 Wenjing Lang
Wenjing Lang Junjie Wang1,2
Junjie Wang1,2 Xiaofeng Ma
Xiaofeng Ma He Li
He Li Pan Cui
Pan Cui Junwei Hao
Junwei Hao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 28 March 2019
Sec. Neurogenomics
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00297
Ischemic stroke (IS) and Parkinson's disease (PD) are two neurological diseases that often strike individuals of advanced age. Although thought of as a disease of old age, PD can occur in younger patients. In many of these cases, genetic mutations underlie the disease. As with PD, stroke can also have a genetic component. Although many of the risk factors for IS are considered to be modifiable, a significant portion is not, suggesting that some of stroke risk factors may have a genetic origin. Large-scale genome-wide association studies (GWAS) have identified several IS and PD gene variants recently. Converging epidemiologic and pathological evidence suggests that IS and PD may be linked. However, it is still unclear whether these two conditions share a common mechanism. Here, we sought to determine the genetic mechanism underlying the possible association between IS and PD. We conducted a multi-step systemic analysis comprising (1) identification of IS and PD variants validated by known GWAS, (2) two separate gene-based tests using Versatile Gene-based Association Study 2 (VEGAS2) and PLINK, (3) a transcriptome-wide association study (TWAS), and (4) analyses of gene expression using an online tool in Gene Expression Omnibus. Our investigation revealed that IS and PD have in common five shared genes: GPX7, LBH, ZCCHC10, DENND2A, and NUDT14, which pass gene-based tests. Functionally, these genes are expressed differentially in IS and PD patients compared to neurologically healthy control subjects. This genetic overlap may provide clues on how IS and PD are linked mechanistically. This new genetic insight into these two diseases may be very valuable for narrowing the focus of future studies on the genetic basis of IS and PD and for developing novel therapies.
Stroke and Parkinson's disease (PD) are two neurological diseases that have great worldwide impact and that share certain clinical and pathological features. Stroke is a major cause of disability in many western countries. Moreover in 2015, it was the second leading cause of death, accounting for 11% of total deaths (6.3 million) (1, 2). Ischemic stroke (IS) accounts for up to 85% of all stroke cases. PD similarly affects many people and in 2015, it resulted in about 117,400 deaths globally (2, 3).
Both IS and PD have substantial genetic components. Evidence for a substantial genetic contribution for IS risk comes from genome-wide association studies (GWAS) and twin and family history studies (4–7). GWAS have uncovered risk loci for IS (4–7). Falcone et al. reviewed several common genetic variants of certain forms of IS that do not follow a clear Mendelian pattern of inheritance. These variants include ABO, PITX2, ZFHX3, HDAC9, SUPT3H/CDC5L, and CDKN2A/CDKN2B (6). Candidate-gene analyses and GWAS have identified a new locus at chromosome 6p25 (rs12204590, near FOXF2) that is related to all-stroke risk (8). Also, a new locus at chromosome 1p13.2 near TSPAN2 was recently identified; this latter locus is related to large artery atherosclerosis (LAA)-related stroke (9). Many researchers suspect that several other variants are yet to be identified.
As with IS, emerging evidence shows that PD has a substantial genetic component (10–12). GWAS and linkage analysis have confirmed the role of genes involved in familial and sporadic forms of PD (13, 14). Analysis of five large-scale GWAS datasets from Europe and the USA has identified some risk SNPs (p < 5.00E-08) through meta-analysis of PD susceptibility genes, including MAPT, SNCA, HLA-DRB5, BST1, GAK, LRRK2, SYT11, ACMSD, STK39, MCCC1/LAMP3, and CCDC62/HIP1R (13). There is now abundant GWAS data on numerous phenotypes of various diseases. Simultaneous analyses of multiple phenotypes can increase the detection of shared pathways, a procedure that could prove to be fruitful for identifying common genes of IS and PD.
Converging molecular, cellular, genetic, and clinical evidence has been reported for IS and PD. In a large population-based study, Huang et al. (15) and Becker et al. (16) confirmed that PD is related to an increased risk of IS and vice versa, implying that the two diseases may share some pathological mechanisms or processes. One possible link between IS and PD may involve α-synuclein, especially oligomeric forms (17). Abnormal aggregations and form conversion of α-synuclein are thought to result from the induction of oxidative stress and may be the pathological basis for PD (18). α-synuclein appears to be similarly elevated in red blood cells of IS and PD patients, being significantly higher than that in healthy people (17). α-synuclein induces microglia-mediated neuroinflammation, and α-synuclein aggregation indirectly damages neurons (19–21). Taken together, it is reasonable to hypothesize that, although IS and PD are two very different diseases, they may share pathophysiological processes that link them at some level. Building on this hypothesized relationship, one might expect to detect common immune-related genetic risk factors.
While GWAS has been revolutionary in unraveling disease genetics in general, for IS and PD, a large proportion of genetic variants remain undiscovered, serving as a reminder that more work are necessary to identify other genes that contribute to the pathology of these two diseases. We hypothesize that combining analyses of genes identified from different gene-based tests may be a powerful approach for identifying genes shared by IS and PD. We tested this hypothesis by conducting two gene-based meta-analyses using VEGAS2 and PLINK on IS and PD data from GWAS. In addition, we examined the shared genes by TWAS, and further validated the shared gene data with gene expression analyses utilizing GEO datasets.
Pankratz et al. (22) originally analyzed the PD GWAS dataset. They also conducted a large meta-analysis on two new datasets obtained directly from the investigators who performed the original GWAS (23–25) and on publicly available GWAS data obtained from dbGaP (10, 26), PROGRNI/GenePD (23), NIA Phase I (26), NIA Phase II (10), HIHG (24), and NGRC (25). They designed a two-stage study, which comprised a discovery stage and an independent replication stage. All the datasets used in the discovery stage came from Caucasian PD patients who were diagnosed using standard UK Brain Bank criteria for PD (27). Since familial PD cases may have a stronger genetic contribution than sporadic PD cases (22), they additionally included data from cases with a family history of PD. Anyone with a PD onset age younger than 18 years was excluded from the study. They also removed data of cases that had a known pathogenic factor, such as two parkin mutations or single LRRK2 mutations.
In the original publication, each study underwent rigorous quality assessment and data cleanup before performing imputation with MACH1.0 (28). To control population stratification, the researchers used principal component analysis. ProbABEL (https://cran.r-project.org/src/contrib/Archive/GenABEL/), a tool for genome-wide association analysis of imputed data, and METAL (http://www.sph.umich.edu/csg/abecasis/Metal), a tool for meta-analysis, were then used to analyze the data. Finally, we acquired summary PD GWAS statistics data from the discovery samples, which included 2,525,704 SNPs from 4,238 PD cases and 4,239 control cases [for additional details, see the original article (22)].
The IS summary GWAS data was obtained from phase I of the METASTROKE collaboration, which consisted of 10,307 Caucasian IS cases and 19,326 Caucasian control cases (29). These cases came from 12 studies (ASGC, BRAINS, GASROS_affy, GASROS_illumina, GEOS, HPS, ISGS-SWISS, MILANO, VISP, WHI, WTCCC2-D, and WTCCC2-UK) with previously genotyped data. The etiologic stroke subtypes were classified according to the criteria of the Trial of ORG 10172 in Acute Stroke Treatment (TOAST) (30).
For all datasets, the researchers performed genotyping individually and quality controls using methods documented previously (30). Using 1000 Genomes phase I data, the researchers imputed the raw autosomal data following a genome-wide logistic regression analysis and a meta-analysis (31). In order to determine whether the effector alleles were identical, SNPs were analyzed across the cohort in a meta-analysis. Additionally, genomic control was also used for test statistics to correct for incidental inflation. Finally, we obtained summary IS GWAS data, including 9,541,572 SNPs [for additional details, see the original article (29)].
To test the IS and PD GWAS datasets, we calculated gene-based p values with VEGAS2 after assigning variants to genes. We chose a broad reference population group, having a European ancestry (1000G EURO). For gene boundary selection for SNP, we implemented option 5, “0kbldbin” (SNPs within this gene and SNPs in high LD outside of this gene with SNPs within this gene). In gene-based tests, distant SNPs with r2 > 0.8 with associated SNPs were usually not taken into consideration systematically, and yet ignoring LD might lead to deficiency of some valuable information, therefore we chose this larger gene boundary. For every gene definition, the gene-based test statistics were calculated by adding the p values of n SNPs after conversion to upper tail χ2 statistics with one degree of freedom (df). Under the null hypothesis, these should have a χ2 distribution with n df, if SNPs are in linkage equilibrium (LD) (32).
SNP correlation was modeled using ∑, a n × n matrix of LD (r) values estimated from a 1000 Genomes European reference population (32). We used this method because LD for the n SNPs scarcely occurs (32). Significance was calculated by comparing the sum of χ2 statistics for every gene with simulated repeats from a multivariate normal distribution, where the mean = 0 and variance = ∑ (32). The formula p = r+1/m+1 was used to calculate empirical p values for every gene, where r represents the number of instances in which the simulation statistic exceeds the observation data, and m represents the number of simulations (32). This gene-based test included all top SNPs (by default, all SNPs are considered). We submitted IS and PD variants to VEGAS2 separately, and then identified shared genes that were nominally associated in each disease separately (PIS−GWAS < 0.05; PPD−GWAS < 0.05) (33).
To test IS and PD GWAS datasets, we used Fisher's method implemented in PLINK software (SET SCREEN TEST). If the performed tests are independent for each SNP, for a given gene, the combined Fisher's statistic
follows a χ2 distribution, with 2N df under the null hypothesis. In this formula, N represents the number of markers (tests), and pi (i = 1,…, N) represents the corresponding p values. If the tests are not independent, the statistic has mean m = 2N and variance (σ2) is
In the formula above, pi and pj (i, j = 1, …, N) represent the p values for each test. The covariance (cov) can be calculated as
where pij approximates the correlation between SNPi and SNPj. These are the non-negative correlation coefficients between the two variables.
Thus, the significance of a complete set of non-independent tests is calculated as
where x2 follows the central Chi-squared distribution, with 8N2 /σ2 as df.
This method was applied to the PD GWAS dataset and IS GWAS dataset using LD information from the HapMap CEU population. An approximate Fisher's test was used for all the SNPs in genes to combine p values (34). By combining a group of p values that were acquired from independent tests with the same null hypothesis, we found that the Fisher's method was asymptotically optimal to achieve overall significance (34). Genes with many of SNPs are well suited to using this approach (34–36). After performing calculations in PLINK, we identified shared genes that were associated with each disease (PIS−GWAS < 0.05; PPD−GWAS < 0.05).
We combined the two p values of the shared genes of IS and PD derived from VEGAS using the simplest meta-analysis method in GWAS: Fisher's method. We conducted the same meta-analysis for shared genes identified by PLINK. For a given gene, we chose the following formula for the statistic
where in the ith study, Pi is the gene's p value; and k represents the overall number of studies. Under 2k df, x2 follows a χ2 distribution (37). We used the program R (https://www.r-project.org/) to finish the analysis.
TWAS combines gene expression data with GWAS data to identify genes that could regulate the expression of complex traits in cis-action (38). We performed a validation of the shared genes using TWAS in different tissues to determine whether they played significant roles in expression-trait associations. The process of TWAS have been widely described in previous articles (38). Here, TWAS integrated pre-computed gene expression weights of whole blood and brain RNA-seq with GWAS data to estimate the associations of gene to traits. The reference data of whole blood comprised 1,264 samples of Cardiovascular Risk in Young Finns Study (YFS) in Finland (39), and the reference data of brain RNA-seq was collected from the dorsolateral prefrontal cortex of 452 samples from the CommonMind Consortium (CMC) (40).
To bolster our gene-based testing results with biological functional data of shared genes and to further validate the shared genes of IS and PD, we used GEO2R (41), an online tool that can identify differentially expressed genes under different experimental conditions. This tool compares two or more sample groups in the Gene Expression Omnibus (GEO) database.
Gene expression data from analyses of peripheral whole blood of 39 IS patients and 24 non-stroke, neurologically healthy control subjects were obtained from GEO dataset GSE16561. Patients were recruited if they were ≥18 years and diagnosed definite IS by MRI. In addition, patients diagnosed hemorrhage and uncertain IS were excluded from the group. There were no significant differences in gender and race between patients and controls, but more vascular risk factors (such as hypertension, diabetes, etc.) were found in stroke subjects. More details about clinical characteristics were provided in the original report (42). The samples were analyzed on an Illumina HumanRef-8 Expression BeadChip. We also used another publically available stroke RNA expression dataset, GEO dataset GSE58294 (43). These data were derived from analyses of peripheral whole blood of 23 cardioembolic stroke patients and 23 vascular risk factor controls (VRFC). Race was not statistically significantly different between stroke cases and VRFC. More subject demographics were described in the original article (43).
For changes in gene expression in PD patients, global expression data derived from analyses of postmortem brain tissue were acquired from GEO dataset GSE20295 (44, 45). This dataset comprises three subseries (GSE20168, GSE20291, and GSE20292). The tissue blocks which were from three brain areas, prefrontal cortex area 9, the putamen, and the entire substantia nigra, were collected from 15 patients with neuropathologically confirmed PD and 15 controls without major brain disease. PD patients diagnosed with additional neuropathological disease were excluded from the study. Between PD and control groups, some variables, for example gender, which could greatly affect the expression profile of RNA in postmortem brain tissues, were matched closely and had no significant differences (p > 0.05). [for more details, see the original report (44, 45)]. The data were analyzed using an Affymetrix Human Genome U133A Array platform.
NCBI GEO can be used as a public repository for a variety of high-throughput experimental data. Currently, GEO contains nearly 140,000 samples and more than 3,000 different microarray platforms (46). GEO2R uses the GEO query (46) and Linear Models for Microarray Analysis (47) (limma) R packages from the Bioconductor project (https://www.bioconductor.org) to compare the processing data tables provided by the original submitter. Although our main goal was to identify the consensus genes (listed above) in each dataset, we also screened and ranked by significance all of the genes shared by IS or PD. The latter was done by assessing their differential expression relative to control subjects. The permutation allowed a null distribution of gene ranking per experiment. Then we evaluated whether the shared genes deviated significantly from the null distribution.
Using VEGAS2, we assigned variants to genes and calculated p values for IS and PD datasets separately. From those analyses, we obtained 20,946 IS genes and 19,858 PD genes with at least two SNPs. We subsequently identified 75 genes that were nominally associated with each individual disease (PIS−GWAS < 0.05; PPD−GWAS < 0.05) and shared by IS and PD. Following meta-analysis of the 75 genes and Bonferroni correction with p < 3.33E-04 (p = 0.05/75/2), we ultimately identified nine shared genes between IS and PD: GAK (p = 1.24E-06 for IS and PD, 3.55E-02 for IS, and 2.00E-06 for PD); MMRN1 (p = 1.82E-05 for IS and PD, 3.38E-02 for IS, and 3.70E-05 for PD); GPX7 (p = 2.70E-05 for IS and PD, 2.00E-03 for IS, and 9.52E-04 for PD); NUDT14 (p = 3.65E-05 for IS and PD, 3.56E-04 for IS, and 7.40E-03 for PD); LBH (p = 4.55E-05 for IS and PD, 1.57E-03 for IS, and 2.13E-03 for PD); ZCCHC10 (p = 5.68E-05 for IS and PD, 6.30E-03 for IS, and 6.75E-04 for PD); P2RX6 (p = 1.03E-04 for IS and PD, 6.23E-03 for IS, and 1.30E-03 for PD); DENND2A (p = 2.24E-04 for IS and PD, 2.42E-03 for IS, and 7.78E-03 for PD); and LOC101928455 (p = 3.07E-04 for IS and PD, 2.69E-03 for IS, and 9.90E-03 for PD). Detailed information about these genes is provided in Table 1.
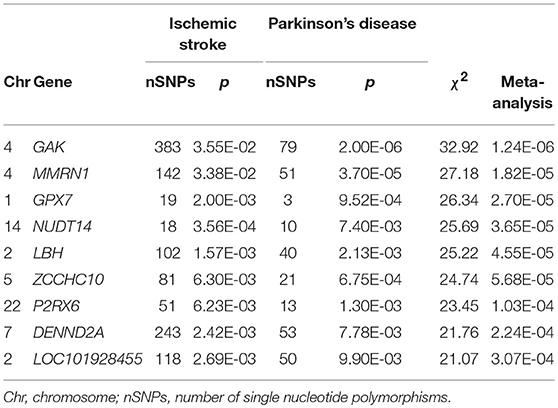
Table 1. Nine genes shared by ischemic stroke and Parkinson's Disease cases identified with VEGAS2 (adjusted p < 3.33E-04).
Using PLINK, we mapped IS and PD SNPs to genes and identified 16,724 IS genes and 16,610 PD genes with at least two SNPs. We subsequently identified 33 genes shared by IS and PD that were nominally associated with each individual disease (PIS−GWAS < 0.05; PPD−GWAS < 0.05). Following meta-analysis of the 33 genes and Bonferroni correction with p < 7.58E-04 (p = 0.05/33/2), we finally identified nine genes associated with the two diseases. Theses nine genes are NUDT14 (p = 3.86E-05 for IS and PD, 3.58E-04 for IS, and 7.82E-03 for PD); PARP3 (p = 5.28E-05 for IS and PD, 2.30E-03 for IS, and 1.71E-03 for PD); GPX7 (p = 8.08E-05 for IS and PD, 2.50E-03 for IS, and 2.49E-03 for PD); ZCCHC10 (p = 8.19E-05 for IS and PD, 7.22E-03 for IS, and 8.75E-04 for PD); MEX3A (p = 2.26E-04 for IS and PD, 4.54E-04 for IS, and 4.20E-02 for PD); DENND2A (p = 5.03E-04 for IS and PD, 5.51E-03 for IS, and 8.30E-03 for PD); CRNN (p = 6.31E-04 for IS and PD, 2.37E-02 for IS, and 2.48E-03 for PD); RGS9BP (p = 6.31E-04 for IS and PD, 3.74E-03 for IS, and 1.57E-02 for PD); and LBH (p = 7.44E-04 for IS and PD, 1.01E-02 for IS, and 7.01E-03 for PD). Detailed information about these genes is provided in Table 2.
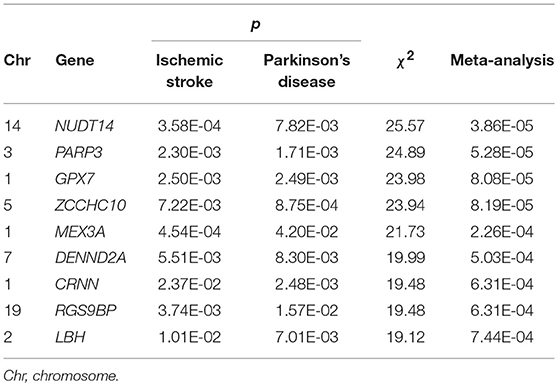
Table 2. Nine genes identified with PLINK shared by cases with ischemic stroke and Parkinson's disease (adjusted p < 7.58E-04).
Due to differences in identifying shared genes with either the VEGAS2 or PLINK method, we expected that shared genes would be more strongly associated with the diseases if they met the gene-based testing criteria of both methods. By determining where the results of statistically significant genes obtained by the two methods intersected, we ultimately obtained five shared genes satisfying the gene-based testing conditions. These genes are GPX7, NUDT14, LBH, ZCCHC10, and DENND2A. Meanwhile 4 of these 5 genes were also shown to be significantly associated with the two diseases in different tissues through TWAS (p < 0.05). Detailed results were summarized in Table 3. We next sought validation through other functional analyses.
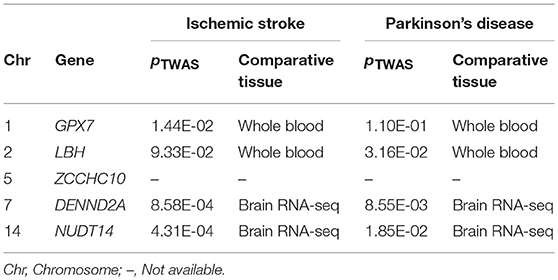
Table 3. Five genes identified with two methods were verified by TWAS.
We further investigated whether these five shared genes were differentially expressed in IS and PD patients compared to neurologically healthy control subjects. We applied the Bonferroni-corrected statistical test at a significance of p < 0.01 (p = 0.05/5) and log2-fold change (logFC) to measure changes in the levels of gene expression. Compared to control subjects, in IS patients we detected significantly altered transcript levels of GPX7 (p = 1.46E-07); NUDT14 (p = 9.00E-03); LBH (p = 5.45E-06) in the GEO dataset GSE16561; and ZCCHC10 (p = 3.87E-08) in the GEO dataset GSE58294 (Table 3). In PD brains, we found significantly altered expression levels of GPX7 (p = 8.81E-03); LBH (p = 7.05E-03); ZCCHC10 (p = 7.01E-03); and DENND2A (p = 9.77E-03) in GEO dataset GSE20295 (Table 4). It is remarkable that transcript expression of GPX7 (GSE16561: logFC = −0.499; GSE20295: logFC = −0.802) and LBH (GSE16561: logFC = −0.553; GSE20295: logFC = −1.058) was significantly decreased in both IS patients and PD patients compared to the corresponding control groups.
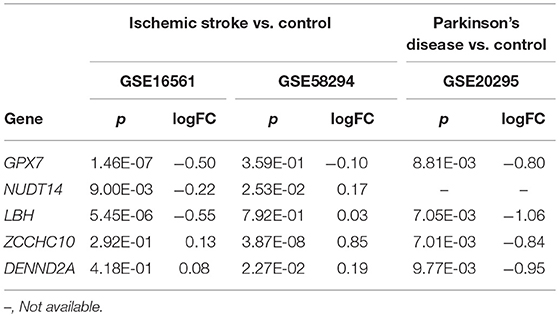
Table 4. Shared genes in patients with ischemic stroke or Parkinson's disease vs. their corresponding healthy controls.
It has been widely reported that IS and PD share various pathological and clinical features (17, 48, 49). However, until now, the underlying genetic relationship between the two diseases has remained unclear. Previous studies have investigated IS and PD susceptibility genes independently and separately through analysis of IS and PD GWAS datasets and by performing independent linkage analyses (6, 8, 9, 13). Despite those efforts, a large proportion of genetic variants related to IS and PD remain undiscovered. What steps can be taken to identify these variants? We hypothesized that combining the findings from both IS and PD GWAS would lead to the identification of new shared variants. In the current study, we performed two gene-based tests on IS and PD GWAS datasets, examined the shared genes by TWAS and then conducted gene-expression analyses for validation. Through our analysis, it is significant to identify that IS and PD have shared genes, which explain shared pathogenesis between them to some extent. What's more, it is also consistent with a previous large study that, not only did PD patients have more frequent history of stroke than matched groups without PD, but the incidence rates of IS were also increased for PD patients compared with PD-free groups (16).
Through the two gene-based meta-analyses, we identified five new genes shared by IS and PD, which were further examined by TWAS. These five genes are GPX7, NUDT14, LBH, ZCCHC10, and DENND2A. Although we have not replicated published findings on IS/PD risk genes, these five shared genes we identified here have been studied extensively, and many experiments show that they play important roles in the pathogenesis of IS and PD (50, 51). GEO2R analyses also confirmed that expression level of these five genes in IS patients was different to that in control subjects. The same was true regarding the expression of these genes in PD patients and control subjects. Taken together, these results suggest that various related genes may underlie certain aspects of the pathogenesis of IS and PD.
GPX7, also known as NPGPx, is a member of the glutathione peroxidase (GPX) family of enzymes, which function to reduce oxidative damage (52). Ectopic expression of GPX7 inhibits H2O2-induced toxic effects, which is consistent with its essential role in reducing oxidative stress (52). GPX7 also transmits endoplasmic reticulum (ER) oxidative stress signals through the formation of disulfide bonds. This signal activates downstream ER glucose-regulated protein 78 (GRP78) and enhances its chaperone activity. Consistently, GPX7-knockout mice have been shown to accumulate reactive oxygen species, and they have a significantly shortened lifespan (53). Thus, it is clear that GPX7 plays an important role as a stress sensor, functionally contributing to the attenuation of ER oxidative stress damage.
Mounting evidence confirms that ER stress contributes to the pathogenesis of PD and IS (51, 54, 55). Coppola-Segovia and collaborators have shown that model mice constructed to develop ER stress exhibit by injection of tunicamycin were induced PD features, such as dopamine neuronal death, increased astroglial reactivity, and extensive oligomerization of α-synuclein (55). These features reinforce the notion that ER stress could play a pivotal role in the pathogenesis of PD (55). ER stress also appears to contribute to the pathogenesis of IS. During stroke, the unfolded protein response (UPR) signaling pathway is initiated by protein misfolding in energy-starved neurons, which is associated with the toxic effects of reperfusion (54). GRP78 mainly regulates the UPR signaling pathway. The UPR signaling pathway plays an important role in attenuating ER stress by lessening protein translation, increasing folding capacity, and promoting ER-associated degradation and expansion of the ER membrane (56, 57). GPX7 has also been shown to enhance the chaperone activity of GRP78 to attenuate ER stress in transgenic animals (53).
In summary, the involvement of ER stress in pathogenesis of both IS and PD has been confirmed in many studies. GPX7 could affect ER stress by direct regulation or by changing GRP78 indirectly, and hence participate in the pathological process of both diseases. We predict that it might be helpful in therapies of both diseases by interfering with GPX7 to change ER stress. We need more validation studies in future.
LBH is a highly conserved, tissue-specific transcriptional regulator that plays a key role in the embryonic development of vertebrates (58–60). In epithelial development and cancer, LBH is a direct target gene for the canonical Wingless/Int (Wnt) signaling pathway (61). The relationship between the Wnt pathway and PD or IS has been widely reported (50, 62).
There is sufficient number of studies now suggesting that the Wnt signaling pathway is critical for the normal functioning of midbrain dopaminergic neurons. A growing number of genes that encode components of the Wnt pathway are involved in the development of dopaminergic neurons in the midbrain (63, 64), a region of early neuronal degeneration in PD, which is a pathological hallmark of the disease (65). In recent years, studies have revealed that the pathogenesis of PD can be traced back to gene mutations. Surprisingly, the Wnt signaling pathway has links with a striking number of PD susceptibility genes, such as LRRK2 (66), PARK2 (67), VPS35 (68), Nurr1 (69), GSK3β (70), and WNT3 (71). LRRK2, for example, has been suggested to play a central role in the canonical Wnt pathway, and mutations in LRRK2 decreases pathway activity (66). Taken together, it is reasonable to think that deregulation of the Wnt pathway might be an important precursor to the pathogenesis of PD. Does deregulation of the Wnt pathway make IS more likely?
Pathologically, stroke is the culmination of various insults to the vasculature. Recent work has shown that the Wnt pathway is involved in the development of central nervous system blood vessels, formation of the blood-brain barrier, and protection of injured endothelial cells (72, 73). In addition, GWAS analysis of IS patients and controls show that gender is implicated in the etiology of stroke (74), and male-specific stroke genes have been shown to be associated with the Wnt pathway (74). Furthermore, the Wnt pathway is involved in neuroinflammation, and it is important for neurogenesis (72, 75); these two processes are involved in PD and stroke. As LBH has been shown to be associated with other inflammatory disorders such as autoimmune diseases, in particular, rheumatoid arthritis (76), it might play a valuable role in IS and PD.
In general, the Wnt pathway has been demonstrated involving in both IS and PD susceptibility pathways by GWAS analysis. Numerous experiments have likewise demonstrated that Wnt signaling pathway is involved in the development of central nervous system, which comprises an important part of the pathological process of IS and PD. While LBH acts as a direct target gene for Wnt pathway, it is possible that LBH plays a role in the common pathogenesis of PD and IS.
ZCCHC10 was found to be closely associated with IS and PD in our analyses. However, to date, the function of ZCCHC10 has revealed few links to IS, PD, or any other human diseases. One study suggested that ZCCHC10 interacts with tumor protein p53 (TP53), LUC7 like 2 (LUC7L2), peptidylprolyl cis/trans isomerase (PIN1), and eukaryotic translation elongation factor 1 alpha 1 (EEF1A1) (77). One of ZCCHC10's interaction partners, EEF1A1, plays an important role in the ability of monocyte locomotion inhibitory factor (MLIF) to protect the brain from ischemic damage. Knockout of EEF1A1 attenuates MLIF's inhibitory effects on the expression of inflammatory molecules and ultimately reduces the protective effect of MLIF on IS (78). EEF1A1 was identified to be an attractive candidate gene for PD as well (79). Furthermore, another ZCCHC10-interacting partner, PIN1, has been found to be dramatically upregulated in the substantia nigra of PD patients and to have a proapoptotic role in the pathophysiological mechanisms of PD (80). Thus, ZCCHC10 could be an attractive gene by interacting with EEF1A1/PIN1 for unraveling the pathophysiological relationship between IS and PD to some extent.
DENND2A is a member of the DEEND2 gene family and has been shown to be a specific guanine nucleotide exchange factor (GEF) for Rab9, which is involved in trafficking between the trans-Golgi network (TGN) and late endosomes (81). Although the function of DENND2A has rarely been reported to be relevant to diseases, Rab9-dependent mitophagy has been shown to contribute to heart disease (82). Recently mitophagy has been reported to play important roles in IS and PD (82–84). DENND2A, therefore, might be a promising gene by playing a part in mitophagy for determining the etiology of PD and IS.
NUDT14 is one member of the 24 Nudix hydrolase genes of the human genome (85). NUDT14 is proposed to be involved in the control of glycogen metabolism, where it modulates UDP-glucose levels during glycolipid and glycoprotein synthesis (86). A recent study showed that NUDT14 could affect viral DNA replication by interacting with human cytomegalovirus RL13 (87). However, the function of NUDT14 has few connections with IS or PD. In our study, NUDT14 was identified as a candidate gene related to the pathogenesis of PD and IS. Additional research is needed in order to confirm this.
As for the gene-based analysis, we used a more liberal cutoff genetic association p < 0.05 as the criterion for determining genes associated with IS or PD, rather than the multiple comparisons according to some considerations. For some complex disorders, the effect size of individual genetic variants is usually modest, which suggests that individual genetic variants could account for a minimal fraction of heritability of complex traits and genetic risk (88). Association signals for complex traits tend to be propagated throughout most of the genome, comprising genes which are not significantly connected to disease (89). In order to capture disease-related genes more comprehensively, we chose genes with nominal associations (p < 0.05) (33, 90). In addition, we obtained 20,946 IS genes and 19,858 PD genes with corresponding p values through VEGAS2. If we select Bonferroni correction for multiple test comparisons, the adjusted p value of IS genes should be < 0.05/20946 = 2.39E-06 and only 1 gene passes the correction, meanwhile, the adjusted p value of PD genes should be < 0.05/19858 = 2.52E-06 and 11 genes are with this significance level. If we take the same correction method for genes and p values obtained from PLINK software, 9 PD genes and 1 IS gene are with the significance level (PIS < 2.99E-06, PPD < 3.01E-06). Thus, we chose genes that were nominally associated in each disease for following analysis. For the obtained share genes, we performed Bonferroni correction for multiple test comparisons and genes with the significance level were verified as associated with both diseases.
The present study has some limitations despite of these interesting results. Since the original datasets were derived from patients who received clinical diagnosis of IS and PD, misdiagnosis could have potentially influenced on our results. In addition, we could not access the original SNP genotype data, we had to use summary data from IS and PD GWAS, which prevented us from using a polygenic risk score or BLUP method to address the shared genetics of complex traits and could have affected our results. We will improve our future work, when the original data is available to us. Besides, we utilized a small size of PD GWAS sample, compared with IS sample, which raised the possibility that the findings might be driven primarily by the IS sample. Moreover, as the multiple testing corrections we used in our statistical analyses may be insufficient to explain all biases, permutation testing should be used to adjust the results at the single SNP level. What's more, the data from different tissues for PD and IS might be a potential limitation to the results. We will further expand the size and tissues of expression data in the future. Furthermore, we lacked transcriptomic and epigenetic data, which may contribute to the identification of more potential causal mechanisms and associations. Finally, we did not further analyze the relationship between IS subtypes and PD. There are still some differences among IS subtypes, even though the pathological processes underlying each subtype have a certain degree of commonality.
In conclusion, for two GWAS datasets for IS and PD, we used two gene-based testing methods and gene-expression analyses to identify several genes that are associated with neuroinflammation and neuro-immunity and that are expressed differentially in IS patients and PD patients. Based on previous work (91, 92), our outcomes support the hypothesis that IS and PD may be linked through shared neuroinflammation- and neuroimmune-related genes.
WL and JH conceived and designed the research for PD and IS; WL analyzed data, and wrote the manuscript; JW revised the manuscript; NZ, HL, and PC collected data and provided technical guidance; XM conceptualized the analysis and supervised the research. The final version was approved for submission by all listed authors.
This work was supported by the National Natural Science Foundation of China (81571600, 81322018, 81273287, and 81100887 to JH); the Youth Topnotch Talent Support Program (to JH); the National Key Clinical Specialty Construction Project of China (to JH); and the Natural Science Foundation of Tianjin (17JCZDJC35500 to JH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. (2016) 388:1459–544. doi: 10.1016/S0140-6736(16)31012-1
3. GBD 2015 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. (2016) 388:1659–724. doi: 10.1016/S0140-6736(16)31679-8
4. Dichgans M. Genetics of ischaemic stroke. Lancet Neurol. (2007) 6:149–61. doi: 10.1016/S1474-4422(07)70028-5
5. Holliday EG, Maguire JM, Evans TJ, Koblar SA, Jannes J, Sturm JW, et al. Common variants at 6p21.1 are associated with large artery atherosclerotic stroke. Nat Genet. (2012) 44:1147–51. doi: 10.1038/ng.2397
6. Falcone GJ, Malik R, Dichgans M, Rosand J. Current concepts and clinical applications of stroke genetics. Lancet Neurol. (2014) 13:405–18. doi: 10.1016/S1474-4422(14)70029-8
7. Holliday EG, Traylor M, Malik R, Bevan S, Falcone G, Hopewell JC, et al. Genetic overlap between diagnostic subtypes of ischemic stroke. Stroke. (2015) 46:615–9. doi: 10.1161/STROKEAHA.114.007930
8. Neurology Working Group of the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium, the Stroke Genetics Network, and the International Stroke Genetics Consortium. Identification of additional risk loci for stroke and small vessel disease: a meta-analysis of genome-wide association studies. Lancet Neurol. (2016) 15:695–707. doi: 10.1016/S1474-4422(16)00102-2
9. NINDS Stroke Genetics Network, International Stroke Genetics Consortium. Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wide association study. Lancet Neurol. (2016) 15:174–84. doi: 10.1016/S1474-4422(15)00338-5
10. Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. (2009) 41:1308–12. doi: 10.1038/ng.487
11. Keller MF, Saad M, Bras J, Bettella F, Nicolaou N, Simon-Sanchez J, et al. Using genome-wide complex trait analysis to quantify 'missing heritability' in Parkinson's disease. Hum Mol Genet. (2012) 21:4996–5009. doi: 10.1093/hmg/dds335
12. Liu G, Liu Y, Jiang Q, Jiang Y, Feng R, Zhang L, et al. Convergent genetic and expression datasets highlight TREM2 in Parkinson's disease susceptibility. Mol Neurobiol. (2016) 53:4931–8. doi: 10.1007/s12035-015-9416-7
13. International Parkinson Disease, Genomics C, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, et al. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. (2011) 377:641–9. doi: 10.1016/S0140-6736(10)62345-8
14. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. (2014) 46:989–93. doi: 10.1038/ng.3043
15. Huang YP, Chen LS, Yen MF, Fann CY, Chiu YH, Chen HH, et al. Parkinson's disease is related to an increased risk of ischemic stroke-a population-based propensity score-matched follow-up study. PLoS ONE. (2013) 8:e68314. doi: 10.1371/journal.pone.0068314
16. Becker C, Jick SS, Meier CR. Risk of stroke in patients with idiopathic Parkinson disease. Parkinsonism Relat Disord. (2010) 16:31–5. doi: 10.1016/j.parkreldis.2009.06.005
17. Zhao HQ, Li FF, Wang Z, Wang XM, Feng T. A comparative study of the amount of alpha-synuclein in ischemic stroke and Parkinson's disease. Neurol Sci. (2016) 37:749–54. doi: 10.1007/s10072-016-2485-1
18. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
19. Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. (2005) 19:533–42. doi: 10.1096/fj.04-2751com
20. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. (2007) 8:57–69. doi: 10.1038/nrn2038
21. Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, et al. MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci. (2013) 33:9592–600. doi: 10.1523/JNEUROSCI.5610-12.2013
22. Pankratz N, Beecham GW, DeStefano AL, Dawson TM, Doheny KF, Factor SA, et al. Meta-analysis of Parkinson's disease: identification of a novel locus, RIT2. Ann Neurol. (2012) 71:370–84. doi: 10.1002/ana.22687
23. Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. (2009) 124:593–605. doi: 10.1007/s00439-008-0582-9
24. Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. (2010) 74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x
25. Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet. (2010) 42:781–5. doi: 10.1038/ng.642
26. Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, et al. Genome-wide genotyping in Parkinson's disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. (2006) 5:911–6. doi: 10.1016/S1474-4422(06)70578-6
27. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatr. (1992) 55:181–4. doi: 10.1136/jnnp.55.3.181
28. Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. (2010) 34:816–34. doi: 10.1002/gepi.20533
29. Malik R, Traylor M, Pulit SL, Bevan S, Hopewell JC, Holliday EG, et al. Low-frequency and common genetic variation in ischemic stroke: the METASTROKE collaboration. Neurology. (2016) 86:1217–26. doi: 10.1212/WNL.0000000000002528
30. Adams HPJr, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. (1993) 24:35–41. doi: 10.1161/01.STR.24.1.35
31. Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. (2012) 491:56–65. doi: 10.1038/nature11632
32. Mishra A, Macgregor S. VEGAS2: software for more flexible gene-based testing. Twin Res Hum Genet. (2015) 18:86–91. doi: 10.1017/thg.2014.79
33. Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet. (2012) 90:636–47. doi: 10.1016/j.ajhg.2012.02.020
34. Moskvina V, O'Dushlaine C, Purcell S, Craddock N, Holmans P, O'Donovan MC. Evaluation of an approximation method for assessment of overall significance of multiple-dependent tests in a genomewide association study. Genet Epidemiol. (2011) 35:861–6. doi: 10.1002/gepi.20636
35. Tang CS, Ferreira MA. A gene-based test of association using canonical correlation analysis. Bioinformatics. (2012) 28:845–50. doi: 10.1093/bioinformatics/bts051
36. Liu G, Zhang F, Jiang Y, Hu Y, Gong Z, Liu S, et al. Integrating genome-wide association studies and gene expression data highlights dysregulated multiple sclerosis risk pathways. Mult Scler. (2017) 23:205–12. doi: 10.1177/1352458516649038
37. Begum F, Ghosh D, Tseng GC, Feingold E. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. (2012) 40:3777–84. doi: 10.1093/nar/gkr1255
38. Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. (2016) 48:245–52. doi: 10.1038/ng.3506
39. Nuotio J, Oikonen M, Magnussen CG, Jokinen E, Laitinen T, Hutri-Kahonen N, et al. Cardiovascular risk factors in 2011 and secular trends since 2007: the Cardiovascular Risk in Young Finns Study. Scand J Public Health. (2014) 42:563–71. doi: 10.1177/1403494814541597
40. Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. (2016) 19:1442–53. doi: 10.1038/nn.4399
41. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. (2013) 41:D991–995. doi: 10.1093/nar/gks1193
42. Barr TL, Conley Y, Ding J, Dillman A, Warach S, Singleton A, et al. Genomic biomarkers and cellular pathways of ischemic stroke by RNA gene expression profiling. Neurology. (2010) 75:1009–14. doi: 10.1212/WNL.0b013e3181f2b37f
43. Stamova B, Jickling GC, Ander BP, Zhan X, Liu D, Turner R, et al. Gene expression in peripheral immune cells following cardioembolic stroke is sexually dimorphic. PLoS ONE. (2014) 9:e102550. doi: 10.1371/journal.pone.0102550
44. Zhang Y, James M, Middleton FA, Davis RL. Transcriptional analysis of multiple brain regions in Parkinson's disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet. (2005) 137B:5–16. doi: 10.1002/ajmg.b.30195
45. Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. (2010) 2:52ra73. doi: 10.1126/scitranslmed.3001059
46. Davis S, Meltzer PS. GEOquery: a bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics. (2007) 23:1846–7. doi: 10.1093/bioinformatics/btm254
47. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. (2004) 3:Article3. doi: 10.2202/1544-6115.1027
48. Aleyasin H, Rousseaux MW, Phillips M, Kim RH, Bland RJ, Callaghan S, et al. The Parkinson's disease gene DJ-1 is also a key regulator of stroke-induced damage. Proc Natl Acad Sci USA. (2007) 104:18748–53. doi: 10.1073/pnas.0709379104
49. Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson's disease and other age-related disorders. Free Radic Biol Med. (2009) 47:1354–61. doi: 10.1016/j.freeradbiomed.2009.08.003
50. Berwick DC, Harvey K. The regulation and deregulation of Wnt signaling by PARK genes in health and disease. J Mol Cell Biol. (2014) 6:3–12. doi: 10.1093/jmcb/mjt037
51. Mercado G, Castillo V, Soto P, Sidhu A. ER stress and Parkinson's disease: pathological inputs that converge into the secretory pathway. Brain Res. (2016) 1648(Pt B):626–32. doi: 10.1016/j.brainres.2016.04.042
52. Utomo A, Jiang X, Furuta S, Yun J, Levin DS, Wang YC, et al. Identification of a novel putative non-selenocysteine containing phospholipid hydroperoxide glutathione peroxidase (NPGPx) essential for alleviating oxidative stress generated from polyunsaturated fatty acids in breast cancer cells. J Biol Chem. (2004) 279:43522–9. doi: 10.1074/jbc.M407141200
53. Wei PC, Hsieh YH, Su MI, Jiang X, Hsu PH, Lo WT, et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol Cell. (2012) 48:747–59. doi: 10.1016/j.molcel.2012.10.007
54. Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. (2013) 12:105–18. doi: 10.1016/S1474-4422(12)70238-7
55. Coppola-Segovia V, Cavarsan C, Maia FG, Ferraz AC, Nakao LS, Lima MM, et al. ER stress induced by tunicamycin triggers alpha-synuclein oligomerization, dopaminergic neurons death and locomotor impairment: a new model of Parkinson's disease. Mol Neurobiol. (2017) 54:5798–806. doi: 10.1007/s12035-016-0114-x
56. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. (2008) 7:1013–30. doi: 10.1038/nrd2755
57. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89–102. doi: 10.1038/nrm3270
58. Briegel KJ, Joyner AL. Identification and characterization of Lbh, a novel conserved nuclear protein expressed during early limb and heart development. Dev Biol. (2001) 233:291–304. doi: 10.1006/dbio.2001.0225
59. Briegel KJ, Baldwin HS, Epstein JA, Joyner AL. Congenital heart disease reminiscent of partial trisomy 2p syndrome in mice transgenic for the transcription factor Lbh. Development. (2005) 132:3305–16. doi: 10.1242/dev.01887
60. Conen KL, Nishimori S, Provot S, Kronenberg HM. The transcriptional cofactor Lbh regulates angiogenesis and endochondral bone formation during fetal bone development. Dev Biol. (2009) 333:348–58. doi: 10.1016/j.ydbio.2009.07.003
61. Rieger ME, Sims AH, Coats ER, Clarke RB, Briegel KJ. The embryonic transcription cofactor LBH is a direct target of the Wnt signaling pathway in epithelial development and in aggressive basal subtype breast cancers. Mol Cell Biol. (2010) 30:4267–79. doi: 10.1128/MCB.01418-09
62. Lambert C, Cisternas P, Inestrosa NC. Role of Wnt signaling in central nervous system injury. Mol Neurobiol. (2016) 53:2297–311. doi: 10.1007/s12035-015-9138-x
63. Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci. (2010) 11:77–86. doi: 10.1038/nrn2755
64. Alves dos Santos MT, Smidt MP. En1 and Wnt signaling in midbrain dopaminergic neuronal development. Neural Dev. (2011) 6:23. doi: 10.1186/1749-8104-6-23
65. Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. (2003) 39:889–909. doi: 10.1016/S0896-6273(03)00568-3
66. Berwick DC, Harvey K. LRRK2 functions as a Wnt signaling scaffold, bridging cytosolic proteins and membrane-localized LRP6. Hum Mol Genet. (2012) 21:4966–79. doi: 10.1093/hmg/dds342
67. Rawal N, Corti O, Sacchetti P, Ardilla-Osorio H, Sehat B, Brice A, et al. Parkin protects dopaminergic neurons from excessive Wnt/beta-catenin signaling. Biochem Biophys Res Commun. (2009) 388:473–8. doi: 10.1016/j.bbrc.2009.07.014
68. Deng H, Gao K, Jankovic J. The VPS35 gene and Parkinson's disease. Mov Disord. (2013) 28:569–75. doi: 10.1002/mds.25430
69. Kitagawa H, Ray WJ, Glantschnig H, Nantermet PV, Yu Y, Leu CT, et al. A regulatory circuit mediating convergence between Nurr1 transcriptional regulation and Wnt signaling. Mol Cell Biol. (2007) 27:7486–96. doi: 10.1128/MCB.00409-07
70. Kwok JB, Hallupp M, Loy CT, Chan DK, Woo J, Mellick GD, et al. GSK3B polymorphisms alter transcription and splicing in Parkinson's disease. Ann Neurol. (2005) 58:829–39. doi: 10.1002/ana.20691
71. Liu X, Cheng R, Verbitsky M, Kisselev S, Browne A, Mejia-Sanatana H, et al. Genome-wide association study identifies candidate genes for Parkinson's disease in an Ashkenazi Jewish population. BMC Med Genet. (2011) 12:104. doi: 10.1186/1471-2350-12-104
72. Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. (2008) 183:409–17. doi: 10.1083/jcb.200806024
73. Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. (2008) 322:1247–50. doi: 10.1126/science.1164594
74. Tian Y, Stamova B, Jickling GC, Liu D, Ander BP, Bushnell C, et al. Effects of gender on gene expression in the blood of ischemic stroke patients. J Cereb Blood Flow Metab. (2012) 32:780–91. doi: 10.1038/jcbfm.2011.179
75. Zhang L, Yang X, Yang S, Zhang J. The Wnt /beta-catenin signaling pathway in the adult neurogenesis. Eur J Neurosci. (2011) 33:1–8. doi: 10.1111/j.1460-9568.2010.7483.x
76. Ekwall AK, Whitaker JW, Hammaker D, Bugbee WD, Wang W, Firestein GS. The Rheumatoid Arthritis Risk Gene LBH Regulates Growth in Fibroblast-like Synoviocytes. Arthritis Rheumatol. (2015) 67:1193–202. doi: 10.1002/art.39060
77. Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. (2005) 122:957–68. doi: 10.1016/j.cell.2005.08.029
78. Zhang Y, Chen J, Li F, Li D, Xiong Q, Lin Y, et al. A pentapeptide monocyte locomotion inhibitory factor protects brain ischemia injury by targeting the eEF1A1/endothelial nitric oxide synthase pathway. Stroke. (2012) 43:2764–73. doi: 10.1161/STROKEAHA.112.657908
79. George G, Singh S, Lokappa SB, Varkey J. Gene co-expression network analysis for identifying genetic markers in Parkinson's disease - a three-way comparative approach. Genomics. (2018). doi: 10.1016/j.ygeno.2018.05.005. [Epub ahead of print].
80. Ghosh A, Saminathan H, Kanthasamy A, Anantharam V, Jin H, Sondarva G, et al. The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease. J Biol Chem. (2013) 288:21955–71. doi: 10.1074/jbc.M112.444224
81. Yoshimura S, Gerondopoulos A, Linford A, Rigden DJ, Barr FA. Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J Cell Biol. (2010) 191:367–81. doi: 10.1083/jcb.201008051
82. Huang CY, Kuo WW, Ho TJ, Chiang SF, Pai PY, Lin JY, et al. Rab9-dependent autophagy is required for the IGF-IIR triggering mitophagy to eliminate damaged mitochondria. J Cell Physiol. (2018) 233:7080–91. doi: 10.1002/jcp.26346
83. Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. (2010) 13:805–11. doi: 10.1038/nn.2575
84. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. (2015) 85:257–73. doi: 10.1016/j.neuron.2014.12.007
85. McLennan AG. The Nudix hydrolase superfamily. Cell Mol Life Sci. (2006) 63:123–43. doi: 10.1007/s00018-005-5386-7
86. Yagi T, Baroja-Fernandez E, Yamamoto R, Munoz FJ, Akazawa T, Hong KS, et al. Cloning, expression and characterization of a mammalian Nudix hydrolase-like enzyme that cleaves the pyrophosphate bond of UDP-glucose. Biochem J. (2003) 370(Pt 2):409–15. doi: 10.1042/bj20021140
87. Wang G, Ren G, Cui X, Lu Z, Ma Y, Qi Y, et al. Human cytomegalovirus RL13 protein interacts with host NUDT14 protein affecting viral DNA replication. Mol Med Rep. (2016) 13:2167–74. doi: 10.3892/mmr.2016.4778
88. Manolio TA, Collins FS. Genes, environment, health, and disease: facing up to complexity. Hum Hered. (2007) 63:63–6. doi: 10.1159/000099178
89. Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. (2017) 169:1177–86. doi: 10.1016/j.cell.2017.05.038
90. Ikeda M, Okahisa Y, Aleksic B, Won M, Kondo N, Naruse N, et al. Evidence for shared genetic risk between methamphetamine-induced psychosis and schizophrenia. Neuropsychopharmacology. (2013) 38:1864–70. doi: 10.1038/npp.2013.94
91. Zhang J, Malik A, Choi HB, Ko RW, Dissing-Olesen L, MacVicar BA. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron. (2014) 82:195–207. doi: 10.1016/j.neuron.2014.01.043
Keywords: ischemic stroke, Parkinson's disease, genome-wide association studies, gene-based test, gene expression analyses
Citation: Lang W, Wang J, Ma X, Zhang N, Li H, Cui P and Hao J (2019) Identification of Shared Genes Between Ischemic Stroke and Parkinson's Disease Using Genome-Wide Association Studies. Front. Neurol. 10:297. doi: 10.3389/fneur.2019.00297
Received: 20 September 2018; Accepted: 07 March 2019;
Published: 28 March 2019.
Edited by:
Michael F. Miles, Virginia Commonwealth University, United StatesReviewed by:
Emma Johnson, Washington University School of Medicine in St. Louis, United StatesCopyright © 2019 Lang, Wang, Ma, Zhang, Li, Cui and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junwei Hao, aGp3QHRtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.