 Kang Wang
Kang Wang Dengchang Wu
Dengchang Wu Baorong Zhang
Baorong Zhang Guohua Zhao
Guohua Zhao- 1Department of Neurology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Department of Neurology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 3Department of Neurology, The Fourth Affiliated Hospital, Zhejiang University School of Medicine, Yiwu, China
Familial cerebral cavernous malformations (CCMs) are autosomal dominant disorders characterized by hemorrhagic strokes, recurrent headache, epilepsy, and focal neurological deficits. Genetic variants in KRIT1/CCM1, MGC4607/CCM2, and PDCD10/CCM3 genes contribute to CCMs. The clinical information of two Chinese families with CCMs was collected. MRI and video-electroencephalography were performed. Genetic variants of CCM1, CCM2, and CCM3 genes were investigated by exome sequencing. The patients were presented with recurrent epilepsy or headache. Susceptibility-weighted images of brains showed many dark dots, while video-electroencephalography revealed many spikes from multiple brain regions of patients. Exome sequencing revealed a novel CCM1 genetic variant (c.1599_1601TGAdel, p.Asp533del) and a novel CCM2 genetic variant (c.773delA, p.K258fsX34) in Family one and Family two, respectively; cosegregation existed in these two families. The two family members presented typical CCMs symptoms. These two novel genetic variants in CCM1 and CCM2 genes were the causation of CCM in the two Chinese families, and our data enriched the genetic variant spectrum of CCM genes.
Introduction
Cerebral cavernous malformations (CCMs) are vascular anomalies characterized by densely packed tortuous microvessels outlined with deficient interstitial brain parenchyma. They are the second most prevalent type of vascular malformation, accounting for 10–15% of all vascular malformations in the central nervous system (CNS). These dysplastic capillaries are mainly present in the brain and spine cord, but they may also affect the skin and liver, with increased propensity to leak and rupture, causing hemorrhagic strokes, recurrent headache, epilepsy, and focal neurological deficits (1). CCMs can occur in a sporadic or autosomal dominant fashion (Familial cerebral cavernous malformations, FCCM), and the latter tends to have multiple lesions as a consequence of inherited loss-of-function genetic variants, predominantly in any of the KRIT1/CCM1 (Krev interaction trapped 1), MGC4607/CCM2 (cerebral cavernous malformation 2), or PDCD10/CCM3 (programmed cell death protein 10) genes (2–4). The prevalence of genetic variants in the three genes is unknown, although there are several cases reported in different populations like Italian (5, 6), French (7), Spanish (8), Germany (9, 10), and Japanese (11). There are a few studies reporting CCM1 genetic variants, but only one CCM2 genetic variant was reported in Chinese patients (12–24).
In this study, we described the clinical and neuroradiological findings in two Chinese families with CCMs and identified two novel genetic variants in CCM1 and CCM2 genes, respectively.
Material and Methods
Subjects
Two pedigrees of the two Chinese families with CCMs were enrolled. This study was approved by the Institutional Review Board of Zhejiang University School of Medicine, and written informed consent was received from all subjects or their family members. The detailed medical history of the family members from the two pedigrees was obtained. Brain MRIs, including susceptibility-weighted images (SWI) and video-electroencephalography (VEEG), were performed in some cases.
Genetic Analysis
Blood samples were collected from all members of these two families, and genomic DNA was extracted using commercial kits and then stored at −20°C. Exome sequencing was performed for CCM1, CCM2, and CCM3 genes, which were sequenced in the two probands by Axeq Technologies (Precision Gene Technologies, Beijing, China). Briefly, Agilent SureSelect v6 reagents were used to capture exons, and Illumina HiSeq X Ten platform was used for sequencing as previously reported (25). Variants identified by sequencing were analyzed for cosegregation in the family members by Sanger sequencing, and polymorphism was excluded by 100 normal controls.
Results
Clinical Investigation
Family One
The proband II:1 (Figure 1A) was a 59-year-old woman who visited our clinic for recurrent episodes with consciousness loss followed by whole body stiffness and shaking movements for 3 years. These attacks occurred 5 to 8 times per year without any medication treatments because the seizures happened during the sleeps. No minor episodic events were reported. SWI of brain MRI taken on July 3, 2017 showed hundreds of dark dots and patches in different sizes distributed across the cerebral hemispheres, cerebellum, and brainstem, which indicates multiple cavernous malformations (Figure 2, upper row). VEEG showed abundant spikes that originated from multiple brain regions (data not shown). Her memory declined, and she suffered insomnia over the past 2 years. Habitual nasal bleeding was denied. On physical exams, no vascular malformation was found on the skin. Routine blood examinations were normal.
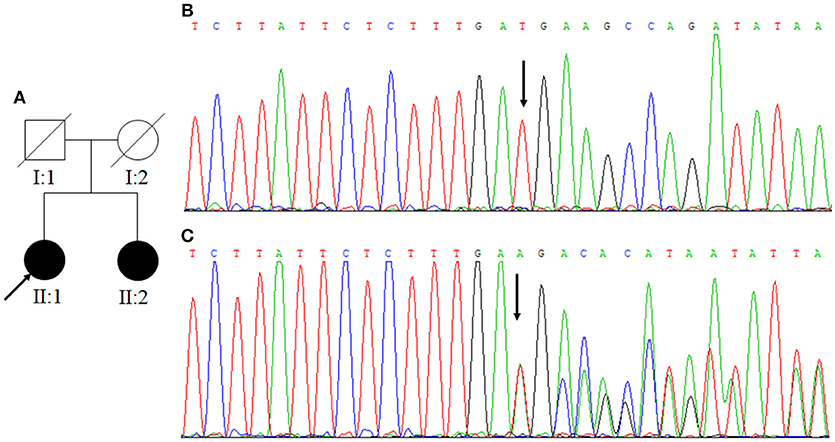
Figure 1. (A) Family One. Arrow indicates the proband; square represents a male, while circles represent females. The filled symbols mean CCM and slash indicates deceased. (B) The normal control sequences of CCM1 gene. (C) The genetic variant of c.1599_1601TGAdel (p.533Aspdel). Affected nucleotide is indicated with an arrow.
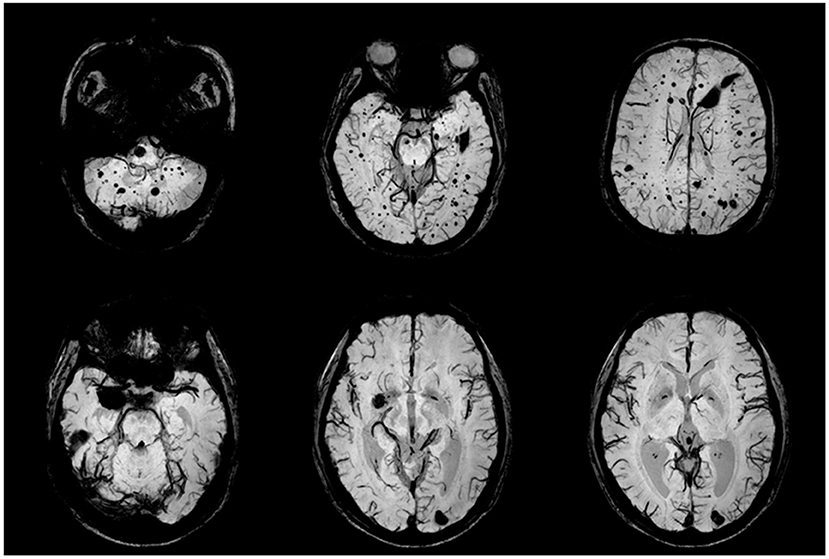
Figure 2. CCM lesions in Family One detected by SWI-MRI. The proband's SWIs (II:1) revealed dot and patchy low intensity lesions on temporal lobe, cerebellum and thalamus (upper row). SWI (II:2) showed 2 popcorn-like lesions in left occipital region and right temporal pole, which were very dark, suggesting chronic bleeding.
The proband's younger sister II:2 complained of chronic intensified and aggravated headaches for over 10 years. T2-weighted images of MRI showed 2 popcorn-like lesions in left occipital region and right temporal pole, and these lesions were hypo-intensities on SWI; those findings suggested chronic bleeding (Figure 2, lower row). No other illnesses or discomforts were reported. The proband's father (I:1) passed away due to a lethal heart attack at the age of 52 years, and her mother died of severe head trauma in a car accident at the age of 60 years. No cutaneous abnormalities, headache, stroke, or convulsion were recorded in the proband's parents.
Family Two
The proband II:1 (Figure 3A) was a 34-year-old woman, presented with paroxysmal brief staring and loss of contact proceeded by uncontrollable weird thoughts for 2 years. Her minor seizures rarely evolved to generalize tonic-clonic seizures. She was administered multiple anti-seizure medications and several combinations, but the seizure frequency remained over 5 times per month. No positive neurologic sign was found. Whole brain MRI on March 6, 2014 showed three ultra-low intensity blooming lesions in the right cerebellar hemisphere and left basal ganglion on SWI, suggesting CCMs, and the “caput medusae”-like lesion on the left temporal-occipital cortex is compatible with venous malformation (Figure 4, upper row). VEEG showed abundant interictal spikes and slow waves over the left temporal region with phase reversals at T1 (data not shown). The patient received gamma knife radiosurgery targeting the left temporal lesion; however, the symptoms were not obviously improved. She was administered a combination of carbamazepine and topiramate. Comprehensive blood examinations including liver function, renal function, blood lipid, blood sugar, and blood electrolyte were normal.
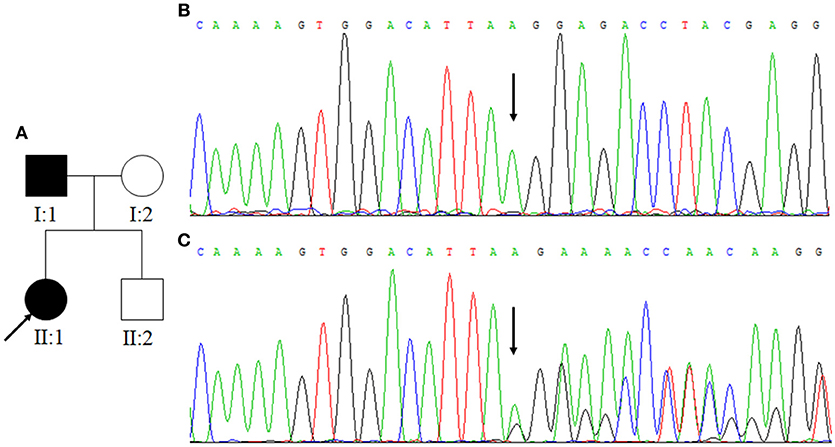
Figure 3. (A) Family Two. Arrow indicates the proband. Squares indicate males while circles indicate females. The filled symbols represent CCM. (B) The normal control sequences of CCM2 gene. (C) The genetic variant of c.773delA. Affected nucleotide is indicated with an arrow.
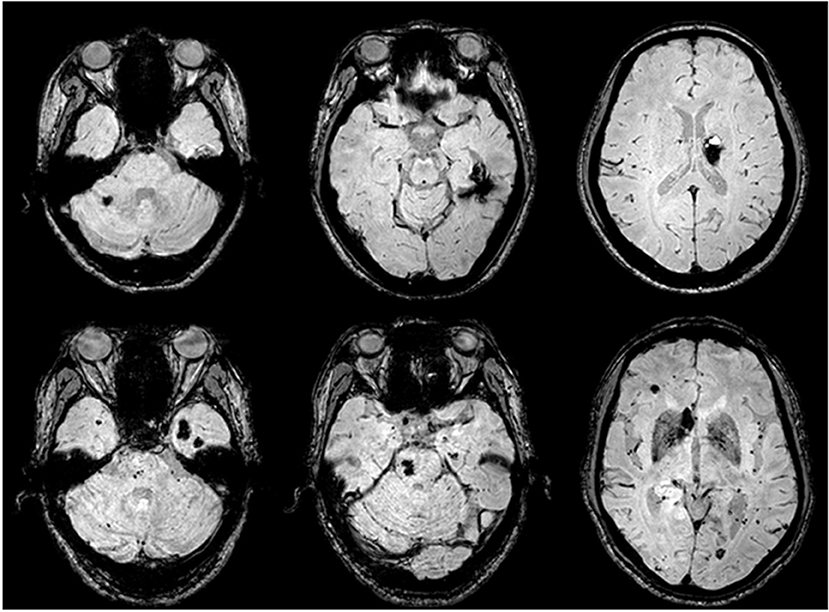
Figure 4. CCM lesions detected by SWI-MRI. The proband's SWIs (II:1) revealed dot and patchy low intensity lesions on temporal lobe, cerebellum, and thalamus (upper row). SWIs of the proband's father (I:1) demonstrated more widely-distributed multiple CCMs throughout the cerebrum, cerebellum, and pons (lower row).
The proband's father I:1 was 60-years-old and began to develop episodic fears followed by confusion and motionlessness about 20 years ago; these episodes lasted 2 to 4 min each time and occurred 3 to 5 times per year, but it progressed to 5 to 10 times per month during the past 5 years. Although he took multiple medications, the compliance was poor. Sudden falls accompanied by extreme convulsion and tongue-bites occurred occasionally. He was a heavy alcohol consumer. Neurological examination and routine blood examinations were normal. SWI sequence of brain MRI revealed eight visible CCMs on the left temporal lobe, bilateral frontal lobe, pons, and cerebellum (Figure 4, lower row). VEEG revealed frequent independent spikes originating from the bilateral temporal lobe with left side predominance (data not shown).
Genetic Analysis
In Family One, a novel CCM1 genetic variant, c.1599_1601TGAdel, was found (Figures 1B,C). This genetic variant was present in family members II:1 and II:2 and is predicted to cause a p.Asp533del. Alignment of CCM1 protein sequences showed that the p.Asp533 is quite conserved in different mammalians, suggesting that the amino acid is important for CCM1 (Figure 5). No DNA samples were obtained from family members I:1 and I:2. In Family Two, a novel CCM2 genetic variant, c.773delA (p.K258fsX34), was identified (Figures 3B,C) in family members I:1 and II:1, but not in family members I:2 and II:2. This genetic variant is predicted to cause a premature stop codon (TAG) at nucleotide 873 to generate a non-sense-mediated decay. The two genetic variants were not present in 100 normal controls, suggesting that they are causative genes for the clinical phenotype. These two genetic variants were not collected by the Human Gene Mutation Database (HGMD), ClinVar, and Gnomad database.

Figure 5. Alignment of the sequences of CCM1 protein in different mammalians.
Discussion
Familial CCMs exhibit more aggressive manifestations and an earlier age of onset when compared with sporadic cases, but familial CCMs are rare, accounting for ~6% of all CCMs. The disease is commonly presented with seizure, headache, and cerebral hemorrhage, but 11–44% patients might be asymptomatic for a long time, sometimes even lifelong (1). Depending on the symptoms and the findings in the proband, we justified that the proband's father or mother in Family One had CCM despite the absence of imaging evidence. CCM can be readily detected by MRI, particularly, the SWI with the characteristics of multiple dot or patchy lesions with markedly signal loss suggesting hemosiderin deposition. Other forms of vascular malformations, such as developmental venous anomalies, are thought to be closely associated with CCM and might co-exist with CCM as seen in the proband in Family Two.
Familial CCMs follow the autosomal dominant inheritance, and genetic variants of CCM1, CCM2, and CCM3 genes are found in 53–65%, 15–19%, and 10–22% of the CCM families (26), respectively. In this study, we investigated two pedigrees with CCMs and identified one novel CCM1 genetic variant (c.1599_1601TGAdel) and one novel CCM2 gene genetic variant (c.773delA). These genetic variants cosegregated with the clinical phenotypes and were not present in the HGMD, ClinVar, and Gnomad database, nor in the 100 normal controls. Therefore, we can propose that these two novel genetic variants may account for the etiology. Our study expands the genetic variant spectrum of the patients with CCMs.
Currently, over 200 distinct germline genetic variants have been identified as causative genetic variants in the CCM1, CCM2, and CCM3 genes (6). Most of the genetic variants are premature termination codons or large deletions (27, 28). Pathogenic deep intronic genetic variants of CCM1-3 remain a controversial topic (29). Current evidence suggests that three CCM-gene-encoding proteins interact with each other to form a complex, the CCM signaling complex (CSC), which further interacts with other proteins (1). CSC can regulate various cellular signaling, including the signaling involved in cell-cell junctions (30). CCM proteins can also interact with other proteins through FERM domain, and subsequently be involved in various signaling pathways such as integrin signaling, Notch signaling, VEGF signaling, etc (31, 32). In addition to loss of function in CCM genes, non-genetic factors are also believed to play an important role in the pathogenesis of CCMs (32).
In Family One, the CCM1 genetic variant, c.1599_1601TGAdel, is predicted to cause p.533Asp deletion. The proband and her sister had similar symptoms and radiological changes. Unfortunately, we did not get much information nor DNA samples from the proband's parents since they died early. Until now, more than 90 different CCM1 genetic variants have been reported, with most of them causing premature stop codons; this suggests a loss-of-function mechanism (2, 33), which is responsible for CCM pathogenesis (31). The p.533Aspdel genetic variant is located in the FERM domain of CCM1 protein at the C-terminus (residues 420–736). Thus, the deletion might affect the binding of CCM1 protein to GTPase Rap1 through the FERM domain, and consequently interfere with the signal pathways involving in cell-cell junctions (30).
In Family Two, the CCM2 genetic variant, c.773delA (p.K258fsX34), is predicted to cause non-sense-mediated decay. The patients showed typical CCM phenotypes, while different clinical manifestations were observed between the proband II:1 and her father I:1; the father I:1 had more severe clinical manifestations and CCMs lesions. This suggests that other unidentified CCM genes; for example, the putative CCM4 gene; epigenetic factors; or other unknown variants of non-CCM genes may contribute to these differences in clinical penetrance between these two familial members. Thus, further studies in gene knockout animals are required to explore genotype-phenotype correlations of these CCM genetic variants and phenotype variability.
In conclusion, two novel genetic variants in CCM1 and CCM2 gene were identified in two Chinese families of CCMs, suggesting a causation of CCM genetic variant in CCMs. Our data enriched the genetic variant spectrum of CCM genes, which is useful for the early diagnosis of CCMs cases.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
Written informed consent to publish the report was obtained from each member of the family.
Author Contributions
KW, DW, BZ, and GZ have actively participated in the data acquisition and interpretation, and they all commented and approved the final version of the manuscript. KW analyzed the data and drafted the manuscript. GZ designed the study and revised the manuscript.
Funding
This study was partly supported by the National Key R&D Program of China (2017YFC0907700), the National Scientific Foundations of China (81000484), Natural Scientific Foundation of Zhejiang Province (LY17H090002), and the Research Foundation of Zhejiang Health (2008QN017, 2014-KY1-001-072, 2016KYB118). We thank the family members for participating in the study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the patients and their family for their contributions to the study.
References
1. Batra S, Lin D, Recinos PF, Zhang J, Rigamonti D. Cavernous malformations: natural history, diagnosis and treatment. Nat Rev Neurol. (2009) 5:659–70. doi: 10.1038/nrneurol.2009.177
2. Laberge-le Couteulx S, Jung HH, Labauge P, Lescoat C, Cecillon M, Marechal E, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. (1999) 23:189–93. doi: 10.1038/13815
3. Liquori CL, Berg MJ, Siegel AM, Zawistowski JS, Stoffer T, Verlaan D, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. (2003) 73:1459–64. doi: 10.1086/380314
4. Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. (2004) 74:326–37. doi: 10.1086/381718
5. D'Angelo R, Marini V, Rinaldi C, Origone P, Dorcaratto A, Avolio M, et al. Mutation analysis of CCM1, CCM2 and CCM3 genes in a cohort of Italian patients with cerebral cavernous malformation. Brain Pathol. (2011) 21:215–24. doi: 10.1111/j.1750-3639.2010.00441.x
6. Scimone C, Bramanti P, Alafaci C, Granata F, Piva F, Rinaldi C, et al. Update on novel CCM gene mutations in patients with cerebral cavernous malformations. J Mol Neurosci. (2017) 61:189–98. doi: 10.1007/s12031-016-0863-z
7. Sirvente J, Enjolras O, Wassef M, Tournier-Lasserve E, Labauge P. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol. (2009) 23:1066–72. doi: 10.1111/j.1468-3083.2009.03263.x
8. Mondéjar R, Solano F, Rubio R, Delgado M, Pérez-Sempere A, González-Meneses A, et al. Mutation prevalence of cerebral cavernous malformation genes in Spanish patients. PLoS ONE (2014) 9: e86286. doi: 10.1371/journal.pone.0086286
9. Stahl S, Gaetzner S, Voss K, Brackertz B, Schleider E, Sürücü O, et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum Mutat. (2008) 29:709–17. doi: 10.1002/humu.20712
10. Rath M, Jenssen SE, Schwefel K, Spiegler S, Kleimeier D, Sperling C, et al. High-throughput sequencing of the entire genomic regions of CCM1/KRIT1, CCM2 and CCM3/PDCD10 to search for pathogenic deep-intronic splice mutations in cerebral cavernous malformations. Eur J Med Genet. (2017) 60:479–84. doi: 10.1016/j.ejmg.2017.06.007
11. Tsutsumi S, Ogino I, Miyajima M, Ikeda T, Shindo N, Yasumoto Y, et al. Genomic causes of multiple cerebral cavernous malformations in a Japanese population. J Clin Neurosci. (2013) 20:667–69. doi: 10.1016/j.jocn.2012.05.041
12. Chen DH, Lipe HP, Qin Z, Bird TD. Cerebral cavernous malformation: novel mutation in a Chinese family and evidence for heterogeneity. J Neurol Sci. (2002) 196:91–6. doi: 10.1016/S0022-510X(02)00031-X
13. Xu YL, Zhao JZ, Wu BQ, Zhong HH, Wang S, Heng WJ. A novel Krit-1 mutation in Han family with cerebral cavernous malformation. Zhonghua Bing Li Xue Za Zhi (2003) 32:220–5.
14. Mao Y, Zhao Y, Zhou LF, Zhong HH, Wang S, Heng WJ. Identification of a novel inheritable CCM1 gene mutation of 671del AT in a Chinese family with cerebral cavernous malformation. Zhonghua Yi Xue Za Zhi (2003) 83:1572–5.
15. Ji BH, Qin W, Sun T, Feng GY, He L, Wang YJ. A novel deletion mutation in CCM1 gene (krit1) is detected in a Chinese family with cerebral cavernous malformations. Yi Chuan Xue Bao (2006) 33:105–10. doi: 10.1016/S0379-4172(06)60028-0
16. Zhao Y, Xie L, Li P, Song J, Qu T, Fan W, et al. A novel CCM1 gene mutation causes cerebral cavernous malformation in a Chinese family. J Clin Neurosci. (2011) 18:61–5. doi: 10.1016/j.jocn.2010.04.051
17. Wang X, Liu XW, Lee N, Liu QJ, Li WN, Han T, et al. Features of a Chinese family with cerebral cavernous malformation induced by a novel CCM1 gene mutation. Chin Med J. (2013) 126:3427–32. doi: 10.3760/cma.j.issn.0366-6999.20131598
18. Zhu H, Guo Y, Feng X, Zhang R, Zhou C, Li G, et al. Familial cerebral cavernous angiomas: clinical and genetic features in a Chinese family with a frame-shift mutation in the CCM1 gene (krit1). J Mol Neurosci. (2014) 54:790–5. doi: 10.1007/s12031-014-0415-3
19. Mao CY, Yang J, Zhang SY, Luo HY, Song B, Liu YT, et al. Exome capture sequencing identifies a novel CCM1 mutation in a Chinese family with multiple cerebral cavernous malformations. Int J Neurosci. (2016) 126:1071–6. doi: 10.3109/00207454.2015.1118628
20. Huang WQ, Lu CX, Zhang Y, Yi KH, Cai LL, Li ML, et al. A novel CCM2 gene mutation associated with familial cerebral cavernous malformation. Front Aging Neurosci. (2016) 8:220. doi: 10.3389/fnagi.2016.00220
21. Yang C, Nicholas VH, Zhao J, Wu B, Zhong H, Li Y, et al. A novel CCM1/KRIT1 heterozygous nonsense mutation (c.1864C>T) associated with familial cerebral cavernous malformation: a genetic insight from an 8-year continuous observational study. J Mol Neurosci. (2017) 61:511–23. doi: 10.1007/s12031-017-0893-1
22. Wang H, Pan Y, Zhang Z, Li X, Xu Z, Suo Y, et al. A novel KRIT1/CCM1 gene insertion mutation associated with cerebral cavernous malformations in a Chinese family. J Mol Neurosci. (2017) 61:221–6. doi: 10.1007/s12031-017-0881-5
23. Yang C, Zhao J, Wu B, Zhong H, Li Y, Xu Y. Identification of a novel deletion mutation (c.1780delG) and a novel splice-site mutation (c.1412-1G>A) in the CCM1/KRIT1 gene associated with familial cerebral cavernous malformation in the Chinese population. J Mol Neurosci. (2017) 61:8–15. doi: 10.1007/s12031-016-0836-2
24. Yang C, Wu B, Zhong H, Zhong H, Li Y, Xu Y. A novel CCM1/KRIT1 heterozygous deletion mutation (c.1919delT) in a Chinese family with familial cerebral cavernous malformation. Clin Neurol Neurosurg. (2018) 164:44–6. doi: 10.1016/j.clineuro.2017.11.005
25. Zhao G, Song J, Yang M, Song X, Liu X. A novel mutation of LRSAM1 in a Chinese family with Charcot-Marie-Tooth disease. J Periph Nerv Syst. (2018) 23:55–9. doi: 10.1111/jns.12247
26. Spiegler S, Najm J, Liu J, Gkalympoudis S, Schröder W, Borck G. High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: one-third of probands are minors. Mol Genet Genomic Med. (2014) 2:176–85. doi: 10.1002/mgg3.60
27. Draheim KM, Fisher OS, Boggon TJ, Calderwood DA. Cerebral cavernous malformation proteins at a glance. J Cell Sci. (2014) 127:701–7. doi: 10.1242/jcs.138388
28. Kumar A, Bhandari A, Goswami C. Surveying genetic variants and molecular phylogeny of cerebral cavernous malformation gene, CCM3/PDCD10. Biochem Biophys Res Commun. (2014) 455:98–106. doi: 10.1016/j.bbrc.2014.10.105
29. Spiegler S, Rath M, Paperlein C, Felbor U. Cerebral cavernous malformations: an update on prevalence, molecular genetic analyses, and genetic counselling. Mol Syndromol. (2018) 9:60–9. doi: 10.1159/000486292
30. Zhang J, Dubey P, Padarti A, Zhang A, Patel R, Patel V, et al. Novel functions of CCM1 delimit the relationship of PTB/PH domains. Biochim Biophys Acta Proteins Proteom. (2017) 1865:1274–86. doi: 10.1016/j.bbapap.2017.07.002
31. Mondejar R, Lucas M. Molecular diagnosis in cerebral cavernous malformations. Neurologia (2017) 32:540–5. doi: 10.1016/j.nrl.2015.07.001
32. Padarti A, Zhang J. Recent advances in cerebral cavernous malformation research. Vessel Plus (2018) 2:21. doi: 10.20517/2574-1209.2018.34
Keywords: cerebral cavernous malformation, CCM1, CCM2, novel, genetic variant
Citation: Wang K, Wu D, Zhang B and Zhao G (2018) Novel KRIT1/CCM1 and MGC4607/CCM2 Gene Variants in Chinese Families With Cerebral Cavernous Malformations. Front. Neurol. 9:1128. doi: 10.3389/fneur.2018.01128
Received: 04 August 2018; Accepted: 10 December 2018;
Published: 21 December 2018.
Edited by:
Paul Pavlidis, University of British Columbia, CanadaReviewed by:
Jun Zhang, Texas Tech University Health Sciences Center, United StatesClaudia Vianna Maurer-Morelli, Universidade Estadual de Campinas, Brazil
Copyright © 2018 Wang, Wu, Zhang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guohua Zhao, Z3poYW9Aemp1LmVkdS5jbg==.