 Wenbin Wan
Wenbin Wan Lan Cao2†
Lan Cao2† Bill Kalionis
Bill Kalionis- 1Department of Neurology, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2State Key Laboratory of Medical Neurobiology, Institutes of Brain Science, Shanghai Medical College, Fudan University, Shanghai, China
- 3Department of Maternal-Fetal Medicine, Pregnancy Research Centre, University of Melbourne, Parkville, VIC, Australia
- 4Department of Obstetrics and Gynecology, Royal Women's Hospital, Parkville, VIC, Australia
- 5Department of Obstetrics and Gynecology, University of Melbourne, Parkville, VIC, Australia
- 6Shanghai Institute of Geriatrics, Huadong Hospital, Fudan University, Shanghai, China
Iron deposition in the brain is an early issue in Alzheimer's disease (AD). However, the pathogenesis of iron-induced pathological changes in AD remains elusive. Insulin resistance in brains is an essential feature of AD. Previous studies determined that insulin resistance is involved in the development of pathologies in AD. Tau pathology is one of most important hallmarks in AD and is associated with the impairment of cognition and clinical grades of the disease. In the present study, we observed that ferrous (Fe2+) chloride led to aberrant phosphorylation of tau, and decreased tyrosine phosphorylation levels of insulin receptor β (IRβ), insulin signal substrate 1 (IRS-1) and phosphoinositide 3-kinase p85α (PI3K p85α), in primary cultured neurons. In the in vivo studies using mice with supplemented dietary iron, learning and memory was impaired. As well, hyperphosphorylation of tau and disrupted insulin signaling in the brain was induced in iron-overloaded mice. Furthermore, in our in vitro work we identified the activation of insulin signaling following exogenous supplementation of insulin. This was further attenuated by iron-induced hyperphosphorylation of tau in primary neurons. Together, these data suggest that dysfunctional insulin signaling participates in iron-induced abnormal phosphorylation of tau in AD. Our study highlights the promising role of insulin signaling in pathological lesions induced by iron overloading.
Introduction
Alzheimer's disease (AD) is a devastating brain disorder and is the most common cause of dementia in the elderly. AD is characterized clinically by cognitive impairment, and pathologically by amyloid beta (Aβ) deposition as well as aberrant phosphorylation of tau in the brain (1). Familial AD, also known as the early onset AD, affects <5% of all AD individuals and is modulated by genetic mutations (2). However, the majority (i.e., 95%) of AD cases are due to sporadic or late onset AD (2).
Neurofibrillary tangles (NFTs) comprise paired helical filaments of hyperphosphorylated tau. In neurons, NFTs lead to cytoskeletal disruption that subsequently results in neural damage and loss of function (3). Previous studies hypothesized that in the amyloid cascade, Aβ was the igniter, leading to pathological changes including hyperphosphorylation of tau in sporadic AD (SAD) (4). However, the role of Aβ remains controversial since other evidence suggests that tau is a necessary factor in Aβ-induced neurotoxicity (5). Indeed, tauopathy is well-correlated with cognitive impairment and clinical grades of AD, but without significant senile plaque (SP) deposition (6, 7), suggesting the importance of tauopathy in the development of AD. Physiologically, the microtubule-associated protein tau carries out multiple critical roles in the neuron, such as morphogenesis, polarity, plasticity, and cytoskeleton stabilization (8, 9). Despite extensive research, the precise etiopathogenesis and pathophysiologic mechanism of SAD is still unclear and disease-modifying therapeutic management is not currently available (10).
Metals such as iron, copper, and zinc are essential elements that are indispensable for all normal cellular activity (11). Accumulating evidence indicates that brain homeostasis of transition metals is significantly disrupted in AD, and in particular, iron dyshomeostasis is an early factor the disease progression (12–15). Iron is a critical factor that participates in the physiological function of neurons (16). Investigations based on magnetic resonance imaging (MRI) and anatomical analysis reveal elevated iron deposition in the brains of AD patients (17, 18). The excessive accumulation of iron in the hippocampus is positively correlated with the deposition of SP, and is negatively associated with memory loss in AD (19–21). Importantly, the iron chelating agent deferomine (DFO) alleviates the pathological lesions including Aβ accumulation, hyperphosphorylation of tau, and behavioral disruption, in an iron-overloaded AD model (22, 23), which indicates the essential role of iron in the pathogenesis of AD progression.
In peripheral tissue, the insulin signaling pathway also plays a pivotal role in the modulation of energy homeostasis, and in maintaining the normal neurological function in the central nervous system (CNS) (24–26). Insulin receptor and its target molecules are wildly distributed and this receptor acts as a crucial neuromodulator in the brain (26). Because of the association between AD and diabetes, studies were undertaken on the effect of dysfunctional insulin signaling in AD progress, which led to subsequent studies that showed brain insulin resistance is a hallmark of AD (27–29). Interestingly, it was previously reported that in AD patients, insulin receptor sensitivity is reduced, and the levels of phosphorylation of the insulin receptor, and its substrates, are decreased in the brain. However, the content of insulin in the cerebrospinal fluid (CSF) does not change, or is mildly elevated in age-matched healthy individuals (30–32). Furthermore, it was demonstrated that the treatment with intranasal insulin improves learning and memory in individuals with mild cognitive impairment (MCI) and AD, suggesting the involvement of dysfunctional insulin signaling as a key player in the pathogenic mechanism of AD progression.
The relationship between insulin signaling and tau phosphorylation has been reported in previous studies, which suggested a biphasic role of insulin signaling in the aberrant phosphorylation of tau (33–35). Treatment of insulin within a short time led to a rapid and transient hyperphosphorylation of tau (33, 34). Contrastly, prolonged exposure to insulin resulted in a significant decrease in tau phosphorylation (34, 35). Furthermore, investigations also reveal that insulin resistance in neurons promotes the phosphorylation level of tau (36–38), and in turn hyperphosphorylation of tau also causes dysfunctional insulin signaling (39, 40). However, whether insulin resistance is linked with iron overloading and the pathological changes in AD, remains to be further investigated. Therefore, in this current study, we investigated the effect of iron on the phosphorylation of tau, and on insulin in neurons. We also analyzed these lesions in the brains of mice treated with high-iron chow. Using an in vitro model, we then investigated the effect of iron-overloaded neurons following exogenous supplementation of insulin to evaluate the potential role of insulin signaling in iron-induced aberrant phosphorylation of tau in AD progression.
Materials and Methods
Cell Culture and Treatments
Primary neurons were isolated, purified, and then cultured as previously described, with some modifications to the methodologies described previously (41, 42). Briefly, on embryonic day 17, pregnant Sprague-Dawley (SD) rats were anesthetized and the fetuses collected to isolate cortices for digestion using trypsin (Gibco, USA). Cell suspensions were filtered, centrifuged, and then plated onto poly-L-lysine-coated dishes or plates. Cells were cultured in a 60 mm dish for protein determination using Western blot, in 24-well plates for immunofluorescence staining, and in 96-well plates for the cell viability assay. After 4 h, the medium was replaced with Neurobasal medium (Gibco, USA) containing B27 and GlutaMAX (Gibco, USA). The cultures were maintained at 37°C in a humidified 5% CO2 atmosphere for 12 days before treatment.
Ferrous (Fe2+) chloride (Sigma, USA) was used to achieve iron overloading in cultures. Ferrous (Fe2+) chloride powder was dissolved in the solvent of sterilized water containing 0.01 N HCl as previously described, and a final concentration of 20 μM in the cultures was used, as we reported elsewhere (42), where an isometric solvent was delivered into the cultures as a normal control.
For treatment with insulin, bovine insulin (Sigma, USA) was solubilized at 1 mg/ml as a stock solution and frozen at −20°C in single-use aliquots. The final concentration of insulin was 1 μg/ml as previously reported (43) and incubation was for 24 h, after which the cells were analyzed.
Cell Viability Detection
To determine cell viability, the CKK-8 assay kit (Dojindo, Japan) was used and the procedures were conducted according to the manufacturer's instructions. A microplate reader set at a wavelength of 450 nm (Thermo Fisher, USA) was used to measure the absorbance.
Animals and Treatments
Animal procedures were approved by the Medical Experimental Animal Administrative Committee of Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Two-month-old male C57BL/6 mice were purchased from Shanghai SLAC (China). Mice were housed with unrestricted access to food and water in a 22°C environment that maintained a 12–12 h light-dark cycle. For the iron overloading model, mice were fed with high-iron chow (3,000 mg carbonyl iron/kg diet, TROPHIC, China) as previously reported (44, 45). Control mice were fed standard chow (50 mg iron/kg diet, TROPHIC, China). Six weeks later, the mice were sacrificed for experimental investigation.
Morris Water Maze Test
The Morris water maze (MWM) was conducted as previously reported (46–48). Initially, mice (n = 8/group for each independent experiment) were trained twice each day to find the concealed platform in the maze. The trial was ended if the mice successfully climbed onto the escape platform, or after 60 s. Each mouse could remain on the platform for 15 s. The training persisted for 5 days and the platform was removed on the sixth day. In the probe test, mice were tracked, and parameters were recorded including escape latency, cross time, target quadrant, and percentage of time in target quadrant.
Brain Tissue Preparation
Mice were deeply anesthetized by inhaling isoflurane and transcardially perfused with normal saline. For Western analysis and iron level detection, mice (n = 3/group for each independent experiment) were then decapitated, and the cerebral cortex of each mouse was quickly collected and frozen for further analysis. For immunofluorescence staining, mice (n = 3/group for each independent experiment) were then fixed with a freshly prepared solution of 4% paraformaldehyde (PFA; Sigma, USA) in 0.1 M PB (pH 7.2). After decapitation, the skull was removed and the whole brain was successively immersed in PFA and then sucrose, successively each for 24 h. Serial 20 μm thick coronal tissue sections were obtained using a freezing microtome (Leica, USA).
Detection of Iron in the Brain
Iron levels in mouse brains were detected using an Enzyme Linked Immunosorbent Assay kit from Abcam Company as previously reported (49). The process was conducted according to the manufacturer's protocol.
Co-immunoprecipitation
IRβ and IRS-1 protein interaction was determined using co-immunoprecipitation (Co-IP) as previously described (50). Briefly, lysates from neural cultures were incubated with the primary antibodies overnight on a rotator at 4°C, followed by addition of Protein A and G Agarose (Roche, CH) to capture the immune-complexes. Then, the agarose-antibody-complexes were eluted and centrifuged for collection. Subsequently, the complexes were mixed with 50 μL of 2 × SDS loading buffer. Finally, Western blot analysis was performed to detect the presence of targeted proteins.
Western Blot
The procedure of Western blotting (51, 52) was carried out as we described previously (48, 53). Briefly, the protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo fisher, USA) and protein samples (30 μg total protein per lane) were loaded onto a 10% sodium dodecyl sulfate–polyacrylamide gel and electrophoresed (SDS-PAGE). Then, fractionated proteins were transferred onto nitrocellulose (NC) membrane, followed by incubation overnight at 4°C with the indicated antibodies (Table 1). Following washing of the membranes, they were incubated with a secondary antibody (IRDye®680LT, IRDye®800CW, 1:10,000 dilution, LI-COR) for 1 h at room temperature. Images were captured, and band intensities measured using the Odyssey infrared fluorescence imaging system (LI-COR, USA).
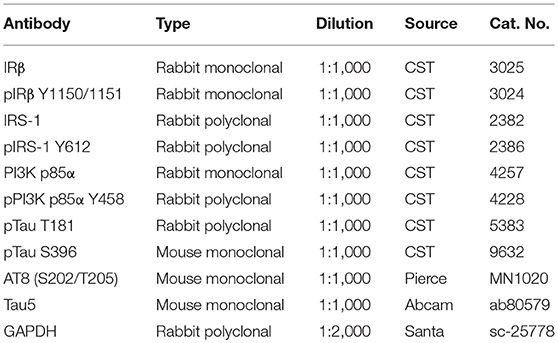
Table 1. Primary antibodies employed in this study.
Immunofluorescence Staining
For brain tissue, the sections were washed with PBS, and this was followed by incubation with 10% goat serum in PBS containing 0.5% Triton-X 100 for 1 h at 37°C. Subsequently, the sections were incubated with AT8 antibody (mouse monoclonal, 1:1,000, Pierce, USA) for 1 h at 37°C followed by overnight at 4°C. After incubation for 1 h at 37°C with the secondary antibodies (Alexa goat anti-rabbit 555 and Alexa goat anti-mouse 555, Invitrogen, USA), the sections were stained with DAPI (1:10,000, Invitrogen, USA) to detect nuclei. Images were acquired with a fluorescence microscope (Nikon, Japan). Fluorescence intensity was measured using Image-Pro Plus, Version 6.0 (MediaCybernetics, Inc., USA) and was normalized to the number of neurons.
For cells maintained on a slide, the slide was washed with 0.01 M PBS and then the cells were fixed with 4% PFA as we described elsewhere (54). After washing with PBS, the cells were then incubated with 10% goat serum in PBS containing 0.5% Triton-X 100 for 1 h at 37°C. Then the subsequent procedures were as described for brain sections above and the fluorescence intensity was normalized to the brain surface.
Statistical Analyses
Results are shown as the mean ± SD. Statistical analysis was carried out using GraphPad Prism 5 (GraphPad Software, Inc. USA). Experiments were repeated on three independent occasions. In the test of MWM, the number of mice was eight in each group for each independent experiment. In the biochemical detection and fluorescence staining, the number of mice was three in group for each independent experiment. The statistical significance of differences among the various groups was examined using one-way analysis of variance (ANOVA) or the student t-test. A value of p < 0.05 was considered to be statistically significant.
Results
Elevated Levels of Phosphorylated Tau in Iron-Treated Neurons
Iron deposition results in a decrease in tau phosphorylation (55). However, the phosphorylation level of tau was increased in the brains of dietary iron-treated mouse (22) and in neurons exposed to ferrous (Fe2+) chloride (56). In neurons, iron is delivered across the neural membrane primarily by transferrin–transferrin receptor system-mediated uptake and efflux, via ferroportin (Fpn) in the form of ferrous (Fe2+) chloride (14). Therefore, in our in vitro work neurons were treated with ferrous (Fe2+) chloride, and in the in vivo study the mice were administered a diet containing carbonyl iron (Fe0). As previously described (42), ferrous (Fe2+) chloride was employed to model the microenvironment of iron deposition. After a 24 h treatment with either 50 μM or 100 μM ferrous (Fe2+) chloride, the viability of neurons was significantly reduced by 23 and 11%, respectively (Figure 1A, p < 0.05). At a concentration of 20 μM, the ferrous (Fe2+) chloride treatment led to a decrease of 5% in cell viability after treatment for 24 h in primary cultured neurons, but this decrease was not statistically significant (Figures 1A,B).
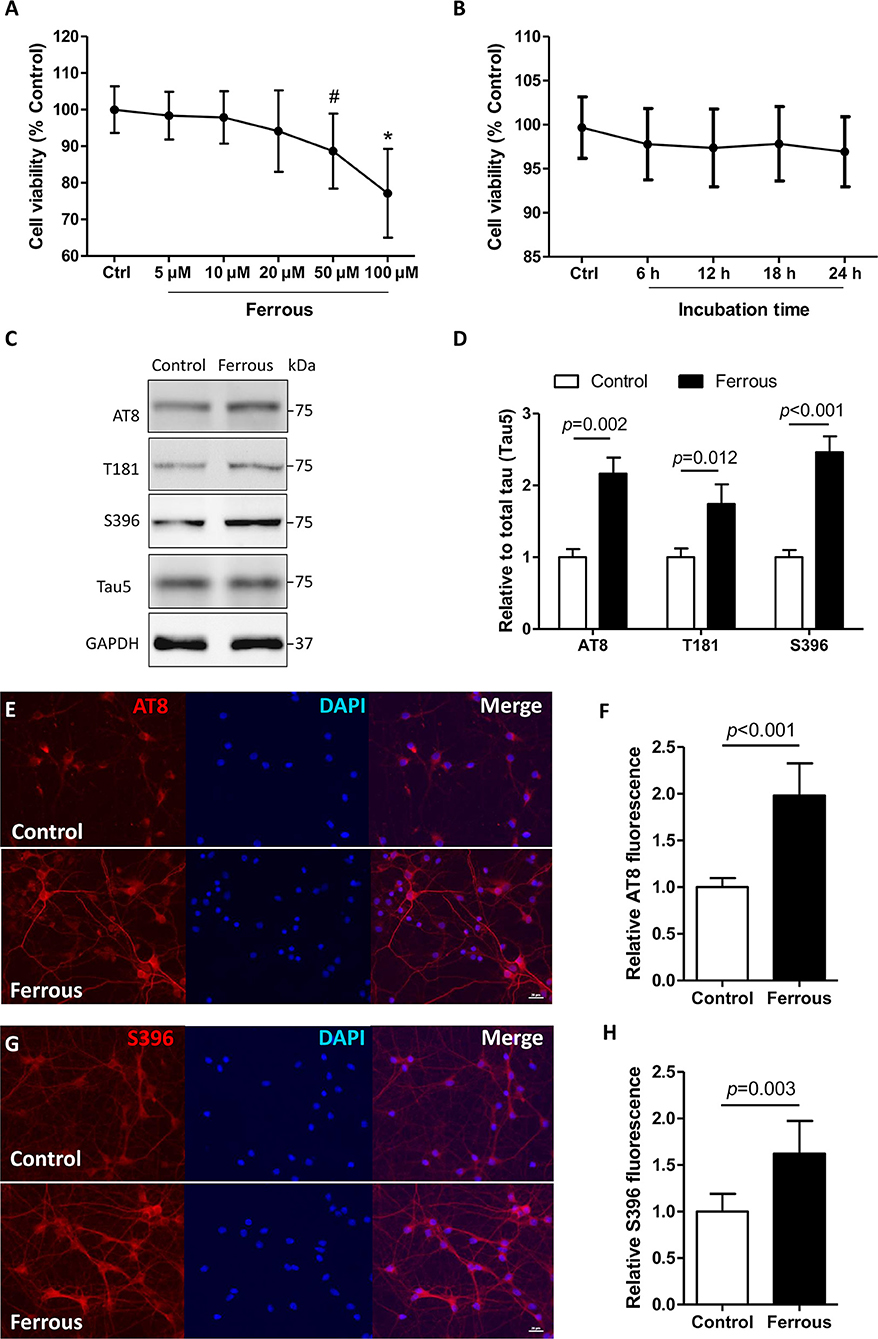
Figure 1. Phosphorylation levels of tau are increased in iron-treated neurons. In primary cultures, neurons were treated with ferrous (Fe2+) chloride for 24 h and then analyzed by various of assays. (A) Shows the cell viability data after treatment with different concentrations of (Fe2+) chloride for 24 h. (B) Shows the cell viability result when neurons were treated 20 μM ferrous (Fe2+) chloride at different time points. (C) The levels of tau protein phosphorylation at epitopes of AT8, T181, and S396 were determined using Western blots. (E,G) Show immunofluorescence staining revealing an increase in the level of phosphorylation at epitopes AT8 and S396 in neurons, respectively. (D,F,H) Show the quantitation of the results presented in (C,E,G), respectively. Results are shown as mean ± SD. Student's t-test was employed to determine the significance. NS, no significant difference. *vs. Control, p < 0.01, #vs. Control, p < 0.05.
In AD, multiple characteristic epitopes of tau protein phosphorylation have been reported including Ser202/Thr205 (AT8), Thr181 (T181), and Ser396 (S396) (57, 58). In iron-treated neurons, the levels of tau phosphorylation at epitopes of AT8, T181, and S396 were elevated by 1.2-, 0.7-, and 1.5-fold, respectively (Figures 1C,D, p < 0.05, p < 0.01). Using immunofluorescence staining, we also observed increased levels of phosphorylation at epitopes of AT8 and S396 in ferrous (Fe2+) chloride-incubated neurons (Figures 1E–H, p < 0.01).
Treatment With Iron Disrupts Insulin Signaling in Neurons
Previous studies suggested an essential role of insulin resistance in the hyperphosphorylation of tau, but whether insulin resistance contributes to iron-induced tauopathy has not been determined. Here, we evaluated the changes in insulin signaling in iron-treated neurons. In physiological conditions, extracellular insulin combines with, and activates, insulin receptor α (IRα). This successively leads to tyrosine phosphorylation of both insulin receptor β (IRβ) and insulin signaling substrates (IRS), which activates and engages the insulin signaling pathway (59).
We were unable to detect any changes in the level of unphosphorylated IRβ (i.e., total IRβ protein content) in iron-treated neurons, but the tyrosine phosphorylation content of the IRβ kinase regulatory domain (pIRβ Y1150/1151) was significantly decreased by 55% as compared to the control (Figures 2A,D, p < 0.01). With regard to downstream factors of insulin signaling, the tyrosine phosphorylation levels of both IRS-1 (pIRS-1 Y612) and phosphoinositide 3-kinase p85α (pPI3K p85α Y458) were also significantly decreased by 70 and 28%, respectively (Figures 2B,C,E,F, p < 0.01), but the total protein content of either of the two targets was not changed after 24 h incubation with iron (Figures 2A–C).
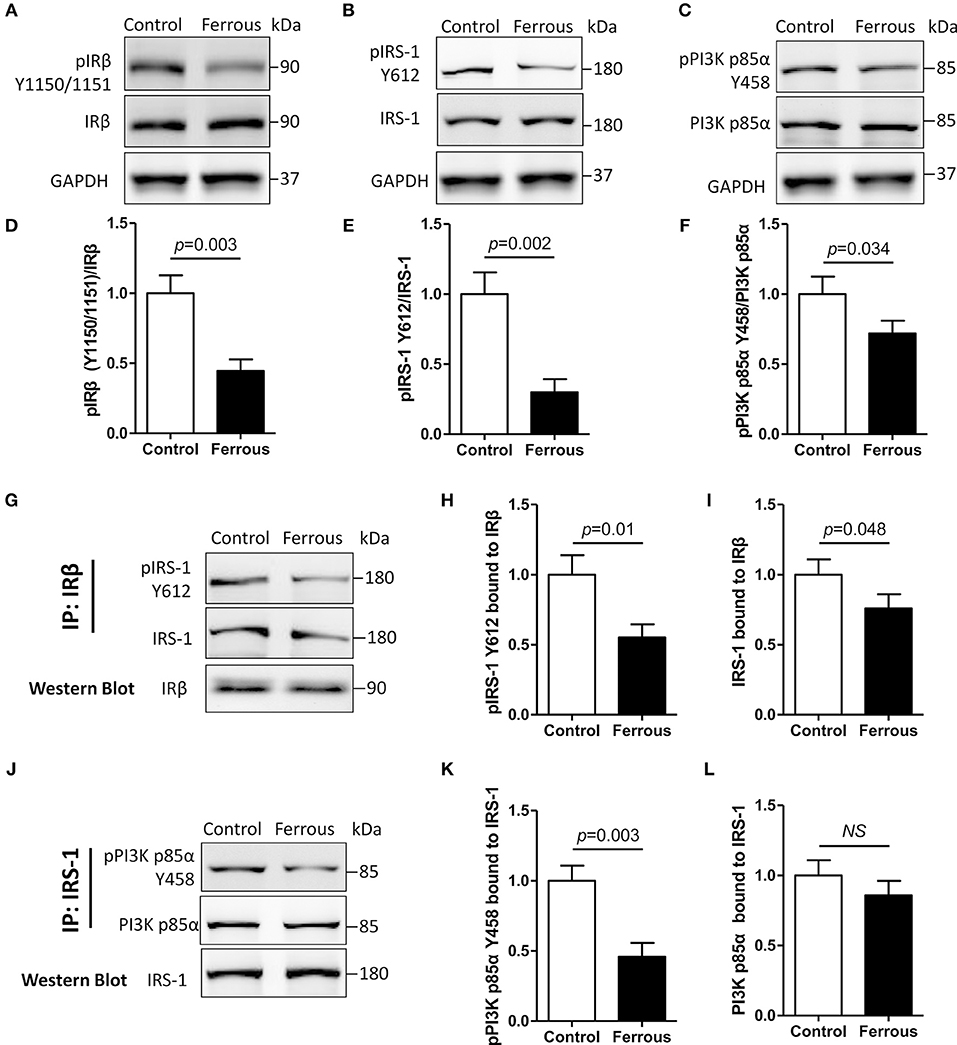
Figure 2. Dysfunctional insulin signaling is induced by the treatment of neurons with iron. Changes of in tyrosine phosphorylation, and total IRβ, IRS-1, and PI3K p85α protein, were evaluated using Western blot (A–C). Antibodies recognizing IRβ (G) or IRS-1 (J) were utilized for co-immunoprecipitation with total proteins, and then the predicated proteins were determined using Western blot. (D–F) Show the quantitation of the results presented in (A–C), respectively. (H,I) Show the quantitation of the results presented in (G). (K,L) Show the quantitation of the results presented in (J). Results are shown as Mean ± SD. Student's t-test was employed to determine the significance. NS, no significant difference.
We then evaluated protein-protein interactions in insulin signaling using the co-immunoprecipitation (Co-IP) assay (50). IRβ-bound total protein, and tyrosine phosphorylation levels of IRS-1, were decreased by 45 and 24%, respectively, in iron-treated neurons (Figures 2G–I, p < 0.05, p < 0.01). Treatment with iron caused a reduction in the level of IRS-1-bound total PI3K p85α, which was not significant (Figures 2J,L), but also led to a significant 54% decrease in IRS-1-bound tyrosine phosphorylation of PI3K p85α (Figures 2J,K, p < 0.01).
Supplemental Dietary Iron Impairs Cognition and Causes Abnormal Phosphorylation of Tau in Mice
To achieve iron overloading in the brain, mice were administered a high-iron diet as previously reported (44, 45). As shown in Figure 3A, the contents of mouse brains were increased by 0.71-fold after treatment with a high-iron diet for 6 weeks (p < 0.05). We evaluated the learning and memory function in iron-overloaded mice. In the MWM test, the escape latency in iron-overloaded mice was increased by 2.7-fold, and the cross time was significantly decreased as compared to the control (Figures 3B,C, p < 0.01). Additionally, the data representing the time to the target quadrant, and the percentage of time in the target quadrant, showed impaired learning and memory in iron-overloaded mice (Figures 3C,D, p < 0.01).
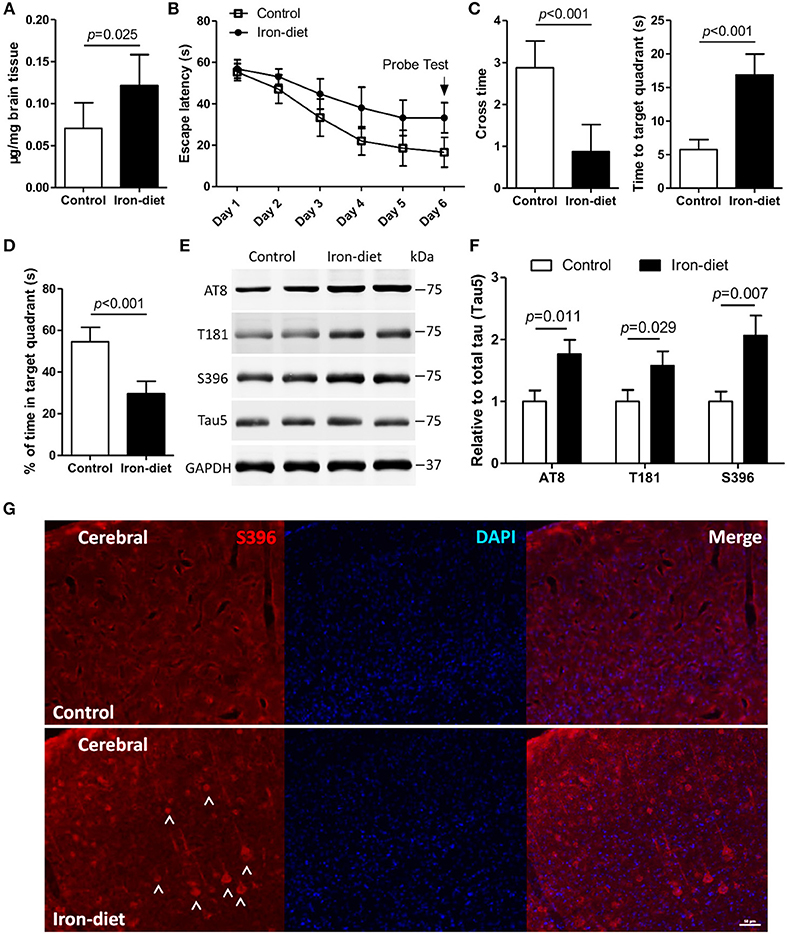
Figure 3. Cognition is impaired, and there is aberrant phosphorylation of tau in iron-overloaded mouse brains. In the MWM test, mice were trained for 5 days and then a probe test (i.e., the platform was removed) was performed on the sixth day. The content of iron in the mouse brains was determined (A). The escape latency (B), cross time and time to target quadrant (C), and the percentage of time in target quadrant (D) were evaluated. The levels of tau phosphorylation in iron-overloaded mouse brains was determined using Western blot analysis (E), and immunofluorescence staining (G). In the MWM test, the number of mice was eight in each group for each independent experiment. In biochemical detection and fluorescence staining, the number of mice was three in group for each independent experiment. (F) shows the quantitation of the results presented in (E). Results are shown as mean ± SD. Student's t-test was employed to determine the significance.
We subsequently investigated whether the pathology associated with aberrant tau phosphorylation was induced in mice fed with high-iron chow. In iron-treated mice, the content of total tau, determined with Tau5 antibody using Western blot, was not changed. However, the levels of tau phosphorylation at multiple epitopes, including AT8, T181, and S396, were increased by 0.76-, 0.57-, and 1.1-fold, respectively, in the brains of iron-overloaded mice (Figures 3E,F, p < 0.05, p < 0.01). These results revealed that alteration of epitopes AT8 and S396 was significant after treatment with a high-iron diet (Figures 3E,F, p < 0.05, p < 0.01). An independent, immunofluorescence staining assay provided evidence of an increase in the level of phosphorylation at epitope S396 in iron-overloaded mice brains (Figure 3G), which is consistent with increased S396 phosphorylation detected in iron-treated neurons (Figures 1C–H).
Insulin Signaling Is Disrupted in the Brains of Mice Fed With High-Iron Chow
As previously mentioned, insulin signaling was disrupted by the treatment of iron in vitro. We then evaluated the phosphorylation changes of IRβ, IRS-1, and PI3K p85α in vivo. As shown in Figure 4, the levels of unphosphorylated IRβ, IRS-1, and PI3K p85α were equivalent in iron-overloaded mouse brains compared with controls. Tyrosine phosphorylation levels of IRβ (pIRβ Y1150/1151) and IRS-1 (pIRS-1 Y612) were decreased by 32 and 52%, respectively (Figures 4A–D, p < 0.05, p < 0.01). We also determined the levels of PI3K p85α in iron-overloaded mouse brains and found that the total level of PI3K p85α was not altered (Figure 4E). However, there was a 15% reduction of tyrosine phosphorylated PI3K p85α (pPI3K p85α Y458), but this was not statistically significant (Figures 4E,F). Furthermore, we also determined the change of blood glucose in iron-overloaded mice and the result showed that there was also no significant difference between the levels of blood glucose in the two groups (Figure 4G).
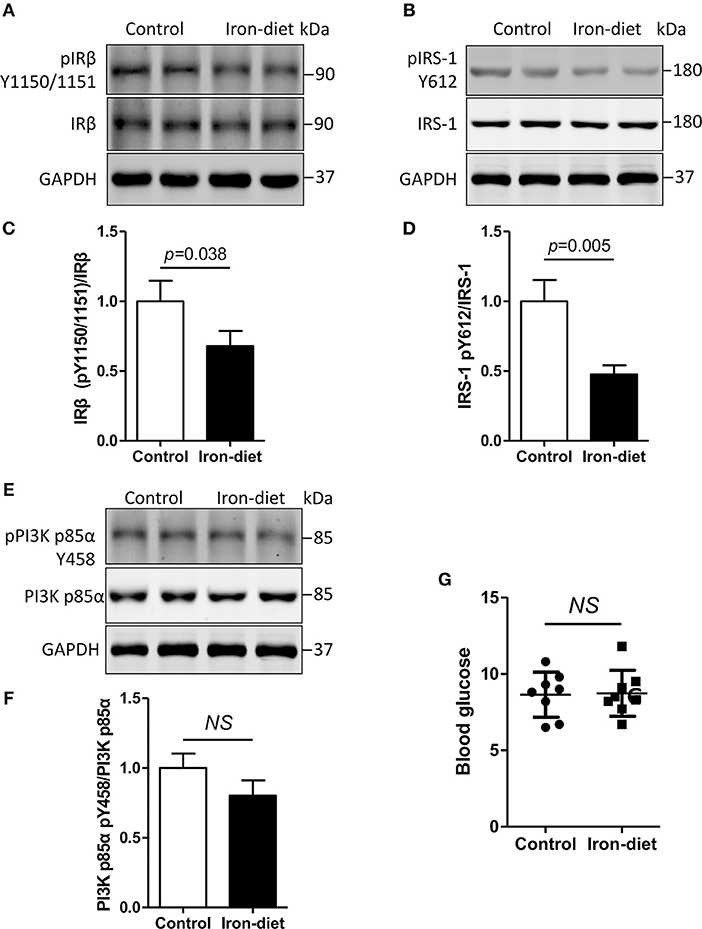
Figure 4. Insulin signaling is disturbed in iron-overloaded mouse brains. The changes of insulin signaling in mouse cerebral cortexes were investigated using Western blot analysis. The total protein content of IRβ and IRS-1 was not altered. However, the tyrosine phosphorylation levels of IRβ and IRS-1 were decreased in iron-overloaded mouse brains (A,B,E). Total PI3K p85α protein in iron-overloaded mouse brains was not changed. A decrease in tyrosine phosphorylated PI3K p85α Y458 was observed but was not significant (E). In the analysis, the number of mice was three in group for each independent experiment. (C,D,F) Show the quantitation of the results presented in (A,B,E), respectively. (G) Shows the blood glucose levels that were determined using a glucometer. Results are shown as mean ± SD. Student's t-test was employed to determine the significance.
Abnormal Phosphorylation of Tau Due to Iron Overloading Is Attenuated in Insulin-Treated Neurons
To further investigate the role of dysfunctional insulin signaling resulting from iron-induced changes to tau protein, neurons were incubated with supplementary insulin, with or without the treatment of ferrous (Fe2+) chloride. The effect of supplementary insulin on insulin signaling was analyzed by determining the tyrosine phosphorylation levels of targets. Western blot analysis showed that supplementary insulin did not alter the total protein levels of IRβ, IRS-1, andPI3K p85α (Figures 5A,C,E), but supplementary insulin raised the phosphorylation levels of pIRβ Y1150/1151, pIRS-1 Y612, and pPI3K p85α Y458 in neurons incubated with or without ferrous (Fe2+) chloride (Figures 5A–E). However, the increase in tyrosine phosphorylation levels in iron-treated neurons remained reduced compared to the controls (Figures 5A–F).
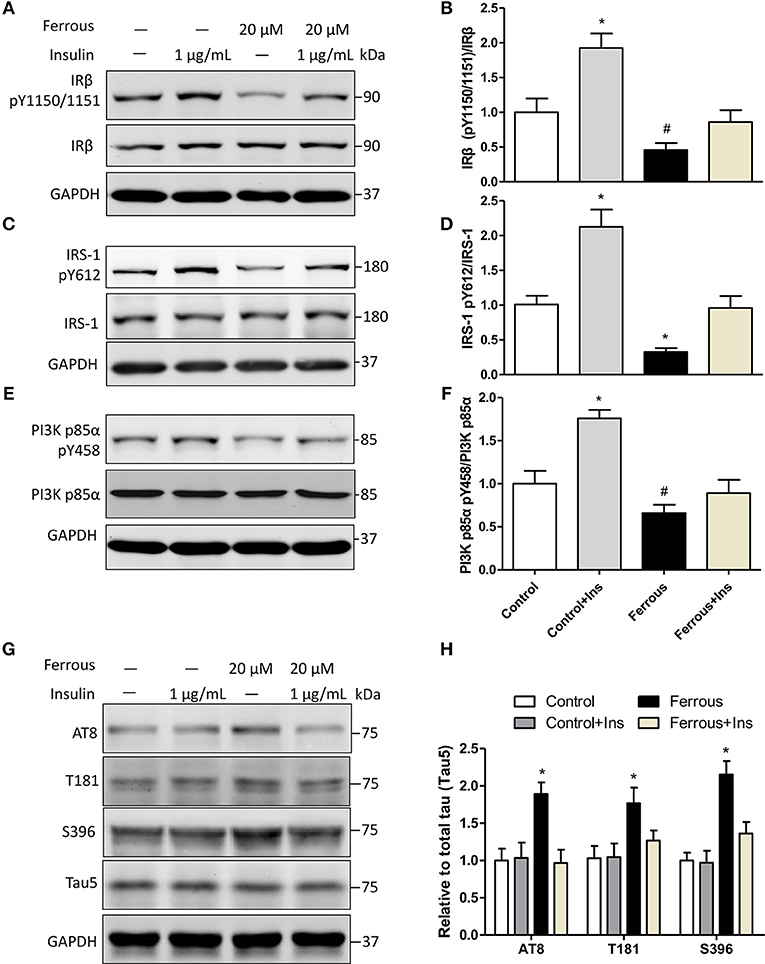
Figure 5. Activation of insulin signaling attenuates the effect of iron-induced hyperphosphorylation tau in primary neurons. Exogenous 1 μg/ml insulin was added to neurons, together with exposure to iron. Twenty-four hours later, neurons were collected and analyzed by Western blot. (A,C,E) show the changes in insulin signaling in neurons, consistent with activated insulin signaling. Phosphorylation levels of tau were determined (G). (B,D,F,H) show the quantitation of the results presented in (A,C,E,G), respectively. Results are shown as mean ± SD. One-way ANOVA was employed to determine the significance. *vs. Ctrl, p < 0.01, #vs. Ctrl, p < 0.05.
Then, we determined the changes of taupathy after exposure to insulin. We found that insulin signaling was activated by exogenous addition of insulin, but the levels of phosphorylated tau were not changed in neurons without iron treatment (Figures 5G,H). This suggested that 1 μg/ml insulin was a suitable concentration in our work and the insulin resistance was not induced in neurons. In iron-treated neurons, increased phosphorylation at AT8, T181, and S396 was ameliorated by supplementation with insulin (Figures 5G,H).
Discussion
Almost all organisms and cells require iron to act as a cofactor in cellular processes such as energy metabolism, DNA synthesis, and repair, oxygen transport, signal transduction, electron transport, and neurotransmitter transmission (16, 60). The constant balance of uptake and elimination is critical for iron homeostasis not only at the whole human body level, but also in each organ and cell (61). Disruption of iron metabolism is implicated in the etiology of many diseases including neurodegenerative disorders. In the brain, iron in the blood circulation is captured mainly via the transferrin- transferrin receptor 1 (TFR1) system, and then crosses the endothelial cells of blood-brain barrier (BBB), to then be taken up by other cells such as neurons and astrocytes (62). In neurons, the influx of iron is mediated mostly by TFR, and efflux via ferroportin (Fpn) (62), but the detailed mechanism is not fully known.
In the past few decades, studies have provided increasing support for a correlation between iron deposition in the brain and neurodegenerative diseases such as AD (63–65). By MRI detection, the level of iron in the brain correlates with cognitive impairment (66). The current study investigated the potential role of intracerebral iron-overloading in taupathology and alteration of insulin signaling. In this study, we first evaluated cognitive function and observed that iron overloading impaired learning and memory in mice. However, patients with AD do not generally adhere to iron-rich diets and iron levels in the blood are normal (67), which suggests that perturbed iron distribution is due to a neuronal imbalance between the influx and efflux of iron.
In AD, iron overloading in the brain is a key pathological feature, together with hyperphosphorylation of tau (20), but the underlying mechanism is not yet fully understood. Previous studies show oxidative stress is involved in iron-induced pathologies (68). Oxidative stress is caused by an imbalance between the generation of reactive oxygen species (ROS) and the detoxifying system in cell (69). Elevated iron levels in neurons lead to the generation of ROS and cause oxidative stress injury (70). Nevertheless, there is controversy regarding the role of oxidative stress, and recent investigations provide evidence that oxidative stress might not be the critical process in iron-induced pathological damage (42, 71). Thus, other mechanisms might also be involved.
Perturbed brain metabolism is an early and invariant characteristic of AD (72, 73). Mounting evidence suggests that cerebral hypometabolism is involved in the development of AD and contributes to the formation of its hallmarks (74–76). Determining the key factors and pathways that lead to dysfunctional cerebral metabolism is essential to determining the mechanisms of AD and for identifying possible targets for intervention. Insulin signaling is the prominent modulator of cellular activities through its regulation of energy metabolism, mitogenesis, development, synaptic plasticity, and other processes (24, 28). Accumulating evidence reveals that dysfunctional insulin signaling is a major risk factor for the onset of AD, particularly the sporadic type (77). Currently, there is no direct evidence of a role for iron in neural insulin signaling. In our study, we observed that tyrosine phosphorylation of particular proteins involved in insulin signaling was decreased in ferrous (Fe2+) chloride-exposed neurons, and in iron-overloaded mouse brains. Moreover, aberrant phosphorylation of tau in neurons as a result of exposure to ferrous (Fe2+) chloride was reduced by supplementary insulin.
In patients, biomarkers of insulin resistance in the CNS are universally and progressively increased from MCI to AD (50). Furthermore, the severity of insulin resistance is also negatively correlated with cognition, and positively associated with amyloid deposition (50). Insulin resistance markedly impacts on the generation of Aβ and the status of phosphorylation in rodent models of AD (78–81). Targeting the improvement of insulin signaling may ameliorate learning and memory, and inhibit Aβ plaque and NFT formation (38, 82, 83). In patients with amnestic MCI or AD, delivery of insulin intranasally improved delayed memory and functional ability (84).
However, previous work revealed that Ca2+ signaling was also modulated by the treatment of iron, which in turn leading to the activation of MAPK/ERK pathway (85). Both Ca2+ signaling and MAPK/ERK pathway have been revealed to involved in the progress of AD (86, 87), but their accurate effect in iron-induced tau pathology is still unclear. Thus, further investigations needed to better understand the role of iron deposition in AD.
The present work revealed that dysfunctional insulin signal is involved in the hyperphosphorylation of tau, as a result of iron-overloading. However, we did not further evaluate the effect of aberrant phosphorylation of tau on insulin signal. The relationship of tau pathology and insulin signal is complicated and more information could be found in Gratuze's review which considered the mutual relationship between hyperphosphorylation tau and dysfunctional insulin signal in AD (88). These findings suggest a potential role for insulin signaling in pathological lesions induced by iron accumulation. However, there are limitations in our current work and further investigations are needed. For example, the role of dysfunctional insulin signaling in the in vivo tau pathology is unclear and needs further elucidation. Additionally, the effect of iron chelators should be evaluated in iron-overloaded mice, and the mechanisms underlying iron-induced aberrant phosphorylation of tau also need to be determined.
Ethics Statement
Animal procedures were approved by the Medical Experimental Animal Administrative Committee of Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Author Contributions
WW and LC carried out the experiment and wrote the manuscript with support from YG and BK. YG helped supervise the project. BK, PM, and SX participated in the English language correction.
Funding
This study was funded by National Natural Science Foundation of China (Grant Nos. 81801043, 81771295, 81870044) and Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant Support (Grant Nos. YS2017ZY050083, TM201508).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Zhu X, Raina AK, Perry G, Smith MA. Alzheimer's disease: the two-hit hypothesis. Lancet Neurol. (2004) 3:219–26. doi: 10.1016/s1474-4422(04)00707-0
2. Piaceri I, Nacmias B, Sorbi S. Genetics of familial and sporadic Alzheimer's disease. Front Biosci. (2013) 5:167–77. doi: 10.2741/e605
3. Jadhav S, Cubinkova V, Zimova I, Brezovakova V, Madari A, Cigankova V, et al. Tau-mediated synaptic damage in Alzheimer's disease. Transl Neurosci. (2015) 6:214–26. doi: 10.1515/tnsci-2015-0023
4. Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: a crucial factor in Alzheimer's disease. Med Princ Pract. (2015) 24:1–10. doi: 10.1159/000369101
5. Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. (2007) 316:750–4. doi: 10.1126/science.1141736
6. Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. (1992) 42:631–9.
7. Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. (1997) 41:17–24. doi: 10.1002/ana.410410106
8. Scholz T, Mandelkow E. Transport and diffusion of tau protein in neurons. Cell Mol Life Sci. (2014) 71:3139–50. doi: 10.1007/s00018-014-1610-7
9. Bandyopadhyay B, Li G, Yin H, Kuret J. Tau aggregation and toxicity in a cell culture model of tauopathy. J Biol Chem. (2007) 282:16454–64. doi: 10.1074/jbc.M700192200
10. Frozza RL, Lourenco MV, De Felice FG. Challenges for Alzheimer's disease therapy: insights from novel mechanisms beyond memory defects. Front Neurosci. (2018) 12:37. doi: 10.3389/fnins.2018.00037
11. Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA. The role of metallobiology and amyloid-beta peptides in Alzheimer's disease. J Neurochem. (2012) 120(Suppl. 1):149–66. doi: 10.1111/j.1471-4159.2011.07500.x
12. Bush AI. The metal theory of Alzheimer's disease. J Alzheimers Dis. (2013) 33(Suppl 1):S277–81. doi: 10.3233/jad-2012-129011
13. Smith MA, Zhu X, Tabaton M, Liu G, McKeel DW Jr, Cohen ML, et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis. (2010) 19:363–72. doi: 10.3233/jad-2010-1239
14. Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. (2014) 13:1045–60. doi: 10.1016/s1474-4422(14)70117-6
15. Tao Y, Wang Y, Rogers JT, Wang F. Perturbed iron distribution in Alzheimer's disease serum, cerebrospinal fluid, and selected brain regions: a systematic review and meta-analysis. J Alzheimers Dis. (2014) 42:679–90. doi: 10.3233/jad-140396
16. Salvador GA. Iron in neuronal function and dysfunction. Biofactors. (2010) 36:103–10. doi: 10.1002/biof.80
17. Ding B, Chen KM, Ling HW, Sun F, Li X, Wan T, et al. Correlation of iron in the hippocampus with MMSE in patients with Alzheimer's disease. J Magn Reson Imaging. (2009) 29:793–8. doi: 10.1002/jmri.21730
18. Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer's disease. J Neurosci Res. (1992) 31:327–35. doi: 10.1002/jnr.490310214
19. Meadowcroft MD, Connor JR, Smith MB, Yang QX. MRI and histological analysis of beta-amyloid plaques in both human Alzheimer's disease and APP/PS1 transgenic mice. J Magn Reson Imaging. (2009) 29:997–1007. doi: 10.1002/jmri.21731
20. Peters DG, Connor JR, Meadowcroft MD. The relationship between iron dyshomeostasis and amyloidogenesis in Alzheimer's disease: two sides of the same coin. Neurobiol Dis. (2015) 81:49–65. doi: 10.1016/j.nbd.2015.08.007
21. Becerril-Ortega J, Bordji K, Freret T, Rush T, Buisson A. Iron overload accelerates neuronal amyloid-beta production and cognitive impairment in transgenic mice model of Alzheimer's disease. Neurobiol Aging. (2014) 35:2288–301. doi: 10.1016/j.neurobiolaging.2014.04.019
22. Guo C, Wang P, Zhong ML, Wang T, Huang XS, Li JY, et al. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem Int. (2013) 62:165–72. doi: 10.1016/j.neuint.2012.12.005
23. Guo C, Wang T, Zheng W, Shan ZY, Teng WP, Wang ZY. Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer's disease. Neurobiol Aging. (2013) 34:562–75. doi: 10.1016/j.neurobiolaging.2012.05.009
24. Zhang Q, Xiao XH, Li M, Li WH, Yu M, Zhang HB, et al. Chromium-containing traditional chinese medicine, Tianmai Xiaoke tablet improves blood glucose through activating insulin-signaling pathway and inhibiting PTP1B and PCK2 in diabetic rats. J Integr Med. (2014) 12:162–70. doi: 10.1016/s2095-4964(14)60020-0
25. Okon UA, Umoren IU. Comparison of antioxidant activity of insulin, Ocimum gratissimum L., and Vernonia amygdalina L. in type 1 diabetic rat model. J Integr Med. (2017) 15:302–9. doi: 10.1016/s2095-4964(17)60332-7
26. Derakhshan F, Toth C. Insulin and the brain. Curr Diabetes Rev. (2013) 9:102–16. doi: 10.2174/1573399811309020002
27. Cai Z, Xiao M, Chang L, Yan LJ. Role of insulin resistance in Alzheimer's disease. Metab Brain Dis. (2015) 30:839–51. doi: 10.1007/s11011-014-9631-3
28. Dineley KT, Jahrling JB, Denner L. Insulin resistance in Alzheimer's disease. Neurobiol Dis. (2014) 72(Pt A):92–103. doi: 10.1016/j.nbd.2014.09.001
29. Medhi B, Chakrabarty M. Insulin resistance: an emerging link in Alzheimer's disease. Neurol Sci. (2013) 34:1719–25. doi: 10.1007/s10072-013-1454-1
30. Molina JA, Jimenez-Jimenez FJ, Vargas C, Gomez P, de Bustos F, Gomez-Escalonilla C, et al. Cerebrospinal fluid levels of insulin in patients with Alzheimer's disease. Acta Neurol Scand. (2002) 106:347–50. doi: 10.1034/j.1600-0404.2002.01326.x
31. Fujisawa Y, Sasaki K, Akiyama K. Increased insulin levels after OGTT load in peripheral blood and cerebrospinal fluid of patients with dementia of Alzheimer type. Biol Psychiatry. (1991) 30:1219–28.
32. Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm. (1998) 105:423–38. doi: 10.1007/s007020050068
33. Lesort M, Johnson GV. Insulin-like growth factor-1 and insulin mediate transient site-selective increases in tau phosphorylation in primary cortical neurons. Neuroscience. (2000) 99:305–16. doi: 10.1016/S0306-4522(00)00200-1
34. Lesort M, Jope RS, Johnson GV. Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3beta and fyn tyrosine kinase. J Neurochem. (1999) 72:576–84.
35. Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. (1997) 272:19547–53.
36. van der Harg JM, Eggels L, Bangel FN, Ruigrok SR, Zwart R, Hoozemans JJM, et al. Insulin deficiency results in reversible protein kinase A activation and tau phosphorylation. Neurobiol Dis. (2017) 103:163–73. doi: 10.1016/j.nbd.2017.04.005
37. Gratuze M, Julien J, Petry FR, Morin F, Planel E. Insulin deprivation induces PP2A inhibition and tau hyperphosphorylation in hTau mice, a model of Alzheimer's disease-like tau pathology. Sci Rep. (2017) 7:46359. doi: 10.1038/srep46359
38. Chen Y, Zhao Y, Dai CL, Liang Z, Run X, Iqbal K, et al. Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces abeta level and microglia activation in the brains of 3xTg-AD mice. Exp Neurol. (2014) 261:610–9. doi: 10.1016/j.expneurol.2014.06.004
39. Rodriguez-Rodriguez P, Sandebring-Matton A, Merino-Serrais P, Parrado-Fernandez C, Rabano A, Winblad B, et al. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain. (2017) 140:3269–85. doi: 10.1093/brain/awx256
40. Peng D, Pan X, Cui J, Ren Y, Zhang J. Hyperphosphorylation of tau protein in hippocampus of central insulin-resistant rats is associated with cognitive impairment. Cell Physiol Biochem. (2013) 32:1417–25. doi: 10.1159/000356579
41. Xu Y, Wang D, Luo Y, Li W, Shan Y, Tan X, et al. Beta amyloid-induced upregulation of death receptor 6 accelerates the toxic effect of N-terminal fragment of amyloid precursor protein. Neurobiol Aging. (2015) 36:157–68. doi: 10.1016/j.neurobiolaging.2014.07.027
42. Wan W, Jin L, Wang Z, Wang L, Fei G, Ye F, et al. Iron deposition leads to neuronal α-synuclein pathology by inducing autophagy dysfunction. Front. Neurol. (2017) 8:1. doi: 10.3389/fneur.2017.00001
43. Rhee YH, Choi M, Lee HS, Park CH, Kim SM, Yi SH, et al. Insulin concentration is critical in culturing human neural stem cells and neurons. Cell Death Dis. (2013) 4:e766. doi: 10.1038/cddis.2013.295
44. Heidari M, Johnstone DM, Bassett B, Graham RM, Chua AC, House MJ, et al. Brain iron accumulation affects myelin-related molecular systems implicated in a rare neurogenetic disease family with neuropsychiatric features. Mol Psychiatry. (2016) 21:1599–607. doi: 10.1038/mp.2015.192
45. Adams BD, Lazova R, Andrews NC, Milstone LM. Iron in skin of mice with three etiologies of systemic iron overload. J Invest Dermatol. (2005) 125:1200–5. doi: 10.1111/j.0022-202X.2005.23949.x
46. Li Y, Pan K, Chen L, Ning JL, Li X, Yang T, et al. Deferoxamine regulates neuroinflammation and iron homeostasis in a mouse model of post-operative cognitive dysfunction. J Neuroinflammation. (2016) 13:268. doi: 10.1186/s12974-016-0740-2
47. Dong XH, Gao WJ, Kong WN, Xie HL, Peng Y, Shao TM, et al. Neuroprotective effect of the active components of three chinese herbs on brain iron load in a mouse model of Alzheimer's disease. Exp Ther Med. (2015) 9:1319–27. doi: 10.3892/etm.2015.2234
48. Wan W, Zhang C, Danielsen M, Li Q, Chen W, Chan Y, et al. EGb761 improves cognitive function and regulates inflammatory responses in the APP/PS1 mouse. Exp Gerontol. (2016) 81:92–100. doi: 10.1016/j.exger.2016.05.007
49. Yu SY, Cao CJ, Zuo LJ, Chen ZJ, Lian TH, Wang F, et al. Clinical features and dysfunctions of iron metabolism in Parkinson disease patients with hyper echogenicity in Substantia nigra: a cross-sectional study. BMC Neurol. (2018) 18:9. doi: 10.1186/s12883-018-1016-5
50. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. (2012) 122:1316–38. doi: 10.1172/jci59903
51. Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate–polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. (1981) 112:195–203. doi: 10.1016/0003-2697(81)90281-5
52. Hnasko TS, Hnasko RM. The western blot. Methods Mol Biol. (2015) 1318:87–96. doi: 10.1007/978-1-4939-2742-5_9
53. Wan W, Cao L, Liu L, Zhang C, Kalionis B, Tai X, et al. Abeta1−42 oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of rage and metalloproteinases, and down-regulation of tight junction scaffold proteins. J Neurochem. (2015) 134:382–93. doi: 10.1111/jnc.13122
54. Hoover JL, Bond CE, Hoover DB, Defoe DM. Effect of neurturin deficiency on cholinergic and catecholaminergic innervation of the murine eye. Exp Eye Res. (2014) 122:32–9. doi: 10.1016/j.exer.2014.03.002
55. Egana JT, Zambrano C, Nunez MT, Gonzalez-Billault C, Maccioni RB. Iron-induced oxidative stress modify tau phosphorylation patterns in hippocampal cell cultures. Biometals. (2003) 16:215–23. doi: 10.1023/A:1020727218493
56. Xie L, Zheng W, Xin N, Xie JW, Wang T, Wang ZY. Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem Int. (2012) 61:334–40. doi: 10.1016/j.neuint.2012.05.016
57. Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of tau and generates a pathological (MC-1) conformation. J. Biol. Chem. (2008) 283:32066–76. doi: 10.1074/jbc.M805300200
58. Bibow S, Ozenne V, Biernat J, Blackledge M, Mandelkow E, Zweckstetter M. Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J Am Chem Soc. (2011) 133:15842–5. doi: 10.1021/ja205836j
59. Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. (2004) 10:65–71. doi: 10.2119/2005-00029.Saltiel
60. Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. (2011) 434:365–81. doi: 10.1042/bj20101825
61. Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci. (2014) 19:164–74.
62. Moos T, Morgan EH. Transferrin and transferrin receptor function in brain barrier systems. Cell Mol Neurobiol. (2000) 20:77–95. doi: 10.1023/A:1006948027674
63. Kruer MC. The neuropathology of neurodegeneration with brain iron accumulation. Int Rev Neurobiol. (2013) 110:165–94. doi: 10.1016/b978-0-12-410502-7.00009-0
64. Lingor P, Carboni E, Koch JC. Alpha-synuclein and iron: two keys unlocking Parkinson's disease. J Neural Transm. (2017) 124:973–81. doi: 10.1007/s00702-017-1695-x
65. Carboni E, Tatenhorst L, Tonges L, Barski E, Dambeck V, Bahr M, et al. Deferiprone rescues behavioral deficits induced by mild iron exposure in a mouse model of alpha-synuclein aggregation. Neuromolecular Med. (2017) 19:309–21. doi: 10.1007/s12017-017-8447-9
66. Zeineh MM, Chen Y, Kitzler HH, Hammond R, Vogel H, Rutt BK. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol Aging. (2015) 36:2483–500. doi: 10.1016/j.neurobiolaging.2015.05.022
67. Wang ZX, Tan L, Wang HF, Ma J, Liu J, Tan MS, et al. Serum iron, zinc, and copper levels in patients with Alzheimer's disease: a replication study and meta-analyses. J Alzheimers Dis. (2015) 47:565–81. doi: 10.3233/jad-143108
68. Lovell MA, Xiong S, Xie C, Davies P, Markesbery WR. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J Alzheimers Dis. (2004) 6:659–71. doi: 10.3233/JAD-2004-6610
69. Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. (2013) 3:461–91. doi: 10.3233/jpd-130230
70. Lu Q, Black SM. Iron metabolism, oxidative stress, and neonatal brain injury. Neural Regen Res. (2016) 11:725–6. doi: 10.4103/1673-5374.182691
71. Li W, Jiang H, Song N, Xie J. Oxidative stress partially contributes to iron-induced alpha-synuclein aggregation in SK-N-SH cells. Neurotox Res. (2011) 19:435–42. doi: 10.1007/s12640-010-9187-x
72. Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci USA. (2010) 107:7036–41. doi: 10.1073/pnas.1000645107
73. Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann N Y Acad Sci. (2008) 1147:180–95. doi: 10.1196/annals.1427.007
74. Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, et al. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. (1996) 334:752–8. doi: 10.1056/NEJM199603213341202
75. Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, et al. Apolipoprotein-E type-4 allele and cerebral glucose-metabolism in relatives at risk for familial Alzheimer-disease. JAMA. (1995) 273:942–7. doi: 10.1001/jama.273.12.942.
76. Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol. (1997) 42:85–94. doi: 10.1002/ana.410420114
77. de la Monte SM. Therapeutic targets of brain insulin resistance in sporadic Alzheimer's disease. Front Biosci. (2012) 4:1582–605. doi: 10.2741/e482
78. Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. (2007) 27:13635–48. doi: 10.1523/jneurosci.3949-07.2007
79. Zhao WQ, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer's disease. Biochim Biophys Acta. (2009) 1792:482–96. doi: 10.1016/j.bbadis.2008.10.014
80. Son SM, Song H, Byun J, Park KS, Jang HC, Park YJ, et al. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy. (2012) 8:1842–4. doi: 10.4161/auto.21861
81. Grossman H. Does diabetes protect or provoke Alzheimer's disease? insights into the pathobiology and future treatment of Alzheimer's disease. CNS Spectr. (2003) 8:815–23. doi: 10.1017/S1092852900019258
82. Yang Y, Ma D, Wang Y, Jiang T, Hu S, Zhang M, et al. Intranasal insulin ameliorates tau hyperphosphorylation in a rat model of type 2 diabetes. J Alzheimers Dis. (2013) 33:329–38. doi: 10.3233/jad-2012-121294
83. Mao YF, Guo Z, Zheng T, Jiang Y, Yan Y, Yin X, et al. Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice. Aging Cell. (2016) 15:893–902. doi: 10.1111/acel.12498
84. Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. (2012) 69:29–38. doi: 10.1001/archneurol.2011.233
85. Munoz P, Zavala G, Castillo K, Aguirre P, Hidalgo C, Nunez MT. Effect of iron on the activation of the MAPK/ERK pathway in PC12 neuroblastoma cells. Biol Res. (2006) 39:189–90. doi: 10.4067/S0716-97602006000100021
86. Min D, Guo F, Zhu S, Xu X, Mao X, Cao Y, et al. The alterations of Ca2+/calmodulin/CaMKII/CaV1.2 signaling in experimental models of Alzheimer's disease and vascular dementia. Neurosci Lett. (2013) 538:60–5. doi: 10.1016/j.neulet.2013.02.001
87. Guillot F, Kemppainen S, Lavasseur G, Miettinen PO, Laroche S, Tanila H, et al. Brain-specific basal and novelty-induced alternations in PI3K-Akt and MAPK/ERK signaling in a middle-aged AbetaPP/PS1 mouse model of Alzheimer's disease. J Alzheimers Dis. (2016) 51:1157–73. doi: 10.3233/jad-150926
Keywords: Alzheimer's disease, tau, phosphorylation, iron deposition, insulin resistance
Citation: Wan W, Cao L, Kalionis B, Murthi P, Xia S and Guan Y (2019) Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling. Front. Neurol. 10:607. doi: 10.3389/fneur.2019.00607
Received: 25 January 2019; Accepted: 22 May 2019;
Published: 19 June 2019.
Edited by:
Jingyun Yang, Rush University Medical Center, United StatesReviewed by:
Maud Gratuze, Washington University in St. Louis, United StatesZhihou Liang, Wuhan Union Hospital, China
Copyright © 2019 Wan, Cao, Kalionis, Murthi, Xia and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yangtai Guan, Z3Vhbnlhbmd0YWkmI3gwMDA0MDtyZW5qaS5jb20=; Shijin Xia, eGlhc2hpamluaGQmI3gwMDA0MDsxNjMuY29t
†These authors have contributed equally to this work