 Yana Yunusova
Yana Yunusova Emily K. Plowman4
Emily K. Plowman4 Jordan R. Green
Jordan R. Green Carolina Barnett
Carolina Barnett Peter Bede
Peter Bede- 1Department of Speech Language Pathology, University of Toronto, Toronto, ON, Canada
- 2Hurvitz Brain Sciences Research Program, Sunnybrook Research Institute, Toronto, ON, Canada
- 3Volcal Tract Visualization Lab, Toronto Rehabilitation Institute, University Health Network, Toronto, ON, Canada
- 4Swallowing Systems Core, Department of Speech, Language, and Hearing Sciences, University of Florida, Gainesville, FL, United States
- 5Department of Communication Sciences and Disorders, MGH Institute of Health Professions, Boston, MA, United States
- 6Speech and Hearing Biosciences and Technology Program, Harvard University, Cambridge, MA, United States
- 7Division of Neurology, Department of Medicine, University of Toronto and University Health Network, Toronto, ON, Canada
- 8Institute of Health Policy, Management and Evaluation, Dalla Lana School of Public Health, University of Toronto, Toronto, ON, Canada
- 9Computational Neuroimaging Group, Academic Unit of Neurology, Trinity College Dublin, Dublin, Ireland
Bulbar impairment represents a hallmark feature of Amyotrophic Lateral Sclerosis (ALS) that significantly impacts survival and quality of life. Speech and swallowing dysfunction are key contributors to the clinical heterogeneity of ALS and require well-timed and carefully coordinated interventions. The accurate clinical, radiological and electrophysiological assessment of bulbar dysfunction in ALS is one of the most multidisciplinary aspects of ALS care, requiring expert input from speech-language pathologists (SLPs), neurologists, otolaryngologists, augmentative alternative communication (AAC) specialists, dieticians, and electrophysiologists—each with their own evaluation strategies and assessment tools. The need to systematically evaluate the comparative advantages and drawbacks of various bulbar assessment instruments and to develop integrated assessment protocols is increasingly recognized. In this review, we provide a comprehensive appraisal of the most commonly utilized clinical tools for assessing and monitoring bulbar dysfunction in ALS based on the COnsensus-based Standards for the selection of health Measurement INstruments (COSMIN) evaluation framework. Despite a plethora of assessment tools, considerable geographical differences exist in bulbar assessment practices and individual instruments exhibit considerable limitations. The gaps identified in the literature offer unique opportunities for the optimization of existing and development of new tools both for clinical and research applications. The multicenter validation and standardization of these instruments will be essential for guideline development and best practice recommendations.
Introduction
ALS is a relentlessly progressive neurodegenerative disease with considerable clinical heterogeneity compared to other neurodegenerative conditions. Bulbar impairment (oro-motor, dysarthria and dysphagia) is a hallmark feature of the disease and has been associated with the condition since its earliest descriptions (1). While only approximately 30% of patients exhibit bulbar symptoms at onset, the majority of patients develop speech and swallowing difficulties with disease progression. Bulbar signs and symptoms play an important role in the diagnosis of ALS and pose unique management challenges. Bulbar presentation has been associated with shorter survival (2, 3), faster functional decline (4), reduced quality of life (5–7) and increased multidisciplinary support needs (8, 9). Dysarthria has been consistently associated with low mood, withdrawal from activities and social isolation (10, 11). Dysphagia in ALS may lead to weight loss, malnutrition, dehydration, aspiration pneumonia, hospitalization and reduced quality of life (12, 13). Despite these important sequelae, bulbar impairment in ALS is relatively understudied, and the research literature is sparse (14). Proxies of bulbar impairment are underrepresented among outcome measures in clinical trials (15). Validated diagnostic, monitoring and prognostic markers of bulbar dysfunction are lacking and clinical assessment practices vary considerably across various centers (16).
Assessment measures are broadly classified as “diagnostic” when their primary purpose is to confirm the diagnosis, exclude mimics, or classify individual patient according to disease-onset. Some measures have been optimized to characterize symptom severity, while other indices are primarily used to monitor longitudinal change. Depending on the primary purpose of a measure, it is subject to a specific set of requirements defined by the COnsensus-based Standards for the selection of health Measurement INstruments (COSMIN) guidelines (17). These require that, in order to reliably integrate assessment tools into clinical practice, their measurement properties need to be firmly established relative to their primary purpose. All tests need to be assessed for validity and reliability (reproducibility). Diagnostic and screening tests should also be evaluated for their detection abilities (i.e., sensitivity/specificity). Discriminative measures need to be able to detect group differences and measures proposed to track longitudinal change need to be assessed for their ability to capture progressive changes.
The objective of this paper is to provide a review of established bulbar measures in ALS from a diagnostic, screening and disease monitoring perspective. This work is not intended as an exhaustive review of all available measures of bulbar impairment in ALS but as a summary of the current state of the field and its most pressing needs.
Tools for Diagnosing and Screening for Bulbar ALS
Table 1 provides a summary of tools primarily used for the diagnosis of bulbar dysfunction in ALS highlighting their main advantages and limitations.
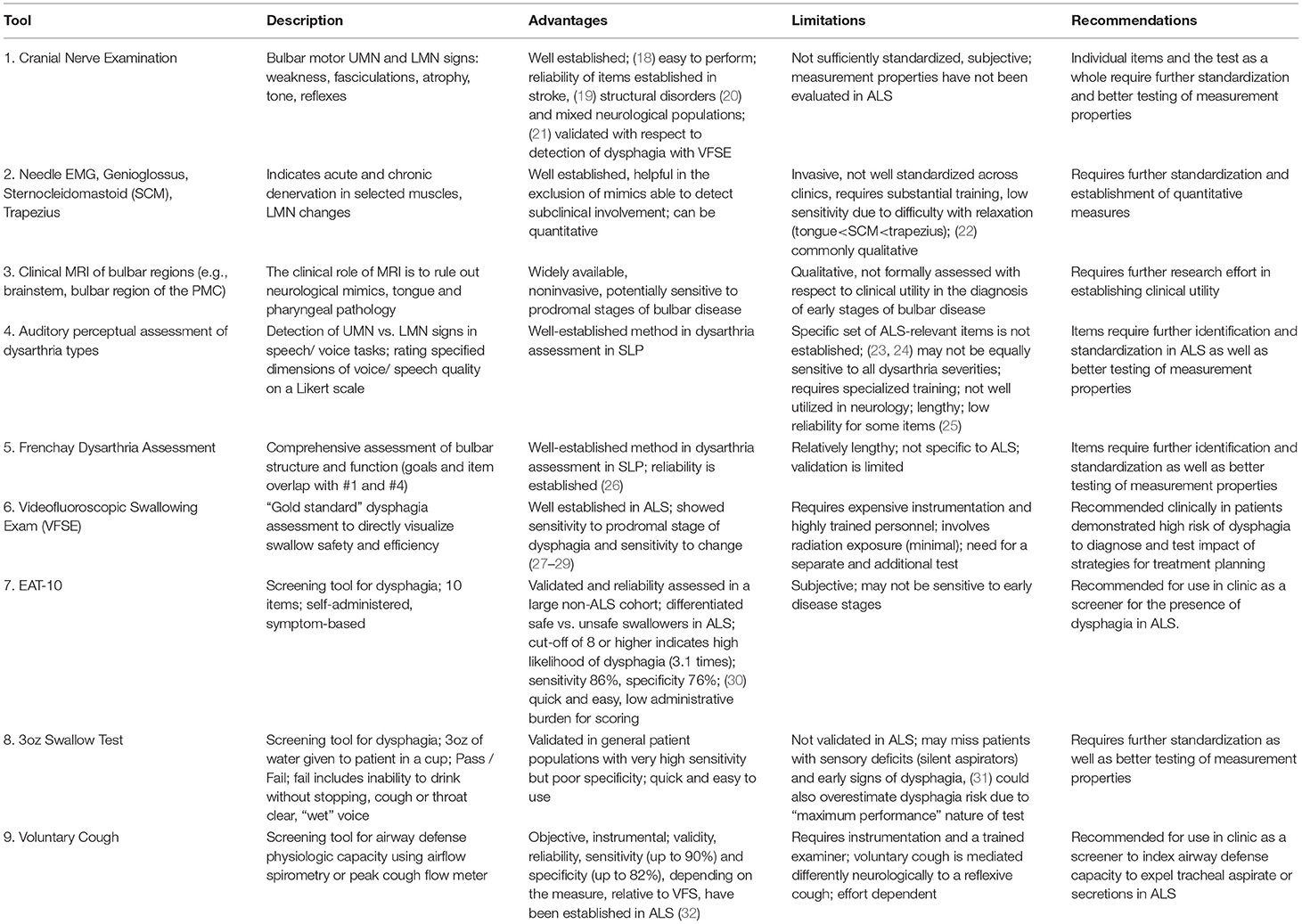
Table 1. Tools for diagnosis of bulbar signs or ALS.
Cranial Nerve Exam (CNE)
Clinical evidence of upper motor neuron (UMN) and lower motor neuron (LMN) degeneration is required for the diagnosis of ALS. With regards to bulbar impairment, clinical UMN signs include pathological reflexes (e.g., brisk jaw jerk, gag, and other facial reflexes) (18) and LMN signs encompass muscle weakness, atrophy and fasciculations in the jaw, face, tongue and palate (33). Although the clinical neurological examination remains “the best way to localize neurodegeneration in vivo and to follow the process in real time,” (34) and the reliability of CNE has been evaluated in various neurologic populations (21, 35), the quantitative psychometric profile of CNE i.e., inter and intra-later reliability, sensitivity, specificity, and responsiveness, have not been systematically evaluated in ALS to date. This represents a research priority for the standardization of assessments.
Needle EMG
The role of electromyography (EMG) in ALS is the confirmation of acute and chronic denervation. The former may be evidenced by fibrillations, positive sharp waves and fasciculation potentials, which in the tongue are not readily detectable since complete relaxation is difficult to achieve (22). Polyphasic motor unit potentials (MUPs) with prolonged duration, increased amplitude and decreased recruitment are suggestive of chronic denervation. Quantitative motor unit action potential analysis in subclinical bulbar involvement is thought to be superior to peak ratio interference pattern analysis (36). Depending on local protocols, the genioglossus is the most commonly assessed muscle (37), but the evaluation of the sternocleidomastoid (38), masseter, temporalis, frontalis (39), mentalis (40), and trapezius (22) muscles have also been proposed to resolve diagnostic uncertainty. While Motor Unit Number Estimation (MUNE) techniques (41, 42), such as MUNIX (43) have been extensively utilized to quantify motor neuron loss in the limbs, they have only been relatively recently adopted to assess the denervation of the tongue (44) and further development is required for their acceptance to clinical practice.
Clinical Neuroimaging
While brain imaging is not required to establish the diagnosis of ALS, MRI is commonly used as part of the diagnostic work-up to rule out alternative neurological conditions which may mimic ALS (45, 46). In bulbar onset patients the careful evaluation of the brain stem for structural, neoplastic, vascular, inflammatory and infiltrative processes is particularly important. Pathological processes superior to the brainstem; demyelination, neurovascular syndromes, neurosarcoidosis, leukodystrophies, malignancies, and neurodegenerative conditions may also manifest in bulbar symptoms if involving the corticobulbar tracts or the bulbar segments of the motor cortex. A number of extrapyramidal and cerebellar conditions may also present with localization-specific (ataxic, hypokinetic, hyperkinetic) dysarthria and imaging has a role to rule out gross striatal, nigral and cerebellar pathologies. The incidental identification of tongue tumors on MRI in patients with suspected ALS has also been reported (47). A number of radiological cues have been associated with ALS, such as high signal along the pyramidal tracts on T2 weighted or FLAIR imaging, low signal in the precentral gyrus on GRE/SWI, isolated motor cortex atrophy on T1W, but these qualitative visual cues are not specific to ALS and are not sensitive for diagnostic or monitoring purposes (48). Quantitative imaging studies of ALS on the other hand have successfully captured the cortical (UMN) components of bulbar dysfunction in a somatotopic distribution (49, 50) and characterized the pathological substrate of pseudobulbar affect (51, 52). With relentless methodological (53) and conceptual advances in neuroimaging (54), the establishment of multicenter data repositories (55) and the increasing availability of 7 Tesla systems (56), the anatomical underpinnings of bulbar dysfunction are likely to be characterized in further detail.
Auditory-Perceptual Dysarthria Evaluation and Frenchay Dysarthria Assessment
“The Mayo Clinic” method of dysarthria categorization involves auditory-perceptual evaluation of specific voice and speech features during a passage reading, phonation of /a/, and oral dysdiadochokinesis (DDK) with /pa, /ta/, /ka/, and /pataka/ (57–59). The identification of “harsh,” “strained,” or “strangled” voice quality, slow speaking rate and “excess and equal” stress pattern during passage reading and DDK are typically linked to UMN dysfunction and “spastic dysarthria.” “Breathy” or weak voice, hypernasality, nasal emissions, and articulatory imprecision without changes in speaking rate are classically associated with LMN dysfunction and “flaccid dysarthria.” ALS is typically characterized by mixed spastic-flaccid dysarthria presenting with articulatory imprecision, hypernasality, harshness, slow rate and prosodic abnormalities. Although the reliability of observational assessments have been repeatedly questioned (10), protocol standardization, assessor training, and reference samples are thought to improve assessment reliability (60). Despite these efforts, auditory-perceptual assessment remains surprisingly underutilized, requiring standardization of practices, psychometric evaluation and multi-center validation in ALS.
Tools like the Frenchay Dysarthria Assessment (FDA) (26) are particularly well-suited for diagnostic purposes as they can comprehensively assess both structure and function of the bulbar musculature through a combination of CNE items and the auditory-perceptual dysarthria assessment. However, FDA was not specifically developed for ALS, and the evaluation of its measurement properties in ALS is lacking. DDK, which is included in CNE, FDA and perceptual dysarthria assessments, is commonly used to track disease progression, and has shown high sensitivity but low specificity for detecting bulbar signs in the prodromal phase of bulbar ALS (61, 62). With further optimization, DDK may have a diagnostic potential, particularly if certain performance constrains are imposed or its complexity is increased (63, 64).
Dysphagia Diagnosis and Screening
Videofluoroscopic swallowing evaluation (VFSE) remains the gold standard of dysphagia assessment in most neurological conditions allowing the direct visualization of swallowing safety and efficiency i.e., aspiration and the presence of residue, respectively. In ALS however, VFSE is underutilized (16) due to a number of factors such as the presumed lack of therapeutic relevance, lack of access to equipment or perceived patient burden etc. A number of screening tools have been recently evaluated for the early identification of those at risk for dysphagia in ALS. Currently, the Eating Assessment Tool-10 (EAT-10) demonstrated good sensitivity and adequate specificity for detecting aspiration in ALS (30), while instrumental measures of airflow during voluntary cough showed excellent sensitivity and specificity to detect aspiration (32). The bedside 3oz water swallow test is also extensively utilized, but its measurement properties in ALS are still unknown. There is a general consensus among SLPs that patients who fail dysphagia screening should be further evaluated by instrumental techniques to directly visualize the swallowing process using VFSE or fiberoptic evaluation of swallowing (FEES) techniques (65). This is an important consideration given the high incidence of “silent” aspiration in this patient population. Instrumental assessments, not only confirm the diagnosis of dysphagia, but inform on swallowing safety, help to identify the specific etiology of dysphagia, and guide therapeutic strategies that can be tested during the instrumental exam by directly visualizing their impact.
Tools for Disease Monitoring—Staging and Longitudinal Tracking
Certain bulbar measures have been optimized to track the decline of bulbar function in individual patients and entire cohorts. Table 2 summarizes proposed bulbar monitoring tools in ALS.
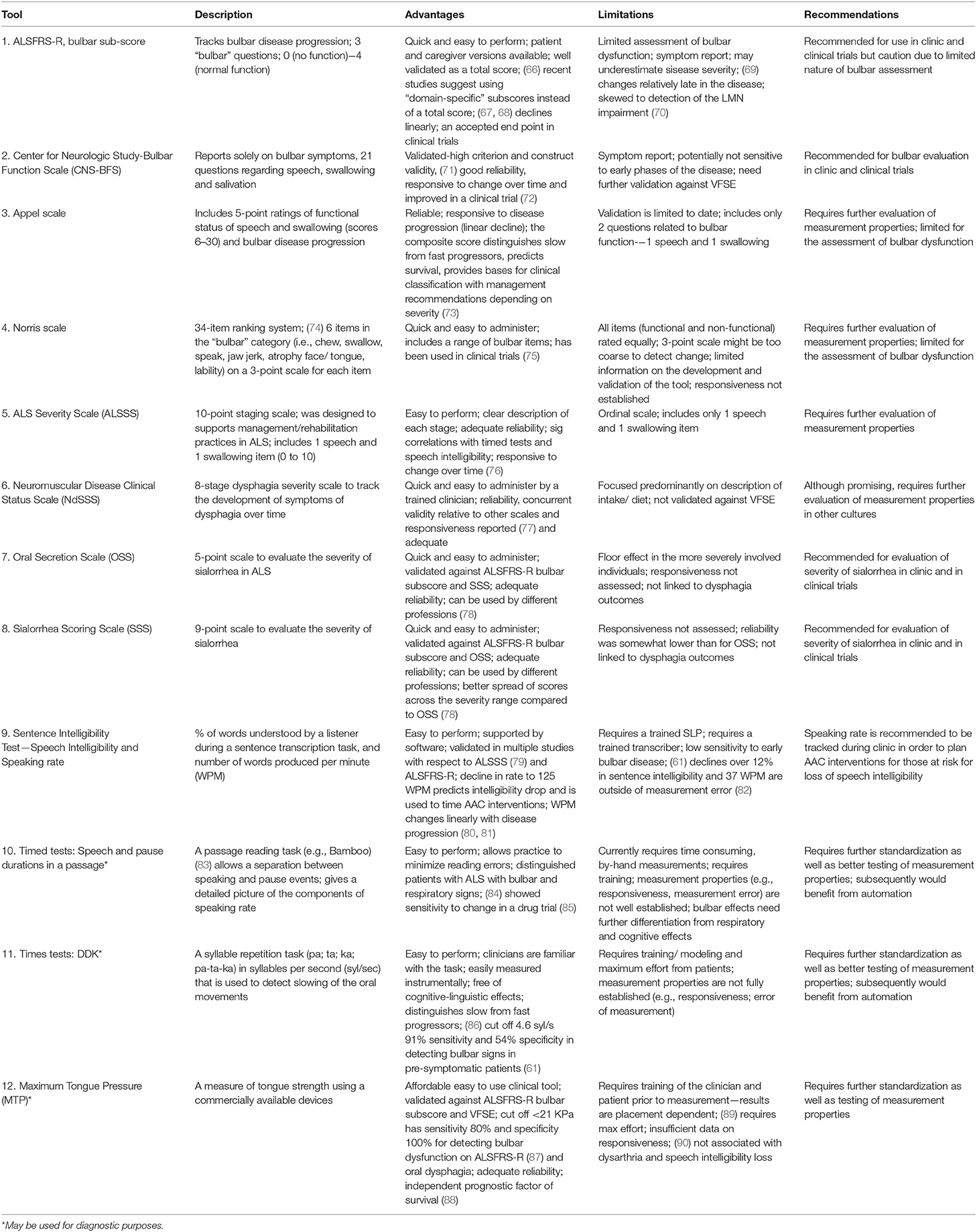
Table 2. Tools to measure bulbar dysfunction severity and disease progression.
Bulbar Monitoring (Overall)
A recent clinical practice survey of ALS care in the United States revealed that the Revised ALS—Functional Rating Scale (ALSFRS-R) bulbar sub-score, clinician or patient administered, represented the only measure routinely used to evaluate bulbar dysfunction in the clinical setting (16). It contains only 3 questions to address changes in speech, swallowing and salivation that are each merely rated on a four-point ordinal scale. While the total ALSFRS-R score is thought to have excellent reliability, the measurement properties of the individual sub-scores (e.g., bulbar) have not been specifically evaluated to date (66, 67, 91, 92). The Center for Neurologic Study-Bulbar Function Scale (CNS-BFS) is a 15-item questionnaire of bulbar involvement which has recently been validated against the ALSFRS-R and “timed” speech and swallowing tasks, and has already been successfully utilized in a clinical trial (71, 72). However, the CNS-BFS still needs to be validated against VFSE.
The Appel scale is one of the best characterized tools to track ALS-associated impairment and functional decline (73). Other clinician-administered instruments include the Norris (74), Tuffs (93), and Charing Cross (94) scales, but their original development, optimization and validation studies can be difficult to acquire and subsequently, their performance is relatively difficult to judge. The ability of these instruments to represent specific stages and their potential to track progressive bulbar impairment is largely unknown. A number of global ALS staging systems have been developed recently, such as the King's clinical staging system, the Milano-Torino (MiToS) functional staging, the Fine'til 9 (FT9) framework (95, 96), but bulbar impairment is just a small component of these instruments. Among the staging tools, the ALS Severity Scale (ALSSS) is particularly noteworthy, as it uses a 10-point scale for two bulbar functions, speech and swallowing. It was designed to guide rehabilitation efforts in ALS and, and pending formal psychometric evaluation, it may prove to be particularly useful (97).
Functional Monitoring of Dysphagia and Oral Secretion Scales
The Neuromuscular Disease Clinical Status Scale (NdSSS), which focuses solely on dysphagia, underwent one of the most rigorous psychometric evaluations to date. This tool exhibited excellent inter- and intra- rater reliability and correlated well with the functional oral intake scale (77). It has not been validated against VFSE yet, and given the potential for considerable geographical differences in oral intake, it is unclear how this tool may be validated around the world. While there are several tools to assess sialorrhea in ALS, such as the Oral Secretion Scale (OSS) and Sialorrhea Scoring Scale (SSS) available (78), these also need comprehensive psychometric evaluation and validation.
Functional Monitoring of Dysarthria
“Speech intelligibility” refers to the degree to which a speaker is understood by a listener, and “speaking rate” refers to speaking speed. Although both of these measures can be assessed on a 5 or 7-point Likert scale (49), the Sentence Intelligibility Test (SIT) is often preferred by SLPs, as it provides a more fine-grained estimate of speech intelligibility (i.e., percent of words transcribed correctly) and speaking rate (i.e., number of words produced per minute) (98). Speech intelligibility is considered abnormal when it falls below 97%, and speaking rate is considered abnormal below 160 words-per-minute (WPM) (99, 100). Speech intelligibility is a general indicator of the severity of dysarthria and it declines relatively late in the course of the disease (101). Speaking rate typically declines prior to significant changes in speech intelligibility, and it changes more linearly with symptom duration than speech intelligibility. Therefore, speaking rate is particularly useful in monitoring bulbar impairment longitudinally (102, 103). A speaking rate of 125 WPM or less is the recommended cut off for to trigger referral to the augmentative and alternative communication services (99).
Digital speech recordings and automated analyses can provide new opportunities for in-depth, observer-independent evaluations, especially during a passage reading and syllable repetition (DDK) tasks. In passage reading tasks, such as the Bamboo Passage, which has been specifically developed to support automatic analyses, certain phrases are semi-automatically identified, and speech duration and pause intervals can be accurately quantified (83). The measures derived from this analysis e.g., percentage pause time, mean phrase duration etc. have been identified to be sensitive to the prodromal stages of bulbar dysfunction (61) and also showed to detect response to pharmaceutical interventions such as dextromethorphan/quinidine (Nuedexta) therapy (85). A recent longitudinal study suggested that the main advantage of the DDK tasks may be in their ability to reliably distinguish slow- and fast-progressors (86).
Physiological Monitoring
Muscle strength testing in ALS has been initially performed using force transducers (strain gauges) (104, 105) and later with pressure bulbs via the Iowa Oral Performance Instrument (IOPI) (IOPI Medical LLC) or TPM-01 (JMS, Hiroshima). Lingual pressure testing using the IOPI revealed adequate reliability of a maximum tongue pressure estimate (MTP, or maximum anterior isometric pressure, MAIP) but not for the measure of endurance (89). Only one study assessed longitudinal changes in MTP in ALS to date (90) and reported its decline in patients with bulbar onset within 3 months and for those with spinal onset within 6 months. Tongue strength has also been shown to be an independent predictor of survival (88); however, formal psychometric evaluation is awaited to determine the MTP's utility to measure progressive changes over time.
Discussion
In order to firmly establish the clinical utility of specific bulbar instruments and their potential as outcome measures in clinical trials, their measurement properties need to be comprehensively characterized. Among the diagnostic dysphagia instruments, screening tools, such as EAT-10 and voluntary cough (30, 32) have been well evaluated. Among speech measures, only DDK rate came close to demonstrating diagnostic utility (61). The remaining tools require extensive evaluation with regards to their diagnostic accuracy. While a large number of novel assessment tools have been proposed to track the progression of bulbar impairment, only the ALSFRS-R, the CNS-BFS and some bulbar staging systems (e.g., NdSSS, OSS, SSS) meet at least basic measurement requirements. Most existing disease monitoring tools lack the ability to capture subtle progressive changes, which is indispensable for disease tracking tools. Robust systematic psychometric evaluation is needed to improve the currently available clinical, academic and pharmacological-trial assessment tools.
Despite the gaps in the current literature and the limitations of current clinical trial designs, we are likely to witness considerable advances in standardized bulbar assessments and the emergence of purpose-designed, disease-specific, well-validated bulbar assessment tools. Emerging technologies such as quantitative neuroimaging, muscle ultrasound, electrical impedance myography (EIM), high-resolution manometry, videomanofluoroscopy, and speech acoustic monitoring are likely to soon complement our armamentarium of clinical tools. A number of promising imaging techniques have already been utilized to characterize the pathological substrate of bulbar impairment in ALS including diffusion tensor imaging (106, 107), cortical thickness measurements (50, 108, 109), morphometry-type analyses (49, 110), magnetization transfer ratio imaging (106), MR spectroscopy (111), MRI intensitometry (112), and task-based functional MRI (113, 114). Despite these advances, MRI-derived metrics remain underutilized in the clinical setting and as outcome measures in pharmacological trials. This is in sharp contrast with clinical trials in Multiple Sclerosis, where MRI plays an established role as a key outcome measure in phase III clinical trials (115). Muscle ultrasound may capture tongue fasciculations in the absence of fasciculation potentials on EMG and the combination of ultrasound and EMG may help the detection of early denervation (116). Likewise, EIM shows promise in detecting changes in the structural composition of the tongue in ALS and may evolve into an important tool to detect early bulbar involvement (117, 118). High-resolution manometry and videomanofluoroscopy may provide unique insights into the dynamics of bolus movement and swallowing pressures enabling early detection of bulbar dysfunction and thus, timely interventions (119, 120). Acoustic analysis of speech has been proposed as a means for the objective assessment of bulbar impairment for over two decades, but until recently extracting these measure has been extremely time consuming. Recent developments in automatic audio and video analysis methods and smart phone technologies make speech analysis technologically feasible, enabling observer-independent multiparametric analyses (121–123). These emerging methodologies will need careful development, optimization and evaluation according to established methodological guidelines (e.g., COSMIN framework).
Conclusions
Recent advances in neuroimaging, development of staging systems, patient-reported outcome measures and the emergence of novel instrumental speech and swallowing assessment techniques promise novel insights into bulbar dysfunction in ALS. However, in order for these methods to be integrated into routine clinical practice and pharmacological trials, they have to be rigorously evaluated with respect to their measurement properties, diagnostic performance and longitudinal tracking abilities. The establishment of large international collaborations and relentless biomarker research efforts give cause for optimism for the development of validated bulbar assessments, which in turn will contribute to best practice recommendations, enable well-timed clinical interventions and facilitate accurate patient stratification in clinical trials.
Author Contributions
YY and PB reviewed references, drafted the manuscript, and generated the tables. EP provided expert opinion on dysphagia assessments and edited the drafts of the manuscript. JG provided expert opinion on dysarthria assessments and edited the drafts of the manuscript. CB provided expert opinion on measurement development and edited the drafts of the manuscript.
Funding
This work was supported by NIH R01DC01729 grant. Peter Bede is supported by the Health Research Board (HRB–Ireland; HRB EIA-2017-019), the Irish Institute of Clinical Neuroscience IICN–Novartis Ireland Research Grant, the Iris O'Brien Foundation, and the Research Motor Neuron (RMN-Ireland) Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Nicholas Wasylyk for his help with literature search and data extraction.
Abbreviations
AAC, augmentative alternative communication; ALS, Amyotrophic Lateral Sclerosis; ALSFRS-R, Amyotrophic Lateral Sclerosis—Functional Rating Scale—Revised; ALSSS, ALS Severity Scale; CNS-BFS, Center for Neurologic Study-Bulbar Function Scale; COSMIN, COnsensus-based Standards for the selection of health Measurement Instruments; CNE, Cranial nerve exam; DDK, dysdiadochokinesis; EAT-10, Eating Assessment Tool-10; EIM, electrical impedance myography; EMG, electromyography; FLAIR, Fluid-attenuated inversion recovery; FDA, Frenchay Dysarthria Assessment; FEES, fiberoptic evaluation of swallowing; FSE, Videofluoroscopic swallowing evaluation; FT9, Fine'til 9; GRE/SWI, gradient recalled echo/susceptibility weighted imaging; IOPI, Iowa Oral Performance Instrument; KPa, kilopascal; LMN, lower motor neuron; MAIP, maximum anterior isometric pressure; MiToS, Milano-Torino staging; MR, magnetic resonance; MRI, magnetic resonance imaging; MUNE, Motor Unit Number Estimation; MTP, maximum tongue pressure; MUNIX, Motor unit number index; MUPs, motor unit potentials; NdSSS, Neuromuscular Disease Clinical Status Scale; OSS, Oral Secretion Scale; SCM, sternocleidomastoid; SLPs, speech-language pathologists; SSS, Sialorrhea Scoring Scale; SIT, Sentence Intelligibility Test; syl/sec, syllables per second; T1 W, T1 weighted; UMN, upper motor neuron; WPM, words-per-minute.
References
1. Clarke JL, Jackson JH. On a case of muscular atrophy, with disease of the spinal cord and medulla oblongata. Med Chirurg Transac. (1867) 50:489–98.1. doi: 10.1177/095952876705000122
2. Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. (2009) 10:310–23. doi: 10.3109/17482960802566824
3. Elamin M, Bede P, Montuschi A, Pender N, Chio A, Hardiman O. Predicting prognosis in amyotrophic lateral sclerosis: a simple algorithm. J Neurol. (2015) 262:1447–54. doi: 10.1007/s00415-015-7731-6
4. Daghlas I, Lever TE, Leary E. A retrospective investigation of the relationship between baseline covariates and rate of ALSFRS-R decline in ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:206–11. doi: 10.1080/21678421.2017.1418001
5. Hecht M, Hillemacher T, Grasel E, Tigges S, Winterholler M, Heuss D, et al. Subjective experience and coping in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. (2002) 3:225–31. doi: 10.1080/146608202760839009
6. Mitsumoto H, Del Bene M. Improving the quality of life for people with ALS: the challenge ahead. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:329–36. doi: 10.1080/146608200300079464
7. Bach JR. Amyotrophic lateral sclerosis. Communication status and survival with ventilatory support. Am J Phys Med Rehabil. (1993) 72:343–9. doi: 10.1097/00002060-199312000-00002
8. Rooney J, Byrne S, Heverin M, Tobin K, Dick A, Donaghy C, et al. A multidisciplinary clinic approach improves survival in ALS: a comparative study of ALS in Ireland and Northern Ireland. J Neurol Neurosurg Psychiatry (2014) 86:496–501. doi: 10.1136/jnnp-2014-309601
9. Van den Berg JP, Kalmijn S, Lindeman E, Veldink JH, de Visser M, Van der Graaff MM, et al. Multidisciplinary ALS care improves quality of life in patients with ALS. Neurology (2005) 65:1264–7. doi: 10.1212/01.wnl.0000180717.29273.12
10. Tomik B, Guiloff RJ. Dysarthria in amyotrophic lateral sclerosis: a review. Amyotroph Lateral Scler. (2010) 11:4–15. doi: 10.3109/17482960802379004
11. Watts CR, Vanryckeghem M. Laryngeal dysfunction in Amyotrophic Lateral Sclerosis: a review and case report. BMC Ear Nose Throat Disord. (2001) 1:1. doi: 10.1186/1472-6815-1-1
12. Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers (2017) 3:17071. doi: 10.1038/nrdp.2017.72
13. Tabor L, Gaziano J, Watts S, Robison R, Plowman EK. Defining swallowing-related quality of life profiles in individuals with amyotrophic lateral sclerosis. Dysphagia (2016) 31:376–82. doi: 10.1007/s00455-015-9686-2
14. Waito AA, Valenzano TJ, Peladeau-Pigeon M, Steele CM. Trends in research literature describing dysphagia in motor neuron diseases (MND): a scoping review. Dysphagia (2017) 32:734–47. doi: 10.1007/s00455-017-9819-x
15. Miller RG, Munsat TL, Swash M, Brooks BR. Consensus guidelines for the design and implementation of clinical trials in ALS. World Federation of Neurology committee on Research. J Neurol Sci. (1999) 169:2–12. doi: 10.1016/S0022-510X(99)00209-9
16. Plowman EK, Tabor LC, Wymer J, Pattee G. The evaluation of bulbar dysfunction in amyotrophic lateral sclerosis: survey of clinical practice patterns in the United States. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:351–7. doi: 10.1080/21678421.2017.1313868
17. Mokkink LB, Terwee CB, Patrick DL, Alonso J, Stratford PW, Knol DL, et al. The COSMIN checklist for assessing the methodological quality of studies on measurement properties of health status measurement instruments: an international Delphi study. Q Life Res. (2010) 19:539–49. doi: 10.1007/s11136-010-9606-8
18. Tremolizzo L, Susani E, Lunetta C, Corbo M, Ferrarese C, Appollonio I. Primitive reflexes in amyotrophic lateral sclerosis: prevalence and correlates. J Neurol. (2014) 261:1196–202. doi: 10.1007/s00415-014-7342-7
19. McCullough GH, Wertz RT, Rosenbek JC, Mills RH, Ross KB, Ashford JR. Inter- and intrajudge reliability of a clinical examination of swallowing in adults. Dysphagia (2000) 15:58–67. doi: 10.1007/s004550010002
20. Chow W, Brandt M, Dworschak-Stokan A, Doyle P, Matic D, Husein M. Validation of the mirror-fogging test as a screening tool for velopharyngeal insufficiency. Open Otorhinolaryngol J. (2015) 8:15–21. doi: 10.2174/1874428101508010015
21. Koch I, Ferrazzi A, Busatto C, Ventura L, Palmer K. Cranial nerve examination for neurogenic dysphagia patients. Otolaryngology (2017) 7:4. doi: 10.4172/2161-119X.1000319
22. Sonoo M, Kuwabara S, Shimizu T, Komori T, Hirashima F, Inaba A, et al. Utility of trapezius EMG for diagnosis of amyotrophic lateral sclerosis. Muscle Nerve (2009) 39:63–70. doi: 10.1002/mus.21196
23. Van der Graaff M, Kuiper T, Zwinderman A, Van de Warrenburg B, Poels P, Offeringa A, et al. Clinical identification of dysarthria types among neurologists, residents in neurology and speech therapists. Eur Neurol. (2009) 61:295–300. doi: 10.1159/000206855
24. Kearns KP, Simmons NN. Interobserver reliability and perceptual ratings: More than meets the ear. J Speech Lang Hear Res. (1988) 31:131–6. doi: 10.1044/jshr.3101.131
25. Bunton K, Kent RD, Duffy JR, Rosenbek JC, Kent JF. Listener agreement for auditory-perceptual ratings of dysarthria. J Speech Lang Hear Res. (2007) 50:1481–95. doi: 10.1044/1092-4388(2007/102)
26. Enderby P. Frenchay dysarthria assessment. Br J Disord Commun. (1980) 15:165–73. doi: 10.3109/13682828009112541
27. Briani C, Marcon M, Ermani M, Costantini M, Bottin R, Iurilli V, et al. Radiological evidence of subclinical dysphagia in motor neuron disease. J Neurol. (1998) 245:211–6. doi: 10.1007/s004150050207
28. Wright R, Jordan C. Videofluoroscopic evaluation of dysphagia in motor neurone disease with modified barium swallow. Palliat Med. (1997) 11:44–8. doi: 10.1177/026921639701100105
29. Higo R, Tayama N, Nito T. Longitudinal analysis of progression of dysphagia in amyotrophic lateral sclerosis. Auris Nasus Larynx (2004) 31:247–54. doi: 10.1016/j.anl.2004.05.009
30. Plowman EK, Tabor LC, Robison R, Gaziano J, Dion C, Watts SA, et al. Discriminant ability of the Eating Assessment Tool-10 to detect aspiration in individuals with amyotrophic lateral sclerosis. Neurogastroenterol Motility (2016) 28:85–90. doi: 10.1111/nmo.12700
31. Mari F, Matei M, Ceravolo MG, Pisani A, Montesi A, Provinciali L. Predictive value of clinical indices in detecting aspiration in patients with neurological disorders. J Neurol Neurosurg Psychiatry (1997) 63:456–60. doi: 10.1136/jnnp.63.4.456
32. Plowman EK, Watts SA, Robison R, Tabor L, Dion C, Gaziano J, et al. Voluntary cough airflow differentiates safe versus unsafe swallowing in amyotrophic lateral sclerosis. Dysphagia (2016) 31:383–90. doi: 10.1007/s00455-015-9687-1
33. Brooks BR, Miller RG, Swash M, Munsat TL. El escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
34. Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology (2009) 73:805–11. doi: 10.1212/WNL.0b013e3181b6bbbd
35. McCullough GH, Rosenbek JC, Wertz RT, McCoy S, Mann G, McCullough K. Utility of clinical swallowing examination measures for detecting aspiration post-stroke. J Speech Lang Hear Res. (2005) 48:1280–93. doi: 10.1044/1092-4388(2005/089)
36. Finsterer J, Erdorf M, Mamoli B, Fuglsang-Frederiksen A. Needle electromyography of bulbar muscles in patients with amyotrophic lateral sclerosis: evidence of subclinical involvement. Neurology (1998) 51:1417–22. doi: 10.1212/WNL.51.5.1417
37. Tankisi H, Otto M, Pugdahl K, Fuglsang-Frederiksen A. Spontaneous electromyographic activity of the tongue in amyotrophic lateral sclerosis. Muscle Nerve (2013) 48:296–8. doi: 10.1002/mus.23781
38. Li J, Petajan J, Smith G, Bromberg M. Electromyography of sternocleidomastoid muscle in ALS: a prospective study. Muscle Nerve (2002) 25:725–8. doi: 10.1002/mus.10115
39. Pan H, Jian F, Lin J, Chen N, Zhang Z, Wang Y, et al. Needle electromyography of the frontalis muscle in patients with amyotrophic lateral sclerosis. Muscle Nerve (2016) 54:1093–6. doi: 10.1002/mus.25236
40. Preston DC, Shapiro BE, Raynor EM, Kothari MJ. The relative value of facial, glossal, and masticatory muscles in the electrodiagnosis of amyotrophic lateral sclerosis. Muscle Nerve (1997) 20:370–2. doi: 10.1002/(SICI)1097-4598(199703)20:3<370::AID-MUS18>3.0.CO;2-4
41. Gooch CL, Pullman SL, Shungu DC, Ulug AM, Chane S, Gordon PH, et al. Motor unit number estimation (MUNE) in diseases of the motor neuron: utility and comparative analysis in a multimodal biomarker study. Suppl Clin Neurophysiol. (2009) 60:153–62. doi: 10.1016/S1567-424X(08)00015-9
42. de Carvalho M, Barkhaus PE, Nandedkar SD, Swash M. Motor unit number estimation (MUNE): where are we now? Clin Neurophysiol. (2018) 129:1507–16. doi: 10.1016/j.clinph.2018.04.748
43. Neuwirth C, Barkhaus PE, Burkhardt C, Castro J, Czell D, de Carvalho M, et al. Tracking motor neuron loss in a set of six muscles in amyotrophic lateral sclerosis using the Motor Unit Number Index (MUNIX): a 15-month longitudinal multicentre trial. J Neurol Neurosurg Psychiatry (2015) 86:1172–9. doi: 10.1136/jnnp-2015-310509
44. McIlduff C, Boegle A, Qi K, Rutkove S. Motor unit number estimation recording from the tongue: a pilot study. Neurology (2017) 88(Suppl. 16):P4.113.
45. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol. (2000) 57:109–13. doi: 10.1001/archneur.57.1.109
46. Turner MR, Talbot K. Mimics and chameleons in motor neurone disease. Pract Neurol. (2013) 13:153–64. doi: 10.1136/practneurol-2013-000557
47. Volanti P, Mannino M, Piccoli T, La Bella V. Carcinoma of the tongue and bulbar-onset amyotrophic lateral sclerosis: unusual differential diagnosis. Neurol Sci. (2007) 28:151–3. doi: 10.1007/s10072-007-0809-x
48. Bede P, Hardiman O. Lessons of ALS imaging: Pitfalls and future directions—a critical review. NeuroImage Clin. (2014) 4:436–43. doi: 10.1016/j.nicl.2014.02.011
49. Bede P, Bokde A, Elamin M, Byrne S, McLaughlin RL, Jordan N, et al. Grey matter correlates of clinical variables in amyotrophic lateral sclerosis (ALS): a neuroimaging study of ALS motor phenotype heterogeneity and cortical focality. J Neurol Neurosurg Psychiatry. (2013) 84:766–73. doi: 10.1136/jnnp-2012-302674
50. Schuster C, Kasper E, Machts J, Bittner D, Kaufmann J, Benecke R, et al. Focal thinning of the motor cortex mirrors clinical features of amyotrophic lateral sclerosis and their phenotypes: a neuroimaging study. J Neurol. (2013) 260:2856–64. doi: 10.1007/s00415-013-7083-z
51. Christidi F, Karavasilis E, Ferentinos P, Xirou S, Velonakis G, Rentzos M, et al. Investigating the neuroanatomical substrate of pathological laughing and crying in amyotrophic lateral sclerosis with multimodal neuroimaging techniques. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:12–20. doi: 10.1080/21678421.2017.1386689
52. Bede P, Finegan E. Revisiting the pathoanatomy of pseudobulbar affect: mechanisms beyond corticobulbar dysfunction. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:4–6. doi: 10.1080/21678421.2017.1392578
53. Querin G, El Mendili MM, Bede P, Delphine S, Lenglet T, Marchand-Pauvert V, et al. Multimodal spinal cord MRI offers accurate diagnostic classification in ALS. J Neurol Neurosurg Psychiatry (2018) 89:1220–1. doi: 10.1136/jnnp-2017-317214
54. Bede P, Querin G, Pradat PF. The changing landscape of motor neuron disease imaging: the transition from descriptive studies to precision clinical tools. Curr Opin Neurol. (2018) 31:431–8. doi: 10.1097/WCO.0000000000000569
55. Muller HP, Turner MR, Grosskreutz J, Abrahams S, Bede P, Govind V, et al. A large-scale multicentre cerebral diffusion tensor imaging study in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2016) 87:570–9. doi: 10.1136/jnnp-2015-311952
56. Atassi N, Xu M, Triantafyllou C, Keil B, Lawson R, Cernasov P, et al. Ultra high-field (7tesla) magnetic resonance spectroscopy in Amyotrophic Lateral Sclerosis. PLoS ONE (2017) 12:e0177680. doi: 10.1371/journal.pone.0177680
57. Darley FL, Aronson AE, Brown JR. Clusters of deviant speech dimensions in the dysarthrias. J Speech Lang Hear Res. (1969) 12:462–96. doi: 10.1044/jshr.1203.462
58. Darley FL, Aronson AE, Brown JR. Differential diagnostic patterns of dysarthria. J Speech Lang Hear Res. (1969) 12:246–69. doi: 10.1044/jshr.1202.246
59. Duffy J. Motor Speech Disorders: Substrates, Differential Diagnosis, and Management. St. Louis, MO: Elsevier (2013).
60. Kent RD. Hearing and believing: some limits to the auditory-perceptual assessment of speech and voice disorders. Am J Speech Lang Pathol. (1996) 5:7–23. doi: 10.1044/1058-0360.0503.07
61. Allison KM, Yunusova Y, Campbell TF, Wang J, Berry JD, Green JR. The diagnostic utility of patient-report and speech-language pathologists' ratings for detecting the early onset of bulbar symptoms due to ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:358–66. doi: 10.1080/21678421.2017.1303515
62. Rong P, Yunusova Y, Richburg B, Green JR. Automatic extraction of abnormal lip movement features from the alternating motion rate task in amyotrophic lateral sclerosis. Int J Speech Lang Pathol. (2018). doi: 10.1080/17549507.2018.1485739. [Epub ahead of print].
63. Mefferd AS, Green JR, Pattee G. A novel fixed-target task to determine articulatory speed constraints in persons with amyotrophic lateral sclerosis. J Commun Disord. (2012) 45:35–45. doi: 10.1016/j.jcomdis.2011.09.002
64. Goonetilleke A, Guiloff RJ. Accuracy, reproducibility and variability of quantitative assessments of bulbar and respiratory function in motor neurone disease. J Neurol Sci. (1994) 124(Suppl.):64–6. doi: 10.1016/0022-510X(94)90181-3
65. Onesti E, Schettino I, Gori MC, Frasca V, Ceccanti M, Cambieri C, et al. Dysphagia in amyotrophic lateral sclerosis: impact on patient behavior, diet adaptation, and riluzole management. Front Neurol. (2017) 8:94. doi: 10.3389/fneur.2017.00094
66. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci. (1999) 169:13–21. doi: 10.1016/S0022-510X(99)00210-5
67. Franchignoni F, Mora G, Giordano A, Volanti P, Chio A. Evidence of multidimensionality in the ALSFRS-R Scale: a critical appraisal on its measurement properties using Rasch analysis. J Neurol Neurosurg Psychiatry (2013) 84:1340–5. doi: 10.1136/jnnp-2012-304701
68. Rooney J, Burke T, Vajda A, Heverin M, Hardiman O. What does the ALSFRS-R really measure? A longitudinal and survival analysis of functional dimension subscores in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2017) 88:381–5. doi: 10.1136/jnnp-2016-314661
69. Voustianiouk A, Seidel G, Panchal J, Sivak M, Czaplinski A, Yen A, et al. ALSFRS and appel ALS scores: discordance with disease progression. Muscle Nerve (2008) 37:668–72. doi: 10.1002/mus.20977
70. Gordon PH, Cheng B, Katz IB, Mitsumoto H, Rowland LP. Clinical features that distinguish PLS, upper motor neuron–dominant ALS, and typical ALS. Neurology (2009) 72:1948–52. doi: 10.1212/WNL.0b013e3181a8269b
71. Smith RA, Macklin EA, Myers KJ, Pattee GL, Goslin KL, Meekins GD, et al. Assessment of bulbar function in amyotrophic lateral sclerosis: Validation of a self-report scale (Center for Neurologic Study Bulbar Function Scale). Eur J Neurol. (2018) 25:907–e66. doi: 10.1111/ene.13638
72. Smith R, Pioro E, Myers K, Sirdofsky M, Goslin K, Meekins G, et al. Enhanced bulbar function in amyotrophic lateral sclerosis: the Nuedexta treatment trial. Neurotherapeutics (2017) 14:762–72. doi: 10.1007/s13311-016-0508-5
73. Appel V, Stewart S, Smith G, Appel SH. A rating scale for amyotrophic lateral sclerosis: description and preliminary experience. Ann Neurol. (1987) 22:328–33. doi: 10.1002/ana.410220308
74. Norris FH, Calanchini PR, Fallat RJ, Panchari S, Jewett B. The administration of guanidine in amyotrophic lateral sclerosis. Neurology (1974) 24:721–8. doi: 10.1212/WNL.24.8.721
75. Lacomblez L, Bouche P, Bensimon G, Meininger V. A double-blind, placebo-controlled trial of high doses of gangliosides in amyotrophic lateral sclerosis. Neurology (1989) 39:1635–7. doi: 10.1212/WNL.39.12.1635
76. Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain (1995) 118:707–19. doi: 10.1093/brain/118.3.707
77. Wada A, Kawakami M, Liu M, Otaka E, Nishimura A, Liu F, et al. Development of a new scale for dysphagia in patients with progressive neuromuscular diseases: the Neuromuscular Disease Swallowing Status Scale (NdSSS). J Neurol. (2015) 262:2225–31. doi: 10.1007/s00415-015-7836-y
78. Abdelnour-Mallet M, Tezenas Du Montcel S, Cazzolli PA, Assouline A, Pointon C, Leveque N, et al. Validation of robust tools to measure sialorrhea in amyotrophic lateral sclerosis: a study in a large French cohort. Amyotroph Lateral Scler Frontotemporal Degener. (2013) 14:302–7. doi: 10.3109/21678421.2012.735238
79. Ball LJ, Willis A, Beukelman DR, Pattee GL. A protocol for identification of early bulbar signs in amyotrophic lateral sclerosis. J Neurol Sci. (2001) 191:43–53. doi: 10.1016/S0022-510X(01)00623-2
80. Makkonen T, Ruottinen H, Puhto R, Helminen M, Palmio J. Speech deterioration in amyotrophic lateral sclerosis (ALS) after manifestation of bulbar symptoms. Int J Lang Commun Disord. (2018) 53:385–92. doi: 10.1111/1460-6984.12357
81. Nishio M, Niimi S. Changes over time in dysarthric patients with amyotrophic lateral sclerosis (ALS): A study of changes in speaking rate and maximum repetition rate (MRR). Clin Linguist Phonet. (2009) 14:485–97. doi: 10.1080/026992000750020323
82. Stipancic KL, Yunusova Y, Berry JD, Green JR. Minimally detectable change and minimal clinically important difference of a decline in sentence intelligibility and speaking rate for individuals with amyotrophic lateral sclerosis. J Speech Lang Hear Res. (2018) 61:2757–71. doi: 10.1044/2018_JSLHR-S-17-0366
83. Green JR, Beukelman DR, Ball LJ. Algorithmic estimation of pauses in extended speech samples of dysarthric and typical speech. J Med Speech Lang Pathol. (2004) 12:149–54.
84. Yunusova Y, Graham NL, Shellikeri S, Phuong K, Kulkarni M, Rochon E, et al. Profiling speech and pausing in amyotrophic lateral sclerosis (als) and frontotemporal dementia (ftd). PLoS ONE (2016) 11:e0147573. doi: 10.1371/journal.pone.0147573
85. Green JR, Allison KM, Cordella C, Richburg BD, Pattee GL, Berry JD, et al. Additional evidence for a therapeutic effect of dextromethorphan/quinidine on bulbar motor function in patients with amyotrophic lateral sclerosis: a quantitative speech analysis. Br J Clin Pharmacol. (2018) 84:2849–56. doi: 10.1111/bcp.13745
86. Rong P, Yunusova Y, Green JR (eds). Speech intelligibility decline in individuals with fast and slow rates of ALS progression. In: INTERSPEECH. Dresden (2015).
87. Hiraoka A, Yoshikawa M, Nakamori M, Hosomi N, Nagasaki T, Mori T, et al. Maximum tongue pressure is associated with swallowing dysfunction in ALS patients. Dysphagia (2017) 32:542–7. doi: 10.1007/s00455-017-9797-z
88. Weikamp JG, Schelhaas HJ, Hendriks JC, de Swart BJ, Geurts AC. Prognostic value of decreased tongue strength on survival time in patients with amyotrophic lateral sclerosis. J Neurol. (2012) 259:2360–5. doi: 10.1007/s00415-012-6503-9
89. Adams V, Mathisen B, Baines S, Lazarus C, Callister R. Reliability of measurements of tongue and hand strength and endurance using the Iowa Oral Performance Instrument with elderly adults. Disabil Rehabil. (2015) 37:389–95. doi: 10.3109/09638288.2014.921245
90. Easterling C, Antinoja J, Cashin S, Barkhaus PE. Changes in tongue pressure, pulmonary function, and salivary flow in patients with amyotrophic lateral sclerosis. Dysphagia (2013) 28:217–25. doi: 10.1007/s00455-012-9436-7
91. Kaufmann P, Levy G, Montes J, Buchsbaum R, Barsdorf AI, Battista V, et al. Excellent inter-rater, intra-rater, and telephone-administered reliability of the ALSFRS-R in a multicenter clinical trial. Amyotroph Lateral Scler. (2007) 8:42–6. doi: 10.1080/17482960600888156
92. Gordon PH, Miller RG, Moore DH. ALSFRS-R. Amyotroph Lateral Scler Other Motor Neuron Disord. (2004) 5(Suppl 1):90–3. doi: 10.1080/17434470410019906
93. Smith RA, Melmed S, Sherman B, Frane J, Munsat TL, Festoff BW. Recombinant growth hormone treatment of amyotrophic lateral sclerosis. Muscle Nerve (1993) 16:624–33. doi: 10.1002/mus.880160608
94. Guiloff RJ, Goonetilleke A. Natural history of amyotrophic lateral sclerosis. Observations with the charing cross amyotrophic lateral sclerosis rating scales. Adv Neurol. (1995) 68:185–98.
95. Fang T, Al Khleifat A, Stahl DR, Lazo La Torre C, Murphy C, Young C, et al. Comparison of the King's and MiToS staging systems for ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2017) 18:227–32. doi: 10.1080/21678421.2016.1265565
96. Thakore NJ, Lapin BR, Kinzy TG, Pioro EP. Deconstructing progression of amyotrophic lateral sclerosis in stages: a Markov modeling approach. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:483–94. doi: 10.1080/21678421.2018.1484925
97. Hillel AD, Miller RM, Yorkston K, McDonald E, Norris FH, Konikow N. Amyotrophic lateral sclerosis severity scale. Neuroepidemiology (1989) 8:142–50. doi: 10.1159/000110176
98. Beukelman D, Yorkston K, Hakel M, Dorsey M. Speech Intelligibility Test. Lincoln, NE: Madonna Rehabilitation Hospital (2007).
99. Beukelman D, Fager S, Nordness A. Communication support for people with ALS. Neurol Res Int. (2011) 2011:714693. doi: 10.1155/2011/714693
100. Yorkston KM, Hammen VL, Beukelman DR, Traynor CD. The effect of rate control on the intelligibility and naturalness of dysarthric speech. J Speech Hear Disord. (1990) 55:550–60. doi: 10.1044/jshd.5503.550
101. Yorkston K, Strande E, Miller R, Hillel A, Smith K. Speech deterioration in amyotrophic lateral sclerosis: Implications for the timing of intervention. J Med Speech Lang Pathol. (1993) 1:35–46.
102. Ball LJ, Beukelman DR, Pattee GL. Timing of speech deterioration in people with amyotrophic lateral sclerosis. J Med Speech Lang Pathol. (2002) 10:231–5.
103. Rong P, Yunusova Y, Wang J, Green JR. Predicting early bulbar decline in amyotrophic lateral sclerosis: a speech subsystem approach. Behav Neurol. (2015) 2015:183027. doi: 10.1155/2015/183027
104. Langmore SE, Lehman ME. Physiologic deficits in the orofacial system underlying dysarthria in amyotrophic lateral sclerosis. J Speech Lang Hear Res. (1994) 37:28–37. doi: 10.1044/jshr.3701.28
105. DePaul R, Brooks BR. Multiple orofacial indices in amyotrophic lateral sclerosis. J Speech Lang Hear Res. (1993) 36:1158–67. doi: 10.1044/jshr.3606.1158
106. Borsodi F, Culea V, Langkammer C, Khalil M, Pirpamer L, Quasthoff S, et al. Multimodal assessment of white matter tracts in amyotrophic lateral sclerosis. PLoS ONE (2017) 12:e0178371. doi: 10.1371/journal.pone.0178371
107. Schuster C, Elamin M, Hardiman O, Bede P. The segmental diffusivity profile of amyotrophic lateral sclerosis associated white matter degeneration. Eur J Neurol. (2016) 23:1361–71. doi: 10.1111/ene.13038
108. Chen ZY, Liu MQ, Ma L. Cortical thinning pattern of bulbar- and spinal-onset amyotrophic lateral sclerosis: a surface-based morphometry study. Chin Med Sci J. (2018) 33:100–6. doi: 10.24920/11812
109. Walhout R, Westeneng HJ, Verstraete E, Hendrikse J, Veldink JH, van den Heuvel MP, et al. Cortical thickness in ALS: towards a marker for upper motor neuron involvement. J Neurol Neurosurg Psychiatry (2014) 86:288–94. doi: 10.1136/jnnp-2013-306839
110. Chen ZY, Liu MQ, Ma L. Gray matter volume changes over the whole brain in the bulbar- and spinal-onset amyotrophic lateral sclerosis: a voxel-based morphometry study. Chin Med Sci J. (2018) 33:20–8. doi: 10.24920/11804
111. Vora M, Kumar S, Sharma S, Sharma S, Makhaik S, Sood RG. Advanced magnetic resonance neuroimaging in bulbar and limb onset early amyotrophic lateral sclerosis. J Neurosci Rural Pract. (2016) 7:102–8. doi: 10.4103/0976-3147.165423
112. Hartung V, Prell T, Gaser C, Turner MR, Tietz F, Ilse B, et al. Voxel-based MRI intensitometry reveals extent of cerebral white matter pathology in amyotrophic lateral sclerosis. PLoS ONE (2014) 9:e104894. doi: 10.1371/journal.pone.0104894
113. Kollewe K, Munte TF, Samii A, Dengler R, Petri S, Mohammadi B. Patterns of cortical activity differ in ALS patients with limb and/or bulbar involvement depending on motor tasks. J Neurol. (2011) 258:804–10. doi: 10.1007/s00415-010-5842-7
114. Mohammadi B, Kollewe K, Samii A, Krampfl K, Dengler R, Munte TF. Decreased brain activation to tongue movements in amyotrophic lateral sclerosis with bulbar involvement but not Kennedy syndrome. J Neurol. (2009) 256:1263–9. doi: 10.1007/s00415-009-5112-8
115. Filippi M, Horsfield MA, Ader HJ, Barkhof F, Bruzzi P, Evans A, et al. Guidelines for using quantitative measures of brain magnetic resonance imaging abnormalities in monitoring the treatment of multiple sclerosis. Ann Neurol. (1998) 43:499–506. doi: 10.1002/ana.410430414
116. Misawa S, Noto Y, Shibuya K, Isose S, Sekiguchi Y, Nasu S, et al. Ultrasonographic detection of fasciculations markedly increases diagnostic sensitivity of ALS. Neurology (2011) 77:1532–7. doi: 10.1212/WNL.0b013e318233b36a
117. Shellikeri S, Yunusova Y, Green JR, Pattee GL, Berry JD, Rutkove SB, et al. Electrical impedance myography in the evaluation of the tongue musculature in amyotrophic lateral sclerosis. Muscle Nerve (2016) 52:584–91. doi: 10.1002/mus.24565
118. McIlduff CE, Yim SJ, Pacheck AK, Rutkove SB. Optimizing electrical impedance myography of the tongue in amyotrophic lateral sclerosis. Muscle Nerve (2017) 55:539–43. doi: 10.1002/mus.25375
119. Noh EJ, Park MI, Park SJ, Moon W, Jung HJ. A case of amyotrophic lateral sclerosis presented as oropharyngeal dysphagia. J Neurogastroenterol Motility (2010) 16:319–22. doi: 10.5056/jnm.2010.16.3.319
120. Takasaki K, Umeki H, Enatsu K, Kumagami H, Takahashi H. Evaluation of swallowing pressure in a patient with amyotrophic lateral sclerosis before and after cricopharyngeal myotomy using high-resolution manometry system. Auris Nasus Larynx (2010) 37:644–7. doi: 10.1016/j.anl.2010.02.003
121. Norel R, Pietrowicz M, Agurto C, Rishoni S, Cecchi G. Detection of amyotrophic lateral sclerosis (ALS) via acoustic analysis. bioRxiv [preprint] (2018). doi: 10.21437/Interspeech.2018-2389
122. Wang J, Kothalkar PV, Cao B, Heitzman D. (eds). Towards automatic detection of amyotrophic lateral sclerosis from speech acoustic and articulatory samples. In: INTERSPEECH. San Francisco, CA (2016). doi: 10.21437/Interspeech.2016-1542
123. Bandini A, Green JR, Taati B, Orlandi S, Zinman L, Yunusova Y. Automatic detection of amyotrophic lateral sclerosis (ALS) from video-based analysis of facial movements: speech and non-speech tasks. In: 13th IEEE International Conference on Automatic Face & Gesture Recognition (FG 2018). X'ian (2018). doi: 10.1109/FG.2018.00031
Keywords: amyotrophic lateral sclerosis, Bulbar ALS, outcome assessment (Health Care), dysphagia, dysarthria, COSMIN
Citation: Yunusova Y, Plowman EK, Green JR, Barnett C and Bede P (2019) Clinical Measures of Bulbar Dysfunction in ALS. Front. Neurol. 10:106. doi: 10.3389/fneur.2019.00106
Received: 20 November 2018; Accepted: 28 January 2019;
Published: 19 February 2019.
Edited by:
Francesca Trojsi, Università degli Studi della Campania Luigi Vanvitelli Caserta, ItalyReviewed by:
Patrizia Longone, Fondazione Santa Lucia (IRCCS), ItalyRodolfo Gabriel Gatto, University of Illinois at Chicago, United States
Copyright © 2019 Yunusova, Plowman, Green, Barnett and Bede. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yana Yunusova, eWFuYS55dW51c292YUB1dG9yb250by5jYQ==