 Mariana Dutra Brito1†‡
Mariana Dutra Brito1†‡ Gustavo Ferro Gomes da Silva1†
Gustavo Ferro Gomes da Silva1† Erick Mutti Tilieri1‡
Erick Mutti Tilieri1‡ Beatriz Grisolia Araujo1‡
Beatriz Grisolia Araujo1‡ Michele Longoni Calió2‡
Michele Longoni Calió2‡ Tatiana Rosado Rosenstock1*‡
Tatiana Rosado Rosenstock1*‡- 1Department of Physiological Science, Santa Casa de São Paulo School of Medical Science, São Paulo, Brazil
- 2Department of Physiology, Federal University of São Paulo, São Paulo, Brazil
Background: The development of strategies that could not only efficiently detect the onset of Amyotrophic Lateral Sclerosis (ALS), a fatal neurodegenerative disorder with no cure but also predict its development and evaluate therapeutic intervention would be of great value. In this respect, the metabolic status of ALS patients has called attention. Hence, this study aimed to investigate the potential correlation between changes in ALS's metabolic parameters with the disease outcome in a systematic review.
Methods: The manuscripts were manually searched within different databases (PubMed, Web of Science and Cochrane). The inclusion criteria were original articles and reviews about individuals with ALS and its survival, disease prognosis and metabolism (weight, cholesterol, hypertension, BMI, and glycaemia). The authors also established three different exclusion criteria: studies including ALS and other degenerative disorders, works including animal models and published before the year 2000.
Results: In total, 29 papers were selected. From all manuscripts, only 82.8% ensured the participation of sALS patients. Also, 27.6% of selected studies described the presence of a genetic mutation. Regarding ALS prognosis, patient's age, the age of ALS onset, ALS duration and survival, <50% of the papers addressed these issues. Specifically, regarding metabolism, 65.5% of articles mentioned BMI, 20.7% mentioned any data concerning hypertension, 6.89% cardiovascular risk, 10.3% obesity, 13.78% diabetes and 10.3% glycaemia. Concerning lipid metabolism, more results were gathered, but still, they did not suffice to establish a correlation with ALS development.
Conclusions: Altogether, the authors concluded that available information is not enough to establish a link between ALS and metabolism. In reality, less than half of the manuscripts evaluated show an association between both factors. Nonetheless, it is worth mentioning that metabolism does influence ALS, but not in a unique manner. There is a debate about patients' hypo- and hypermetabolism. Thus, to provide a reliable record, a public policy in which all research and clinical centers might assess the parameters discussed herein is suggested. Accordingly, this systematic review attempts to provide a comprehensible database to facilitate multicentered collaboration, validation, and clinical translation.
Introduction
The Amyotrophic Lateral Sclerosis (ALS) is a progressive and fatal neurodegenerative disorder (1–3). ALS, also known as Lou Gehrig's disease, has an incidence of 2.5 cases per 100,000 people/year and a prevalence of 4-6:100,000; definitely, it is the most common motor neuron disorder (4). Therefore, ALS has not being considered a rare disease once its developing risk is 1/400-1/700 (5). Currently, it is well-known for the involvement of more than 24 genes in ALS. However, these mutations account for 68% of familial cases (fALS) and just for 11% of sporadic ALS (sALS) (6). Oddly, 90% of all ALS cases are sporadic. ALS onset, regardless of its form, occurs in the fifth decade of life. The survival rate is 3–5 years after diagnosis, being men more affected than women (1.5:1) (7).
Along ALS occurs a specific and progressive degeneration of upper motor neurons of the corticospinal tract, motor cortex, and motor neurons from the lower brainstem and spinal cord (8–12). However, ALS is a multifactorial disorder and, consequently, not only neurons are afflicted, but also it presents reactive astrocytes, dysfunctional oligodendrocytes and activated microglia committed (13–20). Moreover, several mechanisms seem to be related to the onset and the evolution of ALS, including excitotoxicity, oxidative stress, mitochondrial dysfunction, protein aggregation, genetic mutations, diminishment in the axonal transport, modifications in RNA metabolism, and neuroinflammation (21–32).
Notably, there is neither a cure for ALS, nor even an effective therapy, although there are drugs used to attenuate ALS' symptoms and bring significant benefits to patients, such as antioxidants, anti-inflammatory, antiapoptotic, and anticytotoxic agents (4, 25, 33). To date, one of the most used medications in the USA is Rilutek™ (riluzole), a glutamate release inhibitor (34). Several clinical trials have demonstrated an increase in the survival rate of ALS patients after Rilutek™ (35–37). More recently, the U.S. Food and Drug Administration (FDA) approved Radicava™ (edaravone), a powerful antioxidant that is not restricted to eliminate hydroxyl radicals and reactive oxygen but counteracts the increase in prostacyclin production (38). Because these new findings are promising, attention has been focused on prognostic factors and biomarkers for ALS. Indeed, biomarkers discovery may enable a reliable diagnosis and a predictable follow-up; nowadays, ALS's diagnosis and prognosis are mainly based on physical exams (39). Furthermore, the discrepancies of signs among patients interpret clinical trials somewhat dubious (40).
A variety of clinical prognostic factors and several ALS-related biological biomarkers have been listed (41–45), including some associated with metabolism (42–58). With interest, metabolic changes in ALS animal models were also observed (46, 59–61). However, there is no consensus on whether these metabolic parameters are indeed related to the disorder prognosis itself or just represent a specific dataset from a single-center and population. In this context, lipid content, cholesterol, and BMI are often contradicting. Additionally, genetic mutation, glycaemia, hypertension, TGL, LDL, HDL, and even medication are seldom mentioned in ALS manuscripts, which make it more difficult to collect clinical investigation outcomes.
Thus, to investigate the potential correlation of metabolic status with ALS's outcome and survival, authors have decided to put together recent data regarding BMI, hypertension, cardiovascular risk, obesity, diabetic, glycaemia, hyperlipidaemia, triglycerides (TGL), LDL, HDL, and cholesterol. In addition, further information directly and indirectly related to metabolism were included, such as the age of onset, smoking habits, disease duration (years), survival (years), population/ethnic group, mutation, family members with ALS (%), family members with motor disorders (n), first motor signs (%), ALSFRS and ALS medication (including medicine intake period). Therefore, this study presents a systematic review regarding the possible relationship between ALS subjects' survival and the course of the disease, with metabolic-related factors.
Methods
Eligibility Criteria
To perform our systematic review, we established inclusions and exclusion criteria. Clear inclusion criteria were original articles and reviews concerning ALS patients and their survival, disease prognosis and metabolism (Figure 1). Five different exclusion criteria were endorsed: (i) studies including ALS and other degenerative disorders, such as Alzheimer disease, Frontotemporal dementia, Myotonic Dystrophy, and Muscular Atrophy, (ii) articles including animal models, (iii) manuscripts published before the year 2000, due to the fact that several papers are issued every year, and epidemiology approaches and biochemical methodologies are constantly changing. Importantly, authors also excluded, (iv) studies in which there is a mix of ALS-FDT patients and ALS-AD subjects without any kind of discrimination, and (v) articles that despite having the keyword of our research is about guidance on the management and care of ALS patients (Figure 2).
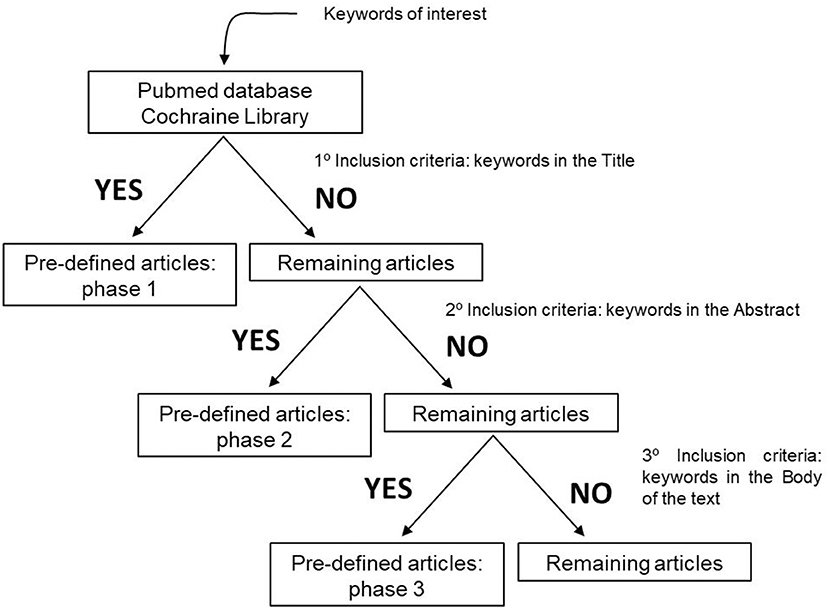
Figure 1. Inclusion criteria used to define the articles that would be evaluated and submitted to this systematic review.
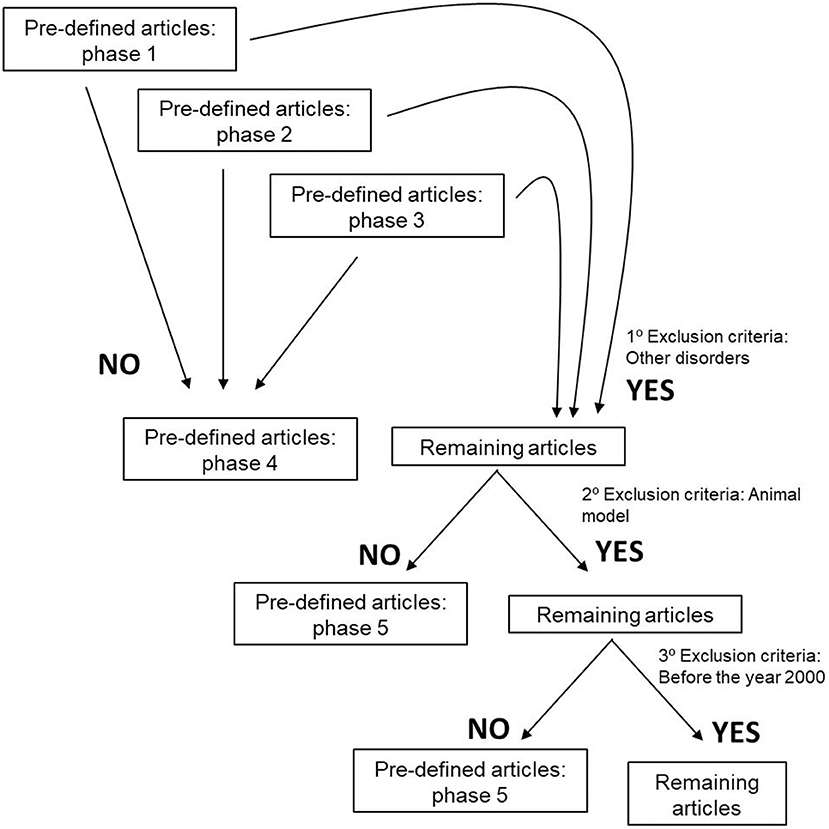
Figure 2. Exclusion criteria used to define the articles that would be evaluated and submitted to this systematic review.
Search Strategy
The keywords and expression of interest were manually searched in various databases, i.e., PubMed, Web of Science and Cochrane. The databanks were selected for topics assurance and for including journals of authors' interest. Specifically, the following strategy was used to establish the inclusion/exclusion criteria: (i) “Amyotrophic Lateral Sclerosis” (not the term ALS) AND “patient” AND “cohort” AND “mutation” AND “mitochondria” OR “metabolism”; (ii) Amyotrophic Lateral Sclerosis” AND “patient” AND “cohort” AND “aging” AND “mitochondria” OR “metabolism”; (iii) “Amyotrophic Lateral Sclerosis” AND “patient” AND “cohort” AND “mutation” AND “glycaemia” OR “hypertension” OR “weight” OR “cholesterol”; (iv) “Amyotrophic Lateral Sclerosis” AND “patient” AND “cohort” AND “aging” AND “glycaemia” OR “hypertension” OR “weight” OR “cholesterol”; (v) “Amyotrophic Lateral Sclerosis” AND “patient” AND “cohort” AND “mutation” AND “aging” AND “metabolism” OR “mitochondria”; (vi) “Amyotrophic Lateral Sclerosis” AND “patient” AND “cohort” AND “mutation” AND “aging” AND “glycaemia” OR “hypertension” OR “weight” OR “cholesterol”; (vii) “Amyotrophic Lateral Sclerosis” AND “patient” AND “glycaemia” AND “weight” OR “hypertension” OR “cholesterol”; (viii) “Amyotrophic Lateral Sclerosis” AND “patient” AND “weight” AND “hypertension” OR “cholesterol”; (ix) “Amyotrophic Lateral Sclerosis” AND “patient” AND “hypertension” AND “cholesterol”; (x) “Amyotrophic Lateral Sclerosis” AND “patient” AND “glycaemia” AND “weight” AND “hypertension” OR “cholesterol”; (xi) “Amyotrophic Lateral Sclerosis” AND “patient” AND “glycaemia” AND “hypertension” AND “cholesterol”; (xii) “Amyotrophic Lateral Sclerosis” AND “patient” AND “weight” AND “hypertension” AND “cholesterol”; (xiii) “Amyotrophic Lateral Sclerosis” AND “patient” AND “glycaemia” AND “weight” AND “hypertension” AND “cholesterol”; (xiv) “Amyotrophic Lateral Sclerosis” AND “patient” AND “cohort” AND “glycaemia” OR “weight” OR “hypertension” OR “cholesterol”; (xv) “Amyotrophic Lateral Sclerosis” AND “patient” AND “cohort” AND “glycaemia” OR “weight” OR “hypertension” OR “cholesterol” AND “mitochondria” OR “metabolism.”
Outcome
Data outcome was categorized as follows: year of publication, total number of ALS patients (n), familial ALS (fALS) patients (n), sporadic ALS (sALS) patients (n), male (M)/female (F) (%) (ratio), El Escorial, age of subject (years), age of onset (symptoms), disease duration (years), survival (years), population/ethnic group, mutation, relatives with ALS (%), relatives with motor disorders (n), first motor signs (%), ALSFRS, BMI, smoking, hypertension, cardiovascular risk, obesity, diabetic, glycaemia, hyperlipidaemia, triglycerides (TGL), LDL, HDL, cholesterol, ALS medication, and treatment extent.
Risk of Bias
The authors did not perform any assessment for the risk of bias. Such tools are used mainly for randomized controlled trials (RCTs), and consequently, this tool would not be appropriate. As an alternative, data was collected and the limitation of each study was recorded. The results are presented herein.
Statistical Analysis
Results for descriptive analyses were expressed in absolute numbers (n) and percentages (%), and the data acquired were demonstrated in different tables. No sufficient data were gathered to perform a meaningful meta-analysis.
Results
The search resulted in the collection of 924 manuscripts from PubMed, 54 from Web of Science and 52 from Cochrane (including original articles and reviews). After applying inclusion and exclusion criteria (Figures 1, 2), followed by a more in-depth and comprehensive evaluation of the pre-selected articles, 29 articles were considered for the study. Tables 1–8 summarize the collected data. As we can observe, from all manuscripts, 17.2% excluded fALS patients, which means that 82.8% of all articles ensured the participation of sALS subjects only. Besides, 93.1% of the studies reported M/F ratio. Despite the significant percentage of studies mentioning this parameter, discrepancies among reported M/F were noticed (Table 1).
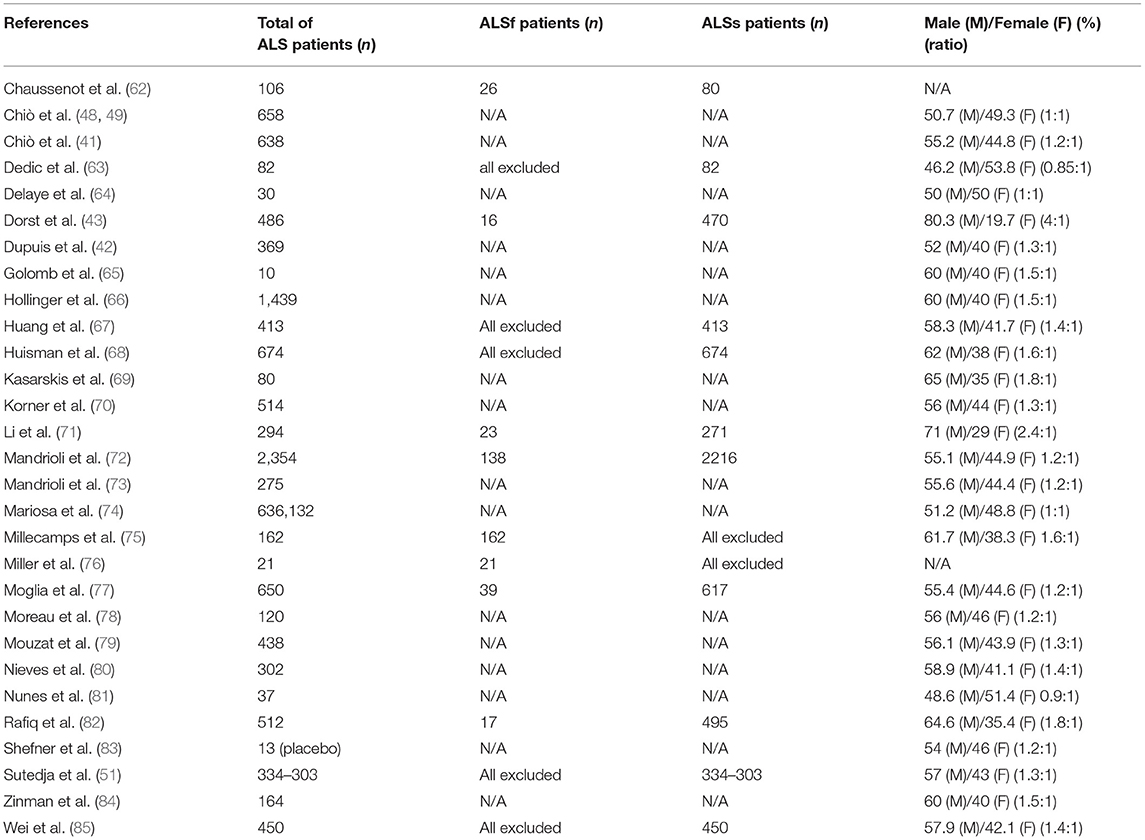
Table 1. Number of ALS patients per study, the percentage of SALs and FALs patients, and the distribution of ALS subjects between sex in general population [male (M)/female (F) ratio].
To better understand ALS prognosis, information regarding fALS and mutations were assembled; amongst which, the most cited were SOD1, FUS, TARDBP, VAPB, ANG, C9orf72, and CHCHD10 (Table 2). However, only 62.5% of the articles certified the inclusion of fALS patients only (27.6% of all selected studies described the genetic mutation).

Table 2. Identification of ALS mutations and the percentage of ALS patients' relatives with ALS and motor disorders.
To further evaluate ALS symptoms, as well as ALS prognosis, patient's age, the age of ALS onset, ALS duration, and survival, data were tabulated in Table 3. The authors could observe that 58.6% of the manuscripts specified the age of ALS onset (mean 59.8 years). For ALS duration and survival, 31 and 44.8% of studies, respectively, addressed this issue. Precisely, the mean of ALS duration is about 2.15 years, while the mean of ALS survival is 2.88 years. Moreover, to investigate the influence of ethnicity on ALS prognosis, few data were acquired; only two studies mentioned the word “caucasian” and only one mentioned the word “white.” No information is known regarding the socio-economic status of ALS individuals.
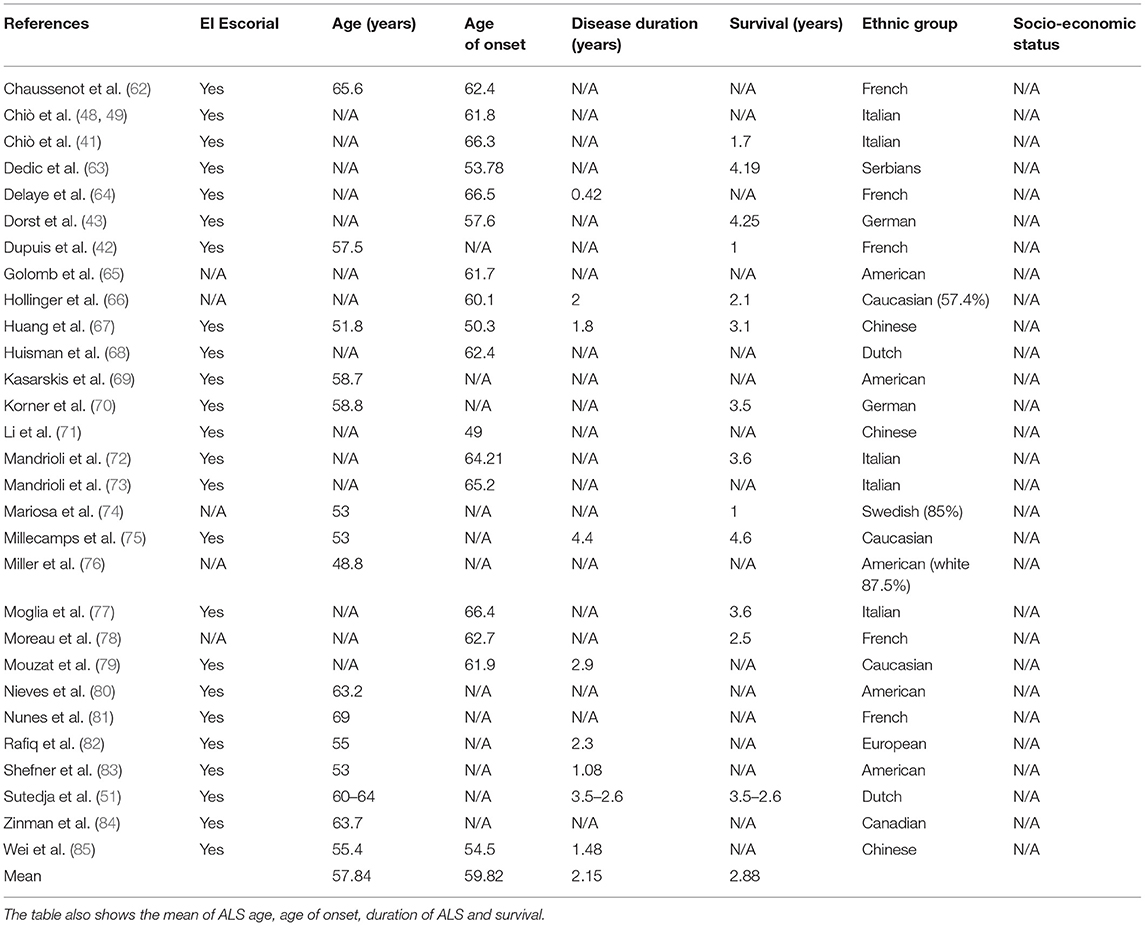
Table 3. El Escorial and general data of ALS patients: age, age of onset (symptoms), disease duration (years), survival (years), population/ethnic group, and socio-economic status.
Considering that the motor symptoms' onset, as well as ALS's phenotypic features, can be related to the disease development and brain pathology, first motor signs and ALSFRS-R were also evaluated (Table 4). The most prevalent motor alteration among ALS patients was related to the upper or lower limbs (71.66%), followed by symptoms occurring at the bulbar level (28%). Specifically, about monitoring ALS progression, 48.3% of the studies used ALSFRS-R; the mean ratio of it was 36.4.

Table 4. First motor signs of ALS patients and ALSFRS scale.
Concerning smoking habits (Table 5), only 17.2% of the evaluated manuscripts pointed out this factor among individuals with ALS. Explicitly, just one study excluded all smokers from statistics. About metabolism (Tables 5, 6), 65.5% of articles mentioned BMI (mean, 24.4). Moreover, only 20.7% declared any data in the matter of hypertension, 6.89% of cardiovascular risk, 10.3% of obesity, 13.78% of diabetes and 10.3% of glycaemia (Table 5). Regarding lipid metabolism (Table 6), more results were collected. Specifically, 31, 34.48, 34.48, and 41.4%, of the manuscripts show, respectively, TGL (mean, 4.13 mmol/L), LDL (mean, 5.40 mmol/L), HDL (mean, 2.21 mmol/L), and cholesterol (mean, 23.25 mmol/L). But, this information did not suffice to establish a correlation between ALS development and/or progression and metabolism alterations. In fact, 48.28% of the manuscripts evaluated show an association between factors (Table 7).
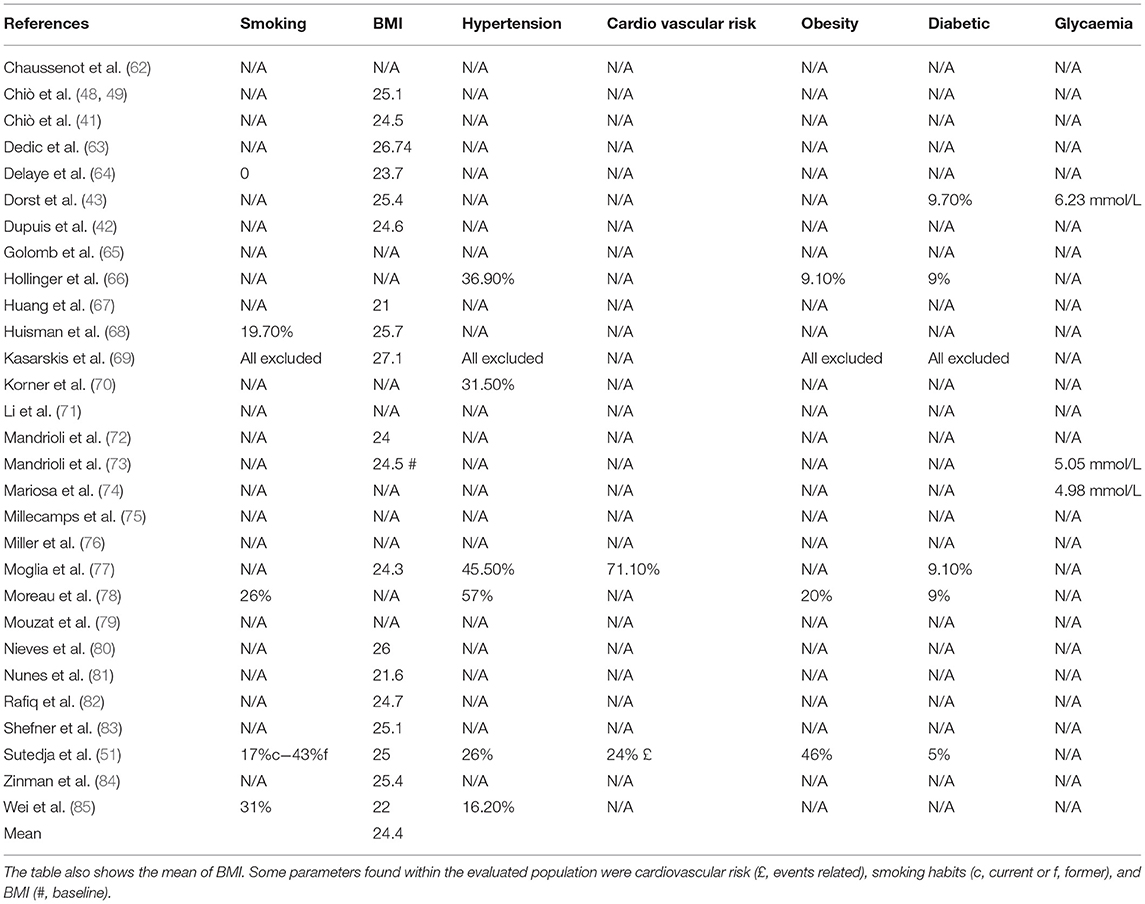
Table 5. Smoking habits, BMI and metabolic parameters of ALS patients.

Table 6. Blood tests outcome of ALS patients.
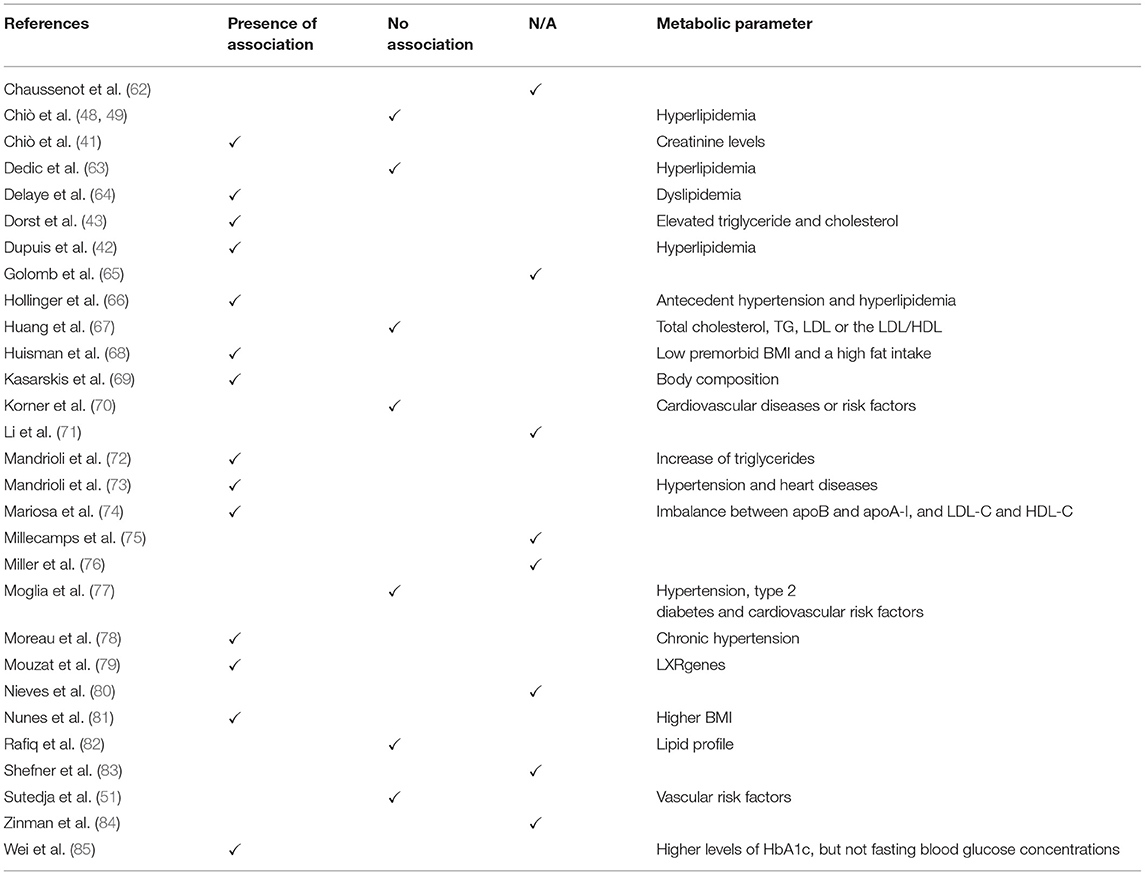
Table 7. Summary of the correlation between metabolic parameters and ALS course and prognosis.
Additionally, to understand if the data outcome could be influenced by medication intake, prescriptions to ALS subjects were also investigated (Table 8). Around 20.7% of the studies mentioned that patients were under Riluzole™ treatment. Only one study stated that patients had been taking the medicine since diagnosis. About other prescriptions, 13.8% of the articles declared that ALS individuals were under cholesterol and hyperlipidaemia lowering agent therapy, or they were taking antioxidants taking antioxidants, antihypertensive, and anti-diabetic agents.
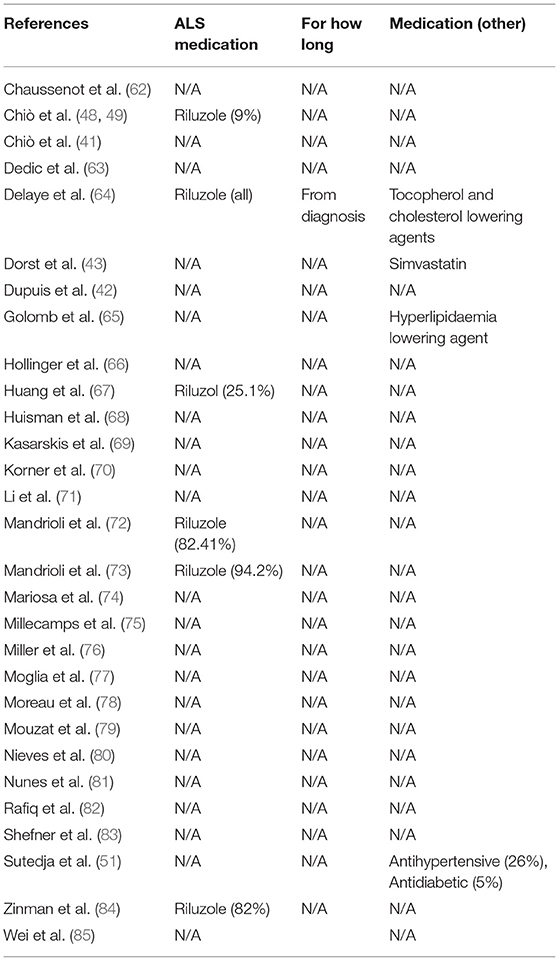
Table 8. Medication taken by ALS patients.
Discussion
Although several mechanisms are being related to ALS, its diagnosis occurs relatively late and, by then, 50% of neurons degenerated, and its prognosis is hard to predict (86). The lack of knowledge regarding secondary mechanisms related to the disease progression along with the limitation concerning studies enclosing individuals presenting ALS represents a struggle for the academic community. The development of strategies that could not only efficiently detect ALS onset but also predict its development and evaluate the therapeutic intervention, would be of great value. In this respect, metabolic status has called attention. Hence, this study aims to put together, in a systematic review, data from ALS patients, which is directly related to their metabolic status i.e., hypertension, cardiovascular risk, obesity, diabetic, glycaemia, hyperlipidaemia, triglycerides (TGL), LDL, HDL, and cholesterol. Further, authors recorded information related to the age of onset, disease duration (years), survival (years), population/ethnic group, mutation, relatives with ALS (%), relatives with motor disorders (n), first motor signs (%), ALSFRS, smoking habits, and ALS medication. Altogether, we indicated that while there is not an assertive data to establish any direct link between changes in metabolic parameters and the progression and survival of ALS, metabolism does impact on ALS. This means that some pathways can be related to it and can modulate ALS outcome, but the complete via and its regulation are not well-defined yet. Accordingly, several studies assume that it is very difficult to establish a correlation between those factors. Indeed, only 48.28% of the articles used to perform this systematic review show an association, meaning that this topic is still a matter of debate.
One of the first data that called the authors' attention due to their strangeness was the presence (or not) of genetic mutation in ALS subjects; only 6.8% of the studies excluded fALS (Table 1). This means that 93.2% of articles put together all types of ALS patients in the same group. Such data indicate that clinical studies did not take into consideration the genetic background of investigated individuals. Hence, considering all ALS individuals as equals (fALS and sALS) could generate a considerable bias.
It is known that ALS patients have a distinct outcome and different age of onset and survival rate (48, 49). For that reason, it was suggested that such differences could be associated to environmental factors that, in turn, might influence ALS genesis and duration (prognosis) (8, 87–95). Such aspects seem to include extreme physical activities, herbicide/pesticide exposure, neurotoxins, viral infection, prion disease, immune response, vascular risk factors and type 1 diabetes (8, 94, 95). On the other hand, diabetes type 2 in elderly, moderate physical activity, fat accumulation, and variations in glucose metabolism, high BMI and hypertension are described to be protective (94). Nevertheless, studies about the relationship between diabetes and ALS have produced conflicting results. In young patients, diabetes is a consistent risk factor for ALS; in older patients, diabetes protects against ALS in Europe but increases the risk of ALS in Asia. Intriguingly, both environment and genotype might subsidize this discrepancy (94, 95).
Other parameters that are often mentioned in ALS patients' studies, in an attempt to correlate ALS prognosis with survival, are patients' age, age of onset and survival rate (that is calculated considering both previous factors) (Table 3). As previously shown, half of the manuscripts depicted patients' age and the age of onset, making it hard to come to any conclusion concerning these factors. Besides, the exact meaning of “age of onset” contrast among works; in this review, two evaluated manuscripts declared its definition. Thus, future studies would be undoubtedly favored if the designation of onset was clear. Likewise, data interpretation would become easier, since results from different study centers could be comparable. Importantly, other groups also suggested that not only the onset but also sex ratio, age at diagnosis, comorbidities, and survival could be due to genetic background, predisposition, socio-economic status and patient's ethnicity (89, 94, 96–100). Actually, we consider that ethnic group is essential for the interpretation of prognostic factors, as it relies (at least partially) on genetics and because it is known that several polymorphisms are population/ethnic-dependent and can modulate metabolism (96, 101–103). Oddly, only two of the evaluated manuscripts mentioned the words “Caucasian” and “white,” indicating that the ethnicity of ALS patients may not have been evaluated. Furthermore, in most reports, the study group is from the same country as the authors. Thus, some favoritism in the interpretation of clinical trials could be assumed.
It is well-known that ALS symptoms appear according to the group of affected neurons. For this reason, one can show changes in the upper or lower limbs (known as the spinal form of ALS) and/or can present dysphagia, dysphonia, or dysarthria (symptoms correlated to the bulbar form). We demonstrated that 71.66% of ALS subjects presented the spinal form and 28% bulbar (Table 4). Although almost 30% of the manuscripts did not mention this factor, the results seem rather consistent among all research centers. Surprisingly, 48.3% of the studies monitored ALSFRS-R; such scale infers about the deterioration caused by the disorder and estimates disease progression per month (104). Considering that ALSFRS-R is world-wide accepted and that more than 50% of clinical evaluations did not cogitate them, the real estimation of ALS patients' evolution is jeopardized, indicating that conclusions regarding any parameter based on clinical studies should be reviewed.
Despite the lack of homogeneity among the examined articles, and consequently, the absence of data that could support an accurate ALS evaluation, the authors further considered the smoking habits (one of the factors that often influence metabolism and patient survival) (Table 5). As already mentioned, 17.2% of all articles included smoking habits in their analysis. Considering that ALS subjects suffer from shortness of breath and that they must be under respiratory therapy throughout their lives, smoking should be determinant to patient's quality of life, response to treatment, and survival.
Because metabolism is the central target of our review, specific parameters are shown in Tables 5–7. For BMI, 65.5% of all articles reported their values. On the other hand, hypertension, cardiovascular risk, obesity, diabetes, and glycaemia were less mentioned (<20%) (Table 5). About lipid metabolism, more results were gathered; around 35% of manuscripts described TGL, LDL, HDL and cholesterol levels (Table 6). However, available information is not enough to establish a direct link between ALS and metabolism alterations. In reality, less than half of the manuscripts evaluated show an association between both factors (Table 7). Nonetheless, it is worth mentioning that metabolism does influence ALS, but not in a unique manner. In fact, in literature, there is a debate about patients' hypo- and hypermetabolism. The so-called hypermetabolic condition is correlated with an augmentation in the energy expenditure to maintain mobility and ventilation avoiding, as a consequence, weakness and exhaustion of the remaining innervated muscles (105–108). Moreover, in an attempt to avoid an increase in oxidative stress, it is suggested that mitochondria become uncoupled ensuing in hypermetabolism (109, 110). Thus, the high resting metabolism could be activated to maintain energetic status. Because of that, hyperlipidaemia and increments in gluconeogenesis, lipolysis and ketogenesis pathways are sighted as neuroprotective (106–108, 111, 112). Interestingly, it was observed a hypolipidemia state in the pre-symptomatic ALS mouse model, in addition to changes in complex lipids during the first phase of motor symptoms (113, 114). Nonetheless, it was also reported that alterations in metabolism, exactly in the lumbar spinal cord in the SOD1G93A mice model before motor symptoms, are primarily caused by the mutation itself than a function of ALS's course (115).
However, it is important to stress that this is not a consensus in the literature and it is defiance for the knowledge of ALS neuropathology (116). Indeed, Chiò et al. described that there was not any effect of metabolism on survival (48, 49), although they suggested that dietetic habits could account for differences in phenotypes and disease progression (48, 49). This hypothesis, by the way, is in accordance with several reports that suggest that hypermetabolism seems to be interconnected with a predisposition of ALS subjects to malnutrition as a result of fear of choking and aspiration, dysphagia and inability to feed themselves (58, 117, 118). It was also shown in vivo FDG-PET study performed with C9orf72 mutation's patients that brain hypometabolism was consistent with ALS clinical phenotypes (119). Moreover, Cedarbaum et al. indicated that such inconsistencies may also be related to the insufficiency relevant information of ALSFRS-R subscale to estimate physical activity (120). Hence, no conclusion involving these parameters should be regarded either.
Because it is known that some medication also modifies cellular metabolism, we also considered ALS subjects' prescriptions in our analysis (Table 8). We observed that (i) 13.8% of articles declared that patients were taking antioxidants, antihypertensive and/or anti-diabetic drugs, in addition to cholesterol and hyperlipidaemia lowering agents, and (ii) 20.7% of them stated that patients were under Riluzole™ treatment (only one study specified that it has been taken since diagnosis). This is very intriguingly data since most of the patients are under a disease-modifying therapy with Riluzole. Riluzole (2-amino-6-trigluoromethoxy benzothiazole) is a wide-spectrum agent ranging from being an anti-glutamatergic drug to increase glial glutamate reuptake and to modulate post-synaptic receptor-mediated effects and excitotoxic pathways (121–126). Moreover, it is known that Riluzole influences, depending on the disease stage, distinct paths, making the intracellular signals of this drug hard to follow (127). Nevertheless, to the context of this review, it is important to mention that Riluzole can interfere with calcium buffering capacity, mitochondrial membrane potential (ΔΨm), sodium currents, voltage-dependent calcium channels and calcium-dependent potassium currents (128). Curiously, Riluzole can also act as a free radical scavenger, blocking reactive oxygen species production through electron transport chain, or by inhibition of calcium efflux at synapse sites (128). Thus, Riluzole, per se might change cellular metabolism.
Limitations of the Study
Several factors might have contributed to the data generated and showed in this review. In fact, numerous methodological limitations of previous works must be listed, i.e., system used by primary centers to acquire evidence, population evaluated (representative vs. available), single-centered vs. multicentric study, sample size, study duration, gene mutation, familial vs. sporadic ALS cases, ethnicity, genetic background, environmental exposure to risk factors, incomplete ALSFRS-R (or the absence of it) and type of study (prospective vs. retrospective; the last can be inaccurate due to lack of data and even lack of patient, in addition to an indisposition of relatives to corroborate with some information) (73). Likewise, in the most of published studies, there were also significant discrepancies regarding age of onset, sex ratio and dose/time of pharmacological therapies. Still, authors believe that there is no sufficient data to perform a meaningful meta-analysis and, therefore, establish a correlation with ALS. Also, a prospective study with an in-depth examination of ALS patients could be supportive.
Conclusion
Our review points to two main conclusions based on recent developments, (i) there is no assertive data available in the literature to establish a specific link between metabolic parameters and the progression and survival of ALS, and (ii) metabolism does influence ALS, but not in a unique manner. This means that some pathways can be related to it and can modulate ALS outcome, but the complete via and its regulation are not well-defined yet. Furthermore, our conclusions can be an alert about how researchers conduct their investigation and how epidemiology studies are designed, since the effort that is being made toward the establishment and validation of a clinical biomarker seems to occur in vain, given the data inconsistency, database divergences, and the absence, in most papers, of relevant information. Essentially, which are the factors, other than the ones mentioned herein, which should be revisited? We believe that investigators should consider exploring other factors, as food habits, neurodevelopment deficits, complications at birth and mother's infection and hypertension (all aspects already correlated to brain development and function). It is worth mentioning that all the topics above can induce modifications in cellular homeostasis through fluctuations in mitochondrial function, ATP synthesis, gene transcription and/or epigenetic regulation. Considering that all these features can also be modulated by genotype background (mutations, polymorphisms), ethnic group, behavioral habits, and environmental factors, ALS's etiology is far more complicated and heterogeneous than we presume (8, 92, 94, 95, 129). Moreover, we should also consider the role of microbiota. Indeed, it was already demonstrated in recent reports that gut microbiota modulates inflammation through short-chain fatty acids and endotoxin synthesis. The microbiota is correlated not only with Alzheimer's disease, neuromyelitis optica, multiple sclerosis, and Parkinson's disease but also ALS (130, 131).
Thus, to provide a reliable record, it is suggested a public policy in which all research and clinical centers might assess the parameters discussed herein. In accordance, Martin et al. in 2016 also recommended a collaborative study involving a wide international consortium to investigate, using a standard methodology, the link between ancestry, environment and ALS phenotype (129). Moreover, the authors genuinely suggest that all conclusions based on clinical trials and patients' evaluation to date should be reconsidered. Accordingly, this systematic review attempts to provide a comprehensible database to facilitate multicentered collaboration, validation, and ultimately, clinical translation.
Data Availability Statement
All data generated or analyzed during this study are included in the article/supplementary material.
Author Contributions
TR and MB analyzed all the articles' titles and abstracts. The defined articles were randomly divided by all authors (GS, MB, ET, BA, and TR), and TR and MC double read all of them. TR wrote the manuscript, and both TR and MC revised it. All authors approved the final version.
Funding
This work was supported by São Paulo Research Foundation (FAPESP) (ref. 2015/02041-1) and Research Support Foundation of Santa Casa de São Paulo School of Medical Science (FCMSCSP) (2017–2019). MB was supported by Master CAPES and FAPESP (2016/12039-7). GS was an undergraduate medical student. ET was under a CAPES Master fellowship program. BA was supported by IC/FAPESP (2018/09084-6).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. D'Alessandro G, Calcagno E, Tartari S, Rizzardini M, Ivernizzi RW, Cantoni L. Glutamate and glutathione interplay in a motor neuronal model of amyotrophic lateral sclerosis reveals altered energy metabolismo. Neurobiol Dis. (2011) 43:346–55. doi: 10.1016/j.nbd.2011.04.003
2. Bozzo F, Mirra A, Carri MT. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: new perspectives. Neurosci Lett. (2017) 636:3–8. doi: 10.1016/j.neulet.2016.04.065
3. Saberi S, Stauffer JE, Schulte DJ, Ravits J. Neuropathology of amyotrophic lateral sclerosis and its variants. Neuro Clin. (2015) 33:855–76. doi: 10.1016/j.ncl.2015.07.012
4. Cozzolino M, Carri MT. Mitochondrial dysfunction in ALS. Prog Neurobiol. (2012) 97:54–6. doi: 10.1016/j.pneurobio.2011.06.003
5. Johnston CA, Stanton BR, Turner MR, Gray R, Blunt AH, Butt D, et al. Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J Neurol. (2006) 253:1642–3. doi: 10.1007/s00415-006-0195-y
6. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expan-sion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
7. Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. (2015) 6:171. doi: 10.4103/2152-7806.169561
8. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. (2001) 344:1688–700. doi: 10.1056/NEJM200105313442207
9. Kunst CB. Complex genetics of amyotrophic lateral sclerosis. Am J Hum Genet. (2004) 75:933–47. doi: 10.1086/426001
10. Martin LJ, Liu Z. Opportunities for neuroprotection in ALS using cell death mechanism rationales. Drug Discov Today Dis Model. (2004) 1:135–43. doi: 10.1016/j.ddmod.2004.09.004
11. Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. (2012) 124:339–52. doi: 10.1007/s00401-012-1022-4
12. Vandoorne T, De Bock K, Van Den Bosch L. Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathol. (2018) 135:489–509. doi: 10.1007/s00401-018-1835-x
13. Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR. Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J Neurochem. (2008) 107:1271–83. doi: 10.1111/j.1471-4159.2008.05699.x
14. Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. (2011) 29:824–8. doi: 10.1038/nbt.1957
15. Brites D, Vaz AR. Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci. (2014) 8:117. doi: 10.3389/fncel.2014.00117
16. Nikodemova M, Small AL, Smith SM, Mitchell GS, Watters JJ. Spinal but not cortical microglia acquire an atypical phenotype with high VEGF, galectin-3 and osteopontin, and blunted inflammatory responses in ALS rats. Neurobiol Dis. (2014) 69:43–53. doi: 10.1016/j.nbd.2013.11.009
17. Beeldman E, Raaphorst J, Klein Twennaar M, de Visser M, Schmand BA, de Haan RJ. The cognitive profile of ALS: a systematic review and meta-analysis update. J Neurol Neurosurg Psychiatry. (2016) 87:611–9. doi: 10.1136/jnnp-2015-310734
18. Matías-Guiu JA, Pytel V, Cabrera-Martín MN, Galán L, Valles-Salgado M, Guerrero A, et al. Amyloid- and FDG-PET imaging in amyotrophic lateral sclerosis. Eur J Nucl Med Mol Imaging. (2016) 43:2050–60. doi: 10.1007/s00259-016-3434-1
19. Martinez-Merino L, Iridoy M, Galbete A, Roldán M, Rivero A, Acha B, et al. Evaluation of chitotriosidase and cc-chemokine ligand 18 as biomarkers of microglia activation in Amyotrophic Lateral Sclerosis. Neurodegener Dis. (2018) 18:208–15. doi: 10.1159/000490920
20. Nonneman A, Criem N, Lewandowski SA, Nuyts R, Thal DR, Pfrieger FW, et al. Astrocyte-derived Jagged-1 mitigates deleterious Notch signaling in amyotrophic lateral sclerosis. Neurobiol Dis. (2018) 119:26–40. doi: 10.1016/j.nbd.2018.07.012
21. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. (1994) 264:1772–5. doi: 10.1126/science.8209258
22. Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. (1995) 92:689–93. doi: 10.1073/pnas.92.3.689
23. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. (1995) 14:1105–16. doi: 10.1016/0896-6273(95)90259-7
24. Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA. (2002) 99:1604–9. doi: 10.1073/pnas.032539299
25. Cozzolino M, Ferri A, Carri MT. Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxid Redox Signal. (2008) 10:405–43. doi: 10.1089/ars.2007.1760
26. Shi P, Gal J, Kwinter DM, Liu X, Zhu H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta. (2010) 1802:45–51. doi: 10.1016/j.bbadis.2009.08.012
27. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. (2011) 377:942–55. doi: 10.1016/S0140-6736(10)61156-7
28. Allen SP, Rajan S, Duffy L, Mortiboys H, Higginbottom A, Grierson AJ, et al. Superoxide dismutase 1 mutation in a cellular model of amyotrophic lateral sclerosis shifts energy generation from oxidative phosphorylation to glycolysis. Neurobiol Aging. (2014) 35:1499–509. doi: 10.1016/j.neurobiolaging.2013.11.025
29. Ladd AC, Keeney PM, Govind MM, Bennett JP. Mitochondrial oxidative phosphorylation transcriptome alterations in human amyotrophic lateral sclerosis spinal cord and blood. Neuromolecular Med. (2014) 16:714–26. doi: 10.1007/s12017-014-8321-y
30. Onesto E, Colombrita C, Gumina V, Borghi MO, Dusi S, Doretti A, et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol Commun. (2016) 4:47. doi: 10.1186/s40478-016-0316-5
31. Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H, et al. C9orf72 hexanucleotide expansions are associated with altered ER calcium homeostasis and stress granule formation in iPSC-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells. (2016) 34:2063–78. doi: 10.1002/stem.2388
32. Carri MT, D'Ambrosi N, Cozzolino M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem Biophys Res Commun. (2017) 483:1187–93. doi: 10.1016/j.bbrc.2016.07.055
33. Vieira FG, Ping Q, Moreno AJ, Kidd JD, Thompson K, Jiang B. Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS ONE. (2015) 10:e0135570. doi: 10.1371/journal.pone.0135570
34. Martin D, Thompson MA, Nadler JV. The neuroprotectivea agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. (1993) 250:473–6. doi: 10.1016/0014-2999(93)90037-I
35. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. (1994) 330:585–91. doi: 10.1056/NEJM199403033300901
36. Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. (1996) 347:1425–31. doi: 10.1016/S0140-6736(96)91680-3
37. Tripathi VB, Al-Chalabi A. Molecular insights and therapeutic targets in amyotrophic lateral sclerosis. CNS Neurol Disord Drug Targets. (2008) 7:11–9. doi: 10.2174/187152708783885110
38. Sawada H. Clinical efficacy of ederavone for the treatment of amyotrophic lateral sclerosis. Expert Opin Pharmacother. (2017) 18:735–8. doi: 10.1080/14656566.2017.1319937
39. Shook SJ, Pioro EP. Racing against the clock: recognizing, differentiating, diagnosing, and referring the amyotrophic lateral sclerosis patient. Ann Neurol. (2009) 65:S10–6. doi: 10.1002/ana.21545
40. Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol. (2014) 13:1127–38. doi: 10.1016/S1474-4422(14)70129-2
41. Chiò A, Calvo A, Bovio G, Canosa A, Bertuzzo D, Galmozzi F, et al. Amyotrophic lateral sclerosis outcome measures and the role of albumin and creatinine: a population-based study. JAMA Neurol. (2014) 71:1134–42. doi: 10.1001/jamaneurol.2014.1129
42. Dupuis L, Corcia P, Fergani A, Gonzalez De Aguilar JL, Bonnefont-Rousselot D, Bittar R, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. (2008) 70:1004–9. doi: 10.1212/01.wnl.0000285080.70324.27
43. Dorst J, Kuhnlein P, Hendrich C, Kassubek J, Sperfeld AD, Ludolph AC. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J Neurol. (2011) 258:613–7. doi: 10.1007/s00415-010-5805-z
44. Ikeda K, Hirayama T, Takazawa T, Kawabe K, Iwasaki Y. Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in Japanese patients with amyotrophic lateral sclerosis: a cross-sectional study. Disabil Rehabil. (2012) 51:1501–8. doi: 10.2169/internalmedicine.51.7465
45. Paillisse C, Lacomblez L, Dib M, Bensimon G, Garcia-Acosta S, Meininger V. Prognostic factors for survival in amyotrophic lateral sclerosis patients treated with riluzole. Amyotroph Lateral Scler Other Motor Neuron Disord. (2005) 6:37–44. doi: 10.1080/14660820510027035
46. Gonzalez de Aguilar JL, Dupuis L, Oudart H, Loeffler JP. The metabolic hypothesis in amyotrophic lateral sclerosis: insights from mutant Cu/Zn-superoxide dismutase mice. Biomed Pharmacother. (2005) 59:190–6. doi: 10.1016/j.biopha.2005.03.003
47. Yang JW, Kim SM, Kim HJ, Kim JE, Park KS, Kim SH, et al. Hypolipidemia in patients with amyotrophic lateral sclerosis: a possible gender difference? J Clin Neurol. (2013) 9:125–9. doi: 10.3988/jcn.2013.9.2.125
48. Chiò A, Calvo A, Ilardi A, Cavallo E, Moglia C, Mutani R, et al. Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology. (2009) 73:1681–5. doi: 10.1212/WNL.0b013e3181c1df1e
49. Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. (2009) 10:310–23. doi: 10.3109/17482960802566824
50. Paganoni S, Zhang M, Quiroz Zárate A, Jaffa M, Yu H, Cudkowicz ME, et al. Uric acid levels predict survival in men with amyotrophic lateral sclerosis. J Neurol. (2012) 259:1923–8. doi: 10.1007/s00415-012-6440-7
51. Sutedja NA, van der Schouw YT, Fischer K, Sizoo EM, Huisman MH, Veldink JH, et al. Beneficial vascular risk profile is associated with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2011) 82:638–42. doi: 10.1136/jnnp.2010.236752
52. Saffer D, Morley J, Bill PL. Carbohydrate metabolism in motor neuron disease. J Neurol Neurosurg Psychiatry. (1977) 40:533–7. doi: 10.1136/jnnp.40.6.533
53. Murai A, Miyahara T, Tanaka T, Kaneko T, Sako Y, Kameyama M. Abnormalities of lipoprotein and carbohydrate metabolism in degenerative diseases of the nervous system–motor neuron disease and spinocerebellar degeneration. Tohoku J Exp Med. (1983) 139:365–76. doi: 10.1620/tjem.139.365
54. Krentz AJ, Williams AC, Nattrass M. Abnormal regulation of carbohydrate metabolism in motor neurone disease. Diabetes Res. (1991) 16:93–9.
55. Creemers H, Grupstra H, Nollet F, van den Berg LH, Beelen A. Prognostic factors for the course of functional status of patients with ALS: a systematic review. J Neurol. (2015) 262:1407–23. doi: 10.1007/s00415-014-7564-8
56. Shimizu T, Nakayama Y, Matsuda C, Haraguchi M, Bokuda K, Ishikawa-Takata K, et al. Prognostic significance of body weight variation after diagnosis in ALS: a single-centre prospective cohort study. J Neurol. (2019) 266:1412–20. doi: 10.1007/s00415-019-09276-2
57. Steyn FJ, Ioannides ZA, van Eijk RPA, Heggie S, Thorpe KA, Ceslis A, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J Neurol Neurosurg Psychiatry. (2018) 89:1016–23. doi: 10.1136/jnnp-2017-317887
58. Jésus P, Fayemendy P, Nicol M, Lautrette G, Sourisseau H, Preux PM, et al. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur J Neurol. (2018) 25:97–104. doi: 10.1111/ene.13468
59. Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a highenergy diet in a transgenic mouse model. Proc Natl Acad Sci USA. (2004) 101:11159–64. doi: 10.1073/pnas.0402026101
60. Mattson MP, Cutler RG, Camandola S. Energy intake and amyotrophic lateral sclerosis. Neuromolecular Med. (2007) 9:17–20. doi: 10.1385/NMM:9:1:17
61. Pedersen WA, Mattson MP. No benefit of dietary restriction on disease onset or progression in amyotrophic lateral sclerosis Cu/Zn-superoxide dismutase mutant mice. Brain Res. (1999) 833:117–20. doi: 10.1016/S0006-8993(99)01471-7
62. Chaussenot A, Le Ber I, Ait-El-Mkadem S, Camuzat A, de Septenville A, Bannwarth S, et al. Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol Aging. (2014) 35:2884. doi: 10.1016/j.neurobiolaging.2014.07.022
63. Dedic SI, Stevic Z, Dedic V, Stojanovic VR, Milicev M, Lavrnic D. Is hyperlipidemia correlated with longer survival in patients with amyotrophic lateral sclerosis? Neurol Res. (2012) 34:576–80. doi: 10.1179/1743132812Y.0000000049
64. Delaye JB, Patin F, Piver E, Bruno C, Vasse M, Vourc'h P, et al. Low IDL-B and high LDL-1 subfraction levels in serum of ALS patients. J Neurol Sci. (2017) 380:124–7. doi: 10.1016/j.jns.2017.07.019
65. Golomb BA, Kwon EK, Koperski S, Evans MA. Amyotrophic lateral sclerosis-like conditions in possible association with cholesterol-lowering drugs: an analysis of patient reports to the University of California, San Diego (UCSD) Statin Effects Study. Drug Saf. (2009) 32:649–61. doi: 10.2165/00002018-200932080-00004
66. Hollinger SK, Okosun IS, Mitchell CS. Antecedent disease and amyotrophic lateral sclerosis: what is protecting whom? Front Neurol. (2016) 7:47. doi: 10.3389/fneur.2016.00047
67. Huang R, Guo X, Chen X, Zheng Z, Wei Q, Cao B, et al. The serum lipid profiles of amyotrophic lateral sclerosis patients: a study from south-west China and a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:359–65. doi: 10.3109/21678421.2015.1047454
68. Huisman MH, Seelen M, van Doormaal PT, de Jong SW, de Vries JH, van der Kooi AJ, et al. Effect of presymptomatic body mass index and consumption of fat and alcohol on amyotrophic lateral sclerosis. JAMA Neurol. (2015) 72:1155–62. doi: 10.1001/jamaneurol.2015.1584
69. Kasarskis EJ, Mendiondo MS, Matthews DE, Mitsumoto H, Tandan R, Simmons Z, et al. Estimating daily energy expenditure in individuals with amyotrophic lateral sclerosis. Am J Clin Nutr. (2014) 99:792–803. doi: 10.3945/ajcn.113.069997
70. Korner S, Kollewe K, Ilsemann J, Muller-Heine A, Dengler R, Krampfl K, et al. Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur J Neurol. (2013) 20:647–54. doi: 10.1111/ene.12015
71. Li XL, Shu S, Li XG, Liu Q, Liu F, Cui B, et al. CHCHD10 is not a frequent causative gene in Chinese ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:458–60. doi: 10.3109/21678421.2016.1170151
72. Mandrioli J, Rosi E, Fini N, Fasano A, Raggi S, Fantuzzi AL, et al. Changes in routine laboratory tests and survival in amyotrophic lateral sclerosis. Neurol Sci. (2017) 38:2177–82. doi: 10.1007/s10072-017-3138-8
73. Mandrioli J, Ferri L, Fasano A, Zucchi E, Fini N, Moglia C, et al. Cardiovascular diseases may play a negative role in the prognosis of amyotrophic lateral sclerosis. Eur J Neurol. (2018) 25:861–8. doi: 10.1111/ene.13620
74. Mariosa D, Hammar N, Malmstrom H, Ingre C, Jungner I, Ye W, et al. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: a more than 20-year follow-up of the Swedish AMORIS cohort. Ann Neurol. (2017) 81:718–28. doi: 10.1002/ana.24936
75. Millecamps S, Salachas F, Cazeneuve C, Gordon P, Bricka B, Camuzat A, et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet. (2010) 47:554–60. doi: 10.1136/jmg.2010.077180
76. Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. (2013) 12:435–42. doi: 10.1016/S1474-4422(13)70061-9
77. Moglia C, Calvo A, Canosa A, Bertuzzo D, Cugnasco P, Solero L, et al. Influence of arterial hypertension, type 2 diabetes and cardiovascular risk factors on ALS outcome: a population-based study. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:590–7. doi: 10.1080/21678421.2017.1336560
78. Moreau C, Brunaud-Danel V, Dallongeville J, Duhamel A, Laurier-Grymonprez L, de Reuck J, et al. Modifying effect of arterial hypertension on amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2012) 13:194–201. doi: 10.3109/17482968.2011.610110
79. Mouzat K, Molinari N, Kantar J, Polge A, Corcia P, Couratier P, et al. Liver X receptor genes variants modulate ALS phenotype. Mol Neurobiol. (2018) 55:1959–65. doi: 10.1007/s12035-017-0453-2
80. Nieves JW, Gennings C, Factor-Litvak P, Hupf J, Singleton J, Sharf V, et al. Association between dietary intake and function in amyotrophic lateral sclerosis. JAMA Neurol. (2016) 73:1425–32. doi: 10.1001/jamaneurol.2016.3401
81. Nunes G, Santos CA, Grunho M, Fonseca J. Enteral feeding through endoscopic gastrostomy in amyotrophic lateral sclerosis patients. Nutr Hosp. (2016) 33:561. doi: 10.20960/nh.561
82. Rafiq MK, Lee E, Bradburn M, McDermott CJ, Shaw PJ. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: Insights from the olesoxime clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 16:478–84. doi: 10.3109/21678421.2015.1062517
83. Shefner JM, Watson ML, Meng L, Wolff AA. A study to evaluate safety and tolerability of repeated doses of tirasemtiv in patients with amitrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. (2013) 14:574–81. doi: 10.3109/21678421.2013.822517
84. Zinman L, Sadeghi R, Gawel M, Patton D, Kiss A. Are statin medications safe in patients with ALS? Amyotroph Lateral Scler. (2008) 9:223–8. doi: 10.1080/17482960802031092
85. Wei QQ, Chen Y, Cao B, Ou RW, Zhang L, Hou Y, et al. Blood hemoglobin A1c levels and amyotrophic lateral sclerosis survival. Mol Neurodegener. (2017) 12:69. doi: 10.1186/s13024-017-0211-y
86. Laferriere F, Polymenidou M. Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis. Swiss Med Wkly. (2015) 145:w14054. doi: 10.4414/smw.2015.14054
87. Bozzoni V, Pansarasa O, Diamanti L, Nosari G, Cereda C, Ceroni M. Amyotrophic lateral sclerosis and environmental factors. Funct Neurol. (2016) 31:7–19. doi: 10.11138/FNeur/2016.31.1.007
88. Oskarsson B, Horton DK, Mitsumoto H. Potential environmental factors in amyotrophic lateral sclerosis. Neuro Clin. (2015) 33:877–88. doi: 10.1016/j.ncl.2015.07.009
89. Brown RH Jr, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. (2017) 377:1602. doi: 10.1056/NEJMra1603471
90. Paez-Colasante X, Figueroa-Romero C, Sakowski SA, Goutman SA, Feldman EL. Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nat Rev Neurol. (2015) 11:266–79. doi: 10.1038/nrneurol.2015.57
91. Sher RB. The interaction of genetics and environmental toxicants in amyotrophic lateral sclerosis: results from animal models. Neural Regen Res. (2017) 12:902–5. doi: 10.4103/1673-5374.208564
92. Das K, Nag C, Ghosh M. Familial, environmental, and occupational risk factors in development of amyotrophic lateral sclerosis. N Am J Med Sci. (2012) 4:350–5. doi: 10.4103/1947-2714.99517
93. Su FC, Goutman SA, Chernyak S, Mukherjee B, Callaghan BC, Batterman S, et al. Association of environmental toxins with amyotrophic lateral sclerosis. JAMA Neurol. (2016) 73:803–11. doi: 10.1001/jamaneurol.2016.0594
94. Logroscino G. Motor neuron disease: Are diabetes and amyotrophic lateral sclerosis related? Nat Rev Neurol. (2015) 11:488–90. doi: 10.1038/nrneurol.2015.145
95. Sun Y, Lu CJ, Chen RC, Hou WH, Li CY. Risk of amyotrophic lateral sclerosis in patients with diabetes: a nationwide population-based cohort study. J Epidemiol. (2015) 25:445–51. doi: 10.2188/jea.JE20140176
96. Cronin S, Hardiman O, Traynor BJ. Ethnic variation in the incidence of ALS: a systematic review. Neurology. (2007) 68:1002–7. doi: 10.1212/01.wnl.0000258551.96893.6f
97. Henry KA, Fagliano J, Jordan HM, Rechtman L, Kaye WE. Geographic variation of amyotrophic lateral sclerosis incidence in New Jersey, 2009–2011. Am J Epidemiol. (2015) 182:512–9. doi: 10.1093/aje/kwv095
98. Roberts AL, Johnson NJ, Chen JT, Cudkowicz ME, Weisskopf MG. Race/ethnicity, socioeconomic status, and ALS mortality in the United States. Neurology. (2016) 87:2300–8. doi: 10.1212/WNL.0000000000003298
99. Martin S, Al Khleifat A, Al-Chalabi A. What causes amyotrophic lateral sclerosis? F1000 Res. (2017) 6: 371. doi: 10.12688/f1000research.10476.1
100. Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol. (2015) 7:181–93. doi: 10.2147/CLEP.S37505
101. Weber WW. Populations and genetic polymorphisms. Mol Diagn. (1999) 4:299–307. doi: 10.1016/S1084-8592(99)80006-X
102. Pontoriero AC, Trinks J, Hulaniuk ML, Caputo M, Fortuny L, Pratx LB, et al. Influence of ethnicity on the distribution of genetic polymorphisms associated with risk of chronic liver disease in South American populations. BMC Genetics. (2015) 16:93. doi: 10.1186/s12863-015-0255-3
103. Xu L, Li J, Tian D, Chen L, Tang L, Fan D. The rs696880 polymorphism in the Nogo-A receptor gene (RTN4R) is associated with susceptibility to sporadic amyotrophic lateral sclerosis in the chinese population. Front Aging Neurosci. (2018) 10:108. doi: 10.3389/fnagi.2018.00108
104. Kollewe K, Mauss U, Krampfl K, Petri S, Dengler R, Mohammadi B. ALSFRS-R score and its ratio: a useful predictor for ALS-progression. J Neurol Sci. (2008) 275:69–73. doi: 10.1016/j.jns.2008.07.016
105. Bouteloup C, Desport JC, Clavelou P, Guy N, Derumeaux-Burel H, Ferrier A, et al. Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol. (2009) 256:1236–42. doi: 10.1007/s00415-009-5100-z
106. Shoesmith CL, Strong MJ. Amyotrophic lateral sclerosis. Update for family physicians. Can Fam Physician. (2006) 52:1562–9.
107. Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, Beaufrère B, et al. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr. (2001) 74:328–34. doi: 10.1093/ajcn/74.3.328
108. Rosenfeld J, Ellis A. Nutrition and dietary supplements in motor neuron disease. Phys Med Rehabil Clin N Am. (2008) 19:573. doi: 10.1016/j.pmr.2008.03.001
109. Funalot B, Desport JC, Sturtz F, Camu W, Couratier P. Highmetabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2009) 10:113–7. doi: 10.1080/17482960802295192
110. Barber SC, Mead RJ, Shaw PJ. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta. (2006) 1762:1051–67. doi: 10.1016/j.bbadis.2006.03.008
111. Dupuis L, Pradat PF, Ludolph AC, Loeffler JP. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. (2011) 10:75–82. doi: 10.1016/S1474-4422(10)70224-6
112. Fergani A, Oudart H, Gonzalez de Aguilar JL, Fricker B, René F, Hocquette JF, et al. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J Lipid Res. (2007) 48:1571–80. doi: 10.1194/jlr.M700017-JLR200
113. Kim SM, Kim H, Kim JE, Park KS, Sung JJ, Kim SH, et al. Amyotrophic lateral sclerosis is associated with hypolipidemia at the presymptomatic stage in mice. PLoS ONE. (2011) 6:e17985. doi: 10.1371/journal.pone.0017985
114. Henriques A, Croixmarie V, Bouscary A, Mosbach A, Keime C, Boursier-Neyret C, et al. Sphingolipid metabolism is dysregulated at transcriptomic and metabolic levels in the spinal cord of an animal model of amyotrophic lateral sclerosis. Front Mol Neurosci. (2018) 10:433. doi: 10.3389/fnmol.2017.00433
115. Pharaoh G, Sataranatarajan K, Street K, Hill S, Gregston J, Ahn B, et al. Metabolic and stress response changes precede disease onset in the spinal cord of mutant SOD1 ALS mice. Front Neurosci. (2019) 13:487. doi: 10.3389/fnins.2019.00487
116. Ferri A, Coccurello R. What is “Hyper” in the ALS Hypermetabolism? Mediators Inflamm. (2017) 2017:7821672. doi: 10.1155/2017/7821672
117. Couratier P, Desport JC, Antonini MT, Mabrouk T, Perna A, Vincent F, et al. Review of nutritional and respiratory support in ALS. Rev Neurol. (2004) 160:243–50. doi: 10.1016/S0035-3787(04)70898-X
118. Desport JC, Torny F, Lacoste M, Preux PM, Couratier P. Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener Dis. (2005) 2:202–7. doi: 10.1159/000089626
119. Castelnovo V, Caminiti SP, Riva N, Magnani G, Silani V, Perani D. Heterogeneous brain FDG-PET metabolic patterns in patients with C9orf72 mutation. Neurol Sci. (2019) 40:515–21. doi: 10.1007/s10072-018-3685-7
120. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (phase III). J Neurol Sci. (1999) 169:13–21. doi: 10.1016/S0022-510X(99)00210-5
121. Umemiya M, Berger AJ. Inhibition by riluzole of glycinergic postsynaptic currents in rat hypoglossal motoneurones. Br J Pharmacol. (1995) 116:3227–30. doi: 10.1111/j.1476-5381.1995.tb15128.x
122. Centonze D, Calabresi P, Pisani A, Marinelli S, Marfia GA, Bernardi G. Electrophysiology of the neuroprotective agent riluzole on striatal spiny neurons. Neuropharmacology. (1998) 37:1063–70. doi: 10.1016/S0028-3908(98)00081-1
123. Zona C, Cavalcanti S, De Sarro G, Siniscalchi A, Marchetti C, Gaetti C, et al. Kainate-induced currents in rat cortical neurons in culture are modulated by riluzole. Synapse. (2002) 43:244–51. doi: 10.1002/syn.10040
124. Pereira AC, Lambert HK, Grossman YS, Dumitriu D, Waldman R, Jannetty SK, et al. Glutamatergic regulation prevents hippocampal dependent age-related cognitive decline through dendritic spine clustering. Proc Natl Acad Sci USA. (2014) 111:18733–8. doi: 10.1073/pnas.1421285111
125. Frizzo ME, Dall'Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. (2004) 24:123–8. doi: 10.1023/B:CEMN.0000012717.37839.07
126. Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. (2008) 578:171–6. doi: 10.1016/j.ejphar.2007.10.023
127. Dash RP, Babu RJ, Srinivas NR. Two decades-long journey from riluzole to edaravone: revisiting the clinical pharmacokinetics of the only two amyotrophic lateral sclerosis therapeutics. Clin Pharmacokinet. (2018) 57:1385–98. doi: 10.1007/s40262-018-0655-4
128. Jaiswal MK. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. (2019) 39:733–48. doi: 10.1002/med.21528
129. Marin B, Logroscino G, Boumédiene F, Labrunie A, Couratier P, Babron MC, et al. Clinical and demographic factors and outcome of amyotrophic lateral sclerosis in relation to population ancestral origin. Eur J Epidemiol. (2016) 31:229–45. doi: 10.1007/s10654-015-0090-x
130. Castillo-Álvarez F, Marzo-Sola ME. Role of the gut microbiota in the development of various neurological diseases. Neurologia. (2019). doi: 10.1016/j.nrl.2019.03.017. [Epub ahead of print].
Keywords: amyotrophic lateral sclerosis, patients, metabolism, prognostic factor, systematic review
Citation: Brito MD, da Silva GFG, Tilieri EM, Araujo BG, Calió ML and Rosenstock TR (2019) Metabolic Alteration and Amyotrophic Lateral Sclerosis Outcome: A Systematic Review. Front. Neurol. 10:1205. doi: 10.3389/fneur.2019.01205
Received: 17 August 2019; Accepted: 29 October 2019;
Published: 20 November 2019.
Edited by:
Angelo Schenone, University of Genoa, ItalyReviewed by:
Christian Lunetta, Nemo Clinical Center, ItalyPaola Sandroni, Mayo Clinic, United States
Copyright © 2019 Brito, da Silva, Tilieri, Araujo, Calió and Rosenstock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatiana Rosado Rosenstock, dGF0aS5mYXJtJiN4MDAwNDA7Z21haWwuY29t; dGF0aWFuYS5yb3NhZG8mI3gwMDA0MDtmY21zYW50YWNhc2FzcC5lZHUuYnI=
†These authors have contributed equally to this work
‡ORCID: Mariana Dutra Brito orcid.org/0000-0002-3161-3294
Erick Mutti Tilieri orcid.org/0000-0002-3072-5526
Beatriz Grisolia Araujo orcid.org/0000-0003-1161-4123
Michele Longoni Calió orcid.org/0000-0002-4367-2283
Tatiana Rosado Rosenstock orcid.org/0000-0003-1153-7412