 Barry W. Festoff
Barry W. Festoff Bruce A. Citron
Bruce A. Citron- 1pHLOGISTIX LLC, Fairway, KS, United States
- 2Department of Neurology, University of Kansas Medical Center, Kansas City, KS, United States
- 3Laboratory of Molecular Biology Research & Development, VA New Jersey Health Care System, East Orange, NJ, United States
- 4Department of Pharmacology, Physiology & Neuroscience, Rutgers New Jersey Medical School, Newark, NJ, United States
This review details our current understanding of thrombin signaling in neurodegeneration, with a focus on amyotrophic lateral sclerosis (ALS, Lou Gehrig's disease) as well as future directions to be pursued. The key factors are multifunctional and involved in regulatory pathways, namely innate immune and the coagulation cascade activation, that are essential for normal nervous system function and health. These two major host defense systems have a long history in evolution and include elements and regulators of the coagulation pathway that have significant impacts on both the peripheral and central nervous system in health and disease. The clotting cascade responds to a variety of insults to the CNS including injury and infection. The blood brain barrier is affected by these responses and its compromise also contributes to these detrimental effects. Important molecules in signaling that contribute to or protect against neurodegeneration include thrombin, thrombomodulin (TM), protease activated receptor 1 (PAR1), damage associated molecular patterns (DAMPs), such as high mobility group box protein 1 (HMGB1) and those released from mitochondria (mtDAMPs). Each of these molecules are entangled in choices dependent upon specific signaling pathways in play. For example, the particular cleavage of PAR1 by thrombin vs. activated protein C (APC) will have downstream effects through coupled factors to result in toxicity or neuroprotection. Furthermore, numerous interactions influence these choices such as the interplay between HMGB1, thrombin, and TM. Our hope is that improved understanding of the ways that components of the coagulation cascade affect innate immune inflammatory responses and influence the course of neurodegeneration, especially after injury, will lead to effective therapeutic approaches for ALS, traumatic brain injury, and other neurodegenerative disorders.
Introduction
In humans, the coagulation system or cascade was conceptualized over the past five to six decades to consist of five serine proteases (factor VII, FVII; factor IX, FIX; factor X, FX; protein C, PC and prothrombin, PT) that act with five cofactors (tissue factor, TF; factor V, FV; factor VIII, FVIII; thrombomodulin, TM; and protein S, PS) to control the generation of fibrin, which is subsequently cross-linked by Factor XIII (FXIII), a transglutaminase (1). This system is essentially conserved throughout mammalian species (schematically shown in Figure 1), but the system's endpoint, hemostasis, has been around for 450 million years. Hemostasis consists of three activities that are closely regulated; vasoconstriction, platelet aggregation, and clotting factor activation. Two different pathways, the intrinsic (contact) and extrinsic (TF), exist to activate clotting and the principal difference is the role of TF in the extrinsic pathway, which works very rapidly. With blood vessel damage, inactive FVII comes in contact with TF, a protein on the endothelial cell (EC), and activates it to a protease (2). Activated Factor VII then proteolytically activates FX that then binds activated FV to form prothrombinase. So, recapping, TF release is very rapid and generated by damaged blood vessels and surrounding tissues, which is especially high in brain, and initiates the extrinsic pathway.
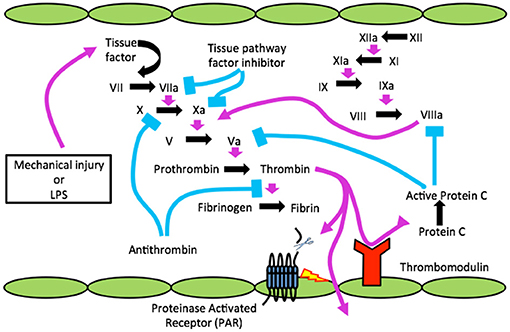
Figure 1. The clotting cascade, TBI, infection (LPS) and the blood brain barrier. Cartoon shows injury or infection, lipopolysaccharide (LPS) releasing tissue factor (TF) to activate clotting resulting in active α-thrombin, which cleaves a receptor(s) known as PAR on endothelial cells (ECs) to disrupt BBB. Through disrupted BBB thrombin gains access to CNS and can cleave to activate PARs on microglia/astrocytes for neuroinflammation and neurons to form neurofibrillary tangles (NFTs).
Since endothelial cell damage is the principal mechanism for clotting factor activation via TF generation it invariably occurs with systemic microbial infection, as in sepsis, where the innate immune system is activated (2). Indicating sepsis in Figure 1 is bacterial LPS (lipopolysaccharide or endotoxin). It was subsequently found that trauma, a sterile injury, also produces TF-generated coagulation (Figure 1) (3).
The clotting system is involved in host defense, and arose with and is linked to innate immunity or inflammation at very early evolutionary stages. TF is the key actor and common generator providing the critical nexus between these two major host defense systems (4). TF belongs to the cytokine receptor superfamily and is a type I integral membrane glycoprotein (5). Thrombin, the ultimate serine protease in the cascade, is the key downstream product of TF-initiated coagulation. Not only does it play a central role in hemostasis but more recent studies have revealed its fundamental and intense proinflammatory effects (6). These latter attributes of thrombin, just as its role in causing platelet aggregation, were subsequently ascribed to its non-coagulation actions as a ligand for cell-surface receptors, now known as protease-activated receptors (PARs) (7–9).
Although these thrombin-mediated, PAR-activated cellular effects involve thrombin's roles in cell proliferation and modulation, cytoprotection and apoptosis, its role as a proinflammatory mediator is key that further brings together coagulation and inflammation—the coag-inflamm nexus. Furthermore, it incorporates innate immune pathways such as toll-like receptors (TLRs) and complement, exosomes/microparticles (MPs) into this nexus. With cellular activation thrombin also recruits other systems to provide a balance for this coag-inflammatory pressure, and this includes the protein C (PC)–thrombomodulin (TM) natural anticoagulant/anti-inflammatory machinery along with activation and monitoring of the fibrinolytic system.
In the 1980's a few studies began to explore the direct effects of thrombin on cultured neural cells (10–13). Those initial reports ushered in a number of successive studies of thrombin, the coagulation and fibrinolytic cascades, TM, PARs in the CNS that continues to the present time. More recent efforts at translation of tissue culture and animal studies to neurologic diseases are now chronicled in other reports in this Frontiers in Neurology collection.
Amyotrophic Lateral Sclerosis (ALS) and Neurodegenerative Disorders
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder exemplified clinically by muscle weakness and wasting and neuropathologically by degeneration of upper and lower motor neurons in the spinal cord, brain and brainstem (14–16). More recent evidence indicates that a number of endophenotypes exist for ALS beyond what was considered 30–50 years ago: the four classic motor neuron disorders. These are: classical ALS (upper and lower motor neuron and bulbar involvement), progressive muscular atrophy (PMA; only lower motor neuron), progressive bulbar palsy (PBP; brainstem with little if any extremity features) and primary lateral sclerosis (PLS; only upper motor) if it is actually part of the spectrum. As a distinct nosologic disorder ALS has been known in the medical literature since Charcot first described it 150 years ago in the late nineteenth century (17).
It is a fatal and currently enigmatic disease with death usually resulting from the inexorable progression of diaphragmatic and intercostal muscle weakness ultimately causing paralysis and respiratory failure typically within 5 years of diagnosis. The incidence of ALS has changed only slightly since the 1970 s and is ~1.5–3 per 100,000 in Western Europe and North America with little variation. It is overwhelmingly a sporadic disease (sALS), but genetic variants exist (fALS) accounting for no more than 10% of all cases (see below), although newer information may be changing this. ALS has an estimated lifetime risk of 1 in 400, is an adult-onset illness that is rare before the age of 40 years increasing exponentially with age. There are no known treatments that impact progression of the disease. Until 2017, the last Food and Drug Administration (FDA) approved drug was Riluzole™, licensed in 1996 and that only extended survival of ALS patients 3 months. In May 2017 the FDA approved edaravone (Radicava™) to treat ALS patients based on a 2nd Phase 3 study after the first was negative (18). As the authors wrote: the drug “….showed efficacy in a small subset of people with ALS who met criteria identified in post-hoc analysis of a previous phase 3 study, showing a significantly smaller decline of ALSFRS-R score compared with placebo.”
As discussed in detail below, our laboratory at the Kansas City VA Medical Center began studies of the coagulation system in ALS in the 1980's (see Table 1).

Table 1. Partial history of thrombin signaling in CNS and PNS: Key processes involving neural and neuromuscular health and the thrombin signaling pathway.
Genetics and ALS: familial ALS (fALS)
Although it was considered a sporadic illness beginning in the 1990 s interest in the ~5–10% of ALS cases that had family history began. Identification of mutations in the superoxide dismutase 1 gene (SOD1) was reported in 1992 (65, 66). Over the next 25 years remarkable progress in our understanding of SOD1 and fALS has occurred (67–73).
Even amongst otherwise sALS cases about 1–3% possess missense mutations in SOD1 (74) and even more, about 5–10% of sporadic ALS cases are caused by intronic expansions in C9orf72, the open reading frame (ORF 72) on chromosome 9 (75–77). This indicates that 1 in 20 cases of sALS and about 40% of fALS are due to C9orf72 hexanucleotide repeats.
With SOD1 and C9orf72 more than 20 mutated genes have now been found to be specifically associated with fALS (78) that include TARDBP (79–81) and FUS (82, 83), the fused in sarcoma gene on chromosome 16p11.2, that is involved with RNA processing, which together with SOD1 and C9orf72, are the four most common genes involved in causing ALS clinically. TARDP encodes a protein, TAR DNA binding protein (TDP-43) that accumulates in most sALS motor neurons but not SOD1 fALS neurons (84). These genes have been numbered now as ALS1-ALS22, along with FTDALS1, FTDALS 2, FTDALS 3, and FTDALS 4 (78). Genome-wide association studies (GWAS) may be changing the role of genetics in ALS including what we now consider sALS (85–87).
The changing viewpoint results from studies of the relatively uncommon genetic cases of this enigmatic and fatal neurodegenerative disorder that have revealed some fundamental clues that might uncover novel therapeutic targets. Amongst these are more recently identified endophenotypes beyond the classical motor sub-types. Endophenotypes are inherited traits identified using clinical or laboratory measures including electroencephalographic or electromyographic abnormalities, neurocognitive deficiencies, and other modalities that identify impairment. Until recently they have been largely used in psychiatric and psychopathology-related research. Originally conceived by Gottesman and Shields (88), they were proposed to appear not only in patients but also in their unaffected relatives. The presumption of endophenotypes is that they are more proximate to gene action than the clinical diagnoses (89, 90). In neurodegenerative diseases such as Parkinson's (PD) and Alzheimer's (AD) diseases, in addition to ALS, they might provide dual positives such as improving diagnoses and initiating therapy in preclinical stages (91–93).
ALS Endophenotypes Beyond the Motor System
ALS is now recognized as a multi-system neurodegeneration rather than a disease limited to motor neurons (94–97). Although 40 years ago if a patient was clinically diagnosed with ALS but exhibited cognitive symptoms that patient was not considered to have classical ALS and typically was removed from consideration. In fact, in the original El Escorial criteria (98) and El Escorial revisited (99, 100), the presence of dementia essentially ruled out ALS as diagnosis. This action was taken despite the fact that descriptions of cognitive and behavioral symptoms resembling frontotemporal dementia (FTD) in otherwise typical ALS motor phenotypes date back to the 1880 s. The neurologic giant, Arnold Pick, whose name is eponymic for a subgroup of FTD known as Pick's disease, was aware that Charcot had considered that non-motor brain regions might also be involved in the neurodegeneration of what is now known as Maladie de Charcot or la sclérose latérale amyotrophique (SLA) by francophones.
One of the first descriptions of FTD associated with ALS in the modern era was provided by the late Canadian neurologist, Arthur Hudson (101), who also described mixed types of ALS with parkinsonism as well as with dementia and other clinical features, reminiscent of the ALS-parkinsonism-dementia complex of Guam (102, 103). Since then increasing interest in FTD-like symptoms in ALS patients appeared and it is now thought that about 10% of patients with one of the four classic motor-neuron disorders: classical ALS, PMA, PBP, and PLS, have cognitive features.
Subsequent reviews of the ALS/FTD complex have appeared (104, 105) that now also include associations with C9orf72 expansions (77, 96, 106, 107). In fact, the seminal discovery of a GGGGCC hexanucleotide repeat expansion (HRE) within the chromosome 9 ORF 72 (75–77, 108), has been established as cause for the most common form of ALS/FTD (107).
Given this common cause of sALS with FTD, 10% of sALS and an additional 10% of FTD, the next question, given that it has taken more than 25 years with SOD1 mutations, is just how cytotoxicity occurs with the GGGGCC (G4C2) HRE within the C9ORF72 gene? Several recent studies have shed light on this: it has been reported that HRE RNA forms hairpin and G-quadruplex structures that bind and sequester RNA-binding proteins (RBPs). The GGGGCC are translated into specific dipeptide-repeat (DPR) proteins, and these form toxic aggregates, particularly the arginine-rich dipeptides, specifically proline-arginine (PR), that possess potent neurotoxicity forming aggregates in nuclei and nucleoli, and stress granule formation, with likely effects on translation (109). These authors used inducible pluripotent stem cells (iPSCs) to differentiate into human motor neurons (iMNs), including those from ALS patients carrying the repeat-expanded C9ORF72. These studies revealed that C9ORF72 in the ALS patients was haploinsufficient. Thus, the ALS/FTD gene had only one functional copy, causing a loss-of-function mutation. Using blood cells from healthy individuals they used gene-editing techniques to delete the C9ORF72 or from ALS patients with the abnormal gene. They found that C9ORF72 cooperated with endosomes, was involved in vesicle trafficking and formation of lysosomes in motor neurons. When they repeated the nucleotide expansion this reduced C9ORF72 expression, and with this process, neurodegeneration was triggered via both gain- and loss-of-function mechanisms. The former produced a buildup of glutamate receptors, causing excitotoxicity, while the latter weakened neurotoxic dipeptide repeat proteins clearance derived from the repeat expansion. This cooperative action led to neurodegeneration. These and other researchers have begun using the gene-editing tool, CRISPR, specifically the CRISPR–Cas9 system, to perform genome-wide gene-knockout screens similar to studies in cancer (110).
Frontototemporal lobar degeneration (FTLD) is the 2nd most common cause of dementia in elderly (over age 65) individuals and is actually a broad spectrum of neurological disorders. FTD is a variant of FTLD and from GWAS studies now appears to share a number of genetic as well as clinical and neuropathological features. In a recent GWAS study of more than 120,000 neurodegenerative diseases and controls unique genetic overlap between ALS and FTD spectrum diseases was found (111). Of interest, the H1 haplotype of the tau protein gene (MAPT) appeared to confer risk for ALS, as did BNIP1, a mitophagy-associated, proapoptotic protein.
If an endophenotype strategy in ALS should be implemented, as has since been undertaken in several neurodegenerative studies, it will depend both on quality and properties of a specific trait. It will be necessary to critically evaluate the trait(s) to determine if it truly can capture pre-diagnosis features of ALS/FTD. However, no consistency has yet appeared for endophenotypes or intermediate traits or even biomarkers, but some encouraging signs have appeared (112, 113). When such validated intermediate traits or biomarkers are considered, it will be necessary to forgo requiring that they be absolutely specific for ALS or FTD. Consequently, application of endophenotypes to future analyses of ALS and FTD seems more than justified.
With consideration of the ALS spectrum as a non-cell autonomous condition (114, 115), it brought to the picture the evidence that glial cells, including astrocytes, oligodendrocytes, and even microglia play important roles in the pathogenesis of ALS (116–118). Prior to the last decade it was widely assumed that motoneuronal cell death proceeded by cell autonomous mechanisms. However, information gained initially from using SOD1 transgenic mice and subsequently with other genetic models, the non-cell autonomous position evolved. In terms of SOD1 more than 170 different mutations have been shown to cause fALS. When SOD1 mutations were expressed only in neurons neurodegeneration did not occur in the mice (119). But just how these mutations in SOD1 result in cytotoxicity is still unclear, despite more than 25 years of study. In fact, no consensus has emerged as to the principal mechanism for neurotoxicity or even how cells might protect themselves from it. Cleveland and colleagues proposed that ALS was just the tip of the iceberg and that non-cell autonomy will be shown to be the mode in other neurodegenerative diseases (114, 115, 120, 121).
Along these lines the multi-faceted roles of astrocytes have now become prominent for investigation in ALS (115, 118, 122–124). Discussed in more detailed below, reactive astrocytosis also known as astrogliosis, is a classic glial response to CNS injury and scar-forming reactive astrocytes are usually viewed as detrimental to clinical outcome (125), but not always (126). Astrocytes are also hallmarks of neurodegeneration (127) and using the ME7 prion mouse model Cunningham and colleagues showed that neurodegeneration primed astrocytes to produce exaggerated chemokine responses when stimulated with acute proinflammatory cytokines (128). The usual neuropathologic means to characterize reactive astrocytes is by using antibodies to the intermediate filament glial fibrillary acidic protein (GFAP). However, all phenotypes of astrocytes including reactive astrocytes and scar-forming astrocytes strongly express GFAP. In fact, being able to modulate extent and phenotypes of reactive astrocyte function (129) is potentially attractive as novel targets to enhance the functional outcomes after spinal cord injury (SCI) (130) or in ALS and other neurodegenerative diseases might be revealed (116, 131).
Connecting Dots To Neurodegeneration: Neuroinflammation, Coagulation, BBB
Inflammatory, and Innate Immune Aspects of ALS
Reviewing numerous studies of the past two decades has divulged previously held concepts that upper and lower motor neurons were the focus of ALS disease burden have now been replaced by non-cell autonomous mechanisms. Such non-cell autonomous mechanisms, particularly neuroinflammation, may not only contribute to the disease process but may initiate it, as detailed below.
Based on several lines of evidence within the last 20 years both sALS and fALS have had numerous proinflammatory markers associated with them (132–135). More than two decades ago, Appel et al. emphasized potential autoimmunity in ALS (136–138), and several different approaches revealed that immunoglobulin G (IgG) from ALS patients' sera caused toxicity in cultured motor neurons and in mouse models (138–141), with activation of L-type Ca2+-channels.
As exception proving the rule or standing in apparent contradiction, since it was present in the context of immunodeficiency, was our earlier report documenting ALS in a young homosexual male patient in whom HTLV-III (subsequently re-named HIV) was isolated (142). This initial observation was later confirmed in more recent accounts (143–147), suggesting that ALS, if truly autoimmune, may also be associated with immune deficiency disorders such as AIDs.
By definition, neuroinflammation is inflammation of nervous tissue and is characterized by proliferation and activation of glial cells, primarily microglia, and astrocytes, as well as transmigration of circulating immune cells, including polymorphonuclear neutrophils (PMNs), monocyte/macrophages, and T lymphocytes (T-cells) into the parenchyma across the blood-brain barrier (BBB) (148–152). In addition to these cellular characteristics, neuroinflammation includes humoral features such as proinflammatory cytokine and chemokine overproduction, along with their respective receptors (151). Of relevance here are the numerous reports of neuroinflammation in both sALS and fALS including its appearance in pre-symptomatic phases in transgenic mice. However, confusion has developed from these data since both deleterious and beneficial effects have been found especially when focusing on motor neuron survival and also depending on what disease stage was examined (153, 154).
Microglia
The understanding of these complex interactions largely centers on microglia, considered the brain's resident macrophages, and their dual roles or Janus faces, in neurodegeneration in general and ALS in particular (107, 153). In essence, although microglial phenotypes were classified as either M1 (“classically activated”) or M2 (“alternatively activated”), similar to circulating macrophages, phenotypic diversity of microglia is actually a spectrum (155). M1 microglia could be considered more proinflammatory while M2 more anti-inflammatory, possibly viewed as “deactivated” after phagocytosis of apoptotic cells. Clearly, a therapeutic strategy in ALS or in AD or PD for that matter, might be to selectively modulate microglial phenotypes, such as inhibiting or blocking M1 or enhancing M2. That may be too simple, although it is a strategy worth evaluating. However, this should not be done with pre-clinical animal models due to known differences in inflammatory responses compared to humans, but in human iPSC ex vivo models that incorporate elements of the blood-spinal cord barrier (BSCB)BBB/NVU along with neurons (156, 157).
Astrocytes
Of the several types of glial cells in the CNS astrocytes are the most abundant. Classically considered “supportive” cells for neurons astrocytes have recently been shown to be critical in regulating CNS immunity, but exactly how they do this is largely unknown. Astrocytes have been shown to be regionally diverse within the brain and in the spinal cord. Regions where astrocytes may be involved in regulating CNS immunity are at their “end-feet” localized to where they are contiguous with ECs of the BBB and neurovascular unit (NVU) as well as perivascular end-feet that form the glia limitans. All astrocytes are ramified and have processes that terminate on basal lamina impacting the perivascular compartment with their end-feet (127).
Of interest, one molecule highly concentrated in astrocytic end-feet is the gap junction protein, connexin 43 (Cx43) (158). Cx43 may have roles in the non-cell autonomous pathogenesis of sALS, implicating toxic mitochondria transferring from astrocytes to motor neurons at the BSCB (159, 160), as detailed below.
By analogy to macrophages, the M1/M2 macrophage and microglial nomenclature (161), although with caveats for its potential simplicity researchers have also applied these to reactive astrocytes (125, 127) into A1 and A2 sub-classes (162), whether caused by neuroinflammation or ischemia, respectively. As with microglial M1 and M2 sub-classes the macrophage phenotypic literature clearly indicates that these circulating immune cells display more than two polarization states (155, 163, 164). Chronic neurodegeneration also produces changes in the secretory profile of astrocytes in terms of what cytokines and chemokines are produced (128).
As M1 macrophages were considered destructive, so, too, are A1 astrocytes. Conversely, since M2 were considered reparative and protective as a macrophage or microglial phenotype, so were the A2 astrocytes. Liddelow, in the late Ben Barres' group, further showed that A1 were induced by reactive microglia (165).
Innate Immune Activation in ALS
Over the past two decades our thinking about the brain and spinal cord as being an immunologically privileged site has changed. It was previously thought that the CNS could not mount an immune response nor process antigens. More recent studies have reversed that indicating that immune surveillance does take place in the CNS, and glial cells of all types act as immune effector cells within the CNS (149, 166). We now know, for example that the primary function of the CNS innate immune system is to provide neuroprotection against invading pathogens. However, in addition to infectious agents it is also protective for injury stimuli, and by so doing maintains CNS homeostasis.
Pattern Recognition Receptors, Pathogen-Associated Molecular Patterns in ALS
Knowledge of how membrane and intracellular receptors respond to pathogenic components dramatically increased with identification of pattern recognition receptors (PRRs) to identify pathogen-associated molecular patterns (PAMPs), the prototype for which is lipopolysaccharide (LPS) or endotoxin, from Gram-negative bacteria. The effect of LPS in the CNS is to cause sickness behavior, a coordinated set of adaptive behavioral changes to LPS and others (166–168) that includes: fever, anorexia, social withdrawal, lethargy, and decreased rapid-eye movement sleep (REMS). Major innate immune system PRRs such as the Toll-like receptors (TLRs) and the receptor for advanced glycosylation endproducts (RAGE) are expressed in the CNS. Most TLRs, now 15 members of the family, and RAGE, are expressed in all neural cells (167–174).
Damage-Associated Molecular Patterns (DAMPs)
The danger-damage theory expressed in 1994 by Polly Matzinger (175–177) changed the concept of immunology from simply detecting self vs. non-self. Her thesis was that the immune system's driving force is the need to recognize danger and prevent destruction. This theory evolved with publication of the proceedings of the EMBO Workshop on Innate Danger Signals and HMGB1 that took place February 2006 in Milan, Italy (organized by M. Bianchi, K. Tracey, and U. Andersson) (178). In keeping with this concept a group of endogenous molecules that signaled damage or danger were developed and were referred to as alarmins, a sub-category of DAMPs. Subsequent studies indicated that the PRRs recognized and responded to DAMPs in essentially the same manner as their response to PAMPs (177, 179, 180) and that the CNS also participated (181). The comparison of PAMPs and DAMPs and list of both is shown in Table 2.
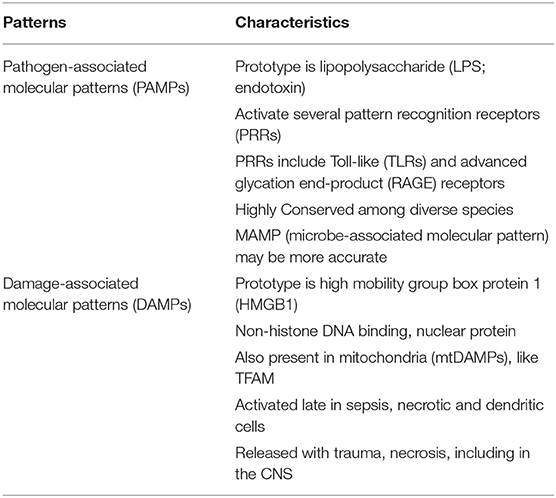
Table 2. PAMPs and DAMPs: danger/damage recognition systems extrinsic and intrinsic.
In fact, as an example that science, certainly more in the pre-cloning era, was guilty of the blind men describing the elephant parable, Finnish workers had identified a protein that guided early neuroblasts to their final locations in developing mouse brain and called this protein, amphoterin (182, 183). Amphoterin, also called P30 protein, was subsequently found to be identical to HMGB1 (170), the prototypic alarmin/DAMP. The structure of the alarmin/DAMP HMGB1 is shown in Figure 2A and its known signaling in Figure 2B. The relationship between HMGB1 and thrombin is interesting. Both are prototypes of ancient host defense systems, inflammation and coagulation (60), but in addition, as shown in Figure 2B, thrombin can cleave HMGB1 at its –NH2 end and does so when bound to TM (184). Of interest, since then HMGB1 has been shown to be involved in a number of neuropathologic processes in the CNS and is also essential for brain development (181, 185).
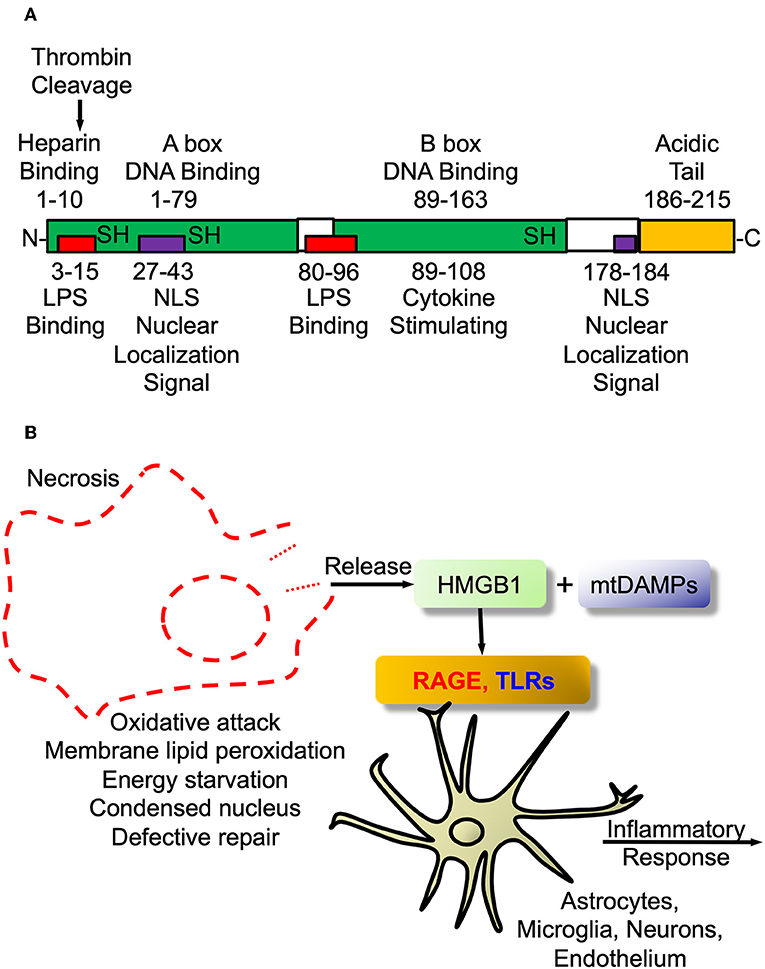
Figure 2. High mobility group box protein 1 (HMGB1). (A) HMGB1 Structure. Schematic of HMGB1's two binding motifs, A-box and B-box. Also showing critical cysteines that determine whether it is proinflammatory when outside cell or DNA binding when inside nucleus. In addition, the C-terminal acidic tail appears to confer protein stability and DNA bending in vitro. (B) HMGB1 signaling. The damage-associated molecular pattern (DAMP) HMGB1 is passively released from necrotic or exploding cells such as with infection or trauma. Within the CNS it engages pattern recognition receptors (PRRs) such as toll-like receptors (TLRs2/4) and receptor for advanced glycation end products (RAGE) to initiate proinflammatory signaling as part of innate immune activation.
As mentioned above PRRs, especially TLRs and RAGE, expressed by immune cells are also expressed by neural cells, particularly astrocytic and microglial, to mediate resident immune cell activation (169, 172). As described for AD and other neurodegenerative diseases (63, 148, 181, 186), DAMPs are probable candidates to partake in, and possibly initiate, ALS neurodegenerative activities. HMGB1 is over-expressed in SOD1 mutant mouse spinal cord and motor cortex and from patients with ALS (187). TLRs were also found to be over-expressed in ALS patients' spinal cords (188), as was RAGE, along with its proinflammatory ligands, including HMGB1, S100B and calgranulin (189). Furthermore, a number of groups have focused on levels of circulating soluble RAGE (sRAGE) in various diseases including diabetes mellitus, cardiovascular and neurodegenerative diseases (190, 191), recently including ALS (174, 189, 192). As opposed to sRAGE being “specific” for any of those diseases it is clear that it is implicated in their pathogenesis and contributes to our understanding of innate immunity in these conditions. Furthermore, it might be useful as therapeutic strategy in one or more of them. Additionally, sRAGE has been considered a “decoy receptor” to block the cellular membrane receptor to block RAGE-mediated signaling. In this regard, sRAGE is decreased in blood while increased in affected CNS in ALS and other neurodegenerative diseases (193).
Mitochondria, ALS, and mtDAMPs
A key mechanism whereby motor neurons degenerate in ALS is by influence of dysfunctional mitochondria (194–196). As in PD and AD and other neurodegenerative diseases, studies in SOD1 transgenics as well as in sALS cells have been performed that show such mitochondrial defects, with an eye to novel therapeutics (197–199). Almost 20 years have elapsed since the close temporal relationship of the onset of motor neuronal degeneration with initiation of astrogliosis in the SOD1 mouse model was first identified (200). With further understanding of the non-cell autonomous, specifically astrocytic, aspect of ALS pathogenesis abnormalities in astrocyte mitochondria have been found (201–205). In particular, the demonstration that “positive” aspects of mitochondria can be shifted to neurons in transcelluar organelle transfer (159) indicates that negative or toxic aspects of astrocytic mitochondria might be transferred to motor neurons in sALS or fALS (206, 207), possibly via connexin 43 (159, 160).
Of interest, aligned with the “danger theory” is the endosymbiotic theory that mitochondria originated from protobacteria that entered into an endosymbiotic relationship with phagocytic, unicellular anaerobes at least a billion years ago (208), prior to the accumulation of oxygen in the atmosphere. Mitochondrial DAMPs (mtDAMPs), are protein DAMPs, coded for by mitochondrial or nuclear genes, that when released from mitochondria are potently proinflammatory (209). Most mtDAMPs are encoded by nuclear genes that after transcription translocate from nuclei to mitochondria. These mtDAMPs are then released into the circulation with infection (sepsis), trauma and/or systemic inflammatory response syndrome (SIRS). In support of this being relevant in the CNS we showed that mtDNA, a nucleic acid mtDAMP, was potently proinflammatory for neurons and microglia (210). Of interest, PCR-amplified purified mtDNA was not proinflammatory, rather only brain isolated mtDNA in the form of nucleoids bound to transcription factor of mitochondria A (TFAM), itself a mtDAMP (211, 212), was proinflammatory (211, 212). Such studies support the prediction that mitochondrial dysfunction in neurodegeneration, and neurotrauma, is tightly linked to neuroinflammation (151, 213), especially with mtDAMP involvement in neurodegeneration (214). The potential roles of mtDAMPs in neurodegeneration are shown in Figure 3.
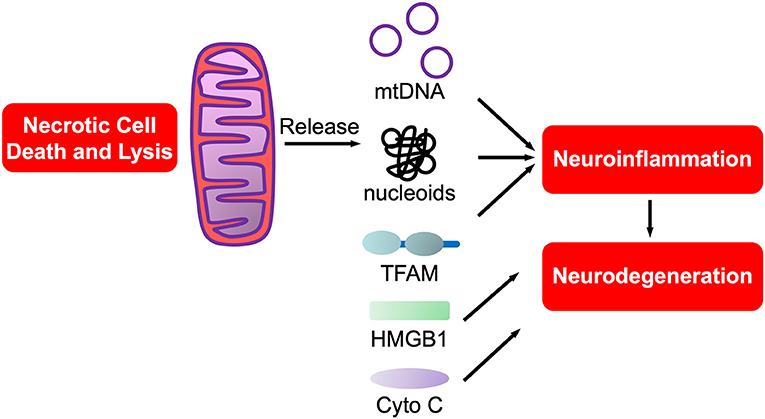
Figure 3. mtDAMPs. A sub-category of DAMPs, mtDAMPs are components of mitochondria released from necrotic or exploding cells. Comprised of mtDNA bound to mitochondrial proteins such as transcription factor of mitochondria A (TFAM), a homolog of HMGB1, in nucleoids. Also includes n-formyl peptides, cytochrome C and others (not shown).
Blood-brain Barrier (BBB)
Just as the CNS responds to PAMPs like LPS so does it respond to DAMPs, both initiating proinflammatory signaling and for the bridging or disruption of the BBB (63, 64). The history of a perceived association between ALS and BBB dysfunction actually dates back to the 1940 s when Robert Aird began a 40 year involvement with the BBB in neurologic diseases (215–219). However, it took several more decades before technology caught up with the concept that ALS was associated with BBB or blood-spinal cord barrier (BSCB) dysfunction, perhaps even at the onset of the disorder (220–231).
Evans blue extravasation from capillaries into spinal cord parenchyma was found in early symptomatic SOD1 transgenic (G93A) mice but it was uncertain whether BBB/BSCB disruption was cause or effect of motor neuron degeneration (220, 221, 223, 232, 233). More evidence suggestive of causative influences have since appeared with further studies of SOD1 transgenic mice with an eye toward therapy as well (233–235). This situation is fundamentally the same for neuroinflammation in ALS—that is, is it cause or effect? The findings that the C9orf72 expansions are also associated with myeloid cell abnormalities and early BBB dysfunction supports the role of these processes in pathogenesis (107, 236). The concept of the BBB being both target of circulating coag-inflammatory molecules as well as the source of pro-(neuro)inflammatory mediators is shown by Festoff et al. (63).
Neurotrauma and ALS
The progression of neurodegenerative disease following neurotrauma is both anecdotal and supported by epidemiologic statistics. Dementia, including AD, and microvascular dementia (mVAD), is now considered to have increased risk following traumatic brain injury (TBI), while the specific mechanistic details are still under study. Similarly, ALS occurs at an increased risk following TBI, more so, in fact than after typical SCI. Numerous mechanisms have been suggested for this association of neurotrauma and neurodegeneration including increased interest for almost 30 years in the nexus of inflammation, BBB disruption and coagulation.
Early reviews of mechanical and other forms of spinal and CNS injury associated with the development of ALS have appeared, some positive while others were negative (237, 238). Case-control studies, however, are few and those, such as that published from Olmsted County, Minnesota by Kurland and colleagues were not supportive (239). However, more recent larger population-based case control investigations, such as the Danish study, have shown an association especially with trauma at an early age (240, 241). Even broader studies such as the European EURALS consortium study (242) are giving credence to a role for trauma in ALS pathogenesis. This large study showed that more than 2 head injuries was associated with >3-fold increased risk of ALS. Although the site of injury was not important the risk was only ~2-fold when trauma occurred between 35 and 54 years of age. Certainly the age at first trauma might help to explain discrepancies in results of past studies of trauma and ALS.
In addition, studies of chronic traumatic encephalopathy (CTE) with repeated mild traumatic brain injury (mTBI) or concussions (243–245), indicate that there may be methods to identify, monitor and treat and/or prevent neurodegenerative disease development in the context of neurotrauma. With more thorough investigation into CTE and former professional athletes, an increased incidence of clinical ALS diagnosis has been reported. A recent study of CTE and CTE associated with ALS (CTE-ALS) confirmed that molecular changes co-existed pathologically. Specifically, these were phosphorylation of tau at threonine 175 (Thr175) and at Thr231 along with GSK3β were found in these tauopathies (246). Furthermore, similar findings were present in rats subjected to moderate TBI in a controlled cortical impact (CCI) model (246). These findings suggest that comparable underlying molecular mechanisms for abnormal tau phosphorylation associated with CTE neuropathologic aspects may be mimicked in a rat moderate TBI model. However, they do not provide evidence for a neurotraumatic basis for sALS.
What neurotrauma does tell us for ALS is that there is a distinct relationship between trauma, BBB disruption and neuroinflammation (247–250), all potential contributing pathogenetic factors in ALS. From the BBB disruption perspective, mechanisms involved with TBI include impact-induced shear force stress that causes initial vascular injury followed by escape of proteins along with extravasation from brain to blood as well macromolecule leakage and cell transmigration from blood to brain.
Coagulation Aspects of ALS and Other Neurodegenerative Diseases
Beginning in the 1980s through the 2000's our laboratory focused its attention on thrombin, the ultimate serine protease in the coagulation cascade, its inhibitors and receptors as specific mediators of either toxic or trophic effects on the nervous system. Our studies utilized in vitro tissue culture to probe the effects of thrombin, and inhibitors of thrombin, on neurons and glial cells. Once the thrombin receptor, subsequently named protease-activated receptor 1 (PAR1), was identified and sequenced by Coughlin's group in the early 1990 s, our studies also involved the expression of PAR1 in parts of the nervous system, in particular, the spinal cord as well as in the brain and neuromuscular system. Our translational interest was primarily in SCI and ALS, since the initial studies in neuronal types found exquisite sensitivity of spinal cord motor neurons that lead to apoptotic motor neuronal cell death in culture by thrombin cleavage of the G-protein coupled receptor (GPCR), PAR1. We also explored PARs in AD and PD as well. A number of other groups in Switzerland, Germany, Israel, Italy, Korea, China and Japan, amongst others, as well as the U.S. also began exploring coagulation and fibrinolytic proteases and inhibitors in the nervous system, especially after publication of The Maratea Meeting Proceedings in 1990 (251).
Although the emphasis of this review is on ALS similar results and concepts have been found for other neurodegenerative diseases including AD, PD, and multiple sclerosis (MS) and numerous reviews are available (252). Of interest, until recent evidence for biased signaling (see below) through PAR1 by APC was discovered (253), the previous data indicated that high thrombin concentrations were neurotoxic and pathologic in brain while low thrombin concentrations could induce neuronal and astrocytic survival after various brain insults. Interestingly, thrombin-mediated cell death and cell survival shared initial signaling proteins (48, 254).
Thrombomodulin (TM) in CNS Development, Neurotrauma, and Neurodegeneration
TM was discovered by Esmon and Owen in the 1970 s and reported in 1982 (255, 256). This discovery came after a decade or more of research that resulted in discovery of Protein C (PC), a vitamin-K dependent factor that is activated by thrombin that results in activated protein C (APC), a serine protease. Initially, the principal role of APC was thought to be its anticoagulant function whereby it proteolytically inactivated FV and FVIII (Figure 1). However, this multi-molecular system, now termed the PC–TM-EPCR (endothelial PC receptor) pathway (257, 258), is a natural mechanism to regulate hemostasis and to integrate it with other host defense system such as innate immunity, inflammation, and to control cell proliferation. Since its cloning, sequencing and chromosomal localization (259), the bulk of studies on TM have also been in terms of its role as a natural anticoagulant. However, as important as this action is, the integration by TM of hemostasis and innate immunity may determine its even greater future in disease processes that affect the CNS.
Of interest, shortly after the discovery of TM a report indicated the presence of a surface marker protein in developing mouse parietal endoderm that was modulated by cAMP (260). Shortly thereafter, fetomodulin was found to be identical to TM by contemporary gene cloning techniques (261). Thus, TM or fetomodulin (FM) is present at very early developmental stages and in adults TM expression is greatest in ECs, more predominant in small, microvascular than in large vessel ECs, and was found in essentially all ECs (262). However, the first article concerning TM and CNS vasculature was negative reporting its absence in brain ECs (263). This was not correct since it was subsequently reported that bovine as well-human brain capillaries expressed TM (264, 265), again suggesting its role as a microvascular EC marker. Not surprisingly given its early developmental appearance in parietal endoderm (FM), TM is also expressed in a number of other cells including keratinocytes, osteoblasts, monocytes, neutrophils and chondrocytes, amongst others. We first found that TM was expressed in mouse brain astrocytes, where it was functionally identical to its role in ECs (266). Subsequently, TM was found to be a novel marker of injury-induced astrogliosis, and identified the involvement of thrombin-activated PAR1 (267). This finding suggested its involvement in nervous system injury, i.e., neurotrauma. The TM gene (THBD) is intronless and is structurally separated into five distinct domains (see Figure 4). Biochemically, TM is a chondroitin sulfate proteoglycan (CSPG), and consistent with its role as a CNS injury-related CSPG would be increased in the “glial scar” and assemble along with other CSPGs such as neurocan and phosphacan that are also expressed in reactive astrocytes (268).
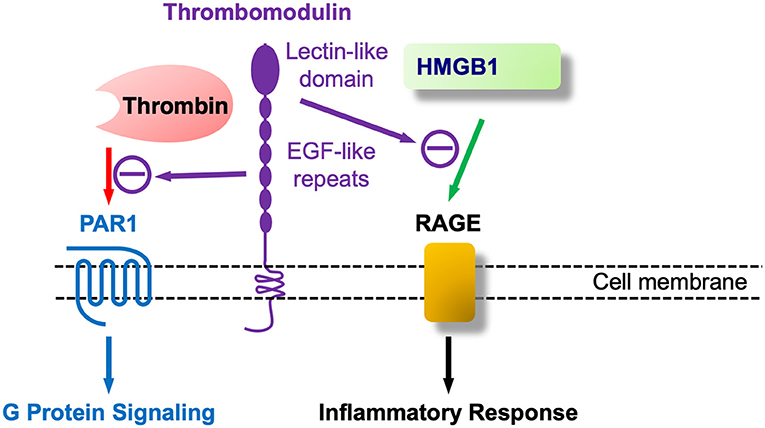
Figure 4. Thrombomodulin (TM) at nexus of coagulation and inflammation (innate immunity): TM as at least a bifunctional molecule that binds coagulation (thrombin) and proinflammatory (HMGB1) agents.
As shown in Figure 4 TM's additional role in regulating inflammation, apart from coagulation and thrombin's proinflammatory role by PAR1 activation, is largely encoded at its –NH2 terminal, known as the C-type lectin like domain (TM-CTLD). The CTLD is involved in a host of inflammatory diseases, as described in the treatise by Conway (258), one of the leaders in this field. A mechanism for the CTLD in these conditions was provided by the pioneering work of Maruyama's group that discovered that the TM-CTLD bound and neutralized the DAMP alarmin HMGB1 (269). This same group found that HMGB1 was upregulated in spinal cord parenchyma following SCI (270). Beyond its sequestering and neutralization of HMGB1 the TM-CTLD also interferes with complement activation and binds to LPS/endotoxin, and, in Gram-negative bacterial infections, to the Lewis Y antigen (271). Of interest, transgenic mice lacking the NH2-terminal CTLD (TMLeD/LeD) have heightened susceptibility to treatment with LPS (258) and should be more vulnerable to weight-drop SCI than wild type mice. The deposition of HMGB1 in the injured spinal cord was shown in rats (270), along with its release into the circulation.
The relationship between the BBB, more precisely the BSCB, and the coag-inflammation nexus in ALS merits comment. As mentioned above, although BBB/BSCB dysfunction in ALS was discussed as far back as the 1940 s, it took many decades and newer technology to establish its actual existence. The identification by Garbuzova-Davis and her colleagues that BSCB dysfunction occurred in both ALS patients and fALS SOD1 mice, prior to motor neuron degeneration (220, 221, 226–229), has been confirmed by other groups (222–225, 230, 231). Furthermore, there is a connection between BSCB dysfunction and the PC–TM-EPCR pathway, as shown by the amelioration of motor problems in SOD1 mice by treatment with non-proteolytic/non-anticoagulant APC analogs (224).
A simultaneous activation of the coagulation cascade after injury, as occurs in sepsis, is an ancient host response dating back very early in the evolution of eukaryotes. The contemporary clinical correlate happens in sepsis and injury where excessive thrombin activation develops in disseminated intravascular coagulation (DIC) associated with sepsis and sterile traumatic SIRS (272, 273). A phylogenetic clue into the nexus of clotting and inflammation comes from studies of the omnipresent East Coast North American horseshoe crab, Limulus polyphemus, with its open circulatory system containing the hemolymph, and single cell, the amoebocyte, with properties of both platelets and phagocytes. Limulus has survived for >250 million years exposed to LPS or endotoxin in the ocean from Cyanobacteria or blue-green algae where they have been for the past 2 billion years (274). The Limulus lysate detection kit for LPS in blood has been in use worldwide for over 30 years. Coagulopathy also commonly develops with TBI since the brain is a rich source of TF and thromboplastin (275).
It should be noted at this point that the thrombin→PAR1→BBB dysfunction pathogenetic pathway is not specific for ALS but occurs in all situations where intravascular prothrombin activation to α-thrombin exceeds its neutralization either by circulating anti-thrombin (AT) or EC-bound TM and the EPCR (276, 277). This dysfunction pathway would be applicable to AD, PD, ALS and neurodegeneration, in general, especially in those situations associated with antecedent trauma. In this regard, all PARs are expressed on ECs and brain microvascular ECs are no exception. However, PAR1 and PAR4 are also expressed on brain pericytes, which appear to be the most thrombin-sensitive perivascular cells to release membrane metalloprotease-9 (MMP-9) (278, 279). MMP-9 has been shown to cause BBB disruption by proteolyzing tight junction (TJ) proteins (280, 281).
Recombinant APC (rAPC; drotrecogin alfa, activated; Xigris™) was the first agent shown to stimulate PAR1-mediated cytoprotection approved for human use (in severe sepsis). However, it was voluntarily removed from the market by Eli Lilly in 2011. A number of studies have emphasized the cytoprotective role of APC, encompassing anti-apoptotic and anti-inflammatory activities, as well as significant stabilization of endothelial barriers including the BBB and BSCB. Most studies indicated this was mediated by PAR1 or PAR3 (282). All PARs are expressed on ECs (9, 283, 284) and brain microvascular ECs of the BBB should be no exception. The evidence that thrombin, via PAR1 activation, caused vascular leakage and disruption across various vascular barriers (285–287), including the BBB (288) while APC activation of PAR1 did the reverse, i.e., protection and prevention of leakage (253, 282), was a conundrum. However, as reviewed by Griffin et al. (282) this lead PAR/APC researchers to the notion of “biased signaling”, a phenomenon found in other GPCRs, a group to which PARs belong. Biased signaling through PAR1 for thrombin and APC, as conceived by Griffin et al. is shown in Figure 5: thrombin cleaves PAR1 at ARG41 in the extracellular –NH2 domain, while APC does so at ARG46 (282). At PAR1 thrombin signals through the small GTPase RhoA and ERK1/2 to disrupt, while APC through RAC1, β-arrestin and P13k/Akt to protect. This puts PAR1 on BBB ECs in a very significant position and its different proteolytic ligands to destroy or save BBB function. APC is effective in compression SCI (289) and we found that recombinant TM is also neuroprotective in rat weight-drop contusion SCI (61). More recently, Noble-Haeusslein and colleagues reported that APC biased signaling through PAR1 enhanced locomotor recovery in rat SCI (290). Zlokovic and colleagues showed that treatment with non-proteolytic/non-anticoagulant APC analogs (224) improved motor functions in SOD1 mice.
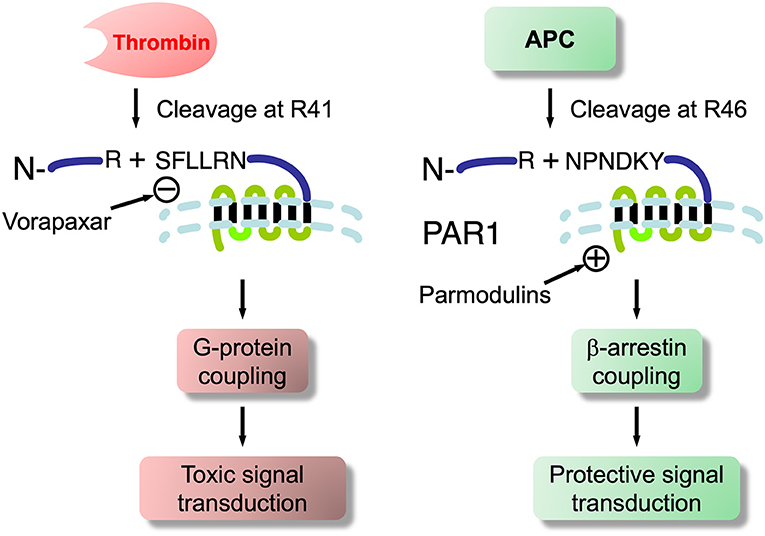
Figure 5. Biased signaling at PAR1 showing thrombin and APC cleavage sites- basis for toxicity and cytoprotection at this G-protein coupled receptor. Also shows synthetic PAR1 antagonist, vorapaxar, an FDA approved therapeutic for cardiovascular conditions. It has been shown to induce endothelial injury, which is associated with BBB/BSCB dysfunction in SOD1 mice and ALS patients. Whether ALS may be triggered in susceptible individuals so treated requires evaluation. Also shown are novel parmodulins that do not have endothelial injury side effects of vorapaxar and, thus, may have therapeutic application to ALS and neurodegenerations.
PARs in ALS and Other Neurodegenerative Diseases
We found that nM thrombin concentrations induced tau neurofibrillary tangle-like aggregates (NFTs) in murine hippocampal neurons and that this required PAR activation that was followed by delayed synaptophysin reduction and apoptotic neuronal death (291). Subsequently, others showed that the initial fragmentation of tau, necessary to then cause aggregation into NFTs, was due to a thrombin-like protease (292). These authors wrote that fragmentation by a thrombin-like protease was a “prelude” to aggregation, although phosphorylation was not. HIV-associated neurodegeneration (HAND) was also shown to require thrombin and PAR1 expression in astrocytes, as subsequently reviewed (293). McGeer and colleagues then showed that thrombin, as well as prothrombin, accumulated with NFTs in the brains of AD patients (294). Additional evidence for thrombin and PAR1 in neurodegeneration was provided by others in AD (295, 296) and PD (297) and then reviewed as well (50).
Our preliminary data indicated that PARs were increased and active in several murine ALS models in which microglia express increased monocyte chemoattractant protein 1 (MCP-1) and other markers. In regards to neurotrauma, we found that SCI was accompanied by an early and significant upregulation of neurotoxic serine proteases, prothrombin, and PAR1 in the rat spinal cord (298). It was subsequently reported that thrombin-recruited microglia also express MCP-1 (now CCL2) and that PAR1 activation is required for this (299).
The wobbler mouse is a model of motor neuron disease sharing many features with ALS, including loss of spinal motor neurons, neuromuscular loss of function over time, and TDP-43 aggregates and C-terminal fragments identical to those seen in the sporadic form of ALS (300). By optimizing transcription and quantitative PCR procedures to facilitate rapid copy number determination in small RNA samples, we documented a 5-fold greater level of PAR1 mRNA in the cervical spinal cord of wobbler (wr/wr) compared to wild type mice (301). Then we subsequently confirmed and extended these results showing that PAR1 mRNA was dramatically increased in spinal cord alpha motor neurons in homozygous, spontaneously mutant autosomal recessive wr/wr mice (302). The gene for wobbler mutation is located on mouse chromosome 11 (303) and was since shown to be a point mutation on Vps54 (vacuolar protein sorting 54) involved with the Golgi apparatus (304). Even before the gene was determined an informative marker at the wobbler locus, the glutamine synthase 1 (glns-1) pseudogene, permitted genotyping mice prior to phenotype development as previously described (305). Using this technique, we found that homozygotes expressed an 8-fold increase in PAR1 message by P8, more than 2 weeks prior to phenotype detection and this appeared primarily in motor neurons (301–303, 305, 306). These earlier studies focused attention on potential roles of coagulation proteases and PARs in the nervous system but took some time before they had generated additional interest in pursuing direct connections between them, neurotrauma, neuroinflammation and neurodegeneration.
Following our earlier reviews (47, 48, 251, 307) more recent efforts have emphasized the participation of coagulation in various neuroinflammatory diseases of the CNS (64, 308–310). Connections between tissue transglutaminase (tTG), in the same family as the clotting cascade Factor XIII, cross-linking and neuroinflammation in ALS also exist. SCI has been shown to upregulate cytokines, microglia and tTG (311). In addition, we found that SCI induced a “switch” from a GTPase function of tTG to a novel GTP-independent cross-linking isoform in the spinal cord (311). More recently, tTG has been implicated in promoting neuroinflammation in SOD1 mice (312). It would be of interest to determine whether tTG upregulation was present in ALS spinal cord and if alternative transcription to a short isoform existed.
Epilog, and Possibly, Prolog (to the Next Phase)
Vorapaxar is a natural product-based orthosteric antagonist of thrombin-induced PAR1 that inhibits all signaling downstream (313). The FDA approved it for post-myocardial infarction following success in two large pivotal multi-center Phase III outcome clinical trials in patients with coronary atherothrombosis. It has a low molecular weight (590.7) and a long effective half-life (3–4 days).
Surprisingly, the FDA review of the adverse events for both Phase III clinical trials revealed an increased number of ALS diagnoses in the vorapaxar arm compared to the placebo arm (314). This adverse event was not mentioned in the publication of the results and this vorapaxar-ALS association may recall the studies we and others carried out with thrombin, PAR1, thrombospondin, TM and related components of the coag-inflamm system in development, neurotrauma, ALS and other neurodegenerative disorders, as described above (see Table 1). Since vorapaxar appears to inhibit all signaling downstream of the PAR1 GPCR it would seem that is where attention should be paid for clues to ALS pathogenesis related to it. Although it is still a relatively rare occurrence after vorapaxar we would hope that knowledge of this small but surprising ALS signal after vorapaxar will uncover novel therapeutic targets for this enigmatic and fatal neurodegenerative disorder and related disorders where synapse retraction may be the earliest pathophysiologic sign of the disease (22, 24–26) and where thrombin→PAR1 activation may well play a role (51, 52).
In this regard, recent development of small molecule PAR antagonists termed parmodulins (315, 316) are based on findings that biased signaling peptides developed around APC are cytoprotective at PAR1 and not anticoagulant (282). We hope that such research will help advance whether or not potential neuronal degeneration and/or impaired neuromuscular activity is a class-specific adverse effect after PAR antagonists (317).
Author Contributions
BF conceived of the review, and was lead on the evaluation of the literature and writing. BC also contributed to the reviewing, writing, and figures.
Conflict of Interest Statement
BF is the Founder of PHLOGISTIX LLC, a startup biotech company.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. (2007) 27:1687–93. doi: 10.1161/ATVBAHA.107.141911
3. Gando S. Disseminated intravascular coagulation in trauma patients. Semi. Thrombosis Hemostasis (2001) 27:585–92. doi: 10.1055/s-2001-18864
4. Opal SM. The nexus between systemic inflammation and disordered coagulation in sepsis. J. Endotoxin Res. (2004) 10:125–9. doi: 10.1177/09680519040100020901
5. Kirchhofer D, Nemerson Y. Initiation of blood coagulation: the tissue factor/factor VIIa complex. Curr Opin Biotechnol. (1996) 7:386–91. doi: 10.1016/S0958-1669(96)80112-1
6. Shavit Stein E, Ben Shimon M, Artan Furman A, Golderman V, Chapman J, Maggio N. Thrombin inhibition reduces the expression of brain inflammation markers upon systemic LPS treatment. Neural Plast. (2018) 2018:7692182. doi: 10.1155/2018/7692182
7. Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci USA. (1999) 96:11023–7. doi: 10.1073/pnas.96.20.11023
8. Derian CK, Damiano BP, D'Andrea MR, Andrade-Gordon P. Thrombin regulation of cell function through protease-activated receptors: implications for therapeutic intervention. Biochemistry (2002) 67:56–64. doi: 10.1023/A:1013900130415
9. Hollenberg MD. Proteinase-mediated signaling: proteinase-activated receptors (PARs) and much more. Life Sci. (2003) 74:237–46. doi: 10.1016/j.lfs.2003.09.010
10. Snider RM, Richelson E. Thrombin stimulation of guanosine 3′,5′-monophosphate formation in murine neuroblastoma cells (clone N1E-115). Science (1983) 221:566–8. doi: 10.1126/science.6306770
11. Means ED, Anderson DK. Thrombin interactions with central nervous system tissue and implications of these interactions. Ann N Y Acad Sci. (1986) 485:314–22. doi: 10.1111/j.1749-6632.1986.tb34593.x
12. Snider RM. Thrombin effects on cultured nerve cells: clinical implications and evidence for a novel mechanism of neuronal activation. Ann N Y Acad Sci. (1986) 485:310–3. doi: 10.1111/j.1749-6632.1986.tb34592.x
13. Gurwitz D, Cunningham DD. Thrombin modulates and reverses neuroblastoma neurite outgrowth. Proc Natl Acad Sci USA. (1988) 85:3440–4. doi: 10.1073/pnas.85.10.3440
14. Rowland LP. Diagnosis of amyotrophic lateral sclerosis. J Neurol Sci. (1998) 160 (Suppl. 1):S6–24. doi: 10.1016/S0022-510X(98)00193-2
15. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. (2001) 344:1688–700. doi: 10.1056/NEJM200105313442207
16. Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. (2004) 27:723–49. doi: 10.1146/annurev.neuro.27.070203.144244
17. Charcot JM. Sclerose des cordons lateraux de la moelle epiniere chez femme hysterique atteinte de contracture permanente des quatre membres. Bull Soc Med Hop Paris (1865) 2(Suppl. 2):24–42.
18. Writing G, Edaravone ALSSG. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2017) 16:505–12. doi: 10.1016/S1474-4422(17)30115-1
19. Fernandez HL, Duell MJ, Festoff BW. Neurotrophic control of 16S acetylcholinesterase at the vertebrate neuromuscular junction. J Neurobiol. (1979) 10:441–54. doi: 10.1002/neu.480100503
20. Fernandez HL, Duell MJ, Festoff BW. Cellular distribution of 16S acetylcholinesterase. J Neurochem. (1979) 32:581–5. doi: 10.1111/j.1471-4159.1979.tb00387.x
21. Fernandez HL, Duell MJ, Festoff BW. Bidirectional axonal transport of 16S acetylcholinesterase in rat sciatic nerve. J Neurobiol. (1980) 11:31–9. doi: 10.1002/neu.480110105
22. Festoff BW. Neuromuscular junction macromolecules in the pathogenesis of amyotrophic leteral sclerosis. Med Hypotheses. (1980) 6:121–31. doi: 10.1016/0306-9877(80)90078-X
23. Festoff BW, Fernandez HL. Plasma and red blood cell acetylcholinesterase in amyotrophic lateral sclerosis. Muscle Nerve (1981) 4:41–7. doi: 10.1002/mus.880040108
24. Festoff BW. Release of acetylcholinesterase in amyotrophic lateral sclerosis. Adv Neurol. (1982) 36:503–17.
25. Campanari ML, Garcia-Ayllon MS, Ciura S, Saez-Valero J, Kabashi E. Neuromuscular junction impairment in amyotrophic lateral sclerosis: reassessing the role of acetylcholinesterase. Front Mol Neurosci. (2016) 9:160. doi: 10.3389/fnmol.2016.00160
26. Cappello V, Francolini M. Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int J Mol Sci. (2017) 18:E2092. doi: 10.3390/ijms18102092
27. Festoff BW, Hantai D, Soria J, Thomaidis A, Soria C. Plasminogen activator in mammalian skeletal muscle: characteristics of effect of denervation on urokinase-like and tissue activator. J Cell Biol. (1986) 103:1415–21. doi: 10.1083/jcb.103.4.1415
28. Festoff BW. Proteases, their inhibitors and the extracellular matrix: factors in nerve-muscle development and maintenance. Adv Exp Med Biol. (1987) 209:25–39. doi: 10.1007/978-1-4684-5302-7_6
29. Festoff BW, Rao JS, Maben C, Hantai D. Plasminogen activators and their inhibitors in the neuromuscular system: I. developmental regulation of plasminogen activator isoforms during in vitro myogenesis in two cell lines. J Cell Physiol. (1990) 144:262–71. doi: 10.1002/jcp.1041440212
30. Festoff BW, Rao JS, Rayford A, Hantai D. Plasminogen activators and their inhibitors in the neuromuscular system: II. serpins and serpin: protease complex receptors increase during in vitro myogenesis. J Cell Physiol. (1990) 144:272–9. doi: 10.1002/jcp.1041440213
31. Festoff BW, Rao JS, Hantai D. Plasminogen activators and inhibitors in the neuromuscular system: III. The serpin protease nexin I is synthesized by muscle and localized at neuromuscular synapses. J Cell Physiol. (1991) 147:76–86. doi: 10.1002/jcp.1041470111
32. Hantai D, Rao JS, Festoff BW. Rapid neural regulation of muscle urokinase-like plasminogen activator as defined by nerve crush. Proc Natl Acad Sci USA. (1990) 87:2926–30. doi: 10.1073/pnas.87.8.2926
33. Hantai D, Rao JS, Kahler C, Festoff BW. Decrease in plasminogen activator correlates with synapse elimination during neonatal development of mouse skeletal muscle. Proc Natl Acad Sci USA. (1989) 86:362–6. doi: 10.1073/pnas.86.1.362
34. Tian WH, Festoff BW, Blot S, Diaz J, Hantai D. Synaptic transmission blockade increases plasminogen activator activity in mouse skeletal muscle poisoned with botulinum toxin type A. Synapse (1995) 20:24–32. doi: 10.1002/syn.890200105
35. Rao JS, Chen M, Festoff BW. Plasminogen activator inhibitor 1, the primary regulator of fibrinolysis, in normal human cerebrospinal fluid. J Neurosci Res. (1993) 34:340–5. doi: 10.1002/jnr.490340311
36. Festoff BW, Rao JS, Chen M. Protease nexin I, thrombin- and urokinase-inhibiting serpin, concentrated in normal human cerebrospinal fluid. Neurology (1992) 42:1361–6. doi: 10.1212/WNL.42.7.1361
37. Blondet B, Barlovatz-Meimon G, Festoff BW, Soria C, Soria J, Rieger F, et al. Plasminogen activators in the neuromuscular system of the wobbler mutant mouse. Brain Res. (1992) 580:303–10. doi: 10.1016/0006-8993(92)90958-C
38. Magnusson C, Hogklint L, Libelius R, Tagerud S. Expression of mRNA for plasminogen activators and protease nexin-1 in innervated and denervated mouse skeletal muscle. J Neurosci Res. (2001) 66:457–63. doi: 10.1002/jnr.10000
39. Smirnova IV, Ma JY, Citron BA, Ratzlaff KT, Gregory EJ, Akaaboune M, et al. Neural thrombin and protease nexin I kinetics after murine peripheral nerve injury. J Neurochem. (1996) 67:2188–99. doi: 10.1046/j.1471-4159.1996.67052188.x
40. Festoff BW, Reddy RB, VanBecelaere M, Smirnova I, Chao J. Activation of serpins and their cognate proteases in muscle after crush injury. J Cell Physiol. (1994) 159:11–8. doi: 10.1002/jcp.1041590103
41. Citron BA, Suo Z, SantaCruz K, Davies PJ, Qin F, Festoff BW. Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int. (2002) 40:69–78. doi: 10.1016/S0197-0186(01)00062-6
42. Festoff BW, Suo Z, Citron BA. Plasticity and stabilization of neuromuscular and CNS synapses: interactions between thrombin protease signaling pathways and tissue transglutaminase. Int Rev Cytol. (2001) 211:153–77. doi: 10.1016/S0074-7696(01)11018-1
43. Citron BA, Gregory EJ, Steigerwalt DS, Qin F, Festoff BW. Regulation of the dual function tissue transglutaminase/Galpha(h) during murine neuromuscular development: gene and enzyme isoform expression. Neurochem Int. (2000) 37:337–49. doi: 10.1016/S0197-0186(00)00044-9
44. Turgeon VL, Lloyd ED, Wang S, Festoff BW, Houenou LJ. Thrombin perturbs neurite outgrowth and induces apoptotic cell death in enriched chick spinal motoneuron cultures through caspase activation. J Neurosci. (1998) 18:6882–91. doi: 10.1523/JNEUROSCI.18-17-06882.1998
45. Chou SM, Taniguchi A, Wang HS, Festoff BW. Serpin = serine protease-like complexes within neurofilament conglomerates of motoneurons in amyotrophic lateral sclerosis. J Neurol Sci. (1998) 160 (Suppl. 1):S73–9. doi: 10.1016/S0022-510X(98)00202-0
46. Houenou LJ, Turner PL, Li L, Oppenheim RW, Festoff BW. A serine protease inhibitor, protease nexin I, rescues motoneurons from naturally occurring and axotomy-induced cell death. Proc Natl Acad Sci USA. (1995) 92:895–9. doi: 10.1073/pnas.92.3.895
47. Smirnova IV, Ho GJ, Fenton JW II, Festoff BW. Extravascular proteolysis and the nervous system: serine protease/serpin balance. Semi Thrombosis Hemostasis (1994) 20:426–32. doi: 10.1055/s-2007-1001931
48. Ho GJ, Smirnova IV, Akaaboune M, Hantai D, Festoff BW. Serine proteases and their serpin inhibitors in Alzheimer's disease. Biomed Pharmacother. (1994) 48:296–304. doi: 10.1016/0753-3322(94)90175-9
49. Almonte AG, Sweatt JD. Serine proteases, serine protease inhibitors, and protease-activated receptors: roles in synaptic function and behavior. Brain Res. (2011) 1407:107–22. doi: 10.1016/j.brainres.2011.06.042
50. Krenzlin H, Lorenz V, Danckwardt S, Kempski O, Alessandri B. The importance of thrombin in cerebral injury and disease. Int J Mol Sci. (2016) 17:E84. doi: 10.3390/ijms17010084
51. Liu Y, Fields RD, Fitzgerald S, Festoff BW, Nelson PG. Proteolytic activity, synapse elimination, and the Hebb synapse. J Neurobiol. (1994) 25:325–35. doi: 10.1002/neu.480250312
52. Zoubine MN, Ma JY, Smirnova IV, Citron BA, Festoff BW. A molecular mechanism for synapse elimination: novel inhibition of locally generated thrombin delays synapse loss in neonatal mouse muscle. Dev Biol. (1996) 179:447–57. doi: 10.1006/dbio.1996.0274
53. Watkins SC, Lynch GW, Kane LP, Slayter HS. Thrombospondin expression in traumatized skeletal muscle. correlation of appearance with post-trauma regeneration. Cell Tissue Res. (1990) 261:73–84. doi: 10.1007/BF00329440
54. Smirnova IV, Festoff BW. Alterations in serum thrombospondin in patients with amyotrophic lateral sclerosis. J Neurol Sci. (1994) 127:207–13. doi: 10.1016/0022-510X(94)90074-4
55. Rao JS, Hantai D, Festoff BW. Thrombospondin, a platelet alpha-granule and matrix glycoprotein, is increased in muscle basement membrane of patients with amyotrophic lateral sclerosis. J Neurol Sci. (1992) 113:99–107. doi: 10.1016/0022-510X(92)90271-L
56. Hantai D, Rao JS, Reddy BR, Festoff BW. Developmental appearance of thrombospondin in neonatal mouse skeletal muscle. Eur J Cell Biol. (1991) 55:286–94.
57. Ho GJ, Gregory EJ, Smirnova IV, Zoubine MN, Festoff BW. Cross-linking of beta-amyloid protein precursor catalyzed by tissue transglutaminase. FEBS Lett. (1994) 349:151–4. doi: 10.1016/0014-5793(94)00663-6
58. Vanhoutte D, Schips TG, Kwong JQ, Davis J, Tjondrokoesoemo A, Brody MJ, et al. Thrombospondin expression in myofibers stabilizes muscle membranes. Elife (2016) 5:e17589. doi: 10.7554/eLife.17589
59. Wang B, Guo W, Huang Y. Thrombospondins and synaptogenesis. Neural Regen Res. (2012) 7:1737–43. doi: 10.3969/j.issn.1673-5374.2012.22.009
60. Festoff BW. Designing drugs that encourage spinal cord injury healing. Exp Opin Drug Discov. (2014) 9:1151–65. doi: 10.1517/17460441.2014.941350
61. Festoff BW, Ameenuddin S, Santacruz K, Morser J, Suo Z, Arnold PM, et al. Neuroprotective effects of recombinant thrombomodulin in controlled contusion spinal cord injury implicates thrombin signaling. J Neurotrauma. (2004) 21:907–22. doi: 10.1089/0897715041526168
62. Festoff BW, Li C, Woodhams B, Lynch S. Soluble thrombomodulin levels in plasma of multiple sclerosis patients and their implication. J Neurol Sci. (2012) 323:61–5. doi: 10.1016/j.jns.2012.08.008
63. Festoff BW, Sajja RK, van Dreden P, Cucullo L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer's disease. J Neuroinflam. (2016) 13:194. doi: 10.1186/s12974-016-0670-z
64. Festoff BW, Sajja RK, Cucullo L. Proximate mediators of microvascular dysfunction at the blood-brain barrier: neuroinflammatory pathways to neurodegeneration. BioMed Res Int. (2017) 2017:1549194. doi: 10.1155/2017/1549194
65. Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature (1993) 364:362. doi: 10.1038/364362c0
67. Brown RH Jr. Amyotrophic lateral sclerosis. insights from genetics. Arch Neurol. (1997) 54:1246–50. doi: 10.1001/archneur.1997.00550220050013
68. Ajroud-Driss S, Siddique T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta. (2015) 1852:679–84. doi: 10.1016/j.bbadis.2014.08.010
69. Borel F, Gernoux G, Cardozo B, Metterville JP, Toro Cabrera GC, Song L, et al. Therapeutic rAAVrh10 mediated SOD1 silencing in adult SOD1(G93A) mice and nonhuman primates. Hum Gene Ther. (2016) 27:19–31. doi: 10.1089/hum.2015.122
70. Browne EC, Abbott BM. Recent progress towards an effective treatment of amyotrophic lateral sclerosis using the SOD1 mouse model in a preclinical setting. Eur J Med Chem. (2016) 121:918–25. doi: 10.1016/j.ejmech.2016.02.048
71. van Zundert B, Brown RH Jr. Silencing strategies for therapy of SOD1-mediated ALS. Neurosci Lett. (2017) 636:32–9. doi: 10.1016/j.neulet.2016.07.059
72. Zhong Y, Wang J, Henderson MJ, Yang P, Hagen BM, Siddique T, et al. Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. Elife (2017) 6:023759. doi: 10.7554/eLife.23759
73. Bali T, Self W, Liu J, Siddique T, Wang LH, Bird TD, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry (2017) 88:99–105. doi: 10.1136/jnnp-2016-313521
74. Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ. Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol. (2012) 8:518–30. doi: 10.1038/nrneurol.2012.156
75. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
76. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
77. Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain (2012) 135:751–64. doi: 10.1093/brain/awr365
78. Alsultan A, Waller R, Heath PR, Kirby J. The genetics of amyotrophic lateral sclerosis: current insights. Degen Neurol Neuromusc Dis. (2016) 6:49–64. doi: 10.2147/DNND.S84956
79. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science (2008) 319:1668–72. doi: 10.1126/science.1154584
80. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. (2008) 40:572–4. doi: 10.1038/ng.132
81. Gendron TF, Rademakers R, Petrucelli L. TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J Alzheimer's Dis. (2013) 33 (Suppl. 1):S35–45. doi: 10.3233/JAD-2012-129036
82. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. (2009) 323:1208–11. doi: 10.1126/science.1165942
83. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (2009) 323:1205–8. doi: 10.1126/science.1166066
84. Robertson J, Sanelli T, Xiao S, Yang W, Horne P, Hammond R, et al. Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett. (2007) 420:128–32. doi: 10.1016/j.neulet.2007.03.066
85. Ramanan VK, Saykin AJ. Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer's disease, Parkinson's disease, and related disorders. Am J Neurodegener Dis. (2013) 2:145–75.
86. Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. (2015) 1607:75–93. doi: 10.1016/j.brainres.2014.10.009
87. Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron (2018) 97:1268–83 e6. doi: 10.1016/j.neuron.2018.02.027
88. Gottesman II, Shields J. Genetic theorizing and schizophrenia. Br J Psychiatry (1973) 122:15–30. doi: 10.1192/bjp.122.1.15
89. Miller GA, Rockstroh B. Endophenotypes in psychopathology research: where do we stand? Annu Rev Clin Psychol. (2013) 9:177–213. doi: 10.1146/annurev-clinpsy-050212-185540
90. Rosen AM, Spellman T, Gordon JA. Electrophysiological endophenotypes in rodent models of schizophrenia and psychosis. Biol Psychiatry (2015) 77:1041–9. doi: 10.1016/j.biopsych.2015.03.021
91. Reitz C, Mayeux R. Endophenotypes in normal brain morphology and Alzheimer's disease: a review. Neuroscience (2009) 164:174–90. doi: 10.1016/j.neuroscience.2009.04.006
92. Berti V, Nacmias B, Bagnoli S, Sorbi S. Alzheimer's disease: genetic basis and amyloid imaging as endophenotype. Q J Nucl Med Mol Imaging (2011) 55:225–36.
93. Looi JC, Walterfang M, Velakoulis D, Macfarlane MD, Svensson LA, Wahlund LO. Frontotemporal dementia as a frontostriatal disorder: neostriatal morphology as a biomarker and structural basis for an endophenotype. Aust N Z J Psychiatry (2012) 46:422–34. doi: 10.1177/0004867411432076
94. Pan XD, Chen XC. Clinic, neuropathology and molecular genetics of frontotemporal dementia: a mini-review. Transl Neurodegener. (2013) 2:8. doi: 10.1186/2047-9158-2-8
95. Cooper-Knock J, Shaw PJ, Kirby J. The widening spectrum of C9ORF72-related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathologica (2014) 127:333–45. doi: 10.1007/s00401-014-1251-9
96. Prado LGR, Bicalho ICS, Magalhaes D, Caramelli P, Teixeira AL, de Souza LC. C9ORF72 and the FTD-ALS spectrum: a systematic review of neuroimaging studies. Dement Neuropsychol. (2015) 9:413–21. doi: 10.1590/1980-57642015DN94000413
97. Ji AL, Zhang X, Chen WW, Huang WJ. Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J Med Genet. (2017) 54:145–54. doi: 10.1136/jmedgenet-2016-104271
98. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. subcommittee on motor neuron diseases/amyotrophic lateral sclerosis of the world federation of neurology research group on neuromuscular diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. (1994) 124 (Suppl):96–107. doi: 10.1016/0022-510X(94)90191-0
99. Belsh JM. ALS diagnostic criteria of El Escorial Revisited: do they meet the needs of clinicians as well as researchers? Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) (Suppl. 1):S57–60. doi: 10.1080/14660820052415925
100. Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
101. Hudson AJ. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain (1981) 104:217–47. doi: 10.1093/brain/104.2.217
102. Lee SE. Guam dementia syndrome revisited in 2011. Curr Opin Neurol. (2011) 24:517–24. doi: 10.1097/WCO.0b013e32834cd50a
103. McGeer PL, Steele JC. The ALS/PDC syndrome of Guam: potential biomarkers for an enigmatic disorder. Progress Neurobiol. (2011) 95:663–9. doi: 10.1016/j.pneurobio.2011.04.001
104. Strong MJ. The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotrop Lateral Sclerosis. (2008) 9:323–38. doi: 10.1080/17482960802372371
105. Achi EY, Rudnicki SA. ALS and frontotemporal dysfunction: a review. Neurol Res Int. (2012) 2012:806306. doi: 10.1155/2012/806306
106. Liscic RM. Molecular basis of ALS and FTD: implications for translational studies. Arh Hig Rada Toksikol. (2015) 66:285–90. doi: 10.1515/aiht-2015-66-2679
107. Lall D, Baloh RH. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J Clin Invest. (2017) 127:3250–8. doi: 10.1172/JCI90607
108. Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS ONE (2013) 8:e83450. doi: 10.1371/journal.pone.0083450
109. Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. (2018) 24:313–25. doi: 10.1038/nm.4490
110. Kramer NJ, Haney MS, Morgens DW, Jovicic A, Couthouis J, Li A, et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat Genet. (2018) 50:603–12. doi: 10.1038/s41588-018-0070-7
111. Karch CM, Wen N, Fan CC, Yokoyama JS, Kouri N, Ross OA, et al. Selective genetic overlap between amyotrophic lateral sclerosis and diseases of the frontotemporal dementia spectrum. JAMA Neurol. (2018) 75:860–75. doi: 10.1001/jamaneurol.2018.0372
112. Mendez EF, Sattler R. Biomarker development for C9orf72 repeat expansion in ALS. Brain Res. (2015) 1607:26–35. doi: 10.1016/j.brainres.2014.09.041
113. Vu LT, Bowser R. Fluid-based biomarkers for amyotrophic lateral sclerosis. Neurotherapeutics (2017) 14:119–34. doi: 10.1007/s13311-016-0503-x
114. Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron (2006) 52:39–59. doi: 10.1016/j.neuron.2006.09.018
115. Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. (2009) 187:761–72. doi: 10.1083/jcb.200908164
116. Rizzo F, Riboldi G, Salani S, Nizzardo M, Simone C, Corti S, et al. Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell Mol Life Sci. (2014) 71:999–1015. doi: 10.1007/s00018-013-1480-4
117. Brites D, Vaz AR. Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci. (2014) 8:117. doi: 10.3389/fncel.2014.00117
118. Yamanaka K, Komine O. The multi-dimensional roles of astrocytes in ALS. Neurosci Res. (2018) 126:31–8. doi: 10.1016/j.neures.2017.09.011
119. Lino MM, Schneider C, Caroni P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci. (2002) 22:4825–32. doi: 10.1523/JNEUROSCI.22-12-04825.2002
120. Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. (2007) 10:1355–60. doi: 10.1038/nn1988
121. Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. (2008) 11:251–3. doi: 10.1038/nn2047
122. Puentes F, Malaspina A, van Noort JM, Amor S. Non-neuronal cells in ALS: Role of glial, immune cells and blood-CNS barriers. Brain Pathol. (2016) 26:248–57. doi: 10.1111/bpa.12352
123. Qosa H, Lichter J, Sarlo M, Markandaiah SS, McAvoy K, Richard JP, et al. Astrocytes drive upregulation of the multidrug resistance transporter ABCB1 (P-Glycoprotein) in endothelial cells of the blood-brain barrier in mutant superoxide dismutase 1-linked amyotrophic lateral sclerosis. Glia (2016) 64:1298–313. doi: 10.1002/glia.23003
124. Serrano A, Donno C, Giannetti S, Peric M, Andjus P, D'Ambrosi N, et al. The astrocytic S100B protein with its receptor RAGE is aberrantly expressed in SOD1(G93A) models, and its inhibition decreases the expression of proinflammatory Genes. Mediators Inflamm. (2017) 2017:1626204. doi: 10.1155/2017/1626204
125. Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist (2005) 11:400–7. doi: 10.1177/1073858405278321
126. Lukovic D, Stojkovic M, Moreno-Manzano V, Jendelova P, Sykova E, Bhattacharya SS, et al. Concise review: reactive astrocytes and stem cells in spinal cord injury: good guys or bad guys? Stem Cells (2015) 33:1036–41. doi: 10.1002/stem.1959
127. Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. (2015) 16:249–63. doi: 10.1038/nrn3898
128. Hennessy E, Griffin EW, Cunningham C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1beta and TNF-alpha. J Neurosci. (2015) 35:8411–22. doi: 10.1523/JNEUROSCI.2745-14.2015
129. Radulovic M, Yoon H, Wu J, Mustafa K, Scarisbrick IA. Targeting the thrombin receptor modulates inflammation and astrogliosis to improve recovery after spinal cord injury. Neurobiol Dis. (2016) 93:226–42. doi: 10.1016/j.nbd.2016.04.010
130. Okada S, Hara M, Kobayakawa K, Matsumoto Y, Nakashima Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci Res. (2018) 126:39–43. doi: 10.1016/j.neures.2017.10.004
131. Ahmed S, Gull A, Khuroo T, Aqil M, Sultana Y. Glial cell: A potential target for cellular and drug based therapy in various CNS diseases. Curr Pharm Des. (2017) 23:2389–99. doi: 10.2174/1381612823666170316124500
132. Almer G, Guegan C, Teismann P, Naini A, Rosoklija G, Hays AP, et al. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol. (2001) 49:176–85. doi: 10.1002/1531-8249(20010201)49:2<176::AID-ANA37>3.0.CO;2-X
133. Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem. (2001) 77:132–45. doi: 10.1046/j.1471-4159.2001.t01-1-00231.x
134. Hensley K, Floyd RA, Gordon B, Mou S, Pye QN, Stewart C, et al. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J Neurochem. (2002) 82:365–74. doi: 10.1046/j.1471-4159.2002.00968.x
135. McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve (2002) 26:459–70. doi: 10.1002/mus.10191
136. Appel SH, Smith RG, Engelhardt JI, Stefani E. Evidence for autoimmunity in amyotrophic lateral sclerosis. J Neurol Sci. (1994) 124 (Suppl):14–9. doi: 10.1016/0022-510X(94)90171-6
137. Smith RG, Alexianu ME, Crawford G, Nyormoi O, Stefani E, Appel SH. Cytotoxicity of immunoglobulins from amyotrophic lateral sclerosis patients on a hybrid motoneuron cell line. Proc Natl Acad Sci USA. (1994) 91:3393–7. doi: 10.1073/pnas.91.8.3393
138. Smith RG, Appel SH. Immunosuppression and anti-inflammatory agents in ALS. Amyotroph Later Scler Motor Neuron Disord. (2000) 1 (Suppl. 4):33–42. doi: 10.1080/14660820050515692
139. Engelhardt JI, Siklos L, Komuves L, Smith RG, Appel SH. Antibodies to calcium channels from ALS patients passively transferred to mice selectively increase intracellular calcium and induce ultrastructural changes in motoneurons. Synapse (1995) 20:185–99. doi: 10.1002/syn.890200302
140. Appel SH, Smith RG, Alexianu MF, Engelhardt JI, Stefani E. Autoimmunity as an etiological factor in sporadic amyotrophic lateral sclerosis. Adv Neurol. (1995) 68:47–57.
141. Kimura F, Smith RG, Delbono O, Nyormoi O, Schneider T, Nastainczyk W, et al. Amyotrophic lateral sclerosis patient antibodies label Ca2+ channel alpha 1 subunit. Ann Neurol. (1994) 35:164–71. doi: 10.1002/ana.410350207
142. Hoffman PM, Festoff BW, Giron LT Jr, Hollenbeck LC, Garruto RM, Ruscetti FW. Isolation of LAV/HTLV-III from a patient with amyotrophic lateral sclerosis. N Engl J Med. (1985) 313:324–5. doi: 10.1056/NEJM198508013130511
143. Galassi G, Gentilini M, Ferrari S, Ficarra G, Zonari P, Mongiardo N, et al. Motor neuron disease and HIV-1 infection in a 30-year-old HIV-positive heroin abuser: a causal relationship? Clin Neuropathol. (1998) 17:131–5.
144. Ahmad K. HIV may underlie ALS-like condition. Lancet Infect Dis. (2001) 1:217. doi: 10.1016/S1473-3099(01)00108-6
145. MacGowan DJ, Scelsa SN, Waldron M. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology (2001) 57:1094–7. doi: 10.1212/WNL.57.6.1094
146. von Giesen HJ, Kaiser R, Koller H, Wetzel K, Arendt G. Reversible ALS-like disorder in HIV infection. an ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology (2002) 59:474; author reply:5.
147. Cone LA, Nazemi R, Cone MO. Reversible ALS-like disorder in HIV infection. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology (2002) 59:474; author reply:5.
148. Griffiths M, Neal JW, Gasque P. Innate immunity and protective neuroinflammation: new emphasis on the role of neuroimmune regulatory proteins. Int Rev Neurobiol. (2007) 82:29–55. doi: 10.1016/S0074-7742(07)82002-2
149. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. (2012) 122:1164–71. doi: 10.1172/JCI58644
150. Ransohoff RM, Schafer D, Vincent A, Blachere NE, Bar-Or A. Neuroinflammation: ways in which the immune system affects the brain. Neurotherapeutics (2015) 12:896–909. doi: 10.1007/s13311-015-0385-3
151. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science (2016) 353:777–83. doi: 10.1126/science.aag2590
152. Masgrau R, Guaza C, Ransohoff RM, Galea E. Should we stop saying ‘Glia’ and ‘Neuroinflammation’? Trends Mol Med. (2017) 23:486–500. doi: 10.1016/j.molmed.2017.04.005
153. Geloso MC, Corvino V, Marchese E, Serrano A, Michetti F, D'Ambrosi N. The dual role of microglia in ALS: mechanisms and therapeutic approaches. Front Aging Neurosci. (2017) 9:242. doi: 10.3389/fnagi.2017.00242
154. Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG, et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci. (2018) 21:329–40. doi: 10.1038/s41593-018-0083-7
155. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
156. Yuva-Aydemir Y, Almeida S, Gao FB. Insights into C9ORF72-Related ALS/FTD from Drosophila and iPSC Models. Trends Neurosci. (2018) 41:457–69. doi: 10.1016/j.tins.2018.04.002
157. Tang BL. Patient-Derived iPSCs and iNs-shedding new light on the cellular etiology of neurodegenerative diseases. Cells (2018) 7:E38. doi: 10.3390/cells7050038
158. Boulay AC, Gilbert A, Oliveira Moreira V, Blugeon C, Perrin S, Pouch J, et al. Connexin 43 controls the astrocyte immunoregulatory phenotype. Brain Sci. (2018) 8:E50. doi: 10.3390/brainsci8040050
159. Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature (2016) 535:551–5. doi: 10.1038/nature18928
160. Torralba D, Baixauli F, Sanchez-Madrid F. Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front Cell Dev Biol. (2016) 4:107. doi: 10.3389/fcell.2016.00107
161. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. (2007) 8:57–69. doi: 10.1038/nrn2038
162. Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity (2017) 46:957–67. doi: 10.1016/j.immuni.2017.06.006
163. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
164. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. (2015) 16:358–72. doi: 10.1038/nrn3880
165. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
166. Perry VH, Andersson PB. The inflammatory response in the CNS. Neuropathol Appl Neurobiol. (1992) 18:454–9. doi: 10.1111/j.1365-2990.1992.tb00811.x
167. Lee SJ, Lee S. Toll-like receptors and inflammation in the CNS. Curr Drug Targets Inflamm Allergy (2002) 1:181–91. doi: 10.2174/1568010023344698
168. Carty M, Bowie AG. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol. (2011) 81:825–37. doi: 10.1016/j.bcp.2011.01.003
169. Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. (2006) 83:711–30. doi: 10.1002/jnr.20767
170. Rauvala H, Rouhiainen A. RAGE as a receptor of HMGB1 (Amphoterin): roles in health and disease. Curr Mol Med. (2007) 7:725–34. doi: 10.2174/156652407783220750
171. Ilzecka J. Serum-soluble receptor for advanced glycation end product levels in patients with amyotrophic lateral sclerosis. Acta Neurol Scand. (2009) 120:119–22. doi: 10.1111/j.1600-0404.2008.01133.x
172. Heiman A, Pallottie A, Heary RF, Elkabes S. Toll-like receptors in central nervous system injury and disease: a focus on the spinal cord. Brain Behav Immun. (2014) 42:232–45. doi: 10.1016/j.bbi.2014.06.203
173. Barichello T, Generoso JS, Goularte JA, Collodel A, Pitcher MR, Simoes LR, et al. Does infection-induced immune activation contribute to dementia? Aging Dis. (2015) 6:342–8. doi: 10.14336/AD.2015.0521
174. Ray R, Juranek JK, Rai V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci Biobehav Rev. (2016) 62:48–55. doi: 10.1016/j.neubiorev.2015.12.006
175. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
176. Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. (2001) 13:114–9. doi: 10.1016/S0952-79150000191-6
177. Matzinger P. The evolution of the danger theory. interview by lauren constable, commissioning editor. Exp Rev Clin Immunol. (2012) 8:311–7. doi: 10.1586/eci.12.21
178. Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. (2006) 7:774–8. doi: 10.1038/sj.embor.7400759
179. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. (2007) 81:1–5. doi: 10.1189/jlb.0306164
180. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. (2011) 29:139–62. doi: 10.1146/annurev-immunol-030409-101323
181. Fang P, Schachner M, Shen YQ. HMGB1 in development and diseases of the central nervous system. Mol Neurobiol. (2012) 45:499–506. doi: 10.1007/s12035-012-8264-y
182. Merenmies J, Pihlaskari R, Laitinen J, Wartiovaara J, Rauvala H. 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. amino acid sequence and localization in the filopodia of the advancing plasma membrane. J Biol Chem. (1991) 266:16722–9.
183. Parkkinen J, Raulo E, Merenmies J, Nolo R, Kajander EO, Baumann M, et al. Amphoterin, the 30-kDa protein in a family of HMG1-type polypeptides. enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J Biol Chem. (1993) 268:19726–38.
184. Ito T, Kawahara K, Okamoto K, Yamada S, Yasuda M, Imaizumi H, et al. Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arterioscler Thromb Vasc Biol. (2008) 28:1825–30. doi: 10.1161/ATVBAHA.107.150631
185. Zhao X, Kuja-Panula J, Rouhiainen A, Chen YC, Panula P, Rauvala H. High mobility group box-1 (HMGB1; amphoterin) is required for zebrafish brain development. J Biol Chem. (2011) 286:23200–13. doi: 10.1074/jbc.M111.223834
186. Hoarau JJ, Krejbich-Trotot P, Jaffar-Bandjee MC, Das T, Thon-Hon GV, Kumar S, et al. Activation and control of CNS innate immune responses in health and diseases: a balancing act finely tuned by neuroimmune regulators (NIReg). CNS Neurol Disord Drug Targets (2011) 10:25–43. doi: 10.2174/187152711794488601
187. Lo Coco D, Veglianese P, Allievi E, Bendotti C. Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of ALS. Neurosci Lett. (2007) 412:73–7. doi: 10.1016/j.neulet.2006.10.063
188. Casula M, Iyer AM, Spliet WG, Anink JJ, Steentjes K, Sta M, et al. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience (2011) 179:233–43. doi: 10.1016/j.neuroscience.2011.02.001
189. Juranek JK, Daffu GK, Wojtkiewicz J, Lacomis D, Kofler J, Schmidt AM. Receptor for advanced glycation end products and its inflammatory ligands are upregulated in amyotrophic lateral sclerosis. Front Cell Neurosci. (2015) 9:485. doi: 10.3389/fncel.2015.00485
190. Yan SF, Ramasamy R, Schmidt AM. Soluble RAGE: therapy and biomarker in unraveling the RAGE axis in chronic disease and aging. Biochem Pharmacol. (2010) 79:1379–86. doi: 10.1016/j.bcp.2010.01.013
191. Schmidt AM. Soluble RAGEs - Prospects for treating & tracking metabolic and inflammatory disease. Vascul Pharmacol. (2015) 72:1–8. doi: 10.1016/j.vph.2015.06.011
192. Juranek JK, Daffu GK, Geddis MS, Li H, Rosario R, Kaplan BJ, et al. Soluble RAGE treatment delays progression of amyotrophic lateral sclerosis in SOD1 mice. Front Cell Neurosci. (2016) 10:117. doi: 10.3389/fncel.2016.00117
193. Derk J, MacLean M, Juranek J, Schmidt AM. The receptor for advanced glycation endproducts (RAGE) and mediation of inflammatory neurodegeneration. J Alzheimers Dis Parkinson. (2018) 8:421. doi: 10.4172/2161-0460.1000421
194. Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. (2010) 131:517–26. doi: 10.1016/j.mad.2010.05.003
195. Mathis S, Couratier P, Julian A, Corcia P, Le Masson G. Current view and perspectives in amyotrophic lateral sclerosis. Neural Regen Res. (2017) 12:181–4. doi: 10.4103/1673-5374.200794
196. Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. (2017) 39:73–82. doi: 10.1080/01616412.2016.1251711
197. Lezi E, Swerdlow RH. Mitochondria in neurodegeneration. Adv Exp Med Biol. (2012) 942:269–86. doi: 10.1007/978-94-007-2869-1_12
198. Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. (2017) 143:418–31. doi: 10.1111/jnc.14037
199. Wilkins HM, Morris JK. New therapeutics to modulate mitochondrial function in neurodegenerative disorders. Curr Pharm Des. (2017) 23:731–52. doi: 10.2174/1381612822666161230144517
200. Levine JB, Kong J, Nadler M, Xu Z. Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS). Glia (1999) 28:215–24. doi: 10.1002/(SICI)1098-1136(199912)28:3<215::AID-GLIA5>3.0.CO;2-C
201. Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, West M, et al. On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal (2006) 8:2075–87. doi: 10.1089/ars.2006.8.2075
202. Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR. Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J Neurochem. (2008) 107:1271–83. doi: 10.1111/j.1471-4159.2008.05699.x
203. Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de Leon A, et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. (2008) 28:4115–22. doi: 10.1523/JNEUROSCI.5308-07.2008
204. Rojas F, Gonzalez D, Cortes N, Ampuero E, Hernandez DE, Fritz E, et al. Reactive oxygen species trigger motoneuron death in non-cell-autonomous models of ALS through activation of c-Abl signaling. Front Cell Neurosci. (2015) 9:203. doi: 10.3389/fncel.2015.00203
205. Agarwal A, Wu PH, Hughes EG, Fukaya M, Tischfield MA, Langseth AJ, et al. Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron (2017) 93:587–605 e7. doi: 10.1016/j.neuron.2016.12.034
206. Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. (2011) 29:824–8. doi: 10.1038/nbt.1957
207. Qian K, Huang H, Peterson A, Hu B, Maragakis NJ, Ming GL, et al. Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Rep. (2017) 8:843–55. doi: 10.1016/j.stemcr.2017.03.003
208. Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science (1999) 283:1476–81. doi: 10.1126/science.283.5407.1476
209. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. (2011) 32:157–64. doi: 10.1016/j.it.2011.01.005
210. Wilkins HM, Carl SM, Weber SG, Ramanujan SA, Festoff BW, Linseman DA, et al. Mitochondrial lysates induce inflammation and Alzheimer's disease-relevant changes in microglial and neuronal cells. J Alzheimer's Dis. (2015) 45:305–18. doi: 10.3233/JAD-142334
211. Little JP, Simtchouk S, Schindler SM, Villanueva EB, Gill NE, Walker DG, et al. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol Cell Neurosci. (2014) 60:88–96. doi: 10.1016/j.mcn.2014.04.003
212. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Landi F, et al. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int J Mol Sci. (2017) 18:E933. doi: 10.3390/ijms18050933
213. Wilkins HM, Carl SM, Greenlief AC, Festoff BW, Swerdlow RH. Bioenergetic dysfunction and inflammation in Alzheimer's disease: a possible connection. Front Aging Neurosci. (2014) 6:311. doi: 10.3389/fnagi.2014.00311
214. Wilkins HM, Weidling IW, Ji Y, Swerdlow RH. Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front Immunol. (2017) 8:508. doi: 10.3389/fimmu.2017.00508
215. Aird RB. Trypan red therapy of amyotrophic lateral sclerosis; preliminary report. Arch Neurol Psychiatry. (1948) 59:779–89. doi: 10.1001/archneurpsyc.1948.02300410090005
216. Aird RB. The role of tissue permeability with particular reference to the blood-brain barrier in diseases of the central nervous system. Calif Med. (1948) 69:360–3.
217. Aird RB, Becker RA. The blood-brain barrier in clinical disease: a review. J Nerv Ment Dis. (1963) 136:517–26. doi: 10.1097/00005053-196306000-00002
219. Aird RB. Significance of the blood-brain barrier in neurological disorders of toxic and metabolic origin. Rev Neuropsiquiatr. (1964) 27:248–59.
220. Garbuzova-Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, Sanberg PR. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res. (2007) 1157:126–37. doi: 10.1016/j.brainres.2007.04.044
221. Garbuzova-Davis S, Saporta S, Haller E, Kolomey I, Bennett SP, Potter H, et al. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE (2007) 2:e1205. doi: 10.1371/journal.pone.0001205
222. Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O'Banion MK, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. (2008) 11:420–2. doi: 10.1038/nn2073
223. Nicaise C, Mitrecic D, Demetter P, De Decker R, Authelet M, Boom A, et al. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. (2009) 1301:152–62. doi: 10.1016/j.brainres.2009.09.018
224. Zhong Z, Ilieva H, Hallagan L, Bell R, Singh I, Paquette N, et al. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest. (2009) 119:3437–49. doi: 10.1172/JCI38476
225. Nicaise C, Soyfoo MS, Delporte C, Pochet R. Aquaporin-4 as a potential marker of BBB disruption in ALS models. Amyotrophic Later Sclerosis (2010) 11:253–4. doi: 10.3109/17482960902803457
226. Garbuzova-Davis S, Woods RL III, Louis MK, Zesiewicz TA, Kuzmin-Nichols N, Sullivan KL, et al. Reduction of circulating endothelial cells in peripheral blood of ALS patients. PLoS ONE (2010) 5:e10614. doi: 10.1371/journal.pone.0010614
227. Garbuzova-Davis S, Rodrigues MC, Hernandez-Ontiveros DG, Louis MK, Willing AE, Borlongan CV, et al. Amyotrophic lateral sclerosis: a neurovascular disease. Brain Res. (2011) 1398:113–25. doi: 10.1016/j.brainres.2011.04.049
228. Garbuzova-Davis S, Hernandez-Ontiveros DG, Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, et al. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. (2012) 1469:114–28. doi: 10.1016/j.brainres.2012.05.056
229. Rodrigues MC, Hernandez-Ontiveros DG, Louis MK, Willing AE, Borlongan CV, Sanberg PR, et al. Neurovascular aspects of amyotrophic lateral sclerosis. Int Rev Neurobiol. (2012) 102:91–106. doi: 10.1016/B978-0-12-386986-9.00004-1
230. Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathologica (2013) 125:111–20. doi: 10.1007/s00401-012-1039-8
231. Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci USA. (2014) 111:E1035–42. doi: 10.1073/pnas.1401595111
232. Milane A, Fernandez C, Dupuis L, Buyse M, Loeffler JP, Farinotti R, et al. P-glycoprotein expression and function are increased in an animal model of amyotrophic lateral sclerosis. Neurosci Lett. (2010) 472:166–70. doi: 10.1016/j.neulet.2010.01.078
233. Chan GN, Evans RA, Banks DB, Mesev EV, Miller DS, Cannon RE. Selective induction of P-glycoprotein at the CNS barriers during symptomatic stage of an ALS animal model. Neurosci Lett. (2017) 639:103–13. doi: 10.1016/j.neulet.2016.12.049
234. Chen L, Watson C, Morsch M, Cole NJ, Chung RS, Saunders DN, et al. Improving the delivery of SOD1 antisense oligonucleotides to motor neurons using calcium phosphate-lipid nanoparticles. Front Neurosci. (2017) 11:476. doi: 10.3389/fnins.2017.00476
235. Stamenkovic S, Pavicevic A, Mojovic M, Popovic-Bijelic A, Selakovic V, Andjus P, et al. In vivo EPR pharmacokinetic evaluation of the redox status and the blood brain barrier permeability in the SOD1(G93A) ALS rat model. Free Radic Biol Med. (2017) 108:258–69. doi: 10.1016/j.freeradbiomed.2017.03.034
236. Philips T, Rothstein JD. Glial cells in amyotrophic lateral sclerosis. Exp Neurol. (2014) 262:111–20. doi: 10.1016/j.expneurol.2014.05.015
237. Kurtzke JF, Beebe GW. Epidemiology of amyotrophic lateral sclerosis: 1. a case-control comparison based on ALS deaths. Neurology (1980) 30:453–62. doi: 10.1212/WNL.30.5.453
238. Kondo K, Tsubaki T. Case-control studies of motor neuron disease: association with mechanical injuries. Arch Neurol. (1981) 38:220–6. doi: 10.1001/archneur.1981.00510040046007
239. Kurland LT, Radhakrishnan K, Smith GE, Armon C, Nemetz PN. Mechanical trauma as a risk factor in classic amyotrophic lateral sclerosis: lack of epidemiologic evidence. J Neurol Sci. (1992) 113:133–43. doi: 10.1016/0022-510X(92)90241-C
240. Kioumourtzoglou MA, Rotem RS, Seals RM, Gredal O, Hansen J, Weisskopf MG. Diabetes mellitus, obesity, and diagnosis of amyotrophic lateral sclerosis: a population-based study. JAMA Neurol. (2015) 72:905–11. doi: 10.1001/jamaneurol.2015.0910
241. Seals RM, Hansen J, Gredal O, Weisskopf MG. Physical Trauma and amyotrophic lateral sclerosis: a population-based study using danish National registries. Am J Epidemiol. (2016) 183:294–301. doi: 10.1093/aje/kwv169
242. Pupillo E, Poloni M, Bianchi E, Giussani G, Logroscino G, Zoccolella S, et al. Trauma and amyotrophic lateral sclerosis: a european population-based case-control study from the EURALS consortium. Amyotrophic Later Sclerosis Frontotemporal Degener. (2018) 19:118–25. doi: 10.1080/21678421.2017.1386687
243. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain (2013) 136:43–64. doi: 10.1093/brain/aws307
244. Stein TD, Alvarez VE, McKee AC. Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther. (2014) 6:4. doi: 10.1186/alzrt234
245. Kenney K, Iacono D, Edlow BL, Katz DI, Diaz-Arrastia R, Dams-O'Connor K, et al. Dementia after moderate-severe traumatic brain injury: coexistence of multiple proteinopathies. J Neuropathol Exp Neurol. (2018) 77:50–63. doi: 10.1093/jnen/nlx101
246. Moszczynski AJ, Strong W, Xu K, McKee A, Brown A, Strong MJ. Pathologic Thr(175) tau phosphorylation in CTE and CTE with ALS. Neurology. (2018) 90:e380–e7. doi: 10.1212/WNL.0000000000004899
247. Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. (2011) 2:492–516. doi: 10.1007/s12975-011-0125-x
248. Alves JL. Blood-brain barrier and traumatic brain injury. J Neurosci Res. (2014) 92:141–7. doi: 10.1002/jnr.23300
249. Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. (2015) 51:966–79. doi: 10.1007/s12035-014-8752-3
250. Logsdon AF, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW. Role of microvascular disruption in brain damage from traumatic brain injury. Compr Physiol. (2015) 5:1147–60. doi: 10.1002/cphy.c140057
251. Festoff BW, Hantaï D, North Atlantic Treaty Organization. Scientific Affairs Division, editors. Serine Proteases and Their Serpin Inhibitors in the Nervous System: Regulation in Development and in Degenerative and Malignant Disease. New York, NY: Plenum Press (1990).
252. Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem. (2003) 84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x
253. Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood (2012) 120:5237–46. doi: 10.1182/blood-2012-08-452169
254. Luo W, Wang Y, Reiser G. The role of thrombin and thrombin receptors in the brain. In: Maragoudakis ME, Tsopanoglou N, editors. Thrombin: Physiology and Disease. New York, NY: Springer (2010). p. 133–59.
255. Esmon CT, Esmon NL, Harris KW. Complex formation between thrombin and thrombomodulin inhibits both thrombin-catalyzed fibrin formation and factor V activation. J Biol Chem. (1982) 257:7944–7.
256. Esmon CT, Owen WG. The discovery of thrombomodulin. J Thromb Haemost. (2004) 2:209–13. doi: 10.1046/j.1538-7933.2003.00537.x
257. Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science (2002) 296:1880–2. doi: 10.1126/science.1071699
258. Conway EM. Thrombomodulin and its role in inflammation. Semin Immunopathol. (2012) 34:107–25. doi: 10.1007/s00281-011-0282-8
259. Wen DZ, Dittman WA, Ye RD, Deaven LL, Majerus PW, Sadler JE. Human thrombomodulin: complete cDNA sequence and chromosome localization of the gene. Biochemistry (1987) 26:4350–7. doi: 10.1021/bi00388a025
260. Imada M, Imada S, Iwasaki H, Kume A, Yamaguchi H, Moore EE. Fetomodulin: marker surface protein of fetal development which is modulatable by cyclic AMP. Dev Biol. (1987) 122:483–91. doi: 10.1016/0012-1606(87)90312-5
261. Imada S, Yamaguchi H, Nagumo M, Katayanagi S, Iwasaki H, Imada M. Identification of fetomodulin, a surface marker protein of fetal development, as thrombomodulin by gene cloning and functional assays. Dev Biol. (1990) 140:113–22. doi: 10.1016/0012-1606(90)90058-Q
262. Maruyama I, Bell CE, Majerus PW. Thrombomodulin is found on endothelium of arteries, veins, capillaries, and lymphatics, and on syncytiotrophoblast of human placenta. J Cell Biol. (1985) 101:363–71. doi: 10.1083/jcb.101.2.363
263. Ishii H, Salem HH, Bell CE, Laposata EA, Majerus PW. Thrombomodulin, an endothelial anticoagulant protein, is absent from the human brain. Blood (1986) 67:362–5.
264. Wong VL, Hofman FM, Ishii H, Fisher M. Regional distribution of thrombomodulin in human brain. Brain Res. (1991) 556:1–5. doi: 10.1016/0006-8993(91)90540-C
265. Wang L, Tran ND, Kittaka M, Fisher MJ, Schreiber SS, Zlokovic BV. Thrombomodulin expression in bovine brain capillaries. anticoagulant function of the blood-brain barrier, regional differences, and regulatory mechanisms. Arterioscler Thromb Vasc Biol. (1997) 17:3139–46. doi: 10.1161/01.ATV.17.11.3139
266. Pindon A, Hantai D, Jandrot-Perrus M, Festoff BW. Novel expression and localization of active thrombomodulin on the surface of mouse brain astrocytes. Glia (1997) 19:259–68. doi: 10.1002/(SICI)1098-1136(199703)19:3<259::AID-GLIA8>3.0.CO;2-U
267. Pindon A, Berry M, Hantai D. Thrombomodulin as a new marker of lesion-induced astrogliosis: involvement of thrombin through the G-protein-coupled protease-activated receptor-1. J Neurosci. (2000) 20:2543–50. doi: 10.1523/JNEUROSCI.20-07-02543.2000
268. Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron (2014) 81:229–48. doi: 10.1016/j.neuron.2013.12.034
269. Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. (2005) 115:1267–74. doi: 10.1172/JCI22782
270. Kawabata H, Setoguchi T, Yone K, Souda M, Yoshida H, Kawahara K, et al. High mobility group box 1 is upregulated after spinal cord injury and is associated with neuronal cell apoptosis. Spine (2010) 35:1109–15. doi: 10.1097/BRS.0b013e3181bd14b6
271. Shi CS, Shi GY, Hsiao HM, Kao YC, Kuo KL, Ma CY, et al. Lectin-like domain of thrombomodulin binds to its specific ligand Lewis Y antigen and neutralizes lipopolysaccharide-induced inflammatory response. Blood (2008) 112:3661–70. doi: 10.1182/blood-2008-03-142760
272. Iba T, Gando S, Murata A, Kushimoto S, Saitoh D, Eguchi Y, et al. Predicting the severity of systemic inflammatory response syndrome (SIRS)-associated coagulopathy with hemostatic molecular markers and vascular endothelial injury markers. J Trauma. (2007) 63:1093–8. doi: 10.1097/01.ta.0000251420.41427.d3
273. Ogura H, Gando S, Iba T, Eguchi Y, Ohtomo Y, Okamoto K, et al. SIRS-associated coagulopathy and organ dysfunction in critically ill patients with thrombocytopenia. Shock (2007) 28:411–7. doi: 10.1097/shk.0b013e31804f7844
274. Opal SM. Phylogenetic and functional relationships between coagulation and the innate immune response. Crit Care Med. (2000) 28(Suppl. 9):S77–80. doi: 10.1097/00003246-200009001-00017
275. Wada T, Gando S, Maekaw K, Katabami K, Sageshima H, Hayakawa M, et al. Disseminated intravascular coagulation with increased fibrinolysis during the early phase of isolated traumatic brain injury. Critical Care (2017) 21:219. doi: 10.1186/s13054-017-1808-9
276. Grammas P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer's disease. J Neuroinflam. (2011) 8:26. doi: 10.1186/1742-2094-8-26
277. Ott BR, Jones RN, Daiello LA, de la Monte SM, Stopa EG, Johanson CE, et al. Blood-cerebrospinal fluid barrier gradients in mild cognitive impairment and alzheimer's disease: relationship to inflammatory cytokines and chemokines. Front Aging Neurosci. (2018) 10:245. doi: 10.3389/fnagi.2018.00245
278. Machida T, Takata F, Matsumoto J, Takenoshita H, Kimura I, Yamauchi A, et al. Brain pericytes are the most thrombin-sensitive matrix metalloproteinase-9-releasing cell type constituting the blood-brain barrier in vitro. Neurosci Lett. (2015) 599:109–14. doi: 10.1016/j.neulet.2015.05.028
279. Machida T, Dohgu S, Takata F, Matsumoto J, Kimura I, Koga M, et al. Role of thrombin-PAR1-PKCtheta/delta axis in brain pericytes in thrombin-induced MMP-9 production and blood-brain barrier dysfunction in vitro. Neuroscience (2017) 350:146–57. doi: 10.1016/j.neuroscience.2017.03.026
280. Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, et al. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. (2011) 114:92–101. doi: 10.3171/2010.6.JNS10207
281. Wang Z, Meng CJ, Shen XM, Shu Z, Ma C, Zhu GQ, et al. Potential contribution of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 to blood-brain barrier disruption and brain edema after experimental subarachnoid hemorrhage. J Mol Neurosci. (2012) 48:273–80. doi: 10.1007/s12031-012-9769-6
282. Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C: biased for translation. Blood (2015) 125:2898–907. doi: 10.1182/blood-2015-02-355974
283. Rezaie AR. Protease-activated receptor signalling by coagulation proteases in endothelial cells. Thromb Haemost. (2014) 112:876–82. doi: 10.1160/th14-02-0167
284. Alberelli MA, De Candia E. Functional role of protease activated receptors in vascular biology. Vascul Pharmacol. (2014) 62:72–81. doi: 10.1016/j.vph.2014.06.001
285. Garcia JG, Pavalko FM, Patterson CE. Vascular endothelial cell activation and permeability responses to thrombin. Blood Coagul Fibrinol. (1995) 6:609–26. doi: 10.1097/00001721-199510000-00001
286. Bogatcheva NV, Garcia JG, Verin AD. Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry (2002) 67:75–84. doi: 10.1023/A:1013904231324
287. van Nieuw Amerongen GP, Musters RJ, Eringa EC, Sipkema P, van Hinsbergh VW. Thrombin-induced endothelial barrier disruption in intact microvessels: role of RhoA/Rho kinase-myosin phosphatase axis. Am J Physiol Cell Physiol. (2008) 294:C1234–41. doi: 10.1152/ajpcell.00551.2007
288. Guan JX, Sun SG, Cao XB, Chen ZB, Tong ET. Effect of thrombin on blood brain barrier permeability and its mechanism. Chin Med J. (2004) 117:1677–81.
289. Taoka Y, Okajima K, Uchiba M, Murakami K, Harada N, Johno M, et al. Activated protein C reduces the severity of compression-induced spinal cord injury in rats by inhibiting activation of leukocytes. J Neurosci. (1998) 18:1393–8. doi: 10.1523/JNEUROSCI.18-04-01393.1998
290. Whetstone WD, Walker B, Trivedi A, Lee S, Noble-Haeusslein LJ, Hsu JC. Protease-Activated Receptor-1 supports locomotor recovery by biased agonist activated protein c after contusive spinal cord injury. PLoS ONE (2017) 12:e0170512. doi: 10.1371/journal.pone.0170512
291. Suo Z, Wu M, Citron BA, Palazzo RE, Festoff BW. Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J Biol Chem. (2003) 278:37681–9. doi: 10.1074/jbc.M301406200
292. Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, et al. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. (2006) 281:1205–14. doi: 10.1074/jbc.M507753200
293. Rohatgi T, Sedehizade F, Reymann KG, Reiser G. Protease-activated receptors in neuronal development, neurodegeneration, and neuroprotection: thrombin as signaling molecule in the brain. Neuroscientist (2004) 10:501–12. doi: 10.1177/1073858404269955
294. Arai T, Miklossy J, Klegeris A, Guo JP, McGeer PL. Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J Neuropathol Exp Neurol. (2006) 65:19–25. doi: 10.1097/01.jnen.0000196133.74087.cb
295. Mhatre M, Nguyen A, Kashani S, Pham T, Adesina A, Grammas P. Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol Aging (2004) 25:783–93. doi: 10.1016/j.neurobiolaging.2003.07.007
296. Grammas P, Martinez JM. Targeting thrombin: an inflammatory neurotoxin in Alzheimer's disease. J Alzheimer's Dis. (2014) 42(Suppl. 4):S537–44. doi: 10.3233/JAD-141557
297. Ishida Y, Nagai A, Kobayashi S, Kim SU. Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J Neuropathol Exp Neurol. (2006) 65:66–77. doi: 10.1097/01.jnen.0000195941.48033.eb
298. Citron BA, Smirnova IV, Arnold PM, Festoff BW. Upregulation of neurotoxic serine proteases, prothrombin, and protease-activated receptor 1 early after spinal cord injury. J Neurotrau. (2000) 17:1191–203. doi: 10.1089/neu.2000.17.1191
299. Chen D, Carpenter A, Abrahams J, Chambers RC, Lechler RI, McVey JH, et al. Protease-activated receptor 1 activation is necessary for monocyte chemoattractant protein 1-dependent leukocyte recruitment in vivo. J Exp Med. (2008) 205:1739–46. doi: 10.1084/jem.20071427
300. Dennis JS, Citron BA. Wobbler mice modeling motor neuron disease display elevated transactive response DNA binding protein. Neuroscience (2009) 158:745–50. doi: 10.1016/j.neuroscience.2008.10.030
301. Salcedo RM, Festoff BW, Citron BA. Quantitative reverse transcriptase PCR to gauge increased protease-activated receptor 1 (PAR-1) mRNA copy numbers in the Wobbler mutant mouse. J Mol Neurosci. (1998) 10:113–9. doi: 10.1007/BF02737122
302. Festoff BW, D'Andrea MR, Citron BA, Salcedo RM, Smirnova IV, Andrade-Gordon P. Motor neuron cell death in wobbler mutant mice follows overexpression of the G-protein-coupled, protease-activated receptor for thrombin. Mol Med. (2000) 6:410–29. doi: 10.1007/BF03401784
303. Wichmann H, Jockusch H, Guenet JL, Gallwitz D, Kaupmann K. The mouse homolog to the ras-related yeast gene YPT1 maps on chromosome 11 close to the wobbler (wr) locus. Mamm Genome (1992) 3:467–8. doi: 10.1007/BF00356159
304. Schmitt-John T, Drepper C, Mussmann A, Hahn P, Kuhlmann M, Thiel C, et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet. (2005) 37:1213–5. doi: 10.1038/ng1661
305. des Portes V, Coulpier M, Melki J, Dreyfus PA. Early detection of mouse wobbler mutation: a model of pathological motoneurone death. Neuroreport (1994) 5:1861–4. doi: 10.1097/00001756-199410000-00005
306. Wedemeyer N, Lengeling A, Ronsiek M, Korthaus D, Baer K, Wuttke M, et al. YAC contigs of the Rab1 and wobbler (wr) spinal muscular atrophy gene region on proximal mouse chromosome 11 and of the homologous region on human chromosome 2p. Genomics (1996) 32:447–54. doi: 10.1006/geno.1996.0140
307. Festoff BW, Smirnova IV, Ma J, Citron BA. Thrombin, its receptor and protease nexin I, its potent serpin, in the nervous system. Semi Thrombosis Hemostasis (1996) 22:267–71. doi: 10.1055/s-2007-999018
308. Chapman J. Coagulation in inflammatory diseases of the central nervous system. Semi Thrombosis Hemostasis (2013) 39:876–80. doi: 10.1055/s-0033-1357482
309. De Luca C, Virtuoso A, Maggio N, Papa M. Neuro-coagulopathy: blood coagulation factors in central nervous system diseases. Int J Mol Sci. (2017) 18:E2128. doi: 10.3390/ijms18102128
310. Petersen MA, Ryu JK, Akassoglou K. Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Neurosci. (2018) 19:283–301. doi: 10.1038/nrn.2018.13
311. Festoff BW, SantaCruz K, Arnold PM, Sebastian CT, Davies PJ, Citron BA. Injury-induced “switch” from GTP-regulated to novel GTP-independent isoform of tissue transglutaminase in the rat spinal cord. J Neurochem. (2002) 81:708–18. doi: 10.1046/j.1471-4159.2002.00850.x
312. Oono M, Okado-Matsumoto A, Shodai A, Ido A, Ohta Y, Abe K, et al. Transglutaminase 2 accelerates neuroinflammation in amyotrophic lateral sclerosis through interaction with misfolded superoxide dismutase 1. J Neurochem. (2014) 128:403–18. doi: 10.1111/jnc.12441
313. Hashemzadeh M, Arreguin JM, Roberts T, Movahed MR. A Novel Inhibitor of Protease-activated Receptor 1: a review of chemical structure and mode of action. Rev Cardiovasc Med. (2015) 16:68–73. doi: 10.3909/ricm0754
314. Serebruany VL, Fortmann SD, Hanley DF, Kim MH. Vorapaxar and amyotrophic lateral sclerosis: coincidence or adverse association? Am J Ther. (2017) 24:e139–e43. doi: 10.1097/MJT.0000000000000395
315. Aisiku O, Peters CG, De Ceunynck K, Ghosh CC, Dilks JR, Fustolo-Gunnink SF, et al. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood (2015) 125:1976–85. doi: 10.1182/blood-2014-09-599910
316. De Ceunynck K, Peters CG, Jain A, Higgins SJ, Aisiku O, Fitch-Tewfik JL, et al. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc Natl Acad Sci USA. (2018) 115:E982–E91. doi: 10.1073/pnas.1718600115
Keywords: thrombin, thrombomodulin, PAR1, DAMPs, HMGB1, blood brain barrier, ALS, neurodegeneration
Citation: Festoff BW and Citron BA (2019) Thrombin and the Coag-Inflammatory Nexus in Neurotrauma, ALS, and Other Neurodegenerative Disorders. Front. Neurol. 10:59. doi: 10.3389/fneur.2019.00059
Received: 09 October 2018; Accepted: 17 January 2019;
Published: 05 February 2019.
Edited by:
Tatiana Koudriavtseva, Istituto Nazionale del Cancro Regina Elena, ItalyReviewed by:
Michele Papa, Università degli Studi della Campania Luigi Vanvitelli Caserta, ItalyJoab Chapman, Tel Aviv University, Israel
Copyright © 2019 Festoff and Citron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barry W. Festoff, bwfestoff@mac.com