 Yanjun Tian
Yanjun Tian Ruijiao Chen1†
Ruijiao Chen1†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol., 03 March 2020
Sec. Stroke
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00075
The orphan receptor APJ and its endogenous ligand apelin, which are expressed in the brain, are the major components of the apelin/APJ system. Growing evidence shows that the apelin/APJ system plays a vital role in the pathophysiology of cerebral ischemic injury. Targeting the apelin/APJ system may have protective effects on cerebral ischemic injury. In this review, we sum up the latest research progress relating to the actions and therapeutic potential of the apelin/APJ system in ischemic stroke. An in-depth knowledge of the pathophysiological effects of the apelin/APJ system and the underlying mechanisms will help to develop novel therapeutic interventions for ischemic stroke.
In 1993, O'Dowd discovered the orphan G protein–coupled receptor (GPCR) APJ while searching for vasopressin receptor subtypes (1). APJ is encoded by a gene located on chromosome 11q12. Although APJ shares 54% homology with angiotensin II receptor type-1 (AT1R) in the hydrophobic transmembrane region, there's no binding site for angiotensin II (1). Tatemoto et al. isolated apelin, the cognate ligand for APJ receptor, from bovine stomach tissue extracts in 1998 (2). The apelin gene, which is located on chromosome Xq25-26.1, encodes the preproapelin of 77 amino acids (apelin-77). Various bioactive isoforms of apelin are derived from apelin-77, including apelin-55, apelin-36, apelin-17, apelin-13, and apelin-12 (3, 4). Recently Chng et al. discovered apela, another endogenous ligand for APJ, which is encoded by a gene located on chromosome 11 and is critical in embryonic development (5, 6). However, in humans, the apela is only expressed in pluripotent cells and kidney (7). The apelin/APJ system mainly refers to APJ and its endogenous ligand, apelin.
Stroke, which is mainly caused by cerebral vascular occlusion and cerebral blood supply disorder, is one of the leading causes of death and disability worldwide, and 87% of cases are ischemic stroke (8). The cerebral infarction area is composed of the ischemic core and penumbra; apoptosis is the main cause of neuronal damage in the penumbra region, which also provides an opportunity for the treatment of ischemic stroke and makes it possible to use drugs to alleviate neuronal injury since the apoptosis is delayed and reversible (9, 10). Neuronal apoptosis in ischemic penumbra is triggered by diffusion of toxic substances released by the dead neurons of the ischemic core in the acute stage of ischemia, while ischemia injury is aggravated after reperfusion, namely ischemia/reperfusion (I/R) injury, which contributes to the neuron apoptosis in the penumbra via numerous biological mechanisms, including excitotoxicity, oxidative and nitrative stress, inflammatory responses, endoplasmic reticulum stress (ERS), and so on (11–14).
The apelin/APJ system is widely expressed in the central nervous system, especially in neurons and oligodendrocytes (15, 16). Growing evidence indicates that the apelin/APJ system is involved in the pathophysiology of ischemic stroke (17, 18). Targeting the apelin/APJ system may have protective effects on cerebral ischemic injury. In this review, we mainly focus on the latest research progress related to the biological functions and therapeutic potential of the apelin/APJ system in ischemic stroke.
The apelin/APJ system mediates signal transduction mainly by coupling to G protein (Figure 1). Gαi participates in the activation of phosphatidylinositide 3-kinase (PI3K)/protein kinase B (PKB, also known as AKT) or contributes to protein kinase C (PKC) activation, initiating the mitogenic extracellular signal-regulated kinase (ERK) signaling pathway; it can also inhibit the activation of protein kinase A (PKA) by inhibiting adenylyl cyclase (AC) to produce cyclic adenosine monophosphate (cAMP) (19, 20). Meanwhile, Gαq activates phospholipase C beta (PLCβ), inducing the production of diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3), facilitating the activation of PKC cascade and the release of intracellular Ca2+ respectively (19). Ca2+ release in turn activates calmodulin, which subsequently exerts vasodilatory effects via activating nitrous oxide synthase (NOS). Besides that, endothelial NOS (eNOS) can be induced by AKT activation via Gαi, also presenting a vasodilation effect of the apelin/APJ system (21). In addition to the canonical pathways, Kang et al. found a novel APJ signaling route in endothelial cells through Gα13, leading to phosphorylation and cytoplasmic translocation of class II histone deacetylases (HDACs) 4 and 5, thereby activating myocyte enhancer factor-2 (MEF2), which induces the expression of MEF2 target gene Kruppel-like factor 2 (KLF2) (Figure 1) (22).
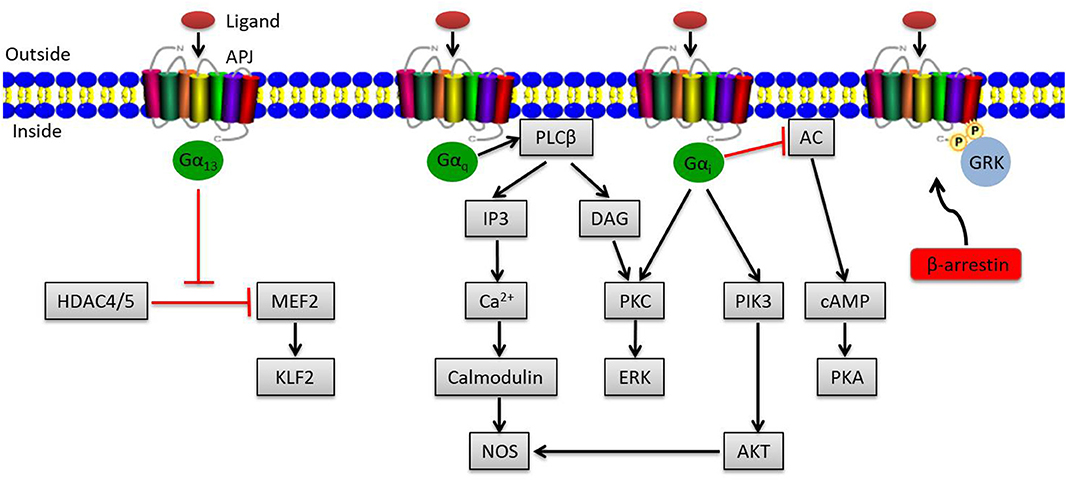
Figure 1. Overview of apelin/APJ system–mediated signaling pathways. Canonical ligand-dependent APJ signaling via Gαi and Gαq leads to the activation of protein kinase C (PKC), phosphatidylinositide 3-kinase (PI3K), and nitrous oxide synthase (NOS) pathways and the inhibition of adenylyl cyclase (AC) (19). In endothelial cells, ligand activates a Gα13-dependent pathway that allows the transcription of myocyte enhancer factor-2 (MEF2). Moreover, the G protein–independent pathway of the apelin/APJ system is mediated by G protein–coupled receptor kinase (GRK) and β-arrestin. Arrowheads indicate activation, and blunted arrows indicate inhibition.
Apart from the canonical G protein–dependent pathways, stimulation of APJ by apelin leads to phosphorylation of APJ via G protein–coupled receptor kinase (GRK) and the subsequent recruitment of β-arrestin, causing desensitization and internalization of APJ, which can activate G protein–independent signaling pathways (Figure 1) (23).
All apelin isoforms share the same 12 C-terminal residues (RPRLSHKGPMPF), which are required for bioactivity (24–28). The smaller apelin isoforms play a unique role in activating certain downstream signaling of APJ, which often induces APJ to be quickly recycled back to the cell surface, while the larger ones with higher affinity for APJ usually account for increased intracellular retention or even degradation (20, 29).
The expression of the apelin/APJ system components in different stages of ischemic stroke is temporally altered (18). A number of transcription factors, including Sp1 transcription factor (SP1), hypoxia inducible factor 1 alpha (HIF-1α), activating transcription factor 4 (ATF4), signal transducer and activator of transcription 3 (STAT3), and so on, are involved in regulating the expression of the apelin/APJ system (30–34).
Oxygen and glucose deprivation are the main consequences of ischemia, which are related to the abnormal expression of the apelin/APJ system (32, 35). During cerebral ischemia phase, the expression of the apelin/APJ system is upregulated, which is induced by HIF-1α and SP1 (30–32). Growing evidence shows that HIF-1α can bind to hypoxia response elements located in the promoter region of apelin and APJ genes under hypoxic conditions, which then significantly enhances the expression of the apelin/APJ system (32, 36). Moreover, the expressions of apelin and APJ in neurons in the early stage of ischemia are induced by increased SP1, which is possibly mediated by HIF-1α (30, 31, 37, 38).
However, the apelin/APJ system expression is downregulated during the reperfusion phase. Compared with the normoxia control group, the expression of APJ in the mouse hippocampus is significantly reduced after 4 weeks of chronic normobaric hypoxia treatment, which can be reversed by apelin-13 (39). Oxidative stress, ERS, autophagy, and inflammatory responses and the interaction between them may be related to the downregulated expression of the apelin/APJ system (18, 40, 41). For example, ERS, which is activated only during the reperfusion phase rather than the ischemic phase, plays critical roles in cerebral I/R injury (42–44). As an important regulator of ERS, ATF4 negatively regulates the expression of the apelin gene via the pro-apoptotic p38 mitogen-activated protein kinase (MAPK) pathway, which indicates that ERS may promote the repression of apelin at the reperfusion stage (34). Consistently, one recent study showed that apelin-12 could reduce neuron apoptosis of the ischemic penumbra in middle cerebral artery occlusion (MCAO)–induced ischemic mice by inhibiting the C-Jun N-terminal kinase (JNK) and P38MAPK signaling pathways (45).
To sum up, the expression of the apelin/APJ system in ischemic stroke is altered in different phases with complex mechanisms, indicating that targeting the apelin/APJ system may provide novel therapeutic interventions for ischemic stroke.
Recently, numerous studies show that the apelin/APJ axis possesses neuroprotective effects by inhibiting neuronal apoptosis and improving functional recovery through diverse pathways in ischemic stroke (Table 1).
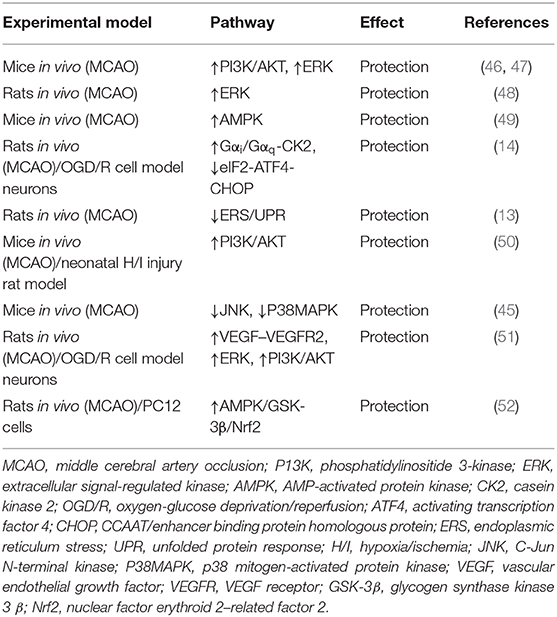
Table 1. Effects of apelin/APJ signaling on ischemic stroke.
Excitotoxicity occurs immediately after the onset of ischemia. During the excitotoxic phase with energy being depleted, membrane potential is lost, and neurons and glia depolarize, which is followed by the activation of somatodendritic and presynaptic voltage-dependent Ca2+ channels and the diffusion of excitatory amino acids in the extracellular space (53, 54). Meanwhile, the extracellular accumulation of excitatory amino acids (especially glutamate) was further increased, since the presynaptic reuptake of excitatory amino acids is obstructed, leading to over-activation of two distinct ionotropic receptors, namely the N-methyl-D-aspartate (NMDA) receptor and the α-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA) receptor, which facilitates an excessive Ca2+ influx into neurons and initiates neuronal damage or death (55).
Substantial research showed that apelin could protect neurons against excitotoxicity induced by quinolinic acid (QUIN) and HIV-infected human macrophages through activating the Raf/ERK1/2 and AKT pathways (56–58). Cook et al. found that apelin/APJ signaling could prevent neuronal excitotoxic signaling by activating pro-survival pathways, including IP3, PKC, mitogen-activated protein kinase kinase 1/2 (MEK1/2), and ERK1/2, and concurrently by inhibiting NMDA receptor activity via regulating NMDA-induced ionic currents as well as Ca2+ accumulation, calpain activation, and NMDA receptor subunit NR2B phosphorylation at S1480 in cerebrocortical neurons (59). Similarly, studies from Zeng et al. indicated that NMDA-induced excitotoxicity in cortical neurons could be attenuated by apelin-13 (60). All of this evidence suggests that NMDA receptor can be used as a therapeutic target for ischemic stroke.
If the production of free radicals, mainly referring to reactive oxygen/nitrogen species (ROS/RNS), exceeds the intrinsic scavenging capacity of the antioxidative system, oxidative and nitrative stresses will occur, which play deleterious roles in cerebral ischemia (61–63). These stresses are part of the downstream consequences of neuronal excitotoxicity due to increased generation of free radicals via several oxidases, which is influenced by Ca2+ overload (64, 65). Substantial experiments indicate that the formation of free radicals increases in all types of stroke (66, 67).
Considerable research has shown that the apelin/APJ system can promote neuron survival by reducing oxidative and nitrative stresses. Apelin-13 can reduce I/R injury–induced oxidative stress by decreasing malondialdehyde (MDA) level and increasing superoxide dismutase (SOD) activity, which may be associated with the ERK1/2 signaling pathway (48). In a recent study, apelin-13 intervention significantly reduced the levels of ROS and MDA and increased the antioxidant proteins' expressions at the same time [glutathione (GSH), GSH-Px, catalase (CAT), and SOD] in a dose-dependent manner by activating adenosine monophosphate (AMP)-activated protein kinase (AMPK)/glycogen synthase kinase 3 β (GSK-3β)/nuclear factor erythroid 2–related factor 2 (Nrf2) signaling (52). The aforementioned evidence strongly suggests that the novel protective effect of the apelin/APJ system on cell death induced by oxidative stress may be achieved by inhibiting production of ROS and facilitating scavenging of ROS.
Nitric oxide (NO) plays dual roles in ischemic injury: when generated by eNOS, it exerts vasodilation and neuroprotective effects, but when produced by neuronal NOS (nNOS) and inducible NOS (iNOS), it is the main mediator of oxidative/nitrosative injury (61, 62, 68). Similarly, apelin has dual vascular effects. For example, activating the apelin/APJ axis induces endothelium- and NO-dependent peripheral arterial relaxation (69, 70). However, apelin/APJ signal can inhibit NO-induced cerebral artery relaxation by blocking calcium-activated K (BKCa) channels in male rats, which can be mediated by a PI3K/AKT-dependent signaling pathway (71, 72). Meanwhile, the specific effect of apelin on oxidative/nitrosative stresses in ischemic stroke remains to be further determined.
Inflammation plays a key role in the pathophysiological process of ischemic stroke, which may contribute to ischemic brain injury. Soon after the ischemic onset, inflammatory cells (e.g., microglia, astrocytes) are activated by multiple factors, including ROS, necrotic cells, and damaged tissues, which trigger inflammatory responses (73–76). Moreover, a number of studies have suggested that postischemic neuroinflammation plays a crucial role in the long-term prognosis of ischemia (77, 78).
The apelin/APJ system can inhibit inflammatory responses after ischemic stroke via reducing the generation of inflammatory mediators. For example, Chen et al. found that apelin-13 could reduce the expressions of chemokines and proinflammatory cytokines, including monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 1α (MIP-1α), tumor necrosis factor α (TNF-α), and interleukin 1β (IL-1β), while the anti-apoptotic cytokine IL-10 was increased by apelin-13 in adult male C57/BL6 mice with ischemic stroke (79). Consistently, when compared with the I/R group, the expressions of many inflammatory cytokines such as TNF-α, IL-1β, and as well as intercellular adhesion molecule 1 (ICAM-1) are significantly reduced by apelin-13 treatment in a dose-dependent way, which is mediated by activating the AMPK/GSK-3β/Nrf2 pathway (12, 52). Additionally, apelin-13 remarkably decreases the activation and recruitment to the ischemic penumbra of microglia, which is related to the increase of cell survival (79). The most recent research indicates that apelin-13 ameliorated neuroinflammation by shifting N9 microglial M1 polarization toward the M2 phenotype, which may be related to the STAT3 signaling pathway (80). The above data indicate that apelin may be a potential target for regulating inflammation in ischemic stroke.
Neuronal apoptosis induced by cerebral I/R injury includes many mechanisms, in which ERS and subsequent unfolded protein response (UPR) play important roles (13, 44, 81). Under conditions of ERS, glucose-regulated protein 78 (GRP78) is isolated from three membrane proteins, i.e., inositol-requiring enzyme 1 (IRE1), phosphorylation of protein kinase–like endoplasmic reticulum kinase (PERK), and ATF6, which initiates UPR, resulting in cell apoptosis (14, 44, 82).
Apelin gene expression is repressed by ATF4 via a p38 MAPK–dependent pathway under ERS (34). Meanwhile, apelin treatment can protect cells from apoptosis by preventing ERS induced by I/R injury in the brain (13, 14). For example, Qiu et al. reported that apelin-36 could inhibit the activation of ERS/UPR by markedly reducing CCAAT/enhancer binding protein homologous protein (CHOP) and GRP78 expression induced by cerebral I/R injury in the rat cortex (13). Moreover, apelin-13 can suppress eukaryotic translation initiation factor 2 (eIF2)-ATF4-CHOP–induced neuronal apoptosis via activating Gαi/Gαq-casein kinase 2 (CK2) signaling (14). Notably, ERS is initiated only in the reperfusion phase rather than in the ischemic phase, indicating that apelin intervention in the reperfusion phase may protect neurons from ERS-mediated apoptosis (42, 43).
Like ERS, autophagy is also activated after ischemic stroke, and excessive ERS can induce autophagy and ultimately leads to neuronal apoptosis (83). Growing evidence indicates that the apelin/APJ system can inhibit apoptosis by regulating autophagy. First, apelin-13 pretreatment attenuates glucose deprivation-induced cardiomyocyte injury and decreases the autophagosome number and the ratio of microtubule-associated protein 1 light chain 3 (LC3)-II/LC3-I, which may be mediated by the PI3K/AKT/mechanistic target of rapamycin (mTOR) signaling pathway (84). Consistently, activating the PI3K/AKT/mTOR pathway by exogenous apelin can prevent hypoxia-induced autophagy and decrease cell proliferation and migration in an APJ receptor–dependent manner (85). In addition, apelin-13 suppresses traumatic brain injury (TBI) by decreasing autophagy-associated protein levels, such as LC3-II/I, Beclin-1, and Beclin-1/Bcl-2, which are expressed in the injured cortex and hippocampus in mice (86).
Up to now, there is no report about the role of apelin/APJ signaling in autophagy in ischemic stroke. However, the regulation of the apelin/APJ system in autophagy in the above diseases suggests that the apelin/APJ system may participate in autophagy induced by brain I/R injury.
Post-stroke angiogenesis facilitates the recovery of cerebral blood flow (CBF) and the supply of substances and energy for neurogenesis (87). A number of studies have shown that vascular endothelial growth factor (VEGF), which is known to have neuroprotective effects in stroke, can improve the neuronal survival and the number of microvessels after cerebral ischemia (88–90). Chen et al. reported that apelin-13 could dramatically accelerate the expression of VEGF and matrix metalloproteinase-9 (MMP-9), which contribute to angiogenesis in mice with focal cerebral ischemic stroke (79). Recent studies suggest that apelin-13 protects the neurovascular unit against ischemic injuries, which is dependent on an increase of VEGF–VEGF receptor 2 (VEGFR2) signaling, possibly by activating ERK and PI3K/AKT pathways (51). Moreover, the apelin/APJ system may work in coordination with VEGF to trigger vascular sprouting and CBF recovery from human urinary kallidinogenase (HUK)–treated stroke patients in an ERK1/2-dependent manner (91, 92). On the contrary, the expression of VEGF in cultured endothelial cells is partially suppressed by the apelin/APJ system inhibitor, suggesting that the apelin/APJ system may cooperate with VEGF to promote vascular growth in ischemic stroke (92). Similarly, apelin deficiency causes compromised angiogenesis and functional recovery and increased susceptibility to ischemic injury (93). Apelin-13 protects the blood–brain barrier (BBB) from ischemic injury by upregulating aquaporin-4 (AQP4) and VEGF via the ERK and PI3K/AKT pathways (46). In conclusion, the apelin/APJ system not only protects neurons from ischemic injury but also facilitates angiogenesis and prognosis of stroke, which may be mediated by the VEGF–VEGFR2 signaling pathway.
Growing evidence has indicated that genetic factors exert important roles in stroke and that people with a family history of stroke are more likely to suffer from stroke (17, 94, 95). Data from genomewide association studies suggest that the heritability in all types of ischemic stroke is 37.9% (96). Hata et al. reported that rs9943582, a single-nucleotide polymorphism (SNP) which is located in the promoter region of the APJ gene and can enhance the expression level of APJ mRNA, could increase the risk of ischemic stroke in Japanese, and that the risk of ischemic stroke in the GG genotype was significantly higher than other genotypes (97–99). Meanwhile, there was no significant correlation between rs9943582 and ischemic stroke in the Chinese Han GeneID population in a case–control study including 1,158 patients and 1,265 controls (100). Consistently, in another study on the Chinese population from Zhang et al. no associations were detected between rs9943582 and the age of onset and prognosis of ischemic stroke (101). Further intensive study and repeated validation of the relationship between gene polymorphism of apelin/APJ system and ischemic stroke can predict the risk of ischemic stroke, which will provide strategies for prevention and treatment of ischemic stroke.
The apelin/APJ system is widely distributed in the central nervous system, participating in many pathophysiological regulations of some brain diseases, including ischemic stroke (4, 15, 16). The apelin/APJ system shows a neuroprotective effect by blocking cell apoptosis or death and improving behavioral performance via various mechanisms including suppressing excitotoxicity, inflammatory responses, ERS, and oxidative and nitrative stresses, and at the same time modulating autophagy, promoting angiogenesis. Recently, researchers have found another endogenous ligand of APJ, apela, which enriches the functions of the system and complicates it as well (5, 6).
Ischemic stroke, known to be a devastating cerebrovascular disease with high morbidity, includes cerebral thrombosis, cerebral embolism, lacunar infarction, transient ischemic attack, and other types. The MCAO model is the most commonly used cerebral ischemia model (Table 1). Electroacupuncture-induced expression of apelin/APJ mRNA and protein of cerebral vascular endothelial cells in rats with cerebral infarction has an important role in the establishment of blood vessel regeneration and collateral circulation (102). In a transient model of focal cerebral ischemia, apelin-13 reduces brain injuries and postischemic cerebral edema in a dose-dependent manner, likely through inhibiting neuronal apoptosis, which may be mediated by many mechanisms, such as activating AMPK, PI3K/AKT, and ERK1/2 signaling pathways (47, 49, 50, 103). However, the specific effects of the apelin/APJ system on other types of ischemic stroke are rarely reported. And at the same time, due to the diversity of the active forms and signal pathways of apelin, it is a long way to go before the apelin/APJ system can be used in clinical practice. Further in-depth study of the physiological and pathological effects of the apelin/APJ system on ischemic stroke and the potential mechanisms will help to guide clinical prevention of and intervention in ischemic stroke and develop a series of drugs targeting different subtypes and phases of the disease, providing good news for patients, which will reduce family and social burdens.
YT, RC, YJ, and BB wrote the manuscript. TY and HL formulated and revised the manuscript.
This work was funded by the National Nature Science Foundation of China (21807041, 81870948, 81901341), Natural Science Foundation of Shandong Province of China (ZR2018PC011, ZR2017LC009), Science and Technology Bureau Foundation of Shandong Province (2019GSF107047), Medical and Health Science and Technology Planning Project of Shandong Province of China (2018WS463, 2017WS653), and Supporting Fund for Teachers' Research of Jining Medical University (JY2017KJ039, JY2017KJ038, JY2017KJ035).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. O'Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, et al. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. (1993) 136:355–60. doi: 10.1016/0378-1119(93)90495-O
2. Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. (1998) 251:471–6. doi: 10.1006/bbrc.1998.9489
3. Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, et al. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. (2000) 74:34–41. doi: 10.1046/j.1471-4159.2000.0740034.x
4. Shin K, Kenward C, Rainey KJ. Apelinergic system structure and function. Compr Physiol. (2017) 8:407–50. doi: 10.1002/cphy.c170028
5. Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell. (2013) 27:672–80. doi: 10.1016/j.devcel.2013.11.002
6. Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. (2014) 343:1248636. doi: 10.1126/science.1248636
7. Wang Z, Yu D, Wang M, Wang Q, Kouznetsova J, Yang R, et al. Elabela-apelin receptor signaling pathway is functional in mammalian systems. Sci Rep. (2015) 5:8170. doi: 10.1038/srep08170
8. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. (2018) 137:e67–492. doi: 10.1161/CIR.0000000000000558
9. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. (1999) 79:1431–568. doi: 10.1152/physrev.1999.79.4.1431
10. Lo HE. A new penumbra: transitioning from injury into repair after stroke. Nat Med. (2008) 14:497–500. doi: 10.1038/nm1735
11. Zhang S, Zis O, Ly PT, Wu Y, Zhang S, Zhang M, et al. Down-regulation of MIF by NFkappaB under hypoxia accelerated neuronal loss during stroke. FASEB J. (2014) 28:4394–407. doi: 10.1096/fj.14-253625
12. Xin Q, Cheng B, Pan Y, Liu H, Yang C, Chen J, et al. Neuroprotective effects of apelin-13 on experimental ischemic stroke through suppression of inflammation. Peptides. (2015) 63:55–62. doi: 10.1016/j.peptides.2014.09.016
13. Qiu J, Wang X, Wu F, Wan L, Cheng B, Wu Y, et al. Low dose of apelin-36 attenuates ER stress-associated apoptosis in rats with ischemic stroke. Front Neurol. (2017) 8:556. doi: 10.3389/fneur.2017.00556
14. Wu F, Qiu J, Fan Y, Zhang Q, Cheng B, Wu Y, et al. Apelin-13 attenuates ER stress-mediated neuronal apoptosis by activating Galphai/Galphaq-CK2 signaling in ischemic stroke. Exp Neurol. (2018) 302:136–44. doi: 10.1016/j.expneurol.2018.01.006
15. Medhurst AD, Jennings CA, Robbins MJ, Davis RP, Ellis C, Winborn KY, et al. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J Neurochem. (2003) 84:1162–72. doi: 10.1046/j.1471-4159.2003.01587.x
16. De Mota N, Lenkei Z, Llorens-Cortes C. Cloning, pharmacological characterization and brain distribution of the rat apelin receptor. Neuroendocrinology. (2000) 72:400–7. doi: 10.1159/000054609
17. Tian Y, Wang S, Jiao F, Kong Q, Liu C, Wu Y. Telomere length: a potential biomarker for the risk and prognosis of stroke. Front Neurol. (2019) 10:624. doi: 10.3389/fneur.2019.00624
18. Wu Y, Wang X, Zhou X, Cheng B, Li G, Bai B. Temporal expression of apelin/apelin receptor in ischemic stroke and its therapeutic potential. Front Mol Neurosci. (2017) 10:1. doi: 10.3389/fnmol.2017.00001
19. Chapman NA, Dupre DJ, Rainey KJ. The apelin receptor: physiology, pathology, cell signalling, and ligand modulation of a peptide-activated class A GPCR. Biochem Cell Biol. (2014) 92:431–40. doi: 10.1139/bcb-2014-0072
20. Masri B, Morin N, Pedebernade L, Knibiehler B, Audigier Y. The apelin receptor is coupled to Gi1 or Gi2 protein and is differentially desensitized by apelin fragments. J Biol Chem. (2006) 281:18317–26. doi: 10.1074/jbc.M600606200
21. Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. (2003) 284:R1–12. doi: 10.1152/ajpregu.00323.2002
22. Kang Y, Kim J, Anderson JP, Wu J, Gleim SR, Kundu RK, et al. Apelin-APJ signaling is a critical regulator of endothelial MEF2 activation in cardiovascular development. Circ Res. (2013) 113:22–31. doi: 10.1161/CIRCRESAHA.113.301324
23. Loot AE, Fleming I. A novel APJ signaling cascade that regulates cardiovascular development. Circ Res. (2013) 113:4–6. doi: 10.1161/CIRCRESAHA.113.301632
24. Tatemoto K, Takayama K, Zou MX, Kumaki I, Zhang W, Kumano K, et al. The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul Pept. (2001) 99:87–92. doi: 10.1016/S0167-0115(01)00236-1
25. Maguire JJ, Kleinz MJ, Pitkin SL, Davenport PA. [Pyr1]apelin-13 identified as the predominant apelin isoform in the human heart: vasoactive mechanisms and inotropic action in disease. Hypertension. (2009) 54:598–604. doi: 10.1161/HYPERTENSIONAHA.109.134619
26. El Messari S, Iturrioz X, Fassot C, De Mota N, Roesch D, Llorens-Cortes C. Functional dissociation of apelin receptor signaling and endocytosis: implications for the effects of apelin on arterial blood pressure. J Neurochem. (2004) 90:1290–301. doi: 10.1111/j.1471-4159.2004.02591.x
27. De Mota N, Reaux-Le Goazigo A, El Messari S, Chartrel N, Roesch D, Dujardin C, et al. Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc Natl Acad Sci USA. (2004) 101:10464–9. doi: 10.1073/pnas.0403518101
28. Kawamata Y, Habata Y, Fukusumi S, Hosoya M, Fujii R, Hinuma S, et al. Molecular properties of apelin: tissue distribution and receptor binding. Biochim Biophys Acta. (2001) 1538:162–71. doi: 10.1016/S0167-4889(00)00143-9
29. Lee DK, Ferguson SS, George SR, O'Dowd FB. The fate of the internalized apelin receptor is determined by different isoforms of apelin mediating differential interaction with beta-arrestin. Biochem Biophys Res Commun. (2010) 395:185–9. doi: 10.1016/j.bbrc.2010.03.151
30. Lv XR, Zheng B, Li SY, Han AL, Wang C, Shi JH, et al. Synthetic retinoid Am80 up-regulates apelin expression by promoting interaction of RARalpha with KLF5 and Sp1 in vascular smooth muscle cells. Biochem J. (2013) 456:35–46. doi: 10.1042/BJ20130418
31. O'Carroll AM, Lolait SJ, Howell MG. Transcriptional regulation of the rat apelin receptor gene: promoter cloning and identification of an Sp1 site necessary for promoter activity. J Mol Endocrinol. (2006) 36:221–35. doi: 10.1677/jme.1.01927
32. He L, Xu J, Chen L, Li L. Apelin/APJ signaling in hypoxia-related diseases. Clin Chim Acta. (2015) 451:191–8. doi: 10.1016/j.cca.2015.09.029
33. Han S, Wang G, Qi X, Englander EW, Greeley GH Jr. Involvement of a Stat3 binding site in inflammation-induced enteric apelin expression. Am J Physiol Gastrointest Liver Physiol. (2008) 295:G1068–78. doi: 10.1152/ajpgi.90493.2008
34. Jeong K, Oh Y, Kim SJ, Kim H, Park KC, Kim SS, et al. Apelin is transcriptionally regulated by ER stress-induced ATF4 expression via a p38 MAPK-dependent pathway. Apoptosis. (2014) 19:1399–410. doi: 10.1007/s10495-014-1013-0
35. Zhang Z, Yu B, Tao ZG. Apelin protects against cardiomyocyte apoptosis induced by glucose deprivation. Chin Med J. (2009) 122:2360–5.
36. Ronkainen VP, Ronkainen JJ, Hanninen SL, Leskinen H, Ruas JL, Pereira T, et al. Hypoxia inducible factor regulates the cardiac expression and secretion of apelin. FASEB J. (2007) 21:1821–30. doi: 10.1096/fj.06-7294com
37. Yeh SH, Yang WB, Gean PW, Hsu CY, Tseng JT, Su TP, et al. Translational and transcriptional control of Sp1 against ischaemia through a hydrogen peroxide-activated internal ribosomal entry site pathway. Nucleic Acids Res. (2011) 39:5412–23. doi: 10.1093/nar/gkr161
38. Woo SK, Kwon MS, Geng Z, Chen Z, Ivanov A, Bhatta S, et al. Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J Cereb Blood Flow Metab. (2012) 32:525–36. doi: 10.1038/jcbfm.2011.159
39. Fan J, Ding L, Xia D, Chen D, Jiang P, Ge W, et al. Amelioration of apelin-13 in chronic normobaric hypoxia-induced anxiety-like behavior is associated with an inhibition of NF-kappaB in the hippocampus. Brain Res Bull. (2017) 130:67–74. doi: 10.1016/j.brainresbull.2017.01.005
40. Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, Zhang XY, et al. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. (2012) 8:310–25. doi: 10.4161/auto.18673
41. Mei Y, Thompson MD, Cohen RA, Tong X. Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys Acta. (2015) 1852:243–51. doi: 10.1016/j.bbadis.2014.05.005
42. Morimoto N, Oida Y, Shimazawa M, Miura M, Kudo T, Imaizumi K, et al. Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience. (2007) 147:957–67. doi: 10.1016/j.neuroscience.2007.04.017
43. Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res. (2010) 17:189–202. doi: 10.1007/s12640-009-9110-5
44. Xin Q, Ji B, Cheng B, Wang C, Liu H, Chen X, et al. Endoplasmic reticulum stress in cerebral ischemia. Neurochem Int. (2014) 68:18–27. doi: 10.1016/j.neuint.2014.02.001
45. Liu DR, Hu W, Chen ZG. Apelin-12 exerts neuroprotective effect against ischemia-reperfusion injury by inhibiting JNK and P38MAPK signaling pathway in mouse. Eur Rev Med Pharmacol Sci. (2018) 22:3888–95. doi: 10.26355/eurrev_201806_15273
46. Chu H, Yang X, Huang C, Gao Z, Tang Y, Dong Q. Apelin-13 protects against ischemic blood-brain barrier damage through the effects of aquaporin-4. Cerebrovasc Dis. (2017) 44:10–25. doi: 10.1159/000460261
47. Yang Y, Zhang X, Cui H, Zhang C, Zhu C, Li L. Apelin-13 protects the brain against ischemia/reperfusion injury through activating PI3K/Akt and ERK1/2 signaling pathways. Neurosci Lett. (2014) 568:44–9. doi: 10.1016/j.neulet.2014.03.037
48. Wu G, Li L, Liao D, Wang Z. Protective effect of Apelin-13 on focal cerebral ischemia-reperfusion injury in rats. Nan Fang Yi Ke Da Xue Xue Bao. (2015) 35:1335–9.
49. Yang Y, Zhang XJ, Li LT, Cui HY, Zhang C, Zhu CH, et al. Apelin-13 protects against apoptosis by activating AMP-activated protein kinase pathway in ischemia stroke. Peptides. (2016) 75:96–100. doi: 10.1016/j.peptides.2015.11.002
50. Gu Q, Zhai L, Feng X, Chen J, Miao Z, Ren L, et al. Apelin-36, a potent peptide, protects against ischemic brain injury by activating the PI3K/Akt pathway. Neurochem Int. (2013) 63:535–40. doi: 10.1016/j.neuint.2013.09.017
51. Huang C, Dai C, Gong K, Zuo H, Chu H. Apelin-13 protects neurovascular unit against ischemic injuries through the effects of vascular endothelial growth factor. Neuropeptides. (2016) 60:67–74. doi: 10.1016/j.npep.2016.08.006
52. Duan J, Cui J, Yang Z, Guo C, Cao J, Xi M, et al. Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3beta/Nrf2 signaling. J Neuroinflammation. (2019) 16:24. doi: 10.1186/s12974-019-1406-7
53. Katsura K, Kristian T, Siesjo KB. Energy metabolism, ion homeostasis, and cell damage in the brain. Biochem Soc Trans. (1994) 22:991–6. doi: 10.1042/bst0220991
54. Dirnagl U, Iadecola C, Moskowitz AM. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. (1999) 22:391–7. doi: 10.1016/S0166-2236(99)01401-0
55. Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. (2000) 403:316–21. doi: 10.1038/35002090
56. Foster AC, Collins JF, Schwarcz R. On the excitotoxic properties of quinolinic acid, 2,3-piperidine dicarboxylic acids and structurally related compounds. Neuropharmacology. (1983) 22:1331–42. doi: 10.1016/0028-3908(83)90221-6
57. O'Donnell LA, Agrawal A, Sabnekar P, Dichter MA, Lynch DR, Kolson LD. Apelin, an endogenous neuronal peptide, protects hippocampal neurons against excitotoxic injury. J Neurochem. (2007) 102:1905–17. doi: 10.1111/j.1471-4159.2007.04645.x
58. O'Donnell LA, Agrawal A, Jordan-Sciutto KL, Dichter MA, Lynch DR, Kolson LD. Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the NMDA receptor subtypes. J Neurosci. (2006) 26:981–90. doi: 10.1523/JNEUROSCI.4617-05.2006
59. Cook DR, Gleichman AJ, Cross SA, Doshi S, Ho W, Jordan-Sciutto KL, et al. NMDA receptor modulation by the neuropeptide apelin: implications for excitotoxic injury. J Neurochem. (2011) 118:1113–23. doi: 10.1111/j.1471-4159.2011.07383.x
60. Zeng XJ, Yu SP, Zhang L, Wei L. Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Exp Cell Res. (2010) 316:1773–83. doi: 10.1016/j.yexcr.2010.02.005
61. Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam FH. Pathogenic mechanisms following ischemic stroke. Neurol Sci. (2017) 38:1167–86. doi: 10.1007/s10072-017-2938-1
62. Love S. Oxidative stress in brain ischemia. Brain Pathol. (1999) 9:119–31. doi: 10.1111/j.1750-3639.1999.tb00214.x
63. Kontos AH. Oxygen radicals in cerebral ischemia: the 2001 Willis lecture. Stroke. (2001) 32:2712–6. doi: 10.1161/hs1101.098653
64. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. (2007) 87:315–424. doi: 10.1152/physrev.00029.2006
65. Chamorro A, Dirnagl U, Urra X, Planas MA. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. (2016) 15:869–81. doi: 10.1016/S1474-4422(16)00114-9
66. Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, et al. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol. (2008) 64:654–63. doi: 10.1002/ana.21511
67. Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. (2009) 4:461–70. doi: 10.1111/j.1747-4949.2009.00387.x
68. Iadecola C, Zhang F, Casey R, Nagayama M, Ross EM. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. (1997) 17:9157–64. doi: 10.1523/JNEUROSCI.17-23-09157.1997
69. Salcedo A, Garijo J, Monge L, Fernandez N, Luis Garcia-Villalon A, Sanchez Turrion V, et al. Apelin effects in human splanchnic arteries. Role of nitric oxide and prostanoids. Regul Pept. (2007) 144:50–5. doi: 10.1016/j.regpep.2007.06.005
70. Japp AG, Cruden NL, Amer DA, Li VK, Goudie EB, Johnston NR, et al. Vascular effects of apelin in vivo in man. J Am Coll Cardiol. (2008) 52:908–13. doi: 10.1016/j.jacc.2008.06.013
71. Mughal A, Sun C, O'Rourke TS. Apelin reduces nitric oxide-induced relaxation of cerebral arteries by inhibiting activation of large-conductance, calcium-activated K channels. J Cardiovasc Pharmacol. (2018) 71:223–32. doi: 10.1097/FJC.0000000000000563
72. Modgil A, Guo L, O'Rourke ST, Sun C. Apelin-13 inhibits large-conductance Ca2+-activated K+ channels in cerebral artery smooth muscle cells via a PI3-kinase dependent mechanism. PLoS ONE. (2013) 8:e83051. doi: 10.1371/journal.pone.0083051
73. Becker JK. Inflammation and acute stroke. Curr Opin Neurol. (1998) 11:45–9. doi: 10.1097/00019052-199802000-00008
74. Stanimirovic DB, Wong J, Shapiro A, Durkin PJ. Increase in surface expression of ICAM-1, VCAM-1 and E-selectin in human cerebromicrovascular endothelial cells subjected to ischemia-like insults. Acta Neurochir. (1997) 70(Suppl.):12–6. doi: 10.1007/978-3-7091-6837-0_4
75. Kriz J. Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol. (2006) 18:145–57. doi: 10.1615/CritRevNeurobiol.v18.i1-2.150
76. Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti TM. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J. (2009) 276:13–26. doi: 10.1111/j.1742-4658.2008.06766.x
77. Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. (2007) 20:334–42. doi: 10.1097/WCO.0b013e32813ba151
78. McColl BW, Allan SM, Rothwell JN. Systemic infection, inflammation and acute ischemic stroke. Neuroscience. (2009) 158:1049–61. doi: 10.1016/j.neuroscience.2008.08.019
79. Chen D, Lee J, Gu X, Wei L, Yu PS. Intranasal delivery of apelin-13 is neuroprotective and promotes angiogenesis after ischemic stroke in mice. ASN Neuro. (2015) 7:1759091415605114. doi: 10.1177/1759091415605114
80. Zhou S, Guo X, Chen S, Xu Z, Duan W, Zeng B. Apelin-13 regulates LPS-induced N9 microglia polarization involving STAT3 signaling pathway. Neuropeptides. (2019) 76:101938. doi: 10.1016/j.npep.2019.101938
81. Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. (2004) 11:403–15. doi: 10.1038/sj.cdd.4401365
82. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89–102. doi: 10.1038/nrm3270
83. Feng D, Wang B, Wang L, Abraham N, Tao K, Huang L, et al. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J Pineal Res. (2017) 62:e12395. doi: 10.1111/jpi.12395
84. Jiao H, Zhang Z, Ma Q, Fu W, Liu Z. Mechanism underlying the inhibitory effect of Apelin-13 on glucose deprivation-induced autophagy in rat cardiomyocytes. Exp Ther Med. (2013) 5:797–802. doi: 10.3892/etm.2013.902
85. Zhang H, Gong Y, Wang Z, Jiang L, Chen R, Fan X, et al. Apelin inhibits the proliferation and migration of rat PASMCs via the activation of PI3K/Akt/mTOR signal and the inhibition of autophagy under hypoxia. J Cell Mol Med. (2014) 18:542–53. doi: 10.1111/jcmm.12208
86. Bao HJ, Zhang L, Han WC, Dai KD. Apelin-13 attenuates traumatic brain injury-induced damage by suppressing autophagy. Neurochem Res. (2015) 40:89–97. doi: 10.1007/s11064-014-1469-x
87. Zhang Q, Zhao HY. Therapeutic angiogenesis after ischemic stroke: Chinese medicines, bone marrow stromal cells (BMSCs) and their combinational treatment. Am J Chin Med. (2014) 42:61–77. doi: 10.1142/S0192415X14500049
88. Yang JP, Liu HJ, Liu FX. VEGF promotes angiogenesis and functional recovery in stroke rats. J Invest Surg. (2010) 23:149–55. doi: 10.3109/08941930903469482
89. Chu H, Tang Y, Dong Q. Protection of vascular endothelial growth factor to brain edema following intracerebral hemorrhage and its involved mechanisms: effect of aquaporin-4. PLoS ONE. (2013) 8:e66051. doi: 10.1371/journal.pone.0066051
90. Sanchez A, Wadhwani S, Grammas P. Multiple neurotrophic effects of VEGF on cultured neurons. Neuropeptides. (2010) 44:323–31. doi: 10.1016/j.npep.2010.04.002
91. Li J, Chen Y, Zhang X, Zhang B, Zhang M, Xu Y. Human urinary kallidinogenase improves outcome of stroke patients by shortening mean transit time of perfusion magnetic resonance imaging. J Stroke Cerebrovasc Dis. (2015) 24:1730–7. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.032
92. Han L, Li J, Chen Y, Zhang M, Qian L, Chen Y, et al. Human urinary kallidinogenase promotes angiogenesis and cerebral perfusion in experimental stroke. PLoS ONE. (2015) 10:e0134543. doi: 10.1371/journal.pone.0134543
93. Wang W, McKinnie SM, Patel VB, Haddad G, Wang Z, Zhabyeyev P, et al. Loss of Apelin exacerbates myocardial infarction adverse remodeling and ischemia-reperfusion injury: therapeutic potential of synthetic Apelin analogues. J Am Heart Assoc. (2013) 2:e000249. doi: 10.1161/JAHA.113.000249
94. Flossmann E, Schulz UG, Rothwell MP. Systematic review of methods and results of studies of the genetic epidemiology of ischemic stroke. Stroke. (2004) 35:212–27. doi: 10.1161/01.STR.0000107187.84390.AA
95. Tian T, Jin G, Yu C, Lv J, Guo Y, Bian Z, et al. China kadoorie biobank collaborative: family history and stroke risk in China: evidence from a large cohort study. J Stroke. (2017) 19:188–95. doi: 10.5853/jos.2016.01270
96. Bevan S, Traylor M, Adib-Samii P, Malik R, Paul NL, Jackson C, et al. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke. (2012) 43:3161–7. doi: 10.1161/STROKEAHA.112.665760
97. Hata J, Kubo M, Kiyohara Y. Genome-wide association study for ischemic stroke based on the Hisayama study. Nihon Eiseigaku Zasshi. (2011) 66:47–52. doi: 10.1265/jjh.66.47
98. Hata J, Matsuda K, Ninomiya T, Yonemoto K, Matsushita T, Ohnishi Y, et al. Functional SNP in an Sp1-binding site of AGTRL1 gene is associated with susceptibility to brain infarction. Hum Mol Genet. (2007) 16:630–9. doi: 10.1093/hmg/ddm005
99. Kubo M. Genetic risk factors of ischemic stroke identified by a genome-wide association study. Brain Nerve. (2008) 60:1339–46.
100. Wang P, Wang C, Li S, Wang B, Xiong L, Tu X, et al. Lack of association between the APLNR variant rs9943582 with ischemic stroke in the Chinese Han GeneID population. Oncotarget. (2017) 8:107678–84. doi: 10.18632/oncotarget.22588
101. Zhang H, Sun L, Wang H, Cai H, Niu G, Bai Y, et al. A study of GWAS-supported variants of rs9943582 in a Chinese Han Population with Ischemic Stroke: No Associations with Disease Onset and Clinical Outcomes. J Stroke Cerebrovasc Dis. (2017) 26:2294–9. doi: 10.1016/j.jstrokecerebrovasdis.2017.05.013
102. Yang LH, Du YH, Li J. Effect of electroacupuncture on expression of apelin-APJ system of cerebral vascular endothelial cell in rats with cerebral infarction. Zhen Ci Yan Jiu. (2017) 42:9–13. doi: 10.13702/j.1000-0607.2017.01.002
Keywords: apelin/APJ system, ischemic stroke, signaling pathways, neuroprotection, molecular mechanisms
Citation: Tian Y, Chen R, Jiang Y, Bai B, Yang T and Liu H (2020) The Protective Effects and Mechanisms of Apelin/APJ System on Ischemic Stroke: A Promising Therapeutic Target. Front. Neurol. 11:75. doi: 10.3389/fneur.2020.00075
Received: 17 October 2019; Accepted: 22 January 2020;
Published: 03 March 2020.
Edited by:
Simone Beretta, San Gerardo Hospital, ItalyCopyright © 2020 Tian, Chen, Jiang, Bai, Yang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tongju Yang, emNzcm15eXlqa0AxNjMuY29t; Haiqing Liu, bGl1LmhhaXFpbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.