 Keli Hočevar
Keli Hočevar Smiljana Ristić
Smiljana Ristić Borut Peterlin
Borut Peterlin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Neurol. , 26 February 2019
Sec. Neuropharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00134
Background: Over the past two decades, various novel disease-modifying drugs for multiple sclerosis (MS) have been approved. However, there is high variability in the patient response to the available medications, which is hypothesized to be partly attributed to genetics.
Objectives: To conduct a systematic review of the current literature on the pharmacogenomics of MS therapy.
Methods: A systematic literature search was conducted using PubMed/MEDLINE database searching for articles investigating a role of genetic variation in response to disease-modifying MS treatments, published in the English language up to October 9th, 2018. PRISMA guidelines for systematic reviews were applied. Studies were included if they investigated response or nonresponse to MS treatment defined as relapse rate, by expanded disability status scale score or based on magnetic resonance imaging. The following data were extracted: first author's last name, year of publication, PMID number, sample size, ethnicity of patients, method, genes, and polymorphisms tested, outcome, significant associations with corresponding P-values and confidence intervals, response criteria, and duration of the follow-up period.
Results: Overall, 48 articles published up to October 2018, evaluating response to interferon-beta, glatiramer acetate, mitoxantrone, and natalizumab, met our inclusion criteria and were included in this review. Among those, we identified 42 (87.5%) candidate gene studies and 6 (12.5%) genome-wide association studies. Existing pharmacogenomic evidence is mainly based on the results of individual studies, or on results of multiple studies, which often lack consistency. In recent years, hypothesis-free approaches identified novel candidate genes that remain to be validated. Various study designs, including the definition of clinical response, duration of the follow-up period, and methodology as well as moderate sample sizes, likely contributed to discordances between studies. However, some of the significant associations were identified in the same genes, or in the genes involved in the same biological pathways.
Conclusions: At the moment, there is no available clinically actionable pharmacogenomic biomarker that would enable more personalized treatment of MS. More large-scale studies with uniform design are needed to identify novel and validate existing pharmacogenomics findings. Furthermore, studies investigating associations between rare variants and treatment response in MS patients, using next-generation sequencing technologies are warranted.
Multiple sclerosis (MS) is a chronic autoimmune disease characterized by the progressive infiltration of inflammatory cells to the central nervous system (CNS), demyelination and axonal damage. Although MS is affecting nearly 2.5 million individuals worldwide (1), its etiology remains largely unexplained. The clinical course of MS is highly heterogeneous, with current evidence suggesting that the combination of environmental and genetic risk factors is involved (1, 2). In general, three types of MS have been characterized, including a relapsing-remitting form of multiple sclerosis (RRMS) (80-85% of MS patients), which might evolve into secondary progressive MS (SPMS), and primary progressive MS (PPMS) manifesting in 15% of patients (3). Also, the response to the existing therapies largely varies between individuals, with estimated non-responder rates ranging up to 50% for interferon beta (IFN-beta) and glatiramer acetate (GA) (4, 5). Although the reasons for that variability remain unclear, several previous studies have implicated the role of genetics in response to MS treatment (6, 7). While RRMS is the main focus of current pharmacogenomic research, as it is the most common and the most responsive to current treatment options, only a few treatments are licensed to slow the progressive form of the disease (3, 8). Approved medicines for MS include immunomodulatory and immunosuppressive drugs and monoclonal antibodies, including subcutaneous and intramuscular interferons, subcutaneous GA, intravenous (iv) natalizumab, oral fingolimod, teriflunomide and dimethyl fumarate, iv mitoxantrone, iv alemtuzumab, and iv ocrelizumab, most of them clinically proven to be effective mainly in reducing annualized relapse rate (ARR) in the early stages of the disease (9). Among most widely prescribed first-line treatments worldwide remain IFN-beta and GA (10), which reduce frequency and severity of relapses in RRMS patients, decrease disease progression rate and improve magnetic resonance imaging outcomes with minimal side effects. Those are the characteristics that are beneficial; however, these drugs are only partially effective, and the response of individual patients to these therapies is highly unpredictable. Current literature suggests that approximately 30-50% of patients do not respond well to first-line therapies (depending on the response criteria used) (5), which is hypothesized to be in part attributed to inter-individual genetic variability. In clinical practice it is often the case that patients should fail to respond to beta-interferons or GA before receiving a second-line treatment (9); moreover, clinical evaluation of response to the therapy requires 1-2 year follow-up (11). It has previously been shown that there is a limited time window for effective intervention, during which the development of early brain atrophy, and thus cognitive and physical deficits, can be minimized more effectively (12). Therefore, the biomarkers that would predict the responsiveness to therapy are indispensable to reduce adverse events and provide the maximized efficacy and safety early in the disease course.
Although pharmacogenomics in clinical practice is increasingly available, there is currently no established genetic or any other clinical biomarker that would reliably predict a response of an individual to selected MS therapy. However, with a growing number of approved treatment options for MS patients in recent years and rapid advances in genomic technologies, personalized medicine has an opportunity to optimize treatment for an individual.
In the present article, we report the results of the conducted systematic review of currently published data on the pharmacogenomics of MS to review the current status of potential pharmacogenomic biomarkers and discuss their future potential in providing the most effective treatment for an individual.
The systematic review was conducted according to Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Statement guidelines1
Articles on the pharmacogenomics of MS therapy published up to October 9th, 2018 were searched in the PubMed/MEDLINE database using the combinations of following keywords: multiple sclerosis, pharmacogenomics, pharmacogenetics, therapy response, genome-wide association study (GWAS), genome-wide, gene association study, candidate gene study, polymorphism/s, allele/s, and genetic variants. Search details are given in the Box 1. The search was limited to articles published in the English language. Firstly, articles were screened by title and abstract, next the full content was evaluated for their eligibility. The selection of articles and eligibility evaluation were carried out independently by the first two authors (KH and SR). We discussed discrepancies between authors and reached an agreement on the selection of articles for systematic review. Finally, the main review articles were screened for possible additional publications.
Box 1. Search details using PubMed database
((“multiple sclerosis”[All Fields] OR “interferon beta”[All Fields] OR “glatiramer acetate”[All Fields] OR “natalizumab”[All Fields] OR “fingolimod”[All Fields] OR “teriflunomide”[All Fields] OR “dimethyl fumarate”[All Fields] OR “mitoxantrone”[All Fields] OR “alemtuzumab”[All Fields] OR “ocrelizumab”[All Fields]) AND (“pharmacogenomics”[All Fields] OR “pharmacogenetics”[All Fields] OR “pharmacogenetic”[All Fields] OR “pharmacogenomic”[All Fields]))
OR
((“multiple sclerosis”[All Fields] OR “interferon beta”[All Fields] OR “glatiramer acetate”[All Fields] OR “natalizumab”[All Fields] OR “fingolimod”[All Fields] OR “teriflunomide”[All Fields] OR “dimethyl fumarate”[All Fields] OR “mitoxantrone”[All Fields] OR “alemtuzumab”[All Fields] OR “ocrelizumab”[All Fields]) AND (“GWAS”[All Fields] OR “genome-wide”[All Fields] OR “gene association study”[All Fields] OR “polymorphism”[All Fields] OR “polymorphisms”[All Fields] OR “allele”[All Fields] OR "gene variant"[All Fields] OR “alleles”[All Fields]) AND (“treatment response”[All Fields] OR “therapy response”[All Fields] OR “response to therap”[All Fields] OR (response[All Fields] AND ("interferon-beta"[MeSH Terms] OR “interferon-beta”[All Fields] OR ("interferon"[All Fields] AND “beta”[All Fields]) OR “interferon beta”[All Fields])) OR (response[All Fields] AND ("glatiramer acetate"[MeSH Terms] OR (“glatiramer”[All Fields] AND "acetate"[All Fields]) OR "glatiramer acetate"[All Fields])) OR (response[All Fields] AND (“mitoxantrone”[MeSH Terms] OR “mitoxantrone”[All Fields])) OR (response[All Fields] AND ("teriflunomide"[Supplementary Concept] OR “teriflunomide”[All Fields])) OR (response[All Fields] AND (“fingolimod hydrochloride”[MeSH Terms] OR (“fingolimod”[All Fields] AND “hydrochloride”[All Fields]) OR “fingolimod hydrochloride”[All Fields] OR “fingolimod”[All Fields])) OR (response[All Fields] AND (“dimethyl fumarate”[MeSH Terms] OR (“dimethyl”[All Fields] AND “fumarate”[All Fields]) OR “dimethyl fumarate”[All Fields])) OR (response[All Fields] AND (“natalizumab”[MeSH Terms] OR “natalizumab”[All Fields])))).
Studies were included if they investigated response or nonresponse to treatment, defined as relapse rate, by expanded disability status scale (EDSS) score or the definition was based on magnetic resonance imaging (MRI), in the association to genetic variability. We included available studies investigating the pharmacogenomics of all currently approved disease-modifying treatment options for MS patients. We excluded articles that: (1) were not written in the English language, (2) were article evaluations, case reports, reviews, study protocols, (3) were using animal model, cell lines, in silico studies, (4) investigated response by measuring NAbs/IFN-beta antibodies or studies evaluating therapeutic response by other biochemical tests, (5) were gene expression studies, and (6) investigated adverse drug reactions, such as liver and cardiac injury, acute leukemia and progressive multifocal leukoencephalopathy.
Two authors (KH and SR) independently extracted the following data from articles: first author's last name, year of publication, PMID number, sample size, ethnic backgrounds of patients, method, genes, and polymorphisms tested, outcome, significant associations with corresponding P-values and confidence intervals, response criteria, the duration of the follow-up period and medication investigated. Finally, The Pharmacogenomics Knowledgebase (PharmGKB) was reviewed for possible clinically actionable variants in MS treatments and to search for the level of evidence of the existing MS pharmacogenomic biomarkers. Genes with detected significant associations were annotated for Gene Ontology (GO) molecular functions and biological process using the online PANTHERTM tool version 13.1 (13).
In the primary search, we identified a total of 297 articles in the PubMed database. After reviewing titles and abstracts, 229 articles were excluded for the reasons presented in Figure 1. Additional 20 studies were excluded after full-length review, because they investigated treatment response by gene expression (n = 9), by measuring Nab/IFN-beta antibodies or by other biochemical tests (n = 6), investigated adverse drug reactions (n = 3), the sample size was small (less than 20) (n = 1), and investigated response to intravenous immunoglobulin (IVIG) (n = 1). In total, 48 publications investigating the association between genetic variation and treatment response met our inclusion criteria and were included in the systematic review: 40 (83 %) studies investigated treatment response to IFN-beta (5 GWAS and 35 candidate gene studies), 9 (19 %) studies investigated treatment response to GA (one GWAS and 8 candidate gene studies). Among them, four studies investigated the response to both medications; IFN-beta and GA. In addition, we identified two candidate gene studies investigating the response to mitoxantrone and one response to natalizumab. No studies on the pharmacogenomics of newest classes of agents, such as dimethyl fumarate, teriflunomide or fingolimod were identified. Eleven variants with the level of evidence 3 and influence on treatment efficacy were found in the PharmGKB database. Results from candidate gene studies were mostly not replicated, and studies were performed in different populations. Furthermore, genes previously assessed in candidate gene studies showed very little overlap with the significant GWAS associations. Nevertheless, few consistent significant findings (P < 0.05) were reported in the candidate gene studies.
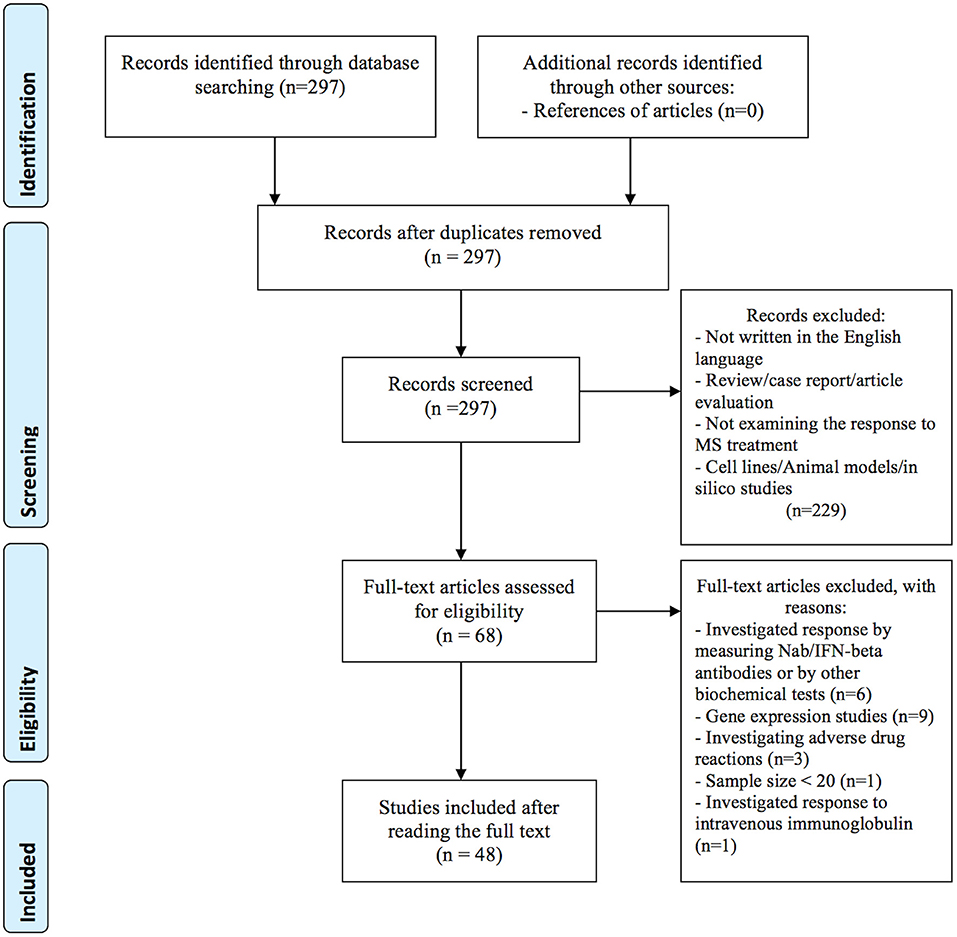
Figure 1. Flow diagram of identification and selection of studies.
Interferon-beta 1 is one of the most commonly prescribed disease-modifying therapies for patients with MS. Interferons are endogenous regulatory cytokines that bind to specific IFN alpha/beta receptors found on the surface of the cells of the immune system and consequently change the expression of many genes, depending on cell type - the inflammatory cytokine synthesis is inhibited (IL-12, IL-17, IL-23), while the production of anti-inflammatory cytokines (IL-4, IL-10) increases, which provokes differentiation toward a CD4+ T helper cell type phenotype -Th2 immune response (14). Additionally, interferon reduces the expression of matrix metalloproteases, affects the expression of cell adhesion molecules located on the endothelial surface and on the activated T-cell surface, which results in reduced T-cell activation and reduced lymphocyte migration across the blood-brain barrier (BBB). The potential antiviral activity of IFN-beta has also been proposed (15).
We identified 35 studies investigating the association between genetic variability and response to IFN-beta, four of them also investigating the response to GA. The details of the included studies are presented in Supplementary Table 1. The selection of candidate genes in these studies was mainly based on the proposed mechanisms of action of IFN-beta, and in recent years, studies have also been designed to validate the significant results obtained from genome-wide studies. Some examples of candidate genes investigated were: HLA class II genes, MXA, genes coding for interferon receptors IFNAR1, IFNAR2 and other interferon-stimulated response elements (ISREs), interferon gamma IFNG, chemokine receptor CCR5, genes related to the type I IFN and TLR pathways, genes coding for GABA and glutamate receptors, genes encoding cytokines and their receptors, innate pattern recognition receptors, antigens CD46 and CD58, CTLA4, HAPLN1, ACE and APOE gene.
There are a limited number of studies conducted on the same polymorphisms. Furthermore, among those, the results were largely inconsistent. Sixteen (46%) of included IFN-beta candidate gene studies failed to identify any significant association comparing genetic variation between responders to non-responders. Non-significant associations were repeatedly reported within the HLA locus of class I and/or II (six times) (4, 16–20), in IFNAR1 and INFAR2 genes (two times) (21, 22), in APOE gene (two times) (23, 24), in IRF5 gene (25, 26), and NLRP3 gene (27, 28). Other non-significant associations included MXA (29), HAPLN (30), IFNL3 (31), IRF8 (26), and GPC5 (26) genes. However, some reproducible significant associations between IFN-beta response and genetic variability have also been detected.
Despite the negative association results between polymorphisms located in the promoter region of the MXA gene and IFN-beta response reported by Weinstock-Guttman et al. (29), the significant association was repeatedly demonstrated by two independent studies, which together comprised three different SNPs in MXA gene, including rs464138 AA (P < 0.0001, OR = 6.23 [95% CI, 2.77–14.03]), rs2071430 G allele (P = 0.015, OR = 3.4 [95% CI, 1.1-11.4]), and rs17000900 GG (P = 0.018, OR = 2.4 [95% CI, 1.1-5.4]) (32, 33). One of those studies, which investigated 100 ISREs-containing genes in association to IFN-beta response heterogeneity, additionally identified significant associations between IFNAR1 rs55884088 (GT)n repeat (P = 0.036), LMP7 rs2071543 C allele (P = 0.002, OR = 6.4 [95% CI, 1.8-24.1]), and CTSS rs1136774 C allele (P = 0.02, OR = 0.4 [95% CI, 0.2-0.8]) (32). Another SNP located in the third intron of the IFNAR1 gene was additionally associated with response to IFN-beta in the study of Sriram et al. (21), suggesting a modest association of rs1012334 A allele with relapse-free status (P = 0.030, OR = 0.9 [95% CI, 0.2-1.2]). Furthermore, IFNAR1 rs1012335 G allele was associated with positive IFN-beta treatment response (34) and was additionally, in allelic combinations, suggested as a marker of choice for IFN-beta treatment over GA (6).
Another repeatedly studied variation is a 32-base pair (bp) deletion of the CCR5 gene (CCR5*d, rs333). A significant association between CCR5 deletion allele and IFN-beta treatment response in MS patients was confirmed by three independent analyses. In the study of Kulakova et al. (6), CCR5*d was more frequently found in Russian MS patients with optimal response to IFN-beta and GA non-responders, while CCR5*w/w was enriched in IFN-beta non-responders and GA responders. In the related study, allelic combinations of (CCR5*d + IFNAR1*G + IFNB1*T/T) or (CCR5*d + IFNAR1*G + IFNG*T) were proven to be beneficial for IFN-beta treatment efficacy (34). A significant association between CCR5*d and IFN-beta treatment response in MS patients was also detected in the Egyptian population by Karam et al. (35) (P = 0.01, OR = 3.2 [95%-CI, 1.1–8.8]).
Certain genotypes of IRF5 gene polymorphisms (rs2004640 TT, P = 0.0006, and rs47281420 AA, P = 0.0023) were reported to exert a poor pharmacological response to IFN-beta, with more T2 lesions detected (36). In terms of particular polymorphic loci, the finding IRF5 rs2004640 was replicated in an independent population within the same study (P = 0.037). The study of Vandenbroeck et al. (25) identified the trend toward a greater T allele frequency for the variant of IRF5 rs3807306 polymorphism in responders (P = 0.09), whereas no evidence of an association for IRF5 rs4728142 was detected. Evidence that an AA genotype of IRF8 rs17445836 polymorphism influences event-free survival in IFN-beta treated subjects was also found (P = 0.017, OR = 0.45 [95% CI, 0.2-0.9]) (19). Contrary, the study conducted in a Danish cohort of patients by Sellebjerg et al. (26), failed to identify any association between polymorphisms located in IRF5 (rs2004640, rs3807306, rs4728142) and in IRF8 (rs13333054 and rs17445836) genes.
The number of studies attempting to validate or further investigate the results of GWAS studies is limited. Polymorphous loci in the GPC5 gene were reproducible with candidate-gene study of Cénit et al. (rs10492503 AA, P = 0.018, OR = 3.0 [95% CI, 1.3-6.6]; rs1411751 GG, P = 0.012, OR = 3.7 [95% CI, 1.5-9.4]), and GWAS by Byun et al. (rs10492503, rs9301789) (37), while the candidate gene study conducted by Sellebjerg et al. (26) yielded non-significant result. The aim of the study conducted by Bustamante et al. (38) was to further investigate the results of two GWAS studies that highlighted the importance of genes playing role in toll-like receptor (TLR) pathways, type I interferon (IFN)-induced genes, and genes coding for GABA and glutamate receptors. An investigation of 384 polymorphisms located in those genes, detected only two significant polymorphisms (rs2277302 in PELI3 gene, P = 0.008, and rs832032 in GABRR3, P = 0.006 gene). Overall association of polymorphisms located in these pathways was therefore not confirmed (38).
As the evidence of polygenic nature of IFN-beta treatment response, allelic combinations (JAK2-IL10RB-GBP1-PIAS1 and JAK2-IL10-CASP3) were detected as significant, while no significant association of tested individual polymorphisms was found (39). In another study, MS patients with non-GCC haplotypes (rs1800896, rs1800871, rs1800872) of the IL10 gene experienced fewer new MRI T1-contrast enhancing lesions than patients with the GCC haplotype (40).
Other positive associations included: intronic polymorphism rs2542109 of the USP18 gene, TGFB1 rs1800469, TRAILR-1 rs20576, CD46 rs2724385, GPC5 rs10492503 and rs1411751 polymorphisms, polymorphic microsatellite located in the first intron of the IFNG gene, and CD58 rs12044852 polymorphism.
Significant associations (P < 0.05) between treatment response and IFN-beta, identified in at least one study, are presented in Table 1.
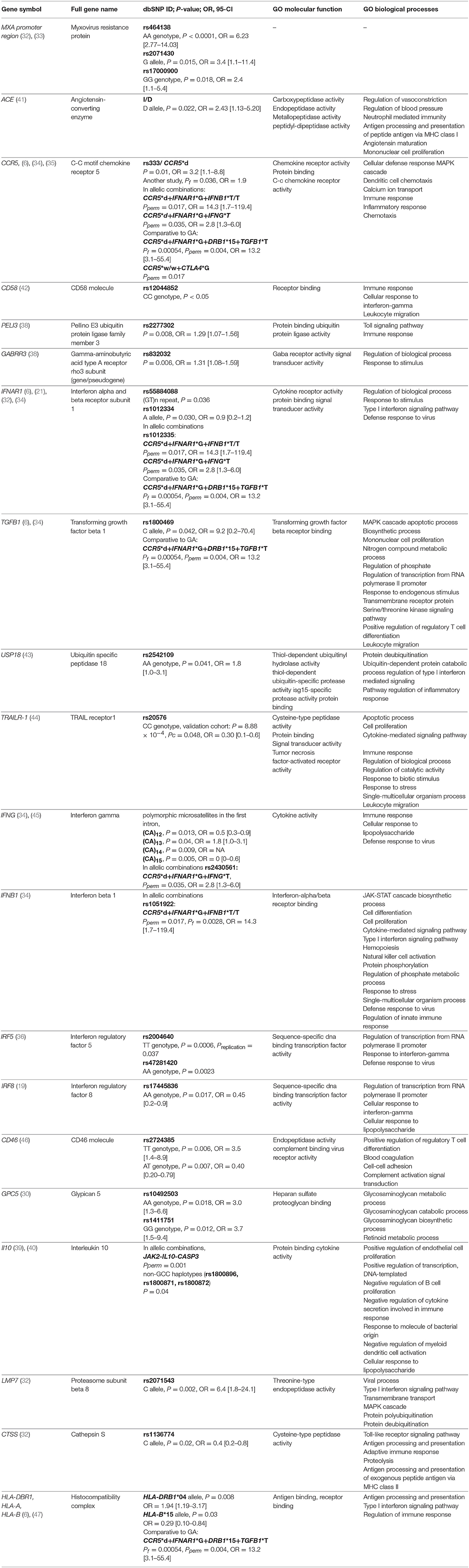
Table 1. Significant associations from candidate gene studies and IFN-beta MS treatment response along with selected gene ontology (GO) annotations. Pperm, P-value permutation test; Pf, P-value exact Fisher's test.
Currently, five GWAS studies investigating an association between IFN-beta treatment response and genetic variation were carried out. GWAS study, which investigated SNPs in HLA- and non-HLA genes in association with the development of antibodies to IFN-beta therapy, was excluded from this review (48). None of the GWAS studies reported similar results, but they uniquely suggested that multiple genes influence the treatment response to IFN-beta. Furthermore, on the level of genes, most of the results were in deviation with previously conducted candidate-gene studies, thus providing novel candidate genes that might be involved in response to IFN-beta treatment. However, it is important to note that some potential candidate genes reported by independent GWAS studies were involved in the same biological pathways.
In the first GWAS study, conducted in 2008 by Byun et al. (37), authors found out that many of the detected differences between responders and non-responders were located in genes involved in ion channels and signal transduction pathways. Additionally, the authors also suggested that genetic variants in heparan sulfate proteoglycan genes (HAPLN1) might be useful as possible clinical predictors of response to MS therapy. Results of the second GWAS study conducted by Comabella et al. (11), indicated the importance of the glutamatergic system (GRIA3 gene) in patients response to IFN-beta therapy. The GWAS study, conducted by Esposito and colleagues followed in 2015 and reported candidate intronic polymorphism rs9828519 in the SLC9A9 gene encoding for sodium/hydrogen exchanger found in endosomes. For this gene, a broader role in MS pathogenesis, beyond treatment with IFN-beta, was also proposed. The gene product was functionally characterized to inhibit the development of pro-inflammatory CD4+ T cells (7).
In the study of Mahurkar et al. (49), none of the SNPs reached the level of genome-wide significance. The strongest associations were observed for FHIT gene and followed by variants in GAPVD1 and near ZNF697 gene. In the discovery stage of this study, samples were individually genotyped using Illumina® arrays, which distinguishes it from previously published GWAS studies where pooled genotyping was performed. A recent GWAS study conducted by Clarelli et al. (50) investigated long-term treatment response considering a 4-year follow-up study period and included only patients with extreme phenotypes of treatment responses. In contrast, all of the previous GWAS studies have taken into account 2-year follow up period. In summary, alterations in the genes involved in immunoregulatory processes, the glutamatergic system (GRIK2 and GRM3), and signal transduction (GAPVD1) reached the highest significance.
Lack of overlap between GWAS studies likely reflects the differences in definitions of responders or non-responders, furthermore, GWAS studies covered populations of various ethnicities, including Italian, German, Spanish, and Australian. Also, the methodology was based on different genotyping platforms - first two GWAS studies used Affymetrix genotyping platforms, covering 100 000 and 428 867 SNPs, respectively, while Illumina arrays were used in all of the later studies (Illumina® Human 660-Quad platform, Illumina® 2.5M platform, Illumina® OmniExpress BeadChip and Illumina® OMNI-5M array). However, we showed that genes identified by GWAS studies were significantly enriched for ionotropic glutamate receptor signaling pathway (GO:0035235). Top-ranking results from GWAS studies are summarized in Table 2.
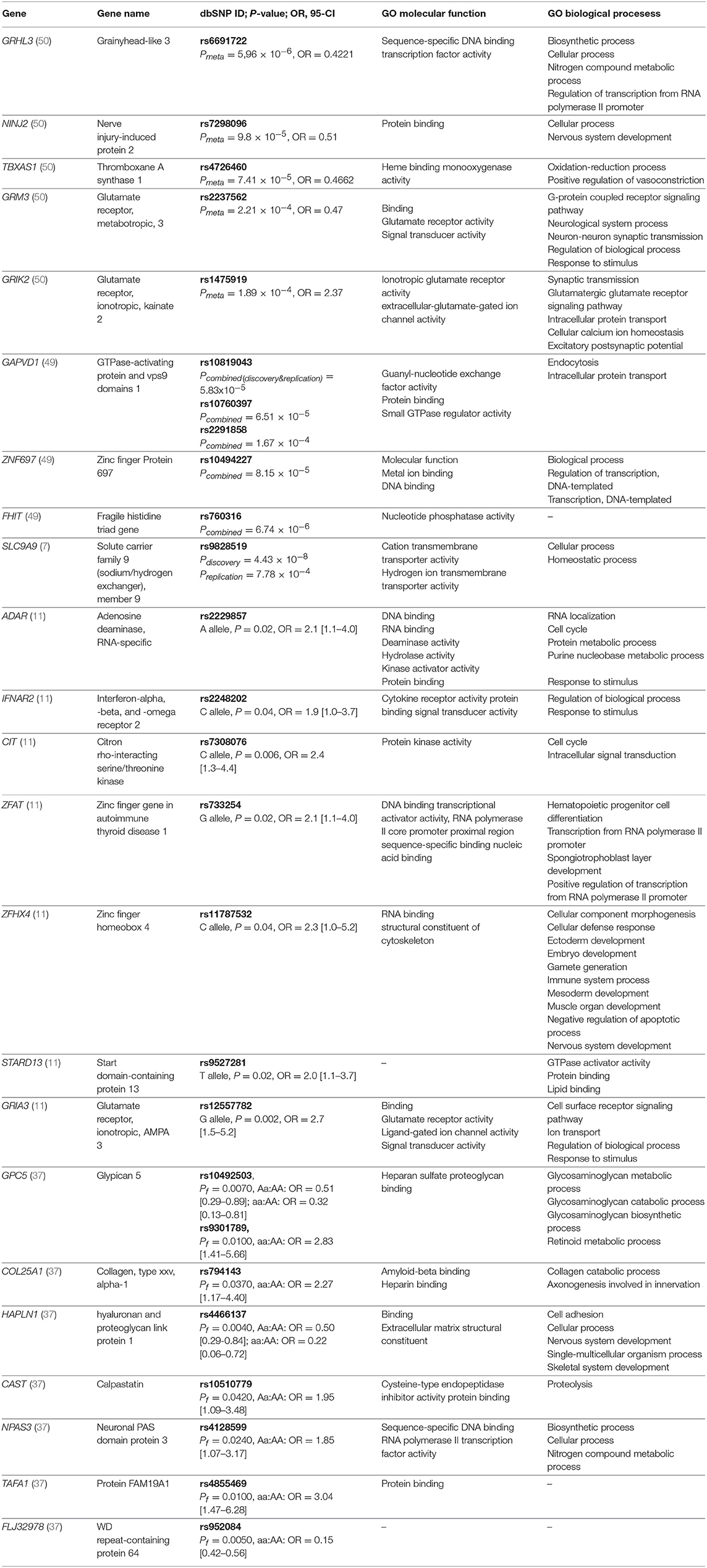
Table 2. Genes with detected significant associations with response to IFN-beta in at least one study and top-ranking results from GWAS studies along with selected gene ontology (GO) biological processes and molecular functions.
Glatiramer acetate is another widely prescribed disease-modifying therapy for patients with MS, with a complex and yet not fully understood mechanism of action. GA is a heterogeneous mixture of synthetic polymers made of random sequences of four amino acids (51). It acts through immunomodulatory actions to the cells of innate and acquired immune response. Through binding to MHC Class II molecules, it participates in the generation of GA-specific T-cells and shifts their phenotype from pro-inflammatory helper-T types 1 and 17 (Th1/Th17) to anti-inflammatory regulatory T cells (Tregs) and helper-T type 2 (Th2) cells. Additionally, GA-specific T-cells are able to migrate through the BBB, where they induce local secretion of anti-inflammatory cytokines at the site of the lesions (51).
Eight candidate gene studies investigating an association between polymorphisms and treatment response to GA met our inclusion criteria. Four of them were mentioned above, as they also investigated IFN-beta treatment response. Detailed data on studies is summarized in Supplementary Table 1. Hypothesis-driven approaches have primarily investigated genes involved in the mechanism of action of GA. Contrary to IFN-beta response, the HLA class I /II genes have been repeatedly positively associated with GA treatment response. The HLA DRB1 *1501 allele was demonstrated to influence the response to GA therapy in the cohort of 44 Italian RRMS patients (P = 0.008) (4) and in the cohort of 332 American patients, HLA-DBR1*1501/1501 genotype was significantly enriched among GA-responders (rs3135388, PAG/AA = 0.015, OR = 2.7 [95% CI, 1.2-6.0]) (19). The related study conducted in 64 American subjects with RRMS, showed that the presence of HLA DR15 or DQ6 alleles or the absence of DR17 and DQ2 alleles is nominally associated with a favorable clinical response (52). The authors also demonstrated that the presence of the DR15-DQ6 haplotype and the absence of the DR17-DQ2 haplotype is significantly associated with positive treatment response (52). Furthermore, in a cohort of 296 Russian patients, the nominally significant association of HLA-DRB1*4 allele with a positive response to GA was detected comparing responders to nonresponders and intermediate responders, Pf = 0.015, OR = 2.02 [95% CI, 1.11–3.67] (53).
One of the first pharmacogenetics candidate-gene studies on GA, reported a significant association between GA response and two SNPs, rs71878 in a T-cell receptor beta (TCRB) gene (P = 0.015, OR = 6.85 [95% CI, 1.45–31.9]) and rs2275235 in the cathepsin S (CTSS) (P = 0.014, OR = 11.59 [95% CI, 1.6–81.9]) (54). Nominally significant associations were shown for additional genes MBP, CD86, FAS, IL1R1, and IL12RB2. However, in the same experiment, no significant association for the HLA-DRB1*1501 allele was identified, suggesting the genetic heterogeneity of this region among the different populations as the possible reason (54).
Using comparative pharmacogenomics approach investigating allelic combinations of CCR5, IFNAR1, TGFB1, DRB, and CTLA4 genes, the CCR5*w/w genotype was the most enriched in GA responders compared to IFN-beta responders (6). In the most recent study examining association between GWAS identified MS susceptibility loci and efficacy of GA therapy in a Russian population of 296 RRMS patients, five SNPs were associated by themselves with event-free phenotype: EOMES rs2371108 T allele, CLEC16A rs6498169 A allele, IL22RA2 rs202573 GG genotype, PVT1 rs2114358 A allele, and HLA-DBR1*4 (Pf = 0.032-0.00092). Authors demonstrated increased significance levels when taking into account biallelic and triallelic combinations of these genes with additionally included polymorphic variants of TYK2, CD6, IL7RA and IRF8 genes (53).
Genes with at least one detected significant association (P < 0.05), along with GO biological processes and molecular functions, are presented in Table 3.
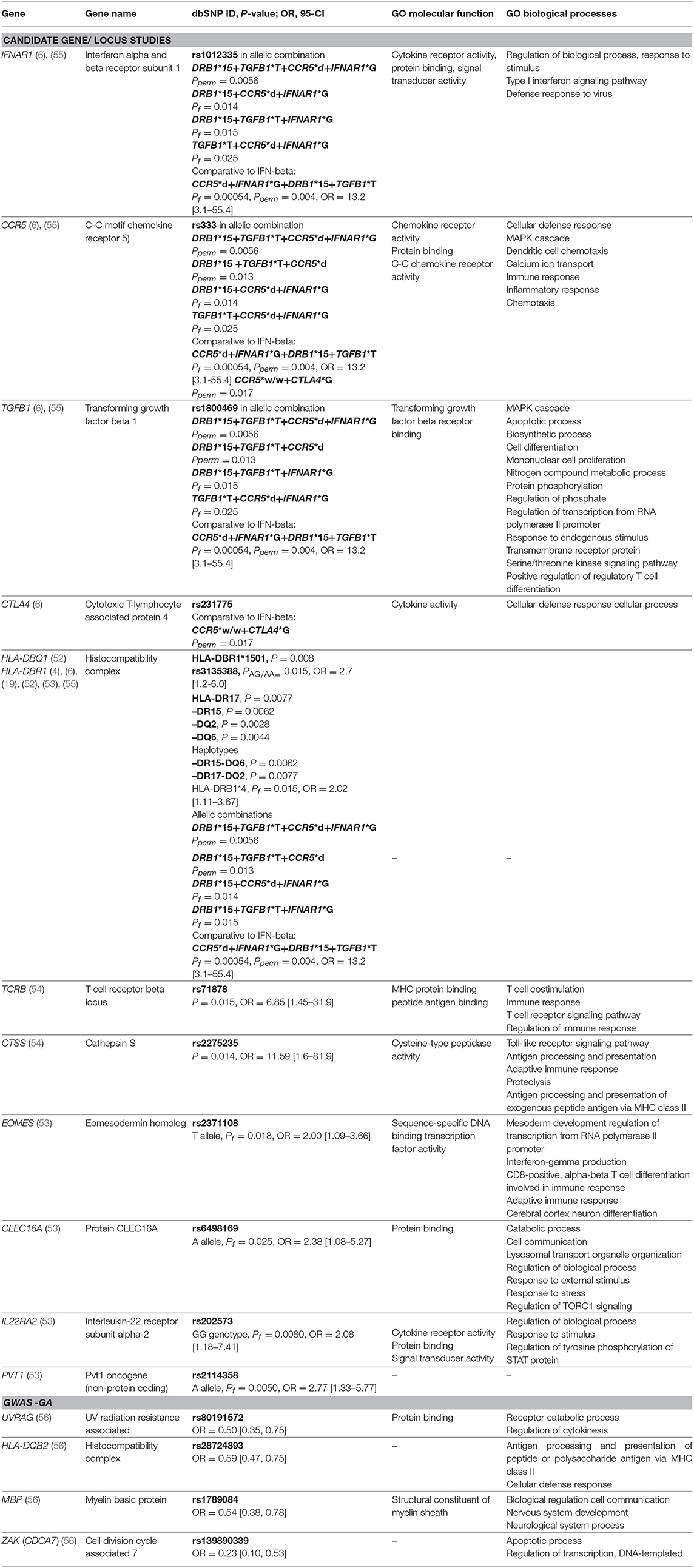
Table 3. Genes with detected significant associations with response to GA from candidate gene studies and GWAS studies along with selected gene ontology (GO) biological processes and molecular functions. Pperm, P-value permutation test; Pf, P-value exact Fisher's test.
To date, one GWAS study investigating the research question of pharmacogenomics and GA has been published (56). Patients with extreme phenotypes were included in the analysis, considering a 4-year follow-up period. Genotyping was conducted using Illumina OMNI-5M genome-wide array® covering 4301331 SNPs. Significant associations with treatment response were identified in the following genes: UVRAG (rs80191572), HLA-DQB2 (rs28724893), MBP (rs1789084), and ZAK (rs139890339). Marginal association with another polymorphism (rs470929) located in the MBP gene has previously been reported in the candidate gene study conducted by Grossman et al. (54). The MBP gene encodes the autoantigen myelin basic protein, which is attacked by the immune system in MS patients. Furthermore, GA was designed as an MBP mimetic (51). The results from the GWAS study warrant further confirmation in independent studies. Significant results are presented in Table 3.
Mitoxantrone is synthetic anthracenedione – a cytotoxic agent that inhibits DNA repair via inhibition of topoisomerase II leading to a suppressed proliferation of T cells, B cells, and macrophages, decreased pro-inflammatory cytokine secretion, enhanced suppressor T cell function, and suppressed macrophage-mediated myelin degradation (57). Two studies investigating an association between genetic polymorphisms and mitoxantrone were published to date, providing conflicting results (58, 59). In the first study, authors proposed that SNPs in ABC-transporter genes (ABCB1 and ABCG2) might serve as pharmacogenetic markers associated with clinical response to mitoxantrone therapy in patients with RRMS or SPMS forms of the disease. The second study failed to confirm the association in PPMS form of the disease (59).
Natalizumab is a humanized monoclonal antibody that inhibits the migration of lymphocytes via the BBB by inhibiting an adhesive molecule of anti-integrin-α4 (57). Currently, only one pharmacogenetic study in association to treatment response was conducted (60). Authors investigated an association between polymorphisms in NQO1 and GSTP1 genes and treatment efficacy. In a combined analysis, it was found that patients who carried the wild-type genotype or only one non-wild polymorphism for either gene have possibly a better clinical outcome after receiving the natalizumab therapy.
According to PharmGKB levels of evidence for variant-drug associations, no clinically actionable variants with the level of evidence 1A or 1B exist for MS (October 9th, 2018). We identified eleven variant-drug combinations associated with treatment efficacy and with the level of evidence 3, which stands for an annotation based on a single significant (not yet replicated) result or annotation for a variant-drug combination evaluated in multiple studies but lacking clear evidence of an association (61). The PharmGKB variants are presented in Table 4.
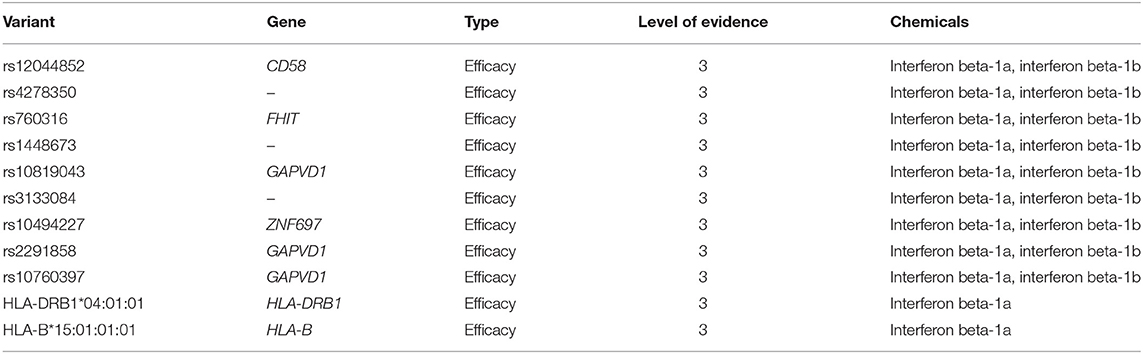
Table 4. Variant/gene-drug pairs currently listed in PharmGKB database (October 9th, 2018).
Part of the unresponsiveness to IFN-beta can also be explained by the development of neutralizing antibodies (NAbs) that reduce the drug efficacy. These develop in up to third of patients, depending on the IFN-beta product administered (62). However, the use of NAbs, as an early pharmacogenetic biomarker is limited because NAbs develop only after 6-24 months from initiation of treatment and patients may even revert to NAbs-negative over time (63). Additionally, NAbs-positivity explains the unresponsiveness to IFN-beta treatment only in a small proportion of patients (64). Nevertheless, it might be useful to include the information on NAbs in the pharmacogenetic studies of MS. Furthermore, the genetic markers that influence the development of NAbs have also been identified in patients with MS (candidate gene studies and GWAS) (48, 65–68).
Although beyond the scope of this paper, several studies indicate that gene expression signatures could prove useful in predicting long-term treatment response in patients with MS (69–71). These studies revealed differences in the expression of genes related to IFN-beta signaling, TLR-4 signaling in monocytes, as well as increased overall molecular response to IFN-beta in non-responders (72). Recently, RNA-sequencing in whole-blood showed that expression of a ribosomal protein S6 was reduced in IFN-beta responders compared to non-responders (73). In another RNA-sequencing study, the different pre-treatment gene expression signature in peripheral blood mononuclear cells (PBMCs) was revealed in MS patients responsive to fingolimod compared to non-responders (74). However, most of the currently proposed transcriptomic biomarkers have only moderate discriminative power and have yet not been validated (75, 76). Additionally, gene expression is more variable than genetic status and largely depends on various environmental factors, drugs co-administered (such as corticosteroids), specific cell populations studied (whole blood, PBMCs, T-cells), and differences in sampling times. Divergent findings can also be explained by the heterogeneity of technical protocols and clinical assessment of treatment response.
In recent years, several actionable pharmacogenomics biomarkers have been identified, comprising many areas of medicine. The implementation of pharmacogenomics in clinical practice has, therefore, a great potential to enable more personalized treatment with several benefits for patients and society. However, despite the increasing number of treatment options available to patients with MS and a high degree of variability in response to these treatments, there is still no reliable pharmacogenomic biomarker that would differentiate between MS-treatment responders and non-responders. Since MS is a chronic progressive disorder, which requires life-long treatment, an early decision for the right therapy may have a high clinical utility for MS patients. By choosing the right treatment for a particular individual early in the disease course, we can slow down the progression of the disease, avoid possible adverse events and improve the efficiency of treatment.
Comprehensive systematic analysis of pharmacogenomic studies showed that the majority of the included studies (87.5%) are limited to candidate genes, mostly hypothesized to be involved in pathways of drug actions. We have observed that candidate gene studies largely lack the replication and confirmation of the results. However, we have identified some genes, the variability of which has been investigated repeatedly, such as MXA, CCR5, GPC5, IFNAR1, IFNAR2, IRF5, NLRP3 genes, and HLA-region. The results of currently published candidate gene studies were mostly inconsistent, which may in part reflect the various study designs, including the inconsistent approach of defining response to treatment, as well as limited sample sizes with insufficient effect size. Nevertheless, it is evident that biological processes defined by statistically significant genes implicated in IFN-beta response are mostly immune-related and include regulation of interleukins production, positive regulation of regulatory T cell differentiation, negative regulation of cytokine production, type I interferon signaling pathway, mononuclear cell proliferation, cellular response to lipopolysaccharide, cellular response to interferon-gamma, regulation of cytokine-mediated signaling pathway, defense response to virus, leukocyte migration, and regulation of innate immune response.
Despite the proposed distinct immunomodulatory mechanisms of actions of IFN-beta and GA, we have observed that some of the significant associations were identified in the same genes, or in genes involved in the same biological pathways. As an example, it has been found that the polymorphisms of the cathepsin S (CTSS) gene are associated with a response to the treatment of both IFN-beta and GA. Cathepsin S has cysteine-type peptidase activity and is involved in several biological processes, including Toll-like receptor signaling pathway, antigen processing and presentation of exogenous peptide antigen via MHC class II, adaptive immune response, and proteolysis, also of human myelin basic protein (MBP) (77). Furthermore, it has been suggested that discriminative allelic variants of the CCR5, IFNAR1, and TGFB1 genes, which are involved in MAPK cascade, defense response, type I IFN signaling pathway, regulatory T cell differentiation, and apoptotic processes, may direct the treatment decision between IFN-beta or GA (6).
In recent years, GWAS studies identified novel candidate genes, which remain to be validated. Moreover, there was no overlap between the top-ranked results of GWAS studies, which suggests that response to existing therapies is influenced by numerous polymorphisms in multiple genes. However, among potential candidate genes identified in GWAS studies of IFN-beta, we detected significant enrichment for genes involved in the glutamate receptor-signaling pathway. Therefore, in the future, more global approaches, such as GWAS or next-generation sequencing (NGS), are required to gain further insight into the pharmacogenomics of MS.
It is important to acknowledge the methodological heterogeneity between the studies included in the present systematic review, such as variability of clinical characterization of the patients, differently defined clinical response, the varying duration of follow-up period among studies, and different genotyping platforms used in GWAS studies. It has previously been reported that the proportion of non-responders varies depending on the definition of treatment response used (78). The clinical criteria for phenotypic classification of patients (responders/non-responders) included: (1) relapse rate (with different thresholds between studies), (2) disease progression, which was most often measured by EDSS score, and (3) changes in MRI activity, such as increase in T2 lesion burden or T1 gadolinium (Gd) enhancing lesions on MRI. The detailed data on the definition of responders/non-responders for each study are presented in Supplementary Table 1. Similarly, the follow-up period ranged from six months (in one study) to 1 year in one study, two years in the majority of the included studies and to four years in two recent studies.
More independent studies investigating the association between already proposed polymorphisms in genes, such as GRHL3, NINJ2, TBXAS1, GRM3, GRIK2, and SLC9A9 and treatment response are warranted to establish reliable and accurate pharmacogenomics predictors. Future studies need to include a larger number of subjects of various ethnicities. It is also crucial to use uniform and precise definitions of treatment response, standardized duration of the follow-up period, and comprehensive clinical characterization of patients.
Also, the GWAS studies are limited to common variants. Of note, rare variants contribute a major part of pharmacogenetic variability (79). In recent years, many important advances in sequencing technologies have been achieved that will in future enable a more comprehensive picture of pharmacogenetic variability in MS patients. Further studies should also consider rare variants obtained by NGS technologies, such as exome or genome sequencing data. To the best of our knowledge, no study investigating the rare variation in exome or genome sequencing data of MS patients in association to treatment response has been published to date.
Furthermore, we suppose that the phenotype of the response to an immunomodulatory pleiotropic therapy, such as IFN-beta and GA, is a sum of numerous contributing genetic factors that were not sufficiently simultaneously and combinatorially assessed by current study designs and methodologies. More studies investigating cumulative effects of polymorphisms in multiple genes (additive effects or epistatic interactions), such as studies of Kulakova et al. (34, 53), are needed to gain a more comprehensive insight into genetic variability in association to the efficacy of treatment.
Another important aspect of future pharmacogenomics, especially for the interpretation of rare variants, are publicly available and easily updatable databases of pharmacogenomic variation, such as PharmGKB, CPIC, ClinVar, as well as population-specific databases. Further standardized dosing recommendations and guidelines based on the patient's genomic test results are required, ideally integrated with demographic, phenotypic and clinical data.
Furthermore, most of the collected studies (94%) were conducted on patients treated with IFN-beta or GA. Lack of pharmacogenomic studies conducted on drugs approved in recent years, such as dimethyl fumarate (Tecidifera®), teriflunomide (Aubagio®), and fingolimod (Gilenya®) limits the implementation of personalized medicine into clinical practice. An increasing number of new treatment options will in future enable more personalized treatment approaches; however, many genome-wide studies carried out on large sample sizes and in different populations are needed to reach reliable pharmacogenomics biomarkers for implementation into daily clinical practice.
In conclusion, current literature data suggests that genetic variability can significantly contribute to the response to treatment in patients with MS. In the future, it is necessary to systematically evaluate the polymorphisms that were previously proposed to influence the response to treatment as well as assess the importance of rare variants and their effects on the treatment of MS. Additional studies and larger ethnically homogenous cohorts are necessary to provide new insides and optimized use of MS drugs. More combinatorial study designs are needed to assess the effect of several combinations of polymorphisms in various genes simultaneously to provide more relevant information for the clinical use of pharmacogenomics. Studies investigating the pharmacogenomics of newer medicines for MS are also necessary, using the clear and uniform criteria of defining treatment response. We believe that all of the above, along with the rapid development of new medications and advances in genomic technologies, will in future enable a more personalized approach to MS treatment.
BP, SR, and KH contributed conception and design of the study; KH and SR searched the database for potentially eligible articles and extracted the data. KH wrote the original draft. SR and BP contributed to manuscript revision. All authors reviewed the final version of the manuscript prior to submission for publication.
This study was funded by the Slovenian Research Agency (ARRS), grant no. P3-0326
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00134/full#supplementary-material
ARR, annualized relapse rate; BBB, blood-brain barrier; CNS, central nervous system; CPIC, Clinical Pharmacogenetics Implementation Consortium; EDSS, expanded disability status scale; GA, glatiramer acetate; GO, gene ontology; GWAS, genome-wide association study; IFN-beta, interferon beta; MHC, major histocompatibility complex; MRI, magnetic resonance imaging; MS, multiple sclerosis; Nabs, neutralizing antibodies; PBMCs, peripheral blood mononuclear cells; PharmGKB, The Pharmacogenomics Knowledgebase; PPMS, primary progressive multiple sclerosis; PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses; RRMS, relapsing-remitting multiple sclerosis; SPMS, secondary progressive MS.
1. Sawcer S, Franklin RJM, Ban M. Multiple sclerosis genetics. Lancet Neurol. (2014) 13:700–9. doi: 10.1016/S1474-4422(14)70041-9
2. Belbasis L, Bellou V, Evangelou E, Ioannidis JPA, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol. (2015) 14:263–73. doi: 10.1016/S1474-4422(14)70267-4
3. Carlson RJ, Doucette JR, Nazarali AJ. Current developments in pharmacogenomics of multiple sclerosis. Cell Mol Neurobiol. (2014) 34:1081–5. doi: 10.1007/s10571-014-0095-0
4. Fusco C, Andreone V, Coppola G, Luongo V, Guerini F, Pace E, et al. HLA-DRB1*1501 and response to copolymer-1 therapy in relapsing-remitting multiple sclerosis. Neurology (2001) 57:1976–9. doi: 10.1212/WNL.57.11.1976
5. Río J, Nos C, Tintoré M, Téllez N, Galán I, Pelayo R, et al. Defining the response to interferon-β in relapsing-remitting multiple sclerosis patients. Ann Neurol. (2006) 59:344–52. doi: 10.1002/ana.20740
6. Kulakova OG, Tsareva EY, Lvovs D, Favorov AV, Boyko AN, Favorova OO. Comparative pharmacogenetics of multiple sclerosis: IFN-beta versus glatiramer acetate. Pharmacogenomics (2014) 15:679–85. doi: 10.2217/pgs.14.26
7. Esposito F, Sorosina M, Ottoboni L, Lim ET, Replogle JM, Raj T, et al. A pharmacogenetic study implicates SLC9a9 in multiple sclerosis disease activity. Ann Neurol. (2015) 78:115-27. doi: 10.1002/ana.24429
8. Tur C, Moccia M, Barkhof F, Chataway J, Sastre-Garriga J, Thompson AJ, et al. Assessing treatment outcomes in multiple sclerosis trials and in the clinical setting. Nat Rev Neurol. (2018) 14:75–93. doi: 10.1038/nrneurol.2017.171
9. Ransohoff R, Hafler D, Lucchinetti C. Multiple sclerosis - a quiet revolution. Nat Rev Neurol. (2015) 11:134–42. doi: 10.1038/nrneurol.2015.14
10. Tsareva E, Kulakova O, Boyko A, Favorova O. Pharmacogenetics of multiple sclerosis: personalized therapy with immunomodulatory drugs. Pharmacogenet Genomics (2016) 26:103–15. doi: 10.1097/FPC.0000000000000194
11. Comabella M, Craig DW, Morcillo-Suarez C, Rio J, Navarro A, Fernandez M, et al. Genome-wide scan of 500 000 single-nucleotide polymorphisms among responders and nonresponders to interferon beta therapy in multiple sclerosis. Arch Neurol. (2009) 66:972–8. doi: 10.1001/archneurol.2009.150
12. Duquette P, Giacomini PS, Bhan V, Hohol M, Schecter R. Balancing early aggression against risk of progression in multiple sclerosis. Can J Neurol Sci. (2015) 43:33–43. doi: 10.1017/cjn.2015.302
13. Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, et al. PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. (2017) 45:D183–9. doi: 10.1093/nar/gkw1138
14. McGraw CA, Lublin FD. Interferon beta and glatiramer acetate therapy. Neurotherapeutics (2013) 10:2–18. doi: 10.1007/s13311-012-0163-4
15. Dhib-Jalbut S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology (2002) 58:S3–9. doi: 10.1212/WNL.58.8_suppl_4.S3
16. Villoslada P, Barcellos LF, Rio J, Begovich AB, Tintore M, Sastre-Garriga J, et al. The HLA locus and multiple sclerosis in Spain. Role in disease susceptibility, clinical course and response to interferon-beta. J Neuroimmunol. (2002) 130:194–201. doi: 10.1016/S0165-5728(02)00215-1
17. Fernández O, Fernández V, Mayorga C, Guerrero M, León A, Tamayo JA, et al. HLA class II and response to interferon-beta in multiple sclerosis. Acta Neurol Scand (2005) 112:391–4. doi: 10.1111/j.1600-0404.2005.00415.x
18. Comabella M, Fernández-Arquero M, Río J, Guinea A, Fernández M, Cenit MC, et al. HLA class I and II alleles and response to treatment with interferon-beta in relapsing-remitting multiple sclerosis. J Neuroimmunol. (2009) 210:116–9. doi: 10.1016/j.jneuroim.2009.01.012
19. Gross R, Healy BC, Cepok S, Chitnis T, Khoury SJ, Hemmer B, et al. Population structure and HLA DRB1*1501 in the response of subjects with multiple sclerosis to first-line treatments. J Neuroimmunol. (2011) 233:168–74. doi: 10.1016/j.jneuroim.2010.10.038
20. Samadzadeh S, Tabibian E, Sabokbar T, Shakoori A, Dehgolan SR, Armaki SA, et al. HLA-DRB1 does not have a role in clinical response to interferon-beta among Iranian multiple sclerosis patients. J Neurol Sci. (2015) 352:37–40. doi: 10.1016/j.jns.2015.03.004
21. Sriram U, Barcellos LF, Villoslada P, Rio J, Baranzini SE, Caillier S, et al. Pharmacogenomic analysis of interferon receptor polymorphisms in multiple sclerosis. Genes Immun. (2003) 4:147–52. doi: 10.1038/sj.gene.6363946
22. Leyva L, Fernández O, Fedetz M, Blanco E, Fernández VE, Oliver B, et al. IFNAR1 and IFNAR2 polymorphisms confer susceptibility to multiple sclerosis but not to interferon-beta treatment response. J Neuroimmunol. (2005) 163:165–71. doi: 10.1016/j.jneuroim.2005.02.010
23. Carmona O, Masuet C, Alía P, Moral E, Alonso-Magdalena L, Casado V, et al. Apolipoprotein alleles and the response to interferon-β-1b in multiple sclerosis. Eur Neurol. (2011) 65:132–7. doi: 10.1159/000323982
24. Guerrero AL, Tejero MA, Gutiérrez F, Martín-Polo J, Iglesias F, Laherran E, et al. Influence of APOE gene polymorphisms on interferon-beta treatment response in multiple sclerosis. Neurol (English Ed (2011) 26:137–142. doi: 10.1016/j.nrl.2010.06.003
25. Vandenbroeck K, Alloza I, Swaminathan B, Antigüedad A, Otaegui D, Olascoaga J, et al. Validation of IRF5 as multiple sclerosis risk gene: putative role in interferon beta therapy and human herpes virus-6 infection. Genes Immun. (2011) 12:40–45. doi: 10.1038/gene.2010.46
26. Sellebjerg F, Søndergaard HB, Koch-Henriksen N, Sørensen PS, Oturai AB. Prediction of response to interferon therapy in multiple sclerosis. Acta Neurol Scand. (2014) 130:268–75. doi: 10.1111/ane.12269
27. Malhotra S, Sorosina M, Río J, Peroni S, Midaglia L, Villar LM, et al. NLRP3 polymorphisms and response to interferon-beta in multiple sclerosis patients. Mult Scler J. (2018) 24:1507–10. doi: 10.1177/1352458517739137
28. Malhotra S, Rio J, Urcelay E, Nurtdinov R, Bustamante MF, Fernandez O, et al. NLRP3 inflammasome is associated with the response to IFN-beta in patients with multiple sclerosis. Brain (2015) 138:644–52. doi: 10.1093/brain/awu388
29. Weinstock-Guttman B, Tamaño-Blanco M, Bhasi K, Zivadinov R, Ramanathan M. Pharmacogenetics of MXA SNPs in interferon-β treated multiple sclerosis patients. J Neuroimmunol. (2007) 182:236–9. doi: 10.1016/j.jneuroim.2006.10.011
30. Cénit MDC, Blanco-Kelly F, de las Heras V, Bartolomé M, de la Concha EG, Urcelay E, et al. Glypican 5 is an interferon-beta response gene: a replication study. Mult Scler (2009) 15:913–7. doi: 10.1177/1352458509106509
31. Malhotra S, Morcillo-Suárez C, Brassat D, Goertsches R, Lechner-Scott J, Urcelay E, et al. IL28B polymorphisms are not associated with the response to interferon-beta in multiple sclerosis. J Neuroimmunol. (2011) 239:101–4. doi: 10.1016/j.jneuroim.2011.08.004
32. Cunningham S, Graham C, Hutchinson M, Droogan A, O'Rourke K, Patterson C, et al. Pharmacogenomics of responsiveness to interferon IFN-β treatment in multiple sclerosis: a genetic screen of 100 type I interferon-inducible genes. Clin Pharmacol Ther. (2005) 78:635-46. doi: 10.1016/j.clpt.2005.08.018
33. Sayad A, Ghafouri-Fard S, Omrani MD, Noroozi R, Taheri M. Myxovirus resistance protein A (MxA) polymorphism is associated with IFNβ response in Iranian multiple sclerosis patients. Neurol Sci. (2017) 38:1093–9. doi: 10.1007/s10072-017-2935-4
34. Kulakova OG, Tsareva EY, Boyko AN, Shchur SG, Gusev EI, Lvovs D, et al. Allelic combinations of immune-response genes as possible composite markers of IFN-β efficacy in multiple sclerosis patients. Pharmacogenomics (2012) 13:1689–700. doi: 10.2217/pgs.12.161
35. Karam RA, Rezk NA, Amer MM, Fathy HA. Immune response genes receptors expression and polymorphisms in relation to multiple sclerosis susceptibility and response to INF-β therapy. IUBMB Life (2016) 68:727–34. doi: 10.1002/iub.1530
36. Vosslamber S, van der Voort LF, van den Elskamp IJ, Heijmans R, Aubin C, Uitdehaag BMJ, et al. Interferon regulatory factor 5 gene variants and pharmacological and clinical outcome of Interferonβ therapy in multiple sclerosis. Genes Immun. (2011) 12:466–72. doi: 10.1038/gene.2011.18
37. Byun E, Caillier SJ, Montalban X, Villoslada P, Fernández O, Brassat D, et al. Genome-wide pharmacogenomic analysis of the response to interferon beta therapy in multiple sclerosis. Arch Neurol. (2008) 65:337–44. doi: 10.1001/archneurol.2008.47
38. Bustamante MF, Morcillo-Suarez C, Malhotra S, Rio J, Leyva L, Fernandez O, et al. Pharmacogenomic study in patients with multiple sclerosis: responders and nonresponders to IFN-β. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e154. doi: 10.1212/NXI.0000000000000154
39. O'Doherty C, Favorov A, Heggarty S, Graham C, Favorova O, Ochs M, et al. Genetic polymorphisms, their allele combinations and IFN-beta treatment response in Irish multiple sclerosis patients. Pharmacogenomics (2009) 10:1177–86. doi: 10.2217/pgs.09.41
40. Wergeland S, Beiske A, Nyland H, Hovdal H, Jensen D, Larsen JP, et al. IL-10 promoter haplotype influence on interferon treatment response in multiple sclerosis. Eur J Neurol. (2005) 12:171–175. doi: 10.1111/j.1468-1331.2004.01102.x
41. Ristić S, Starčević Cizmarević N, Lavtar P, Lovrečić L, Perković O, Sepčić J, et al. Angiotensin-converting enzyme insertion/deletion gene polymorphism and interferon-β treatment response in multiple sclerosis patients. Pharmacogenet Genomics (2017) 27:232–5. doi: 10.1097/FPC.0000000000000283
42. Torbati S, Karami F, Ghaffarpour M, Zamani M. Association of CD58 polymorphism with multiple sclerosis and response to interferon ß therapy in a subset of Iranian population. Cell J. 16:506–13. doi: 10.22074/cellj.2015.505
43. Malhotra S, Morcillo-Suárez C, Nurtdinov R, Rio J, Sarro E, Moreno M, et al. Roles of the ubiquitin peptidase USP18 in multiple sclerosis and the response to interferon-β treatment. Eur J Neurol. (2013) 20:1390–7. doi: 10.1111/ene.12193
44. López-Gómez C, Pino-Ángeles A, Órpez-Zafra T, Pinto-Medel MJ, Oliver-Martos B, Ortega-Pinazo J, et al. Candidate gene study of TRAIL and TRAIL receptors: association with response to interferon beta therapy in multiple sclerosis patients. PLoS O NE(2013) 8:e62540. doi: 10.1371/journal.pone.0062540
45. Martínez A, De Las Heras V, Mas Fontao A, Bartolomé M, De La Concha EG, Urcelay E, et al. An IFNG polymorphism is associated with interferon-beta response in Spanish MS patients. J Neuroimmunol. (2006) 173:196–9. doi: 10.1016/j.jneuroim.2005.12.002
46. Alvarez-Lafuente R, Blanco-Kelly F, Garcia-Montojo M, Martínez A, De Las Heras V, Dominguez-Mozo MI, et al. CD46 in a Spanish cohort of multiple sclerosis patients: genetics, mRNA expression and response to interferon-beta treatment. Mult Scler. (2011) 17:513–20. doi: 10.1177/1352458510393263
47. Mazdeh M, Taheri M, Sayad A, Bahram S, Omrani MD, Movafagh A, et al. HLA genes as modifiers of response to IFN-beta-1a therapy in relapsing-remitting multiple sclerosis. Pharmacogenomics (2016) 17:489–98. doi: 10.2217/pgs.16.2
48. Weber F, Cepok S, Wolf C, Berthele A, Uhr M, Bettecken T, et al. Single-nucleotide polymorphisms in HLA- and non-HLA genes associated with the development of antibodies to interferon-β therapy in multiple sclerosis patients. Pharmacogenomics J. (2012) 12:238–45. doi: 10.1038/tpj.2011.14
49. Mahurkar S, Moldovan M, Suppiah V, Sorosina M, Clarelli F, Liberatore G, et al. Response to interferon-beta treatment in multiple sclerosis patients: a genome-wide association study. Pharmacogenomics J. (2017) 17:312–8. doi: 10.1038/tpj.2016.20
50. Clarelli F, Liberatore G, Sorosina M, Osiceanu AM, Esposito F, Mascia E, et al. Pharmacogenetic study of long-term response to interferon-beta treatment in multiple sclerosis. Pharmacogenomics J. (2017) 17:84–91. doi: 10.1038/tpj.2015.85
51. Hasson T, Kolitz S, Towfic F, Laifenfeld D, Bakshi S, Beriozkin O, et al. Functional effects of the antigen glatiramer acetate are complex and tightly associated with its composition. J Neuroimmunol. (2016) 290:84–95. doi: 10.1016/j.jneuroim.2015.11.020
52. Dhib-Jalbut S, Valenzuela RM, Ito K, Kaufman M, Ann Picone M, Buyske S. HLA DR and DQ alleles and haplotypes associated with clinical response to glatiramer acetate in multiple sclerosis. Mult Scler Relat Disord. (2013) 2:340–8. doi: 10.1016/j.msard.2013.02.005
53. Kulakova O, Bashinskaya V, Kiselev I, Baulina N, Tsareva E, Nikolaev R, et al. Pharmacogenetics of glatiramer acetate therapy for multiple sclerosis: the impact of genome-wide association studies identified disease risk loci. Pharmacogenomics (2017) 18:1563–74. doi: 10.2217/pgs-2017-0058
54. Grossman I, Avidan N, Singer C, Goldstaub D, Hayardeny L, Eyal E, et al. Pharmacogenetics of glatiramer acetate therapy for multiple sclerosis reveals drug-response markers. Pharmacogenet Genomics (2007) 17:657–66. doi: 10.1097/FPC.0b013e3281299169
55. Tsareva EY, Kulakova OG, Boyko AN, Shchur SG, Lvovs D, Favorov AV, et al. Allelic combinations of immune-response genes associated with glatiramer acetate treatment response in Russian multiple sclerosis patients. Pharmacogenomics (2012) 13:43–53. doi: 10.2217/pgs.11.136
56. Ross CJ, Towfic F, Shankar J, Laifenfeld D, Thoma M, Davis M, et al. A pharmacogenetic signature of high response to Copaxone in late-phase clinical-trial cohorts of multiple sclerosis. Genome Med. (2017) 9:1–15. doi: 10.1186/s13073-017-0436-y
57. Auricchio F, Scavone C, Cimmaruta D, Di Mauro G, Capuano A, Sportiello L, et al. Drugs approved for the treatment of multiple sclerosis: review of their safety profile. Expert Opin Drug Saf. (2017) 16:1359–71. doi: 10.1080/14740338.2017.1388371
58. Cotte S, Von Ahsen N, Kruse N, Huber B, Winkelmann A, Zettl UK, et al. ABC-transporter gene-polymorphisms are potential pharmacogenetic markers for mitoxantrone response in multiple sclerosis. Brain (2009) 132:2517–30. doi: 10.1093/brain/awp164
59. Grey Née Cotte S, Salmen Née Stroet A, von Ahsen N, Starck M, Winkelmann A, Zettl UK, et al. Lack of efficacy of mitoxantrone in primary progressive Multiple Sclerosis irrespective of pharmacogenetic factors: a multi-center, retrospective analysis. J Neuroimmunol. (2015) 278:277–9. doi: 10.1016/j.jneuroim.2014.11.017
60. Alexoudi A, Zachaki S, Stavropoulou C, Gavrili S, Spiliopoulou C, Papadodima S, et al. Possible implication of GSTP1 and NQO1 polymorphisms on natalizumab response in multiple sclerosis. Ann Clin Lab Sci. (2016) 46:586–91.
61. Whirl-Carrillo M, McDonogh E, Herbet J, Gong L, Sangkuhl K, Thotn C, et al. Pharmacogenomics knowledge for personlized medicine. Clin Pharmacol Ther. (2012) 92:414–7. doi: 10.1038/clpt.2012.96
62. Bertolotto A, Deisenhammer F, Gallo P, Sölberg Sørensen Per. Immunogenicity of interferon beta: differences among products. J Neurol. (2004) 251 (Suppl. 2):II15–II24. doi: 10.1007/s00415-004-1204-7
63. Hartung HP, Freedman MS, Polman CH, Edan G, Kappos L, Miller DH, et al. Interferon β-1b-neutralizing antibodies 5 years after clinically isolated syndrome. Neurology (2011) 77:835–43. doi: 10.1212/WNL.0b013e31822c90d7
64. Sbardella E, Tomassini V, Gasperini C, Bellomi F, Cefaro LA, Morra VB, et al. Neutralizing antibodies explain the poor clinical response to Interferon beta in a small proportion of patients with Multiple Sclerosis: a retrospective study. BMC Neurol. (2009) 9:54. doi: 10.1186/1471-2377-9-54
65. Hoffmann S, Cepok S, Grummel V, Lehmann-Horn K, Hackermueller J, Stadler PF, et al. HLA-DRB1*0401 and HLA-DRB1*0408 are strongly associated with the development of antibodies against interferon-β therapy in multiple sclerosis. Am J Hum Genet. (2008) 83:219–27. doi: 10.1016/j.ajhg.2008.07.006
66. Buck D, Cepok S, Hoffmann S, Grummel V, Jochim A, Berthele A, et al. Influence of the HLA-DRB1 genotype on antibody development to interferon beta in multiple sclerosis. Arch Neurol. (2011) 68:480–7. doi: 10.1001/archneurol.2011.65
67. Núñez C, Cénit MC, Alvarez-Lafuente R, Río J, Fernández-Arquero M, Arroyo R, et al. HLA alleles as biomarkers of high-titre neutralising antibodies to interferon-β therapy in multiple sclerosis. J Med Genet. (2014) 51:395–400. doi: 10.1136/jmedgenet-2014-102348
68. Link J, Ryner ML, Fink K, Hermanrud C, Lima I, Brynedal B, et al. Human leukocyte antigen genes and interferon beta preparations influence risk of developing neutralizing anti-drug antibodies in multiple sclerosis. PLoS ONE (2014) 9:e90479. doi: 10.1371/journal.pone.0090479
69. van Baarsen LGM, Vosslamber S, Tijssen M, Baggen JMC, van der Voort LF, Killestein J, et al. Pharmacogenomics of interferon-β therapy in multiple sclerosis: baseline IFN signature determines phamacological differences between patients. PLoS ONE (2008) 3:e1927. doi: 10.1371/journal.pone.0001927
70. Comabella M, Lünemann JD, Río J, Sánchez A, López C, Julià E, et al. A type i interferon signature in monocytes is associated with poor response to interferon-β in multiple sclerosis. Brain (2009) 132:3353–65. doi: 10.1093/brain/awp228
71. Goertsches RH, Zettl UK, Hecker M. Sieving treatment biomarkers from blood gene-expression profiles: A pharmacogenomic update on two types of multiple sclerosis therapy. Pharmacogenomics (2011) 12:423–32. doi: 10.2217/pgs.10.190
72. Rudick RA, Rani MRS, Xu Y, Lee JC, Na J, Shrock J, et al. Excessive biologic response to IFNβ is associated with poor treatment response in patients with multiple sclerosis. PLoS ONE (2011) 6:e19262. doi: 10.1371/journal.pone.0019262
73. Parnell GP, Gatt PN, McKay FC, Schibeci S, Krupa M, Powell JE, et al. Ribosomal protein S6 mRNA is a biomarker upregulated in multiple sclerosis, downregulated by interferon treatment, and affected by season. Mult Scler J. (2014) 20:675–85. doi: 10.1177/1352458513507819
74. Moreno-Torres I, González-García C, Marconi M, García-Grande A, Rodríguez-Esparragoza L, Elvira V, et al. Immunophenotype and transcriptome profile of patients with multiple sclerosis treated with fingolimod: Setting up a model for prediction of response in a 2-year translational study. Front Immunol. (2018) 9:1693. doi: 10.3389/fimmu.2018.01693
75. Hecker M, Paap BK, Goertsches RH, Kandulski O, Fatum C, Koczan D, et al. Reassessment of blood gene expression markers for the prognosis of relapsing-remitting multiple sclerosis. PLoS ONE (2011) 6:e29648. doi: 10.1371/journal.pone.0029648
76. Martire S, Navone ND, Montarolo F, Perga S, Bertolotto A. A gene expression study denies the ability of 25 candidate biomarkers to predict the interferon-beta treatment response in multiple sclerosis patients. J Neuroimmunol. (2016) 292:34–9. doi: 10.1016/j.jneuroim.2016.01.010
77. Beck H, Schwarz G, Schröter CJ, Deeg M, Baier D, Stevanovic S, et al. Cathepsin S and an asparagine-specific endoprotease dominate the proteolytic processing of human myelin basic protein in vitro. Eur J Immunol. (2001) 31:3726–36. doi: 10.1002/1521-4141(200112)31:12<3726::AID-IMMU3726>3.0.CO;2-O
78. Río J, Comabella M, Montalban X. Predicting responders to therapies for multiple sclerosis. Nat Rev Neurol. (2009) 5:553–60. doi: 10.1038/nrneurol.2009.139
Keywords: Systematic review, multiple sclerosis, pharmacogenomics, treatment response, personalized treatment
Citation: Hočevar K, Ristić S and Peterlin B (2019) Pharmacogenomics of Multiple Sclerosis: A Systematic Review. Front. Neurol. 10:134. doi: 10.3389/fneur.2019.00134
Received: 13 October 2018; Accepted: 01 February 2019;
Published: 26 February 2019.
Edited by:
Hector J. Caruncho, University of Victoria, CanadaReviewed by:
Joan Guàrdia-Olmos, University of Barcelona, SpainCopyright © 2019 Hočevar, Ristić and Peterlin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Borut Peterlin, Ym9ydXQucGV0ZXJsaW5AZ3Vlc3QuYXJuZXMuc2k=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.