
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Neurol. , 13 August 2019
Sec. Stroke
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00871
 Hanne Stotesbury1*
Hanne Stotesbury1* Jamie M. Kawadler1
Jamie M. Kawadler1 Patrick W. Hales1
Patrick W. Hales1 Dawn E. Saunders1,2
Dawn E. Saunders1,2 Christopher A. Clark1
Christopher A. Clark1 Fenella J. Kirkham1,3,4,5
Fenella J. Kirkham1,3,4,5It is well-established that patients with sickle cell disease (SCD) are at substantial risk of neurological complications, including overt and silent stroke, microstructural injury, and cognitive difficulties. Yet the underlying mechanisms remain poorly understood, partly because findings have largely been considered in isolation. Here, we review mechanistic pathways for which there is accumulating evidence and propose an integrative systems-biology framework for understanding neurological risk. Drawing upon work from other vascular beds in SCD, as well as the wider stroke literature, we propose that macro-circulatory hyper-perfusion, regions of relative micro-circulatory hypo-perfusion, and an exhaustion of cerebral reserve mechanisms, together lead to a state of cerebral vascular instability. We suggest that in this state, tissue oxygen supply is fragile and easily perturbed by changes in clinical condition, with the potential for stroke and/or microstructural injury if metabolic demand exceeds tissue oxygenation. This framework brings together recent developments in the field, highlights outstanding questions, and offers a first step toward a linking pathophysiological explanation of neurological risk that may help inform future screening and treatment strategies.
Sickle cell disease (SCD) refers to a group of inherited hemoglobinopathies that affect ~20–25 million people globally (1, 2). The condition is caused by a single-base substitution that leads to the production of mutant hemoglobin type S (HbS). When oxygen tension is low, HbS polymerizes, giving erythrocytes their characteristic “sickle” shape. The wider pathophysiology is complex and appears to involve a cycle of inter-related processes, including erythrocyte-leukocyte adhesion to the endothelium, endothelial activation, hemolysis, inflammation, and hyper-coagulation (3–8).
In developed countries, medical advances have led to dramatically increased life expectancy for children with SCD (9). The transition from fatal disorder to chronic illness has, however, brought a new set of challenges with regard to the clinical complications that can have major implications for quality of life. Among the most debilitating and poorly understood complications are a number of conditions affecting the brain, including overt and silent stroke, cerebrovascular disease, cognitive impairment, and structural abnormalities (Figure 1). Below, we consider these in turn.
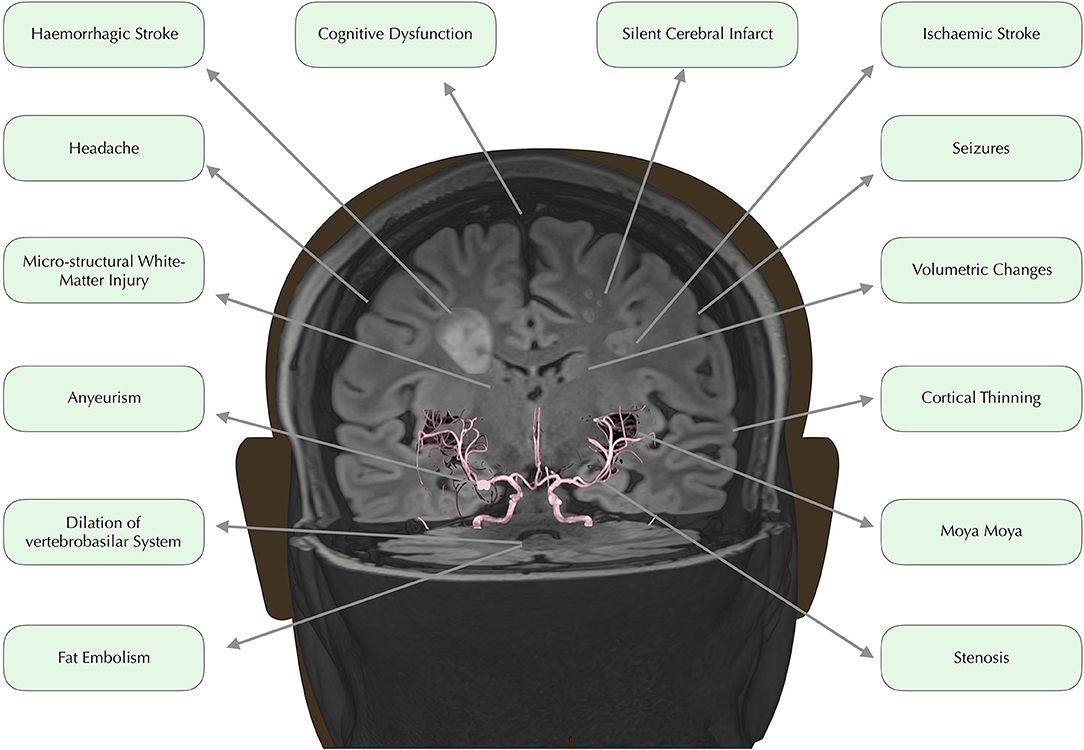
Figure 1. Neurological complications. Time of flight angiography image overlaid on 3D rendered Fluid Attenuated Inversion Recovery (FLAIR) image, edited to depict common neurological complications in SCD.
In the absence of screening and prophylactic treatment (10), ~11% of SCD patients will suffer an overt stroke by their 20th birthday, and 24% by their 45th (11). Ischemic insults are most common, accounting for up to 75% of SCD-related strokes (12, 13). Patients are however at considerable risk of both overt ischemic and hemorrhagic stroke, with the former reported more frequently in children, and the latter more frequently in young adults (14–16). In a recent cohort study, 10% of SCD patient deaths were attributable to overt stroke (17). Whilst overt ischemic stroke is rarely fatal, death may occur following 26% of hemorrhagic cases (11). Without secondary prevention, recurrence rates of up to 70% have been reported for overt ischemic stroke, with the risk greatest within 36 months of the initial event (18). Both types of overt stroke are associated with significant long-term morbidity, including seizures, physical disability, and cognitive impairment (19).
More common than overt stroke is “silent cerebral infarction” [SCI; (20)], where hyperintensities consistent with infarction/ischemia are apparent on brain MRI in the absence of focal neurological symptoms. SCI may occur as early as the 6th month of life (21, 22). There is evidence that prevalence reaches 25% by 6 years of age (23), 39% by 18 years of age (24), and 53% by young adulthood (25), with no reports of a plateau. Although clinically “silent,” evidence of progression was first provided by the co-operative study of SCD (CSSD), where SCI was associated with a 14-fold increase in risk of overt ischemic stroke, and 25% of children with SCI presented with new or enlarged lesions at follow-up (26). In the CSSCD, SCI was also associated with cognitive decline (27). These findings have been replicated in more recent work, including in a study where SCI in patients younger than 5 years old were shown to be associated with later progressive ischemia, vasculopathy, academic difficulties, and a higher risk of overt ischemic stroke (21). Further indicative of progressive ischemia, a recent clinical review of 60 unselected adult cases found that 37% of patients with SCI had more than one lesion (25).
Infarction in the territory of large intracranial vessels is the most common pattern in SCD patients with overt ischemic stroke, but the watershed regions of the deep white-matter are particularly vulnerable (16, 28, 29), whether or not there is concomitant intra-cranial cerebral vasculopathy (30). The distribution of SCI is similar, with up to 90% of SCI reportedly occurring in a relatively small deep watershed white matter region, encompassing only 5.6% of brain volume (31). SCI and overt ischemic stroke are often indistinguishable on MRI (32), and several authors have suggested that it may be differences in lesion size and location, rather than underlying physiological mechanism, that determines whether an ischemic insult is accompanied by focal symptoms (ischemic stroke) or goes undetected [SCI; (33)].
Both in addition to, and in the absence of, overt stroke and SCI, vasculopathy on MR angiography (MRA) is common in SCD patients (32). Although vasculopathy definitions have varied considerably between studies, intra- and extra- cranial steno-occlusive arteriopathy, often involving the distal internal carotid and the proximal anterior and middle cerebral arteries, are frequently reported, particularly in patients with overt ischemic stroke (34, 35), and SCI (24). Incidence of progressive stenosis with compensatory collateral vessel formation is as high as 30–40% in SCD patients with vasculopathy (36, 37). In a multi-center pediatric study in which 37 chronically transfused patients underwent serial MRI, 38% of patients presented with a new vessel segment of stenosis or occlusion at follow-up (38). Despite aggressive hematological management, the children with vasculopathy progression were also 12 times more likely to present with new SCI or overt ischemic stroke than those with no progression.
Some authors have proposed a sequential moyamoya-like model of SCD vasculopathy and stroke (39), in which early ischemic events are associated with stenosis, and later hemorrhagic events with the development and eventual rupturing of friable and maximally dilated collateral vessels. However, the majority of SCD-related intra-cerebral and subarachnoid hemorrhages are associated not with collateral vessel rupture, but with aneurysm rupture (40, 41). Intracerebral aneurysms are also prevalent in SCD patients (25, 41), and tortuosity and ectasia are well-documented in humans and animal models (42–45). Whilst aneurysms are not significantly associated with collateral vessel formation (46) they do appear to form in the context of progressive vasculopathy, with a majority of patients with aneurysms having more than one (47). In a recent clinical case review of children with SCD, five of seven patients with overt hemorrhagic stroke and/or aneurysm presented with evidence of overt ischemic stroke and/or SCI (48). These findings may indicate concurrent development of pathology underlying both ischemia and hemorrhage (49), with shared underlying mechanisms (50). Further support for this notion comes from the identification of a number of common, albeit non-specific, risk factors for both ischemic and hemorrhagic stroke, including anemia, chest syndrome, hypertension, and previous infarction (15, 51).
Overt stroke was originally identified as the primary cause of cognitive impairment in SCD (52). However, subsequent work has indicated that, whilst overt stroke and SCI are typically associated with the greatest impairment, cognitive difficulties may be common even in patients with no observable MRI abnormality (53, 54), manifesting as poorer school-readiness during the preschool years (55, 56), academic difficulties during childhood through adolescence (57–59), and employment difficulties during adulthood (60).
Already in infancy, up to 50% of patients show delay in early markers of cognition and expressive language (61). Throughout development, patients continue to be at risk of impairment across a range of domains including executive function, memory, and processing speed (27, 62–67). Although several authors have highlighted the need to consider SCD in the framework of a neurodevelopmental disorder (68), there have been no comprehensive longitudinal studies modeling raw cognitive trajectories over time. The extent to which later cognitive impairment is causally related to earlier developmental delay, and/or previous/ongoing pathophysiological processes, therefore remains unclear.
Quantitative MRI studies have indicated that the total extent of cerebral tissue injury may go beyond overt stroke, SCI, and large vessel disease in SCD. There have been reports of reduced cortical and subcortical gray matter volumes (69–72) as well as reduced subcortical white matter volumes (73–75). Abnormal patterns of brain maturation have also been described (76–78). Diffusion imaging studies have further revealed significant reductions in white matter integrity, with watershed regions of the centrum-semiovale consistently affected in SCI patients well as in those without MRI-defined lesions (79–83).
Several studies have provided evidence that volumetric and structural integrity alterations contribute to cognitive impairment in patients with and without SCI. Lower gray matter volumes have been associated with worse performance IQ in adults (72), with decline in FSIQ in children (84), and with memory impairment in mice (85). Moreover, decreases in white matter density (75) and reductions in white matter integrity (86), have been associated with worse performance on tests of processing speed, irrespective of presence of SCI. It is therefore possible that cerebrovascular disease represents only the “tip of the iceberg” in terms of functionally significant cerebral tissue injury in SCD.
Although the incidence and impact of neurological complications in SCD are well-described, the underlying mechanisms remain poorly understood. As a result, current treatment strategies are inadequate, with many patients continuing to suffer progressive vasculopathy and/or ischemia despite being on gold-standard transfusion regimes (38, 87). Co-existing and interdependent pathophysiological processes pose significant challenges to understanding the individual impact of each in SCD. Systems and network approaches, which focus on the relationship between processes, have not been comprehensively applied, but may be useful in combination with reductionist approaches in developing an understanding of the complex pathophysiology (88).
Taking a systems-biology approach to neurological complications, we propose a novel framework that emphasizes a role for vascular instability as a linking pathophysiological explanation for the various implicated mechanisms, including vaso-occlusion, hypercoagulability, thrombosis, hemolytic anemia, and hypoxia, as well as the interactions between them (Figure 2). According to this tentative framework, vascular instability is in part a result of operating at the limits of hemodynamic compensation for these physiological mechanisms. In this state, tissue oxygen supply is fragile and easily perturbed by relatively minor changes in clinical condition, with the potential for overt stroke, SCI, and/or micro-structural brain injury if metabolic demand exceeds tissue oxygenation. In the following sections, we review frequently implicated mechanisms, and demonstrate how the proposed framework is able to integrate them with the most current evidence in the field.
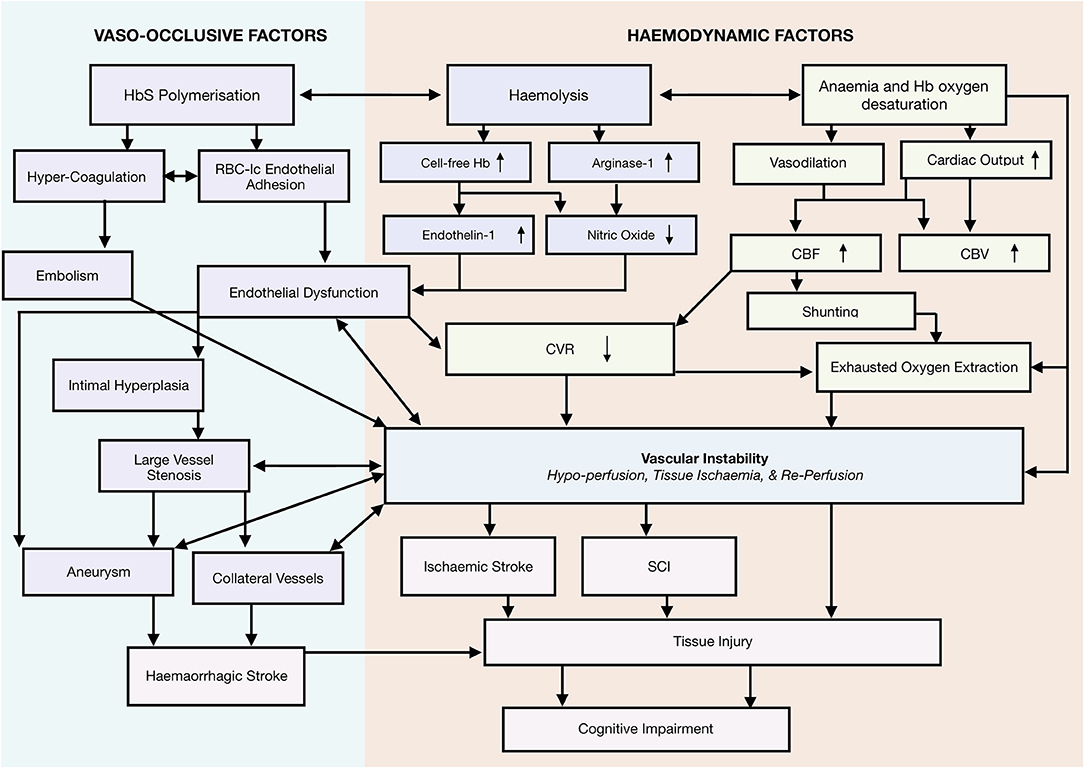
Figure 2. Systems biology framework. Proposed Model of neurological risk emphasizing role for vascular instability. Highlighting several potential mutually enforcing pathways. Different colors used to differentiate different mechanistic pathways, and to distinguish them from outcomes.
Vaso-occlusion was originally proposed to cause progressive vasculopathy and stroke, with erythrocyte adhesion, sickling, sludging, and congestion in small arterioles and venules. When it became clear that patients with SCD and overt stroke had large vessel disease, the suggestion that this process involved the vaso-vasorum network of feeders of large-vessels, gained traction as an explanatory mechanism for both macro- and micro-circulatory pathology (34). However, with the later discovery that large intracranial vessels lack a vaso-vasorum, the notion that vaso-occlusion alone is the proximate cause of macro-circulatory pathology has been challenged (50). Erythrocyte adhesion and congestion in post-capillary venules, with backward propagation and potential vascular pruning [i.e., regression; (89)], nevertheless remain an influential model of micro-circulatory pathology or “cerebral small-vessel disease” [CSVD; (90–93)].
Despite a lack of histological evidence, SCI, structural abnormalities, and cognitive impairment are often described as manifestations of CSVD, secondary to vaso-occlusive pathophysiology in SCD (94, 95). CSVD is typically regarded as a “whole-brain” disease, encompassing not only white-matter hyperintensities, but also other diffuse pathologies including silent micro-hemorrhages, white matter hyperintensities similar to SCI but in non-SCD populations, lacunar infarcts, and prominent perivascular spaces (96). However, the scanty available data do not suggest a high prevalence of silent microhemorrhages in children and young adults with SCD (97, 98). Moreover, whilst susceptibility-weighted MRI (SWI) of the brain has revealed patterns consistent with venular rarefaction in SCD patients (98), which may indicate vascular pruning (89, 93), concerns have been voiced about the potentially confounding effects of decreased hemoglobin and increased cerebral blood flow on SWI signal (98). Moreover, erythrocyte congestion, CSVD, and/or vascular pruning cannot alone account for the disproportionate vulnerability to injury of the deep watershed white matter regions in SCD (30, 31).
Models have also been proposed in which neurological complications are thought to occur as a result of downstream ischemia from progressive large-vessel vasculopathy (16). It has been postulated that endothelial damage, exacerbated by inflammation, hyper-coagulation, and erythrocyte-leukocyte adhesion to the endothelium, may play a cardinal role in progressive large-vessel vasculopathy and perhaps also in CSVD and capillary pruning (16, 51, 93). There is indirect clinical evidence in support, with studies reporting associations between risk of cerebral infarction and leukocyte count (99), leukocyte expression of L-selectin (100), and endothelial expression of VCAM1 variant(-1594) (101).
According to one model of large-vessel vasculopathy (50), endothelial dysfunction may either lead to a reparative response involving intimal thickening and smooth muscle-cell proliferation or to fragmentation of the elastic lamina, with the former resulting in vessel narrowing and the development of stenosis, and the latter in vessel wall dilation and aneurysm formation. It has been suggested that local rheology, shear-stress, and/or tissue characteristics may determine whether endothelial injury leads to focal narrowing or dilation (48). Although the precise mechanisms are not well-defined, this model is able to account for cases in which regional ischemic and hemorrhagic pathology develop concurrently (48).
However, whilst associated with an increased risk of ischemic events, there is evidence that intracranial vasculopathy alone is neither necessary, nor sufficient, for the development of overt ischemic stroke, SCI, reduced integrity, or cognitive impairment in SCD. Neuropathological (49), angiographic (34, 102), and MRA/I (30, 38, 71, 82, 103, 104) studies have consistently described cases of overt ischemic stroke and SCI both in the presence and absence of observable intracranial vasculopathy. Conversely there have been reports of intracranial vasculopathy in the presence and absence of SCI and overt stroke (104–106).
For example, in a large trial (n = 516) in which patients with prior overt stroke or abnormal transcranial-doppler (TCD) screening results were excluded, 84% of children with SCI showed no MRA evidence of intracranial vasculopathy at baseline, and 36% of those with MRA defined vasculopathy showed no evidence of SCI (104). At exit, only 1 of 15 patients with SCI recurrence had baseline vasculopathy (107). Similarly, in a medium-sized trial (n = 150) in SCD children with prior overt stroke, there was no consistent pattern of intracranial vasculopathy associated with SCI, and there were no consistent hematological biomarkers for SCI or vasculopathy (87). Reduced cortical thickness (71), sub-cortical volumes (69), white-matter integrity, and cognitive performance (82, 86) are also well-documented in patients without MRI-defined lesions or MRA-defined vasculopathy. Whilst these studies are plagued by highly heterogeneous samples and use of inconsistent vasculopathy definitions (32), these and other findings have nevertheless encouraged authors to explore alternative etiologies for neurological morbidity in SCD (108).
There is indirect evidence that cerebral embolic events occur in SCD patients, including reports of associations between overt ischemic stroke and thromboemboli (109, 110) as well as of fat-embolism syndrome from bone-marrow necrosis (111–114). Although comprehensive prevalence data are lacking, shunting at intra-pulmonary or intra-cardiac (e.g., through a PFO; patent foramen ovale) level and paradoxical embolism may also be more common in children (33, 115, 116) and adult (117) SCD patients and may be associated with cerebral infarction (118). Although there are few data in SCD, PFO is an established risk-factor for overt stroke in the general population (119–121).
Hypercoagulability may pre-dispose to cerebral thromboembolism and is also a feature of SCD. Activation of the coagulation cascade and fibrinolysis are favored (122) and there is a high risk of venous thromboembolism (123). Risk factors may include genetic predisposition (124), inflammation (122), and splenectomy (125). Phosphotidylserine exposure on red cells and microparticles may play a role, related in part to acquired protein S deficiency (126). Whole blood thrombin generation is increased in SCD, while plasma thrombin generation is decreased, suggesting a cellular component, although this does not appear to be related to phosphotidylserine exposure (126). There is cross-sectional evidence indicating that hypercoagulability may contribute to risk of overt stroke and SCI in SCD patients (127, 128). Proteomic analyses have revealed associations between SCI and the prothrombotic proteins α2-antiplasmin, fibrinogen-γ chain and thrombospondin-4 which are considered to predispose to hypercoagulability (124). Although findings have been mixed, several studies have also shown lower serum levels of coagulation markers [e.g., D-Dimer, Von Willebrand factor, TAT complex; (129)] and lower thrombin generation (130) in SCD children deemed at low risk of overt stroke on the basis of transcranial doppler (TCD) velocities (see page 10). In addition, poor splenic function is associated with SCI (99), although the link to hypercoagulability has not been made for SCD as it has for thalassemia (131, 132).
Upregulation of platelets may exacerbate the hypercoagulable as well as the proinflammatory state associated with SCD. Platelets may also promote endothelial activation and erythrocyte adhesion by stimulating several major endothelial adhesion molecules, including vascular adhesion molecule (VCAM-1) (133) and by forming an increased number of platelet-erythrocyte (134, 135), platelet-monocyte (136) and platelet-neutrophil aggregates (136). Although the underlying mechanisms are unclear, there is cross-sectional evidence that patients with SCI or overt ischemic stroke have higher mean platelet values than patients without lesions on MRI (137). Also, thrombocytosis (platelets > 500*109/L) is associated with cognitive impairment across multiple domains in children with SCD (138), and elevated levels of erythrocyte and platelet derived microparticles have been described in those with a history of overt stroke (139). Whilst there is also evidence that higher mean platelet volume is associated with a global increase in white matter volume in SCD patients, further work is required to determine whether this is adaptive or a reflection of edema (74). Given these data, there is a good case for further investigation into the relationships between platelet activation, hypercoagulability, and neurological complications in SCD. Mechanisms could include embolism through a cardiac or pulmonary shunt from the systemic venous circulation e.g., in the pelvis or the limbs, as well as local thrombosis.
Whilst vaso-occlusive, thrombotic, and/or embolic events may contribute to some ischemic insults in SCD, several authors have argued that the high density of overt and silent infarction and microstructural abnormalities in watershed regions may point to hemodynamic compromise or “brain drain” as a more common contributor (32, 140). Historically, in non-SCD patients, watershed infarcts have been associated with hemodynamic causes, and are sometimes referred to as hemodynamic strokes (141, 142). As the watershed regions lie at the end junctions between adjacent arterial territories, vascular supply is inherently low. Much as the last field on a farm is the area with the least supply of water and therefore the most vulnerable to a reduction in flow, the watershed regions of the brain are believed to be the most vulnerable to a reduction in perfusion (143).
Despite only accounting for 2% of total body weight, the brain has the highest metabolic requirements of any organ, consuming a disproportionate 20% of the body's total oxygen supply. In children, brain oxygen consumption is even higher, reaching 50% during the first decade of life (144). As reflected by these high demands, a baseline cerebral metabolic rate of oxygen utilization (CMRO2) is required to maintain tissue viability (145).
CMRO2 is defined as the product of arterial oxygen content (CaO2), rate of blood delivery (CBF; Cerebral blood flow), and the percentage of oxygen extracted by the tissue (Oxygen extraction fraction; OEF).
The following equations, derived from the Fick principle show their relationship;
Where 1.34 is the oxygen affinity of normal hemoglobin type A, paO2 is the partial pressure of oxygen in arterial blood, SaO2 is the ratio of oxygenated hemoglobin to the sum of oxygenated and deoxygenated hemoglobin in arterial blood, and CvO2 is venous oxygen content (CvO2) defined similarly to CaO2, but with metrics drawn from venous rather than arterial blood.
In normal vascular physiology, CBF is closely coupled to baseline CMRO2, leading to globally uniform OEF (146, 147). By arteriolar dilatation, CBF increases in response to increased metabolic demand related to function, e.g., movement of a limb or response to a visual stimulus. Under conditions in which oxygen delivery is decreased [e.g., hypoxia; (148), carotid artery occlusion; (146)] or CMRO2 is increased beyond normal functional demands [e.g., pyrexia or seizures; (149)] the brain is able to fall back on two reserves; a cerebrovascular dilatory reserve (CVR) and a metabolic reserve. CVR reflects the capacity of smooth muscles to alter vessel caliber in response to fluctuations in arterial blood gases such as carbon dioxide and oxygen (150). The arterioles respond to changing carbon dioxide tension with a positive linear response across the physiological range but flattening at the extremes (151). Although the underlying mechanisms are less well-understood, the metabolic reserve reflects the capacity of the brain to augment CMRO2 via increases in OEF, which may potentially involve changes in effective oxygen diffusibility (152).
In models of hemodynamic stroke there is a disproportionate drop in oxygen delivery relative to baseline CMRO2, and an exhaustion of vascular reserve mechanisms (141). Within the first 48 h of an ischemic insult, a state of hemodynamic compromise known as “misery perfusion” is often observed, involving reductions in regional CBF that are accompanied by increases in regional OEF. Regional OEF increases may serve to maintain CMRO2 up to a point, beyond which tissue injury may ensue (146, 153–155).
There have been reports of hemodynamic changes consistent with a similar model of hemodynamic compromise in patients with SCD, including altered CaO2, CBF (156–161), CVR (162–164), and OEF (165–167). Whilst vaso-occlusion, vasculopathy, and emboli are all flow-restricting phenomena that may contribute to hemodynamic compromise, some hemodynamic changes may represent compensatory responses to physiological stressors associated with SCD pathophysiology (140), including anemia and hypoxia. In the following subsections, we consider research on aspects of CMRO2 in SCD in turn.
In patients with SCD, hemolytic-anemia leads to chronic hemoglobin-driven reductions in CaO2 (6). CaO2 may, however, be further reduced in these patients due to acute, intermittent, and/or chronic daytime, nocturnal, and/or exercise-induced oxyhemoglobin desaturation. Daytime oxyhemoglobin desaturation, when defined by pulse oximetry as SpO2 < 96%, may affect between 30 and 50% of steady-state patients (168–173).
Although a well-described phenomenon, there is no consensus on cause, definition, or treatment of oxyhemoglobin desaturation in SCD (174). Proposed mechanisms include phenomena often considered “hypoxemic” such as abnormal HbS oxygen affinity, elevated levels of dyshemoglobins, and pulse oximeter calibration for HbA rather than HbS (175, 176). “Hypoxic” phenomena, including obstructive and restrictive lung disease, sleep disordered breathing, and shunting, have also been proposed to play a role (176, 177).
The affinity of hemoglobin for oxygen is a fundamental determinant of the oxygen-carrying capacity of blood and is altered in patients with SCD. HbS polymerization has long been known to reduce oxygen affinity, causing a right shift of the oxyhemoglobin dissociation curve [ODC; (178–181)]. Although there is significant heterogeneity, the pO2 at which hemoglobin is 50% saturated (P50) is increased in a majority of SCD patients, meaning that hemoglobin oxygen saturation for any given pO2 is lower (171, 182–185). Whilst this right shift of the ODC is seen in many anemia's, and facilitates unloading of oxygen from blood to tissue (see section below on OEF), it inhibits oxygen loading at the lungs, which may promote oxyhemoglobin desaturation in SCD (186).
Studies using near-infrared spectrophotometry have provided evidence that cerebral oxyhemoglobin tissue desaturation is common and can be severe in steady-state SCD patients (187–189). However, oxygen carrying capacity appears to only partially explain cerebral desaturation, with CaO2, age, and male gender together accounting for 40% of the variance (188).
Cerebral tissue oxygenation is dependent not only upon oxygen availability and the blood's oxygen carrying capacity, but also on tissue perfusion. Whilst CaO2 is chronically decreased in SCD patients, studies have consistently reported compensatory vessel dilation (190), leading to increases in global CBF and CBV (157, 166, 191, 192), which appear to maintain oxygen delivery and metabolism when averaged globally (157, 167).
However, in patients with SCD, compensatory increases in global CBF are associated with reduced CVR (70, 162–164, 193), with the white matter also exhibiting disproportionate delays in CVR response times (194). There is evidence that a majority of patients may approach the upper limit of dilatory capacity, and that a quarter may also exhibit negative reactivity or “steal” (193). Steal refers to blood being “stolen” from one cerebral region and given to another, and occurs when a pressure gradient exists between regions, such as when one region is maximally dilated and unable to respond to a vasodilatory stimulus [e.g., hypercapnic challenge; (195)]. In these instances, blood may be redistributed from regions unable to dilate to regions that are able to. Theoretically, therefore, in a parallel vascular system where there is CVR exhaustion, an increase in perfusion in one region can lead to a relative decrease in perfusion in another.
Studies in healthy populations suggest that some brain regions are more vulnerable to CVR exhaustion and steal than others. CVR appears to be greater in gray matter than white matter (196, 197), as well as in phylogenetically older than phylogenetically younger gray-matter regions of the brain (198, 199), which may be related to the relatively greater vascularization in gray matter regions that perform essential homeostatic functions (198). There is also evidence that watershed white matter regions are disproportionately at risk of steal in young healthy populations during hypercapnia (196), suggesting that these regions may be continuously compensating for low perfusion pressure. Exhausted CVR, alone or in combination with steal, may thus render the watershed white matter regions disproportionately vulnerable to ischemia in settings where there is increased metabolic demand (e.g., infection, pyrexia, seizures) or an acute drop in CaO2 [e.g., acute chest syndrome with acute anemia and hypoxia; (33)], which are common in SCD.
Whilst several MRI studies suggest that global white matter CBF is on average elevated in “steady-state” SCD patients (158, 166, 200), the elevation is lower than that observed for gray matter, and may therefore be insufficient to maintain oxygen delivery regionally. Results from a more recent study are consistent with this notion, and suggest that global white-matter oxygen delivery is significantly reduced in “steady-state” SCD patients without MRA defined vasculopathy compared to controls (201). Using a rigorous partial volume correction, the authors found significantly elevated global gray-matter CBF in patients, but no differences in global white-matter CBF, indicating inadequate compensatory vasodilation in white-matter. Importantly, through t-score maps, the authors showed the reduction in white matter oxygen delivery to be disproportionate in watershed regions vulnerable to SCI and reduced integrity, going beyond that expected due to anatomical constraints and the watershed effect alone (201). These findings suggest that watershed regions are hypo-perfused in SCD patients, and highlight the need for future studies to consider regional perfusion characteristics alongside global averages. In line with this, Positron Emission Tomography [PET; (161, 192, 202)], Single Photon Emission Computed Tomography [SPECT; (203–205)], Xenon-Computed Tomography [CT; (206)] and MRI studies (158, 160, 201) have also reported regions of hypo-metabolism and/or hypo-perfusion in patients with SCD.
The etiology of regional hypo-perfusion in SCD is unclear. In the absence of longitudinal data, it is impossible to determine whether injury in these regions occurs secondary to hypo-perfusion, or whether hypo-perfusion is secondary to the lower metabolic requirements of injured tissue. Given that CBF and CMRO2 are closely coupled, it is possible to speculate that injured regions have lower CMRO2, resulting in hypo-perfusion. However, SCI-burden was relatively low in the Chai et al. (201) sample, with only half of patients showing small lesions. Moreover, there is evidence that even in “steady-state” patients without SCI, PET may find regions of hypo-metabolism and hypo-perfusion (161). Whilst CVR-exhaustion and vascular steal secondary to compensatory increases in gray matter CBF is another plausible explanation, it is also possible that there is a broader vulnerability of vasculature regulation in SCD.
Cerebral autoregulation (CA) refers to the ability of the brain to maintain relatively constant CBF over a broad range of cerebral perfusion pressures (CPP), and is thought to involve a complex interplay of autonomic, myogenic, and neuronal mechanisms (207).
Cerebral perfusion pressure (CPP) is defined as either:
Where MAP is the mean arterial pressure and JVP is the jugular venous pressure.
If blood pressure decreases or increases, CA maintains constant CBF across the autoregulatory range which varies with age and a variety of conditions. Below this range, CBF falls with decreasing CPP, risking ischemia, particularly in the watershed regions. Above this range, CBF rises with increasing CPP, with the risk of edema, particularly in the posterior circulation. There is some evidence indicating impaired CA in SCD patients, with one study showing that patients have a reduced capacity to buffer the transfer of blood pressure surges to the cerebral tissue (208). Whilst CA has traditionally been treated as separate from CVR, both are mechanisms deployed to ensure CBF-CMRO2 coupling in the face of changing physiological conditions, and there are persuasive data indicating synergism and interdependence between them (150, 209, 210). For example, progressive hypotension appears to blunt and abolish the CBF response to hypo and hypercapnia (151, 211), and hypoxia and hypercapnia appear to reduce the ability of the brain to defend against changes in perfusion pressure as well (212, 213), suggesting that CVR and CA may rely on the same underlying flow reserve.
The role of reduced CPP secondary to intra/or extracranial vasculopathy (24, 35), diastolic dysfunction (214), relative systemic hypotension (215), and/or embolism, has received relatively little attention in SCD, but any effect may be compounded by CVR exhaustion. Some SCD patients may thus face a “quadruple jeopardy” of reduced CaO2, systemic hypotension, CBF restricting stenosis/emboli, and exhausted CVR (140). It is unclear whether low flow conditions are further exacerbated by increases in JVP secondary to erythrocyte congestion in post-capillary venules, and/or increases in ICP secondary to acute drops in CaO2 and cerebral oedema [e.g., in acute hypertension; (216) or hypoxia; (217)]. However, it is possible that critical closing pressure, the CPP at which vessels collapse and close completely, is reached during acute illness with relatively small increases in either ICP or JVP or reductions in MAP.
Both CVR and CA must necessarily rely on the same underlying capacity for cerebral vessels to regulate resistance (150), a capacity which is modulated by local metabolites, RBC chemistry, the autonomic nervous system, and blood rheology, all of which are abnormal in SCD (218). Vessel caliber is ultimately dependent on the balance between the myriad of vaso-constricting and vaso-dilating agents derived from the endothelium, neuronal innervations, and physical factors such as shear and stretch (219). Evidence from forearm and renal studies suggests that the vaso-active balance is inherently vulnerable in SCD patients, with concomitant upregulation and exhaustion of vaso-constricting and vasodilating agents (220). For example, low nitric oxide (NO) bioavailability occurs secondary to hemolysis in patients with SCD, and given that NO is a powerful vasodilator that also inhibits the vaso-constrictive effect of endothelin-1, this may increase reliance on other agents and tip the balance in favor of vaso-constriction once alternative agents are exhausted (91, 221).
Although it is unclear how this plays out in the cerebral circulation in SCD, and the molecular mechanisms underlying CVR and CA remain the subject of much debate (150, 222), studies in animals and humans suggest that endothelial NO may play a role in moderating CVR as well as in extending the lower limit of CA (223, 224). In endothelial nitric oxide synthase knockout mice, for example, there is a substantial rightward shift of the CA curve at low perfusion pressures (225). Whilst a right shifted CA may protect the brain from brain-barrier disruption secondary to hyper-perfusion, it may also mean that a higher perfusion pressure is required to prevent hypo-perfusion.
Of note, impaired CA (226–228) and reduced CVR (229, 230), have also been observed following sympathetic stimulation in animals and humans. There is evidence for autonomic nervous system dysfunction in SCD, with enhanced sympathetically mediated vasoconstriction reflexes (218, 231–234), which theoretically, could compound any effect of reduced NO. Although there are no data comparing CVR, CA, and the interaction between them in SCD, a vulnerability in the availability of regulatory agents, either alone or in combination with autonomic nervous system dysfunction, may mean that normal CVR and CA ranges are right-shifted and/or narrower with loss of the plateau. Coupled with the inherent anatomical vulnerability of watershed white matter regions, reduced and/or altered regulatory capacity may further predispose SCD patients to hypoperfusion and/or oxygen supply-demand mismatch in these regions.
Theoretically, in patients with higher hematocrit, either naturally or as a result of transfusion, the increased viscosity of blood containing HbS could exacerbate hypo-perfusion in low-shear watershed regions (51, 235, 236). There is in-vitro evidence, including in patients with SCD, suggesting that a lower hematocrit to viscosity ratio (HVR) measured at high shear rate is associated with poorer cerebral oxyhemoglobin saturation as measured by NIRS (237). However, HVR is a measure that confounds CaO2 and viscosity, meaning that it could be low secondary to either low CaO2 or high viscosity. Moreover, studies in animals using high and low viscosity replacement fluids (238) as well as in humans with other anemia's, polycythemia, and paraproteinemia (148, 239), suggest that once any differences in CaO2 are accounted for, the impact of viscosity on global CBF is negligible. These findings have been replicated in other populations with normal vascular function and hematocrit during isovolumic conditions (240). This apparent contradiction of Poiseuille's law may relate to the physiological conditions of the cerebral vasculature, with turbulent flow, non-Newtonian fluid, and atuoregulation of vessel caliber (224). However, if vessels are less able to dilate in SCD patients, either due to CVR exhaustion secondary to reduced CaO2 and/or a broader vulnerability in regulatory capacity, the ability to compensate for increases in viscosity and/or reductions in deformability may be reduced. Whilst there is some data indicating no independent effect of blood viscosity on global CBF in patients with SCD (157), further work is required to examine the effects of viscosity and deformability in both low and high shear regions of the brain.
Of note, hypo-perfusion reduces shear-stress, and there is evidence that endothelial cells exposed to low-shear conditions show sustained activation of adhesion molecules, tissue factors, and inflammatory agents, as well as decreased production of nitric oxide (241, 242). Hypo-perfusion may also increase the risk of thrombus formation secondary to platelet-aggregation (243). Interestingly, murine studies have demonstrated that pre-conditioning via prior exposure to ischemia can be neuroprotective by reprogramming the genetic response to ischemia, with adaptations including the suppression of thrombus formation (244). Presence or absence, or even the degree and timing, of pre-conditioning may be relevant in determining the nature of acute neurological presentations in SCD where patients are at risk of chronic sustained and intermittent exposure to hypoxia (245).
There is also evidence that high and turbulent shear-stress, which may occur secondary to hyper-perfusion and reduced CaO2, can induce angiogenesis and vascular remodeling (241, 246, 247). Hypoxic exposure may additionally promote angiogenesis through several non-mechanic endothelial pathways (248), although these may be perturbed in SCD. Nevertheless, reports indicate that patients with SCD display a heightened “angiogenetic tone,” with elevated levels of proangiogenic growth factors, which in combination with endothelial dysfunction, could contribute to vasculopathy (4). Taken together, these findings illustrate how mutually enforcing pathophysiological processes may be at play, and suggest that, depending on the extent of any pre-conditioning (e.g., via prior exposure to hypoxia), both global hyperperfusion and regional hypoperfusion could in turn exacerbate erythrocyte-leukocyte adhesion, hypercoagulation, endothelial dysfunction, and vasculopathy (249).
Reports that OEF is abnormal (165, 167), particularly in watershed regions prone to SCI (166), are further indicative of hemodynamic compromise and regional vulnerability in SCD patients (250). There is evidence that changes in global OEF are associated with increases in global CBF, but that only changes in OEF are related to higher levels of clinico-radiological impairment, defined as moderate stenosis >50% in any major vessel, prior overt stroke or SCI, and/or chronic debilitating pain (167). Although there is controversy as to whether oxygen extraction is higher (166, 167, 251) or lower (165, 252), both patterns would be consistent with on-going metabolic stress, with higher or lower OEF potentially either reflecting compensation for, or exacerbation of, hemodynamic compromise.
These paradoxical findings may be explained in the context of preliminary reports of venous hyperintensities on arterial-spin labeling MRI, consistent with arterio-venous shunting (253). One theory, named the “functional shunting hypothesis” (254), postulates that regional shunting pathophysiology, coupled with compensatory increases in global CBF and reductions in arterial transit times and CVR, lead to regions of impaired oxygen unloading and diffusion in SCD, reflected by regional reductions in OEF. In these instances, tissue oxygen delivery may be compromised even though differences between arterial and venous saturation are small. Such shunting could be compensatory in terms of minimizing HbS polymerization and/or metabolic demand [e.g., hibernation; (165)], but could also be a dysfunctional consequence of macrocirculatory hyperperfusion in the setting of reduced and/or shifted CA.
The functional shunting hypothesis has been challenged, however, with one study finding no relationship between venous hyperintensities and global OEF (253, 255). Part of this controversy stems from the need for calibration models when using oxygen-sensitive MRI techniques. In SCD patients, T2 relaxation under spin tagging (TRUST) can yield diametrically opposing results depending on data calibration model [HbA vs. HbS calibration; (165, 256)], and there is no consensus on model validity (255). As a result, global CMRO2 in patients with SCD has been reported to be higher (166, 257, 258), lower (165, 252), and similar (167) to that of controls.
Reports of higher global OEF are broadly consistent with a previous PET study (192) and have been established using two different MRI methods [asymmetric spin echo—(166); TRUST with HbA calibration–(167, 258)]. However, both depend on broad assumptions that may not be valid in SCD patients (165, 259), as demonstrated by a recent study employing a novel susceptibility-based technique, where venous oxygen saturation was found to be elevated in SCD patients, consistent with lower global OEF (259). Further complicating matters, it is possible that there are regional differences in OEF and transit times, or thresholds beyond which increases in CBF begin to impair oxygen unloading. Whilst further work is required to determine whether OEF is higher, lower, or spatially heterogenous in SCD patients, the available data are nevertheless indicative of OEF exhaustion and/or insufficiency, consistent with hemodynamic compromise, and likely exacerbated by hypoxic and anemic exposure.
Of note, shunting pathophysiology has been described in other vascular beds in SCD, including the peripheral [Upper arm; (260)] and pulmonary (118) circulations. Whilst the similarities and differences between vascular beds of various organs have received little attention and are poorly understood, the cardiac description of a “superimposed restrictive and hyperdynamic physiology” (261), and the renal, fingertip, and skeletal muscle descriptions of a “perfusion paradox” (93, 220), are similar to the hemodynamic changes observed in the cerebral circulation, with hyper-perfusion in the macro-circulation, hypo-perfusion in watershed regions of the micro-circulation, and an exhaustion of vascular reserve mechanisms. These findings are suggestive of a state of vascular instability, in which tissue oxygen supply is fragile, and easily perturbed by fluctuations in clinical condition.
Taken together, the reviewed mechanisms are consistent with a tentative systems-biology framework of neurological morbidity with vascular instability at its core (Figure 2). According to this tentative framework, increases in CBF, reductions in CVR, and exhausted/insufficient OEF, may act synergistically to cause vascular instability (Figure 2), a state in which risk of regional hypoperfusion, ischemia, re-perfusion, and the associated inflammatory milieu are high. These factors may contribute either alone, or in combination with acute drops in CaO2, vasculopathy, erythrocyte congestion, and/or thrombo-emboli to perturb tissue oxygenation, leading to overt stroke, SCI, or microstructural tissue injury (e.g., reduced integrity). In this tentative framework, it is differences in the severity, duration, and precise location of a hypoxic-ischemic or hemorrhagic insult, that determine structural and functional tissue outcome.
Importantly, vascular instability provides a linking pathophysiological explanation for the various implicated processes, including vaso-occlusive, coagulative, thrombotic, hypoxic, and hemolytic phenomena, as well as the interactions between them. The framework is consistent with a previous systems-biology model of systemic vasculopathy in SCD, in which ischemia-re-perfusion injury and inflammation are emphasized, along with multiple overlapping and mutually-enforcing mechanistic pathways (88). The current neurological model similarly attempts to provide a parsimonious account of neurological risk and morbidity, in which multiple potential pathways are highlighted, but the most proximate mechanism is emphasized. Below, we consider evidence consistent with the framework.
There are many strands of indirect clinical evidence broadly consistent with a role for vascular instability in neurological morbidity, with decreased hemoglobin and peripheral oxygen saturation, components of CaO2, consistently associated with overt stroke (11, 262, 263), SCI (20, 23, 24, 99, 264), reduced white matter volume and tissue integrity (81, 82), and cognitive impairment (86, 265–267). Several case-series have highlighted acute chest syndrome in patients presenting with overt ischemic stroke in the absence of intracranial large-vessel vasculopathy (11, 103, 268), which may indicate a role for reduced oxygen delivery and hemodynamic failure (269).
Moreover, TCD, which captures the time averaged mean of the maximum velocity of blood, can be high as a compensatory mechanism for reduced CaO2 (270, 271) rather than vessel narrowing, and there is evidence that up to 79% of SCD children with high TCD have either no stenosis or <25% stenosis (272). In an analysis of the STOP trial data, only 2 out of 6 high TCD patients who went on to have a stroke showed evidence of intra-cranial vasculopathy (273). Although extra-cranial vasculopathy may not have been excluded, these findings are consistent with the notion that hemodynamic factors, e.g., reduced CVR associated with high CBF, may be more pertinent to the etiology of overt stroke than vasculopathy alone.
According to one seminal model of hemodynamic stroke in non-SCD patients, transition from misery perfusion to ischemic stroke is a result of perfusion pressure dropping to such an extent that CMRO2 is no longer maintainable by increases in OEF (146). Whilst there are a number of PET studies in support (274, 275), isolated reports of favorable tissue outcome following misery perfusion, termed the “ischemic penumbra,” including in one patient with SCD (276), indicate that there may be additional mechanisms involved in determining transition to observable tissue infarction (146, 277).
Although the hemodynamic underpinnings have not been investigated, a similar, albeit lower level, potentially reversible phenomenon termed “acute silent cerebral event” (ASCIE), has also been observed in SCD patients (278–282). In a prospective case-series, 18% of SCD patients and 7% of non-SCD patients presenting with acute anemia (hemoglobin <5 g/dl and >30% lower than steady state) secondary to infection, acute chest syndrome, and/or fever, showed lesions consistent with ischemia on DWI, termed ASCIE (279). On follow-up MRI, a majority, but not all, patients showed evidence of SCI corresponding to the original ASCIE. In 75% of the SCD patients presenting with ASCIE, there was no evidence of vasculopathy. A more recent multi-center trial established that ASCIE were detectable and prevalent also in “steady-state” SCD patients undergoing MRI screening, with an estimated 10 times greater incidence of ASCIE compared to SCI [47.3 vs. 4.8 per 100 patient years; (282)].
The temporal association of ASCIE with acute anemia, along with the observed transition of some ASCIE to SCI, are consistent with, but do not establish, a role for reduced oxygen delivery in SCI. Given that not all ASCIE progress to permanent lesions (i.e., SCI), these findings may suggest that additional hemodynamic, vaso-occlusive, inflammatory and/or pre-conditioning mechanisms are involved in determining transition from ASCIE to observable tissue infarction.
It is unclear what determines this tipping point in patients with SCD, but in prospective studies of non-SCD patients with carotid occlusion, risk of infarction is highest in patients with both increased OEF and CBV, the former indicative of misery perfusion, and the latter potentially of vasodilation and exhausted CVR (283, 284). These findings are consistent with models of hemodynamic stroke and tissue ischemia in which regional reductions in oxygen delivery (201) along with both exhausted CVR and OEF may play mechanistic roles. Recent SCD research indicating that regions of CBF and CMRO2 nadir overlap with the regions of highest SCI density (31) and highest oxygen extraction (166), provide further indirect evidence for a similar model in SCD (Figure 3). Research on these hemodynamic factors is, however, just beginning. Further work is required to establish whether concomitant measurement of CBF, OEF, and CBV/R may lead to better stratification of neurological risk than TCD in patients with SCD.
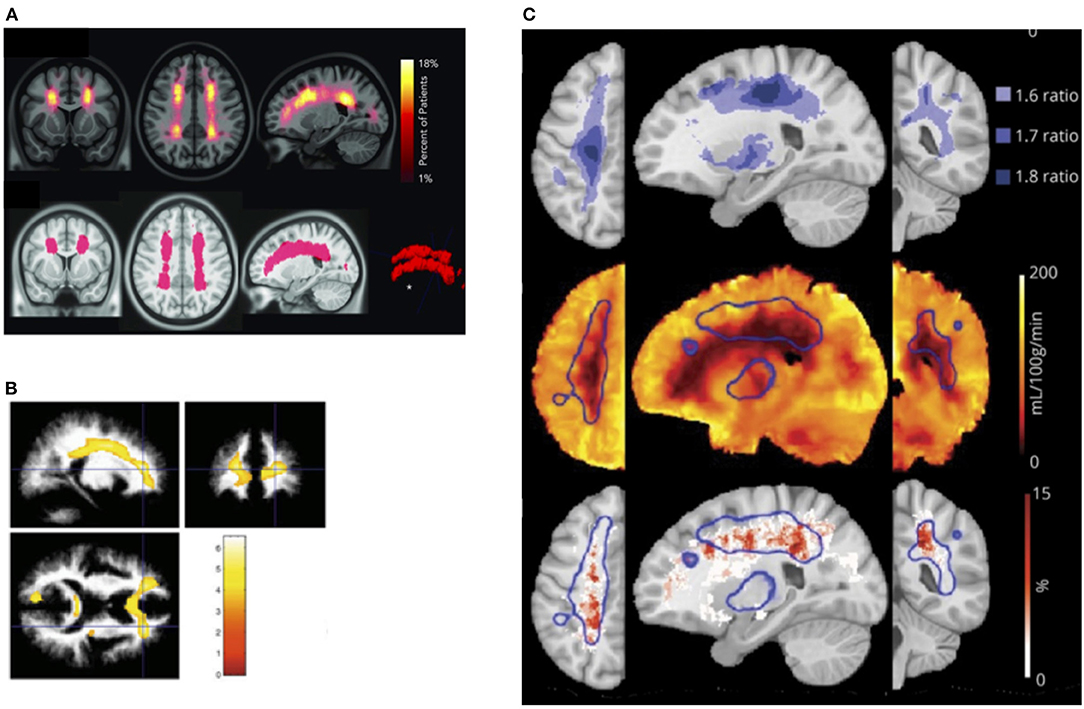
Figure 3. Watershed vulnerability. Results from collection of studies illustrating watershed vulnerability in SCD. (A) Top: SCI density map from 286 SCD children. Bottom: region encompassing 5.6% of brain volume in which 90% of SCI were confined [from Ford et al. (31)]. (B) Regions in which 20 SCD children without SCI demonstrated reduced white-matter density compared to 31 controls [from Baldeweg et al. (73)]. (C) Top: Ratiometric maps showing regions of elevated OEF derived from the ratio of SCD (n = 36) to control (n = 20) OEF values. (C) Middle: Region of high OEF (threshold 1.6, outlined in blue) overlaid on the average CBF map from the SCD cohort. (C) Bottom: Region of elevated OEF overlaid on SCI density map created from an independent cohort of 23 participants with SCD [from Fields et al. (166)].
Whilst the mechanisms by which transfusion reduces risk of stroke are unknown and vigorously debated, there are some reports indicating that a reduction in vascular instability may play a role. Transfusion significantly increases CaO2, and has immediate hemodynamic effects, reflected by reductions in CBF (285) and TCD velocity (286). Post-transfusion reductions in OEF (191, 255) and increases in CVR (162) have also been reported. Similar hemodynamic changes in global OEF and CBF have also been observed following bone marrow transplantation (287), which is the only curative treatment option currently available for SCD. Taken together, these findings provide proof of principle that normalization of CaO2 and hyperemia, along with restoration of vascular reserves, may contribute to the efficacy of transfusion in reducing risk of overt stroke.
Interestingly, post-transfusion reductions in global OEF and CBF are independently associated with improvement in total hemoglobin, but not HbS fraction (191, 255, 288), which suggests that a reduction in vascular instability is primarily achieved via improvement in global oxygen delivery rather than RBC rheology, and has implications for current transfusion strategies with HbS% targets. However, given their interdependence, these effects are difficult to disentangle. In SCD, both the compensatory global increases and post-transfusion reductions in CBF are greater than would be expected from changes in hemoglobin levels alone (289), suggesting that factors beyond correction of CaO2 are at play.
Moreover, transfusion appears to reduce, but not completely normalize, CBF and OEF in SCD patients, with watershed zones continuing to exhibit “at-risk” regions (191). There is also evidence that OEF and CBF responses to transfusion are blunted in adult SCD patients (255). These factors could contribute to continuing risk of morbidity in some patients, and may relate to vaso-occlusive/rheological factors, endothelial dysfunction, concomitant shifts in the oxygen-dissociation curve, and/or reduced regulatory capacity. There is in-vitro evidence that low-shear HVR decreases following simple chronic transfusion therapy in SCD patients, indicating that despite improvement in CaO2, post-transfusion increases in blood viscosity may worsen oxygen delivery in low-flow regions (235, 290). However, these findings are inconsistent with the observation that “at risk” regions of elevated OEF in watershed white matter zones appear to become smaller, rather than larger, following exchange transfusion (191). A possible explanation for this apparent juxtaposition is a difference in flow mechanics following simple and exchange transfusion, with exchange transfusion significantly reducing HbS% without substantially increasing hematocrit and viscosity (291). It is also possible that post-transfusion increases in CaO2 and CVR somewhat restore the ability of the brain to compensate for slight increases in viscosity.
Consistent with this notion, a recent study comparing untreated, chronically exchange transfused, and hydroxyurea (HU)-treated SCD patients, a less invasive treatment strategy based on stimulation of fetal hemoglobin (HbF), found OEF to be lowest in the transfused patients (288). Whilst “at-risk” regions of elevated OEF in watershed zones were detected across all groups, they were larger in the untreated and HU-treated patients than in the transfused patients, respectively. Interestingly, global gray and white matter CBF were similar among all groups, and there were no differences in total hemoglobin or SpO2 between HU-treated and transfused patients, suggesting that the between-group differences in OEF are not explainable by differences in global oxygen delivery. Of note, given that imaging was conducted on the day before scheduled transfusion, and other studies have shown reductions in global CBF and OEF 24 h post-transfusion (191), these findings may suggest that the hemodynamic effects of transfusion are greater near transfusion, compared to far from transfusion, as has been demonstrated for cognitive impairment (292). Nevertheless, the apparent inferiority of HU, even when compared with “late” transfusion effects, may be accounted for by the increased affinity of hemoglobin F for oxygen (293). Whilst the authors found no independent effect of HbF% or HbS% in multivariate models, left shifts in the oxygen dissociation curve are likely to impair oxygen offloading, and may be greater following HU than following transfusion. Whilst the effect on global oxygen metabolism may be balanced by the concomitant improvement in global oxygen delivery, more work is required to establish whether this is the case also for regional oxygen delivery, particularly in view of the finding that “at-risk” regions remain.
Whether vascular instability contributes to structural delay/deterioration not visible using conventional MRI and associated cognitive impairment, is an open question (294). The increased prevalence of ASCIE compared to SCI in acutely ill as well as steady state patients is consistent with the notion of an on-going state of vascular instability, and suggests that risk of ischemic insult may be far higher than previously recognized in SCD. It is unclear whether some of these insults are radiologically reversible or lead to microstructural tissue injury not visible using conventional MRI techniques. In support of the latter possibility, there is evidence that lesion detectability increases with increasing magnet strength in SCD, with one study showing that 3T MRI fails to detect lesions that are visible at 7T (295). Also, glial fibrillary acidic protein (GFAP), a marker of acute stroke and brain trauma, is significantly upregulated and associated with performance IQ, but not verbal IQ in “steady state” SCD patients with and without SCI (296).
Correlations have also been demonstrated between reductions in CVR and cortical thinning in regions of high metabolic activity in children with SCD (70), which may suggest that reduced dilatory capacity is involved in more subtle, and widespread tissue atrophy and/or delayed maturation. This notion is further supported by a recent report demonstrating a disruption in the relationship between CVR and white-matter integrity in SCD patients (297). There is evidence that reduced integrity is more common (82), and potentially also more functionally significant than SCI alone in SCD patients (86). Case reports of deterioration in cognitive function with acute drops in CaO2 in SCD (33) along with studies showing correlations between TCD abnormalities and executive dysfunction (298–300) and between reduced blood-oxygenated dependent (BOLD) MRI responses to visual stimulation and intelligence (301), lend further support to a role for vascular instability in cognitive impairment.
In summary, the pathophysiology of neurological morbidity in SCD is complex, and likely involves multiple mutually enforcing pathways, including vaso-occlusive/rheological, hemolytic, and hypoxic phenomena. Based on existing theories and accumulating evidence, we have proposed an integrative framework which emphasizes a role for vascular instability as a potential linking pathophysiological explanation. This framework brings together recent developments in the field, highlights outstanding questions, and provides mechanistic hypotheses that may guide future research.
Whilst the many strands of indirect evidence presented are broadly consistent with the framework, they do not rule out alternative and/or additional mechanisms of neurological morbidity. In order to interrogate and refine the framework, further advanced MRI studies are required. For this purpose, longitudinal measures of oxygen-metabolism would be most useful. As the framework and reviewed literature demonstrate, aspects of CMRO2, such as oxygen delivery and extraction ought to be considered together, both globally and regionally. Multi-modal, neurodevelopmental approaches that combine structural, diffusion, hemodynamic, and cognitive measures would also be helpful in further addressing outstanding questions.
One of the key challenges with these advanced MRI techniques remains validation in SCD patients, in whom some of the underlying assumptions may not be valid (165, 256, 259, 302–304). Comparison with current clinical gold-standard (e.g., PET for oxygen-extraction) may be useful in this regard. Employment of standardized criteria for detection of SCI and grading of vasculopathy will also be important in facilitating between-study comparisons (32, 164). Given that the vast majority of reviewed studies are based on patients with homozygous SCD, exploring these measures in patients with other genotypes is also vital and may help shed further light on the underlying physiology. Finally, further work is also required to establish the applicability of this framework to other end-organs which are also at risk of damage in SCD.
If fruitful, this line of enquiry has the potential to improve precision medicine in SCD, which is a crucial next step in efforts to screen and intervene. Whilst there are evidence-based strategies for stroke prevention in children (10), treatment is often burdensome (305), the specificity of screening is poor (10), and many patients continue to suffer progressive vasculopathy and/or recurrent insults (38). There are few evidence-based strategies for cognitive dysfunction, and none that tackle microstructural tissue injury. Improved strategies are therefore urgently required.
According to the proposed framework, measures of regional oxygen delivery, CVR, and OEF are likely the most proximate targets for prediction of neurological risk. With further refinement, development of a “vascular instability risk profile” based on these measures may enable selection of patients with sufficiently high-risk for invasive, burdensome, and costly treatment options such as transfusion, bone marrow transplant, or eventually gene therapy. Such a profile may also enable on-going monitoring of risk so that transfusion is not necessarily lifelong. Another implication is the identification of regional oxygen delivery as a potential treatment target for therapies, with several potential avenues for intervention (e.g., anemia, oxygen desaturation, endothelial dysfunction). Crucially, therapies need to balance any increases in oxygen-delivery with any potential reductions in oxygen-unloading (293). With further refinement, this framework may therefore hold promise not only for guiding research, but also for prediction of risk and implementation of tailored preventative strategies before stroke, SCI and/or microstructural injury occurs.
HS and FK: design and conception. HS, FK, and JK: literature review. HS: drafting the article. FK, JK, PH, DS, and CC: critical revision of the article. HS, FK, JK, PH, DS, and CC: final approval of the version to be published.
HS was funded by Action Medical Research (GN2509), JK was funded by Great Ormond Street Children's Charity (V4615) (gosh.org/what-we-do/grant-funding/recently-fundedprojects/national-calls), and PH was funded by Children with Cancer UK (CwCUK-15-203). The National Institute for Health Research (UK; PB-PG1112-29099) and National Heart, Lung, and Blood Institute (USA; R01HL079937) also provided funding, and the work was supported by the NIHR Great Ormond Street Hospital Biomedical Research Center. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Aygun B, Odame I. A global perspective on sickle cell disease. Pediatr Blood Cancer. (2012) 59:386–90. doi: 10.1002/pbc.24175
2. Saraf SL, Molokie RE, Nouraie M, Sable CA, Luchtman-Jones L, Ensing GJ, et al. Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatr Respir Rev. (2014) 15:4–12. doi: 10.1016/j.prrv.2013.11.003
3. Ataga KI, Orringer EP. Hypercoagulability in sickle cell disease: a curious paradox. Am J Med. (2003) 115:721–8. doi: 10.1016/j.amjmed.2003.07.011
4. Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. (2004) 11:129–51. doi: 10.1080/mic.11.2.129.151
5. Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. (2007) 21:37–47. doi: 10.1016/j.blre.2006.07.001
6. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. (2010) 376:2018–31. doi: 10.1016/S0140-6736(10)61029-X
7. Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. (2017) 390:311–23. doi: 10.1016/S0140-6736(17)30193-9
8. Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. (2016) 127:801–9. doi: 10.1182/blood-2015-09-618538
9. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. (1994) 330:1639–44. doi: 10.1056/NEJM199406093302303
10. Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. (1998) 339:5–11. doi: 10.1056/NEJM199807023390102
11. Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. (1998) 91:288–94.
12. Adams RJ, McKie VC, Brambilla D, Carl E, Gallagher D, Nichols FT, et al. Stroke prevention trial in sickle cell anemia. Control Clin Trials. (1998) 19:110–29. doi: 10.1016/S0197-2456(97)00099-8
13. Hart RG, Kanter MC. Hematologic disorders and ischemic stroke. A selective review. Stroke. (1990) 21:1111–21. doi: 10.1161/01.STR.21.8.1111
14. Jabbarli R, Dinger TF, Pierscianek D, Oppong MD, Chen B, Dammann P, et al. Intracranial aneurysms in sickle cell disease. Curr Neurovasc Res. (2019) 16:63–76. doi: 10.2174/1567202616666190131160847
15. Strouse JJ, Hulbert ML, DeBaun MR, Jordan LC, Casella JF. Primary hemorrhagic stroke in children with sickle cell disease is associated with recent transfusion and use of corticosteroids. Pediatrics. (2006) 118:1916–24. doi: 10.1542/peds.2006-1241
16. Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol. (2006) 5:501–12. doi: 10.1016/S1474-4422(06)70469-0
17. de Araujo OMR, Ivo ML, Ferreira Júnior MA, Pontes ERJC, Bispo IMGP, Oliveira, et al. Survival and mortality among users and non-users of hydroxyurea with sickle cell disease. Rev Lat Am Enfermagem. (2015) 23:67–73. doi: 10.1590/0104-1169.3385.2526
18. Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. (1978) 65:461–71. doi: 10.1016/0002-9343(78)90772-6
19. DeBaun MR, Kirkham FJ. Central nervous system complications and management in sickle cell disease. Blood. (2016) 127:829–38. doi: 10.1182/blood-2015-09-618579
20. DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. (2012) 119:4587–96. doi: 10.1182/blood-2011-02-272682
21. Cancio MI, Helton KJ, Schreiber JE, Smeltzer MP, Kang G, Wang WC. Silent cerebral infarcts in very young children with sickle cell anaemia are associated with a higher risk of stroke. Br J Haematol. (2015) 171:120–9. doi: 10.1111/bjh.13525
22. Wang WC, Langston JW, Steen RG, Wynn LW, Mulhern RK, Wilimas JA, et al. Abnormalities of the central nervous system in very young children with sickle cell anemia. J Pediatr. (1998) 132:994–8. doi: 10.1016/S0022-3476(98)70397-X
23. Kwiatkowski JL, Zimmerman RA, Pollock AN, Seto W, Smith-Whitley K, Shults J, et al. Silent infarcts in young children with sickle cell disease. Br J Haematol. (2009) 146:300–5. doi: 10.1111/j.1365-2141.2009.07753.x
24. Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Vasile M, Kasbi F, et al. Chronic and acute anemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood. (2015) 125:1653–61. doi: 10.1182/blood-2014-09-599852
25. Kassim AA, Pruthi S, Day M, Rodeghier M, Gindville MC, Brodsky MA, et al. Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood. (2016) 127:2038–40. doi: 10.1182/blood-2016-01-694562
26. Pegelow CH, Macklin EA, Moser FG, Wang WC, Bello JA, Miller ST, et al. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood. (2002) 99:3014–8. doi: 10.1182/blood.V99.8.3014
27. Wang W, Enos L, Gallagher D, Thompson R, Guarini L, Vichinsky E, et al. Neuropsychologic performance in school-aged children with sickle cell disease: a report from the cooperative study of sickle cell disease. J Pediatr. (2001) 139:391–7. doi: 10.1067/mpd.2001.116935
28. Adams RJ, Nichols FT, McKie V, McKie K, Milner P, Gammal TE. Cerebral infarction in sickle cell anemia: mechanism based on CT and MRI. Neurology. (1988) 38:1012–7. doi: 10.1212/WNL.38.7.1012
29. Pavlakis SG, Bello J, Prohovnik I, Sutton M, Ince C, Mohr JP, et al. Brain infarction in sickle cell anemia: magnetic resonance imaging correlates. Ann Neurol. (1988) 23:125–30. doi: 10.1002/ana.410230204
30. Guilliams KP, Fields ME, Ragan DK, Chen Y, Eldeniz C, Hulbert ML, et al. Large-vessel vasculopathy in children with sickle cell disease: a magnetic resonance imaging study of infarct topography and focal atrophy. Pediatr Neurol. (2017) 69:49–57. doi: 10.1016/j.pediatrneurol.2016.11.005
31. Ford AL, Ragan DK, Fellah S, Binkley MM, Fields ME, Guilliams KP, et al. Silent infarcts in sickle cell disease occur in the border zone region and are associated with low cerebral blood flow. Blood. (2018) 132:1714–23. doi: 10.1182/blood-2018-04-841247
32. Guilliams KP, Fields ME, Dowling MM. Advances in understanding ischemic stroke physiology and the impact of vasculopathy in children with sickle cell disease. Stroke. (2019) 50:266–73. doi: 10.1161/STROKEAHA.118.020482
33. Dowling MM, Kirkham FJ. Stroke in sickle cell anaemia is more than stenosis and thrombosis: the role of anaemia and hyperemia in ischaemia. Br J Haematol. (2017) 176:151–3. doi: 10.1111/bjh.14422
34. Stockman JA, Nigro MA, Mishkin MM, Oski FA. Occlusion of large cerebral vessels in sickle-cell anemia. N Engl J Med. (1972) 287:846–9. doi: 10.1056/NEJM197210262871703
35. Telfer PT, Evanson J, Butler P, Hemmaway C, Abdulla C, Gadong N, et al. Cervical carotid artery disease in sickle cell anemia: clinical and radiological features. Blood. (2011) 118:6192–9. doi: 10.1182/blood-2011-03-337915
36. Dobson SR, Holden KR, Nietert PJ, Cure JK, Laver JH, Disco D, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood. (2002) 99:3144–50. doi: 10.1182/blood.V99.9.3144
37. Seeler RA, Royal JE, Powe L, Goldberg HR. Moyamoya in children with sickle cell anemia and cerebrovascular occlusion. J Pediatr. (1978) 93:808–10. doi: 10.1016/S0022-3476(78)81086-5
38. Hulbert ML, McKinstry RC, Lacey JL, Moran CJ, Panepinto JA, Thompson AA, et al. Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood. (2011) 117:772–9. doi: 10.1182/blood-2010-01-261123
39. Powars D, Adams RJ, Nichols FT, Milner P, Charache S, Sarnaik S. Delayed intracranial hemorrhage following cerebral infarction in sickle cell anemia. J Assoc Acad Minor Phys. (1990) 1:79–82.
40. Kassim AA, Galadanci NA, Pruthi S, DeBaun MR. How I treat and manage strokes in sickle cell disease. Blood. (2015) 125:3401–10. doi: 10.1182/blood-2014-09-551564
41. Yao Z, Li J, He M, You C. Intracranial aneurysm in patients with sickle cell disease: a systematic review. World Neurosurg. (2017) 105:302–13. doi: 10.1016/j.wneu.2017.05.139
42. Buch K, Arya R, Shah B, Nadgir RN, Saito N, Qureshi MM, et al. Quantitative analysis of extracranial arterial tortuosity in patients with sickle cell disease. J Neuroimaging. (2017) 27:421–27. doi: 10.1111/jon.12418
43. Hyacinth HI, Sugihara CL, Spencer TL, Archer DR, Shih AY. Higher prevalence of spontaneous cerebral vasculopathy and cerebral infarcts in a mouse model of sickle cell disease. J Cereb Blood Flow Metab. (2019) 39:342–51. doi: 10.1177/0271678X17732275
44. Steen RG, Langston JW, Ogg RJ, Manci E, Mulhern RK, Wang W. Ectasia of the basilar artery in children with sickle cell disease: relationship to hematocrit and psychometric measures. J Stroke Cerebrovasc Dis. (1998) 7:32–43. doi: 10.1016/S1052-3057(98)80019-0
45. Steen RG, Reddick WE, Glass JO, Wang WC. Evidence of cranial artery ectasia in sickle cell disease patients with ectasia of the basilar artery. J Stroke Cerebrovasc Dis. (1998) 7:330–8. doi: 10.1016/S1052-3057(98)80051-7
46. Birkeland P, Gardner K, Kesse-Adu R, Davies J, Lauritsen J, Rom Poulsen F, et al. Intracranial aneurysms in sickle-cell disease are associated with the hemoglobin SS genotype but not with moyamoya syndrome. Stroke. (2016) 47:1710–3. doi: 10.1161/STROKEAHA.116.012664
47. Preul MC, Cendes F, Just N, Mohr G. Intracranial aneurysms and sickle cell anemia: multiplicity and propensity for the vertebrobasilar territory. Neurosurgery. (1998) 42:971–7. doi: 10.1097/00006123-199805000-00007
48. Kossorotoff M, Brousse V, Grevent D, Naggara O, Brunelle F, Blauwblomme T, et al. Cerebral haemorrhagic risk in children with sickle-cell disease. Dev Med Child Neurol. (2015) 57:187–93. doi: 10.1111/dmcn.12571
49. Koshy M, Thomas C, Goodwin J. Vascular lesions in the central nervous system in sickle cell disease (neuropathology). J Assoc Acad Minor Phys. (1990) 1:71–8.
50. Oyesiku NM, Barrow DL, Eckman JR, Tindall SC, Colohan AR. Intracranial aneurysms in sickle-cell anemia: clinical features and pathogenesis. J Neurosurg. (1991) 75:356–63. doi: 10.3171/jns.1991.75.3.0356
51. Connes P, Verlhac S, Bernaudin F. Advances in understanding the pathogenesis of cerebrovascular vasculopathy in sickle cell anaemia. Br J Haematol. (2013) 161:484–98. doi: 10.1111/bjh.12300
52. Craft S, Schatz J, Glauser TA, Lee B, DeBaun MR. Neuropsychologic effects of stroke in children with sickle cell anemia. J Pediatr. (1993) 123:712–7. doi: 10.1016/S0022-3476(05)80844-3
53. Kawadler JM, Clayden JD, Clark CA, Kirkham FJ. Intelligence quotient in paediatric sickle cell disease: a systematic review and meta-analysis. Dev Med Child Neurol. (2016) 58:672–9. doi: 10.1111/dmcn.13113
54. Prussien KV, Jordan LC, DeBaun MR, Compas BE. Cognitive function in sickle cell disease across domains, cerebral infarct status, and the lifespan: a meta-analysis. J Pediatr Psychol. (2019). doi: 10.1093/jpepsy/jsz031. [Epub ahead of print].
55. Chua-Lim C, Moore RB, McCleary G, Shah A, Mankad VN. Deficiencies in school readiness skills of children with sickle cell anemia: a preliminary report. South Med J. (1993) 86:397–402. doi: 10.1097/00007611-199304000-00005
56. Steen RG, Hu XJ, Elliott VE, Miles MA, Jones S, Wang WC. Kindergarten readiness skills in children with sickle cell disease: evidence of early neurocognitive damage? J Child Neurol. (2002) 17:111–6. doi: 10.1177/088307380201700204
57. Schatz J. Brief report: academic attainment in children with sickle cell disease. J Pediatr Psychol. (2004) 29:627–33. doi: 10.1093/jpepsy/jsh065
58. Schatz J, Brown RT, Pascual JM, Hsu L, DeBaun MR. Poor school and cognitive functioning with silent cerebral infarcts and sickle cell disease. Neurology. (2001) 56:1109–11. doi: 10.1212/WNL.56.8.1109
59. Smith KE, Patterson CA, Szabo MM, Tarazi RA, Barakat LP. Predictors of academic achievement for school age children with sickle cell disease. Adv Sch Ment Health Promot. (2013) 6:5–20. doi: 10.1080/1754730X.2012.760919
60. Sanger M, Jordan L, Pruthi S, Day M, Covert B, Merriweather B, et al. Cognitive deficits are associated with unemployment in adults with sickle cell anemia. J Clin Exp Neuropsychol. (2016) 38:661–71. doi: 10.1080/13803395.2016.1149153
61. Drazen CH, Abel R, Gabir M, Farmer G, King AA. Prevalence of developmental delay and contributing factors among children with sickle cell disease. Pediatr Blood Cancer. (2016) 63:504–10. doi: 10.1002/pbc.25838
62. Armstrong FD, Thompson RJ, Wang W, Zimmerman R, Pegelow CH, Miller S, et al. Cognitive functioning and brain magnetic resonance imaging in children with sickle cell disease. neuropsychology committee of the cooperative study of sickle cell disease. Pediatrics. (1996) 97:864–70.
63. Castro IPS, Viana MB. Cognitive profile of children with sickle cell anemia compared to healthy controls. J Pediatr (Rio J). (2019) 95:451–7. doi: 10.1016/j.jped.2018.04.012
64. Hijmans CT, Fijnvandraat K, Grootenhuis MA, van Geloven N, Heijboer H, Peters M, et al. Neurocognitive deficits in children with sickle cell disease: a comprehensive profile. Pediatr Blood Cancer. (2011) 56:783–8. doi: 10.1002/pbc.22879
65. Prussien KV, DeBaun MR, Yarboi J, Bemis H, McNally C, Williams E, et al. Cognitive function, coping, and depressive symptoms in children and adolescents with sickle cell disease. J Pediatr Psychol. (2018) 43:543–51. doi: 10.1093/jpepsy/jsx141
66. Vichinsky EP, Neumayr LD, Gold JI, Weiner MW, Rule RR, Truran D, et al. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA. (2010) 303:1823–31. doi: 10.1001/jama.2010.562
67. Nunes S, Argollo N, Mota M, Vieira C, de Sena EP. Comprehensive neuropsychological evaluation of children and adolescents with sickle cell anemia: a hospital-based sample. Rev Bras Hematol Hemoter. (2017) 39:32–9. doi: 10.1016/j.bjhh.2016.09.004
68. Schatz J, McClellan CB. Sickle cell disease as a neurodevelopmental disorder. Ment Retard Dev Disabil Res Rev. (2006) 12:200–7. doi: 10.1002/mrdd.20115
69. Kawadler JM, Clayden JD, Kirkham FJ, Cox TC, Saunders DE, Clark CA. Subcortical and cerebellar volumetric deficits in paediatric sickle cell anaemia. Br J Haematol. (2013) 163:373–6. doi: 10.1111/bjh.12496
70. Kim JA, Leung J, Lerch JP, Kassner A. Reduced cerebrovascular reserve is regionally associated with cortical thickness reductions in children with sickle cell disease. Brain Res. (2016) 1642:263–9. doi: 10.1016/j.brainres.2016.03.041
71. Kirk GR, Haynes MR, Palasis S, Brown C, Burns TG, McCormick M, et al. Regionally specific cortical thinning in children with sickle cell disease. Cereb Cortex. (2009) 19:1549–56. doi: 10.1093/cercor/bhn193
72. Mackin RS, Insel P, Truran D, Vichinsky EP, Neumayr LD, Armstrong FD, et al. Neuroimaging abnormalities in adults with sickle cell anemia: associations with cognition. Neurology. (2014) 82:835–41. doi: 10.1212/WNL.0000000000000188
73. Baldeweg T, Hogan AM, Saunders DE, Telfer P, Gadian DG, Vargha-Khadem F, et al. Detecting white matter injury in sickle cell disease using voxel-based morphometry. Ann Neurol. (2006) 59:662–72. doi: 10.1002/ana.20790
74. Choi S, Bush AM, Borzage MT, Joshi AA, Mack WJ, Coates TD, et al. Hemoglobin and mean platelet volume predicts diffuse T1-MRI white matter volume decrease in sickle cell disease patients. NeuroImage Clin. (2017) 15:239–46. doi: 10.1016/j.nicl.2017.04.023
75. Schatz J, Buzan R. Decreased corpus callosum size in sickle cell disease: relationship with cerebral infarcts and cognitive functioning. J Int Neuropsychol Soc. (2006) 12:24–33. doi: 10.1017/S1355617706060085
76. Chen R, Arkuszewski M, Krejza J, Zimmerman RA, Herskovits EH, Melhem ER. A prospective longitudinal brain morphometry study of children with sickle cell disease. AJNR Am J Neuroradiol. (2015) 36:403–10. doi: 10.3174/ajnr.A4101
77. Darbari DS, Eigbire-Molen O, Ponisio MR, Milchenko MV, Rodeghier MJ, Casella JF, et al. Progressive loss of brain volume in children with sickle cell anemia and silent cerebral infarct: a report from the silent cerebral infarct transfusion trial. Am J Hematol. (2018) 93:E406–8. doi: 10.1002/ajh.25297
78. Kawadler JM, Clark CA, McKinstry RC, Kirkham FJ. Brain atrophy in paediatric sickle cell anaemia: findings from the silent infarct transfusion (SIT) trial. Br J Haematol. (2017) 177:151–3. doi: 10.1111/bjh.14039
79. Balci A, Karazincir S, Beyoglu Y, Cingiz C, Davran R, Gali E, et al. Quantitative brain diffusion-tensor MRI findings in patients with sickle cell disease. AJR Am J Roentgenol. (2012) 198:1167–74. doi: 10.2214/AJR.11.7404
80. Chai Y, Coloigner J, Qu X, Choi S, Bush A, Borzage M, et al. Tract specific analysis in patients with sickle cell disease. Proc SPIE Int Soc Opt Eng. (2015) 9681:968108. doi: 10.1117/12.2213617
81. Choi S, Bush AM, Borzage M, Joshi A, Coates TD, Leahy R, et al. Regional susceptibility to chronic anemia in WM microstructure using diffusion tensor imaging. Blood. (2016) 128:3640. Availbale online at: http://www.bloodjournal.org/content/128/22/3640
82. Kawadler JM, Kirkham FJ, Clayden JD, Hollocks MJ, Seymour EL, Edey R, et al. White matter damage relates to oxygen saturation in children with sickle cell anemia without silent cerebral infarcts. Stroke. (2015) 46:1793–9. doi: 10.1161/STROKEAHA.115.008721
83. Sun B, Brown RC, Hayes L, Burns TG, Huamani J, Bearden DJ, et al. White matter damage in asymptomatic patients with sickle cell anemia: screening with diffusion tensor imaging. AJNR Am J Neuroradiol. (2012) 33:2043–9. doi: 10.3174/ajnr.A3135
84. Chen R, Krejza J, Arkuszewski M, Zimmerman RA, Herskovits EH, Melhem ER. Brain morphometric analysis predicts decline of intelligence quotient in children with sickle cell disease: a preliminary study. Adv Med Sci. (2017) 62:151–7. doi: 10.1016/j.advms.2016.09.002
85. Wang L, Almeida LEFF, de Souza Batista CM, Khaibullina A, Xu N, Albani S, et al. Cognitive and behavior deficits in sickle cell mice are associated with profound neuropathologic changes in hippocampus and cerebellum. Neurobiol Dis. (2016) 85:60–72. doi: 10.1016/j.nbd.2015.10.004
86. Stotesbury H, Kirkham FJ, Kölbel M, Balfour P, Clayden JD, Sahota S, et al. White matter integrity and processing speed in sickle cell anemia. Neurology. (2018) 90:e2042–50. doi: 10.1212/WNL.0000000000005644
87. Helton KJ, Adams RJ, Kesler KL, Lockhart A, Aygun B, Driscoll C, et al. Magnetic resonance imaging/angiography and transcranial Doppler velocities in sickle cell anemia: results from the SWiTCH trial. Blood. (2014) 124:891–8. doi: 10.1182/blood-2013-12-545186
88. Hebbel RP, Vercellotti G, Nath KA. A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxsis. Cardiovasc Hematol Disord Drug Targets. (2009) 9:271–92. doi: 10.2174/1871529X10909040271
89. Ricard N, Simons M. When it is better to regress: dynamics of vascular pruning. PLoS Biol. (2015) 13:e1002148. doi: 10.1371/journal.pbio.1002148
90. Adams RJ. Big strokes in small persons. Arch Neurol. (2007) 64:1567–74. doi: 10.1001/archneur.64.11.1567
91. Hillery CA, Panepinto JA. Pathophysiology of stroke in sickle cell disease. Microcirculation. (2004) 11:195–208. doi: 10.1080/10739680490278600
92. Jacob S, Adcock A, Murray A, Kolodney J. More than meets the eye: cerebrovascular disease in sickle cell disease is about more than sickling. Stroke. (2018) 49:e224–7. doi: 10.1161/STROKEAHA.118.021057
93. Detterich JA, Kato R, Bush A, Chalacheva P, Ponce D, De Zoysa M, et al. Sickle cell microvascular paradox-oxygen supply-demand mismatch. Am J Hematol. (2019) 94:678–88. doi: 10.1002/ajh.25476
94. Jorgensen DR, Rosano C, Novelli EM. Can neuroimaging markers of vascular pathology explain cognitive performance in adults with sickle cell anemia? A review of the literature. Hemoglobin. (2016) 40:381–7. doi: 10.1080/03630269.2016.1242493
95. van der Land V, Zwanenburg JJM, Biemond BJ, Hendrikse J, Mutsaerts HJ, Engelen M, et al. Cerebral small vessel disease in patients with sickle cell disease: initial findings with ultra-high field 7T MRI. Blood. (2013) 122:1011. Available online at: http://www.bloodjournal.org/content/122/21/1011
96. Shi Y, Wardlaw JM. Update on cerebral small vessel disease: a dynamic whole-brain disease. Stroke Vasc Neurol. (2016) 1:83–92. doi: 10.1136/svn-2016-000035
97. Kija EN, Saunders DE, Munubhi E, Darekar A, Barker S, Cox TCS, et al. Transcranial Doppler and magnetic resonance in tanzanian children with sickle cell disease. Stroke. (2019) 50:1719–26. doi: 10.1161/STROKEAHA.118.018920
98. Novelli EM, Elizabeth Sarles C, Jay Aizenstein H, Ibrahim TS, Butters MA, Connelly Ritter A, et al. Brain venular pattern by 7T MRI correlates with memory and haemoglobin in sickle cell anaemia. Psychiatry Res. (2015) 233:18–22. doi: 10.1016/j.pscychresns.2015.04.005
99. Kinney TR, Sleeper LA, Wang WC, Zimmerman RA, Pegelow CH, Ohene-Frempong K, et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The cooperative study of sickle cell disease. Pediatrics. (1999) 103:640–5. doi: 10.1542/peds.103.3.640
100. Okpala I, Daniel Y, Haynes R, Odoemene D, Goldman J. Relationship between the clinical manifestations of sickle cell disease and the expression of adhesion molecules on white blood cells. Eur J Haematol. (2002) 69:135–44. doi: 10.1034/j.1600-0609.2002.02775.x
101. Hoppe C, Klitz W, Cheng S, Apple R, Steiner L, Robles L, et al. Gene interactions and stroke risk in children with sickle cell anemia. Blood. (2004) 103:2391–6. doi: 10.1182/blood-2003-09-3015
103. Calvet D, Bernaudin F, Gueguen A, Hosseini H, Habibi A, Galactéros F, et al. First ischemic stroke in sickle-cell disease: are there any adult specificities? Stroke. (2015) 46:2315–7. doi: 10.1161/STROKEAHA.115.010153
104. Thangarajh M, Yang G, Fuchs D, Ponisio MR, McKinstry RC, Jaju A, et al. Magnetic resonance angiography-defined intracranial vasculopathy is associated with silent cerebral infarcts and glucose-6-phosphate dehydrogenase mutation in children with sickle cell anaemia. Br J Haematol. (2012) 159:352–9. doi: 10.1111/bjh.12034
105. Steen RG, Emudianughe T, Hankins GM, Wynn LW, Wang WC, Xiong X, et al. Brain imaging findings in pediatric patients with sickle cell disease. Radiology. (2003) 228:216–25. doi: 10.1148/radiol.2281020943
106. Wilimas J, Goff JR, Anderson HR, Langston JW, Thompson E. Efficacy of transfusion therapy for one to two years in patients with sickle cell disease and cerebrovascular accidents. J Pediatr. (1980) 96:205–8. doi: 10.1016/S0022-3476(80)80803-1
107. Choudhury NA, DeBaun MR, Ponisio MR, Jordan LC, Rodeghier M, Pruthi S, et al. Intracranial vasculopathy and infarct recurrence in children with sickle cell anaemia, silent cerebral infarcts and normal transcranial Doppler velocities. Br J Haematol. (2018) 183:324–26. doi: 10.1111/bjh.14979
108. Dowling MM, Quinn CT, Rogers ZR, Journeycake JM. Stroke in sickle cell anemia: alternative etiologies. Pediatr Neurol. (2009) 41:124–6. doi: 10.1016/j.pediatrneurol.2009.02.011
109. Kumar R, Stanek J, Creary S, Dunn A, O'Brien SH. Prevalence and risk factors for venous thromboembolism in children with sickle cell disease: an administrative database study. Blood Adv. (2018) 2:285–91. doi: 10.1182/bloodadvances.2017012336
110. Rothman SM, Fulling KH, Nelson JS. Sickle cell anemia and central nervous system infarction: a neuropathological study. Ann Neurol. (1986) 20:684–90. doi: 10.1002/ana.410200606
111. Gangaraju R, Reddy VVB, Marques MB. Fat embolism syndrome secondary to bone marrow necrosis in patients with hemoglobinopathies. South Med J. (2016) 109:549–53. doi: 10.14423/SMJ.0000000000000520
112. Nathan CL, Aamodt WW, Yalamarti T, Dogon C, Kinniry P. Cerebral fat embolism syndrome in sickle cell disease without evidence of shunt. ENeurol Sci. (2019) 14:19–20. doi: 10.1016/j.ensci.2018.11.012
113. Targueta EP, Hirano AC de G, de Campos FPF, Martines JA dos S, Lovisolo SM, Felipe-Silva A. Bone marrow necrosis and fat embolism syndrome: a dreadful complication of hemoglobin sickle cell disease. Autops. Case Rep. (2017) 7:42–50. doi: 10.4322/acr.2017.043
114. Tsitsikas DA, Gallinella G, Patel S, Seligman H, Greaves P, Amos RJ. Bone marrow necrosis and fat embolism syndrome in sickle cell disease: increased susceptibility of patients with non-SS genotypes and a possible association with human parvovirus B19 infection. Blood Rev. (2014) 28:23–30. doi: 10.1016/j.blre.2013.12.002
115. Dowling MM, Lee N, Quinn CT, Rogers ZR, Boger D, Ahmad N, et al. Prevalence of intracardiac shunting in children with sickle cell disease and stroke. J Pediatr. (2010) 156:645–50. doi: 10.1016/j.jpeds.2009.10.012
116. Razdan S, Strouse JJ, Naik R, Lanzkron S, Urrutia V, Resar JR, et al. Patent foramen ovale in patients with sickle cell disease and stroke: case presentations and review of the literature. Case Rep Hematol. (2013) 2013:516705. doi: 10.1155/2013/516705
117. Razdan S, Strouse JJ, Reddy A, Resar DF, Hasan RK, Resar JR, et al. Patent foramen ovale in adults with sickle cell disease and stroke. Am J Hematol. (2016) 91:E358–60. doi: 10.1002/ajh.24440
118. Dowling MM, Quinn CT, Ramaciotti C, Kanter J, Osunkwo I, Inusa B, et al. Increased prevalence of potential right-to-left shunting in children with sickle cell anaemia and stroke. Br J Haematol. (2017) 176:300–8. doi: 10.1111/bjh.14391
119. Dowling MM, Ikemba CM. Intracardiac shunting and stroke in children: a systematic review. J Child Neurol. (2011) 26:72–82. doi: 10.1177/0883073810383913
120. Mattle HP, Meier B, Nedeltchev K. Prevention of stroke in patients with patent foramen ovale. Int J Stroke. (2010) 5:92–102. doi: 10.1111/j.1747-4949.2010.00413.x
121. Overell JR, Bone I, Lees KR. Interatrial septal abnormalities and stroke: a meta-analysis of case-control studies. Neurology. (2000) 55:1172–9. doi: 10.1212/WNL.55.8.1172
122. Toledo SLde O, Guedes JVM, Alpoim PN, Rios DRA, Pinheiro Mde B. Sickle cell disease: hemostatic and inflammatory changes, and their interrelation. Clin Chim Acta. (2019) 493:129–37. doi: 10.1016/j.cca.2019.02.026
123. Woods GM, Sharma R, Creary S, O'Brien S, Stanek J, Hor K, et al. Venous thromboembolism in children with sickle cell disease: a retrospective Cohort study. J Pediatr. (2018) 197:186–90.e1. doi: 10.1016/j.jpeds.2018.01.073
124. Tewari S, Renney G, Brewin J, Gardner K, Kirkham F, Inusa B, et al. Proteomic analysis of plasma from children with sickle cell anemia and silent cerebral infarction. Haematologica. (2018) 103:1136–42. doi: 10.3324/haematol.2018.187815
125. Yu TT, Nelson J, Streiff MB, Lanzkron S, Naik RP. Risk factors for venous thromboembolism in adults with hemoglobin SC or Sβ(+) thalassemia genotypes. Thromb Res. (2016) 141:35–8. doi: 10.1016/j.thromres.2016.03.003
126. Whelihan MF, Lim MY, Mooberry MJ, Piegore MG, Ilich A, Wogu A, et al. Thrombin generation and cell-dependent hypercoagulability in sickle cell disease. J Thromb Haemost. (2016) 14:1941–52. doi: 10.1111/jth.13416
127. Faes C, Sparkenbaugh EM, Pawlinski R. Hypercoagulable state in sickle cell disease. Clin Hemorheol Microcirc. (2018) 68:301–18. doi: 10.3233/CH-189013
128. Noubouossie D, Key NS, Ataga KI. Coagulation abnormalities of sickle cell disease: relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev. (2016) 30:245–56. doi: 10.1016/j.blre.2015.12.003
129. Hyacinth HI, Adams RJ, Greenberg CS, Voeks JH, Hill A, Hibbert JM, et al. Effect of chronic blood transfusion on biomarkers of coagulation activation and thrombin generation in sickle cell patients at risk for stroke. PLoS ONE. (2015) 10:e0134193. doi: 10.1371/journal.pone.0134193
130. Noubouossie DCF, Lê PQ, Rozen L, Ziereisen F, Willems D, Demulder A, et al. Thrombin generation in children with sickle cell disease: relationship with age, hemolysis, transcranial Doppler velocity, and hydroxyurea treatment. Eur J Haematol. (2013) 91:46–54. doi: 10.1111/ejh.12113
131. Cappellini MD, Musallam KM, Poggiali E, Taher AT. Hypercoagulability in non-transfusion-dependent thalassemia. Blood Rev. (2012) 26(suppl. 1):S20–3. doi: 10.1016/S0268-960X(12)70007-3
132. Taher AT, Otrock ZK, Uthman I, Cappellini MD. Thalassemia and hypercoagulability. Blood Rev. (2008) 22:283–92. doi: 10.1016/j.blre.2008.04.001
133. Proença-Ferreira R, Brugnerotto AF, Garrido VT, Dominical VM, Vital DM, Ribeiro Mde FR, et al. Endothelial activation by platelets from sickle cell anemia patients. PLoS ONE. (2014) 9:e89012. doi: 10.1371/journal.pone.0089012
134. Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, et al. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol. (2008) 111:474–81. doi: 10.1111/j.1365-2141.2000.02353.x
135. Wun T, Paglieroni T, Tablin F, Welborn J, Nelson K, Cheung A. Platelet activation and platelet-erythrocyte aggregates in patients with sickle cell anemia. J Lab Clin Med. (1997) 129:507–16.
136. Dominical VM, Samsel L, Nichols JS, Costa FF, McCoy JP, Conran N, et al. Prominent role of platelets in the formation of circulating neutrophil-red cell heterocellular aggregates in sickle cell anemia. Haematologica. (2014) 99:e214–7. doi: 10.3324/haematol.2014.108555
137. Celik T, Unal S, Ekinci O, Ozer C, Ilhan G, Oktay G, et al. Mean platelet volume can predict cerebrovascular events in patients with sickle cell anemia. Pakistan J Med Sci. (2015) 31:203–8. doi: 10.12669/pjms.311.4104
138. Bernaudin F, Verlhac S, Fréard F, Roudot-Thoraval F, Benkerrou M, Thuret I, et al. Multicenter prospective study of children with sickle cell disease: radiographic and psychometric correlation. J Child Neurol. (2000) 15:333–43. doi: 10.1177/088307380001500510
139. Tantawy AAG, Adly AAM, Ismail EAR, Habeeb NM, Farouk A. Circulating platelet and erythrocyte microparticles in young children and adolescents with sickle cell disease: relation to cardiovascular complications. Platelets. (2013) 24:605–14. doi: 10.3109/09537104.2012.749397
140. Hulbert ML, Ford AL. Understanding sickle cell brain drain. Blood. (2014) 124:830–1. doi: 10.1182/blood-2014-06-582403
141. Klijn CJM, Kappelle LJ. Haemodynamic stroke: clinical features, prognosis, and management. Lancet Neurol. (2010) 9:1008–17. doi: 10.1016/S1474-4422(10)70185-X
142. Shi J, Meng R, Konakondla S, Ding Y, Duan Y, Wu D, et al. Cerebral watershed infarcts may be induced by hemodynamic changes in blood flow. Neurol Res. (2017) 39:538–44. doi: 10.1080/01616412.2017.1315499
143. Bladin CF, Chambers BR, Donnan GA. Confusing stroke terminology: watershed or borderzone infarction? Stroke. (1993) 24:477–8. doi: 10.1161/01.STR.24.3.477
144. Kennedy C, Sokoloff L. An adaptation of the nitrous oxide method to the study of the cerebral circulation in children; normal values for cerebral blood flow and cerebral metabolic rate in childhood. J Clin Invest. (1957) 36:1130–7. doi: 10.1172/JCI103509
145. Powers WJ, Grubb RL, Darriet D, Raichle ME. Cerebral blood flow and cerebral metabolic rate of oxygen requirements for cerebral function and viability in humans. J Cereb Blood Flow Metab. (1985) 5:600–8. doi: 10.1038/jcbfm.1985.89
146. Baron JC. Pathophysiology of acute cerebral ischemia: PET studies in humans. Cerebrovasc Dis. (1991) 1:22–31. doi: 10.1159/000108877
147. Hyder F, Herman P, Bailey CJ, Møller A, Globinsky R, Fulbright RK, et al. Uniform distributions of glucose oxidation and oxygen extraction in gray matter of normal human brain: no evidence of regional differences of aerobic glycolysis. J Cereb Blood Flow Metab. (2016) 36:903–16. doi: 10.1177/0271678X15625349
148. Brown MM, Wade JP, Marshall J. Fundamental importance of arterial oxygen content in the regulation of cerebral blood flow in man. Brain. (1985) 108:81–93. doi: 10.1093/brain/108.1.81
149. Busija DW, Leffler CW, Pourcyrous M. Hyperthermia increases cerebral metabolic rate and blood flow in neonatal pigs. Am J Physiol Circ Physiol. (1988) 255:H343–6. doi: 10.1152/ajpheart.1988.255.2.H343
150. Willie CK, Tzeng Y-C, Fisher JA, Ainslie PN. Integrative regulation of human brain blood flow. J Physiol. (2014) 592:841–59. doi: 10.1113/jphysiol.2013.268953
151. Harper AM, Glass HI. Effect of alterations in the arterial carbon dioxide tension on the blood flow through the cerebral cortex at normal and low arterial blood pressures. J Neurol Neurosurg Psychiatry. (1965) 28:449–52. doi: 10.1136/jnnp.28.5.449
152. Hayashi T, Watabe H, Kudomi N, Kim KM, Enmi J-I, Hayashida K, et al. A theoretical model of oxygen delivery and metabolism for physiologic interpretation of quantitative cerebral blood flow and metabolic rate of oxygen. J Cereb Blood Flow Metab. (2003) 23:1314–23. doi: 10.1097/01.WCB.0000090506.76664.00
153. Baird AE, Austin MC, McKay WJ, Donnan GA. Changes in cerebral tissue perfusion during the first 48 hours of ischaemic stroke: relation to clinical outcome. J Neurol Neurosurg Psychiatry. (1996) 61:26–9. doi: 10.1136/jnnp.61.1.26
154. Baron JC, Bousser MG, Rey A, Guillard A, Comar D, Castaigne P. Reversal of focal ‘misery-perfusion syndrome' by extra-intracranial arterial bypass in hemodynamic cerebral ischemia. A case study with 15O positron emission tomography. Stroke. (1981) 12:454–9. doi: 10.1161/01.STR.12.4.454
155. Fieschi C. Cerebral blood flow and energy metabolism in vascular insufficiency. Stroke. (1980) 11:431–2. doi: 10.1161/01.STR.11.5.431
156. Behpour AM, Shah PS, Mikulis DJ, Kassner A. Cerebral blood flow abnormalities in children with sickle cell disease: a systematic review. Pediatr Neurol. (2013) 48:188–99. doi: 10.1016/j.pediatrneurol.2012.12.004
157. Bush AM, Borzage MT, Choi S, Václavu L, Tamrazi B, Nederveen AJ, et al. Determinants of resting cerebral blood flow in sickle cell disease. Am J Hematol. (2016) 91:912–7. doi: 10.1002/ajh.24441
158. Helton KJ, Paydar A, Glass J, Weirich EM, Hankins J, Li C-S, et al. Arterial spin-labeled perfusion combined with segmentation techniques to evaluate cerebral blood flow in white and gray matter of children with sickle cell anemia. Pediatr Blood Cancer. (2009) 52:85–91. doi: 10.1002/pbc.21745
159. Kawadler JM, Hales PW, Barker S, Cox TCS, Kirkham FJ, Clark CA. Cerebral perfusion characteristics show differences in younger versus older children with sickle cell anaemia: results from a multiple-inflow-time arterial spin labelling study. NMR Biomed. (2018) 31:e3915. doi: 10.1002/nbm.3915
160. Kirkham FJ, Calamante F, Bynevelt M, Gadian DG, Evans JP, Cox TC, et al. Perfusion magnetic resonance abnormalities in patients with sickle cell disease. Ann Neurol. (2001) 49:477–85. doi: 10.1002/ana.97.abs
161. Powars DR, Conti PS, Wong WY, Groncy P, Hyman C, Smith E, et al. Cerebral vasculopathy in sickle cell anemia: diagnostic contribution of positron emission tomography. Blood. (1999) 93:71–9.
162. Kosinski PD, Croal PL, Leung J, Williams S, Odame I, Hare GMT, et al. The severity of anaemia depletes cerebrovascular dilatory reserve in children with sickle cell disease: a quantitative magnetic resonance imaging study. Br J Haematol. (2017) 176:280–7. doi: 10.1111/bjh.14424
163. Nur E, Kim Y-S, Truijen J, van Beers EJ, Davis SCAT, Brandjes DP, et al. Cerebrovascular reserve capacity is impaired in patients with sickle cell disease. Blood. (2009) 114:3473–8. doi: 10.1182/blood-2009-05-223859
164. Václavu L, Meynart BN, Mutsaerts HJMM, Petersen ET, Majoie CBLM, VanBavel ET, et al. Hemodynamic provocation with acetazolamide shows impaired cerebrovascular reserve in adults with sickle cell disease. Haematologica. (2019) 104:690–9. doi: 10.3324/haematol.2018.206094
165. Bush AM, Coates TD, Wood JC. Diminished cerebral oxygen extraction and metabolic rate in sickle cell disease using T2 relaxation under spin tagging MRI. Magn Reson Med. (2018) 80:294–303. doi: 10.1002/mrm.27015
166. Fields ME, Guilliams KP, Ragan DK, Binkley MM, Eldeniz C, Chen Y, et al. Regional oxygen extraction predicts border zone vulnerability to stroke in sickle cell disease. Neurology. (2018) 90:e1134–42. doi: 10.1212/WNL.0000000000005194
167. Jordan LC, Gindville MC, Scott AO, Juttukonda MR, Strother MK, Kassim AA, et al. Non-invasive imaging of oxygen extraction fraction in adults with sickle cell anaemia. Brain. (2016) 139:738–50. doi: 10.1093/brain/awv397
168. Fowler NO, Smith O, Greenfield JC. Arterial blood oxygenation in sickle cell anemia. Am J Med Sci. (1957) 234:449–58. doi: 10.1097/00000441-195710000-00010
169. Homi J, Levee L, Higgs D, Thomas P, Serjeant G. Pulse oximetry in a cohort study of sickle cell disease. Clin Lab Haematol. (1997) 19:17–22. doi: 10.1046/j.1365-2257.1997.00215.x
170. Quinn CT, Ahmad N. Clinical correlates of steady-state oxyhaemoglobin desaturation in children who have sickle cell disease. Br J Haematol. (2005) 131:129–34. doi: 10.1111/j.1365-2141.2005.05738.x
171. Rackoff WR, Kunkel N, Silber JH, Asakura T, Ohene-Frempong K. Pulse oximetry and factors associated with hemoglobin oxygen desaturation in children with sickle cell disease. Blood. (1993) 81:3422–7.
172. Setty BNY, Stuart MJ, Dampier C, Brodecki D, Allen JL. Hypoxaemia in sickle cell disease: biomarker modulation and relevance to pathophysiology. Lancet. (2003) 362:1450–5. doi: 10.1016/S0140-6736(03)14689-2
173. Waltz X, Romana M, Lalanne-Mistrih M-L, Machado RF, Lamarre Y, Tarer V, et al. Hematologic and hemorheological determinants of resting and exercise-induced hemoglobin oxygen desaturation in children with sickle cell disease. Haematologica. (2013) 98:1039–44. doi: 10.3324/haematol.2013.083576
174. Caboot JB, Allen JL. Hypoxemia in sickle cell disease: significance and management. Paediatr Respir Rev. (2014) 15:17–23. doi: 10.1016/j.prrv.2013.12.004
175. Di Liberto G, Kiger L, Marden MC, Boyer L, Poitrine FC, Conti M, et al. Dense red blood cell and oxygen desaturation in sickle-cell disease. Am J Hematol. (2016) 91:1008–13. doi: 10.1002/ajh.24467
176. Rosen CL, Debaun MR, Strunk RC, Redline S, Seicean S, Craven DI, et al. Obstructive sleep apnea and sickle cell anemia. Pediatrics. (2014) 134:273–81. doi: 10.1542/peds.2013-4223
177. Needleman JP, Franco ME, Varlotta L, Reber-Brodecki D, Bauer N, Dampier C, et al. Mechanisms of nocturnal oxyhemoglobin desaturation in children and adolescents with sickle cell disease. Pediatr. Pulmonol. (1999) 28:418–22. doi: 10.1002/(SICI)1099-0496(199912)28:6<418::AID-PPUL6>3.3.CO;2-4
178. Charache S, Grisolia S, Fiedler AJ, Hellegers AE. Effect of 2,3-diphosphoglycerate on oxygen affinity of blood in sickle cell anemia. J Clin Invest. (1970) 49:806–12. doi: 10.1172/JCI106294
179. Milner PF. Oxygen transport in sickle cell anemia. Arch Intern Med. (1974) 133:565–72. doi: 10.1001/archinte.1974.00320160059006
180. Riggs A, Wells M. The oxygen equilibrium of sickle-cell hemoglobin. Biochim Biophys Acta. (1961) 50:243–8. doi: 10.1016/0006-3002(61)90322-5
181. Seakins M, Gibbs WN, Milner PF, Bertles JF. Erythrocyte Hb-S concentration. An important factor in the low oxygen affinity of blood in sickle cell anemia. J Clin Invest. (1973) 52:422–32. doi: 10.1172/JCI107199
182. Needleman JP, Setty BNN, Varlotta L, Dampier C, Allen JL. Measurement of hemoglobin saturation by oxygen in children and adolescents with sickle cell disease. Pediatr Pulmonol. (1999) 28:423–8. doi: 10.1002/(SICI)1099-0496(199912)28:6andlt;423::AID-PPUL7andgt;3.0.CO;2-C
183. Pianosi P, Charge TD, Esseltine DW, Coates AL. Pulse oximetry in sickle cell disease. Arch Dis Child. (1993) 68:735–8. doi: 10.1136/adc.68.6.735
184. Serjeant G, Serjeant B, Stephens A, Roper D, Higgs D, Beckford M, et al. Determinants of haemoglobin level in steady-state homozygous sickle cell disease. Br J Haematol. (1996) 92:143–9. doi: 10.1046/j.1365-2141.1996.284816.x
185. Young RC, Rachal RE, Del Pilar Aguinaga M, Nelson BL, Kim BC, Winter WP, et al. Automated oxyhemoglobin dissociation curve construction to assess sickle cell anemia therapy. J Natl Med Assoc. (2000) 92:430–5.
186. Safo MK, Kato GJ. Therapeutic strategies to alter the oxygen affinity of sickle hemoglobin. Hematol Oncol Clin North Am. (2014) 28:217–31. doi: 10.1016/j.hoc.2013.11.001
187. Charlot K, Antoine-Jonville S, Moeckesch B, Jumet S, Romana M, Waltz X, et al. Cerebral and muscle microvascular oxygenation in children with sickle cell disease: influence of hematology, hemorheology and vasomotion. Blood Cells Mol Dis. (2017) 65:23–8. doi: 10.1016/j.bcmd.2017.03.015
188. Quinn CT, Dowling MM. Cerebral tissue hemoglobin saturation in children with sickle cell disease. Pediatr Blood Cancer. (2012) 59:881–7. doi: 10.1002/pbc.24227
189. Raj A, Bertolone SJ, Mangold S, Edmonds HL. Assessment of cerebral tissue oxygenation in patients with sickle cell disease: effect of transfusion therapy. J Pediatr Hematol Oncol. (2004) 26:279–83. doi: 10.1097/00043426-200405000-00004
190. Baird RL, Weiss DL, Ferguson AD, French JH, Scott RB. Studies in sickle cell anemia. XXI. Clinico-pathological aspects of neurological manifestations. Pediatrics. (1964) 34:92–100.
191. Guilliams KP, Fields ME, Ragan DK, Eldeniz C, Binkley MM, Chen Y, et al. Red cell exchange transfusions lower cerebral blood flow and oxygen extraction fraction in pediatric sickle cell anemia. Blood. (2018) 131:1012–21. doi: 10.1182/blood-2017-06-789842
192. Herold S, Brozovic M, Gibbs J, Lammertsma AA, Leenders KL, Carr D, et al. Measurement of regional cerebral blood flow, blood volume and oxygen metabolism in patients with sickle cell disease using positron emission tomography. Stroke. (1986) 17:692–8. doi: 10.1161/01.STR.17.4.692
193. Prohovnik I, Hurlet-Jensen A, Adams R, De Vivo D, Pavlakis SG. Hemodynamic etiology of elevated flow velocity and stroke in sickle-cell disease. J Cereb Blood Flow Metab. (2009) 29:803–10. doi: 10.1038/jcbfm.2009.6
194. Leung J, Duffin J, Fisher JA, Kassner A. MRI-based cerebrovascular reactivity using transfer function analysis reveals temporal group differences between patients with sickle cell disease and healthy controls. NeuroImage Clin. (2016) 12:624–30. doi: 10.1016/j.nicl.2016.09.009
195. Cottrell JE, Patel P. Cottrell and Patel's Neuroanesthesia. 6th ed. Elsevier (2017). Available online at: https://books.google.co.uk/books?id=xVboDAAAQBAJ.
196. Mandell DM, Han JS, Poublanc J, Crawley AP, Kassner A, Fisher JA, et al. Selective reduction of blood flow to white matter during hypercapnia corresponds with leukoaraiosis. Stroke. (2008) 39:1993–8. doi: 10.1161/STROKEAHA.107.501692
197. Thomas BP, Liu P, Park DC, van Osch MJP, Lu H. Cerebrovascular reactivity in the brain white matter: magnitude, temporal characteristics, and age effects. J Cereb Blood Flow Metab. (2014) 34:242–7. doi: 10.1038/jcbfm.2013.194
198. Binks AP, Cunningham VJ, Adams L, Banzett RB. Gray matter blood flow change is unevenly distributed during moderate isocapnic hypoxia in humans. J Appl Physiol. (2008) 104:212–7. doi: 10.1152/japplphysiol.00069.2007
199. Pagani M, Salmaso D, Sidiras GG, Jonsson C, Jacobsson H, Larsson SA, et al. Impact of acute hypobaric hypoxia on blood flow distribution in brain. Acta Physiol. (2011) 202:203–9. doi: 10.1111/j.1748-1716.2011.02264.x
200. Fields ME, Guilliams KP, Ragan D, Eldeniz C, Binkley M, Hulbert ML, et al. Elevations in MR measurements of whole brain and regional cerebral blood flow and oxygen extraction fraction suggest cerebral metabolic stress in children with sickle cell disease unaffected by overt stroke. Blood. (2015) 126:69. Available online at: http://www.bloodjournal.org/content/126/23/69
201. Chai Y, Bush AM, Coloigner J, Nederveen AJ, Tamrazi B, Vu C, et al. White matter has impaired resting oxygen delivery in sickle cell patients. Am J Hematol. (2019) 94:467–74. doi: 10.1002/ajh.25423
202. Rodgers GP, Clark CM, Larson SM, Rapoport SI, Nienhuis AW, Schechter AN. Brain glucose metabolism in neurologically normal patients with sickle cell disease. Regional alterations. Arch Neurol. (1988) 45:78–82. doi: 10.1001/archneur.1988.00520250084025
203. Al-Kandari FA, Owunwanne A, Syed GM, Ar Marouf R, Elgazzar AH, Shiekh M, et al. Regional cerebral blood flow in patients with sickle cell disease: study with single photon emission computed tomography. Ann Nucl Med. (2007) 21:439–45. doi: 10.1007/s12149-007-0050-y
204. Deus-Silva L, Bonilha L, Damasceno BP, Costa ALF, Yasuda CL, Costa FF, et al. Brain perfusion impairment in neurologically asymptomatic adult patients with sickle-cell disease shown by voxel-based analysis of SPECT images. Front Neurol. (2013) 4:207. doi: 10.3389/fneur.2013.00207
205. Parsa MA, Mehregany D, Schulz SC. Psychiatric manifestation of sickle cell disease and findings on single photon emission computed tomography. Psychosomatics. (1992) 33:239–41. doi: 10.1016/S0033-3182(92)72011-8
206. Numaguchi Y, Haller JS, Humbert JR, Robinson AE, Lindstrom WW, Gruenauer LM, et al. Cerebral blood flow mapping using stable xenon-enhanced CT in sickle cell cerebrovascular disease. Neuroradiology. (1990) 32:289–95. doi: 10.1007/BF00593047
207. Rivera-Lara L, Zorrilla-Vaca A, Geocadin RG, Healy RJ, Ziai W, Mirski MA. Cerebral autoregulation-oriented therapy at the bedside: a comprehensive review. Anesthesiology. (2017) 126:1187–99. doi: 10.1097/ALN.0000000000001625
208. Kim Y-S, Nur E, van Beers EJ, Truijen J, Davis SCATAT, Biemond BJ, et al. Dynamic cerebral autoregulation in homozygous Sickle cell disease. Stroke. (2009) 40:808–14. doi: 10.1161/STROKEAHA.108.531996
209. Donnelly J, Budohoski KP, Smielewski P, Czosnyka M. Regulation of the cerebral circulation: bedside assessment and clinical implications. Crit Care. (2016) 20:129. doi: 10.1186/s13054-016-1293-6
210. Meng L, Hou W, Chui J, Han R, Gelb AW. Cardiac output and cerebral blood flow: the integrated regulation of brain perfusion in adult humans. Anesthesiology. (2015) 123:1198–208. doi: 10.1097/ALN.0000000000000872
211. Nishimura S, Suzuki A, Hatazawa J, Nishimura H, Shirane R, Yasui N, et al. Cerebral blood-flow responses to induced hypotension and to CO2 inhalation in patients with major cerebral artery occlusive disease: a positron-emission tomography study. Neuroradiology. (1999) 41:73–9. doi: 10.1007/s002340050709
212. Meng L, Gelb AW. Regulation of cerebral autoregulation by carbon dioxide. Anesthesiology. (2015) 122:196–205. doi: 10.1097/ALN.0000000000000506
213. Tzeng YC, Ainslie PN, Cooke WH, Peebles KC, Willie CK, MacRae BA, et al. Assessment of cerebral autoregulation: the quandary of quantification. Am J Physiol Heart Circ Physiol. (2012) 303:H658–71. doi: 10.1152/ajpheart.00328.2012
214. Johnson MC, Kirkham FJ, Redline S, Rosen CL, Yan Y, Roberts I, et al. Left ventricular hypertrophy and diastolic dysfunction in children with sickle cell disease are related to asleep and waking oxygen desaturation. Blood. (2010) 116:16–21. doi: 10.1182/blood-2009-06-227447
215. Pegelow CH, Colangelo L, Steinberg M, Wright EC, Smith J, Phillips G, et al. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med. (1997) 102:171–7. doi: 10.1016/S0002-9343(96)00407-X
216. Coley SC, Porter DA, Calamante F, Chong WK, Connelly A. Quantitative MR diffusion mapping and cyclosporine-induced neurotoxicity. AJNR Am J Neuroradiol. (1999) 20:1507–10.
217. Schoonman GG, Sándor PS, Nirkko AC, Lange T, Jaermann T, Dydak U, et al. Hypoxia-induced acute mountain sickness is associated with intracellular cerebral edema: a 3 T magnetic resonance imaging study. J Cereb Blood Flow Metab. (2008) 28:198–206. doi: 10.1038/sj.jcbfm.9600513
218. Connes P, Coates TD. Autonomic nervous system dysfunction: implication in sickle cell disease. C R Biol. (2013) 336:142–7. doi: 10.1016/j.crvi.2012.09.003
219. Ainslie PN, Ogoh S. Regulation of cerebral blood flow in mammals during chronic hypoxia: a matter of balance. Exp Physiol. (2010) 95:251–62. doi: 10.1113/expphysiol.2008.045575
220. Nath KA, Katusic ZS, Gladwin MT. The perfusion paradox and vascular instability in sickle cell disease. Microcirculation. (2004) 11:179–93. doi: 10.1080/10739680490278592
221. Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med. (2004) 36:707–17. doi: 10.1016/j.freeradbiomed.2003.11.032
222. Ainslie PN, Brassard P. Why is the neural control of cerebral autoregulation so controversial? F1000Prime Rep. (2014). 6:14. doi: 10.12703/P6-14
223. Garry PS, Ezra M, Rowland MJ, Westbrook J, Pattinson KTS. The role of the nitric oxide pathway in brain injury and its treatment–from bench to bedside. Exp Neurol. (2015) 263:235–43. doi: 10.1016/j.expneurol.2014.10.017
224. Hoiland RL, Bain AR, Rieger MG, Bailey DM, Ainslie PN. Hypoxemia, oxygen content, and the regulation of cerebral blood flow. Am J Physiol Regul Integr Comp Physiol. (2016) 310:R398–413. doi: 10.1152/ajpregu.00270.2015
225. Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. (1996) 16:981–7. doi: 10.1097/00004647-199609000-00023
226. Hamner JW, Tan CO, Lee K, Cohen MA, Taylor JA. Sympathetic control of the cerebral vasculature in humans. Stroke. (2010) 41:102–9. doi: 10.1161/STROKEAHA.109.557132
227. Ogoh S, Brothers RM, Eubank WL, Raven PB. Autonomic neural control of the cerebral vasculature: acute hypotension. Stroke. (2008) 39:1979–87. doi: 10.1161/STROKEAHA.107.510008
228. Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD. Autonomic neural control of dynamic cerebral autoregulation in humans. Circulation. (2002) 106:1814–20. doi: 10.1161/01.CIR.0000031798.07790.FE
229. Jordan J, Shannon JR, Diedrich A, Black B, Costa F, Robertson D, et al. Interaction of carbon dioxide and sympathetic nervous system activity in the regulation of cerebral perfusion in humans. Hypertens. (2000) 36:383–8. doi: 10.1161/01.HYP.36.3.383
230. Zhang P, Huang G, Shi X. Cerebral vasoreactivity during hypercapnia is reset by augmented sympathetic influence. J Appl Physiol. (2011) 110:352–8. doi: 10.1152/japplphysiol.00802.2010
231. Chalacheva P, Kato RM, Shah P, Veluswamy S, Denton CC, Sunwoo J, et al. Sickle cell disease subjects have a distinct abnormal autonomic phenotype characterized by peripheral vasoconstriction with blunted cardiac response to head-up tilt. Front Physiol. (2019) 10:381. doi: 10.3389/fphys.2019.00381
232. L'Esperance VS, Cox SE, Simpson D, Gill C, Makani J, Soka D, et al. Peripheral vascular response to inspiratory breath hold in paediatric homozygous sickle cell disease. Exp. Physiol. (2013) 98:49–56. doi: 10.1113/expphysiol.2011.064055
233. Sangkatumvong S, Khoo MCK, Kato R, Detterich JA, Bush A, Keens TG, et al. Peripheral vasoconstriction and abnormal parasympathetic response to sighs and transient hypoxia in sickle cell disease. Am J Respir Crit Care Med. (2011) 184:474–81. doi: 10.1164/rccm.201103-0537OC
234. Shah P, Khaleel M, Thuptimdang W, Sunwoo J, Veluswamy S, Chalacheva P, et al. Mental stress causes vasoconstriction in sickle cell disease and normal controls. Haematologica. (2019). doi: 10.3324/haematol.2018.211391. [Epub ahead of print].
235. Detterich J, Alexy T, Rabai M, Wenby R, Dongelyan A, Coates T, et al. Low-shear red blood cell oxygen transport effectiveness is adversely affected by transfusion and further worsened by deoxygenation in sickle cell disease patients on chronic transfusion therapy. Transfusion. (2013) 53:297–305. doi: 10.1111/j.1537-2995.2012.03822.x
236. Tripette J, Alexy T, Hardy-Dessources M-D, Mougenel D, Beltan E, Chalabi T, et al. Red blood cell aggregation, aggregate strength and oxygen transport potential of blood are abnormal in both homozygous sickle cell anemia and sickle-hemoglobin C disease. Haematologica. (2009) 94:1060–5. doi: 10.3324/haematol.2008.005371
237. Waltz X, Hardy-Dessources M-D, Lemonne N, Mougenel D, Lalanne-Mistrih M-L, Lamarre Y, et al. Is there a relationship between the hematocrit-to-viscosity ratio and microvascular oxygenation in brain and muscle? Clin Hemorheol Microcirc. (2015) 59:37–43. doi: 10.3233/CH-131742
238. Waschke KF, Krieter H, Hagen G, Albrecht DM, Van Ackern K, Kuschinsky W. Lack of dependence of cerebral blood flow on blood viscosity after blood exchange with a Newtonian O2 carrier. J Cereb Blood Flow Metab. (1994) 14:871–6. doi: 10.1038/jcbfm.1994.109
239. Brown MM, Marshall J. Regulation of cerebral blood flow in response to changes in blood viscosity. Lancet. (1985) 1:604–9. doi: 10.1016/S0140-6736(85)92145-2
240. Brown MM, Marshall J. Effect of plasma exchange on blood viscosity and cerebral blood flow. Br Med J. (1982) 284:1733–6. doi: 10.1136/bmj.284.6331.1733
241. Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. (2007) 49:2379–93. doi: 10.1016/j.jacc.2007.02.059
242. Chiu J-J, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. (2011) 91:327–87. doi: 10.1152/physrev.00047.2009
243. Cosemans JMEM, Angelillo-Scherrer A, Mattheij NJA, Heemskerk JWM. The effects of arterial flow on platelet activation, thrombus growth, and stabilization. Cardiovasc Res. (2013) 99:342–52. doi: 10.1093/cvr/cvt110
244. Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. (2003) 362:1028–37. doi: 10.1016/S0140-6736(03)14412-1
245. Kirkham FJ, Datta AK. Hypoxic adaptation during development: relation to pattern of neurological presentation and cognitive disability. Dev Sci. (2006) 9:411–27. doi: 10.1111/j.1467-7687.2006.00507.x
246. Cramer NP, Korotcov A, Bosomtwi A, Xu X, Holman DR, Whiting K, et al. Neuronal and vascular deficits following chronic adaptation to high altitude. Exp Neurol. (2019) 311:293–304. doi: 10.1016/j.expneurol.2018.10.007
247. Wang Y, Qiu J, Luo S, Xie X, Zheng Y, Zhang K, et al. High shear stress induces atherosclerotic vulnerable plaque formation through angiogenesis. Regen Biomater. (2016) 3:257–67. doi: 10.1093/rb/rbw021
248. Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. (2011) 2:1117–33. doi: 10.1177/1947601911423654
249. Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. (1999) 282:2035–42. doi: 10.1001/jama.282.21.2035
250. Croal PL, Serafin MG, Kosinski P, Leung J, Williams S, Kassner A. Quantitative MRI of hemodynamic compromise in children with sickle cell disease: new insights into pathophysiology. Blood. (2015) 126:2168. Available online at: http://www.bloodjournal.org/content/126/23/2168?sso-checked=true%3fsso-checked=true
251. Watchmaker JM, Juttukonda MR, Davis LT, Scott AO, Faraco CC, Gindville MC, et al. Hemodynamic mechanisms underlying elevated oxygen extraction fraction (OEF) in moyamoya and sickle cell anemia patients. J Cereb Blood Flow Metab. (2018) 38:1618–30. doi: 10.1177/0271678X16682509
252. Vaclavu L, Petersen ET, VanBavel ET, Majoie CB, Nederveen AJ, Biemond BJ. Reduced cerebral metabolic rate of oxygen in adults with sickle cell disease. Blood. (2018) 132:11. doi: 10.1182/blood-2018-99-116194
253. Juttukonda MR, Donahue MJ, Davis LT, Gindville MC, Lee CA, Patel NJ, et al. Preliminary evidence for cerebral capillary shunting in adults with sickle cell anemia. J Cereb Blood Flow Metab. (2019) 39:1099–110. doi: 10.1177/0271678X17746808
254. Wood JC. Unwinding the path from anemia to stroke. Blood. (2018) 131:950–52. doi: 10.1182/blood-2018-01-824185
255. Juttukonda MR, Lee CA, Patel NJ, Davis LT, Waddle SL, Gindville MC, et al. Differential cerebral hemometabolic responses to blood transfusions in adults and children with sickle cell anemia. J Magn Reson Imaging. (2019) 49:466–77. doi: 10.1002/jmri.26213
256. Li W, Xu X, Liu P, Jacobs E, Strouse JJ, Casella JF, et al. Quantification of whole-brain oxygenation extraction fraction among people with sickle cell anemia without stroke history. Blood. (2017) 130:3524. Available online at: http://www.bloodjournal.org/content/130/Suppl_1/3524
257. Bush AM, Borzage M, Choi S, Coates T, Wood JC. Elevated cerebral metabolic oxygen consumption in sickle cell disease. Blood. (2014) 124:2706. Available online at: http://www.bloodjournal.org/content/124/21/2706
258. Bush AM, Borzage M, Choi S, Coates T, Wood JC. Elevated cerebral blood oxygen extraction in non-transfused sickle cell disease patients. Blood. (2014) 124:1387. Available online at: http://www.bloodjournal.org/content/124/21/1387
259. Croal PL, Leung J, Phillips CL, Serafin MG, Kassner A. Quantification of pathophysiological alterations in venous oxygen saturation: a comparison of global MR susceptometry techniques. Magn Reson Imaging. (2019) 58:18–23. doi: 10.1016/j.mri.2019.01.008
260. Nahavandi M, Millis RM, Tavakkoli F, Wyche MQ, Perlin E, Winter WP, et al. Arterialization of peripheral venous blood in sickle cell disease. J. Natl. Med. Assoc. (2002) 94:320–6.
261. Niss O, Quinn CT, Lane A, Daily J, Khoury PR, Bakeer N, et al. Cardiomyopathy With Restrictive Physiology in Sickle Cell Disease. JACC Cardiovasc Imaging. (2016) 9:243–52. doi: 10.1016/j.jcmg.2015.05.013
262. Kirkham FJ, Hewes DK, Prengler M, Wade A, Lane R, Evans JP. Nocturnal hypoxaemia and central-nervous-system events in sickle-cell disease. Lancet. (2001) 357:1656–9. doi: 10.1016/S0140-6736(00)04821-2
263. Quinn CT, Sargent JW. Daytime steady-state haemoglobin desaturation is a risk factor for overt stroke in children with sickle cell anaemia. Br J Haematol. (2008) 140:336–9. doi: 10.1111/j.1365-2141.2007.06927.x
264. Henderson JN, Noetzel MJ, McKinstry RC, White DA, Armstrong M, DeBaun MR. Reversible posterior leukoencephalopathy syndrome and silent cerebral infarcts are associated with severe acute chest syndrome in children with sickle cell disease. Blood. (2003) 101:415–9. doi: 10.1182/blood-2002-04-1183
265. Hogan AM, Telfer PT, Kirkham FJ, de Haan M. Precursors of executive function in infants with sickle cell anemia. J Child Neurol. (2013) 28:1197–202. doi: 10.1177/0883073812453495
266. Hollocks MJ, Kok TB, Kirkham FJ, Gavlak J, Inusa BP, DeBaun MR, et al. Nocturnal oxygen desaturation and disordered sleep as a potential factor in executive dysfunction in sickle cell anemia. J Int Neuropsychol Soc. (2012) 18:168–73. doi: 10.1017/S1355617711001469
267. King AA, Strouse JJ, Rodeghier MJ, Compas BE, Casella JF, McKinstry RC, et al. Parent education and biologic factors influence on cognition in sickle cell anemia. Am J Hematol. (2014) 89:162–7. doi: 10.1002/ajh.23604
268. Fridlyand D, Wilder C, Clay ELJ, Gilbert B, Pace BS. Stroke in a child with hemoglobin SC disease: a case report describing use of hydroxyurea after transfusion therapy. Pediatr Rep. (2017) 9:6984. doi: 10.4081/pr.2017.6984
269. Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. (2000) 342:1855–65. doi: 10.1056/NEJM200006223422502
270. Makani J, Kirkham FJ, Komba A, Ajala-Agbo T, Otieno G, Fegan G, et al. Risk factors for high cerebral blood flow velocity and death in Kenyan children with Sickle Cell Anaemia: role of haemoglobin oxygen saturation and febrile illness. Br J Haematol. (2009) 145:529–32. doi: 10.1111/j.1365-2141.2009.07660.x
271. Rankine-Mullings AE, Morrison-Levy N, Soares D, Aldred K, King L, Ali S, et al. Transcranial Doppler velocity among Jamaican children with sickle cell anaemia: determining the significance of haematological values and nutrition. Br J Haematol. (2018) 181:242–51. doi: 10.1111/bjh.15162
272. Abboud MR, Cure J, Granger S, Gallagher D, Hsu L, Wang W, et al. Magnetic resonance angiography in children with sickle cell disease and abnormal transcranial Doppler ultrasonography findings enrolled in the STOP study. Blood. (2004) 103:2822–6. doi: 10.1182/blood-2003-06-1972
273. Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood. (2006) 108:847–52. doi: 10.1182/blood-2005-10-009506
274. Yamauchi H, Fukuyama H, Nagahama Y, Nabatame H, Nakamura K, Yamamoto Y, et al. Evidence of misery perfusion and risk for recurrent stroke in major cerebral arterial occlusive diseases from PET. J Neurol Neurosurg Psychiatry. (1996) 61:18–25. doi: 10.1136/jnnp.61.1.18
275. Yamauchi H, Fukuyama H, Nagahama Y, Nabatame H, Ueno M, Nishizawa S, et al. Significance of increased oxygen extraction fraction in five-year prognosis of major cerebral arterial occlusive diseases. J. Nucl. Med. (1999) 40:1992–8.
276. Connelly A, Chong WK, Johnson CL, Ganesan V, Gadian DG, Kirkham FJ. Diffusion weighted magnetic resonance imaging of compromised tissue in stroke. Arch Dis Child. (1997) 77:38–41. doi: 10.1136/adc.77.1.38
277. Powers WJ, Grubb RL, Baker RP, Mintun MA, Raichle ME. Regional cerebral blood flow and metabolism in reversible ischemia due to vasospasm. Determination by positron emission tomography. J Neurosurg. (1985) 62:539–46. doi: 10.3171/jns.1985.62.4.0539
278. Dowling MM, Quinn CT, Rogers ZR, Buchanan GR. Acute silent cerebral infarction in children with sickle cell anemia. Pediatr Blood Cancer. (2010) 54:461–4. doi: 10.1002/pbc.22242
279. Dowling MM, Quinn CT, Plumb P, Rogers ZR, Rollins NK, Koral K, et al. Acute silent cerebral ischemia and infarction during acute anemia in children with and without sickle cell disease. Blood. (2012) 120:3891–7. doi: 10.1182/blood-2012-01-406314
280. Enninful-Eghan H, Moore RH, Ichord R, Smith-Whitley K, Kwiatkowski JL. Transcranial Doppler ultrasonography and prophylactic transfusion program is effective in preventing overt stroke in children with sickle cell disease. J Pediatr. (2010) 157:479–84. doi: 10.1016/j.jpeds.2010.03.007
281. Lee KH, McKie VC, Sekul EA, Adams RJ, Nichols FT. Unusual encephalopathy after acute chest syndrome in sickle cell disease: acute necrotizing encephalitis. J Pediatr Hematol Oncol. (2002) 24:585–8. doi: 10.1097/00043426-200210000-00021
282. Quinn CT, McKinstry RC, Dowling MM, Ball WS, Kraut MA, Casella JF, et al. Acute silent cerebral ischemic events in children with sickle cell anemia. JAMA Neurol. (2013) 70:58–65. doi: 10.1001/jamaneurol.2013.576
283. Derdeyn CP, Videen TO, Yundt KD, Fritsch SM, Carpenter DA, Grubb RL, et al. Variability of cerebral blood volume and oxygen extraction: stages of cerebral haemodynamic impairment revisited. Brain. (2002) 125:595–607. doi: 10.1093/brain/awf047
284. Yamauchi H, Higashi T, Kagawa S, Nishii R, Kudo T, Sugimoto K, et al. Is misery perfusion still a predictor of stroke in symptomatic major cerebral artery disease? Brain. (2012) 135:2515–26. doi: 10.1093/brain/aws131
285. Hurlet-Jensen AM, Prohovnik I, Pavlakis SG, Piomelli S. Effects of total hemoglobin and hemoglobin S concentration on cerebral blood flow during transfusion therapy to prevent stroke in sickle cell disease. Stroke. 25:1688–92. doi: 10.1161/01.STR.25.8.1688
286. Venketasubramanian N, Prohovnik I, Hurlet A, Mohr JP, Piomelli S. Middle cerebral artery velocity changes during transfusion in sickle cell anemia. Stroke. (1994) 25:2153–8. doi: 10.1161/01.STR.25.11.2153
287. Jordan LC, Juttukonda MR, Kassim AA, DeBaun MR, Davis LT, Pruthi S, et al. Haploidentical bone marrow transplantation improves cerebral hemodynamics in adults with sickle cell disease. Am J Hematol. (2019) 94:E155–8. doi: 10.1002/ajh.25455
288. Fields ME, Guilliams KP, Ragan D, Binkley MM, Mirro A, Fellah S, et al. Hydroxyurea reduces cerebral metabolic stress in patients with sickle cell anemia. Blood. (2019) 133:2436–44. doi: 10.1182/blood-2018-09-876318
289. Prohovnik I, Pavlakis SG, Piomelli S, Bello J, Mohr JP, Hilal S, et al. Cerebral hyperemia, stroke, and transfusion in sickle cell disease. Neurology. (1989) 39:344–8. doi: 10.1212/WNL.39.3.344
290. Coates TD. So what if blood is thicker than water? Blood. (2011) 117:745–6. doi: 10.1182/blood-2010-11-314484
291. Detterich JA. Simple chronic transfusion therapy, a crucial therapeutic option for sickle cell disease, improves but does not normalize blood rheology: what should be our goals for transfusion therapy? Clin Hemorheol Microcirc. (2018) 68:173–86. doi: 10.3233/CH-189006
292. Hood AM, King AA, Fields ME, Ford AL, Guilliams KP, Hulbert ML, et al. Higher executive abilities following a blood transfusion in children and young adults with sickle cell disease. Pediatr Blood Cancer. (2019) e27899. doi: 10.1002/pbc.27899
293. Wood JC. Red cells, iron, and erythropoiesis: brain O2 reserve in sickle cell disease. Blood. (2019) 133:2356. doi: 10.1182/blood-2019-04-901124
294. Quinn CT, Dowling MM. Anemia and ischemia: how low can you go? Blood. (2015) 125:1516–7. doi: 10.1182/blood-2015-01-619981
295. van der Land V, Zwanenburg JJM, Fijnvandraat K, Biemond BJ, Hendrikse J, Mutsaerts HJMM, et al. Cerebral lesions on 7 tesla MRI in patients with sickle cell anemia. Cerebrovasc Dis. (2015) 39:181–9. doi: 10.1159/000373917
296. Savage WJ, Barron-Casella E, Fu Z, Dulloor P, Williams L, Crain BJ, et al. Plasma glial fibrillary acidic protein levels in children with sickle cell disease. Am J Hematol. (2011) 86:427–9. doi: 10.1002/ajh.21995
297. Kapustin D, Leung J, Odame I, Williams S, Shroff M, Kassner A. Hydroxycarbamide treatment in children with Sickle Cell Anaemia is associated with more intact white matter integrity: a quantitative MRI study. Br. J. Haematol. (2019). doi: 10.1111/bjh.16063. [Epub ahead of print].
298. Kral MC, Brown RT. Transcranial Doppler ultrasonography and executive dysfunction in children with sickle cell disease. J Pediatr Psychol. (2004) 29:185–95. doi: 10.1093/jpepsy/jsh020
299. Prussien KV, Salihu A, Abdullahi SU, Galadanci NA, Bulama K, Belonwu RO, et al. Associations of transcranial doppler velocity, age, and gender with cognitive function in children with sickle cell anemia in Nigeria. Child Neuropsychol. (2019) 25:705–20. doi: 10.1080/09297049.2018.1526272
300. Sanchez CE, Schatz J, Roberts CW. Cerebral blood flow velocity and language functioning in pediatric sickle cell disease. J Int Neuropsychol Soc. (2010) 16:326–34. doi: 10.1017/S1355617709991366
301. Zou P, Helton KJ, Smeltzer M, Li C-S, Conklin HM, Gajjar A, et al. Hemodynamic responses to visual stimulation in children with sickle cell anemia. Brain Imaging Behav. (2011) 5:295–306. doi: 10.1007/s11682-011-9133-4
302. Bush A, Chai Y, Choi SY, Vaclavu L, Holland S, Nederveen A, et al. Pseudo continuous arterial spin labeling quantification in anemic subjects with hyperemic cerebral blood flow. Magn Reson Imaging. (2018) 47:137–46. doi: 10.1016/j.mri.2017.12.011
303. Hales PW, Kirkham FJ, Clark CA. A general model to calculate the spin-lattice (T1) relaxation time of blood, accounting for haematocrit, oxygen saturation and magnetic field strength. J Cereb Blood Flow Metab. (2016) 36:370–4. doi: 10.1177/0271678X15605856
304. Juttukonda MR, Jordan LC, Gindville MC, Davis LT, Watchmaker JM, Pruthi S, et al. Cerebral hemodynamics and pseudo-continuous arterial spin labeling considerations in adults with sickle cell anemia. NMR Biomed. (2017) 30:e3681. doi: 10.1002/nbm.3681
Keywords: sickle cell disease, stroke, silent cerebral infarction, cerebral hemodynamics, vascular instability, anemia, oxygen extraction fraction, cerebrovascular reserve
Citation: Stotesbury H, Kawadler JM, Hales PW, Saunders DE, Clark CA and Kirkham FJ (2019) Vascular Instability and Neurological Morbidity in Sickle Cell Disease: An Integrative Framework. Front. Neurol. 10:871. doi: 10.3389/fneur.2019.00871
Received: 09 May 2019; Accepted: 26 July 2019;
Published: 13 August 2019.
Edited by:
Nishant K. Mishra, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Monica Hulbert, Washington University in St. Louis, United StatesCopyright © 2019 Stotesbury, Kawadler, Hales, Saunders, Clark and Kirkham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanne Stotesbury, aGFubmUuc3RvdGVzYnVyeS4xNUB1Y2wuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.