 Atsuko Katsumoto
Atsuko Katsumoto Hideyuki Takeuchi
Hideyuki Takeuchi Fumiaki Tanaka
Fumiaki Tanaka- Department of Neurology and Stroke Medicine, Graduate School of Medicine, Yokohama City University, Yokohama, Japan
Traumatic brain injury (TBI) has been associated with the development of Alzheimer's disease (AD) because these conditions share common pathological hallmarks: amyloid-β and hyperphosphorylated tau accumulation. However, given recent data it is uncertain if a history of TBI leads to the development of AD. Moreover, chronic traumatic encephalopathy (CTE), caused by repetitive mild TBI and characterized by progressive neurodegeneration with hyperphosphorylated tau, has come to be recognized as distinct from AD. Therefore, it is important to elucidate the clinical outcomes and molecular mechanisms underlying tau pathology following TBI. We summarize the histopathological features and clinical course of TBI in CTE, comparing the tau pathology with that in AD. Following brain injury, diffuse axonal injury, and hyperphosphorylated tau aggregates are observed within a shorter period than in AD. Hyperphosphorylated tau deposition usually begins in the perivascular area of the sulci in the cerebral cortex, then spreads unevenly in the cortex in CTE, while AD shows diffuse distribution of hyperphosphorylated tau in the cortical areas. We also highlight the molecular profile of tau and the implications of tau progression throughout the brain in both diseases. Tau contains phosphorylation sites common to both conditions. In particular, phosphorylation at Thr231 triggers a conformational change to the toxic cis form of tau, which is suggested to drive neurodegeneration. Although the mechanism of rapid tau accumulation remains unknown, the structural diversity of tau might result in these different outcomes. Finally, future perspectives on CTE in terms of tau reduction are discussed.
Introduction
Traumatic brain injury (TBI) is defined as damage to the brain caused by an impact such as a blow or jolt to the head. In contrast to the previous view that most people fully recover from mild TBI, a recent scoping review revealed that approximately half of patients experience serious long-term cognitive impairment, including problems with executive function, learning memory, attention, processing speed, and language function (1). A large cohort study using data from 2.8 million medical records showed that a single mild TBI was associated with a 20% greater risk of dementia (2). Moreover, emerging data have indicated that moderate and severe TBIs demonstrate a dose-response trend as risk factors for neurodegenerative diseases, including cerebral atrophy (3, 4), Alzheimer's disease (AD) (5), chronic traumatic encephalopathy (CTE) (6–10), and Parkinson's disease (PD) (11–13). However, the underlying mechanisms between TBI and these neurodegenerative diseases remain unknown. While tauopathy is a common pathological finding and there seems to be a close association between tau pathology following TBI and dementia, it has long been debated whether TBI can specifically lead to AD, or whether CTE following TBI can cause AD. Here we review both the pathological and molecular features of tau in TBI, including prospective therapeutic strategies.
Tau Pathology
Tau is an abbreviated or alternative term for the microtubule-associated protein tau (MAPT). Microtubules are essential for the normal trafficking of cellular cargo in neuronal axonal projections (14). Under normal conditions, MAPT is a soluble protein that facilitates microtubule stabilization in cells, and is found in particularly high concentrations in neurons. In pathological conditions, tau can be more phosphorylated than normal (phosphorylation, Figure 1A). Hyperphosphorylated tau molecules dissociate from microtubules in the axon, translocate to the cell body and proximal dendrites, and aggregate into intracellular inclusions termed neurofibrillary tangles (NFTs), leading to impaired axonal function. Tau hyperphosphorylation itself decreases tau binding to microtubules and promotes tau fibrillization (15, 16). Furthermore, there is growing evidence that tau aggregates can recruit other tau aggregates to themselves and then spread to surrounding regions (17).
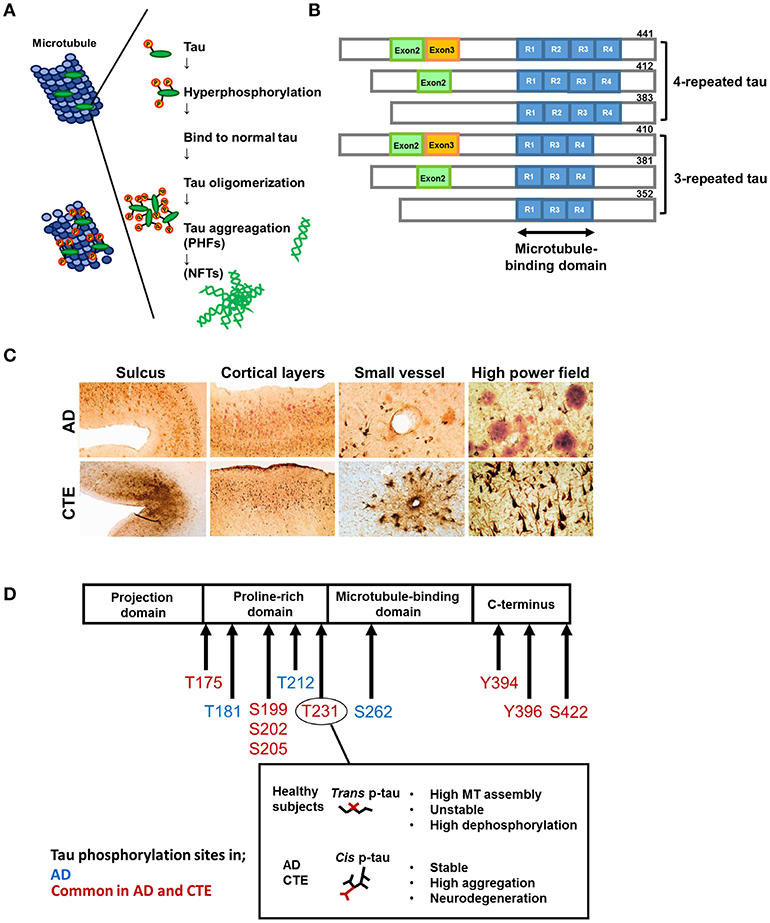
Figure 1. Formation of hyperphosphorylated tau aggregates. Under normal condition, the microtubule-associated protein tau is soluble and abundant in axons of neurons. In pathological conditions, tau can be hyperphosphorylated and dissociates from microtubules. Then hyperphosphorylated tau translocates to the cell body and aggregates into intracellular inclusions termed paired helical filaments (PHFs) and neurofibrillary tangles (NFTs). (B) Tau isoforms. There are six isoforms of tau in human brain. Tau isoforms with four microtubule binding domains, designated as 4R-tau, are accumulated in progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS), whereas tau isoforms with three microtubule binding domains, designated as 3R-tau, are observed in Pick's disease. All six tau isoforms are involved in AD. This pattern is also detected in Down syndrome (DS), and amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam syndrome (ALS/PDC), and chronic traumatic encephalopathy (CTE) following TBI. (C) Characteristic pathology in AD and CTE. Upper panels: AD brains show diffuse cortical distribution of neurofibrillary tangles, preferentially distributed in laminae III and V without accentuation deep within sulci. Few fibrillary tangles exist around the small vessels. Double immunostaining demonstrates the coexsistence of abundant amyloid-β plaques (red) and interspersed PHF-1 neurofibrillary tangles (brown). Lower panels: In CTE, AT8 staining exhibits irregular cortical distribution of p-tau pathology with prominent subpial clusters of p-tau astrocytic tangles, focal accumulation deep within sulci, and neurofibrillary tangles in superficial cortical laminae II–III. Perivascular distribution of astrocytic tangles and neurofibrillary tangles are prominent in the small vessels. Double immunostaining reveals dense neurofibrillary tangles (brown) without amyloid-β plaques (red). Reproduced with permission from McKee et al. (8). (D) Representative tau phosphorylation sites in AD and CTE. CTE shares common phosphorylation sites with AD. Phosphorylation of tau at Thr231 enables the cis-trans conformational change of p-tau Cis and the trans formation of phosphorylated tau. Trans formation of p-tau, observed in healthy subjects, promotes microtubule assembly, and is critical for normal neuronal function. Phosphorylation status is unstable and easy to be dephosphorylated. Cis p-tau is stable and resistant to binding to microtubules, protein phosphatases, and degradation. In AD and TBI, cis p-tau is robustly accumulated and thereby causes and spreads tau aggregation, contributing to the development of neurodegeneration.
The term “tauopathy” was used for the first time in 1997 in the context of familial multiple system tauopathy with presenile dementia, a disease with abundant NFTs. Tauopathy is currently a collective designation of diseases, such as AD, Pick's disease, frontotemporal lobar degeneration (FTLD), progressive supranuclear palsy (PSP), and corticobasal degeneration, in which abnormal accumulation of phosphorylated tau (p-tau) protein in cell bodies is considered to be important in pathogenesis. Accumulated p-tau protein is observed as NFTs in neurons and glial cytoplasmic inclusions in astrocytes and oligodendrocytes. There are six isoforms of tau in the human brain (Figure 1B). Tau isoforms with three microtubule-binding domains, designated as 3-repeat tau (3R-tau), are generated by the splicing of exon 10 and are observed in Pick's disease (18), whereas tau isoforms with four microtubule-binding domains, designated as 4-repeat tau (4R-tau), accumulate in PSP and corticobasal degeneration (19). All 3R- and 4R-tau isoforms are contained in filamentous inclusions in AD, Down syndrome, CTE, and the amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam syndrome following TBI (details below) [(20–22); Table 1].

Table 1. Comparison of pathology between CTE and AD.
Pathological Comparison Between Tau in TBI and Alzheimer's Disease
Single TBI
One of the reasons why TBI has been considered to increase the risk of AD is that they share similar pathological features and a clinical course involving a dementing process. Two main pathological hallmarks of AD are the extracellular deposition of amyloid-β (Aβ) aggregates and hyperphosphorylated tau (37). Diffuse axonal injury (DAI) and Aβ deposition were identified in up to 30% of patients who died acutely following a single TBI (38–41); these findings were present even just a few hours after the TBI occurred (39). By contrast, several studies with a short observation period failed to identify tau pathology (40, 42). More recent studies, however, showed that up to a third of patients with even a single episode of TBI developed NFT pathologies, and there seemed to be an association between even a single TBI and the development of dementia (43, 44). Similarly, severe TBI in wild-type mice induced progressive tau pathology that spread to the contralateral side of the injury (43). A recent proteomics study comparing diffuse and focal TBI patients showed heterogeneity among the different subtypes of TBI (45). In that study, the presence of DAI caused larger global alterations in the cortical tissue than focal TBI, resulting in increased production of proteins related to neurodegeneration and reduced protein production related to antioxidant defense (45). These findings suggest that even a single TBI can induce progressive tau pathology for years after the initial injury, especially in the presence of DAI.
Repetitive TBI
Hyperphosphorylated tau aggregates and NFTs are often detected after TBI in combat-experienced military veterans and professional sports players at high risk of repetitive head injury, such as American football players, boxers, and wrestlers (6–10). These patients present clinically with mood and behavioral disorders and cognitive impairment known as CTE, previously termed “punch-drunk syndrome” or “dementia pugilistica” (7, 23, 46). Comprehensive analysis of post-mortem brains from individuals who experienced repetitive mild TBI revealed a strikingly high frequency of CTE (68/85 cases), with pathological changes distinct from those of other neurodegenerative diseases, including AD [Figure 1C; (8)]. As shown in Table 1, cavum septum pellucidum and enlargement of the lateral and third ventricles are often observed in CTE. Distinct diffuse cortical atrophy, which is characteristic of AD, is not common in CTE (47). Histologically, NFT is one of the most common pathological findings in CTE, and has been observed focally and perivascularly in the cerebral cortex, with a predilection for deep sulci in the superficial neocortical layers (layers II and III) [Figure 1C; (7–9, 24, 48)]. In the later stages of CTE, NFTs typically spread irregularly to the neocortex, medial temporal lobe, diencephalon, basal ganglia, and brainstem. By contrast, NFTs in AD first develop in the brainstem and entorhinal cortex (Braak stage I–II), then in the medial temporal lobe (Braak stage III–IV), and finally in the neocortex (Braak stage V–VI), where NFTs are evenly distributed in layers III and V (8, 25, 37, 49, 50). A previous study using 2-(1-{6-[(2-[F-18]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene) malononitrile ([F-18]FDDNP)-PET imaging, which identifies NFTs in living humans, demonstrated that professional American football players with suspected CTE had fibrillar insoluble protein aggregates in the brainstem white matter tracts, with axonal damage along subcortical and cortical brain areas. This radiological deposition pattern is consistent with paired helical filament (PHF)-tau distribution in the autopsy samples from mild TBI patients. Importantly, these pathological changes in TBI are not consistent with Braak stage in AD (51, 52). Given these findings, it is plausible that CTE may occur frequently in cases of repetitive TBI. Since CTE is characterized by the distinct distribution of hyperphosphorylated tau pathology seen in AD, we discuss the differences in tau levels in biofluids in these diseases, including the possibility of utilizing tau as a biomarker.
Tau Protein as a Biomarker
The clinical features of CTE are wide-ranging, and its diagnosis is made based on pathological findings in brain biopsy samples and at post-mortem examination. Therefore, prognostic biomarkers for CTE and TBI-related AD have been required.
The serum levels of total tau (t-tau) in concussed sports players increased in the acute stage following TBI, and decreased in the subacute stage (53). Similarly, serum p-tau (T231 and S202) and t-tau were elevated in severe human TBI and in a rodent model of repetitive mild TBI during both the acute and subacute periods (54–56). Acute elevation of both p-tau levels and the p-tau/t-tau ratio was found in TBI patients with poor outcomes (55). In addition, the recovery of serum t-tau to pre-injury levels correlated well with good outcomes (57). Elevations of plasma t-tau, p-tau (T231), and the p-tau/t-tau ratio were also observed in the chronic phase (6–18 months) following moderate-to-severe TBI and in military personnel with repetitive TBI (58, 59). While these data can help predict the short-term outcomes of TBI, they cannot clarify the long-term prognosis or identify CTE.
Stern et al. found that higher plasma exosomal tau levels in professional football players were associated with poor memory and psychomotor speed (60). Similar findings were seen in military personnel (61). Since exosomes are very stable, cross the blood–brain barrier, and reflect their cellular origin, exosomal tau may serve as an ideal predictor of CTE. The ultrasensitive tau seed amplification assay detected that seeding activity of tau aggregates in AD and CTE was markedly higher than in other tauopathies, such as Pick's disease, and seed concentrations in the brains of two CTE cases were comparable to the lowest concentrations in AD brains (62). These methods might be useful to distinguish CTE from AD.
Conformational Variations of Hyperphosphorylated Tau
In most TBI cases, tau phosphorylation in the brain occurs as early as a few hours after the injury and then spreads at a high density within a shorter period than in AD (44). The mechanism underlying the rapid protein accumulation following TBI remains unknown. The molecular profile and function of tau might be different between AD and other tau-related disorders, including TBI. Analysis of tau phospho-epitopes in AD, FTLD, PSP, and CTE has shown that each neurodegenerative disease has a specific profile of p-tau formation (63–66). In addition, the misfolding and hyperphosphorylation statuses of tau proteins depend on mutations or different isoforms of tau, which may result in phenotypic differences between AD and other tau-related disorders (67). For example, mutated FTLD-tau proteins change their conformation without hyperphosphorylation (16, 26), which has not been observed in AD (67, 68). Tau phosphorylation sites in AD have been identified on serine (Ser) or threonine (Thr) residues that precede a Pro residue (31, 32). Three sites of tau phosphorylation, namely, Thr231, Thr181, and Ser199, serve as biomarkers for AD (27, 33, 34). In particular, hyperphosphorylation of tau at Ser199/Ser202/Thr205 or Thr212/Thr231/Ser262 is sufficient to induce microtubule instability that results in cell death (33, 34). Additionally, tau phosphorylated at Tyr394 has also been reported in PHFs from AD brains (28). In CTE, phosphorylation at Thr175 and Ser422 has been reported in addition to that at Ser199and Thr231 (29–31). A study in mice reported that TBI triggered calpain-2 activation and resulted in increased tyrosine phosphorylation of kinase c-Abl at Tyr245, which enhanced its kinase activity and p-tau at Tyr394 (69). Other common phosphorylation sites (Ser199, Ser202/Thr205, and Ser396) were also found in both AD and CTE (70). Whole RNA sequencing analysis of post-mortem brain tissue revealed that the expression of PPP3CA, which encodes a calcium-dependent, calmodulin-stimulated protein phosphatase, was decreased in CTE compared to normal controls, and the PPP3CA protein level was inversely correlated to the p-tau (Ser202/Thr205) level (70).
The same tau epitopes were found to map to filamentous tau inclusions in CTE and AD brains, although the abnormal tau proteins from CTE brains did not overlap with the six abnormally phosphorylated tau isoforms in AD (25). Similarly, insoluble tau protein from CTE brains contained all six isoforms, while an AD case contained the three isoforms that comprise PHFs (31). A recent study showed that phosphorylation of tau at Thr231 enabled the cis-trans conformational change of p-tau [Figure 1D; (71)]. The cis conformation of p-tau appears in neurons within hours after TBI, prior to the formation of tau oligomers, pre-fibrillary tangles, and NFTs, and results in axonal disruption (71). In both a TBI mouse model and TBI patients, cis p-tau not only triggered neurotoxic effects, but also spread to other brain regions, including the hemisphere contralateral to the injury, causing cognitive impairment (71). These data suggest that cis p-tau functions as a driver of neurodegeneration. Indeed, p-tau with the cis conformation, but not the healthy, physiological trans form, is detectable in both CTE and AD patients (72). Since the molecular weight of insoluble p-tau (Ser199) differed between CTE and epileptic brains (29), tau conformation may differ depending on disease type. A more precise understanding of the structural diversity of tau might clarify the basis for the heterogeneity of tau-related pathologies and the molecular differences between CTE and AD.
Prion-Like Propagation of Tau Pathology in TBI
In prion disease, misfolded prion proteins act as seeds to initiate the misfolding and aggregation of the native prion protein (73). Eventually, the long polymers undergo fragmentation to release more seeds, which accelerate the rate of prion propagation (73). Tau pathology (especially 4R-tau) in AD appears to develop hierarchically along anatomical connections, which suggests cell-to-cell transfer of toxic tau through neuronal cell contacts (74, 75). Studies of animal models imply that tau transfer may be partly mediated by a prion-like templated misfolding of tau (65, 76–80). Indeed, inoculated misfolded cellular prion protein (PrPc) can promote Aβ aggregation in AD mice by cross-seeding (81), accelerating tau hyperphosphorylation (82, 83). As such, increased levels of total and phosphorylated tau following severe TBI were associated with PrPc production levels (58). Inoculation of brain homogenates from a mouse with severe TBI into the hippocampus and cerebral cortex of wild-type mice enhanced tau propagation and led to memory deficits; similar findings in the same study were observed using brain homogenates obtained contralateral to the side of the TBI, which supports a prion-like propagation mechanism of tau (43). Since misfolded tau aggregates in AD and CTE were replicated in cells expressing both 3R- and 4R-tau isoforms, but not cells expressing either 3R-tau or 4R-tau, the propagation properties of misfolded tau aggregates in AD and CTE are distinct from those in Pick's disease and PSP (22).
It is also plausible that hyperphosphorylated tau deposition promotes the accumulation of other aggregate-prone proteins. In addition to tauopathy, CTE is accompanied by other proteinopathies such as amyloidopathy and TAR DNA-binding protein 43 (TDP-43) proteinopathy (7–9), the latter of which was originally thought to be a specific marker for ALS that forms ubiquitinated inclusions (35, 36). TDP-43 inclusions were found in 85% of CTE cases [(8, 84, 85); Table 1]. Especially in the late stage of CTE, TDP-43 inclusions are severe in the cortex, white matter, diencephalon and brainstem, a distribution that is similar to that of NFTs. Partial co-localization of TDP-43 with tau was detected in the late stage of CTE (8). Like in AD, aggregated tau propagation in TBI might promote TDP-43 accumulation. However, it is not yet fully clear why the pathological tau distribution differs between these diseases.
Does TBI Accelerate the Development of Alzheimer's Disease?
A retrospective analysis of TBI patients demonstrated that the onset of AD was significantly earlier in those who survived from TBI (86, 87), especially in men (88). Examination of autopsy samples in a small cohort showed an AD prevalence of 22% in TBI patients, which was significantly higher than the 14% observed in the general population over age 70 (89). Since the concept of mild cognitive impairment (MCI) was accepted (90), it has been recognized that there is a link between TBI and the early diagnosis of MCI, although no significant association has been observed between TBI and the rapid progression from MCI to AD (91). Similarly, retired American football players with a history of at least three concussions demonstrated a 5-fold greater prevalence of MCI (92). Both moderate and severe TBI in military veterans were also associated with an increased risk of developing AD, with onset accelerated by 2 years (93–95). Accordingly, TBI is associated with increased risks of MCI and AD, which also accelerate AD development. Interestingly, Kanaan et al. have documented the mixed phenotype of AD and CTE (32), where both AD and CTE may be identified in the same patient. However, it remains unclear whether AD develops concurrently with CTE or if instead CTE induces AD.
Future TBI Treatment Prospects Considering TAU Pathology
There is a growing interest in pathogenic tau as a therapeutic target, since ablation or reduction of tau restores cognitive impairment following TBI (96, 97). Different tau kinases have been shown to control the binding ability of tau to microtubules (98). For example, when tau is first phosphorylated by glycogen synthase-3 or cyclin-dependent kinase-5, its binding ability to microtubules is inhibited by 79% (98). In addition, the combination of these kinases accelerates phosphorylation at Thr231 and Ser262. Replacement of these phosphorylation sites or inhibition of phosphorylation kinases has been attempted in vitro and in AD murine models (34, 99, 100). In TBI, the finding that calpain-2 activation by TBI accelerates tau phosphorylation at the Tyr394 site (28) suggests that inhibiting calpain-2 or its pathway may be a promising treatment strategy.
Given that cis p-tau is a precursor of tau pathology, proline isomerase Pin1, which converts p-tau from pathogenic cis to physiologic trans, may be a viable drug target (71, 101, 102). Remarkably, a neutralizing antibody for cis p-tau that acts both extracellularly and intracellularly facilitated tau binding to microtubules, restored normal tau function, and prevented tau spread, axonal damage, and neuronal degradation in mouse models of both AD and CTE (71, 102, 103). The cis to trans transition mediated by Pin1 also enhanced the degradation of p-tau (104, 105). In addition, therapeutic administration of the cis p-tau neutralizing antibody after TBI suppressed cis p-tau induction in a TBI mouse model (72). Further development of therapeutics for clinical application are expected.
Conclusion
We reviewed the distribution, molecular characteristics, and development of tau pathology in TBI, with a particular emphasis on CTE. Although the observed tau isoforms are common between AD and CTE, the brain lesions involving tau are distinct in both diseases, and each tau progression pattern is different. Conformational variance may influence the rapid tau development following TBI. Epidemiological studies have shown that TBI may be a risk factor for AD as well as CTE, and more interestingly, the mixed pathology of AD and CTE coexists in some TBI patients. However, the reasons why clinical and pathological outcomes differ between patients have yet to be fully elucidated. Further investigation is needed to provide an effective treatment for TBI.
Author Contributions
AK wrote the manuscript. HT and FT edited the manuscript.
Funding
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and grants from the Ministry of Health, Labor and Welfare of Japan (18K 07531).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AD, Alzheimer's disease; CTE, chronic traumatic encephalopathy; DAI, diffuse axonal injury; FTLD, frontotemporal lobar degeneration; MCI, mild cognitive impairment; p-tau, phosphorylated tau; TBI, traumatic brain injury; TDP-43, TAR DNA-binding protein 43.
References
1. McInnes K, Friesen CL, MacKenzie DE, Westwood DA, Boe SG, McInnes K, et al. Mild traumatic brain injury (mTBI) and chronic cognitive impairment: a scoping review. PLoS ONE. (2017) 12:e0174847. doi: 10.1371/journal.pone.0174847
2. Fann JR, Ribe AR, Pedersen HS, Fenger-Grøn M, Christensen J, Benros ME, et al. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry. (2018) 5:424–431. doi: 10.1016/S2215-0366(18)30065-8
3. Ding K, Marquez de la Plata C, Wang JY, Mumphrey M, Moore C, Harper C, et al. Cerebral atrophy after traumatic white matter injury: correlation with acute neuroimaging and outcome. J Neurotrauma. (2008) 25:1433–40. doi: 10.1089/neu.2008.0683
4. Warner MA, Youn TS, Davis T, Chandra A, Marquez de la Plata C, Moore C, et al. Regionally selective atrophy after traumatic axonal injury. Arch Neurol. (2010) 67:1336–44. doi: 10.1001/archneurol.2010.149
5. Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, et al. Traumatic brain injury and Alzheimer's disease: the cerebrovascular link. EBio Med. (2018) 28:21–30. doi: 10.1016/j.ebiom.2018.01.021
6. Dale GE, Leigh PN, Luthert P, Anderton BH, Roberts GW. Neurofibrillary tangles in dementia pugilistica are ubiquitinated. J Neurol Neurosurg Psychiatry. (1991) 54:116–8. doi: 10.1136/jnnp.54.2.116
7. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. (2010) 69:918–29. doi: 10.1097/NEN.0b013e3181ee7d85
8. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. (2013) 136:43–64. doi: 10.1093/brain/aws307
9. Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. (2011) 69:173–83; discussion 183. doi: 10.1227/NEU.0b013e318212bc7b
10. Blennow K, Brody DL, Kochanek PM, Levin H, McKee A, Ribbers GM, et al. Traumatic brain injuries. Nat Rev Dis Primers. (2016) 2:16084. doi: 10.1038/nrdp.2016.85
11. Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K. Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol. (2015) 77:987–95. doi: 10.1002/ana.24396
12. Crane PK, Gibbons LE, Dams-O'Connor K, Trittschuh E, Leverenz JB, Keene CD, et al. Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol. (2016) 73:1062–9. doi: 10.1001/jamaneurol.2016.1948
13. Gardner RC, Byers AL, Barnes DE, Li Y, Boscardin J, Yaffe K. Mild TBI and risk of Parkinson disease: a chronic effects of neurotrauma consortium study. Neurology. (2018) 90:e1771–9. doi: 10.1212/WNL.0000000000005522
14. Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat Rev Drug Discov. (2009) 8:783–93. doi: 10.1038/nrd2959
15. Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. (2010) 7:656–64. doi: 10.2174/156720510793611592
16. Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, et al. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. (2006) 281:1205–14. doi: 10.1074/jbc.M507753200
17. Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. (2014) 127:667–83. doi: 10.1007/s00401-014-1254-6
18. Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature. (2018) 561:137–40. doi: 10.1038/s41586-018-0454-y
19. Espinoza M, de Silva R, Dickson DW, Davies P. Differential incorporation of tau isoforms in Alzheimer's disease. J Alzheimers Dis. (2008) 14:1–16. doi: 10.3233/JAD-2008-14101
20. Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. (1992) 8:159–68. doi: 10.1016/0896-6273(92)90117-V
21. Cox K, Combs B, Abdelmesih B, Morfini G, Brady ST, Kanaan NM. Analysis of isoform-specific tau aggregates suggests a common toxic mechanism involving similar pathological conformations and axonal transport inhibition. Neurobiol Aging. (2016) 47:113–26. doi: 10.1016/j.neurobiolaging.2016.07.015
22. Woerman AL, Aoyagi A, Patel S, Kazmi SA, Lobach I, Grinberg LT, et al. Tau prions from Alzheimer's disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci USA. (2016) 113:E8187–96. doi: 10.1073/pnas.1616344113
23. Omalu B. Chronic traumatic encephalopathy. Prog Neurol Surg. (2014) 28:38–49. doi: 10.1159/000358761
24. Geddes JF, Vowles GH, Robinson SF, Sutcliffe JC. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol. (1996) 22:12–6. doi: 10.1046/j.1365-2990.1996.2598025.x
25. Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathol. (2001) 101:518–24. doi: 10.1007/s004010000330
26. Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. (2008) 28:737–48. doi: 10.1523/JNEUROSCI.2824-07.2008
27. Hampel H, Blennow K, Shaw LM, Hoessler YC, Zetterberg H, Trojanowski JQ. Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Exp Gerontol. (2010) 45:30–40. doi: 10.1016/j.exger.2009.10.010
28. Tremblay MA, Acker CM, Davies P. Tau phosphorylated at tyrosine 394 is found in Alzheimer's disease tangles and can be a product of the Abl-related kinase, Arg. J Alzheimers Dis. (2010) 19:721–33. doi: 10.3233/JAD-2010-1271
29. Puvenna V, Engeler M, Banjara M, Brennan C, Schreiber P, Dadas A, et al. Is phosphorylated tau unique to chronic traumatic encephalopathy? Phosphorylated tau in epileptic brain and chronic traumatic encephalopathy. Brain Res. (2016) 1630:225–40. doi: 10.1016/j.brainres.2015.11.007
30. Mufson EJ, Perez SE, Nadeem M, Mahady L, Kanaan NM, Abrahamson EE, et al. Progression of tau pathology within cholinergic nucleus basalis neurons in chronic traumatic encephalopathy: a chronic effects of neurotrauma consortium study. Brain Inj. (2016) 30:1399–413. doi: 10.1080/02699052.2016.1219058
31. Moszczynski AJ, Strong W, Xu K, McKee A, Brown A, Strong MJ. Pathologic Thr(175) tau phosphorylation in CTE and CTE with ALS. Neurology. (2018) 90:e380–7. doi: 10.1212/WNL.0000000000004899
32. Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC. Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. (2016) 75:19–34. doi: 10.1093/jnen/nlv001
33. Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, et al. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci. (2012) 32:2430–41. doi: 10.1523/JNEUROSCI.5927-11.2012
34. Alonso AD, Di Clerico J, Li B, Corbo CP, Alaniz ME, Grundke-Iqbal I, et al. Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J Biol Chem. (2010) 285:30851–60. doi: 10.1074/jbc.M110.110957
35. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. (2006) 351:602–11. doi: 10.1016/j.bbrc.2006.10.093
36. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. (2006) 314:130–3. doi: 10.1126/science.1134108
37. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
38. Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol. (1994) 87:55–62. doi: 10.1007/BF00386254
39. Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. (1994) 57:419–25. doi: 10.1136/jnnp.57.4.419
40. Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. (2007) 208:185–92. doi: 10.1016/j.expneurol.2007.06.018
41. Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. (2009) 19:214–23. doi: 10.1111/j.1750-3639.2008.00176.x
42. Smith C, Graham DI, Murray LS, Nicoll JA. Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. (2003) 29:496–502. doi: 10.1046/j.1365-2990.2003.00488.x
43. Zanier ER, Bertani I, Sammali E, Pischiutta F, Chiaravalloti MA, Vegliante G, et al. Induction of a transmissible tau pathology by traumatic brain injury. Brain. (2018) 141:2685–99. doi: 10.1093/brain/awy193
44. Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. (2012) 22:142–9. doi: 10.1111/j.1750-3639.2011.00513.x
45. Abu Hamdeh S, Shevchenko G, Mi J, Musunuri S, Bergquist J, Marklund N. Proteomic differences between focal and diffuse traumatic brain injury in human brain tissue. Sci Rep. (2018) 8:6807. doi: 10.1038/s41598-018-25060-0
46. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. (2005) 57:128–34; discussion 128–34. doi: 10.1227/01.NEU.0000163407.92769.ED
47. Gavett BE, Stern RA, McKee AC. Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med. (2011) 30:179–88, xi. doi: 10.1016/j.csm.2010.09.007
48. Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. (2015) 130:21–34. doi: 10.1007/s00401-015-1435-y
49. Hof PR, Bouras C, Buée L, Delacourte A, Perl DP, Morrison JH. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer's disease cases. Acta Neuropathol. (1992) 85:23–30. doi: 10.1007/BF00304630
50. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. (2011) 70:960–9. doi: 10.1097/NEN.0b013e318232a379
51. Barrio JR, Small GW, Wong KP, Huang SC, Liu J, Merrill DA, et al. In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc Natl Acad Sci USA. (2015) 112:E2039–47. doi: 10.1073/pnas.1409952112
52. Small GW, Kepe V, Siddarth P, Ercoli LM, Merrill DA, Donoghue N, et al. PET scanning of brain tau in retired national football league players: preliminary findings. Am J Geriatr Psychiatry. (2013) 21:138–44. doi: 10.1016/j.jagp.2012.11.019
53. Shahim P, Blennow K, Zetterberg H. Tau, s-100 calcium-binding protein B, and neuron-specific enolase as biomarkers of concussion-reply. JAMA Neurol. (2014) 71:926–7. doi: 10.1001/jamaneurol.2014.1160
54. Yang Z, Wang P, Morgan D, Lin D, Pan J, Lin F, et al. Temporal MRI characterization, neurobiochemical and neurobehavioral changes in a mouse repetitive concussive head injury model. Sci Rep. (2015) 5:11178. doi: 10.1038/srep15922
55. Rubenstein R, Chang B, Davies P, Wagner AK, Robertson CS, Wang KK. A novel, ultrasensitive assay for tau: potential for assessing traumatic brain injury in tissues and biofluids. J Neurotrauma. (2015) 32:342–52. doi: 10.1089/neu.2014.3548
56. Rubenstein R, Sharma DR, Chang B, Oumata N, Cam M, Vaucelle L, et al. Novel mouse tauopathy model for repetitive mild traumatic brain injury: evaluation of long-term effects on cognition and biomarker levels after therapeutic inhibition of tau phosphorylation. Front Neurol. (2019) 10:124. doi: 10.3389/fneur.2019.00124
57. Shahim P, Tegner Y, Wilson DH, Randall J, Skillbäck T, Pazooki D, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. (2014) 71:684–92. doi: 10.1001/jamaneurol.2014.367
58. Rubenstein R, Chang B, Grinkina N, Drummond E, Davies P, Ruditzky M, et al. Tau phosphorylation induced by severe closed head traumatic brain injury is linked to the cellular prion protein. Acta Neuropathol Commun. (2017) 5:30. doi: 10.1186/s40478-017-0435-7
59. Olivera A, Lejbman N, Jeromin A, French LM, Kim HS, Cashion A, et al. Peripheral total tau in military personnel who sustain traumatic brain injuries during deployment. JAMA Neurol. (2015) 72:1109–16. doi: 10.1001/jamaneurol.2015.1383
60. Stern RA, Tripodis Y, Baugh CM, Fritts NG, Martin BM, Chaisson C, et al. Preliminary study of plasma exosomal tau as a potential biomarker for chronic traumatic encephalopathy. J Alzheimers Dis. (2016) 51:1099–109. doi: 10.3233/JAD-151028
61. Gill J, Mustapic M, Diaz-Arrastia R, Lange R, Gulyani S, Diehl T, et al. Higher exosomal tau, amyloid-beta 42 and IL-10 are associated with mild TBIs and chronic symptoms in military personnel. Brain Inj. (2018) 32:1277–84. doi: 10.1080/02699052.2018.1471738
62. Kraus A, Saijo E, Metrick MA, Newell K, Sigurdson CJ, Zanusso G, et al. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. (2019) 137:585–98. doi: 10.1007/s00401-018-1947-3
63. Duka V, Lee JH, Credle J, Wills J, Oaks A, Smolinsky C, et al. Identification of the sites of tau hyperphosphorylation and activation of tau kinases in synucleinopathies and Alzheimer's diseases. PLoS ONE. (2013) 8:e75025. doi: 10.1371/journal.pone.0075025
64. Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. (2013) 154:103–17. doi: 10.1016/j.cell.2013.05.057
65. Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. (2014) 82:1271–88. doi: 10.1016/j.neuron.2014.04.047
66. Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. (2016) 92:796–812. doi: 10.1016/j.neuron.2016.09.055
67. Dujardin S, Bégard S, Caillierez R, Lachaud C, Carrier S, Lieger S, et al. Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol Commun. (2018) 6:132. doi: 10.1186/s40478-018-0637-7
68. Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, et al. Unique pathological tau conformers from Alzheimer's brains transmit tau pathology in nontransgenic mice. J Exp Med. (2016) 213:2635–54. doi: 10.1084/jem.20160833
69. Wang Y, Hall RA, Lee M, Kamgar-Parsi A, Bi X, Baudry M. The tyrosine phosphatase PTPN13/FAP-1 links calpain-2, TBI and tau tyrosine phosphorylation. Sci Rep. (2017) 7:11771. doi: 10.1038/s41598-017-12236-3
70. Seo JS, Lee S, Shin JY, Hwang YJ, Cho H, Yoo SK, et al. Transcriptome analyses of chronic traumatic encephalopathy show alterations in protein phosphatase expression associated with tauopathy. Exp Mol Med. (2017) 49:e333. doi: 10.1038/emm.2017.56
71. Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. (2015) 523:431–6. doi: 10.1038/nature14658
72. Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. (2017) 8:1000. doi: 10.1038/s41467-017-01068-4
73. Soto C. Transmissible proteins: expanding the prion heresy. Cell. (2012) 149:968–77. doi: 10.1016/j.cell.2012.05.007
74. Goedert M, Spillantini MG. Propagation of Tau aggregates. Mol Brain. (2017) 10:18. doi: 10.1186/s13041-017-0298-7
75. Dujardin S, Lécolle K, Caillierez R, Bégard S, Zommer N, Lachaud C, et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun. (2014) 2:14. doi: 10.1186/2051-5960-2-14
76. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. (2009) 11:909–13. doi: 10.1038/ncb1901
77. Kim W, Lee S, Jung C, Ahmed A, Lee G, Hall GF. Interneuronal transfer of human tau between Lamprey central neurons in situ. J Alzheimers Dis. (2010) 19:647–64. doi: 10.3233/JAD-2010-1273
78. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE. (2012) 7:e31302. doi: 10.1371/journal.pone.0031302
79. de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. (2012) 73:685–97. doi: 10.1016/j.neuron.2011.11.033
80. Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife. (2018) 7:e36584. doi: 10.7554/eLife.36584
81. Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, et al. Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases. J Neurosci. (2010) 30:4528–35. doi: 10.1523/JNEUROSCI.5924-09.2010
82. Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, et al. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. (2004) 24:2304–12. doi: 10.1523/JNEUROSCI.4162-03.2004
83. Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, et al. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci. (2012) 32:16857–71a. doi: 10.1523/JNEUROSCI.1858-12.2012
84. Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. (2007) 171:227–40. doi: 10.2353/ajpath.2007.070182
85. Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. (2007) 61:435–45. doi: 10.1002/ana.21154
86. Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, et al. Traumatic brain injury and time to onset of Alzheimer's disease: a population-based study. Am J Epidemiol. (1999) 149:32–40. doi: 10.1093/oxfordjournals.aje.a009724
87. Schaffert J, LoBue C, White CL, Chiang HS, Didehbani N, Lacritz L, et al. Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed Alzheimer's disease. Neuropsychology. (2018) 32:410–6. doi: 10.1037/neu0000423
88. Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. (2003) 74:857–62. doi: 10.1136/jnnp.74.7.857
89. Jellinger KA, Paulus W, Wrocklage C, Litvan I. Traumatic brain injury as a risk factor for Alzheimer disease. Comparison of two retrospective autopsy cohorts with evaluation of ApoE genotype. BMC Neurol. (2001) 1:3. doi: 10.1186/1471-2377-1-3
90. Petersen RC, Morris JC. Mild cognitive impairment as a clinical entity and treatment target. Arch Neurol. (2005) 62:1160–3; discussion 1167. doi: 10.1001/archneur.62.7.1160
91. LoBue C, Woon FL, Rossetti HC, Hynan LS, Hart J, Cullum CM. Traumatic brain injury history and progression from mild cognitive impairment to Alzheimer disease. Neuropsychology. (2018) 32:401–9. doi: 10.1037/neu0000431
92. Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Cantu RC, Randolph C, et al. Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery. (2005) 57:719–26; discussion 719–26. doi: 10.1227/01.NEU.0000175725.75780.DD
93. Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K. Traumatic brain injury and risk of dementia in older veterans. Neurology. (2014) 83:312–9. doi: 10.1212/WNL.0000000000000616
94. Barnes DE, Byers AL, Gardner RC, Seal KH, Boscardin WJ, Yaffe K. Association of mild traumatic brain injury with and without loss of consciousness with dementia in US military veterans. JAMA Neurol. (2018) 75:1055–61. doi: 10.1001/jamaneurol.2018.0815
95. Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et al. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. (2000) 55:1158–66. doi: 10.1212/WNL.55.8.1158
96. Cheng JS, Craft R, Yu GQ, Ho K, Wang X, Mohan G, et al. Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS ONE. (2014) 9:e115765. doi: 10.1371/journal.pone.0115765
97. Rosenmann H. Immunotherapy for targeting tau pathology in Alzheimer's disease and tauopathies. Curr Alzheimer Res. (2013) 10:217–28. doi: 10.2174/1567205011310030001
98. Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. (1998) 357:299–309. doi: 10.1006/abbi.1998.0813
99. Ando K, Maruko-Otake A, Ohtake Y, Hayashishita M, Sekiya M, Iijima KM. Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by Par-1/MARK contributes to augmentation of AD-related phosphorylation and Abeta42-induced tau toxicity. PLoS Genet. (2016) 12:e1005917. doi: 10.1371/journal.pgen.1005917
100. Stepanov A, Karelina T, Markevich N, Demin O, Nicholas T. A mathematical model of multisite phosphorylation of tau protein. PLoS ONE. (2018) 13:e0192519. doi: 10.1371/journal.pone.0192519
101. Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. (1999) 399:784–8. doi: 10.1038/21650
102. Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ. Potential of the antibody against cis-phosphorylated tau in the early diagnosis, treatment, and prevention of Alzheimer disease and brain injury. JAMA Neurol. (2016) 73:1356–62. doi: 10.1001/jamaneurol.2016.2027
103. Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell. (2012) 149:232–44. doi: 10.1016/j.cell.2012.02.016
104. Nakamura K, Kosugi I, Lee DY, Hafner A, Sinclair DA, Ryo A, et al. Prolyl isomerase Pin1 regulates neuronal differentiation via beta-catenin. Mol Cell Biol. (2012) 32:2966–78. doi: 10.1128/MCB.05688-11
Keywords: tau, traumatic brain injury, chronic traumatic encephalopathy, cis p-tau, prion
Citation: Katsumoto A, Takeuchi H and Tanaka F (2019) Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer's Disease: Similarities and Differences. Front. Neurol. 10:980. doi: 10.3389/fneur.2019.00980
Received: 21 March 2019; Accepted: 28 August 2019;
Published: 10 September 2019.
Edited by:
Leonardo Cruz de Souza, Federal University of Minas Gerais, BrazilReviewed by:
Parmenion P. Tsitsopoulos, Aristotle University of Thessaloniki, GreeceKevin K. W. Wang, University of Florida, United States
Copyright © 2019 Katsumoto, Takeuchi and Tanaka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hideyuki Takeuchi, aHRha2UmI3gwMDA0MDt5b2tvaGFtYS1jdS5hYy5qcA==; Fumiaki Tanaka, ZnRhbmFrYSYjeDAwMDQwO3lva29oYW1hLWN1LmFjLmpw