
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 25 July 2019
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00790
This article is part of the Research Topic Strategies to Fight Exercise Intolerance in Neuromuscular Disorders View all 11 articles
 Carmen Fiuza-Luces1
Carmen Fiuza-Luces1 Pedro L. Valenzuela2Sara Laine-Menéndez1Miguel Fernández-de la Torre1Verónica Bermejo-Gómez1Laura Rufián-Vázquez1Joaquín Arenas1,3Miguel A. Martín1,3
Pedro L. Valenzuela2Sara Laine-Menéndez1Miguel Fernández-de la Torre1Verónica Bermejo-Gómez1Laura Rufián-Vázquez1Joaquín Arenas1,3Miguel A. Martín1,3 Alejandro Lucia4,5
Alejandro Lucia4,5 María Morán1,3*
María Morán1,3*Purpose: Mitochondrial diseases (MD) are among the most prevalent neuromuscular disorders. Unfortunately, no curative treatment is yet available. This study analyzed the effects of exercise training in an animal model of respiratory chain complex I deficiency, the Harlequin (Hq) mouse, which replicates the clinical features of this condition.
Methods: Male heterozygous Harlequin (Hq/Y) mice were assigned to an “exercise” (n = 10) or a “sedentary” control group (n = 11), with the former being submitted to an 8 week combined exercise training intervention (aerobic + resistance training performed five times/week). Aerobic fitness, grip strength, and balance were assessed at the beginning and at the end of the intervention period in all the Hq mice. Muscle biochemical analyses (with results expressed as percentage of reference data from age/sex-matched sedentary wild-type mice [n = 12]) were performed at the end of the aforementioned period for the assessment of major molecular signaling pathways involved in muscle anabolism (mTOR activation) and mitochondrial biogenesis (proliferator activated receptor gamma co-activator 1α [PGC-1α] levels), and enzyme activity and levels of respiratory chain complexes, and antioxidant enzyme levels.
Results: Exercise training resulted in significant improvements in aerobic fitness (−33 ± 13 m and 83 ± 43 m for the difference post- vs. pre-intervention in total distance covered in the treadmill tests in control and exercise group, respectively, p = 0.014) and muscle strength (2 ± 4 g vs. 17 ± 6 g for the difference post vs. pre-intervention, p = 0.037) compared to the control group. Higher levels of ribosomal protein S6 kinase beta-1 phosphorylated at threonine 389 (156 ± 30% vs. 249 ± 30%, p = 0.028) and PGC-1α (82 ± 7% vs. 126 ± 19% p = 0.032) were observed in the exercise-trained mice compared with the control group. A higher activity of respiratory chain complexes I (75 ± 4% vs. 95 ± 6%, p = 0.019), III (79 ± 5% vs. 97 ± 4%, p = 0.031), and V (77 ± 9% vs. 105 ± 9%, p = 0.024) was also found with exercise training. Exercised mice presented with lower catalase levels (204 ± 22% vs. 141 ± 23%, p = 0.036).
Conclusion: In a mouse model of MD, a training intervention combining aerobic and resistance exercise increased aerobic fitness and muscle strength, and mild improvements were found for activated signaling pathways involved in muscle mitochondrial biogenesis and anabolism, OXPHOS complex activity, and redox status in muscle tissue.
Mitochondrial diseases (MD) encompass a heterogeneous group of rare diseases caused by defects in oxidative phosphorylation (OXPHOS) (1, 2), and are among the most common neuromuscular disorders, but showing variable estimated prevalence (3–5). Given the ubiquity of mitochondria and their essential role in ATP production, MD can affect several tissues and those with higher metabolic demands, particularly (but not only) the skeletal muscle, are usually the most affected, with mitochondrial myopathy and poor exercise capacity (as reflected by low levels of aerobic fitness) being frequent features among patients with MD (6, 7).
Although no curative treatment is yet available for MD, preclinical evidence from animal and in vitro studies suggests that promoting the increase in mitochondrial content or mitochondrial biogenesis, through the activation of peroxisome proliferator-activated receptor gamma co-activator 1-alpha (PGC-1α) might be a therapeutic option (8, 9). In this respect, regular endurance exercise is known to be a potent stimulus for muscle mitochondrial biogenesis (10), and in fact previous research has shown that endurance-based exercise interventions can increase the muscle oxidative capacity of patients with MD, as directly reflected by increases in the activity of citrate synthase (CS) and respiratory chain complexes in skeletal muscle biopsies (11–13), or indirectly, by improvements in patients' aerobic fitness (11–18). Another exercise modality, resistance or “strength” training (e.g., weight lifting), is also known to promote muscle mitochondrial biogenesis (19) together with other benefits, notably improvements in muscle mass and strength (20), but scarce data are available in MD patients. One study assessed the effects of a 12 week resistance training program in 8 patients with mitochondrial DNA large scale deletions, showing strength improvements and a decrease in mitochondrial DNA heteroplasmy without side effects (21). Another study found an increase in upper-body muscle strength after 12 weeks of combined resistance and endurance training in 10 mitochondrial patients with biopsy diagnosis of MD but with no genetic confirmation (22). Finally, we recently described significant improvements in upper-, lower-body, and respiratory muscle strength in 12 well-characterized MD patients (all with genetic diagnosis) after 8 weeks of combined resistance, endurance and respiratory training (14).
On the other hand, studying mouse models of MD allows to gain mechanistic insight into the molecular processes underlying exercise benefits in these conditions. In this respect, although previous exercise training studies have been published with mouse models of respiratory chain complex IV deficiency (23) or of a defect in mitochondrial polymerase gamma (i.e., “mutator” mice) (24–26), the effects of exercise-training on a mouse model of the commonest mitochondrial defect, respiratory chain complex I deficiency, have yet not been assessed. Further, the effects of resistance training remain to be determined in mouse models of MD.
The aim of the present study was to determine the effects of a combined exercise intervention (aerobic + resistance exercise) in the aerobic fitness, muscle strength and muscle-tissue adaptations at the molecular level of the Harlequin (Hq) mutant mouse. In this animal, proviral insertion in the X-linked gene encoding Apoptosis Inducing Factor (AIF) produces a large decrement (~80%) in the expression of this mitochondrial protein (27), leading to respiratory chain complex I deficiency (28) and to a phenotype that mimics the clinical features of affected patients, including skeletal muscle myopathy (29–31).
All experimental protocols were approved by the institutional ethics committee (project number 111/15) and were conducted in accordance with European (European convention ETS 123) and Spanish (32/2007 and R.D. 1201/2005) laws on animal protection in research.
Heterozygous 6–8 week-old, male Harlequin mice (n = 21, Hq/Y named “Hq” hereafter, B6CBACa Aw-J/A-Aifm1Hq/J) and gender and age-matched wild-type (WT) mice (n = 12) of the same genetic background (B6CBACa Aw-J/A), were obtained from The Jackson Laboratory (Bar Harbor, ME). Animals were housed at 21°C and 60% humidity with a 12-h light/dark cycle and with free access to food and water in the animal facility of Hospital Universitario 12 de Octubre (Madrid, Spain). To ensure that all mice presented with the same degree of disease development at the moment of their incorporation into the study groups, each Hq mouse was previously evaluated from 8 weeks of age until the first signals of ataxia appeared following a locomotor skill and balance evaluation (Figure 1). The data obtained from the WT animals during the same period were used as a reference for absence of the aforementioned alterations as well as for biochemical analyses (see below).
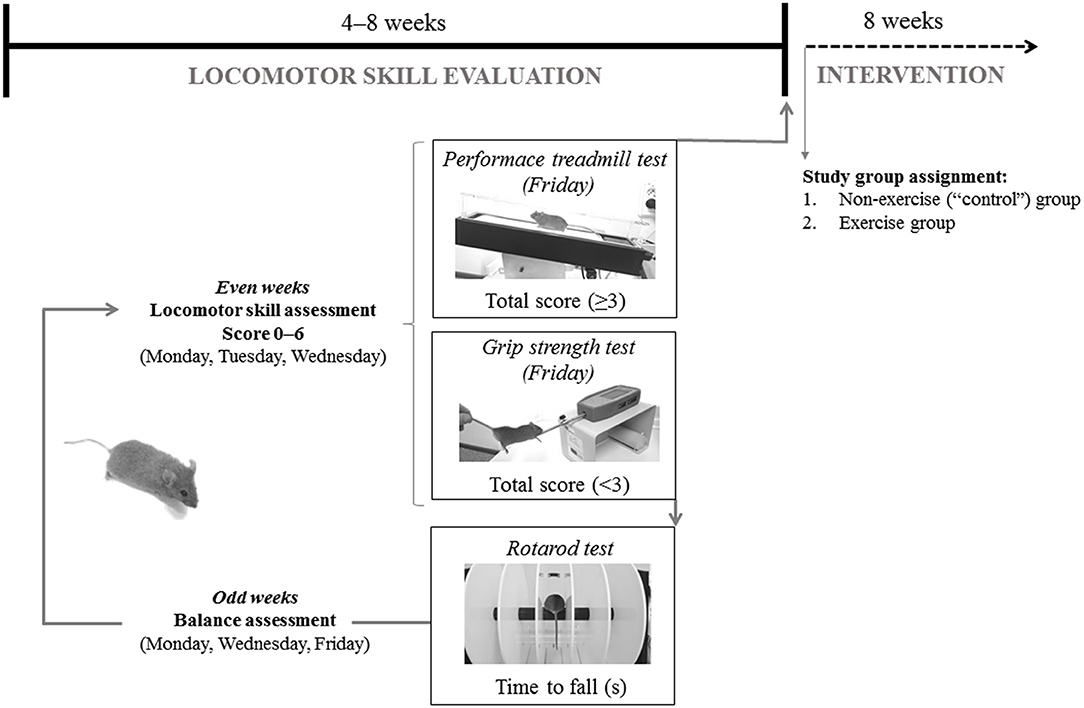
Figure 1. Representation of mice baseline evaluation before group assignment.
Locomotion analysis was assessed while the mouse walked at a speed of 10 cm·s−1 (15 min, 15% inclination) on a treadmill (Harvard Apparatus; Panlab, Barcelona, Spain) during the even weeks of the locomotor skill evaluation phase. All mice were allowed to adapt to the treadmill in three familiarization sessions. An electric grid was placed at the end of the treadmill to deliver electrical shocks of constant intensity to encourage the animals to continue running on the treadmill. These sessions involved a gradual increase in running time and intensity, starting just placing the mice on the treadmill the first day: 0% inclination and 0 cm·s−1 speed for 1 min, with no electrical stimulation, and ending with a 15 min running period at low intensity on the third day (15% and 10 cm·s−1, electrical stimulation 0.1 mA, 1 Hz, 200 ms). Two examiners scored the motor function of each mouse and, if two scores did not match, the lowest score was chosen. Scores ranged from 0 (absence) to 2 (maximal severity grade) in the following manner: (i) walking gait balance, 0, 1, or 2 for smooth gait pattern, mild imbalanced and unstable walking gait, respectively; (ii) 0, 1, or 2 for ability to walk straight, first signs of diagonal walk and gait instability, or inability to walk straight, respectively; (iii) alterations in hind limb gait, 0, 1, or 2 for normal coordination, first signs of alterations in the stride frequency, and lack of hind left-right limb coordination, respectively. A total score (0–6) was generated by summing individual scores. Data from the 3 days was averaged and if the score was <3, the animals continued in the locomotor skill evaluation phase and performed the grip strength test (described below). Conversely, animals with an average score ≥3 were included in the training phase and were subjected to a treadmill performance test (described below).
During the locomotor skill evaluation phase, cerebellar ataxia was also tested, using the rotarod device (Letica Scientific Instruments; Panlab, Barcelona, Spain) on alternative days (odd weeks, 3 days in total) to avoid fatigue and following the protocol described elsewhere (27). To familiarize with the apparatus, mice underwent a training session of three trials (60 s each) in which the rod was kept stationary for the first trial and held at 4 rpm for the last two trials (32).
Hq mice were paired-matched based on their aerobic fitness in the treadmill test (see below for a description of the assessment method) at baseline. Thus, among each pair of Hq mice with similar performance in the pre-intervention aerobic treadmill tests, one mouse was randomly assigned to an exercise group (n = 10), subjected to an 8 week exercise training program, and the other to a non-exercise (control) group (n = 11), allowed to freely move in the cage, but not performing the program (Figure 2). On the other hand, none of the WT mice used as reference underwent exercise training (i.e., same treatment as for the Hq control group).
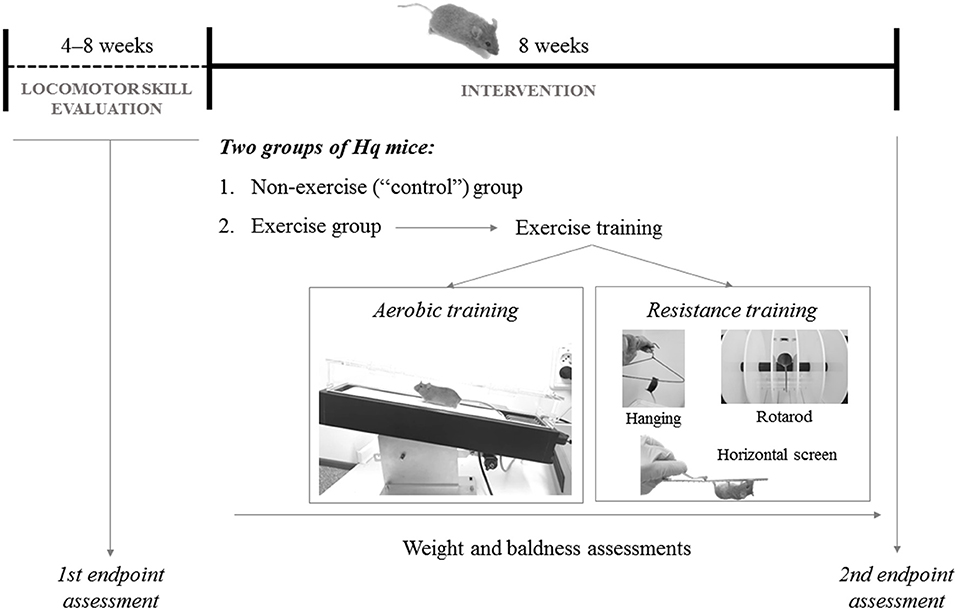
Figure 2. Representation of the study design.
The exercise intervention included 5 weekly sessions (Monday–Friday; session duration of 40–60 min), each including aerobic and resistance training.
Hq mice were allowed to adapt to the treadmill (Harvard Apparatus; Panlab, Barcelona, Spain) before their incorporation into the exercise intervention, as previously described (33). Exercise duration, treadmill speed, and inclination were gradually increased during the program following a defined interval training protocol from our group, with slight modifications (33). Briefly, beginning at very low workloads in the first session (20 min at 35% of the maximal velocity obtained during the aerobic fitness test that is described below) and 0% gradient on the first day, and ending with 40 min at 65–70% of maximal velocity and 15% gradient in the last sessions. Only gentle tail touching was used to prompt the mice to run, and no electrical stimulation was applied during the training sessions.
This followed the aerobic training and included three exercises: (i) horizontal screen exercise (Monday); (ii) hanging exercise with two limbs (Wednesday and Friday); and (iii) rotarod exercise (Tuesday and Thursday). For (i), the Hq mouse was placed inverted on top of a screen and had to climb back over to the top. When this was achieved, the mouse was placed again in the initial position. The number of repetitions was gradually increased (from 1 to 6), each also of increasing duration (from 30 s at the beginning to 90 s at the end of the training phase), with a constant 2 min rest period between repetitions. For (ii), Hq mice were picked up by the tail and placed on a metal cloth hanger taped to a shelf and maintained at 40 cm above a layer of bedding to cushion the falls (32). Mice were allowed to grasp the wire only using the two forepaws for as long as they could during one set of six repetitions, with a 10 min rest period between them. For (iii), each Hq mouse was placed onto the rod in a rotarod device (Letica Scientific Instruments, Panlab; Barcelona, Spain) and walked the rotating rod at speeds that increased from 4 to 40 rpm over a 300 s period. When the first fall occurred, the number of rpm was recorded, and the mouse exercised at this speed during 5 min (32).
Study endpoints were assessed at the start and the end of the exercise intervention except for muscle molecular measurements, which could only be measured at the end. Serum samples were obtained before and after concluding the exercise program. Forty eight hours after the last exercise test, Hq mice were killed by cervical dislocation and the biceps femoris and quadriceps femoris muscles were dissected and immediately snap-frozen in liquid nitrogen before storage at −80°C until molecular analysis. The WT mice were sacrificed with the aforementioned procedure at the same time point.
An incremental treadmill test was used to determine the maximal aerobic fitness (expressed as the total distance [meters] run by the mice) of Hq mice (33). The test was performed after a warm-up period of 10 min at a speed of 10 cm·s−1 (with 15% inclination) and followed a previous protocol from our group with slight modifications in workload increases (33). Thus, the initial velocity was 5 cm·s−1 and was increased in 2 cm·s−1 every 2 min until exhaustion, with a constant treadmill inclination of 15% during the whole test. Mice were considered exhausted when they spent more than 5 continuous s on the electric grid and were unable to continue running at the next speed (33).
Grip strength of Hq mice was measured using an isometric force transducer (Harvard apparatus; Panlab, Barcelona, Spain). Maximum force (grams) exerted by the mouse before losing grip was recorded in three trials (each separated from the next by a 5 min interval). The best score for each animal was recorded as the maximal grip strength.
Cerebellar ataxia in Hq mice was tested on the rotarod device. Each mouse walked the rotating rod at speeds that increased from 4 to 40 rpm over a 300 s period (27). The latency to fall from the rotating rod was recorded in each trial. The best of the 3 day latency was considered for analysis. We additionally evaluated two other hallmarks of the Hq phenotype (27) weekly throughout the study: (i) hair loss, which was assessed as the percentage of body surface area without hair (30), and (ii) weight.
Muscle damage in Hq mice was assessed through the measurement of creatine kinase (CK) activity in serum samples (Creatine Kinase Activity Assay Kit; Sigma Aldrich, MO).
Muscle tissue of both Hq and WT mice was either processed to obtain total homogenates as described previously (33), or processed to obtain mitochondria-enriched fractions with a specific kit (Mitochondria Isolation Kit for Tissue; Abcam, UK) according to manufacturers' instructions.
Aliquots of total homogenates or mitochondria-enriched fractions containing equal amount of protein were resolved by sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) electrophoresis, transferred to polyvinylidene difluoride (PVDF) membranes, blocked, and incubated with primary antibodies for the determination of levels of the following proteins: catalase (CAT), citrate synthase (CS), cytosolic superoxide dismutase (cSOD), gluthathione reductase (GR), mitochondrial superoxide dismutase (mtSOD), mitochondrial transcription factor A (TFAM), NADH-ubiquinone oxidoreductase subunit B8 (NDUFB8) and S1 (NDUFS1), peroxisome proliferator-activated receptor-gamma co-activator 1α (PGC-1α), and ribosomal protein S6 kinase beta-1 total (P70S6K) or phosphorylated at threonine 389 (P70S6K Thr389, or simply “phosphorylated” pP70S6K) (Supplementary Table 1). After incubation with peroxidase-conjugated secondary antibodies, immunoreactive bands were detected with the ECL Prime Western Blotting Detection Reagent (Amersham Biosciences, UK) in a ChemiDoc™ MP Imager (Bio-Rad, Hercules, CA). Band densities were evaluated by densitometric scanning [ImageJ software, National Institutes of Health (34)]. Either glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was immunodetected and used as loading control for total homogenates, or total protein load per lane was determined with Coomassie Blue staining of the PVDF membranes (35). When needed, membrane stripping was performed before immunodetection of GAPDH.
Activities of OXPHOS complexes were determined in muscle total homogenates prepared in 225 mM mannitol, 75 mM sucrose, 0.1 mM EDTA, and 10 mM Tris–HCl pH 7.4, according to standardized protocols for spectrophotometric assays (36), and in mitochondria-enriched fractions by a researcher blinded to the mouse group (control Hq, exercised Hq, or WT) from which the samples were obtained. For complexes II–IV, 20 μg of protein was used in total homogenates, and 6 μg in mitochondria-enriched fractions, whereas 60 μg of protein was added to the assay for complex I in total homogenates, and 10 μg in mitochondria-enriched fractions. Complex V activity was assayed as F1-ATPase using 20 μg of protein according to a previously described method (37). CS activity, was spectrophotometrically determined at 30°C in the presence of 0.1% Triton X-100 following the formation of 5-thio-2-nitrobenzoic acid at 412 nm, as previously described (38), using 60 μg of protein in total homogenates and 6 μg in mitochondria-enriched fractions.
All the aforementioned muscle biochemical variables obtained with western blotting or spectrophotometry in samples of Hq mice were expressed relative to the corresponding mean values for the WT mice.
For muscle histology tibialis anterior muscles were frozen in N2-cooled isopentane, and hematoxylin-eosin staining on transverse sections of muscle was performed on cryostat sections according to standard techniques (39), and cross-sectional area of fibers was determined with Image J software.
Data are presented as mean ± standard error of measurement (SEM). The normal distribution (Shapiro-Wilk test) and homoscedasticity (Levene's test) of the data were checked before any statistical treatment. A two-factor (group [exercise, control] by time) ANOVA with repeated measures on time was used for between-group comparisons of outcomes with more than one measurement over time (i.e., aerobic fitness, muscle strength, balance, serum CK, hair loss and weight). To minimize the risk of type I error, post hoc comparisons were only performed within-groups (with the Bonferroni test) when a significant interaction group by time was found. The non-parametric Mann–Whitney's U-test was used for between-group comparisons of outcomes assessed at one single time point (i.e., muscle biochemical analyses). All statistical analyses were performed with the SPSS 23.0 package (SPSS, Inc., Chicago, IL) setting the significance level at 0.05.
A significant group by time interaction effect was found for aerobic fitness (p = 0.014, effect size [η2] = 0.304, power = 0.728), with post hoc analyses indicating an improvement after the training intervention (p = 0.016) and no change in control mice after the same time period (p = 0.277) (Table 1). A significant group by time interaction effect was also observed for muscle strength (p = 0.037, η2 = 0.209, power = 0.566), which increased in the exercise group (p = 0.003) but did not change in control mice (p = 0.751). Individual data on aerobic fitness and muscle strength by group are shown in Supplementary Figure 1. On the other hand, no significant group by time interaction was found for time to fall (p = 0.544), serum CK (p = 0.290) (Table 1), body weight (p = 0.058), or hair loss (p = 0.406) (Supplementary Figure 2).
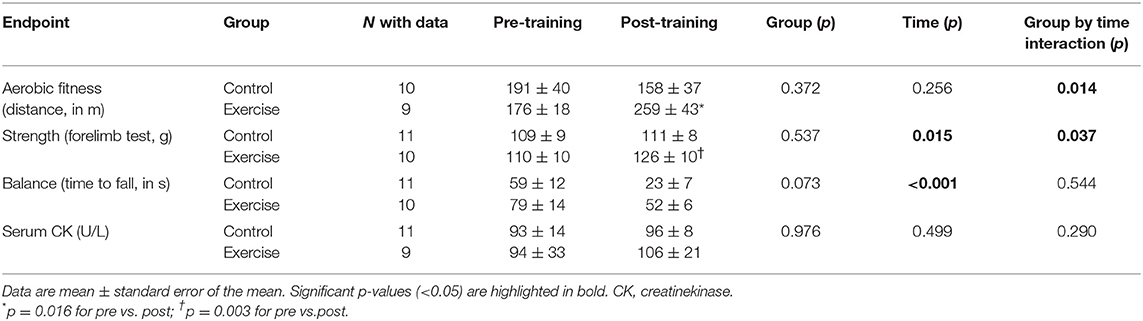
Table 1. Effects of the exercise intervention on study endpoints with repeated measurements before and after the intervention.
Higher levels of total P70S6K and pP70S6K were observed at the end of the training intervention in the exercise group compared with the control group (Table 2; Supplementary Figure 3A), which also showed high levels compared to the wild-type animals, as well as a non-significant trend toward a higher pP70S6K/ P70S6K ratio (Table 2; Supplementary Figure 3A). Exercise resulted in higher levels of PGC-1α and a tendency toward higher levels of TFAM (Table 2; Supplementary Figure 3B).
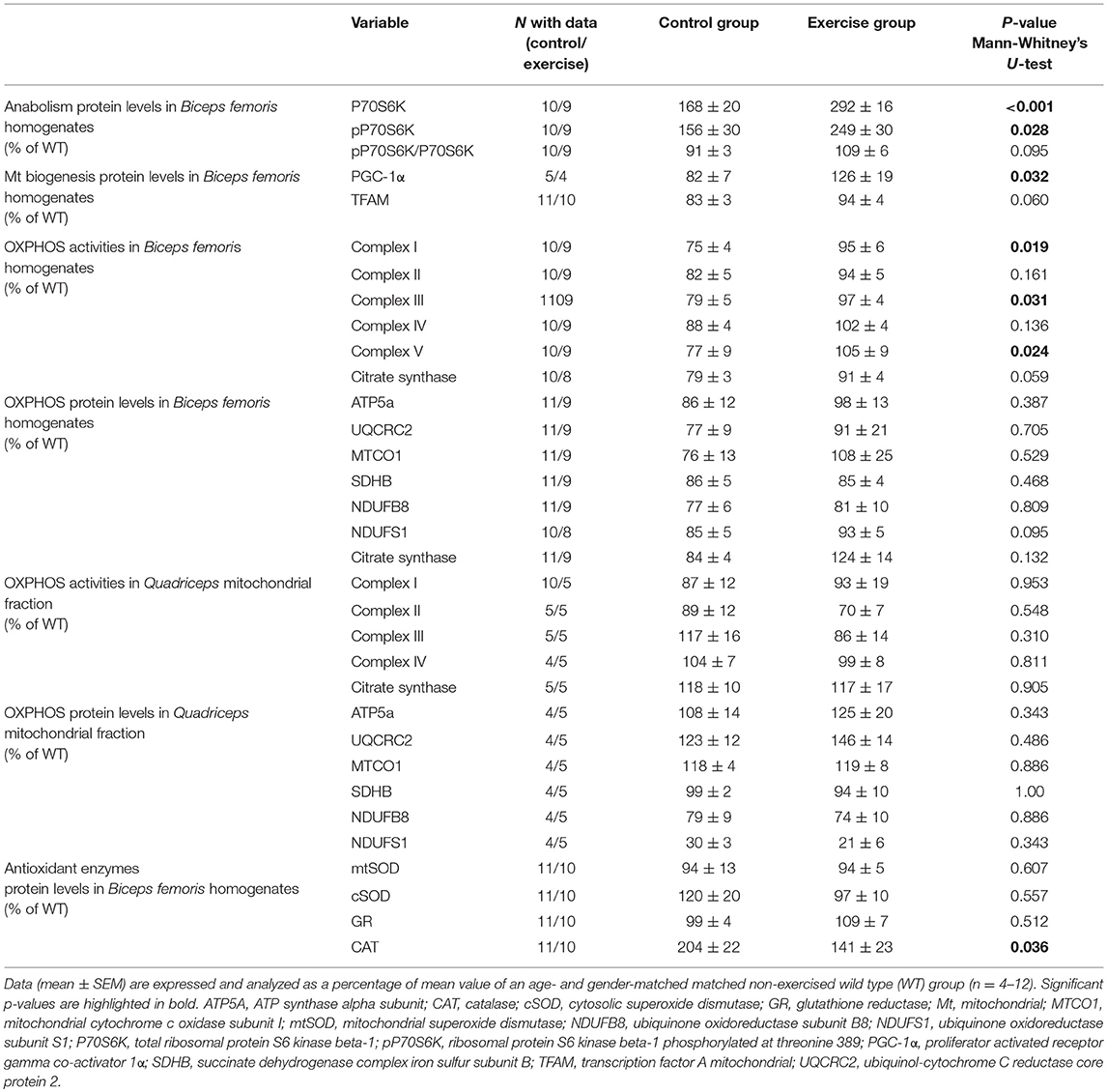
Table 2. Biochemical variables in Hq mice.
The activities of the OXPHOS complexes I, III, and V measured in muscle homogenates were higher in the exercised than in the control group and a trend toward significant differences was also found for CS activity (Table 2). However, no significant between-group differences were found for the muscle protein levels of representative subunits of the different OXPHOS complexes or for CS as measured by western blotting in total homogenates (Table 2; Supplementary Figure 3C), the enzymatic activity of respiratory chain complexes, or the representative OXPHOS proteins measured in mitochondria-enriched fractions (Table 2; Supplementary Figure 3D).
Exercised mice presented with lower CAT levels at the end of the training intervention compared to control mice, which showed abnormally high levels compared to the wild-type mice with no between-group differences for the remainder of antioxidant enzymes (Table 2; Supplementary Figure 3E).
To further assess the time course of the myopathy in the Hq mice, we analyzed forelimb strength in an independent cohort of Hq and WT mice from 1.5 to 3 months of age, with the results showing significantly lower muscle strength in the former since 1.5 months (Supplementary Figure 4). Finally, to study the effects of exercise training and P70S6K activation on skeletal muscle mass, we studied the cross-sectional fiber area of tibialis anterior muscles from some WT and Hq mice. The results indicated lower cross-sectional area in both control and exercise Hq animals compared to WT (Supplementary Figure 5).
The present study shows that an exercise intervention combining aerobic and resistance training induced significant benefits on the aerobic fitness and muscle strength of Hq mice, partially counteracting the myopathy progression that characterizes the time-course of the disease. These improvements were accompanied by mild increases in the activity of OXPHOS complexes I, III, and V, and by a trend to higher CS activity in total homogenates. Further, the intervention did not induce major skeletal muscle damage, as reflected by the lack of differences between groups in a well-accepted marker of this phenomenon, serum CK activity. No benefits were noted on phenotypes not directly related to muscle tissue such as cerebellar ataxia (i.e., balance performance) or growth impairment, which, as opposed to the aforementioned fitness/muscle variables, showed a similar evolution over time in the two groups. Of note, the fact that the exercise training program was initiated when the first signs of ataxia and myopathy were already present might explain, at least partly, why we did not find greater improvements in muscle biomarkers. In this respect, however, a strength of our design is that it mimics a frequent situation in patients, that is, the treatment starts when the disease phenotype is already well-established.
The Hq mouse model replicates the main features of respiratory chain complex I deficiency in humans (30), which is the most common respiratory chain defect (40). Hq mutant mice not only mimic clinical features of this condition in important aspects such as tissue specificity, disease onset and course, or inter-individual variability (30), but also show disturbances frequently found in other diseases associated with respiratory chain deficiencies such as ataxia, myopathy and retinal degeneration (30). Therefore, this model has been considered a valuable tool for assessing potential therapeutic approaches in MD, and previously nutritional, genetic, and pharmacological interventions, have been proven effective to attenuate several disease-related markers (41–44). In this regard, muscular weakness, the main feature of myopathy, is an early finding in the Hq mouse model, in which we observed weakness since 1.5 month of age. In turn, the latter result correlates well with the finding of complex I deficiency in skeletal muscle previously reported in 1 month-old Hq mice, a molecular phenotype trait that remains during the course of the disease (30). The early appearance of myopathy in Hq mice also makes this model a valuable tool for the study of exercise training as therapy for MD. Our results showing physical capacity improvements in trained Hq are in agreement with previous studies analyzing exercise training as therapy in other mouse models of MD. For example, Wenz et al., reported a lower number of falls during a treadmill test after a 1.5 month training program in a mouse model of cytochrome oxidase deficiency (23). After a 1 month training program, “Mutator” mice were able to reach levels of treadmill running performance similar to those of healthy (WT) animals (25). Our results demonstrating improvements in the physical capacity of Hq mice in response to exercise training are novel for this strain, and might allow to gain insight into the molecular underpinnings of exercise-induced adaptations in this model.
The promotion of mitochondrial biogenesis through either genetically or pharmacologically (bezafibrate administration) stimulation of PGC-1α, has been shown to increase OXPHOS capacity in cytochrome c oxidase-deficient mice (9). Accordingly, exercise training-induced promotion of mitochondrial biogenesis through the activation of PGC-1α, has also proven to mediate the enhancement of oxidative capacity in animal (23, 25) and patient studies of MD (11–13). In this regard, in the present study, exercise training slightly improved muscle levels of PGC-1α and TFAM, and complex I, III, and V enzyme activities in muscle homogenates of Hq mice. Additionally, CS activity, which is a common marker of mitochondrial content, also showed a trend to increase in this group. Overall, these results indicate that exercise training was able to slightly promote mitochondrial biogenesis in the Hq mice, leading to partial normalization of the altered oxidative capacity. Moreover, we observed higher activities of OXPHOS complexes in muscle tissue homogenates but not in mitochondria-enriched fractions, reflecting that the results in the homogenates of the exercised Hq were due to a higher content of mitochondria rather than to a higher density of OXPHOS complexes per mitochondria. Therefore, our results support the role of mitochondrial biogenesis as a mediator of the beneficial effects of physical exercise on muscle OXPHOS capacity in the Hq mice. Of note, administration of the PGC-1α agonist bezafibrate has failed to improve mitochondrial biogenesis or muscle strength in Hq mice, with this treatment in fact inducing liver disease as a major adverse effect (41). Thus, up to date exercise training appears as the safest and most effective strategy to induce mitochondrial biogenesis in the Hq mouse model.
One aspect of our study that must be highlighted is the novelty of the exercise intervention, which included both endurance and resistance training. Previous studies in animals and patients with MD have focused on aerobic exercise owing to its potential to promote mitochondrial biogenesis and eventually increase aerobic fitness (11–13, 16–18, 45). Resistance training, however, might not only improve OXPHOS capacity (19) but also provide benefits on muscle mass and strength (20), and indeed we recently observed improvements with this type of intervention not only in the aerobic fitness, but also in the muscle strength and mass of patients with MD (14). Here, we also observed training-induced increases in muscle OXPHOS capacity and a trend to higher CS activity [being a classical marker of muscle aerobic adaptation and upregulated mitochondrial biogenesis (46)], and also in muscle strength. The higher levels of activation of P70S6K (i.e., p70S6K) and the trend toward higher levels of the ratio p70S6K/total P70S6K (a marker of muscle anabolism) observed in the exercise group could also support the role of combined resistance and aerobic exercise in the promotion of muscle anabolism, as P70S6K (a downstream effector of the mammalian target of rapamycin [commonly known as “mTOR”] pathway) is involved in protein synthesis and consequently muscle mass growth (47). Of note, the benefits observed in muscle strength are of particular relevance given that the Hq phenotype is characterized by marked muscle atrophy (i.e., lower muscle cross-sectional area and strength, and lower number of fast-twitch fibers compared to wild-type mice) (29, 48), and we recently showed that patients with MD also present with a lower (−18%) lean mass than their healthy peers (14). Nevertheless, preliminary assessment of fiber cross-sectional area in the tibialis anterior of control and exercised animals suggested no differences between both groups and thus no major occurrence of exercise-promoted skeletal muscle anabolism. In this respect, studies involving patients with MD have also demonstrated improvements in physical performance and strength without increases in the cross-sectional area of muscle fibers in response to training (21).
Another interesting finding of our study were the lower levels of the antioxidant enzyme CAT found in the exercised mice. Hq mice typically present with an 80% decrease in AIF, a protein that might be involved not only in apoptosis and OXPHOS complexes I and IV biogenesis (49, 50), but also in facilitating cellular redox homeostasis (27). Specifically, AIF has been proposed to exert an antioxidant activity as a peroxide scavenger, with reduced AIF levels resulting in increased peroxide sensitivity and consequently in greater levels of oxidative damage and cell death (27). Elevated levels of oxidative stress—as reflected by increased CAT activity in the cerebellum, heart, and skeletal muscle—have been previously reported in Hq mice and in other AIF-deficient mouse models (27, 51), which is in agrement with our results (i.e., the CAT levels of control Hq mice were significantly higher [by ~2-fold] than those of wild-type mice). Our results, therefore, suggest that the exercise training intervention might have contributed to improve redox status, with the antioxidant levels of exercised Hq mice resembling more those of wild-type mice compared to their non-exercise pairs.
The present study has some limitations. First, randomization, allocation concealment, and blinding outcome assessment can reduce the risk of bias in animal studies. In this respect, although we could not adhere to these recommendations, we followed some measures to limit bias. First, we pair-matched mice in both groups based on their baseline aerobic fitness. Second, researchers in charge of muscle biochemical assessments were blinded to the mouse group from which samples were taken. Furthermore, we limited the risk of statistical error type I by using non-parametric statistical tests unpaired comparisons between exercise and control group, respectively. Another limitation is that we did not assess the effects of exercise training on muscle mass, which would have provided additional information, and we only performed preliminary assessment of muscle fiber cross-sectional area in a few mice. In turn, one of the major novelties of our study was the type of intervention applied, as to our knowledge this is the first study in which a mouse model of MD is subjected to resistance training. Moreover, we assessed both functional (i.e., fitness status) and biological (OXPHOS activity) markers and analyzed potential molecular pathways involved in these adaptations, which we consider as a major strength of our study.
In summary, an intervention combining aerobic and resistance exercises improved markers of physical capacity (aerobic fitness and muscle strength) in Hq mutant mice, a model of respiratory chain complex I deficiency. In turn, this intervention induced mild increases in the activation of signaling pathways involved in mitochondrial biogenesis (i.e., PGC-1α) and muscle anabolism (i.e., pP70S6K), muscle OXPHOS activity, and redox status–as reflected by a decrease in CAT levels. Although more research is needed, these results are encouraging and might provide useful information for the management of MD.
Datasets are available on request to the authors.
All experimental protocols were approved by the institutional ethics committee (project number 111/15) and were conducted in accordance with European (European convention ETS 123) and Spanish (32/2007 and R.D. 1201/2005) laws on animal protection in research.
CF-L: study design, experimental procedures, and drafting of the manuscript. PV: data analysis and drafting of the manuscript. SL-M, MF-T, VB-G, and LR-V: experimental procedures. JA, MAM, and AL: data analysis and manuscript edition. MM: study design, drafting of the manuscript, and direction. All authors revised the manuscript critically for important intellectual content and approved the final version.
This study was funded by FIS, Fondo de Investigaciones Sanitarias (grant numbers PI14/01085, PI15/00558, PI15/00431, and PI17/00093), and FEDER funds from the European Union. MM was supported by Miguel Servet contract (CPII16/00023) by the Institute of Health Carlos III (ISCIII) and FEDER funds. CF-L was supported by Miguel Servet contract (CP18/00034) by ISCIII. PV was supported by a contract granted by University of Alcalá (FPI2016). This study was awarded with the 2nd prize of the Premios Liberbank de Investigación en Medicina del Deporte, año 2017. Escuela de Medicina del Deporte. Universidad de Oviedo.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge Belén Fernández Pérez for her kind support with the histochemical analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00790/full#supplementary-material
1. DiMauro S, Schon E, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. (2013) 9:429–44. doi: 10.1038/nrneurol.2013.126
2. Gorman G, Chinnery P, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Prim. (2016) 2:16080. doi: 10.1038/nrdp.2016.80
3. Chinnery PF. Mitochondrial disorders overview. genereviews®. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews®. Seattle,WA: University of Washington. (2000). pp. 1993–2019.
4. Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. (2015) 77:753–9. doi: 10.1002/ana.24362
5. Thornburn DR. Mitochondrial disorders: prevalence, myths, and advances. J Inherit Metab Dis. (2004) 27:349–62. doi: 10.1023/B:BOLI.0000031098.41409.55
6. Fernández J, Montemayor T, Bautista J, Márquez R, Jiménez L, Arenas J, et al. [The use of cardiopulmonary exercise test in patients with mitochondrial myopathies]. Med Clin. (2000) 114:121–7. doi: 10.1016/S0025-7753(00)71216-4
7. Taivassalo T, Jensen TD, Kennaway N, DiMauro S, Vissing J, Haller RG. The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain. (2003) 126:413–23. doi: 10.1093/brain/awg028
8. Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. PGC-1α/β induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum Mol Genet. (2009) 18:1805–12. doi: 10.1093/hmg/ddp093
9. Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1α pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. (2008) 8:249–56. doi: 10.1016/j.cmet.2008.07.006
10. Irrcher I, Adhihetty PJ, Joseph AM, Ljubicic V, Hood DA. Regulation of mitochondrial biogenesis in muscle by endurance exercise. Sport Med. (2003) 33:783–93. doi: 10.2165/00007256-200333110-00001
11. Jeppesen TD, Schwartz M, Olsen DB, Wibrand F, Krag T, Dunø M, et al. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain. (2006) 129:3402–12. doi: 10.1093/brain/awl149
12. Jeppesen TD, Dunø M, Schwartz M, Krag T, Rafiq J, Wibrand F, et al. Short- and long-term effects of endurance training in patients with mitochondrial myopathy. Eur J Neurol. (2009) 16:1336–39. doi: 10.1111/j.1468-1331.2009.02660.x
13. Taivassalo T, Shoubridge EA, Chen J, Kennaway NG, DiMauro S, Arnold DL, et al. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann Neurol. (2001) 50:133–41. doi: 10.1002/ana.1050
14. Fiuza-Luces C, Díez-Bermejo J, Fernández-De La Torre M, Rodríguez-Romo G, Sanz-Ayán P, Delmiro A, et al. Health benefits of an innovative exercise program for mitochondrial disorders. Med Sci Sports Exerc. (2018) 50:1142–51. doi: 10.1249/MSS.0000000000001546
15. Porcelli S, Marzorati M, Morandi L, Grassi B. Home-based aerobic exercise training improves skeletal muscle oxidative metabolism in patients with metabolic myopathies. J Appl Physiol. (2016) 121:699–708. doi: 10.1152/japplphysiol.00885.2015
16. Taivassalo T, De Stefano N, Argov Z, Matthews PM, Chen J, Genge A, et al. Effects of aerobic training in patients with mitochondrial myopathies. Neurology. (1998) 50:1055–60.
17. Taivassalo T, De Stefano N, Chen J, Karpati G, Arnold DL, Argov Z. Short-term aerobic training response in chronic myopathies. Muscle Nerve. (1999) 22:1239–43.
18. Taivassalo T, Gardner JL, Taylor RW, Schaefer AM, Newman J, Barron MJ, et al. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. (2006) 129:3391–401. doi: 10.1093/brain/awl282
19. Groennebaek T, Vissing K. Impact of resistance training on skeletal muscle mitochondrial biogenesis, content, and function. Front Physiol. (2017) 8:713. doi: 10.3389/fphys.2017.00713
20. Kraemer WJ, Ratamess NA, French DN. Resistance training for health and performance. Curr Sports Med Rep. (2002) 1:165–71. doi: 10.1021/jp4081977
21. Murphy JL, Blakely EL, Schaefer AM, He L, Wyrick P, Haller RG, et al. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain. (2008) 131:2832–40. doi: 10.1093/brain/awn252
22. Cejudo P, Bautista J, Montemayor T, Villagómez R, Jiménez L, Ortega F, et al. Exercise training in mitochondrial myopathy: a randomized controlled trial. Muscle Nerve. (2005) 32:342–50. doi: 10.1002/mus.20368
23. Wenz T, Diaz F, Hernandez D, Moraes CT. Endurance exercise is protective for mice with mitochondrial myopathy. J Appl Physiol. (2009) 106:1712–9. doi: 10.1152/japplphysiol.91571.2008
24. Clark-Matott J, Saleem A, Dai Y, Shurubor Y, Ma X, Safdar A, et al. Metabolomic analysis of exercise effects in the POLG mitochondrial DNA mutator mouse brain. Neurobiol Aging. (2015) 36:2972–83. doi: 10.1016/j.neurobiolaging.2015.07.020
25. Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci USA. (2011) 108:4135–40. doi: 10.1073/pnas.1019581108
26. Safdar A, Khrapko K, Flynn JM, Saleem A, De Lisio M, Johnston AP, et al. Exercise-induced mitochondrial p53 repairs mtDNA mutations in mutator mice. Skelet Muscle. (2016) 6:7. doi: 10.1186/s13395-016-0075-9
27. Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. (2002) 419:367–74. doi: 10.1038/nature01034
28. Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. (2004) 23:4679–89. doi: 10.1038/sj.emboj.7600461
29. Armand AS, Laziz I, Djeghloul D, Lécolle S, Bertrand AT, Biondi O, et al. Apoptosis-inducing factor regulates skeletal muscle progenitor cell number and muscle phenotype. PLoS ONE. (2011) 6:227283. doi: 10.1371/journal.pone.0027283
30. Bénit P, Goncalves S, Dassa EP, Brière JJ, Rustin P. The variability of the Harlequin mouse phenotype resembles that of human mitochondrial-complex I-deficiency syndromes. PLoS ONE. (2008) 3:e3208. doi: 10.1371/journal.pone.0003208
31. Irwin MH, Parameshwaran K, Pinkert CA. Mouse models of mitochondrial complex I dysfunction. Int J Biochem Cell Biol. (2013) 45:34–40. doi: 10.1016/j.biocel.2012.08.009
32. Aartsma-Rus A, van Putten M. Assessing functional performance in the mdx mouse model. J Vis Exp. (2014) 85:e51303. doi: 10.3791/51303
33. Fiuza-Luces C, Soares-Miranda L, González-Murillo A, Palacio JM, Colmenero I, Casco F, et al. Exercise benefits in chronic graft versus host disease: a murine model study. Med Sci Sports Exerc. (2013) 45:1703–11. doi: 10.1249/MSS.0b013e31828fa004
34. Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods. (2012) 9:671–5. doi: 10.1038/nmeth.2089
35. Welinder C, Ekblad L. Coomassie staining as loading control in Western blot analysis. J Proteome Res. (2011) 10:1416–9. doi: 10.1021/pr1011476
36. Medja F, Allouche S, Frachon P, Jardel C, Malgat M, Mousson de Camaret B, et al. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion. (2009) 9:331–9. doi: 10.1016/j.mito.2009.05.001
37. Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Methods Cell Biol. (2007) 80:93–119. doi: 10.1016/S0091-679X(06)80004-X
38. Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. (1996) 264:484–509. doi: 10.1016/S0076-6879(96)64044-0
39. Lojda Z, Gossrau R, Schiebler TH. Enzyme Histochemistry: A Laboratory Manual. New York, NY: Springer-Verlag (1979).
40. Haack TB, Haberberger B, Frisch E-M, Wieland T, Iuso A, Gorza M, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet. (2012) 49:277–83. doi: 10.1136/jmedgenet-2012-100846
41. Bénit P, Pelhaître A, Saunier E, Bortoli S, Coulibaly A, Rak M, et al. Paradoxical inhibition of glycolysis by pioglitazone opposes the mitochondriopathy caused by AIF deficiency. EBioMedicine. (2017) 17:75–87. doi: 10.1016/j.ebiom.2017.02.013
42. Lechauve C, Augustin S, Cwerman-Thibault H, Reboussin É, Roussel D, Lai-Kuen R, et al. Neuroglobin gene therapy prevents optic atrophy and preserves durably visual function in Harlequin mice. Mol Ther. (2014) 22:1096–109. doi: 10.1038/mt.2014.44
43. Mekala NK, Kurdys J, Depuydt MM, Vazquez EJ, Rosca MG. Apoptosis inducing factor deficiency causes retinal photoreceptor degeneration. The protective role of the redox compound methylene blue. Redox Biol. (2019) 20:107–17. doi: 10.1016/j.redox.2018.09.023
44. Schiff M, Bénit P, El-Khoury R, Schlemmer D, Benoist J-F, Rustin P. Mouse studies to shape clinical trials for mitochondrial diseases: high fat diet in harlequin mice. PLoS ONE. (2011) 6:e28823. doi: 10.1371/journal.pone.0028823
45. Bates MGD, Newman JH, Jakovljevic DG, Hollingsworth KG, Alston CL, Zalewski P, et al. Defining cardiac adaptations and safety of endurance training in patients with m.3243A>G-related mitochondrial disease. Int J Cardiol. (2013) 168:3599–608. doi: 10.1016/j.ijcard.2013.05.062
46. Powers SK, Criswell D, Lawler J, Martin D, Li Li Ji, Herb RA, et al. Regional training-induced alterations in diaphragmatic oxidative and antioxidant enzymes. Respir Physiol. (1994) 95:227–37. doi: 10.1016/0034-5687(94)90118-X
47. Yoon MS. mTOR as a key regulator in maintaining skeletal muscle mass. Front Physiol. (2017) 8:788. doi: 10.3389/fphys.2017.00788
48. Wischhof L, Gioran A, Sonntag-Bensch D, Piazzesi A, Stork M, Nicotera P, et al. A disease-associated Aifm1 variant induces severe myopathy in knockin mice. Mol Metab. (2018) 13:10–23. doi: 10.1016/j.molmet.2018.05.002
49. Hangen E, Féraud O, Lachkar S, Mou H, Doti N, Fimia GM, et al. Interaction between AIF and CHCHD4 regulates respiratory chain biogenesis. Mol Cell. (2015) 58:1001–14. doi: 10.1016/j.molcel.2015.04.020
50. Meyer K, Buettner S, Ghezzi D, Zeviani M, Bano D, Nicotera P. Loss of apoptosis-inducing factor critically affects MIA40 function. Cell Death Dis. (2015) 6:e1814. doi: 10.1038/cddis.2015.170
Keywords: rare diseases, mitochondrial diseases, OXPHOS, harlequin mutant mouse, resistance training, AIF deficiency, respiratory chain complex I
Citation: Fiuza-Luces C, Valenzuela PL, Laine-Menéndez S, Fernández-de la Torre M, Bermejo-Gómez V, Rufián-Vázquez L, Arenas J, Martín MA, Lucia A and Morán M (2019) Physical Exercise and Mitochondrial Disease: Insights From a Mouse Model. Front. Neurol. 10:790. doi: 10.3389/fneur.2019.00790
Received: 28 March 2019; Accepted: 09 July 2019;
Published: 25 July 2019.
Edited by:
Mauro Marzorati, Institute of Biomedical Technologies (CNR), ItalyReviewed by:
Olimpia Musumeci, University of Messina, ItalyCopyright © 2019 Fiuza-Luces, Valenzuela, Laine-Menéndez, Fernández-de la Torre, Bermejo-Gómez, Rufián-Vázquez, Arenas, Martín, Lucia and Morán. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Morán, bW1vcmFuQGgxMm8uZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.