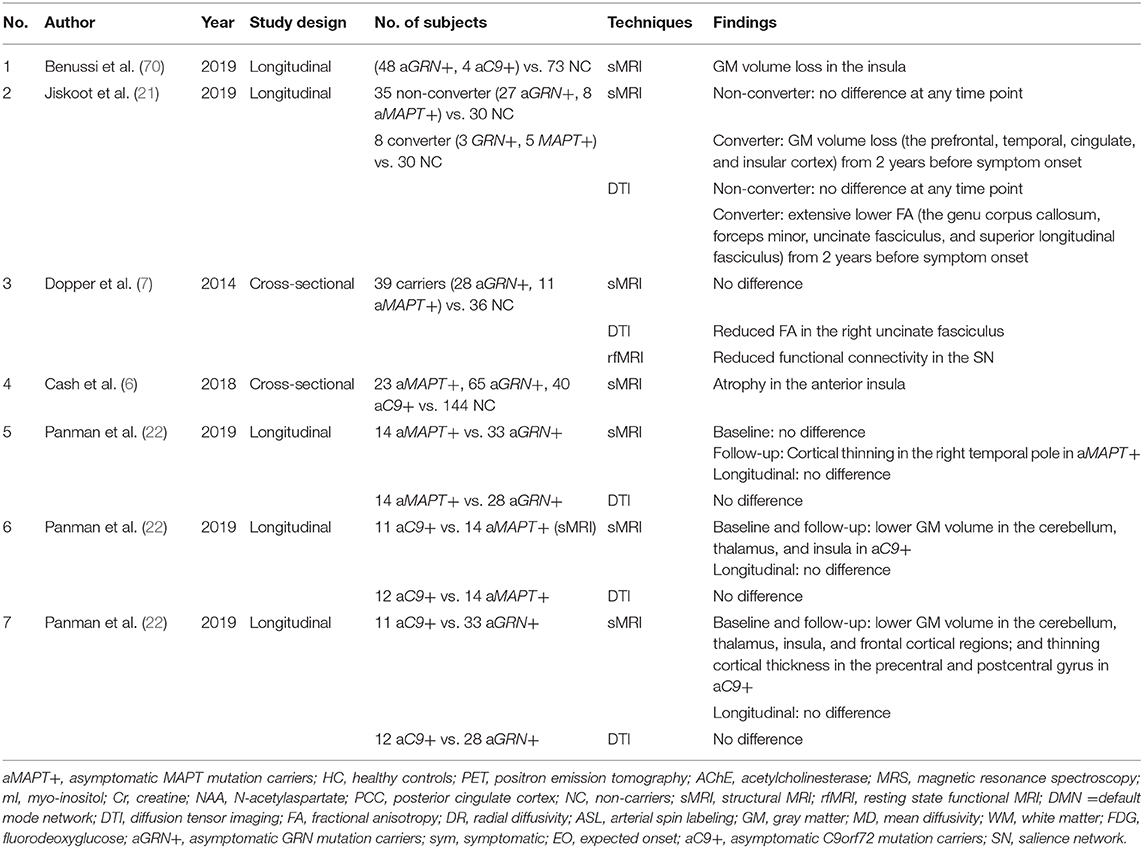
Table 4. Studies investigating multiple different mutations in FTLD.
There has been a comparison of atrophy patterns resulting from mutations in different genes (22); the distinct pattern of atrophy in the early stage of disease course among the MAPT, GRN, and C9orf72 mutation carriers align well with previous studies in symptomatic mutation carriers (67).
Diffusion Tensor Imaging
Diffusion tensor imaging (DTI) is a sensitive technique to detect the white matter microstructure alterations. Damage to the white matter is a common finding in postmortem studies of sporadic and familial FTLD patients (71–73). Loss of white matter integrity on DTI was characterized by increased mean diffusivity (MD), radial diffusivity (RD), and axial diffusivity (AD), as well as reduced fractional anisotropy (FA).
MAPT_DTI
In asymptomatic MAPT mutation carriers, reduced FA and increased diffusivity were reported in bilateral uncinate fasciculus and cingulum in cross-sectional studies (7, 36), while a longitudinal study only found increased MD in entorhinal white matter with accelerated increase in MD during follow-up (38). Furthermore, in a longitudinal study, lower FA in uncinate fasciculus and other white matter tracts were reported in MAPT and GRN mutations 2 years before symptom onset, while no difference in FA was found between the combined group of asymptomatic MAPT and GRN mutation carriers who remained asymptomatic and non-carriers (21). Another recent longitudinal investigation also reported no difference in DTI measurements between asymptomatic MAPT mutation carriers and non-carriers (22).
Compared to FA, the diffusivity parameters such as MD appear to be more sensitive measures of the white matter integrity alterations in asymptomatic familial FTLD (36, 38). One possible explanation is that FA is more influenced by the crossing fibers, which may limit its sensitivity to detect subtle white matter changes (74). The underlying pathologic mechanism of white matter diffusion abnormalities in MAPT mutation carriers remains unclear. It may be due to the Wallerian degeneration secondary to the cortical tau pathology or to the tau pathology observed in the white matter. Future studies are needed to correlate these findings with pathology.
GRN_DTI
White matter involvement from DTI studies in asymptomatic GRN mutation carriers showed reduced FA in the left uncinate fasciculus, left inferior occipitofrontal fasciculus, and the genu of the corpus callosum (55). Reduced FA and increased diffusivity in the internal capsule was also reported (36). Another DTI investigation did not find a difference in FA, MD, and RD, but reported increased AD in the right cingulum, superior longitudinal fasciculus, and corticospinal tract (25). In a longitudinal design, reduced FA was found in bilateral superior longitudinal fasciculus, frontal corpus callosum, and left corticospinal tract in asymptomatic GRN mutation carriers, with greater annualized FA change in frontal corpus callosum and right superior longitudinal fasciculus compared to controls (27). Others did not report any abnormalities of DTI metrics in both cross-sectional (7) and longitudinal investigations (22) of asymptomatic GRN mutation carriers.
As indicated in sMRI, left–right asymmetry was also found in most white matter tracts of GRN mutation carriers. The most consistent asymmetry was reported in the uncinate fasciculus, retrolenticular part, and anterior limb of the internal capsule, as well as external capsule (36). However, the development of asymmetry over time may have different patterns in various white matter tracts. Longitudinal data with larger cohorts are needed for such investigations.
C9orf72_DTI
Investigations in asymptomatic C9orf72 carriers with DTI have shown reduced white matter integrity in the fiber tracts connecting frontal lobe (the uncinate fasciculus and inferior longitudinal fasciculus), motor function-related tracts (corticospinal tracts, corona radiata, and internal/external capsule), thalamic radiation, as well as corpus callosum and cingulum bundles and cerebellar peduncles (30, 31, 36, 69). One cross-sectional investigation reported no DTI abnormality in asymptomatic C9orf72 mutation carriers (29). A recent longitudinal investigation reported a more diffuse white matter involvement in asymptomatic C9orf72 mutation carriers, with lower FA in the frontotemporal tracts and higher MD in the entire white matter skeleton compared to non-carriers. However, these white matter differences remained relatively stable during a 2-year follow-up period (22).
The heterogeneity in clinical phenotypes that ranged from motor neuron disease to FTD and the variation in time of symptom onset within and between families of C9orf72 mutation carriers may be responsible for the variable results from cross-sectional DTI investigations in asymptomatic C9orf72 mutation carriers. Longitudinal follow-up is essential in gaining insight into clinical and neuroimaging characteristics. Therefore, studying families with varying disease phenotypes along the ALS-FTD spectrum would be of particular interest in addressing the issue of clinical heterogeneity in relation to specific early neuroimaging changes.
Functional MRI
Functional MRI can evaluate the alterations from functionally connected networks in spatially distinct subcortical and cortical areas in asymptomatic FTLD mutation carriers. In asymptomatic MAPT mutation carriers, resting-state fMRI demonstrated decreased connectivity in the default mode network (DMN) predominantly between lateral temporal lobe and precuneus (33). Components of the DMN, such as lateral temporal lobes and medial prefrontal cortex, were implicated in functions of semantic memory (75) and theory of mind (76), which show abnormalities in both patients with sporadic bvFTD and symptomatic MAPT mutation carriers. However, another fMRI study found no difference in functional connectivity between asymptomatic MAPT mutation carriers and controls (7). The change of functional connectivity was observed early in the disease course even without gray matter loss in asymptomatic MAPT mutation carriers (33), suggesting that functional abnormalities may precede the occurrence of atrophy in these regions.
In the asymptomatic C9orf72 mutation carriers, the reduced functional connectivity was found prominently in salience network (SN), as well as in medial pulvinar of thalamus-seeded networks, DMN, and sensorimotor network (30). The fMRI alterations can already be distinguished in persons younger than 40 years of age (30). The SN is the main functional network associated with FTLD (77), which plays a role in behavioral functioning and emotion processing (78). Thus, reduced connectivity across central nodes of the SN may lead to some of the clinical features of FTLD.
Increasing evidence from the resting-state fMRI studies demonstrate widely affected networks in asymptomatic GRN mutation carriers, including both increased (56) and reduced connectivity in the SN (7), as well as reduced connectivity in the DMN (7). Functional activation of the SN was inversely related to global reserve index in asymptomatic GRN mutation carriers (60). Furthermore, hypoconnectivity of the left fronto-parietal network (FPN) and a hyper-connectivity within the executive network have been demonstrated (62). The functional impairment of parietal lobes had been thought to be the earliest feature of FTD with GRN mutations (61), while one study reported no difference in functional connectivity between asymptomatic GRN mutation carriers and non-carriers (25). When performing a relational reasoning task, asymptomatic GRN mutation carriers presented lower task-evoked functional activation in anterior and posterior ventrolateral prefrontal cortex compared to controls (64).
As suggested by theoretical concept of molecular nexopathies (79), a specific proteinopathy affects specific networks, while other brain networks could be dynamically involved during the course of the disease. This may possibly explain the heterogeneity in the fMRI findings in asymptomatic FTLD mutation carriers. However, other environmental or genetic factors can also affect the involved networks in a complex way. For example, TMEM106B genetic variations had a modulating effect on the functional connectivity in asymptomatic GRN mutation carriers. For example, the TT genotype of RMEM106B may cause additional neurodegeneration by reduced connectivity in the left fronto-parietal network and ventral SN (62). More studies with longitudinal follow-up are needed to further validate the use of functional connectivity as a potential biomarker for clinical trials.
Proton Magnetic Resonance Spectroscopy
Proton magnetic resonance spectroscopy (1H MRS), which provides non-invasive measurements of brain biochemistry, is a potential imaging marker for early detection of neurodegenerative disease progression in familial FTLD. 1H MRS metabolite measurements have been sensitive biomarkers of early neurodegenerative pathology in FTLD, AD, and Lewy body dementia (80–85). N-acetylaspartate/Creatine (NAA/Cr) is considered a biomarker of neuronal integrity (86), neuronal viability (87, 88), and synaptic integrity (89), while elevated mI may be associated with astrocytic and microglial activation, as well as glial proliferation (90, 91).
In both asymptomatic and symptomatic MAPT mutation carriers, neurochemical alterations from the posterior cingulate voxel were found on single-voxel 1H MRS (92). An elevation in myoinositol (mI) or mI to creatine (mI/Cr) and a decrease in the neuronal integrity marker NAA or NAA/Cr have been found in symptomatic patients with FTLD (83), while only elevated mI/Cr in the posterior cingulate gyrus has been reported in asymptomatic MAPT mutation carriers (8). During longitudinal follow-up in MAPT mutation carriers who converted from the asymptomatic to symptomatic disease, serial 1H MRS from the posterior cingulate voxel demonstrated that metabolite ratio changes, characterized by increasing mI/Cr and decreasing NAA/mI ratios, begin to accelerate ~2 years prior to symptom onset (40).
However, 1H MRS from the medial frontal lobe voxel was more sensitive than 1H MRS from the PCC voxel in the asymptomatic MAPT mutation carriers, characterized by decreased NAA/Cr and NAA/mI ratios (39), suggesting specific regional involvement in neurodegenerative diseases. Regional differences that are apparent in the early stage of disease course may be lost as neurodegenerative pathology spreads to most of the brain regions during the later stages in MAPT mutation carriers.
Potentially, anterior temporal lobe 1H MRS measurements would be the most sensitive to early changes in MAPT mutation carriers. However, this may be offset by a reduction in the quality of the 1H MRS data, since anterior temporal lobes are proximal to the magnetic susceptibility artifacts from the skull base that impact the quality of the 1H MR spectra.
To date, no MRS study was reported in asymptomatic GRN and C9orf72 mutation carriers. In the future, whole-brain and multi-voxel MRS could provide metabolite changes in more brain regions than single-voxel MRS (93). By utilizing more advanced acquisition methods, it would be possible to quantify metabolites, such as glutamine, glutathione, and Scyllo-Inosital (94). For example, an advanced 1H-MRS protocol composed of semi-localization by adiabatic selective refocusing (sLASER) localization and FAST(EST)MAP shimming detected lower glutamate concentration in patients with amnestic mild cognitive impairment than clinically normal controls indicating early Alzheimer's disease pathophysiology (95).
Positron Emission Tomography
Positron emission tomography (PET) imaging is often suggested as a useful biomarker for the earliest stage of FTD and had demonstrated differences in metabolism by different tracers in the presymptomatic stage of familial FTLD.
MAPT_PET
In symptomatic MAPT mutation carriers, [18F]-fluorodeoxyglucose (FDG)-PET has showed asymmetric temporal lobe hypometabolism (46). Frontotemporal hypometabolism on FDG-PET was reported in two asymptomatic MAPT mutation carriers in the same family who had only mild speech change at the time of examination. These participants were in a transitional stage from normal neurological functioning to overt FTLD and one became symptomatic later, suggesting that FDG-PET changes can be detected when asymptomatic MAPT mutation carriers get close to phenoconversion. However, specificity of FDG-PET to differentiate between various neurodegenerative diseases at the early stages remains unknown (96).
Given the heterogeneity of tau pathology observed in MAPT mutations, tau PET (e.g., 18F-AV-1451 PET) was explored as a biomarker for various forms of tau pathology within a similar patient population. Subtypes of MAPT mutations were associated with different AV-1451 uptake patterns due to different types of underlying tauopathies. The mutations inside exon 10 (i.e., S305N, P301L, and N279K) with 4R tau pathology had a low level of AV-1451 binding in Tau PET in symptomatic MAPT mutation carriers. Mutations outside exon 10 (i.e., V337M and R406W) with mixed 3R/4R tau pathology that are more likely to produce AD-like tau pathology presented had a high magnitude of binding of AV-1451 in symptomatic MAPT mutation carriers (37). In symptomatic MAPT V337M mutation carriers, the tau accumulation assessed by [18F] AV1451 tracer was also associated with regional brain atrophy by structural MRI (97). Tau PET studies in asymptomatic MAPT mutation carriers are very limited. One asymptomatic MAPT N279K mutation carrier presented low level of AV-1451 uptake, one of two asymptomatic MAPT R406W mutation carriers had little to no signal and the other one had high level of AV-1451 uptake (37). General conclusions about the tau PET in asymptomatic MAPT mutation carriers remain inconclusive due to small number of cases reported so far.
Tracers associated with dopaminergic function were applied in MAPT mutation carriers, especially in the N279K mutation type, which may present with parkinsonism as the first symptom. Dopaminergic dysfunction was shown early in asymptomatic MAPT N279K mutation carriers via 2b-carbomethoxy-3b-(4-trmethylstannylphenyl) tropane (11C-CFT)-PET (34, 98). Glial activity was elevated in the frontal cortex, the posterior cingulate cortex, and the occipital cortex of asymptomatic MAPT mutation carriers on [11C] DAA1106 PET, and acetylcholinesterase activity was reduced in the temporo-parietal cortex using [11C] N-methylpiperidin-4-yl acetate PET in three asymptomatic MAPT mutation carriers (34). These case studies with small sample sizes will need to be replicated in larger cohorts.
GRN_PET
In asymptomatic GRN mutation carriers, hypometabolism was detected in right medial, ventral frontal cortex, and insula on FDG-PET (59). A longitudinal investigation reported that hypometabolism in the left middle temporal gyrus at baseline is associated with greater decrease in metabolism in the fronto-temporal lobes and thalamus in asymptomatic GRN mutation carriers compared to non-carriers (26).
C9orf72_PET
A radioligand for the microtubule associated protein tau (18F-Flortaucipir) was found to show increased binding in semantic variant primary progressive aphasia (svPPA) (99), which is associated with underlying TDP-43 pathology. In contrast, a recent study reported that none or limited 18F-Flortaucipir retention was found in symptomatic C9orf72 mutation carriers, suggesting that 18F-Flortaucipir binding in svPPA patients is not a general TDP-43 related phenomenon (100). To date, a PET tracer for the in vivo detection of TDP-43 pathology does not exist.
Conclusion
This review underscores the importance of imaging biomarkers for accurate prediction of symptom onset and tracking of disease progression during the presymptomatic stage of familial FTLD. The application of advanced neuroimaging techniques in monogenetic familial FTLD represents a unique model to detect the natural history of specific proteinopathies and clinical phenotypes. They may also provide a direct comparison across different gene groups in the future. Furthermore, multiple MRI and PET modalities may provide information on the different aspects and stages of the neurodegenerative disease process in a single individual. Many of the studies discussed in this review focus on a single imaging biomarker. Thus, it is not possible to compare biomarkers on their accuracy in prediction of onset of clinical symptoms and tracking disease progression in familial FTLD. The potential application of multi-model imaging is to provide evidence of very early asymptomatic alterations by collectively including cortical atrophy, white matter integrity loss, functional alteration, as well as brain metabolic changes. There are several ongoing prospective multisite studies (e.g., GENFI, ARTFL, and LEFFTDS) involving the familial FTLD kindred. These longitudinal investigations in large cohorts on the genotype-specific imaging profiles for MAPT, GRN, and C9orf72 at different time points during the disease course would provide insights into their potential use as prognostic biomarkers for clinical trials of disease-modifying therapies.
Author Contributions
QC data collection, analysis and interpretation of the data, drafting the manuscript. KK design or conceptualization of the study, data collection, analysis and interpretation of the data, drafting the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. (2015) 386:1672–82. doi: 10.1016/S0140-6736(15)00461-4
2. Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. (2011) 24:542–9. doi: 10.1097/WCO.0b013e32834cd442
3. Ingram EM, Spillantini MG. Tau gene mutations: dissecting the pathogenesis of FTDP-17. Trends Mol Med. (2002) 8:555–62. doi: 10.1016/S1471-4914(02)02440-1
4. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393:702–5. doi: 10.1038/31508
5. Bunker JM, Kamath K, Wilson L, Jordan MA, Feinstein SC. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J Biol Chem. (2006) 281:11856–63. doi: 10.1074/jbc.M509420200
6. Cash DM, Bocchetta M, Thomas DL, Dick KM, van Swieten JC, Borroni B, et al. Patterns of gray matter atrophy in genetic frontotemporal dementia: results from the GENFI study. Neurobiol Aging. (2018) 62:191–6. doi: 10.1016/j.neurobiolaging.2017.10.008
7. Dopper EG, Rombouts SA, Jiskoot LC, den Heijer T, de Graaf JR, de Koning I, et al. Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology. (2014) 83:e19–26. doi: 10.1212/WNL.0000000000000583
8. Kantarci K, Boeve BF, Wszolek ZK, Rademakers R, Whitwell JL, Baker MC, et al. MRS in presymptomatic MAPT mutation carriers: a potential biomarker for tau-mediated pathology. Neurology. (2010) 75:771–8. doi: 10.1212/WNL.0b013e3181f073c7
9. McCarthy A, Lonergan R, Olszewska DA, O'Dowd S, Cummins G, Magennis B, et al. Closing the tau loop: the missing tau mutation. Brain. (2015) 138:3100–9. doi: 10.1093/brain/awv234
10. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. (2006) 442:916–9. doi: 10.1038/nature05016
11. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. (2006) 442:920–4. doi: 10.1038/nature05017
12. Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. (2016) 138(Suppl. 1):32–53. doi: 10.1111/jnc.13622
13. Beck J, Rohrer JD, Campbell T, Isaacs A, Morrison KE, Goodall EF, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain. (2008) 131:706–20. doi: 10.1093/brain/awm320
14. Kelley BJ, Haidar W, Boeve BF, Baker M, Graff-Radford NR, Krefft T, et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging. (2009) 30:739–51. doi: 10.1016/j.neurobiolaging.2007.08.022
15. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
16. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
17. Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. (2012) 135:709–22. doi: 10.1093/brain/awr354
18. Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. (2012) 135:765–83. doi: 10.1016/j.jalz.2012.05.2129
19. Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. (2015) 14:253–62. doi: 10.1016/S1474-4422(14)70324-2
20. Fumagalli GG, Basilico P, Arighi A, Bocchetta M, Dick KM, Cash DM, et al. Distinct patterns of brain atrophy in Genetic Frontotemporal Dementia Initiative (GENFI) cohort revealed by visual rating scales. Alzheimers Res Ther. (2018) 10:46. doi: 10.1186/s13195-018-0376-9
21. Jiskoot LC, Panman JL, Meeter LH, Dopper EGP, Donker Kaat L, Franzen S, et al. Longitudinal multimodal MRI as prognostic and diagnostic biomarker in presymptomatic familial frontotemporal dementia. Brain. (2019) 142:193–208. doi: 10.1093/brain/awy288
22. Panman JL, Jiskoot LC, Bouts M, Meeter LHH, van der Ende EL, Poos JM, et al. Gray and white matter changes in presymptomatic genetic frontotemporal dementia: a longitudinal MRI study. Neurobiol Aging. (2019) 76:115–24. doi: 10.1016/j.neurobiolaging.2018.12.017
23. Chen Q, Boeve BF, Senjem M, Tosakulwong N, Lesnick TG, Brushaber D, et al. Rates of lobar atrophy in asymptomatic MAPT mutation carriers. Alzheimers Dement. (2019) 5:338–46. doi: 10.1016/j.trci.2019.05.010
24. Rohrer JD, Warren JD, Barnes J, Mead S, Beck J, Pepple T, et al. Mapping the progression of progranulin-associated frontotemporal lobar degeneration. Nat Clin Pract Neurol. (2008) 4:455–60. doi: 10.1038/ncpneuro0869
25. Pievani M, Paternico D, Benussi L, Binetti G, Orlandini A, Cobelli M, et al. Pattern of structural and functional brain abnormalities in asymptomatic granulin mutation carriers. Alzheimers Dement. (2014) 10(5 Suppl.):S354–63 e1. doi: 10.1016/j.jalz.2013.09.009
26. Caroppo P, Habert MO, Durrleman S, Funkiewiez A, Perlbarg V, Hahn V, et al. Lateral temporal lobe: an early imaging marker of the presymptomatic grn disease? J Alzheimers Dis. (2015) 47:751–9. doi: 10.3233/JAD-150270
27. Olm CA, McMillan CT, Irwin DJ, Van Deerlin VM, Cook PA, Gee JC, et al. Longitudinal structural gray matter and white matter MRI changes in presymptomatic progranulin mutation carriers. Neuroimage Clin. (2018) 19:497–506. doi: 10.1016/j.nicl.2018.05.017
28. Chen Q, Boeve BF, Senjem M, Tosakulwong N, Lesnick T, Brushaber D, et al. Trajectory of lobar atrophy in asymptomatic and symptomatic GRN mutation carriers: a longitudinal MRI study. Neurobiol. Aging. (2019). doi: 10.1016/j.neurobiolaging.2019.12.004. [Epub ahead of print].
29. Walhout R, Schmidt R, Westeneng HJ, Verstraete E, Seelen M, van Rheenen W, et al. Brain morphologic changes in asymptomatic C9orf72 repeat expansion carriers. Neurology. (2015) 85:1780–8. doi: 10.1212/WNL.0000000000002135
30. Lee SE, Sias AC, Mandelli ML, Brown JA, Brown AB, Khazenzon AM, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. (2017) 14:286–97. doi: 10.1016/j.nicl.2016.12.006
31. Bertrand A, Wen J, Rinaldi D, Houot M, Sayah S, Camuzat A, et al. Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol. (2018) 75:236–45. doi: 10.1001/jamaneurol.2017.4266
32. Popuri K, Dowds E, Beg MF, Balachandar R, Bhalla M, Jacova C, et al. Gray matter changes in asymptomatic C9orf72 and GRN mutation carriers. Neuroimage Clin. (2018) 18:591–8. doi: 10.1016/j.nicl.2018.02.017
33. Whitwell JL, Josephs KA, Avula R, Tosakulwong N, Weigand SD, Senjem ML, et al. Altered functional connectivity in asymptomatic MAPT subjects: a comparison to bvFTD. Neurology. (2011) 77:866–74. doi: 10.1212/WNL.0b013e31822c61f2
34. Miyoshi M, Shinotoh H, Wszolek ZK, Strongosky AJ, Shimada H, Arakawa R, et al. In vivo detection of neuropathologic changes in presymptomatic MAPT mutation carriers: a PET and MRI study. Parkinsonism Relat Disord. (2010) 16:404–8. doi: 10.1016/j.parkreldis.2010.04.004
35. Dopper EG, Chalos V, Ghariq E, den Heijer T, Hafkemeijer A, Jiskoot LC, et al. Cerebral blood flow in presymptomatic MAPT and GRN mutation carriers: a longitudinal arterial spin labeling study. Neuroimage Clin. (2016) 12:460–5. doi: 10.1016/j.nicl.2016.08.001
36. Jiskoot LC, Bocchetta M, Nicholas JM, Cash DM, Thomas D, Modat M, et al. Presymptomatic white matter integrity loss in familial frontotemporal dementia in the GENFI cohort: a cross-sectional diffusion tensor imaging study. Ann Clin Transl Neurol. (2018) 5:1025–36. doi: 10.1002/acn3.601
37. Jones DT, Knopman DS, Graff-Radford J, Syrjanen JA, Senjem ML, Schwarz CG, et al. In vivo (18)F-AV-1451 tau PET signal in MAPT mutation carriers varies by expected tau isoforms. Neurology. (2018) 90:e947–e54. doi: 10.1212/WNL.0000000000005117
38. Chen Q, Boeve BF, Schwarz CG, Reid R, Tosakulwong N, Lesnick TG, et al. Tracking white matter degeneration in asymptomatic and symptomatic MAPT mutation carriers. Neurobiol Aging. (2019) 83:54–62. doi: 10.1016/j.neurobiolaging.2019.08.011
39. Chen Q, Boeve BF, Tosakulwong N, Lesnick T, Brushaber D, Dheel C, et al. Frontal lobe (1)H MR spectroscopy in asymptomatic and symptomatic MAPT mutation carriers. Neurology. (2019) 93:e758–65. doi: 10.1212/WNL.0000000000007961
40. Chen Q, Boeve BF, Tosakulwong N, Lesnick T, Brushaber D, Dheel C, et al. Brain MR spectroscopy changes precede frontotemporal lobar degeneration phenoconversion in mapt mutation carriers. J Neuroimaging. (2019) 29:624–9. doi: 10.1111/jon.12642
41. Whitwell JL, Jack CR Jr, Boeve BF, Senjem ML, Baker M, et al. Atrophy patterns in IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M MAPT mutations. Neurology. (2009) 73:1058–65. doi: 10.1212/WNL.0b013e3181b9c8b9
42. Wszolek ZK, Kardon RH, Wolters EC, Pfeiffer RF. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17): PPND family. A longitudinal videotape demonstration. Mov Disord. (2001) 16:756–60. doi: 10.1002/mds.1131
43. Wszolek ZK, Pfeiffer RF, Bhatt MH, Schelper RL, Cordes M, Snow BJ, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol. (1992) 32:312–20. doi: 10.1002/ana.410320303
44. Wszolek ZK, Tsuboi Y, Uitti RJ, Reed L. Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology. (2000) 55:1939. doi: 10.1212/WNL.55.12.1939
45. Arima K, Kowalska A, Hasegawa M, Mukoyama M, Watanabe R, Kawai M, et al. Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology. (2000) 54:1787–95. doi: 10.1212/WNL.54.9.1787
46. Arvanitakis Z, Witte RJ, Dickson DW, Tsuboi Y, Uitti RJ, Slowinski J, et al. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation). Parkinsonism Relat Disord. (2007) 13:230–9. doi: 10.1016/j.parkreldis.2006.10.007
47. Cheshire WP, Tsuboi Y, Wszolek ZK. Physiologic assessment of autonomic dysfunction in pallidopontonigral degeneration with N279K mutation in the tau gene on chromosome 17. Auton Neurosci. (2002) 102:71–7. doi: 10.1016/S1566-0702(02)00205-9
48. Ferman TJ, McRae CA, Arvanitakis Z, Tsuboi Y, Vo A, Wszolek ZK. Early and pre-symptomatic neuropsychological dysfunction in the PPND family with the N279K tau mutation. Parkinsonism Relat Disord. (2003) 9:265–70. doi: 10.1016/S1353-8020(02)00098-6
49. Slowinski J, Dominik J, Uitti RJ, Ahmed Z, Dickson DD, Wszolek ZK. Frontotemporal dementia and Parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology. (2007) 27:73–80. doi: 10.1111/j.1440-1789.2006.00742.x
50. Whitwell JL, Weigand SD, Gunter JL, Boeve BF, Rademakers R, Baker M, et al. Trajectories of brain and hippocampal atrophy in FTD with mutations in MAPT or GRN. Neurology. (2011) 77:393–8. doi: 10.1212/WNL.0b013e318227047f
51. Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, et al. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage. (2010) 53:1070–6. doi: 10.1016/j.neuroimage.2009.12.088
52. Premi E, Cauda F, Costa T, Diano M, Gazzina S, Gualeni V, et al. Looking for neuroimaging markers in frontotemporal lobar degeneration clinical trials: a multi-voxel pattern analysis study in granulin disease. J Alzheimers Dis. (2016) 51:249–62. doi: 10.3233/JAD-150340
53. Whitwell JL, Jack CR Jr, Boeve BF, Senjem ML, Baker M, Rademakers R, et al. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology. (2009) 72:813–20. doi: 10.1212/01.wnl.0000343851.46573.67
54. Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. (2012) 135:794–806. doi: 10.1093/brain/aws001
55. Borroni B, Alberici A, Premi E, Archetti S, Garibotto V, Agosti C, et al. Brain magnetic resonance imaging structural changes in a pedigree of asymptomatic progranulin mutation carriers. Rejuvenation Res. (2008) 11:585–95. doi: 10.1089/rej.2007.0623
56. Borroni B, Alberici A, Cercignani M, Premi E, Serra L, Cerini C, et al. Granulin mutation drives brain damage and reorganization from preclinical to symptomatic FTLD. Neurobiol Aging. (2012) 33:2506–20. doi: 10.1016/j.neurobiolaging.2011.10.031
57. Moreno F, Sala-Llonch R, Barandiaran M, Sanchez-Valle R, Estanga A, Bartres-Faz D, et al. Distinctive age-related temporal cortical thinning in asymptomatic granulin gene mutation carriers. Neurobiol Aging. (2013) 34:1462–8. doi: 10.1016/j.neurobiolaging.2012.11.005
58. Gazzina S, Benussi A, Premi E, Paternico D, Cristillo V, Dell'Era V, et al. Neuroanatomical correlates of transcranial magnetic stimulation in presymptomatic granulin mutation carriers. Brain Topogr. (2018) 31:488–97. doi: 10.1007/s10548-017-0612-9
59. Jacova C, Hsiung GY, Tawankanjanachot I, Dinelle K, McCormick S, Gonzalez M, et al. Anterior brain glucose hypometabolism predates dementia in progranulin mutation carriers. Neurology. (2013) 81:1322–31. doi: 10.1212/WNL.0b013e3182a8237e
60. Premi E, Gazzina S, Bozzali M, Archetti S, Alberici A, Cercignani M, et al. Cognitive reserve in granulin-related frontotemporal dementia: from preclinical to clinical stages. PLoS ONE. (2013) 8:e74762. doi: 10.1371/journal.pone.0074762
61. Premi E, Cauda F, Gasparotti R, Diano M, Archetti S, Padovani A, et al. Multimodal FMRI resting-state functional connectivity in granulin mutations: the case of fronto-parietal dementia. PLoS ONE. (2014) 9:e106500. doi: 10.1371/journal.pone.0106500
62. Premi E, Formenti A, Gazzina S, Archetti S, Gasparotti R, Padovani A, et al. Effect of TMEM106B polymorphism on functional network connectivity in asymptomatic GRN mutation carriers. JAMA Neurol. (2014) 71:216–21. doi: 10.1001/jamaneurol.2013.4835
63. Galimberti D, Fumagalli GG, Fenoglio C, Cioffi SMG, Arighi A, Serpente M, et al. Progranulin plasma levels predict the presence of GRN mutations in asymptomatic subjects and do not correlate with brain atrophy: results from the GENFI study. Neurobiol Aging. (2018) 62:245 e9–12. doi: 10.1016/j.neurobiolaging.2017.10.016
64. Alexander C, Zeithamova D, Hsiung GR, Mackenzie IR, Jacova C. Decreased prefrontal activation during matrix reasoning in predementia progranulin mutation carriers. J Alzheimers Dis. (2018) 62:583–9. doi: 10.3233/JAD-170716
65. van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. (2008) 7:965–74. doi: 10.1016/S1474-4422(08)70194-7
66. Ghidoni R, Paterlini A, Albertini V, Binetti G, Benussi L. Losing protein in the brain: the case of progranulin. Brain Res. (2012) 1476:172–82. doi: 10.1016/j.brainres.2012.01.075
67. Whitwell JL, Boeve BF, Weigand SD, Senjem ML, Gunter JL, Baker MC, et al. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol. (2015) 22:745–52. doi: 10.1111/ene.12675
68. Floeter MK, Bageac D, Danielian LE, Braun LE, Traynor BJ, Kwan JY. Longitudinal imaging in C9orf72 mutation carriers: relationship to phenotype. Neuroimage Clin. (2016) 12:1035–43. doi: 10.1016/j.nicl.2016.10.014
69. Papma JM, Jiskoot LC, Panman JL, Dopper EG, den Heijer T, Donker Kaat L, et al. Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology. (2017) 89:1256–64. doi: 10.1212/WNL.0000000000004393
70. Benussi A, Gazzina S, Premi E, Cosseddu M, Archetti S, Dell'Era V, et al. Clinical and biomarker changes in presymptomatic genetic frontotemporal dementia. Neurobiol Aging. (2019) 76:133–40. doi: 10.1016/j.neurobiolaging.2018.12.018
71. Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R, et al. White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol. (2008) 67:963–75. doi: 10.1097/NEN.0b013e318187a80f
72. Larsson E, Passant U, Sundgren PC, Englund E, Brun A, Lindgren A, et al. Magnetic resonance imaging and histopathology in dementia, clinically of frontotemporal type. Dement Geriatr Cogn Disord. (2000) 11:123–34. doi: 10.1159/000017225
73. Sima AA, Defendini R, Keohane C, D'Amato C, Foster NL, Parchi P, et al. The neuropathology of chromosome 17-linked dementia. Ann Neurol. (1996) 39:734–43. doi: 10.1002/ana.410390609
74. Jeurissen B, Leemans A, Tournier JD, Jones DK, Sijbers J. Investigating the prevalence of complex fiber configurations in white matter tissue with diffusion magnetic resonance imaging. Hum Brain Mapp. (2013) 34:2747–66. doi: 10.1002/hbm.22099
75. Wirth M, Jann K, Dierks T, Federspiel A, Wiest R, Horn H. Semantic memory involvement in the default mode network: a functional neuroimaging study using independent component analysis. Neuroimage. (2011) 54:3057–66. doi: 10.1016/j.neuroimage.2010.10.039
76. Spreng RN, Grady CL. Patterns of brain activity supporting autobiographical memory, prospection, and theory of mind, and their relationship to the default mode network. J Cogn Neurosci. (2010) 22:1112–23. doi: 10.1162/jocn.2009.21282
77. Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain. (2010) 133:1352–67. doi: 10.1093/brain/awq075
78. Seeley WW, Menon V, Schatzberg AF, Keller J, Glover GH, Kenna H, et al. Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci. (2007) 27:2349–56. doi: 10.1523/JNEUROSCI.5587-06.2007
79. Warren JD, Rohrer JD, Schott JM, Fox NC, Hardy J, Rossor MN. Molecular nexopathies: a new paradigm of neurodegenerative disease. Trends Neurosci. (2013) 36:561–9. doi: 10.1016/j.tins.2013.06.007
80. Kantarci K, Knopman DS, Dickson DW, Parisi JE, Whitwell JL, Weigand SD, et al. Alzheimer disease: postmortem neuropathologic correlates of antemortem 1H MR spectroscopy metabolite measurements. Radiology. (2008) 248:210–20. doi: 10.1148/radiol.2481071590
81. Kantarci K. Magnetic resonance spectroscopy in common dementias. Neuroimaging Clin N Am. (2013) 23:393–406. doi: 10.1016/j.nic.2012.10.004
82. Graff-Radford J, Boeve BF, Murray ME, Ferman TJ, Tosakulwong N, Lesnick TG, et al. Regional proton magnetic resonance spectroscopy patterns in dementia with Lewy bodies. Neurobiol Aging. (2014) 35:1483–90. doi: 10.1016/j.neurobiolaging.2014.01.001
83. Ernst T, Chang L, Melchor R, Mehringer CM. Frontotemporal dementia and early Alzheimer disease: differentiation with frontal lobe H-1 MR spectroscopy. Radiology. (1997) 203:829–36. doi: 10.1148/radiology.203.3.9169712
84. Waragai M, Moriya M, Nojo T. Decreased N-Acetyl aspartate/myo-inositol ratio in the posterior cingulate cortex shown by magnetic resonance spectroscopy may be one of the risk markers of preclinical Alzheimer's disease: a 7-year follow-up study. J Alzheimers Dis. (2017) 60:1411–27. doi: 10.3233/JAD-170450
85. Joe E, Medina LD, Ringman JM, O'Neill J. (1)H MRS spectroscopy in preclinical autosomal dominant Alzheimer disease. Brain Imaging Behav. (2019) 13:925–32. doi: 10.1007/s11682-018-9913-1
86. Valenzuela MJ, Sachdev P. Magnetic resonance spectroscopy in AD. Neurology. (2001) 56:592–8. doi: 10.1212/WNL.56.5.592
87. Bates TE, Strangward M, Keelan J, Davey GP, Munro PM, Clark JB. Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. Neuroreport. (1996) 7:1397–400. doi: 10.1097/00001756-199605310-00014
88. Petroff OA, Errante LD, Kim JH, Spencer DD. N-acetyl-aspartate, total creatine, and myo-inositol in the epileptogenic human hippocampus. Neurology. (2003) 60:1646–51. doi: 10.1212/01.WNL.0000068020.85450.8B
89. Murray ME, Przybelski SA, Lesnick TG, Liesinger AM, Spychalla A, Zhang B, et al. Early Alzheimer's disease neuropathology detected by proton MR spectroscopy. J Neurosci. (2014) 34:16247–55. doi: 10.1523/JNEUROSCI.2027-14.2014
90. Brand A, Richter-Landsberg C, Leibfritz D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev Neurosci. (1993) 15:289–98. doi: 10.1159/000111347
91. Yamada T, McGeer E, Schelper R, Wszolek ZK, McGeer P, Pfeiffer R, et al. Histological and biochemical pathology in a family with autosomal dominant Parkinsonism and dementia. Neurology Psychiatry and Brain Research. (1993) 2:26–35.
92. Kizu O, Yamada K, Ito H, Nishimura T. Posterior cingulate metabolic changes in frontotemporal lobar degeneration detected by magnetic resonance spectroscopy. Neuroradiology. (2004) 46:277–81. doi: 10.1007/s00234-004-1167-5
93. Su L, Blamire AM, Watson R, He J, Hayes L, O'Brien JT. Whole-brain patterns of (1)H-magnetic resonance spectroscopy imaging in Alzheimer's disease and dementia with Lewy bodies. Transl Psychiatry. (2016) 6:e877. doi: 10.1038/tp.2016.140
94. Barbagallo G, Morelli M, Quattrone A, Chiriaco C, Vaccaro MG, Gulla D, et al. In vivo evidence for decreased scyllo-inositol levels in the supplementary motor area of patients with Progressive Supranuclear Palsy: a proton MR spectroscopy study. Parkinsonism Relat Disord. (2019) 62:185–91. doi: 10.1016/j.parkreldis.2018.12.008
95. Zeydan B, Deelchand DK, Tosakulwong N, Lesnick TG, Kantarci OH, Machulda MM, et al. Decreased glutamate levels in patients with amnestic mild cognitive impairment: an sLASER proton MR spectroscopy and PiB-PET study. J Neuroimaging. (2017) 27:630–6. doi: 10.1111/jon.12454
96. Arbizu J, Festari C, Altomare D, Walker Z, Bouwman F, Rivolta J, et al. Clinical utility of FDG-PET for the clinical diagnosis in MCI. Eur J Nucl Med Mol Imaging. (2018) 45:1497–508. doi: 10.1007/s00259-018-4039-7
97. Spina S, Schonhaut DR, Boeve BF, Seeley WW, Ossenkoppele R, O'Neil JP, et al. Frontotemporal dementia with the V337M MAPT mutation: Tau-PET and pathology correlations. Neurology. (2017) 88:758–66. doi: 10.1212/WNL.0000000000003636
98. Wu L, Liu J, Feng X, Dong J, Qin W, Liu Y, et al. 11C-CFT-PET in presymptomatic FTDP-17: a potential biomarker predicting onset. J Alzheimers Dis. (2018) 61:613–8. doi: 10.3233/JAD-170561
99. Makaretz SJ, Quimby M, Collins J, Makris N, McGinnis S, Schultz A, et al. Flortaucipir tau PET imaging in semantic variant primary progressive aphasia. J Neurol Neurosurg Psychiatry. (2018) 89:1024–31. doi: 10.1136/jnnp-2017-316409
Keywords: imaging biomarker, presymptomatic, familial frontotemporal lobar degeneration, MAPT, GRN, C9orf72
Citation: Chen Q and Kantarci K (2020) Imaging Biomarkers for Neurodegeneration in Presymptomatic Familial Frontotemporal Lobar Degeneration. Front. Neurol. 11:80. doi: 10.3389/fneur.2020.00080
Received: 30 October 2019; Accepted: 22 January 2020;
Published: 28 February 2020.
Edited by:
Jingyun Yang, Rush University Medical Center, United StatesReviewed by:
Maria Carmela Tartaglia, University of Toronto, CanadaShihui Ying, Shanghai University, China
Copyright © 2020 Chen and Kantarci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kejal Kantarci, a2FudGFyY2kua2VqYWwmI3gwMDA0MDttYXlvLmVkdQ==