 Anelia A. Y. Kassi1
Anelia A. Y. Kassi1 Anil K. Mahavadi1
Anil K. Mahavadi1 Angelica Clavijo2
Angelica Clavijo2 Daniela Caliz2Stephanie W. Lee1
Daniela Caliz2Stephanie W. Lee1 Aminul I. Ahmed3
Aminul I. Ahmed3 Shoji Yokobori4
Shoji Yokobori4 Zhen Hu5Markus S. Spurlock1
Zhen Hu5Markus S. Spurlock1 Joseph M Wasserman1
Joseph M Wasserman1 Karla N. Rivera1Samuel Nodal1Henry R. Powell1Long Di1Rolando Torres1
Karla N. Rivera1Samuel Nodal1Henry R. Powell1Long Di1Rolando Torres1 Lai Yee Leung6,7Andres Mariano Rubiano2
Lai Yee Leung6,7Andres Mariano Rubiano2 Ross M. Bullock1
Ross M. Bullock1 Shyam Gajavelli1*
Shyam Gajavelli1*- 1Department of Neurological Surgery, The Miami Project to Cure Paralysis, University of Miami Miller School of Medicine, Miami, FL, United States
- 2Neurosurgery Service, INUB-MEDITECH Research Group, El Bosque University, Bogotá, CO, United States
- 3Wessex Neurological Centre, University Hospitals Southampton, Southampton, United Kingdom
- 4Department of Emergency and Critical Care Medicine, Nippon Medical School, Tokyo, Japan
- 5Department of Neurosurgery, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China
- 6Branch of Brain Trauma Neuroprotection and Neurorestoration, Center for Military Psychiatry and Neuroscience, Walter Reed Army Institute of Research, Silver Spring, MD, United States
- 7Department of Surgery, Uniformed Services University of the Health Sciences, Bethesda, MD, United States
Traumatic brain injury (TBI) is the largest cause of death and disability of persons under 45 years old, worldwide. Independent of the distribution, outcomes such as disability are associated with huge societal costs. The heterogeneity of TBI and its complicated biological response have helped clarify the limitations of current pharmacological approaches to TBI management. Five decades of effort have made some strides in reducing TBI mortality but little progress has been made to mitigate TBI-induced disability. Lessons learned from the failure of numerous randomized clinical trials and the inability to scale up results from single center clinical trials with neuroprotective agents led to the formation of organizations such as the Neurological Emergencies Treatment Trials (NETT) Network, and international collaborative comparative effectiveness research (CER) to re-orient TBI clinical research. With initiatives such as TRACK-TBI, generating rich and comprehensive human datasets with demographic, clinical, genomic, proteomic, imaging, and detailed outcome data across multiple time points has become the focus of the field in the United States (US). In addition, government institutions such as the US Department of Defense are investing in groups such as Operation Brain Trauma Therapy (OBTT), a multicenter, pre-clinical drug-screening consortium to address the barriers in translation. The consensus from such efforts including “The Lancet Neurology Commission” and current literature is that unmitigated cell death processes, incomplete debris clearance, aberrant neurotoxic immune, and glia cell response induce progressive tissue loss and spatiotemporal magnification of primary TBI. Our analysis suggests that the focus of neuroprotection research needs to shift from protecting dying and injured neurons at acute time points to modulating the aberrant glial response in sub-acute and chronic time points. One unexpected agent with neuroprotective properties that shows promise is transplantation of neural stem cells. In this review we present (i) a short survey of TBI epidemiology and summary of current care, (ii) findings of past neuroprotective clinical trials and possible reasons for failure based upon insights from human and preclinical TBI pathophysiology studies, including our group's inflammation-centered approach, (iii) the unmet need of TBI and unproven treatments and lastly, (iv) present evidence to support the rationale for sub-acute neural stem cell therapy to mediate enduring neuroprotection.
Introduction
TBI is a critical public health problem and one of the leading causes of death and disability around the globe (1–5). The World Health Organization (WHO) and the World Bank estimate that 69 million (95% CI 64–74 million) individuals suffer from TBI every year, with Southeast Asian and Western Pacific regions experiencing the greatest overall burden (6). A recent estimate of the Global Incidence of TBI puts it at ~939 cases per 100,000 people each year with 79% being mild TBI. The calculated incidence of TBI in the Americas (including United States (US) /Canada) is 1,299 cases per 100,000 people each year. The calculated incidence for Latin America is about 909 per 100,000 people each year (7). Worldwide about 90% of all TBI-related deaths occur in developing countries (8). In 2016, road traffic injuries were among the three leading causes of death from injuries independent of gender. The economic status (a surrogate for investment in health care, trauma centers, and road safety) of the country rather than its global location appear to influence trauma outcomes. For example, in the poorest country in the Western Hemisphere, road traffic accidents accounted for >40% of TBI incidence (9). Similarly, African and Eastern Mediterranean regions are above the global average while the rest are on par or below (6, 10). Within the US, road traffic accidents have been on the decline and apart from age related vulnerability to falls, firearm injury has become an increasingly serious problem (11, 12). Overall, TBI affects 1.7 million people in the US with ~50,000 fatalities annually. Timely and aggressive management of acute trauma patients has lowered the fatality rate but does not eliminate the socioeconomic consequences of TBI (13–17). The annual cost of TBI in the US is estimated to be between $168 billion in medical spending and $223 billion in work losses (18). Globally it is estimated at $400 billion (19). Despite the outpouring of resources for TBI management and research, 5.3 million TBI patients in the US continue to live with disabilities, a consequence that is independent of injury severity (20). Improved clinical care has led to increased post injury survival, while return-to-work has remained static for the past five decades (21–24). The current clinical management of severe TBI exploits the limits of physiological interventions and addresses issues mainly at the systemic level and sometimes at the cellular and biochemical levels but rarely at the subcellular organelle dysfunction level. As an example, TBI induced mitochondrial dysfunction has remained intractable (25). Consequently, TBI survivors experience the full wrath of secondary mechanisms (26–30). This “secondary mechanism fueled” histopathology seen in human TBI is recapitulated with preclinical TBI models (31–36) and offers an opportunity to test interventions.
Current TBI Treatment
The Brain Trauma Foundation (BTF), a non-profit group of TBI expert clinicians, has dictated the management of severe TBI since its establishment in 1996 (37, 38). Since that time, there has not been much change in the treatment of TBI despite a better understanding of the destructive events inherent to the disease process. Though adherence to these guidelines decreased overall healthcare costs and improved patient survival (39, 40), the latest fourth edition offers no class I and few class II recommendations in regards to severe TBI management. The major focus of current neurointensive care is (i) metabolic stabilization of the patient, (ii) prevention of further deterioration, and (iii) facilitation of “spontaneous” brain recovery. Along with prompt neurosurgical interventions when warranted, optimizing hemostasis, oxygenation, ventilation, temperature, blood pressure, blood glucose, and acute seizure prophylaxis increased positive outcomes after severe TBI (38, 41, 42). Contrary to previous guidelines, a Glasgow Coma Scale (GCS) of lower than 5 is no longer a contraindication to surgery because of advances in modern surgery and the neurointensive care unit which have improved survival of these patients (43, 44). Early management and proper monitoring of parameters such as intracranial pressure and sodium levels have limited certain types of secondary brain injury (42, 45). Compliance with BTF guidelines is proportional to the strength of evidence (46). For implementation of an efficient trauma system in under-privileged areas, the organization of low cost resources such as trauma registries are required (47–50). For example, Latin American neurosurgeons have advocated for improving clinical research methodologies and topics in the region (51), to better understand implications and relationships between intervention and outcomes. Aggressive surgical therapy seems to be an option for improving survival even in penetrating TBI (PTBI) (48) in developed countries. Intensive critical care management and less aggressive surgical therapy based on the military experience acquired during the 1970's war in Lebanon also produces favorable outcomes especially in pediatric and adult severe TBI (52, 53). Severe TBI patients are treated with a combined medical-surgical approach, managed initially in the intensive care unit (ICU) with neuromonitoring (54, 55), in conjunction with BTF “living” guidelines (updated to incorporate the findings of randomized clinical trials (RCT), the gold standard for proving the efficacy of new treatments) (37, 38, 56). The next section revisits a few trials and identifies TBI pathophysiological processes that may have led to their failure.
Can Insights Into TBI Pathomechanism Explain Failure Of Past Neuroprotection Trials?
Primary injury, which occurs at the time of impact, includes tissue laceration, cerebral contusion, axonal damage and hemorrhage. Following hospital care, TBI patients can also remain disabled, rendering them worse off which led to the conclusion that secondary injury. It was deduced that secondary insults also significantly influenced outcomes (20, 56, 57). Investigations into the secondary injury process revealed several concurrent processes with distinct spatiotemporal peaks occurring within seconds after injury and lasting for years (58, 59). The result is a complex cascade of molecular and cellular damage, which magnifies the primary injury causing delayed and remote secondary injury (60–64). Initial descriptions of secondary mechanisms included clinical parameters necessary for decision-making, which led to the invention and adoption of the Glasgow coma scale (GCS) (57, 65). The list now includes parameters known to influence TBI outcome such as cerebral blood flow (66), hypoxia-ischemia (67), mitochondrial dysfunction (25), cerebral metabolism (68), cell death (69), glutamate excitotoxicity (70, 71), calcium dysregulation, edema (72) culminating in inflammation, the most enduring of the secondary damage mechanisms (73–75). Inflammation has also been linked to depression like symptoms causing depression like symptoms via failure of neurogenesis (76, 77) in multiple CNS conditions including TBI. All these processes have been recapitulated in animals model (Figures 1) (78). In the early post-traumatic period (seconds to days), injured neurons in contusions appear swollen, but over time (days or weeks), they become shrunken and eosinophilic, with pyknosis of the nuclei (79). Neuronal and glial “apoptosis” was observed after TBI in human tissue prior to description of the process (69) and later confirmed (80).
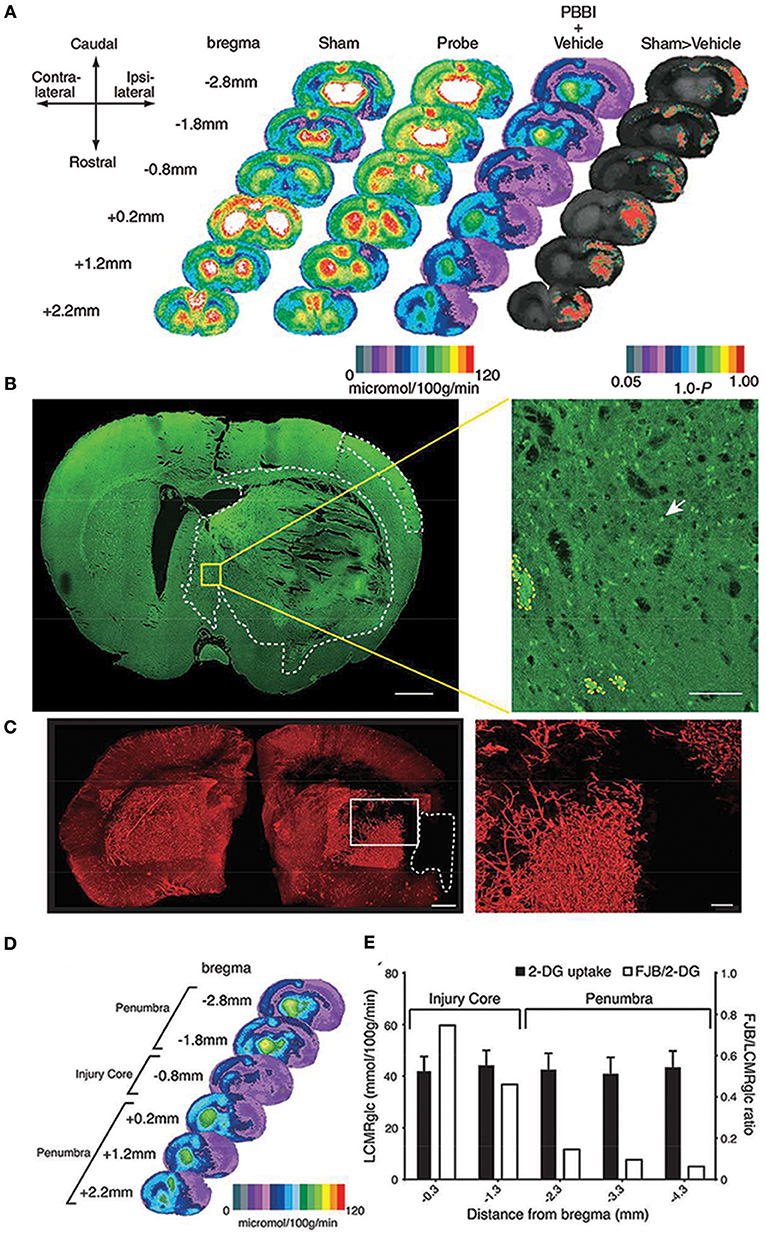
Figure 1. Local cerebral glucose metabolism after penetrating ballistic-like brain injury (PBBI) (A) is shown as color-coded maps of average local cerebral metabolic rate for glucose (LCMRglc) at 2.5 h after injury. Each coronal section is a representation of multiple animals within a group at that particular level. Rat brain atlas levels are given on the left column as millimeters from bregma. Compared with controls (columns 1 and 2) in PBBI (column 3), LCMRglc decreased radially from injury core into perilesional areas and globally across the entire brain. P-maps of average local cerebral glucose utilization were produced by comparing the values of pixels corresponding to the same anatomic position across groups. (B) Confocal image of a Fluorojade B (FJB)-stained coronal section at 0.8 mm distance from bregma shows regions with FJB+ cells (circumscribed by white-dotted line). Greater neurodegeneration was observed in the injury core and peri-injury zone in the ipsilateral than those in the contralateral cerebral cortex. (C) Composite light sheet microscopy image shows ipsi and contralateral hemispheres perfused with fluorescent tomato-lectin at 2.5 h post PBBI. Region with injury induced hypoperfusion is circumscribed by white-dashed line. Surface reconstruction renders the labeled vasculature in 3D. (D) Hypoperfused region overlaps with the 2-deoxy glucose (2-DG) uptake impairment heat map. (E) The incidence of neurodegeneration was proportional to 2-DG uptake impairment at the injury core but not in regions caudal to the injury core. Fluorojade B (FJB)/LCMRglc ratio decreased from injury core toward more caudal regions, decreasing maximally at−2.3 mm from bregma and plateaued (penumbra). Further details are present in the original article (78).
Over the three decades, the improved survival of TBI patients upon management with Glasgow coma score (21, 65) and the adoption of cerebral cardiopulmonary resuscitation (CCPR) protocols based upon quantitation of physiological measures (81) led to RCTs that attempted to block/reverse the TBI pathological processes. Such RCTs mostly failed to yield any class I evidence necessary to improve TBI outcomes. These trials included surgical interventions, which unlike decompressive craniectomy (DC) in stroke (82), did not find benefit and had to be stopped due to adverse effects and low recruitment. For e.g., both Decompressive Craniectomy in Patients with Severe Traumatic Brain Injury (DECRA) and Randomised Evaluation of Surgery with Craniectomy for Uncontrollable Elevation of Intracranial Pressure (RESCUEicp) showed poor outcome (55). DECRA was criticized for excluding second tier treatments often used in “real life,” not representing the “real world population,” and because the duration of high ICP was too short (83, 84). Further negating the DECRA findings, a retrospective analysis revealed benefit of DC and/or barbiturate combination for refractory intracranial pressure management after severe TBI (85). More recently another DC trial (with 80% of patients similar to DECRA and 38% to RESCUE-ICP) showed that the addition of a barbiturate step following DC was more effective than DC alone, barbiturate alone or barbiturate before DC (86). RESCUEicp reported that at 6 months post-decompressive craniectomy, mortality was lowered but at the cost of higher rates of vegetative state, and severe disability. The trial evaluating Early Surgery vs. Initial Conservative Treatment in Patients with Traumatic Intracerebral Hemorrhage was halted after enrolling <20% of the planned number (87, 88). The limited success of surgical intervention is unsurprising as numerous secondary processes (discussed below) are initiated after primary insult and cannot be surgically targeted. More perplexing is the failure of neuroprotective pharmacological RCTs (11, 89–92) including the ProTECT trial (93) which were based on robust preclinical data. Therefore, in the next section we explore the possible reasons that single TBI pathological mechanism targeting RCTs failed.
Mitochondrial Dysfunction/Calcium Dysregulation
TBI induced mitochondrial dysfunction is the rate-limiting step in metabolic restoration of a patient with clinical management (25, 94, 95). Persistently elevated intracellular calcium levels play a central role in activating cellular death mechanisms. Dysfunction of mitochondria (96), production of pro-inflammatory cytokines, as well as axonopathy (97) are all related to calcium dysregulation (98). Upon binding of calcium, the calmodulin-calcineurin complex upregulates the expression of IL-2 by activating the transcription factor NFAT. IL-2 stimulates the proliferation of T lymphocytes, which then recruit more immune cells and amplify the process (99). This pathway is exploited in the treatment of cancer, transplant rejection, and autoimmune diseases. Insights into what constitutes mitochondrial dysfunction came from studies in cardiomyocytes. In these cells low ATP, high calcium caused mitochondrial dysfunction due to the opening of high conductance pores in the inner mitochondrial membrane, uncoupling mitochondrial oxidative phosphorylation and promoting ATP hydrolysis. Cyclosporine A (CsA) was found to prevent such pore opening in isolated mitochondria (100) and cells (101). However, prevention of neurological deterioration was incorrectly attributed to CsA (102), albeit unknown at that time (103). CsA was found to be beneficial in transient forebrain ischemia rodent models upon intracerebral injection provided it could cross the blood brain barrier (BBB) (104). It was therefore given before and after the injury to enable entry to the brain during the opening of BBB and stabilized isolated mitochondria in rodent TBI brains (105). However, despite safety in humans (106, 107), the drug failed to meet the OBTT criteria for advancing to translation (108). This could be in part due to inadequate dosing or possible adverse effects associated with the vehicle Cremaphor. The drug also has a short therapeutic window and needs continuous infusion over the first 3 days post injury to stabilize the mitochondria (109–112). Recent work with a new carrier in gyrencephalic animals is reportedly neuroprotective (113). A related drug, minocycline, although not tested in OBTT and initially used for a different purpose i.e., reducing neuroinflammation via ablation of activated microglia, was tested in human TBI and found to have no benefits (114) though the marker of inflammation was reduced. Minocycline, a well-known antibiotic in the tetracycline family with anti-inflammatory qualities that include inhibition of NFkB prevents the transcription of pro-inflammatory cytokines and activation of M1 microglia (99, 115). Consistent with equivocal data in rodent TBI (116), a clinical trial using minocycline in chronic TBI patients (6 to 142 months post TBI) showed reduced microglial activation but increased neurodegeneration over time (114). Microglial activation, measured with 11C-PBR28 PET was reduced after 12 weeks of treatment with minocycline but plasma axonal neurofilament light levels, a marker of neurodegeneration, increased and white matter atrophy was more prominent on MRI (114). This suggests that mitochondrial function in even activated microglia are necessary to prevent neurodegeneration, and that there is a need to separate the anti-inflammatory regenerative properties e.g., phagocytosis and proinflammatory degenerative properties e.g., pyroptosis that reside within the same activated microglia, rather than merely ablating all microglia. It is possible that attempts to reduce proinflammatory activated microglia via cell ablation does not mitigate their pyroptosis, an inflammatory process that causes damage to intact tissue. Instead there may be a need to ramp up the anti-inflammatory microglia activity (117). Collectively, preclinical data and the minocycline TBI trial suggest that non-specifically targeting activated glia through these corresponding classes of drugs [Consistent with such data mitochondrial uncoupling agents are also capable of tissue sparing (118)] is not sufficient for neuroprotection in neither an acute nor chronic time point when used with certain dosing regimen.
Calcium is also required for the activation of calpain proteases that cause the breakdown of the cytoskeleton and the cessation of axonal transport (97). Neurofilaments and microtubules accumulate and swelling ensues, forming axonal bulbs that eventually separate the axon. Years after injury, extensive axonopathy is associated with atrophy of the brain, expansion of the ventricles, and premature dementia (119). Of the several clinical trials that evaluated the effect of the calcium channel blocker nimodipine in acute severe TBI, only one improved neurological outcome at 6 months (120). However, subgroup analyses from failed studies revealed that the patients with evidence of subarachnoid hemorrhages on CT benefited from the drug (121). Nimodipine is now widely used in patients with SAH from traumatic and non-traumatic etiology as vasospasm prophylaxis.
Glutamate Excitotoxicity
Another contributory mechanism to TBI is a surge of excitotoxic amino acids, primarily glutamate, that occurs which causes irreparable disturbances in ion fluxes (71). Binding of glutamate to the N-methyl-D-aspartate (NMDA) receptor results in depolarization of neurons, followed by a massive influx of calcium, and efflux of (71, 122). Loss of GABAergic inhibition (possibly due to higher numbers of GluR3 subunit containing but GluR2 subunit depleted AMPA receptors which allow influx of calcium mediating greater vulnerability to excitotoxicity), and downregulation of astrocytic glutamate transporters which allow excitatory neurotransmitter accumulation in the synapse culminating in neuronal and glial (oligodendrocyte) cell death (123, 124). The integrity of GABAergic inhibition needed to contend with excitotoxicity may be compromised by reduced glutathione activity. Glutathione activity was reduced in a rodent model of TBI (125), and increased super oxide production decreased parvalbumin expression in parvalbumin (PV+) GABAergic cells (126). PV+ cells have dendritic arborizations receiving fast converging excitatory inputs (127–130) as reviewed earlier (131) and are endowed with a fast spiking phenotype (132). Higher cognitive functions such as perception (133), and the deterrence of epileptiform activity (134, 135) is contingent upon reliable PV+ cell mediated inhibition. As such, pharmacological disruption of glutamatergic signaling onto fast spiking parvalbumin GABAergic cells disinhibits the circuitry mediating gamma oscillations which facilitate information storage and transfer (136). Promising preclinical results with magnesium blockade of the neuronal NMDA receptor (70), did not translate into neuroprotection in clinical trials, in part due to narrow therapeutic windows, adverse side effects, and interference with normal electrical activity of the brain (11, 19, 90, 137). Glutamate NMDA receptor antagonists (competitive receptor antagonists, ion channel blockers, and glycine antagonists)—such as selfotel, aptiganel, eliprodil, licostinel, and gavestinel—failed to show efficacy in clinical trials of stroke or traumatic brain injury. Deficient properties of the molecules used in human trials or inappropriate design of clinical studies may have contributed to failure. It is possible that excitoxicity kills inhibitory GABAergic cells (such as hippocampal parvalbumin neurons) due to their higher amounts of GluR3 containing and GluR2 lacking AMPA receptor subunits than glutamatergic neurons, as mentioned above. The clinical NMDA antagonists dose then targets only the glutamatergic neurons, oligodendrocytes, and astrocytes impairing brain metabolic capacity. An alternative hypothesis suggests that glutamate may be involved in the acute neuro-destructive phase immediately after traumatic or ischaemic injury (excitotoxicity), but later, is required for normal physiological functions except during spreading depolarizations (138). Thus blockade of synaptic transmission mediated by NMDA receptors may hinder neuronal survival (139). Ikonomidou speculated that these drugs could be useful if used just prior to a TBI event, akin to an unexpected decreased in TBI related mortality in alcohol and methamphetamine abusers (140, 141), despite neurotoxicity of the abused substance (142). It remains to be seen what role such drugs can play in TBI management.
Hyperglycolysis
Human brain normally uses glucose as the sole substrate and due to lack of fuel stores; the brain requires a continuous supply. Seymour Kety and Carl Schmidt introduced the first quantitative measurements of human, whole-brain blood flow and metabolism (143). Kety, Sokoloff and their colleagues noted that, while the human brain is only 2% of the body weight, it accounts for 20% of the body's energy consumption. Their technique laid the foundation for studies of brain metabolism in terms of rates of glucose and oxygen consumption (144). When the technique was applied to TBI, both cerebral metabolic rate of oxygen (CMRO2) and cerebral metabolic rate of glucose changes (CMRO2) were evident (66, 145). During the first 6 days after moderate or severe TBI, CMRO2 and arterial lactate levels are the strongest predictors of neurologic outcome (146). Relative to the impaired glucose uptake in the TBI brain, some regions exhibited a high level of glycolysis. This process called “hyperglycolysis” is defined as an increase in glucose utilization two standard deviations above the normal. Hyperglycolysis is thought to be mediated by the release of catecholamines in response to injury to meet increased energy demand upon cells to drive pumping mechanisms in order to restore membrane ionic balance (147). In a study of 28 patients, Hovda et al. reported that hyperglycolysis was observed in 56% of patients on fluorodeoxyglucose-positron emission tomography (FDG-PET within the first week of injury and persisted for several weeks (148, 149). In the context of TBI, it is not clear which cells are undergoing hyperglycolysis. Lactate accumulation in the injured brain can stem from neuronal mitochondrial dysfunction (150) and/or due to massive influx of lactate from peripheral tissues (151). In turn, elevated lactate levels contribute to pan necrosis (plasma membrane damage, cerebral edema, BBB permeability and overall cell breakdown) (152). In preclinical (153) and clinical studies (149) of TBI, hyperglycolysis and related metabolic crisis (non-ischemic high lactate) increased the incidence of spreading depolarization (138), seizures (154) and were associated with poor outcomes (148). Misled by an “insufficient fuel” concept, glucose supplementation was explored in preclinical studies but surprisingly turned out to be harmful (155–157). However, as in the case of mild TBI, mere fasting was found to be neuroprotective (158). Accordingly, there is no consensus on glycemic control after TBI. Novel metabolic imaging techniques (159) or combination of metabolic studies and neuromonitoring with imaging will be key to gaining insights into the TBI metabolic crisis (94, 138, 150, 154). In three clinical trials, intensive insulin therapy—when compared to conventional insulin therapy—consistently increased the risk of hypoglycemia in moderate to severe TBI patients and failed to decrease mortality at 6 months (160–162). Among these studies, only Yang et al. reported better neurological outcome, measured with Glasgow Outcome Scale (GOS), at 6 months. Supply-demand mismatch, generated from increased metabolism in the setting of decreased cerebral blood flow (CBF), provokes an energy crisis that promotes further damage (94).
This led to the search for alternative substrate to improve cellular metabolism. Ketogenic diet (KD) via induction of ketosis is known to increase cerebral metabolism of ketones. Age-dependent neuroprotection after TBI in part could be due to younger animals achieving significant β-hydroxybutyrate levels earlier than adults do. In both juvenile rats subjected to weight drop model and adolescent rats to cortical contusion injury (CCI), KD resulted in decreased brain edema, cytochrome c release, apoptotic and oxidative stress marker expression, mitochondrial calcium loading, improved cellular energetics, increased expression of brain-derived neurotrophic factor, smaller contusion volumes and better motor, and cognitive performances. Ketosis mediated by fasting or calorie restriction was also neuroprotective in adult rats with TBI. One of the prominent mechanisms of KD includes inhibition of glycolysis (and subsequently dependent proinflammatory cytokine synthesis), thus lowering inflammation and upregulating bioenergetics via mitochondrial biogenesis (163). At the organelle level a ketogenic diet was found to reduce onset of seizures by preventing the opening of mitochondrial membrane permeability transition pores (164) effectively acting as a neuroprotective uncoupling agent (165).
In contrast to the beneficial effects of ketone metabolism, poor nutritional support can exacerbate TBI (166). Currently, clinical studies are underway to determine the optimal method to induce cerebral ketone metabolism in the post-injury brain, and to validate the neuroprotective benefits of ketogenic therapy in humans (167). Improvements in the understanding of human brain metabolism (168) led to the documentation of metabolism perturbations in injured brains (94, 169–171) and the ability to test if supplementation can bypass these impairments (172).
Hypoxia
In order to maintain its high metabolic activity, the brain receives a substantial proportion of the cardiac output and is therefore highly susceptible to hypoxia (173, 174). Under normal circumstances, a decrease in arterial partial oxygen tension (pBTiO2) is balanced by increases in cerebral blood flow (CBF) to prevent cerebral ischemia, sometimes at the expense of rising ICP (175). In TBI, loss of autoregulation, as evidenced by concurrent reduction in CBF and pBTiO2, is one of the mechanisms that exacerbate injury. Hypoxia accelerates uncoupling of the electron transport chain and mitochondrial permeabilization, which induces the release of pro-apoptotic signals, such as reactive oxygen species (ROS) and cytochrome c, inside the cytosol (96). Leakage of cytochrome c in conjunction with elevated cytoplasmic calcium activates the caspase cascade, leading to cell apoptosis. HIF-1α is a constitutively expressed protein whose activity depends on oxygen availability (176). In normoxia, HIF-1α is hydroxylated by prolyl hydroxylase (Lee et al.) then tagged with ubiquitin for degradation in proteasomes (177). In contrast, hypoxia decreases the activity of PHD and HIF-1α is translocated to the nucleus where it binds HIF-1β and pyruvate kinase M2. This complex induces the transcription of molecules that stimulate the expression of genes involved in glycolysis, angiogenesis, neurogenesis and the synthesis of proinflammatory cytokines (177–179).
Methods to alleviate TBI-induced brain tissue hypoxia by using blood substitutes, increasing hemoglobin, oxygen saturation, or oxygen tension are currently part of the TBI critical care armament. Attempts to improve cerebral oxygenation with blood substitutes such as perfluorocarbons (PFCs) alleviated hypoxia in animal models of PBBI (78, 180, 181). However, clinical translation of this artificial oxygen carrier was deemed unsafe due to the development of thrombocytopenia, an abnormality that could be detrimental in TBI patients, which led to the cessation of the Safety and Tolerability of Oxycyte in Patients With Traumatic Brain Injury (STOP-TBI) trial (182). Insights into PFC cellular distribution facilitated its repurposing to instead identify injury penumbra: perfluorocarbon enhanced Glasgow Oxygen Level Dependent (GOLD) magnetic resonance metabolic imaging (183).
Because of the prevalence of anemia in TBI patients and associated worse outcomes, administration of erythropoietin (EPO) was evaluated after head injury. The discovery of EPO, a hormone principally produced by the peritubular interstitial cells of the kidney, revolutionized the treatment of anemia in chronic kidney disease patients. Identification of EPO of neuronal and astrocytic origins and the hormone's non-hematopoietic functions (184) have been investigated. Binding to the EPO receptor prevents apoptosis of mature neurons and enhances the proliferation of neural progenitor cells (185). Its anti-apoptotic properties are mediated by the inhibition of pro-apoptotic molecules -including apoptosis regulators bcl-2-like protein 4 (BAX) and cytochrome c, and the activation of the NFKB pathway, which results in the stimulation of the adaptive immune system cells. EPO also promotes proliferation of the endothelium and the production of nitric oxide (NO) (185). In the multicenter international EPO-TBI trial, EPO did not display neuroprotective effects in patients with moderate to severe TBI (186, 187). Perhaps EPO-induced inflammatory NO may have abrogated any beneficial effect as it increased lesion volume after PBBI (188).
Although, it could seem counterintuitive to use hyperoxia in TBI (189) (i.e., to avoid reperfusion injury), researchers found that markedly increasing oxygen (O2) delivery to the traumatized brain, with hyperbaric oxygen therapy (HBOT) or normobaric hyperoxia (NBH), could reverse the lack of O2 for e.g., vascular stenosis (Figure 1) that precipitates cellular energy failure and subsequent neuronal death. A recently published review identified eight phase I and phase II clinical trials evaluating the role of acute and subacute HBOT and/or NBH in severe TBI patients. Overall, HBOT alone or in combination with NBH improved physiologic markers of metabolic function (microdialysate LPR, glycerol, ICP) and decreased long-term morbidity and mortality to a greater extent than NBH alone or standard of care (67, 190–192). The “Hyperbaric Oxygen Brain Injury Treatment” (HOBIT) trial: (193) is a proposed adaptive clinical trial designed to answer questions about dosage and safety parameters of HBO2 and to provide important data to plan a definitive phase III efficacy trial.
Edema
After TBI, edema develops because of cellular dysfunction (cytotoxic edema) and blood brain barrier (BBB) disruption (vasogenic edema). Increased permeability of the cell membrane to Na+ and K+ followed by failure of the Na+/K+ ATPase pump traps osmotically active molecules inside the cell. Mechanical destruction of endothelial cells causes the capillaries to leak a protein-rich exudate into the brain parenchyma. CBF reduction, glutamate excitotoxicity, osmolar gradients additionally participate in extending the edematous state and can contribute to elevations in ICP (194). Different osmotic therapies (mannitol, hypertonic saline, hypertonic lactate, barbiturate) have been examined but none have improved long-term neurological outcome or survival (38, 186). The “BRAIN” trial tried to exploit the role of the kallikrein-kinin system in TBI but it was terminated because Anatibant, an antagonist of the bradykinin B2 receptor, caused more deaths than control 15 days post-injury (195, 196). The kinin family also is known to have a neuroprotective role via the attenuation of microglial proinflammatory secretion through actions of prostaglandin E and microsomal prostaglandin E synthase (197, 198). Bradykinin receptor B1 but not B2 deficiency protects from focal closed head injury in mice by reducing axonal damage and astroglia activation (198, 199). Anatibant may have selectively inhibited the neuroprotective effects while allowing proinflammatory signaling to persist leading to poor outcomes. Nevertheless, the historical failure of acute neuroprotective interventions (11, 114, 137, 200–202) has exposed the limitations of preclinical TBI models in guiding clinical trials in TBI. Similarly, limitations inherent in pre-clinical testing such as insufficient rigor in pre-clinical studies may also have contributed to RCT failure (203). To offset this two groups have come up a similar suggestion regarding data reporting in preclinical studies that would help compare preclinical studies. Use of “delta Score” i.e., summing the change in outcome that may occur in patients in the placebo vs. drug-treated groups over time or effect size used to run meta-analyses are helpful (204, 205). In aggregate, the RCTs failed as they allow for persistence of dual edged inflammation. To provide insights into how unmitigated inflammation underlies progressive tissue loss, our laboratory research uses a rodent model of penetrating TBI (PTBI). PTBI and TBI secondary damage mechanism are similar and may differ only in magnitude. Acute and delayed consequences of human PTBI (64, 206–208) are replicated in Penetrating ballistic-like brain injury (PBBI), a survivable rat PTBI model (34, 35, 78, 209, 210). We detail the results from using this model below.
Why Is The Penumbra Vulnerable?
Our recent study with rat PTBI showed that the ipsilateral cortical region at 48 h post injury is replete with activated microglia (boxed regions in Figures 2A–C) (210). Only the “penumbra” (yellow highlight in Figure 2D) disappears by 10 weeks post injury (35) while regions more dorsal persist despite the presence of proinflammatory activated glia (210). It is possible that a gradient of DAMPS or NAMPs exists with the high concentration at the injury core gradually diminishing radially in a tissue architecture dependent manner. Consistent with this concept, activated microglia in penumbra surrounding the core region may undergo pyroptosis unlike microglia in distal regions. Consequently by 10 weeks post injury the entire penumbra with “critical” levels of DAMPs/NAMPs” disappears while the regions with sub-critical levels survive. This data is consistent with human TBI study where molecular characterization revealed greater numbers of activated microglia in pericontusion (penumbra) than contusion (211). Hence, if reversal of penumbra vulnerability changes TBI outcomes, rescuing the tissue may become a priority.
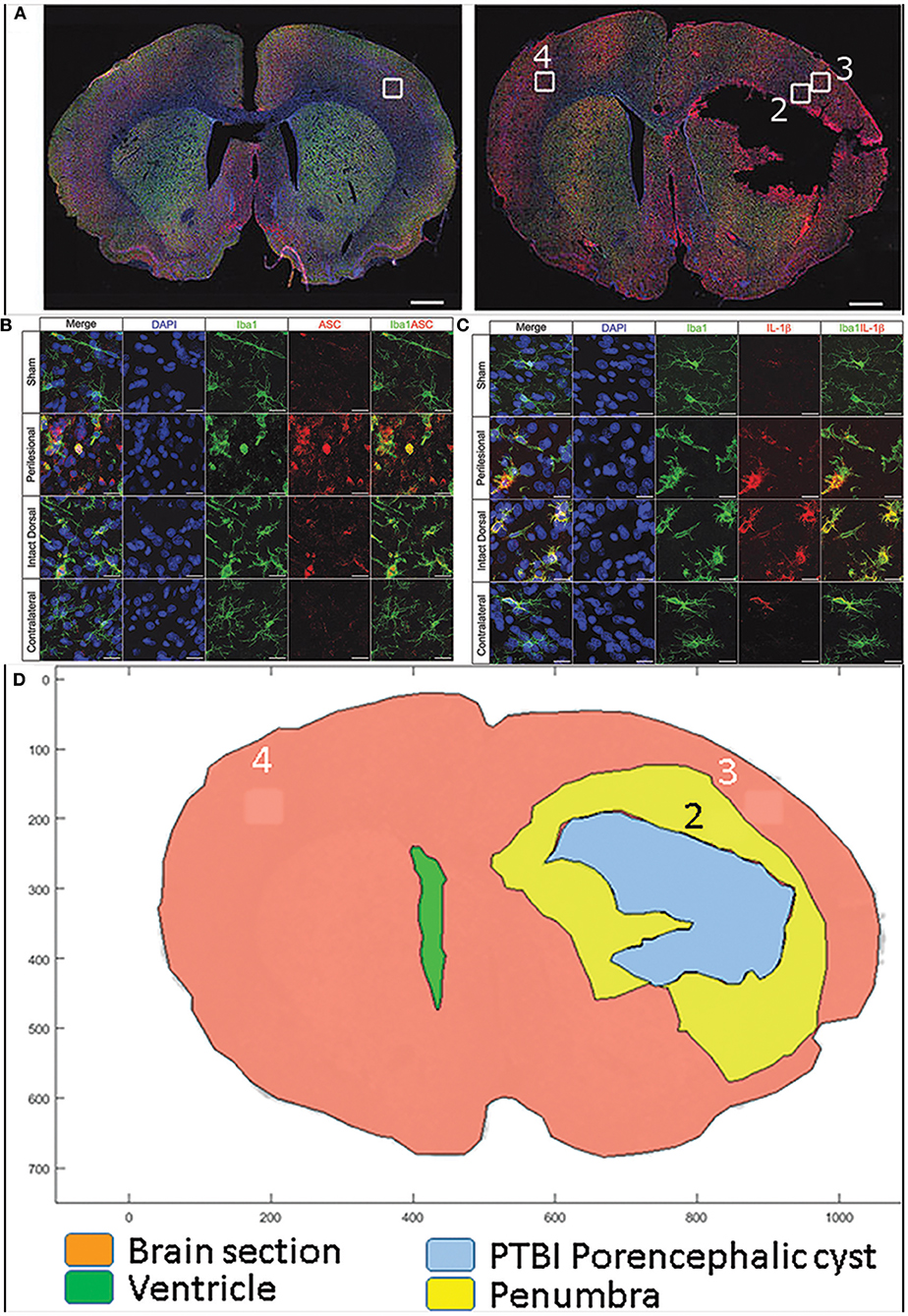
Figure 2. Confocal images of free-floating rat brain sections stained with 2-(4-amidnophenyl)-1H-indole-6-carboxmidine (DAPI; blue), ionized calcium-binding adapter molecule 1 (Iba-1; green), and apoptosis speck-like protein containing caspase-activation and recruitment domain (ASC) or interleukin (IL)-1b (red). Top panels (A) show whole–brain sections from a representative sham (left) and 10% penetrating ballistic-like brain injury (PBBI) animal 48 h after injury (right). Sections show Iba-1+ microglia widely dispersed throughout the brain. White boxes (1–4) in the whole–brain images are shown at 100x magnification in panels below. ASC immunoreactive cells are absent in sham cortex (box 1), numerous ASC positive cells are present in PBBI perilesional area (box 2), but to a lesser extent in PBBI intact ipsilateral dorsal cortex (box 3), and absent in contralateral cortex (box 4) (B) Iba-1 and ASC double positive cells are present in the ipsilateral hemisphere. In (C) Iba-1 and IL-1b co-labeled cells are predominantly present in the ipsilateral cortex. Double positive cells are morphologically large with round/hypertrophied cell bodies and short processes. Additional details are presented in the original publication (210). By subtracting the traces of the brain sections at 48 h post PBBI from those at 10 weeks post PBBI, the PBBI penumbra (box 2 within yellow highlight) is identified (D). The penumbra (box2) was occupied by highly activated microglia at 48 h post injury is lost by 10 weeks post PBBI. In contrast, in box 3 microglial were activated to a lesser extent and at 10 weeks post injury such tissue survives.
Vulnerable TBI Events That Can And Need To Be Targeted By Clinical Therapy To Spare Penumbra
Inflammation
Within minutes of injury, damaged cells release damage-associated molecular patterns (DAMPs)—high extracellular potassium, adenosine triphosphate (ATP), mitochondrial DNA, heat-shock proteins (HSPs), high mobility group binding proteins (HMGB1) molecules, and Amyloid beta. These can activate an inflammatory response in nearby cells (212, 213). Consequentially, the assembly of inflammasomes, activation of complement pathways and local immune cells, and production of pro-inflammatory chemicals (chemokines, cytokines, ROS, NO) trigger inflammatory cell death mechanisms (79, 210, 214). The production of interleukin 1 beta (IL-1β) by activated microglia peaks 48 h post-injury and favors polarization of microglia into the pro-inflammatory type.
Broad anti-inflammatory interventions such as hypothermia (215) or neuropeptide blockade (216) appear to be promising based on biomarker profiles. The anti-apoptotic and anti-inflammatory effects of hypothermia have also been investigated in TBI (217). Use of hypothermia for refractory ICP after TBI was beneficial in some centers (217). Particularly in China where three of the four trials had positive effects (acute reduction in ICP and long-term improvement of neurological deficits and mortality at 6 months), however all other trials failed to show similar results (186) e.g., Eurotherm3235 Trial, POLAR RCT failed to reproduce the benefits and stopped due to adverse effects. In the Eurotherm trial, titration with therapeutic hypothermia successfully reduced ICP in participants with TBI + ICP of >20 mmHg, but also led to a higher mortality rate and worse functional outcome (218). Post-hoc subgroup analysis of the NABIS-HII trial revealed that hypothermia improved outcomes in patients with evacuated subdural hematomas (219). The failure of the latest hypothermia trial in TBI (220) provides insights into the barriers of translating preclinical findings into human TBI and may unfortunately lead to suboptimal use of this potentially powerful therapeutic in potentially treatable severe trauma patients (221). However, anti-inflammation is not the only consequence of hypothermia, as this approach continues to remain controversial in TBI due to its risk of altering mortality or leading to poor outcomes or new pneumonia (222, 223).
Based on anti-inflammatory action in rheumatoid arthritis, anakinra, FDA approved competitive inhibitor of an interleukin 1 (IL-1) receptor, role of such signaling was evaluated in controlled cortical impact (CCI), a TBI model in rodents, injured IL-1R1 null and wild type mice did not differ in respect to brain lymphocyte numbers (224). In a less sever TBI model, ablation of ILR1 signaling or exogenous IL-1Ra was sufficient to reverse TBI induced deficits (225). Both IL1-alpha (IL-1α), IL-1β signal through the same IL1R. Elevated IL1alpha/IL1 beta are associated with favorable outcomes after TBI (226, 227). Off label, use of anakinra for human TBI is reportedly beneficial (228) in that it shift the microglia to less inflammatory phenotype (229). Based on the preclinical data the detrimental effects of IL1R signaling seem to dominate over the beneficial effects in TBI context hence seeking total IL1R blockade in human TBI needs to be tested next. Another inflammatory cytokine of interest is tumor necrosis factor alpha (TNF-alpha), which upon interacting with TNF receptor 1 but not TNF receptor 2 was found detrimental in neurodegenerative disorders (230). This made an FDA approved TNF antagonist Etanercept, an attractive candidate for decreasing microglia activation after human TBI (231). However, further studies are needed to achieve selective blocking of the TNF receptor 1 rather than broad TNF receptor blockade.
Example of an anti-inflammatory not useful in TBI is statin. Statins downregulate the expression of vascular adhesion molecules and chemoattractant molecules, and were thought to be potential candidates in lowering the infiltration by immune cells into injured brain. However, in a clinical trial, administration of rosuvastatin 11 h after injury did not display any differences in terms of disability (amnesia and disorientation time) with the control group at 3 months but increased IL-6 levels were seen 3 days after injury (232). Consistent with these results, the OBTT study found no beneficial effects of simvastatin administration over 2 weeks post-TBI using the oral route of administration in multiple rodent models (233). Statins are known to inhibit mitochondrial complex III (234) and can produce myopathy as a side effect (235). Thus, statins possibly exacerbate TBI mitochondrial dysfuction (95) which may be the reason that they failed to provide any benefit.
Although on the surface it appears that since inflammation after TBI and SCI are mediated by activated microglia these two pathologies could be identical, there is evidence to suggest that the extent of mitochondrial impairment (a measure of inflammation amplification) is different (236), which becomes apparent with aging. For example, a neuroinflammatory modulator, FTY720, which was unable to improve lesion size or functional outcome in both trauma models at either stage, acute vs. chronic, when given as a single dose (237), improved neurological outcome when dosed over 3 days as was seen with CsA (230), and was more effective in SCI even though the inflammation in SCI is different from that in TBI (238).
While microglia are major effectors of inflammation and mediated neuronal death, neutrophils are the first peripheral immune cells to reach the site of injury (239). Over the following hours to days after trauma, neutrophils infiltrate the CNS and migrate across the BBB in response to chemoattractants secreted by the choroid plexus (99). Recruitment of monocyte-derived macrophages and T lymphocytes then follows. Antibody blockade of cluster of differentiation (Cd) Cd11d/CD18, a type of integrin found on both neutrophils and macrophages, reduced systemic inflammatory response syndrome and improved neurological outcomes in rodent models of TBI (240, 241). In contrast to microglia, circulating immune cells such as neutrophils in TBI only produce short duration inflammation that resolves in part due to gasdermin (242). It remains to be shown if this molecule can be exploited to resolve TBI induced microglial inflammatory response.
The role of the adaptive immune system in TBI is still unclear. While infiltration of T cells in the lesion site promoted inflammation in a rat model of SCI, T cell-deficient mice were found to have poorer outcomes than controls after CNS injury (243, 244). Though T cells could have neuroprotective function in TBI, maladaptive response to self-antigens in conjunction with M1-like microglia action can extend damage and maintain a chronic state of low-level inflammation (117).
Inflammation modulation could reduce the loss of neurons, oligodendrocytes, and neural stem cells. In addition, clearance of debris could help resolve and prevent secondary tissue loss. This approach has been found to mitigate injury-induced cognitive decline at 3 weeks post TBI. Inflammation reduction by suppressing polarization into pro-inflammatory microglia (115, 116), promoting anti-inflammatory microglial activity (245) or enhancing clearance of apoptotic cells (246) may confer greater neuroprotection than focusing solely on inhibiting neuronal death mechanisms. Immunomodulatory therapies for TBI need to be developed with a goal to guide inflammation toward the reparative phenotype (99, 247). To better target such therapies, biochemical and imaging biomarkers can been considered to quantitate TBI consequences, validate preclinical research findings, and track effectiveness of therapeutic interventions in humans(201, 248–254).
Preconditioning Penumbra Against Vulnerability To Secondary Injury
Consequences of TBI are not limited to the immediate results of primary and secondary injury mechanisms. Years after initial injury, TBI survivors can develop non-convulsive seizures/post-traumatic epilepsy (255–257) and progressive brain atrophy due to “accelerated brain aging” (258–260) that render them susceptible to further neurodegeneration (261, 262). Recurrent post-traumatic seizures, or “post-traumatic epilepsy” (PTE), are highly prevalent in TBI patients with a history of combat and are a major cause of morbidity in veterans (263). TBI severity, dural penetration, loss of consciousness, and post-traumatic amnesia are some of the risk factors that contribute to the development of PTE (263). In multiple models of TBI, it has been found that the formation of epileptogenic foci stems from excitatory/inhibitory neurotransmitter and receptor imbalances and loss of GABAergic cells (264), and tauopathy (263). Although prevention of acute seizures with anticonvulsants can manage immediate glutamate excitotoxicity, PTE tends to be refractory to current medical treatment (263). Axonal debris generated at impact (62, 209) are interrogated by microglia as early as 6 h post-TBI (265). Failure of activated microglia to phagocytose persistent axonal fragments may lead to development of TBI-induced autoantibodies (266). The persistence of axonal fragments and chronic inflammation has been documented several years after primary injury (207, 259, 267). This suggests that poor clearance of axonal debris may provoke the chronic inflammation that underlies neurodegenerative diseases (262). This may be in part due to the presence of “do not eat me” or the absence of “find me/eat me” signals, as seen in mice without CD47 (a ubiquitously expressed surface glycoprotein that provides “do not eat me” signals) which improved outcomes after TBI compared to wild type (268). Although it is clear that modulation of neuroinflammation may improve outcomes, the pharmacological and molecular tools needed to achieve this goal remain to be determined.
Beta-amyloid is another contributor to the long-term degeneration after TBI. The release of inflammasomes from activated microglia promotes seeding and polymerization of beta-amyloid (34, 210, 269) in the synapses (270). The accumulation of insoluble plaques in the extracellular space and tau neurofibrillary tangles inside neurons is already known to precede clinical symptoms of Alzheimer's disease. Tau protein deposits are also the hallmark of chronic traumatic encephalopathy (CTE), another degenerative brain disease associated with TBI. CTE tends to develop in people with a history of repetitive mild TBI such as military veterans and collision sports athletes. Retired players of the National Football League are three times more likely to die from a neurodegenerative disorder than matched controls (271). Similar to PTE, no TBI therapy has been able to address the neurodegenerative consequences modulated by the chronic inflammation that lingers years after injury.
Adult Neurogenesis (Reparative Endogenous Neurogenesis) As A “Neuroregenerative Therapy”?
Since the discovery of CNS blast cells/neural stem cells in mammalian brain in 1989, the ability of these cells to become neurons became a topic of interest (272). Several lower-order mammals, reptiles, amphibians, and birds continue to experience neurogenesis well into adulthood. However, in adult humans, neurogenesis remains a topic of controversy. In order to understand the role of endogenous neurogenesis, it is desirable to consider how development of the cell types of the brain and the spinal cord occurs. In the mammalian fetus, NSCs are the fundamental ancestor cells for the central nervous system (CNS), as defined by their ability to self-renew and produce all three major CNS cell types: neurons, astrocytes, and oligodendrocytes (272, 273). In humans, these predecessor cells are the first neurons observed at 5-weeks after conception, even before closure of the neural tube which is lined by the neural stem cells of the ventricular zone (274). These cells are found in a layer just below the pial surface of the prospective cortex and migrate tangentially into the telencephalon (275). The two principal colonies of neural stem cells so far identified are located in the subventricular zone (SVZ) and the subgranular zone (SGZ) (276). They give rise to striatal and olfactory bulb neurons (274, 276), and hippocampal neurons, respectively.
Because neuronal loss is the single most important consequence of TBI, efforts to replace neurons have become a fundamental part of TBI research. In rodents, adult neurogenesis is robust even after TBI (277–279). In humans, the evidence for effective reparative adult neurogenesis is controversial, and is probably insignificant at best (280–282). NSCs persist in adult human brains and can produce astrocytes but not neurons (283). Apart from cell death, cell proliferation has also been observed after TBI. This regeneration raises the possibility of therapeutic manipulation of multipotent precursors in situ to repair the injured brain. The poor outcomes after TBI characterized by marked gray and white matter loss (259) do not support the notion that proliferating endogenous cells could replace lost neurons in mammals. However, physiological markers of neurogenesis and cell proliferation, measured in tissue samples one to 16 h after TBI, may indicate that the adult injured brain has the potential to replace lost cells and needs to be correlated to patients' outcomes (284). Complicating this, inflammation also contributes to the apoptosis of neural stem cells (285) and possibly oligodendrocytes (286) Figure 3. Providing neurotrophic factors, such as S100B or FGF, seems to enhance endogenous neurogenesis in experimental animals and correlates with better cognitive function (287). Failure to boost endogenous proliferation of NSCs in clinical studies (288) and the inability to produce mature neurons in vitro from cultured adult human NSCs reiterate that humans are incapable of adult neurogenesis (289). Proneurogenic compounds that have been found to be beneficial in preclinical TBI (290) as well as other CNS dysfunction models (291–294). Although the exact mechanism of action for these compounds is still an area of active research, the effect of proneurogenic compounds in human TBI remains to be explored. In contrast, clinical treatments with exogenous, transplanted NSCs are moving to Phase II trials, for non-TBI indications (295–300).
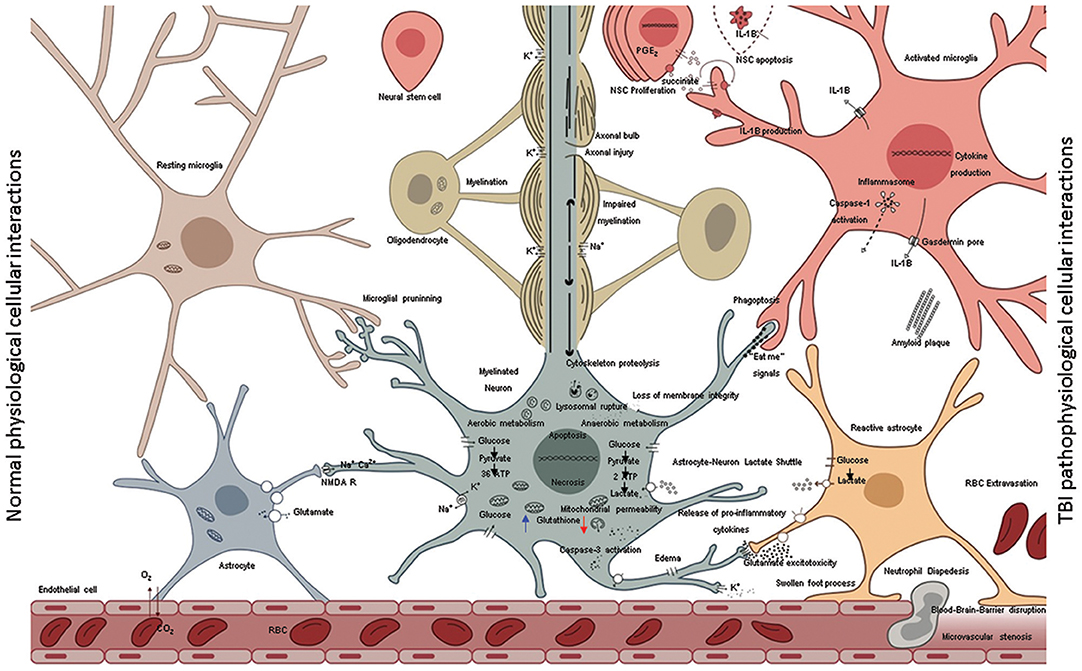
Figure 3. A schematic representation shows the normal cellular interactions in the intact brain (left) in contrast to neurotoxic interactions post injury (right). The intact vasculature (left bottom) is held in place by astrocytic end-feet; astrocytic blood brain barrier keeps immune cells out of the parenchyma but allows diffusion of glucose. Glucose is taken by neural cells metabolized through glycolysis in cytoplasm and oxidative phosphorylation in mitochondria. High neuronal glutathione levels (blue arrow) mitigate oxidative damage due to inherent metabolic activity. The multipartite synapses are pruned by microglia, the axons wrapped by myelin from oligodendrocytes facilitate rapid neurotransmission. Astrocytes soaking up excess glutamate in synapses, which in turns increases glucose uptake from blood. Normal microglia phagocytose various extracellular debris (including amyloid β) produce by metabolic activity. Aberrant cellular interactions after TBI (right) as consequence of mechanical forces disrupt the blood-brain-barrier causing leakage of intravascular contents into the brain parenchyma and facilitate invasion of the CNS with non-resident cells such as RBCs and neutrophils. Initial trauma causes the release of glutamate and other excitatory amino acids and potassium efflux. Lowered neuronal glutathione levels lead to deceased capacity to inhibit excitatory neurotransmission. Ions with their water shells enter astrocytes swelling the cells such that end-feet of astrocytes fail to maintain blood barrier or clear synaptic glutamate. Excess glutamate binds NMDA receptors on neurons and oligodendrocytes. Resulting neuronal depolarization and accumulation of calcium in mitochondria abolishes normal electrochemical gradient required to generate ATP. ATP dependent ion pump activity is required to work against electrochemical gradient to hyperpolarize neurons. Due to irreversible ionic imbalance membrane integrity is lost, unraveling the myelin and death of neurons and oligodendrocytes. This process is called “glutamate excitotoxicity” produces the second wave of TBI related cell death after the primary mechanical injury. Calcium pollution renders mitochondria depolarized, builds up oxidative damage, opening of the permeability transition pore, lipid peroxidation, cytochrome c release, assembly of caspase dependent proteases, and apoptosis. In addition, with injured axons calpain-induced lysosomal rupture, cathepsin-induced cytoskeletal proteolysis set into motion the self-destructive axonal degeneration. Hypoxia stabilizes HIF-1α facilitating expression of pro-inflammatory cytokine genes (IL-1β, IL-18, TNFα). Succinate acts as a signal that positively feeds inflammation. NSC disrupt such feedback and mediate inflammation resolution by rending microglia anti-inflammatory. Pro-inflammatory cytokines are released in the extracellular environment via pores (ex: IL-1β via gasdermin D) and spread inflammation to adjacent cells including mitochondrial dysfunction and secondary death of oligodendrocytes, neural stem cells and neurons. Presence of proinflammatory microglia corrupts astrocytes turning them into agents of neurotoxicity. Transient “eat-me” signals on the surface of neurons activate glial Phagoptosis leading to further loss of tissue that was otherwise intact at the time of primary injury. Proinflammatory microglia and neurons may undergo pyroptosis or other inflammatory cell death further spreading the inflammation.
Anti-Inflammatory Effects Of Transplant vs. Cell Replacement Effects
Several preclinical studies support the hypothesis that TBI-responsive neuroinflammation is a clinically relevant therapeutic target; however, few clinical trials target traumatic inflammation (117). Cell ablation pharmacological inhibition studies (114, 116, 233, 301–305) suggest that neural stem cells (NSCs), astrocytes, and activated microglia stabilize the brain lesion and prevent further neurodegeneration. However, unlike NSCs, reactive astrocytes and activated microglia are also known to exacerbate TBI (210, 270, 306, 307). It is in this context, that exogenous NSC transplantation alone may be a means to reduce neuroinflammation. Anti-inflammatory properties of mesenchyme-derived stem cells have been extensively reviewed (308) and their utility in TBI has been described elsewhere (309). Intra carotid artery delivery of human MSC was in fact found to be safe in stroke patients (310) as well as ALS (311). See Table 1 for MSC use in clinical trails.
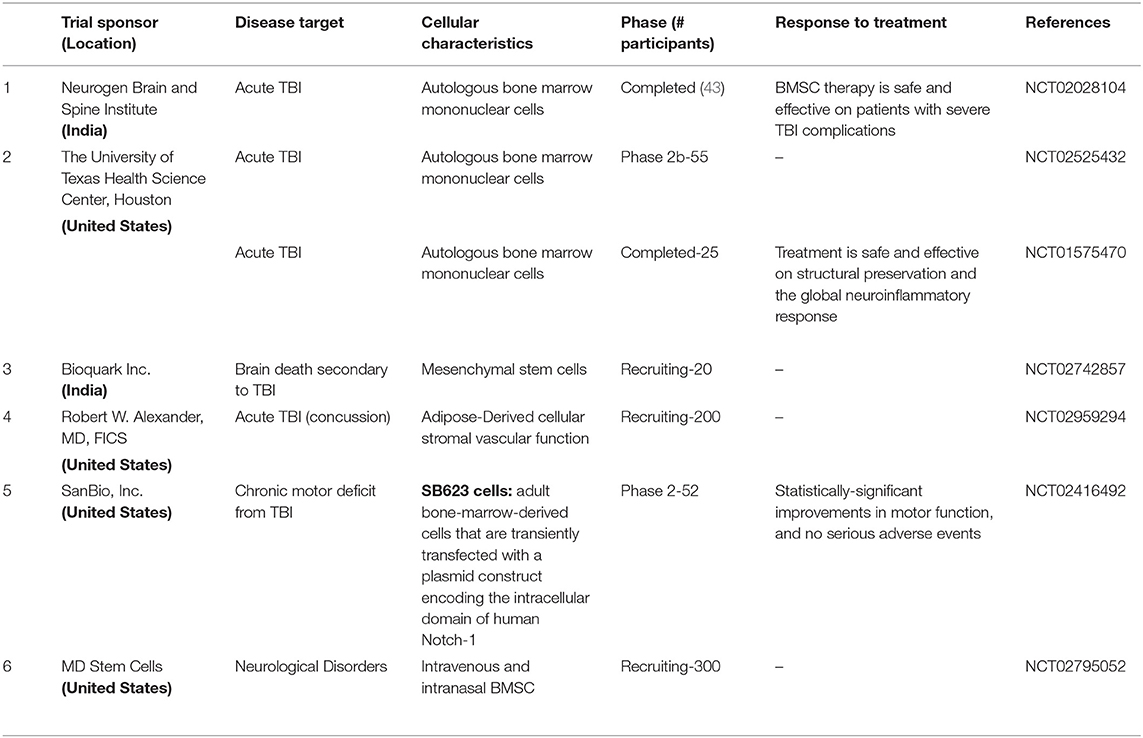
Table 1. MSC trials.
Similar to other chronic inflammatory diseases, addressing the impaired debris clearance by microglia may be essential in converting degeneration into regeneration (162, 312). How does chronic inflammation negatively influence TBI outcomes? As mentioned earlier, microglia are the main phagocytic cells of the brain and are responsible in part for ECF microenvironment homeostasis (313). As shown in Figure 3 following injury, the accumulation of myelin debris, beta-amyloid, and other DAMPs could impair microglia phagocytosis and exclusively activate their proinflammatory phenotype (210, 270, 314). Recent insights into biochemical differences in myelin between normal in comparison to injured subjects show how injury induced autoimmune demyelination may progress (315). This situation could potentially benefit from cell transplantation. Transplantation of human fetal NSCs within 24 h of TBI has been shown to reduce microglial pro-inflammatory activation (316) and can alleviate post-traumatic cognitive deficits (316–319). It is not known if such transplantation after PBBI would produce sustained beneficial effects. Recently, our lab demonstrated robust and durable engraftment of hNSCs when transplanted 7–10 days after the injury, in models of PBBI (309) Furthermore, the FDA has already approved of these cells for clinical trials in other CNS disorders but not yet in TBI (217, 305, 320).
One of the impediments for the long-term implementation of stem cell-based therapies is lack of insight into their mechanism of action (321). However, restorative neuroscience has been energized following the discovery of NSCs (272), their mitogens (322), their ability to be cultured from adult rodent brain (323), and embryonic (324) human brains (289). Recapitulation of human neuronal development after transplantation of human fetal neural stem cells in rodent embryos (325, 326) suggests that transplantation of NSCs could rebuild injured brains by emulating aspects of CNS development, such as tract forming and target cell innervation.
Human Neural Stem Cells as Agents of Neuroprotection After TBI
Successful transplantation of fetal tissue in adult rat brains (327), led to the first neuroprotective fetal cortical tissue transplants in TBI rats (328). The source of NSCs was cortical tissue (329–331). Preclinical studies of TBI showed that transplantation was acutely neuroprotective but not past 2 weeks post injury. The lack of neuronal replacement was attributed to robust host immune response. In order to overcome this limitation, researchers initiated a number of studies, including transplantation of human neuronal precursors, where experimental subjects receive immunosuppression (205). One of the mechanisms of action for hNSC transplants was elucidated in a multiple sclerosis model. In that study, NSCs appeared to sense the extracellular succinate that accumulates in the chronically inflamed CNS and ameliorated neuroinflammation via succinate-SUCNR1-dependent mechanisms (332). Consistent with these findings iPSC derived NSC as well as oligo precursor transplants are reported to spare tissue in rodent spinal cord injury model (333). Such tissue sparing occurs following inflammation resolution. As outlined in Figure 4, in the absence of timely resolution of injury induced activated microglia, the injury is magnified over time and space producing progressive increased tissue destruction in part via microglial pyroptosis (210) and facilitates worse outcomes such as antibody generation against cellular breakdown products (266) and neurodegeneration (Figure 5). Neural stem cell transplants could confer neuroprotection to alleviate such tissue loss and lead to a desired outcome via inhibiting microglial pyroptosis, disrupting the succinate based inflammation amplification, and promoting phagocytosis by surviving activated microglia (Figure 5). Perhaps all stem cells confer neuroprotection via efferocytosis (334).
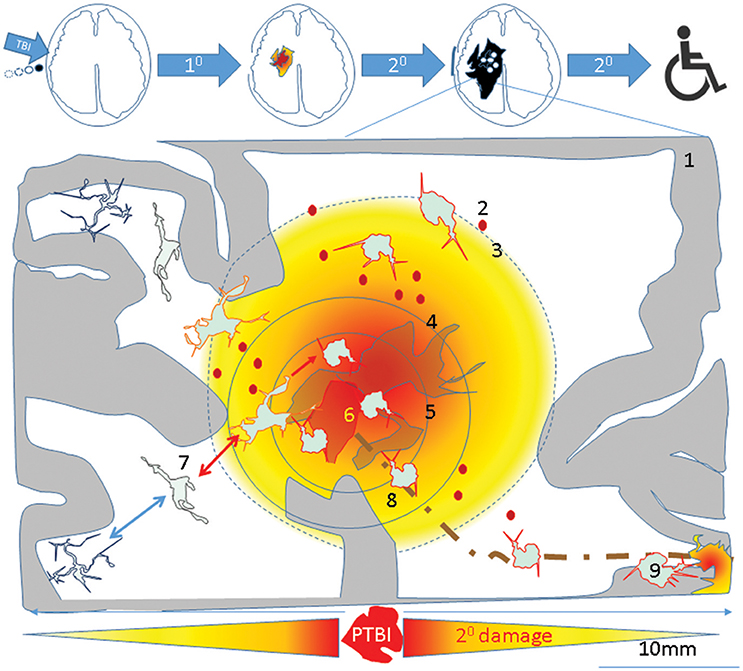
Figure 4. A theoretical schematic (top) shows evolution of the primary PTBI injury into disability. A PTBI brain schematic focused on inflammatory microglia over nine regions outlines possible mechanisms underlying magnification of primary injury and spread to remote sites. 1- intact brain tissue, 2-petechiae in injured brain which perpetuate blood brain barrier compromise, hemorrhage, delayed cell death, 3- a gradient of local DAMPs/NAMPs may combine with circulating inflammatory mediators (via broken BBB) to recruit and activate microglia in penumbra rendering tissue vulnerable to secondary damage, 4- region with neuronal apoptosis/pyroptosis and neural stem cell recruitment and apoptotic death., 5- regions of astrocyte destruction and reactive gliosis which stems from 6-Focal injury. The focal injury turns into a permanent cavity in part due to oncotic cell death and axonal destruction (brown dotted line). 7- Normal surveying microglial acquire various reversible/irreversible activation states as they travel along the DAMP/NAMP gradient. 8. Irreversibly activated microglia could migrate away from injury core toward remote deeper regions. 9-Pyroptosis of activated microglia in remote regions connected to injury site (via pathways that were axotomised by injury) may mediate secondary axotomy and remote neurodegeneration (210, 270).
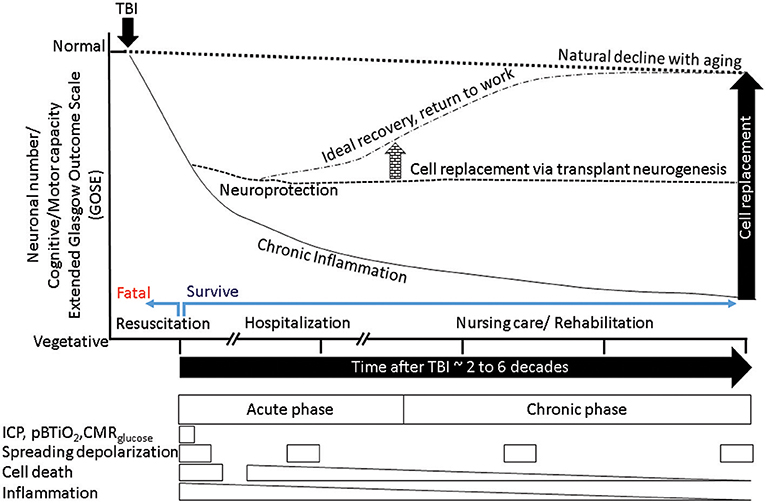
Figure 5. A schematic summarizes the outcome of normal aging, accelerated aging following TBI, putative mitigation of disease progression by neuroprotection, and additional benefits from cell replacement. The x-axis represents time, the y-axis represents neuronal numbers and dependent behaviors ranging from normal at the top to vegetative at the bottom. The normal aging process produces a gradual decline (dotted outline) in cognitive and motor behaviors. Following a TBI (black arrow) the process of aging is accelerated (solid downward line), with chronic inflammation and tissue loss reducing ability. Successful resuscitation can help survive otherwise fatal TBI, if post survival hospitalization produces ideal recovery then return to work is possible (dotted-dashed line), mitigation of chronic inflammation via neuroprotective agents could stem tissue loss and stabilize ability (dashed line). If the neuroprotection is mediated by neural stem cells that have the potential to replace lost cells, the new tissue in conjunction with nursing care and rehabilitation may facilitate sufficient recovery that is indistinguishable from normal aging (arrow elevating the dashed line to dashed-dotted level). The boxes below represent the transient nature of various therapeutic windows. It is evident that therapeutic windows during hospitalization are short, while those associated with disability such as post-traumatic epilepsy (PTE)/seizures depend on incidence, each event if prevented by timely intervention could mitigate further decline in ability. Acute cell death is transient, however chronic inflammation and secondary cell death that are diminishing opportunities. Hence, only acute/sub-acute neuroprotection can afford maximum benefit. However, if the cell replacement can be exploited with rehabilitation in a timely manner, there is no limit to the therapeutic window.
Cell Replacement
Transplanted NSC-derived neurons can integrate and contribute electrophysiologically in both sham as well as injured rodent and primate brains (321, 335–339). However, previous studies have reported limited neuronal replacement after hNSCs transplantation in rat models of TBI (318). Recently our lab and others have shown robust and durable engraftment of hNSCs with delayed differentiation into mature neurons, for as many as 20% of transplanted cells, up to 16 weeks in a rodent PBBI model. Nevertheless, their integration into injured rat CNS and contribution to reversal of TBI induced motor and cognitive deficits has yet to be fully demonstrated (309, 340). Assessing electrophysiological properties and their contribution to amelioration of TBI-induced deficits would provide crucial mechanistic insight. It is not yet known if it is possible and/or necessary to guide transplant-derived neurons to a specific target (such as anterior horn cells, or substantia nigra) and how this can be done. The use of clinically relevant neurogenic compounds could be key to assist in targeting the migration of transplant-derived neurons. In primates with SCI, researchers have found that transplanted NSI 566 cells can be harnessed to restore lost function 9 months after grafting by differentiating into neurons and supportive glia (339). If cell replacement is indeed achieved it could positively steer outcomes (Figure 5).
The current literature suggests that NSC mediated neuroprotection (332, 333) could be achieved more easily than cell replacement, especially in severe TBI. It is true that the incidence of severe TBI is small and cell therapy to treat will not be appealing to mild and moderate TBI patients. However, the costs associated with TBI requiring hospitalization necessitate use of cell therapy to treat sever TBI (18, 341). Following the discovery of the mechanistic insights, “the neuroprotective factor” could be delivered via non-cell transplantation means even for less severe TBI where cell therapy is not warranted.
Rationale For Use Of hNSC To Treat TBI And Guidelines For Cell Therapy
After examining several RCTs and gaining insights into their failure to confer neuroprotection, (11, 90, 114), identification of anti-inflammatory mechanisms as leading neuroprotective agents, the neuroprotective properties sub-acute use of human NSCs (332, 333, 342)is worthy of exploration in TBI. NSCs could potentially mitigate secondary damage by (1) reducing inflammation; (2) promoting regeneration with appropriate pharmacological interventions (e.g., drugs promoting neurogenesis such as NSI-189) and rehabilitative measures; and (3) Slowing down TBI-induced delayed disability. Accomplishing this set of objectives in itself would be an important goal of NSC therapy. As of mid-2018, a total of 316 patients with various reported CNS disorders have received clinical grade hNSC transplants (Table 2). None of these patients had any safety issues yet. Neuralstem Inc., has sponsored phase I and phase II clinical trials evaluating hNSC transplantation as a potential therapy for ALS (354). In these, a post hoc analysis compared ambulatory limb-onset ALS participants who were administered open-label intraspinal hNSC and followed for up to 3 years after transplant. Due to lack of controls, participants in these phase 1 and 2 trials were matched to subjects from the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) and ceftriaxone datasets to provide required analyses in order to inform future clinical trial designs. The ALS Functional Rating Scale revised (ALSFRSR) and a composite statistic combining survival and functional status (ALS/SURV) were assessed to monitor changes in function. Results from These Ph1/2 studies revealed significantly improved survival and function (346) when compared to historical datasets. In another study where non-ambulatory ALS patients received either unilateral or bilateral injections, no increase of disease progression after the transplants was observed for up to 18 months after surgery. Rather, two patients showed a transitory improvement of the subscore ambulation on the ALS-FRS-R scale (from 1 to 2). A third patient showed improvement of the MRC score for tibialis anterior, which persisted for as long as 7 months. Three of the patients died due to disease progression (353). More recently a study of stereotactic, intracerebral injection of CTX0E03 neural stem cells from ReNeuron into patients with moderate to moderately severe disability as a result of an ischemic stroke has progressed from a Phase I to Phase IIb as the clinical endpoints are being met albeit slower than expected (295, 299, 300).
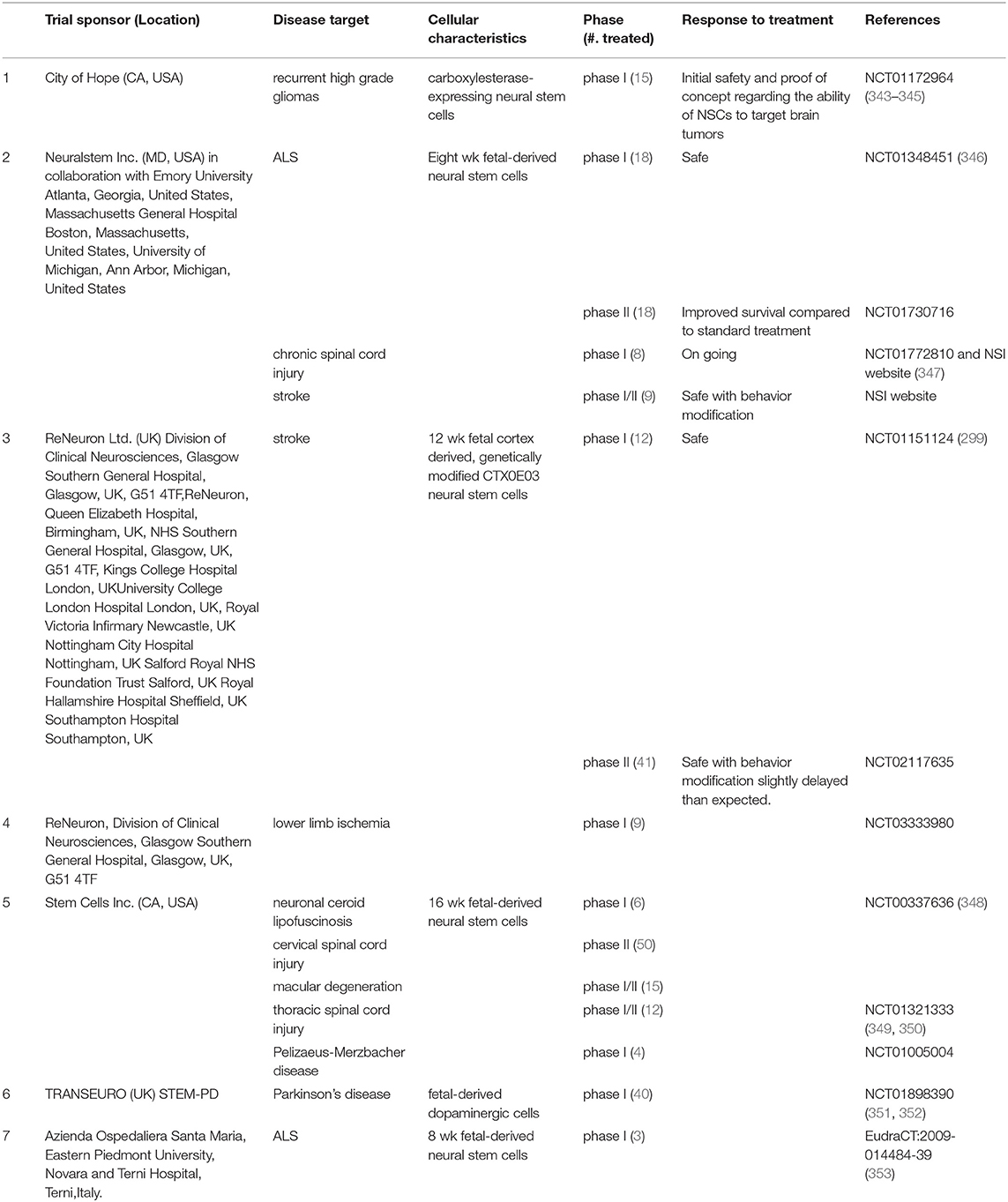
Table 2. Human fetal neural stem cells in Clinical Trials.
Pathway To Address Unmet Patient Need, Clinical Trials, To Arrive At Proven Treatments
The previous sections suggest that the unit of intervention for TBI should be at the cellular level i.e., at the unit of life. However, it is important to be wary of moving too hastily. The compelling unmet TBI medical need and desperation on the part of patients, in the absence of multicenter clinical trials, can lead to unproven therapies being administered to patients. Three such cases of unproven stem cell therapies (mix of multiple fetal NSCs or MSCs and NSCs) have been documented (355–357). Fortunately, all of the issues could be resolved by corrective measures i.e., removal of the transplanted cells. The aforementioned events have their roots in premature and unapproved use of treatments that were initiated by investigator/patient. “Stem cell tourism” that exploits the therapeutic hope of patients and families with incurable neurological diseases can jeopardize the legitimacy of stem cell research. Julian et al posit that an improvement in education, regulation, legislation, and involvement of authorities in global health in neurology and neurosurgery is required to prevent such exploitation (358) (Table 3).
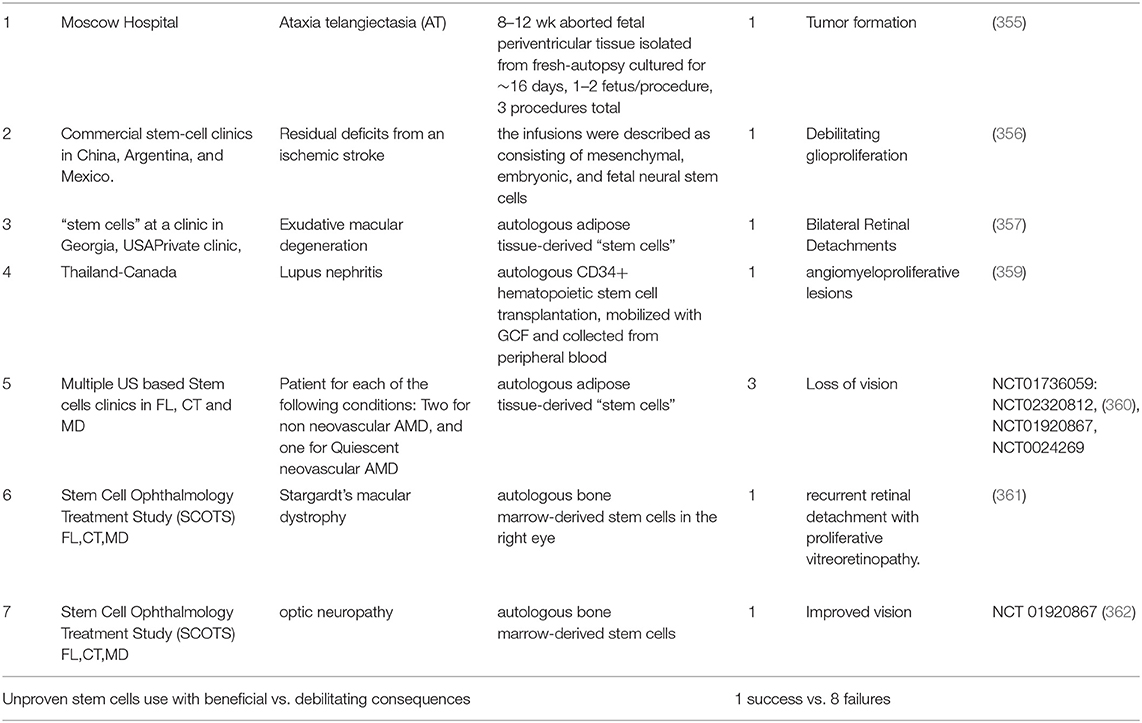
Table 3. Unproven application of cell therapy.
Conclusion
For over 30 years of TBI research, neuroprotection via RCTs has been elusive. Progressive tissue loss in severe TBI is an unmet need that turns TBI into a disease process with no hope for recovery. Analysis of the trial failures has led to insights into the mechanisms that need to be targeted, specifically neuroinflammation. Preclinical animal model studies that recapitulate human severe TBI have led to the identification of mechanisms underlying the vulnerability of the penumbra and evaluating the extent of penumbra sparing will likely give insights into the neuroprotective ability of an intervention. Additionally, the continued exploration of neural stem cells transplantation, which was bolstered by initial efforts with fetal cortical tissue transplants that were neuroprotective, resulted in the discovery that cell transplants can resolve inflammation via disruption of proinflammatory pyroptotic signaling and without interfering with activated glial functions such as phagocytosis. Multiple independent studies in a variety of CNS conditions suggest use of clinical trial grade human neural stem cells which have been found to be safe and meeting the clinical end points. Thus, the rational for using human neural stem cell based transplantation for TBI is well supported as both enduring neuroprotection and cell replacement can be achieved with single agent.
Author Contributions
AK, AC, DC, SL, MS, KR, HP, LD, and JW reviewed the preclinical articles and prepared the draft, AM wrote the section on trial cost. AA, SY, ZH, SL, AR, RB, and RT worked clinical trials articles and prepared draft. SN, HP, and MS generated the data for figures. LL, AA, SY, SG, and RB conceived and revised the draft to produce final manuscript. AK produced original artwork to encapsulate the literature on cellular interactions with RB and SG.
Funding
Department of Defense Grant W81XWH-16-2-0008 and the Miami Project to Cure Paralysis funding to Dr. R. Bullock, Dr. Deborah A. Shear, and Dr. S. Gajavelli.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ng I, Lew TW, Yeo TT, Seow WT, Tan KK, Ong PL, et al. Outcome of patients with traumatic brain injury managed on a standardised head injury protocol. Ann Acad Med Singapore (1998) 27:332–9.
2. Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation (2007) 22:341–53.
3. de Almeida CE, de Sousa Filho JL, Dourado JC, Gontijo PA, Dellaretti MA, Costa BS. Traumatic brain injury epidemiology in Brazil. World Neurosurg. (2016) 87:540–7. doi: 10.1016/j.wneu.2015.10.020
4. Majdan M, Plancikova D, Brazinova A, Rusnak M, Nieboer D, Feigin V, et al. Epidemiology of traumatic brain injuries in Europe: a cross-sectional analysis. Lancet Public Health (2016) 1:e76–e83. doi: 10.1016/S2468-2667(16)30017-2
5. Eaton J, Hanif AB, Grudziak J, Charles A. Epidemiology, management, and functional outcomes of traumatic brain injury in Sub-Saharan Africa. World Neurosurg. (2017) 108:650–5. doi: 10.1016/j.wneu.2017.09.084
6. Safety WHOR. Global Status Report on Road Safety 2015 46:70–7. Washington, DC: World Health Organization (2015).
7. Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg. (2018). doi: 10.3171/2017.10.JNS17352 [Epub ahead of print].
8. Johnson WD, Griswold DP. Traumatic brain injury: a global challenge. Lancet Neurol. (2017) 16:949–50. doi: 10.1016/S1474-4422(17)30362-9
9. Zuraik C, Sampalis J. Epidemiology of traumatic injuries at an urban hospital in Port-au-Prince, Haiti. World J Surg. (2017) 41:2674–80. doi: 10.1007/s00268-017-4088-2
10. Lawrence T, Helmy A, Bouamra O, Woodford M, Lecky F, Hutchinson PJ. Traumatic brain injury in England and Wales: prospective audit of epidemiology, complications and standardised mortality. BMJ Open (2016) 6:e012197. doi: 10.1136/bmjopen-2016-012197
11. Hawryluk GW, Bullock MR. Design of acute neuroprotection studies. Handb Clin Neurol. (2015) 128:761–78. doi: 10.1016/B978-0-444-63521-1.00047-9
12. Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic brain injury-related emergency department visits, hospitalizations, and deaths - United States, 2007 and 2013. MMWR Surveill Summ. (2017) 66:1–16. doi: 10.15585/mmwr.ss6609a1
13. Young NH, Andrews PJ. Developing a prognostic model for traumatic brain injury–a missed opportunity? PLoS Med. (2008) 5:e168. doi: 10.1371/journal.pmed.0050168
14. Gerber LM, Chiu YL, Carney N, Hartl R, Ghajar J. Marked reduction in mortality in patients with severe traumatic brain injury. J Neurosurg. (2013) 119:1583–90. doi: 10.3171/2013.8.JNS13276
15. Hennekens CH, Drowos J, Levine RS. Mortality from homicide among young black men: a new American tragedy. Am J Med. (2013) 126:282–3. doi: 10.1016/j.amjmed.2012.07.007
16. Jarman MP, Haut ER, Curriero FC, Castillo RC. Mapping areas with concentrated risk of trauma mortality: a first step toward mitigating geographic and socioeconomic disparities in trauma. J Trauma Acute Care Surg. (2018) 85:54–61. doi: 10.1097/TA.0000000000001883
17. Kaufman EJ, Ertefaie A, Small DS, Holena DN, Delgado MK. Comparative effectiveness of initial treatment at trauma center vs neurosurgery-capable non-trauma center for severe, isolated head injury. J Am Coll Surg. (2018) 26:741–751.e2. doi: 10.1016/j.jamcollsurg.2018.01.055
18. Zonfrillo MR, Spicer RS, Lawrence BA, Miller TR. Incidence and costs of injuries to children and adults in the United States. Inj Epidemiol. (2018) 5:37. doi: 10.1186/s40621-018-0167-6
19. Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. (2017) 16:987–1048. doi: 10.1016/S1474-4422(17)30371-X
20. Chesnut RM. Secondary brain insults after head injury: clinical perspectives. New Horiz. (1995) 3:366–75.
21. London PS. Some observations on the course of events after severe injury of the head. Hunterian Lecture delivered at the Royal College of Surgeons of England on12th January 1967. Ann R Coll Surg Engl. (1967) 41:460–79.
22. Cifu DX, Keyser-Marcus L, Lopez E, Wehman P, Kreutzer JS, Englander J, et al. Acute predictors of successful return to work 1 year after traumatic brain injury: a multicenter analysis. Arch Phys Med Rehabil. (1997) 78:125–31.
23. Buchanan C. Gun violence, disability and recovery. C. Buchanan, editor. Surviving Gun Violence Project (2013).
24. Dillahunt-Aspillaga C, Nakase-Richardson R, Hart T, Powell-Cope G, Dreer LE, Eapen BC, et al. Predictors of employment outcomes in veterans with traumatic brain injury: a va traumatic brain injury model systems study. J Head Trauma Rehabil. (2017) 32:271–82. doi: 10.1097/HTR.0000000000000275
25. Siesjo BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. (1981) 1:155–85. doi: 10.1038/jcbfm.1981.18
26. Servadei F, Murray GD, Penny K, Teasdale GM, Dearden M, Iannotti F, et al. The value of the “worst” computed tomographic scan in clinical studies of moderate and severe head injury. European brain injury consortium. Neurosurgery (2000) 46:70–5 discussion 75–77. doi: 10.1093/neurosurgery/46.1.70
27. Chang EF, Meeker M, Holland MC. Acute traumatic intraparenchymal hemorrhage: risk factors for progression in the early post-injury period. Neurosurgery (2007) 61(1 Suppl):222–230 discussion 230–221. doi: 10.1227/01.neu.0000279217.45881.69
28. Smith JS, Chang EF, Rosenthal G, Meeker M, von Koch C, Manley GT, et al. The role of early follow-up computed tomography imaging in the management of traumatic brain injury patients with intracranial hemorrhage. J Trauma (2007) 63:75–82. doi: 10.1097/01.ta.0000245991.42871.87
29. Sidaros A, Skimminge A, Liptrot MG, Sidaros K, Engberg AW, Herning M, et al. Long-term global and regional brain volume changes following severe traumatic brain injury: a longitudinal study with clinical correlates. Neuroimage (2009) 44:1–8. doi: 10.1016/j.neuroimage.2008.08.030
30. Bigler ED, Maxwell WL. Neuroimaging and neuropathology of TBI. NeuroRehabilitation (2011) 28:63–74. doi: 10.3233/NRE-2011-0633
31. Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience (1998) 87:359–69.
32. Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. (2002) 103:607–14. doi: 10.1007/s00401-001-0510-8
33. Rodriguez-Paez AC, Brunschwig JP, Bramlett HM. Light and electron microscopic assessment of progressive atrophy following moderate traumatic brain injury in the rat. Acta Neuropathol. (2005) 109:603–16. doi: 10.1007/s00401-005-1010-z
34. Williams AJ, Wei HH, Dave JR, Tortella FC. Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J Neuroinflamm. (2007) 4:17. doi: 10.1186/1742-2094-4-17
35. Shear DA, Lu XC, Bombard MC, Pedersen R, Chen Z, Davis A, et al. Longitudinal characterization of motor and cognitive deficits in a model of penetrating ballistic-like brain injury. J Neurotrauma (2010) 27:1911–23. doi: 10.1089/neu.2010.1399
36. Bramlett HM, Dietrich WD. Long-term consequences of traumatic brain injury: current status of potential mechanisms of injury and neurological outcomes. J Neurotrauma (2015) 32:1834–48. doi: 10.1089/neu.2014.3352
37. Bullock R, Chesnut RM, Clifton G, Ghajar J, Marion DW, Narayan RK, et al. Guidelines for the management of severe head injury. Brain Trauma Foundation Eur J Emerg Med. (1996) 3:109–27.
38. Carney N, Totten AM, O'Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery (2017) 80:6–15. doi: 10.1227/NEU.0000000000001432
39. Faul M, Wald MM, Rutland-Brown W, Sullivent EE, Sattin RW. Using a cost-benefit analysis to estimate outcomes of a clinical treatment guideline: testing the brain trauma foundation guidelines for the treatment of severe traumatic brain injury. J Trauma (2007) 63:1271–8. doi: 10.1097/TA.0b013e3181493080
40. Talving P, Karamanos E, Teixeira PG, Skiada D, Lam L, Belzberg H, et al. Intracranial pressure monitoring in severe head injury: compliance with Brain Trauma Foundation guidelines and effect on outcomes: a prospective study. J Neurosurg. (2013) 119:1248–54. doi: 10.3171/2013.7.JNS122255
41. Joseph B, Aziz H, Pandit V, Kulvatunyou N, O'Keeffe T, Wynne J, et al. Improving survival rates after civilian gunshot wounds to the brain. J Am Coll Surg. (2014) 218:58–65. doi: 10.1016/j.jamcollsurg.2013.08.018
42. Van Wyck DW, Grant GA. Penetrating traumatic brain injury: a review of current evaluation and management concepts. J Neurol Neurophysiol. (2015) 6:1–7. doi: 10.4172/2155-9562.1000336
43. Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet (2012) 380:1088–98. doi: 10.1016/S0140-6736(12)60864-2
44. Weisbrod AB, Rodriguez C, Bell R, Neal C, Armonda R, Dorlac W, et al. Long-term outcomes of combat casualties sustaining penetrating traumatic brain injury. J Trauma Acute Care Surg. (2012) 73:1525–30. doi: 10.1097/TA.0b013e318270e179
45. Bizhan A, Mossop C, Aarabi JA. Surgical management of civilian gunshot wounds to the head. Handb Clin Neurol. (2015) 127:181–93. doi: 10.1016/B978-0-444-52892-6.00012-X
46. Khormi YH, Gosadi I, Campbell S, Senthilselvan A, O'Kelly C, Zygun D. Adherence to brain trauma foundation guidelines for management of traumatic brain injury patients and its effect on outcomes: systematic review. J Neurotrauma (2018) 35:1407–18. doi: 10.1089/neu.2017.5345
47. Rubiano AM, Puyana JC, Mock CN, Bullock MR, Adelson PD. Strengthening neurotrauma care systems in low and middle income countries. Brain Inj. (2013) 27:262–72. doi: 10.3109/02699052.2012.750742
48. Charry JD, Rubiano AM, Puyana JC, Carney N, David Adelson P. Damage control of civilian penetrating brain injuries in environments of low neuro-monitoring resources. Br J Neurosurg. (2016) 30:235–9. doi: 10.3109/02688697.2015.1096905
49. Bonow RH, Barber J, Temkin NR, Videtta W, Rondina C, Petroni G, et al. The outcome of severe traumatic brain injury in Latin America. World Neurosurg. (2018) 111:e82–e90. doi: 10.1016/j.wneu.2017.11.171
50. Hendrickson P, Pridgeon J, Temkin NR, Videtta W, Petroni G, Lujan S, et al. Development of a severe traumatic brain injury consensus-based treatment protocol conference in Latin America. World Neurosurg. (2018) 110:e952–e957. doi: 10.1016/j.wneu.2017.11.142
51. Rubiano AM, Carney N, Chesnut R, Puyana JC. Global neurotrauma research challenges and opportunities. Nature (2015) 527:S193–197. doi: 10.1038/nature16035
52. Carney NA, Petroni GJ, Lujan SB, Ballarini NM, Faguaga GA, du Coudray HE, et al. Postdischarge care of pediatric traumatic brain injury in argentina: a multicenter randomized controlled trial. Pediatr Crit Care Med. (2016) 17:658–66. doi: 10.1097/PCC.0000000000000772
53. Vavilala MS, Lujan SB, Qiu Q, Bell MJ, Ballarini NM, Guadagnoli N, et al. Intensive care treatments associated with favorable discharge outcomes in Argentine children with severe traumatic brain injury: for the South American guideline adherence group. PLoS ONE (2017) 12:e0189296. doi: 10.1371/journal.pone.0189296
54. Mazzeo AT, Gupta D. Monitoring the injured brain. J Neurosurg Sci. (2018) 62:549–62. doi: 10.23736/S0390-5616.18.04465-X
55. Hutchinson PJ, Kolias AG, Timofeev IS, Corteen EA, Czosnyka M, Timothy J, et al. Trial of Decompressive Craniectomy for Traumatic Intracranial Hypertension. N Engl J Med. (2016) 375:1119–30. doi: 10.1056/NEJMoa1605215
56. Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. (1993) 160:139–44.
57. Rose J, Valtonen S, Jennett B. Avoidable factors contributing to death after head injury. Br Med J. (1977) 2:615–8.
58. Bullock R, Maxwell WL, Graham DI, Teasdale GM, Adams JH. Glial swelling following human cerebral contusion: an ultrastructural study. J Neurol Neurosurg Psychiatry (1991) 54:427–34.
60. Povlishock JT. Pathobiology of traumatically induced axonal injury in animals and man. Ann Emerg Med. (1993) 22:980–6.
61. Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma (1994) 11:173–86. doi: 10.1089/neu.1994.11.173
62. Oehmichen M, Meissner C, Konig HG. Brain injury after survived gunshot to the head: reactive alterations at sites remote from the missile track. Forensic Sci Int. (2001) 115:189–97. doi: 10.1016/S0379-0738(00)00335-2
63. Erturk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. (2007) 27:9169–80. doi: 10.1523/JNEUROSCI.0612-07.2007
64. Oehmichen M, Meissner C. Routine techniques in forensic neuropathology as demonstrated by gunshot injury to the head. Leg Med. (2009) 11(Suppl 1):S50–3. doi: 10.1016/j.legalmed.2009.01.113
66. Tabaddor K, Bhushan C, Pevsner PH, Walker AE. Prognostic value of cerebral blood flow (CBF) and cerebral metabolic rate of oxygen (CMRO 2) in acute head trauma. J Trauma (1972) 12:1053–5.
67. Artru F, Chacornac R, Deleuze R. Hyperbaric oxygenation for severe head injuries. Preliminary results of a controlled study Eur Neurol. (1976) 14:310–8. doi: 10.1159/000114753
68. Clifton GL, Robertson CS, Grossman RG, Hodge S, Foltz R, Garza C. The metabolic response to severe head injury. J Neurosurg. (1984) 60:687–96. doi: 10.3171/jns.1984.60.4.0687
69. Petito CK, Kraig RP, Pulsinelli WA. Light and electron microscopic evaluation of hydrogen ion-induced brain necrosis. J Cereb Blood Flow Metab. (1987) 7:625–32. doi: 10.1038/jcbfm.1987.115
70. McIntosh TK, Vink R, Yamakami I, Faden AI. Magnesium protects against neurological deficit after brain injury. Brain Res. (1989) 482:252–60.
71. Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. (1990) 73:889–900. doi: 10.3171/jns.1990.73.6.0889
72. Schoettle RJ, Kochanek PM, Magargee MJ, Uhl MW, Nemoto EM. Early polymorphonuclear leukocyte accumulation correlates with the development of posttraumatic cerebral edema in rats. J Neurotrauma (1990) 7:207–17. doi: 10.1089/neu.1990.7.207
73. Holmin S, Mathiesen T. Biphasic edema development after experimental brain contusion in rat. Neurosci Lett. (1995) 194:97–100.
74. Holmin S, Mathiesen T, Shetye J, Biberfeld P. Intracerebral inflammatory response to experimental brain contusion. Acta Neurochir. (1995) 132:110–9.
75. Holmin S, Soderlund J, Biberfeld P, Mathiesen T. Intracerebral inflammation after human brain contusion. Neurosurgery (1998) 42:291–8; discussion 298–299.
76. Leng L, Zhuang K, Liu Z, Huang C, Gao Y, Chen G, et al. Menin deficiency leads to depressive-like behaviors in mice by modulating astrocyte-mediated neuroinflammation. Neuron (2018) 100:551–63.e7. doi: 10.1016/j.neuron.2018.08.031
77. Syed SA, Beurel E, Loewenstein DA, Lowell JA, Craighead WE, Dunlop BW, et al. Defective inflammatory pathways in never-treated depressed patients are associated with poor treatment response. Neuron (2018) 99:914–24 e913. doi: 10.1016/j.neuron.2018.08.001
78. Gajavelli S, Kentaro S, Diaz J, Yokobori S, Spurlock M, Diaz D, et al. Glucose and oxygen metabolism after penetrating ballistic-like brain injury. J Cereb Blood Flow Metab. (2015) 35:773–80. doi: 10.1038/jcbfm.2014.243
79. Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC. Neuronal cell death. Physiol Rev. (2018) 98:813–80. doi: 10.1152/physrev.00011.2017
80. Dressler J, Hanisch U, Kuhlisch E, Geiger KD. Neuronal and glial apoptosis in human traumatic brain injury. Int J Legal Med. (2007) 121:365–75. doi: 10.1007/s00414-006-0126-6
81. Jennett B, Teasdale G, Braakman R, Minderhoud J, Knill-Jones R. Predicting outcome in individual patients after severe head injury. Lancet (1976) 1:1031–4.
82. Vahedi K, Hofmeijer J, Juettler E, Vicaut E, George B, Algra A, et al. Early decompressive surgery in malignant infarction of the middle cerebral artery: a pooled analysis of three randomised controlled trials. Lancet Neurol. (2007) 6:215–22. doi: 10.1016/S1474-4422(07)70036-4
83. Honeybul S, Ho KM, Lind CR. What can be learned from the DECRA study. World Neurosurg. (2013) 79:159–61. doi: 10.1016/j.wneu.2012.08.012
84. Kitagawa RS, Bullock MR. Lessons from the DECRA study. World Neurosurg. (2013) 79:82–4. doi: 10.1016/j.wneu.2012.10.031
85. Stocchetti N, Zanaboni C, Colombo A, Citerio G, Beretta L, Ghisoni L, et al. Refractory intracranial hypertension and “second-tier” therapies in traumatic brain injury. Intensive Care Med. (2008) 34:461–7. doi: 10.1007/s00134-007-0948-9
86. Wettervik TS, Lenell S, Nyholm L, Howells T, Lewen A, Enblad P. Decompressive craniectomy in traumatic brain injury: usage and clinical outcome in a single centre. Acta Neurochir. (2018) 160:229–37. doi: 10.1007/s00701-017-3418-3
87. Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet (2005) 365:387–97. doi: 10.1016/S0140-6736(05)17826-X
88. Gregson BA, Rowan EN, Francis R, McNamee P, Boyers D, Mitchell P, et al. Surgical Trial In Traumatic intraCerebral Haemorrhage (STITCH): a randomised controlled trial of Early Surgery compared with Initial Conservative Treatment. Health Technol Assess. (2015) 19:1–138. doi: 10.3310/hta19700
89. Bullock MR, Lyeth BG, Muizelaar JP. Current status of neuroprotection trials for traumatic brain injury: lessons from animal models and clinical studies. Neurosurgery (1999) 45:207–17; discussion 217–220.
90. Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma (2002) 19:503–57. doi: 10.1089/089771502753754037
91. Chakraborty S, Skolnick B, Narayan RK. Neuroprotection trials in traumatic brain injury. Curr Neurol Neurosci Rep. (2016) 16:29. doi: 10.1007/s11910-016-0625-x
92. Hawryluk GW, Bullock MR. Past, present, and future of traumatic brain injury research. Neurosurg Clin N Am. (2016) 27:375–96. doi: 10.1016/j.nec.2016.05.002
93. Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med. (2014) 371:2457–66. doi: 10.1056/NEJMoa1404304
94. Carpenter KL, Jalloh I, Gallagher CN, Grice P, Howe DJ, Mason A, et al. (13)C-labelled microdialysis studies of cerebral metabolism in TBI patients. Eur J Pharm Sci. (2014) 57:87–97. doi: 10.1016/j.ejps.2013.12.012
95. Gajavelli S, Sinha VK, Mazzeo AT, Spurlock MS, Lee SW, Ahmed AI, et al. Evidence to support mitochondrial neuroprotection, in severe traumatic brain injury. J Bioenerg Biomembr. (2015) 47:133–48. doi: 10.1007/s10863-014-9589-1
96. Verweij BH, Muizelaar JP, Vinas FC, Peterson PL, Xiong Y, Lee CP. Mitochondrial dysfunction after experimental and human brain injury and its possible reversal with a selective N-type calcium channel antagonist (SNX-111). Neurol Res. (1997) 19:334–9.
97. Gerdts J, Summers DW, Milbrandt J, DiAntonio A. Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron (2016) 89:449–60. doi: 10.1016/j.neuron.2015.12.023
98. Yokobori S, Mazzeo AT, Gajavelli S, Bullock MR. Mitochondrial neuroprotection in traumatic brain injury: rationale and therapeutic strategies. CNS Neurol Disord Drug Targets (2014) 13:606–19.
99. Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron (2017) 95:1246–65. doi: 10.1016/j.neuron.2017.07.010
100. Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. (1987) 19:297–303.
101. Duchen MR, McGuinness O, Brown LA, Crompton M. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc Res. (1993) 27:1790–4.
102. Ryba M, Pastuszko M, Iwanska K, Bidzinski J, Dziewiecki C. Cyclosporine A prevents neurological deterioration of patients with SAH–a preliminary report. Acta Neurochir. (1991) 112:25–7.
103. Borlongan CV, Paul OI, Sanberg R. Immunosuppressant Analogs in Neuroprotection. New York, NY: Humana Press (2003).
104. Uchino H, Elmer E, Uchino K, Li PA, He QP, Smith ML, et al. Amelioration by cyclosporin A of brain damage in transient forebrain ischemia in the rat. Brain Res. (1998) 812:216–26.
105. Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. (1999) 160:226–34. doi: 10.1006/exnr.1999.7197
106. Hatton J, Rosbolt B, Empey P, Kryscio R, Young B. Dosing and safety of cyclosporine in patients with severe brain injury. J Neurosurg. (2008) 109:699–707. doi: 10.3171/JNS/2008/109/10/0699
107. Mazzeo AT, Brophy GM, Gilman CB, Alves OL, Robles JR, Hayes RL, et al. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma (2009) 26:2195–206. doi: 10.1089/neu.2009.1012
108. Dixon CE, Bramlett HM, Dietrich WD, Shear DA, Yan HQ, Deng-Bryant Y, et al. Cyclosporine treatment in traumatic brain injury: operation brain trauma therapy. J Neurotrauma (2016) 33:553–66. doi: 10.1089/neu.2015.4122
109. Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma (1999) 16:783–92. doi: 10.1089/neu.1999.16.783
110. Sullivan PG, Thompson M, Scheff SW. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp Neurol. (2000) 161:631–7. doi: 10.1006/exnr.1999.7282
111. Sullivan PG, Sebastian AH, Hall ED. Therapeutic window analysis of the neuroprotective effects of cyclosporine A after traumatic brain injury. J Neurotrauma (2011) 28:311–8. doi: 10.1089/neu.2010.1646
112. Kulbe JR, Singh IN, Wang JA, Cebak JE, Hall ED. Continuous infusion of phenelzine, cyclosporine A, or their combination: evaluation of mitochondrial bioenergetics, oxidative damage, and cytoskeletal degradation following severe controlled cortical impact traumatic brain injury in rats. J Neurotrauma (2018) 35:1280–93. doi: 10.1089/neu.2017.5353
113. Karlsson M, Pukenas B, Chawla S, Ehinger JK, Plyler R, Stolow M, et al. Neuroprotective effects of cyclosporine in a porcine pre-clinical trial of focal traumatic brain injury. J Neurotrauma (2018) 35:1–11. doi: 10.1089/neu.2018.5706
114. Scott G, Zetterberg H, Jolly A, Cole JH, De Simoni S, Jenkins PO, et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain (2017) 14:459–71. doi: 10.1093/brain/awx339
115. Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. (2013) 4:e525. doi: 10.1038/cddis.2013.54
116. Hanlon LA, Huh JW, Raghupathi R. Minocycline transiently reduces microglia/macrophage activation but exacerbates cognitive deficits following repetitive traumatic brain injury in the neonatal rat. J Neuropathol Exp Neurol. (2016) 75:214–26. doi: 10.1093/jnen/nlv021
117. Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. (2017) 13:171–91. doi: 10.1038/nrneurol.2017.13
118. Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, et al. Post-injury administration of mitochondrial uncouplers increases tissue sparing and improves behavioral outcome following traumatic brain injury in rodents. J Neurotrauma (2007) 24:798–811. doi: 10.1089/neu.2006.3673
119. Mazzeo AT, Beat A, Singh A, Bullock MR. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol. (2009) 218:363–70. doi: 10.1016/j.expneurol.2009.05.026
120. Harders A, Kakarieka A, Braakman R. Traumatic subarachnoid hemorrhage and its treatment with nimodipine. German tSAH Study Group J Neurosurg. (1996) 85:82–9. doi: 10.3171/jns.1996.85.1.0082
121. Injury T.E.S.G.o.N.i.S.H. A multicenter trial of the efficacy of nimodipine on outcome after severe head injury. The European Study Group on Nimodipine in Severe Head Injury. J Neurosurg. (1994) 80:797–804. doi: 10.3171/jns.1994.80.5.0797
122. Bullock R, Zauner A, Woodward JJ, Myseros J, Choi SC, Ward JD, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. (1998) 89:507–18. doi: 10.3171/jns.1998.89.4.0507
123. Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int. (2006) 48:394–403. doi: 10.1016/j.neuint.2005.12.001
124. Doyle S, Hansen DB, Vella J, Bond P, Harper G, Zammit C, et al. Vesicular glutamate release from central axons contributes to myelin damage. Nat Commun. (2018) 9:1032. doi: 10.1038/s41467-018-03427-1
125. Liang J, Wu S, Xie W, He H. Ketamine ameliorates oxidative stress-induced apoptosis in experimental traumatic brain injury via the Nrf2 pathway. Drug Des Devel Ther. (2018) 12:845–53. doi: 10.2147/DDDT.S160046
126. Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science (2007) 318:1645–7. doi: 10.1126/science.1148045
127. Gulyas AI, Megias M, Emri Z, Freund TF. Total number and ratio of excitatory and inhibitory synapses converging onto single interneurons of different types in the CA1 area of the rat hippocampus. J Neurosci. (1999) 19:10082–97.
128. Norenberg A, Hu H, Vida I, Bartos M, Jonas P. Distinct nonuniform cable properties optimize rapid and efficient activation of fast-spiking GABAergic interneurons. Proc Natl Acad Sci USA. (2010) 107:894–9. doi: 10.1073/pnas.0910716107
129. Kubota Y, Karube F, Nomura M, Gulledge AT, Mochizuki A, Schertel A, et al. Conserved properties of dendritic trees in four cortical interneuron subtypes. Sci Rep. (2011) 1:89. doi: 10.1038/srep00089
130. Tukker JJ, Lasztoczi B, Katona L, Roberts JD, Pissadaki EK, Dalezios Y, et al. Distinct dendritic arborization and in vivo firing patterns of parvalbumin-expressing basket cells in the hippocampal area CA3. J Neurosci. (2013) 33:6809–25. doi: 10.1523/JNEUROSCI.5052-12.2013
131. Hu H, Gan J, Jonas P. Interneurons. fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science (2014) 345:1255263. doi: 10.1126/science.1255263
132. Kawaguchi Y, Katsumaru H, Kosaka T, Heizmann CW, Hama K. Fast spiking cells in rat hippocampus (CA1 region) contain the calcium-binding protein parvalbumin. Brain Res. (1987) 416:369–74.
133. Song C, Sandberg K, Andersen LM, Blicher JU, Rees G. Human occipital and parietal GABA selectively influence visual perception of orientation and size. J Neurosci. (2017) 37:8929–37. doi: 10.1523/JNEUROSCI.3945-16.2017
134. Chang WJ, Chang WP, Shyu BC. Suppression of cortical seizures by optic stimulation of the reticular thalamus in PV-mhChR2-YFP BAC transgenic mice. Mol Brain (2017) 10:42. doi: 10.1186/s13041-017-0320-0
135. Drexel M, Romanov RA, Wood J, Weger S, Heilbronn R, Wulff P, et al. Selective silencing of hippocampal parvalbumin interneurons induces development of recurrent spontaneous limbic seizures in mice. J Neurosci. (2017) 37:8166–79. doi: 10.1523/JNEUROSCI.3456-16.2017
136. Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. (1999) 33:523–33.
137. Zoerle T, Carbonara M, Zanier ER, Ortolano F, Bertani G, Magnoni S, et al. Rethinking neuroprotection in severe traumatic brain injury: toward bedside neuroprotection. Front Neurol. (2017) 8:354. doi: 10.3389/fneur.2017.00354
138. Hinzman JM, Wilson JA, Mazzeo AT, Bullock MR, Hartings JA. Excitotoxicity and metabolic crisis are associated with spreading depolarizations in severe traumatic brain injury patients. J Neurotrauma (2016) 33:1775–83. doi: 10.1089/neu.2015.4226
139. Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. (2002) 1:383–6. doi: 10.1016/S1474-4422(02)00164-3
140. O'Phelan K, McArthur DL, Chang CW, Green D, Hovda DA. The impact of substance abuse on mortality in patients with severe traumatic brain injury. J Trauma (2008) 65:674–7. doi: 10.1097/TA.0b013e31817db0a5
141. Kim YJ. A systematic review of factors contributing to outcomes in patients with traumatic brain injury. J Clin Nurs. (2011) 20:1518–32. doi: 10.1111/j.1365-2702.2010.03618.x
142. Gold MS, Kobeissy FH, Wang KK, Merlo LJ, Bruijnzeel AW, Krasnova IN, et al. Methamphetamine- and trauma-induced brain injuries: comparative cellular and molecular neurobiological substrates. Biol Psychiatry (2009) 66:118–27. doi: 10.1016/j.biopsych.2009.02.021
143. Kety SS, Schmidt CF. The nitrous oxide method for the quantitative determination of cerebral blood flow in man: theory, procedure and normal values. J Clin Invest. (1948) 27:476–83. doi: 10.1172/JCI101994
144. Frackowiak RS, Lenzi GL, Jones T, Heather JD. Quantitative measurement of regional cerebral blood flow and oxygen metabolism in man using 15O and positron emission tomography: theory, procedure, and normal values. J Comput Assist Tomogr. (1980) 4:727–36.
145. Andersen BJ, Marmarou A. Post-traumatic selective stimulation of glycolysis. Brain Res. (1992) 585:184–9.
146. Glenn TC, Kelly DF, Boscardin WJ, McArthur DL, Vespa P, Oertel M, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab. (2003) 23:1239–50. doi: 10.1097/01.WCB.0000089833.23606.7F
147. Hovda DA, Becker DP, Katayama Y. Secondary injury and acidosis. J Neurotrauma (1992) 9(Suppl 1):S47–60.
148. Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. (1997) 86:241–51. doi: 10.3171/jns.1997.86.2.0241
149. Vespa P, Bergsneider M, Hattori N, Wu HM, Huang SC, Martin NA, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. (2005) 25:763–74. doi: 10.1038/sj.jcbfm.9600073
150. Carpenter KL, Jalloh I, Hutchinson PJ. Glycolysis and the significance of lactate in traumatic brain injury. Front Neurosci. (2015) 9:112. doi: 10.3389/fnins.2015.00112
151. Brooks GA, Martin NA. Cerebral metabolism following traumatic brain injury: new discoveries with implications for treatment. Front Neurosci. (2014) 8:408. doi: 10.3389/fnins.2014.00408
152. Lama S, Auer RN, Tyson R, Gallagher CN, Tomanek B, Sutherland GR. Lactate storm marks cerebral metabolism following brain trauma. J Biol Chem. (2014) 289:20200–8. doi: 10.1074/jbc.M114.570978
153. Nilsson P, Hillered L, Ponten U, Ungerstedt U. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J Cereb Blood Flow Metab. (1990) 10:631–7. doi: 10.1038/jcbfm.1990.115
154. Vespa P, Tubi M, Claassen J, Buitrago-Blanco M, McArthur D, Velazquez AG, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol. (2016) 79:579–90. doi: 10.1002/ana.24606
155. Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. (1991) 561:106–19.
156. Moro N, Ghavim S, Harris NG, Hovda DA, Sutton RL. Glucose administration after traumatic brain injury improves cerebral metabolism and reduces secondary neuronal injury. Brain Res. (2013) 1535:124–36. doi: 10.1016/j.brainres.2013.08.044
157. Shijo K, Ghavim S, Harris NG, Hovda DA, Sutton RL. Glucose administration after traumatic brain injury exerts some benefits and no adverse effects on behavioral and histological outcomes. Brain Res. (2015) 1614:94–104. doi: 10.1016/j.brainres.2015.04.022
158. Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. (2008) 86:1812–22. doi: 10.1002/jnr.21628
159. Guglielmetti C, Chou A, Krukowski K, Najac C, Feng X, Riparip LK, et al. In vivo metabolic imaging of Traumatic Brain Injury. Sci Rep. (2017) 7:17525. doi: 10.1038/s41598-017-17758-4
160. Bilotta F, Caramia R, Cernak I, Paoloni FP, Doronzio A, Cuzzone V, et al. Intensive insulin therapy after severe traumatic brain injury: a randomized clinical trial. Neurocrit Care (2008) 9:159–66. doi: 10.1007/s12028-008-9084-9
161. Yang M, Guo Q, Zhang X, Sun S, Wang Y, Zhao L, et al. Intensive insulin therapy on infection rate, days in NICU, in-hospital mortality and neurological outcome in severe traumatic brain injury patients: a randomized controlled trial. Int J Nurs Stud. (2009) 46:753–8. doi: 10.1016/j.ijnurstu.2009.01.004
162. Coester A, Neumann CR, Schmidt MI. Intensive insulin therapy in severe traumatic brain injury: a randomized trial. J Trauma (2010) 68:904–11. doi: 10.1097/TA.0b013e3181c9afc2
163. Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. (2006) 60:223–35. doi: 10.1002/ana.20899
164. Kim DY, Simeone KA, Simeone TA, Pandya JD, Wilke JC, Ahn Y, et al. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann Neurol. (2015) 78:77–87. doi: 10.1002/ana.24424
165. Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. (2004) 55:576–80. doi: 10.1002/ana.20062
166. Hoane MR, Swan AA, Heck SE. The effects of a high-fat sucrose diet on functional outcome following cortical contusion injury in the rat. Behav Brain Res. (2011) 223:119–24. doi: 10.1016/j.bbr.2011.04.028
167. Prins M, Matsumoto J. The collective therapeutic potential of cerebral ketone metabolism in Traumatic Brain Injury. J Lipid Res. (2014) 54:2450–7. doi: 10.1194/jlr.R046706
168. Gallagher CN, Carpenter KL, Grice P, Howe DJ, Mason A, Timofeev I, et al. The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain (2009) 132(Pt 10):2839–49. doi: 10.1093/brain/awp202
169. De Fazio M, Rammo R, O'Phelan K, Bullock MR. Alterations in cerebral oxidative metabolism following traumatic brain injury. Neurocrit Care (2011) 14:91–6. doi: 10.1007/s12028-010-9494-3
170. Jalloh I, Carpenter KL, Grice P, Howe DJ, Mason A, Gallagher CN, et al. Glycolysis and the pentose phosphate pathway after human traumatic brain injury: microdialysis studies using 1,2-(13)C2 glucose. J Cereb Blood Flow Metab. (2015) 35:111–20. doi: 10.1038/jcbfm.2014.177
171. Jalloh I, Helmy A, Howe D, Shannon RJ, Grice P, Mason A, et al. A comparison of oxidative lactate metabolism in traumatically injured brain and control brain. J Neurotrauma (2018) 35:2025–35. doi: 10.1089/neu.2017.5459
172. Jalloh I, Helmy A, Howe DJ, Shannon RJ, Grice P, Mason A, et al. Focally perfused succinate potentiates brain metabolism in head injury patients. J Cereb Blood Flow Metab. (2017) 37:2626–38. doi: 10.1177/0271678X16672665
173. Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. (2011) 14:724–38. doi: 10.1016/j.cmet.2011.08.016
174. Beynon C, Kiening KL, Orakcioglu B, Unterberg AW, Sakowitz OW. Brain tissue oxygen monitoring and hyperoxic treatment in patients with traumatic brain injury. J Neurotrauma (2012) 29:2109–23. doi: 10.1089/neu.2012.2365
176. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. (1995) 92:5510–4.
177. Shirato K, Nakajima K, Korekane H, Takamatsu S, Gao C, Angata T, et al. Hypoxic regulation of glycosylation via the N-acetylglucosamine cycle. J Clin Biochem Nutr. (2011) 48:20–5. doi: 10.3164/jcbn.11-015FR
178. Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. (2006) 70:1469–80. doi: 10.1124/mol.106.027029
179. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1beta production and to prevent DSS-induced colitis in mice. Cell Rep. (2017) 19:2331–44. doi: 10.1016/j.celrep.2017.05.065
180. Daugherty WP, Levasseur JE, Sun D, Spiess BD, Bullock MR. Perfluorocarbon emulsion improves cerebral oxygenation and mitochondrial function after fluid percussion brain injury in rats. Neurosurgery (2004) 54:1223–30; discussion 1230.
181. Kwon TH, Sun D, Daugherty WP, Spiess BD, Bullock MR. Effect of perfluorocarbons on brain oxygenation and ischemic damage in an acute subdural hematoma model in rats. J Neurosurg. (2005) 103:724–30. doi: 10.3171/jns.2005.103.4.0724
182. NCT00908063. Safety and Tolerability of Oxycyte in Patients With Traumatic Brain Injury (TBI) (STOP-TBI) (2009).
183. Deuchar GA, Brennan D, Holmes WM, Shaw M, Macrae IM, Santosh C. Perfluorocarbon enhanced glasgow oxygen level dependent (GOLD) magnetic resonance metabolic imaging identifies the penumbra following acute ischemic stroke. Theranostics (2018) 8:1706–22. doi: 10.7150/thno.21685
184. Juul S. Erythropoietin in the central nervous system, and its use to prevent hypoxic-ischemic brain damage. Acta Paediatr. (2002) 91(Suppl):36–42.
185. Noguchi CT, Asavaritikrai P, Teng R, Jia Y. Role of erythropoietin in the brain. Crit Rev Oncol Hematol. (2007) 64:159–71. doi: 10.1016/j.critrevonc.2007.03.001
186. Lu J, Gary KW, Neimeier JP, Ward J, Lapane KL. Randomized controlled trials in adult traumatic brain injury. Brain Inj. (2012) 26:1523–48. doi: 10.3109/02699052.2012.722257
187. Nichol A, French C, Little L, Haddad S, Presneill J, Arabi Y, et al. Erythropoietin in traumatic brain injury (EPO-TBI): a double-blind randomised controlled trial. Lancet (2015) 386:2499–506. doi: 10.1016/S0140-6736(15)00386-4
188. Bramlett HM, Dietrich WD, Dixon CE, Shear DA, Schmid KE, Mondello S, et al. Erythropoietin treatment in traumatic brain injury: operation brain trauma therapy. J Neurotrauma (2016) 33:538–52. doi: 10.1089/neu.2015.4116
189. Nortje J, Coles JP, Timofeev I, Fryer TD, Aigbirhio FI, Smielewski P, et al. Effect of hyperoxia on regional oxygenation and metabolism after severe traumatic brain injury: preliminary findings. Crit Care Med. (2008) 36:273–81. doi: 10.1097/01.CCM.0000292014.60835.15
190. Rockswold GL, Ford SE, Anderson DC, Bergman TA, Sherman RE. Results of a prospective randomized trial for treatment of severely brain-injured patients with hyperbaric oxygen. J Neurosurg. (1992) 76:929–34. doi: 10.3171/jns.1992.76.6.0929
191. Prakash A, Parelkar SV, Oak SN, Gupta RK, Sanghvi BV, Bachani M, et al. Role of hyperbaric oxygen therapy in severe head injury in children. J Pediatr Neurosci. (2012) 7:4–8. doi: 10.4103/1817-1745.97610
192. Rockswold SB, Rockswold GL, Zaun DA, Liu J. A prospective, randomized Phase II clinical trial to evaluate the effect of combined hyperbaric and normobaric hyperoxia on cerebral metabolism, intracranial pressure, oxygen toxicity, and clinical outcome in severe traumatic brain injury. J Neurosurg. (2013) 118:1317–28. doi: 10.3171/2013.2.JNS121468
194. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience (2004) 129:1021–9. doi: 10.1016/j.neuroscience.2004.06.046
195. Shakur H, Andrews P, Asser T, Balica L, Boeriu C, Quintero JD, et al. The BRAIN TRIAL: a randomised, placebo controlled trial of a Bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials (2009) 10:109. doi: 10.1186/1745-6215-10-109
196. Trabold R, Eros C, Zweckberger K, Relton J, Beck H, Nussberger J, et al. The role of bradykinin B(1) and B(2) receptors for secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. (2010) 30:130–9. doi: 10.1038/jcbfm.2009.196
197. Noda M, Kariura Y, Pannasch U, Nishikawa K, Wang L, Seike T, et al. Neuroprotective role of bradykinin because of the attenuation of pro-inflammatory cytokine release from activated microglia. J Neurochem. (2007) 101:397–410. doi: 10.1111/j.1471-4159.2006.04339.x
198. Asraf K, Torika N, Danon A, Fleisher-Berkovich S. Involvement of the bradykinin B1 receptor in microglial activation: in vitro and in vivo studies. Front Endocrinol. (2017) 8:82. doi: 10.3389/fendo.2017.00082
199. Albert-Weissenberger C, Stetter C, Meuth SG, Gobel K, Bader M, Siren AL, et al. Blocking of bradykinin receptor B1 protects from focal closed head injury in mice by reducing axonal damage and astroglia activation. J Cereb Blood Flow Metab. (2012) 32:1747–56. doi: 10.1038/jcbfm.2012.62
200. Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. (2010) 31:596–604. doi: 10.1016/j.tips.2010.09.005
201. Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx CE, Grimes CJ, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma (2014) 31:135–58. doi: 10.1089/neu.2013.3019
202. Hoshide R, Cheung V, Marshall L, Kasper E, Chen CC. Do corticosteroids play a role in the management of traumatic brain injury? Surg Neurol Int. (2016) 7:84. doi: 10.4103/2152-7806.190439
203. DeWitt D, Hawkins BE, Dixon CE, Kochanek PM, Armstead WM, Bass C, et al. Preclinical testing of therapies for traumatic brain injury. J Neurotrauma (2018) 35:2737:54. doi: 10.1089/neu.2018.5778
204. Doppenberg EM, Choi SC, Bullock R. Clinical trials in traumatic brain injury: lessons for the future. J Neurosurg Anesthesiol. (2004) 16:87–94.
205. Chang J, Phelan M, Cummings BJ. A meta-analysis of efficacy in pre-clinical human stem cell therapies for traumatic brain injury. Exp Neurol. (2015) 273:225–33. doi: 10.1016/j.expneurol.2015.08.020
206. Maxwell WL, MacKinnon MA, Stewart JE, Graham DI. Stereology of cerebral cortex after traumatic brain injury matched to the Glasgow outcome score. Brain (2010) 133(Pt 1):139–60. doi: 10.1093/brain/awp264
207. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. (2011) 70:374–83. doi: 10.1002/ana.22455
208. Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, et al. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. (2015) 11:97–106. doi: 10.2147/NDT.S65815
209. Lu XC, Shear DA, Deng-Bryant Y, Leung LY, Wei G, Chen Z, et al. Comprehensive evaluation of neuroprotection achieved by extended selective brain cooling therapy in a rat model of penetrating ballistic-like brain injury. Ther Hypothermia Temp Manag. (2016) 6:30–9. doi: 10.1089/ther.2015.0017
210. Lee SW, Gajavelli S, Spurlock MS, Andreoni C, de Rivero Vaccari JP, Bullock MR, et al. Microglial inflammasome activation in penetrating ballistic-like brain injury. J Neurotrauma (2018) 35:1681–93. doi: 10.1089/neu.2017.5530
211. Harish G, Mahadevan A, Pruthi N, Sreenivasamurthy SK, Puttamallesh VN, Keshava Prasad TS, et al. Characterization of traumatic brain injury in human brains reveals distinct cellular and molecular changes in contusion and pericontusion. J Neurochem. (2015) 134:156–72. doi: 10.1111/jnc.13082
212. Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J Neurosurg. (2012) 117:1119–25. doi: 10.3171/2012.9.JNS12815
213. Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ III, Nonner D, Bullock MR, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab. (2014) 34:621–9. doi: 10.1038/jcbfm.2013.236
214. Loane DJ, Stoica BA, Faden AI. Neuroprotection for traumatic brain injury. Handb Clin Neurol. (2015) 127:343–66. doi: 10.1016/B978-0-444-52892-6.00022-2
215. NCT02064959. To Study the Effect of Early Cooling in Acute Subdural Hematoma Patients (HOPES) (2014).
217. Ahmed AI, Bullock MR, Dietrich WD. Hypothermia in traumatic brain injury. Neurosurg Clin N Am. (2016) 27:489–97. doi: 10.1016/j.nec.2016.05.004
218. Andrews PJ, Sinclair HL, Rodriguez A, Harris B, Rhodes J, Watson H, et al. Therapeutic hypothermia to reduce intracranial pressure after traumatic brain injury: the Eurotherm3235 RCT. Health Technol Assess. (2018) 22:1–134. doi: 10.3310/hta22450
219. Clifton GL, Miller ER, Choi SC, Levin HS, McCauley S, Smith KR Jr, et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. (2001) 344:556–63. doi: 10.1056/nejm200102223440803
220. Cooper DJ, Nichol AD, Bailey M, Bernard S, Cameron PA, Pili-Floury S, et al. Effect of early sustained prophylactic hypothermia on neurologic outcomes among patients with severe traumatic brain injury: the POLAR randomized clinical trial. JAMA (2018) 320:2211–20. doi: 10.1001/jama.2018.17075
221. Dietrich WD, Bramlett HM. Therapeutic hypothermia and targeted temperature management for traumatic brain injury: experimental and clinical experience. Brain Circ. (2017) 3:186–98. doi: 10.4103/bc.bc_28_17
222. Hirst TC, Watzlawick R, Rhodes JK, Macleod MR, Andrews PJ. Study protocol - a systematic review and meta-analysis of hypothermia in experimental traumatic brain injury: why have promising animal studies not been replicated in pragmatic clinical trials? Evid Based Preclin Med. (2016) 3:e00020. doi: 10.1002/ebm2.20
223. Watson HI, Shepherd AA, Rhodes JKJ, Andrews PJD. Revisited: a systematic review of therapeutic hypothermia for adult patients following traumatic brain injury. Crit Care Med. (2018) 46:972–9. doi: 10.1097/CCM.0000000000003125
224. Chung JY, Krapp N, Wu L, Lule S, McAllister L, Edmiston Iii W, et al. Interleukin-1 receptor 1 deletion in focal and diffuse experimental traumatic brain injury in mice. J Neurotrauma (2018) 35:1–10. doi: 10.1089/neu.2018.5659
225. Newell EA, Todd BP, Mahoney J, Pieper AA, Ferguson PJ, Bassuk AG. Combined blockade of interleukin-1alpha and−1beta signaling protects mice from cognitive dysfunction after traumatic brain injury. eNeuro (2018) 5:2. doi: 10.1523/ENEURO.0385-17.2018
226. Hutchinson PJ, O'Connell MT, Rothwell NJ, Hopkins SJ, Nortje J, Carpenter KL, et al. Inflammation in human brain injury: intracerebral concentrations of IL-1alpha, IL-1beta, and their endogenous inhibitor IL-1ra. J Neurotrauma (2007) 24:1545–57. doi: 10.1089/neu.2007.0295
227. Mazzeo AT, Filippini C, Rosato R, Fanelli V, Assenzio B, Piper I, et al. Multivariate projection method to investigate inflammation associated with secondary insults and outcome after human traumatic brain injury: a pilot study. J Neuroinflamm. (2016) 13:157. doi: 10.1186/s12974-016-0624-5
228. Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab. (2014) 34:845–51. doi: 10.1038/jcbfm.2014.23
229. Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist promotes M1 microglia biased cytokines and chemokines following human traumatic brain injury. J Cereb Blood Flow Metab. (2016) 36:1434–48. doi: 10.1177/0271678X15620204
230. Gao C, Qian Y, Huang J, Wang D, Su W, Wang P, et al. A three-day consecutive fingolimod administration improves neurological functions and modulates multiple immune responses of CCI mice. Mol Neurobiol. (2017) 54:8348–60. doi: 10.1007/s12035-016-0318-0
231. Tuttolomondo A, Pecoraro R, Pinto A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: a review of the evidence to date. Drug Des Devel Ther. (2014) 8:2221–38. doi: 10.2147/DDDT.S67655
232. Tapia-Perez J, Sanchez-Aguilar M, Torres-Corzo JG, Gordillo-Moscoso A, Martinez-Perez P, Madeville P, et al. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758). J Neurotrauma (2008) 25:1011–7. doi: 10.1089/neu.2008.0554
233. Kochanek PM, Bramlett HM, Shear DA, Dixon CE, Mondello S, Dietrich WD, et al. Synthesis of findings, current investigations, and future directions: operation brain trauma therapy. J Neurotrauma (2016) 33:606–14. doi: 10.1089/neu.2015.4133
234. Schirris TJ, Renkema GH, Ritschel T, Voermans NC, Bilos A, van Engelen BG, et al. Statin-induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab. (2015) 22:399–407. doi: 10.1016/j.cmet.2015.08.002
235. Mammen AL. Statin-associated autoimmune myopathy. N Engl J Med. (2016) 374:664–9. doi: 10.1056/NEJMra1515161
236. Yonutas HM, Pandya JD, Sullivan PG. Changes in mitochondrial bioenergetics in the brain versus spinal cord become more apparent with age. J Bioenerg Biomembr. (2015) 47:149–54. doi: 10.1007/s10863-014-9593-5
237. Mencl S, Hennig N, Hopp S, Schuhmann MK, Albert-Weissenberger C, Siren AL, et al. FTY720 does not protect from traumatic brain injury in mice despite reducing posttraumatic inflammation. J Neuroimmunol. (2014) 274:125–31. doi: 10.1016/j.jneuroim.2014.07.010
238. Norimatsu Y, Ohmori T, Kimura A, Madoiwa S, Mimuro J, Seichi A, et al. FTY720 improves functional recovery after spinal cord injury by primarily nonimmunomodulatory mechanisms. Am J Pathol. (2012) 180:1625–35. doi: 10.1016/j.ajpath.2011.12.012
239. Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature (2014) 505:223–8. doi: 10.1038/nature12808
240. Bao F, Shultz SR, Hepburn JD, Omana V, Weaver LC, Cain DP, et al. A CD11d monoclonal antibody treatment reduces tissue injury and improves neurological outcome after fluid percussion brain injury in rats. J Neurotrauma (2012) 29:2375–92. doi: 10.1089/neu.2012.2408
241. Weaver LC, Bao F, Dekaban GA, Hryciw T, Shultz SR, Cain DP, et al. CD11d integrin blockade reduces the systemic inflammatory response syndrome after traumatic brain injury in rats. Exp Neurol. (2015) 271:409–22. doi: 10.1016/j.expneurol.2015.07.003
242. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. (2018) 22:2924–36. doi: 10.1016/j.celrep.2018.02.067
243. Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci USA. (2002) 99:15620–5. doi: 10.1073/pnas.232565399
244. Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest. (2015) 125:2547. doi: 10.1172/JCI82458
245. Bachstetter AD, Zhou Z, Rowe RK, Xing B, Goulding DS, Conley AN, et al. MW151 inhibited IL-1beta levels after traumatic brain injury with no effect on microglia physiological responses. PLoS ONE (2016) 11:e0149451. doi: 10.1371/journal.pone.0149451
246. Xing Z, Xia Z, Peng W, Li J, Zhang C, Fu C, et al. Xuefu Zhuyu decoction, a traditional Chinese medicine, provides neuroprotection in a rat model of traumatic brain injury via an anti-inflammatory pathway. Sci Rep. (2016) 6:20040. doi: 10.1038/srep20040
247. Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. (2015) 72:355–62. doi: 10.1001/jamaneurol.2014.3558
248. Kochanek PM, Berger RP, Bayir H, Wagner AK, Jenkins LW, Clark RS. Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: diagnosis, prognosis, probing mechanisms, and therapeutic decision making. Curr Opin Crit Care (2008) 14:135–41. doi: 10.1097/MCC.0b013e3282f57564
249. Kobeissy FH, Guingab-Cagmat JD, Razafsha M, O'Steen L, Zhang Z, Hayes RL, et al. Leveraging biomarker platforms and systems biology for rehabilomics and biologics effectiveness research. PMR (2011) 3(6 Suppl 1):S139–47. doi: 10.1016/j.pmrj.2011.02.012
250. Stein DM, Lindell AL, Murdock KR, Kufera JA, Menaker J, Bochicchio GV, et al. Use of serum biomarkers to predict cerebral hypoxia after severe traumatic brain injury. J Neurotrauma (2012) 29:1140–9. doi: 10.1089/neu.2011.2149
251. Yokobori S, Hosein K, Burks S, Sharma I, Gajavelli S, Bullock R. Biomarkers for the clinical differential diagnosis in traumatic brain injury–a systematic review. CNS Neurosci Ther. (2013) 19:556–65. doi: 10.1111/cns.12127
252. Papa L, Brophy GM, Welch RD, Lewis LM, Braga CF, Tan CN, et al. Time course and diagnostic accuracy of glial and neuronal blood biomarkers GFAP and UCH-L1 in a large cohort of trauma patients with and without mild traumatic brain injury. JAMA Neurol. (2016) 73:551–60. doi: 10.1001/jamaneurol.2016.0039
253. Posti JP, Takala RS, Runtti H, Newcombe VF, Outtrim J, Katila AJ, et al. The levels of glial fibrillary acidic protein and ubiquitin C-terminal hydrolase-L1 during the first week after a traumatic brain injury: correlations with clinical and imaging findings. Neurosurgery (2016) 79:456–64. doi: 10.1227/NEU.0000000000001226
254. Takala RS, Posti JP, Runtti H, Newcombe VF, Outtrim J, Katila AJ, et al. Glial fibrillary acidic protein and ubiquitin C-terminal hydrolase-L1 as outcome predictors in traumatic brain injury. World Neurosurg. (2016) 87:8–20. doi: 10.1016/j.wneu.2015.10.066
255. Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. (1998) 338:20–4. doi: 10.1056/NEJM199801013380104
256. Vespa PM, Nuwer MR, Nenov V, Ronne-Engstrom E, Hovda DA, Bergsneider M, et al. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg. (1999) 91:750–60. doi: 10.3171/jns.1999.91.5.0750
257. Hartings JA, Bullock MR, Okonkwo DO, Murray LS, Murray GD, Fabricius M, et al. Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. Lancet Neurol. (2011) 10:1058–64. doi: 10.1016/S1474-4422(11)70243-5
258. Cole JH, Leech R, Sharp DJ, Alzheimer's Disease Neuroimaging I. Prediction of brain age suggests accelerated atrophy after traumatic brain injury. Ann Neurol. (2015) 77:571–81. doi: 10.1002/ana.24367
259. Cole JH, Jolly A, de Simoni S, Bourke N, Patel MC, Scott G, et al. Spatial patterns of progressive brain volume loss after moderate-severe traumatic brain injury. Brain (2018) 141:822–36. doi: 10.1093/brain/awx354
260. Griesbach GS, Masel BE, Helvie RE, Ashley MJ. The impact of traumatic brain injury on later life: effects on normal aging and neurodegenerative diseases. J Neurotrauma (2018) 35:17–24. doi: 10.1089/neu.2017.5103
261. Weiner MW, Friedl KE, Pacifico A, Chapman JC, Jaffee MS, Little DM, et al. Military risk factors for Alzheimer's disease. Alzheimers Dement. (2013) 9:445–51. doi: 10.1016/j.jalz.2013.03.005
262. Kokiko-Cochran ON, Godbout JP. The inflammatory continuum of traumatic brain injury and alzheimer's disease. Front Immunol. (2018) 9:672. doi: 10.3389/fimmu.2018.00672
263. Lucke-Wold BP, Nguyen L, Turner RC, Logsdon AF, Chen YW, Smith KE, et al. Traumatic brain injury and epilepsy: underlying mechanisms leading to seizure. Seizure (2015) 33:13–23. doi: 10.1016/j.seizure.2015.10.002
264. Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. (2015) 15:27. doi: 10.1007/s11910-015-0545-1
265. Lafrenaye AD, Todani M, Walker SA, Povlishock JT. Microglia processes associate with diffusely injured axons following mild traumatic brain injury in the micro pig. J Neuroinflamm. (2015) 12:186. doi: 10.1186/s12974-015-0405-6
266. Zhang Z, Zoltewicz JS, Mondello S, Newsom KJ, Yang Z, Yang B, et al. Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PLoS ONE (2014) 9:e92698. doi: 10.1371/journal.pone.0092698
267. Tomaiuolo F, Bivona U, Lerch JP, Di Paola M, Carlesimo GA, Ciurli P, et al. Memory and anatomical change in severe non missile traumatic brain injury: approximately 1 vs. approximately 8 years follow-up. Brain Res Bull. (2012) 87:373–82. doi: 10.1016/j.brainresbull.2012.01.008
268. Zhao S, Yu Z, Liu Y, Bai Y, Jiang Y, van Leyen K, et al. CD47 deficiency improves neurological outcomes of traumatic brain injury in mice. Neurosci Lett. (2017) 643:125–30. doi: 10.1016/j.neulet.2016.12.006
269. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer's disease. Nature (2017) 552:355–61. doi: 10.1038/nature25158
270. Donat CK, Scott G, Gentleman SM, Sastre M. Microglial activation in traumatic brain injury. Front Aging Neurosci. (2017) 9:208. doi: 10.3389/fnagi.2017.00208
271. Lehman EJ, Hein MJ, Baron SL, Gersic CM. Neurodegenerative causes of death among retired National Football League players. Neurology (2012) 79:1970–4. doi: 10.1212/WNL.0b013e31826daf50
272. Temple S. Division and differentiation of isolated CNS blast cells in microculture. Nature (1989) 340:471–3. doi: 10.1038/340471a0
273. Temple S, Qian X. Vertebrate neural progenitor cells: subtypes and regulation. Curr Opin Neurobiol. (1996) 6:11–7.
274. Stiles J, Jernigan TL. The basics of brain development. Neuropsychol Rev. (2010) 20:327–48. doi: 10.1007/s11065-010-9148-4
275. Bystron I, Rakic P, Molnar Z, Blakemore C. The first neurons of the human cerebral cortex. Nat Neurosci. (2006) 9:880–6. doi: 10.1038/nn1726
276. Urban N, Guillemot F. Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front Cell Neurosci. (2014) 8:396. doi: 10.3389/fncel.2014.00396
277. Salman H, Ghosh P, Kernie SG. Subventricular zone neural stem cells remodel the brain following traumatic injury in adult mice. J Neurotrauma (2004) 21:283–92. doi: 10.1089/089771504322972077
278. Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, et al. Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci USA. (2008) 105:3581–6. doi: 10.1073/pnas.0709002105
279. Ahmed AI, Shtaya AB, Zaben MJ, Owens EV, Kiecker C, Gray WP. Endogenous GFAP-positive neural stem/progenitor cells in the postnatal mouse cortex are activated following traumatic brain injury. J Neurotrauma (2012) 29:828–42. doi: 10.1089/neu.2011.1923
280. Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, et al. Neurogenesis in the adult human hippocampus. Nat Med. (1998) 4:1313–7. doi: 10.1038/3305
281. Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, et al. Dynamics of hippocampal neurogenesis in adult humans. Cell (2013) 153:1219–27. doi: 10.1016/j.cell.2013.05.002
282. Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature (2018) 555:377–81. doi: 10.1038/nature25975
283. Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature (2004) 427:740–4. doi: 10.1038/nature02301
284. Zheng W, ZhuGe Q, Zhong M, Chen G, Shao B, Wang H, et al. Neurogenesis in adult human brain after traumatic brain injury. J Neurotrauma (2013) 30:1872–80. doi: 10.1089/neu.2010.1579
285. Guadagno J, Swan P, Shaikh R, Cregan SP. Microglia-derived IL-1beta triggers p53-mediated cell cycle arrest and apoptosis in neural precursor cells. Cell Death Dis. (2015) 6:e1779. doi: 10.1038/cddis.2015.151
286. Flygt J, Ruscher K, Norberg A, Mir A, Gram H, Clausen F, et al. Neutralization of interleukin-1beta following diffuse traumatic brain injury in the mouse attenuates the loss of mature oligodendrocytes. J Neurotrauma (2018) 35:2837–49. doi: 10.1089/neu.2018.5660
287. Richardson RM, Singh A, Sun DFillmore HL, Dietrich DW III, Bullock MR. Stem cell biology in traumatic brain injury: effects of injury and strategies for repair. J Neurosurg. (2010) 112:1125–38. doi: 10.3171/2009.4.JNS081087
288. Cramer SC, Hill MD, Investigators R-L. Human choriogonadotropin and epoetin alfa in acute ischemic stroke patients (REGENESIS-LED trial). Int J Stroke (2014) 9:321–7. doi: 10.1111/ijs.12260
289. Arsenijevic Y, Villemure JG, Brunet JF, Bloch JJ, Deglon N, Kostic C, et al. Isolation of multipotent neural precursors residing in the cortex of the adult human brain. Exp Neurol. (2001) 170:48–62. doi: 10.1006/exnr.2001.7691
290. Blaya MO, Wasserman JM, Pieper AA, Sick TJ, Bramlett HM, Dietrich WD. Neurotherapeutic capacity of P7C3 agents for the treatment of Traumatic Brain Injury. Neuropharmacology (2018) 17:S0028-3908(18)30672-5. doi: 10.1016/j.neuropharm.2018.09.024
291. Fava M, Johe K, Ereshefsky L, Gertsik LG, English BA, Bilello JA, et al. A Phase 1B, randomized, double blind, placebo controlled, multiple-dose escalation study of NSI-189 phosphate, a neurogenic compound, in depressed patients. Mol Psychiatry (2016) 21:1372–80. doi: 10.1038/mp.2015.178
292. McIntyre RS, Johe K, Rong C, Lee Y. The neurogenic compound, NSI-189 phosphate: a novel multi-domain treatment capable of pro-cognitive and antidepressant effects. Expert Opin Investig Drugs (2017) 26:767–70. doi: 10.1080/13543784.2017.1324847
293. Tajiri N, Quach DM, Kaneko Y, Wu S, Lee D, Lam T, et al. NSI-189, a small molecule with neurogenic properties, exerts behavioral, and neurostructural benefits in stroke rats. J Cell Physiol. (2017) 232:2731–40. doi: 10.1002/jcp.25847
294. Allen BD, Acharya MM, Lu C, Giedzinski E, Chmielewski NN, Quach D, et al. Remediation of radiation-induced cognitive dysfunction through oral administration of the neuroprotective compound NSI-189. Radiat Res. (2018) 189:345–53. doi: 10.1667/RR14879.1
296. Hassani Z, O'Reilly J, Pearse Y, Stroemer P, Tang E, Sinden J, et al. Human neural progenitor cell engraftment increases neurogenesis and microglial recruitment in the brain of rats with stroke. PLoS ONE (2012) 7:e50444. doi: 10.1371/journal.pone.0050444
297. Smith EJ, Stroemer RP, Gorenkova N, Nakajima M, Crum WR, Tang E, et al. Implantation site and lesion topology determine efficacy of a human neural stem cell line in a rat model of chronic stroke. Stem Cells (2012) 30:785–96. doi: 10.1002/stem.1024
298. Hicks C, Stevanato L, Stroemer RP, Tang E, Richardson S, Sinden JD. In vivo and in vitro characterization of the angiogenic effect of CTX0E03 human neural stem cells. Cell Transplant. (2013) 22:1541–52. doi: 10.3727/096368912X657936
299. Kalladka D, Sinden J, Pollock K, Haig C, McLean J, Smith W, et al. Human neural stem cells in patients with chronic ischaemic stroke (PISCES): a phase 1, first-in-man study. Lancet (2016) 388:787–96. doi: 10.1016/S0140-6736(16)30513-X
300. Sinden JD, Hicks C, Stroemer P, Vishnubhatla I, Corteling R. Human neural stem cell therapy for chronic ischemic stroke: charting progress from laboratory to patients. Stem Cells Dev. (2017) 26:933–47. doi: 10.1089/scd.2017.0009
301. Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. (2006) 9:173–9. doi: 10.1038/nn1620
302. Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain (2006) 129(Pt 10):2761–72. doi: 10.1093/brain/awl165
303. Dixon KJ, Theus MH, Nelersa CM, Mier J, Travieso LG, Yu TS, et al. Endogenous neural stem/progenitor cells stabilize the cortical microenvironment after traumatic brain injury. J Neurotrauma (2015) 32:753–64. doi: 10.1089/neu.2014.3390
304. Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. (2016) 173:692–702. doi: 10.1111/bph.13125
305. Kochanek PM, Jackson TC, Ferguson NM, Carlson SW, Simon DW, Brockman EC, et al. Emerging therapies in traumatic brain injury. Semin Neurol. (2015) 35:83–100. doi: 10.1055/s-0035-1544237
306. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. (2014) 15:209–16. doi: 10.1038/nrn3710
307. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
308. Hasan A, Deeb G, Rahal R, Atwi K, Mondello S, Marei HE, et al. Mesenchymal stem cells in the treatment of traumatic brain injury. Front Neurol. (2017) 8:28. doi: 10.3389/fneur.2017.00028
309. Spurlock MS, Ahmed AI, Rivera KN, Yokobori S, Lee SW, Sam PN, et al. Amelioration of penetrating ballistic-like brain injury induced cognitive deficits after neuronal differentiation of transplanted human neural stem cells. J Neurotrauma (2017) 34:1981–95. doi: 10.1089/neu.2016.4602
310. NCT01273337. “Study of ALD-401 Via Intracarotid Infusion in Ischemic Stroke Subjects”.) (2014).
311. Mazzini L, Ferrero I, Luparello V, Rustichelli D, Gunetti M, Mareschi K, et al. Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: a phase I clinical trial. Exp Neurol. (2010) 223:229–37. doi: 10.1016/j.expneurol.2009.08.007
312. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol. (2017) 18:826–31. doi: 10.1038/ni.3790
313. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. (2014) 32:367–402. doi: 10.1146/annurev-immunol-032713-120240
314. Lampron A, Larochelle A, Laflamme N, Prefontaine P, Plante MM, Sanchez MG, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. (2015) 212:481–95. doi: 10.1084/jem.20141656
315. Micu I, Plemel JR, Caprariello AV, Nave KA, Stys PK. Axo-myelinic neurotransmission: a novel mode of cell signalling in the central nervous system. Nat Rev Neurosci. (2017) 19:58. doi: 10.1038/nrn.2017.166
316. Gao J, Grill RJ, Dunn TJ, Bedi S, Labastida JA, Hetz RA, et al. Human neural stem cell transplantation-mediated alteration of microglial/macrophage phenotypes after traumatic brain injury. Cell Transplant. (2016) 25:1863–77. doi: 10.3727/096368916X691150
317. Gao J, Prough DS, McAdoo DJ, Grady JJ, Parsley MO, Ma L, et al. Transplantation of primed human fetal neural stem cells improves cognitive function in rats after traumatic brain injury. Exp Neurol. (2006) 201:281–92. doi: 10.1016/j.expneurol.2006.04.039
318. Wennersten A, Holmin S, Al Nimer F, Meijer X, Wahlberg LU, Mathiesen T. Sustained survival of xenografted human neural stem/progenitor cells in experimental brain trauma despite discontinuation of immunosuppression. Exp Neurol. (2006) 199:339–47. doi: 10.1016/j.expneurol.2005.12.035
319. Skardelly M, Gaber K, Burdack S, Scheidt F, Hilbig H, Boltze J, et al. Long-term benefit of human fetal neuronal progenitor cell transplantation in a clinically adapted model after traumatic brain injury. J Neurotrauma (2011) 28:401–14. doi: 10.1089/neu.2010.1526
320. Gage FH, Temple S. Neural stem cells: generating and regenerating the brain. Neuron (2013) 80:588–601. doi: 10.1016/j.neuron.2013.10.037
321. Steinbeck JA, Choi SJ, Mrejeru A, Ganat Y, Deisseroth K, Sulzer D, et al. Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson's disease model. Nat Biotechnol. (2015) 33:204–9. doi: 10.1038/nbt.3124
322. Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RD. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. (1996) 10:3129–40.
323. Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (1992) 255:1707–10.
324. Buc-Caron MH. Neuroepithelial progenitor cells explanted from human fetal brain proliferate and differentiate in vitro. Neurobiol Dis. (1995) 2:37–47. doi: 10.1006/nbdi.1995.0004
325. Brustle O, Maskos U, McKay RD. Host-guided migration allows targeted introduction of neurons into the embryonic brain. Neuron (1995) 15:1275–85.
326. Brustle O, Choudhary K, Karram K, Huttner A, Murray K, Dubois-Dalcq M, et al. Chimeric brains generated by intraventricular transplantation of fetal human brain cells into embryonic rats. Nat Biotechnol. (1998) 16:1040–4. doi: 10.1038/3481
327. Stenevi U, Bjorklund A, Svendgaard NA. Transplantation of central and peripheral monoamine neurons to the adult rat brain: techniques and conditions for survival. Brain Res. (1976) 114:1–20.
328. Soares H, McIntosh TK. Fetal cortical transplants in adult rats subjected to experimental brain injury. J Neural Transplant Plast. (1991) 2:207–20. doi: 10.1155/NP.1991.207
329. Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA. (2000) 97:14720–5. doi: 10.1073/pnas.97.26.14720
330. Pollock K, Stroemer P, Patel S, Stevanato L, Hope A, Miljan E, et al. A conditionally immortal clonal stem cell line from human cortical neuroepithelium for the treatment of ischemic stroke. Exp Neurol. (2006) 199:143–55. doi: 10.1016/j.expneurol.2005.12.011
331. Guo X, Johe K, Molnar P, Davis H, Hickman J. Characterization of a human fetal spinal cord stem cell line, NSI-566RSC, and its induction to functional motoneurons. J Tissue Eng Regen Med. (2010) 4:181–93. doi: 10.1002/term.223
332. Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa ASH, Kwok CK, Leonardi T, et al. Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem Cell (2018) 22:355-368 e313. doi: 10.1016/j.stem.2018.01.020
333. Nagoshi N, Khazaei M, Ahlfors JE, Ahuja CS, Nori S, Wang J, et al. Human spinal oligodendrogenic neural progenitor cells promote functional recovery after spinal cord injury by axonal remyelination and tissue sparing. Stem Cells Transl Med. (2018) 7:806–18. doi: 10.1002/sctm.17-0269
334. Galipeau J, Sensebe L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell (2018) 22:824–33. doi: 10.1016/j.stem.2018.05.004
335. Rolfe A, Sun D. Stem cell therapy in brain trauma: implications for repair and regeneration of injured brain in experimental TBI models In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton, FL: CRC Press/Taylor & Francis (2015).
336. Tennstaedt A, Aswendt M, Adamczak J, Collienne U, Selt M, Schneider G, et al. Human neural stem cell intracerebral grafts show spontaneous early neuronal differentiation after several weeks. Biomaterials (2015) 44:143–54. doi: 10.1016/j.biomaterials.2014.12.038
337. Falkner S, Grade S, Dimou L, Conzelmann KK, Bonhoeffer T, Gotz M, et al. Transplanted embryonic neurons integrate into adult neocortical circuits. Nature (2016) 539:248–53. doi: 10.1038/nature20113
338. Grade S, Gotz M. Neuronal replacement therapy: previous achievements and challenges ahead. NPJ Regen Med. (2017) 2:29. doi: 10.1038/s41536-017-0033-0
339. Rosenzweig ES, Brock JH, Lu P, Kumamaru H, Salegio EA, Kadoya K, et al. Restorative effects of human neural stem cell grafts on the primate spinal cord. Nat Med. (2018) 24:484–90. doi: 10.1038/nm.4502
340. Haus DL, Lopez-Velazquez L, Gold EM, Cunningham KM, Perez H, Anderson AJ, et al. Transplantation of human neural stem cells restores cognition in an immunodeficient rodent model of traumatic brain injury. Exp Neurol. (2016) 281:1–16. doi: 10.1016/j.expneurol.2016.04.008
341. Corso P, Finkelstein E, Miller T, Fiebelkorn I, Zaloshnja E. Incidence and lifetime costs of injuries in the United States. Inj Prev. (2006) 12:212–8. doi: 10.1136/ip.2005.010983
342. McGinley LM, Kashlan ON, Bruno ES, Chen KS, Hayes JM, Kashlan SR, et al. Human neural stem cell transplantation improves cognition in a murine model of Alzheimer's disease. Sci Rep. (2018) 8:14776. doi: 10.1038/s41598-018-33017-6
343. Brem SS, Bierman PJ, Black P, Blumenthal DT, Brem H, Chamberlain MC, et al. Central nervous system cancers: clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2005) 3:644–90. doi: 10.6004/jnccn.2005.0038
344. Metz MZ, Gutova M, Lacey SF, Abramyants Y, Vo T, Gilchrist M, et al. Neural stem cell-mediated delivery of irinotecan-activating carboxylesterases to glioma: implications for clinical use. Stem Cells Transl Med. (2013) 2:983–92. doi: 10.5966/sctm.2012-0177
345. Portnow J, Synold TW, Badie B, Tirughana R, Lacey SF, D'Apuzzo M, et al. Neural stem cell-based anticancer gene therapy: a first-in-human study in recurrent high-grade glioma patients. Clin Cancer Res. (2017) 23:2951–60. doi: 10.1158/1078-0432.CCR-16-1518
346. Goutman SA, Brown MB, Glass JD, Boulis NM, Johe K, Hazel T, et al. Long-term Phase 1/2 intraspinal stem cell transplantationoutcomes in ALS. Ann Clin Trans Neurol. (2018) 5:730–40. doi: 10.1002/acn3.567
347. Curtis E, Martin JR, Gabel B, Sidhu N, Rzesiewicz TK, Mandeville R, et al. A first-in-human, phase I study of neural stem cell transplantation for chronic spinal cord injury. Cell Stem Cell (2018) 22:941–50 e946. doi: 10.1016/j.stem.2018.05.014
348. Selden NR, Al-Uzri A, Huhn SL, Koch TK, Sikora DM, Nguyen-Driver MD, et al. Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J Neurosurg Pediatr. (2013) 11:643–52. doi: 10.3171/2013.3.PEDS12397
349. Ghobrial GM, Anderson KD, Dididze M, Martinez-Barrizonte J, Sunn GH, Gant KL, et al. Human neural stem cell transplantation in chronic cervical spinal cord injury: functional outcomes at 12 months in a phase II clinical trial. Neurosurgery (2017) 64(CN_suppl_1):87–91. doi: 10.1093/neuros/nyx242
350. Levi AD, Anderson KD, Okonkwo DO, Park P, Bryce T, Kurpad SN, et al. Clinical outcomes from a multi-center study of human neural stem cell transplantation in chronic cervical spinal cord injury. J Neurotrauma (2018). doi: 10.1089/neu.2018.5843 [Epub ahead of print].
351. Moore SF, Guzman NV, Mason SL, Williams-Gray CH, Barker RA. Which patients with Parkinson's disease participate in clinical trials? One centre's experiences with a new cell based therapy trial (TRANSEURO). J Parkinsons Dis. (2014) 4:671–6. doi: 10.3233/JPD-140432
352. Kirkeby A, Parmar M, Barker RA. Strategies for bringing stem cell-derived dopamine neurons to the clinic: a European approach (STEM-PD). Prog Brain Res. (2017) 230:165–90. doi: 10.1016/bs.pbr.2016.11.011
353. Mazzini L, Gelati M, Profico DC, Sgaravizzi G, Projetti Pensi M, Muzi G, et al. Human neural stem cell transplantation in ALS: initial results from a phase I trial. J Transl Med. (2015) 13:17. doi: 10.1186/s12967-014-0371-2
354. Feldman EL, Boulis NM, Hur J, Johe K, Rutkove SB, Federici T, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann Neurol. (2014) 75:363–73. doi: 10.1002/ana.24113
355. Amariglio N, Hirshberg A, Scheithauer BW, Cohen Y, Loewenthal R, Trakhtenbrot L, et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. (2009) 6:e1000029. doi: 10.1371/journal.pmed.1000029
356. Berkowitz AL, Miller MB, Mir SA, Cagney D, Chavakula V, Guleria I, et al. Glioproliferative lesion of the spinal cord as a complication of “stem-cell tourism”. N Engl J Med. (2016) 375:196–8. doi: 10.1056/NEJMc1600188
357. Saraf SS, Cunningham MA, Kuriyan AE, Read SP, Rosenfeld PJ, Flynn HW Jr, et al. Bilateral retinal detachments after intravitreal injection of adipose-derived 'stem cells' in a patient with exudative macular degeneration. Ophthalmic Surg Lasers Imaging Retina (2017) 48:772–5. doi: 10.3928/23258160-20170829-16
358. Julian K, Yuhasz N, Hollingsworth E, Imitola J. The “growing” reality of the neurological complications of global “stem cell tourism”. Semin Neurol. (2018) 38:176–81. doi: 10.1055/s-0038-1649338
359. Thirabanjasak D, Tantiwongse K, Thorner PS. Angiomyeloproliferative lesions following autologous stem cell therapy. J Am Soc Nephrol. (2010) 21:1218–22. doi: 10.1681/ASN.2009111156
360. Kuriyan AE, Albini TA, Townsend JH, Rodriguez M, Pandya HK, Leonard RE II, et al. Vision loss after intravitreal injection of autologous “stem cells” for AMD. N Engl J Med. (2017) 376:1047–53. doi: 10.1056/NEJMoa1609583
361. Leung EH, Flynn HWJr, Albini TA, Medina CA. Retinal detachment after subretinal stem cell transplantation. Ophthalmic Surg Lasers Imaging Retina (2016) 47:600–1. doi: 10.3928/23258160-20160601-16
Keywords: traumatic brain injury, inflammasome, pyroptosis, neural stem cell, cell transplantation
Citation: Kassi AAY, Mahavadi AK, Clavijo A, Caliz D, Lee SW, Ahmed AI, Yokobori S, Hu Z, Spurlock MS, Wasserman JM, Rivera KN, Nodal S, Powell HR, Di L, Torres R, Leung LY, Rubiano AM, Bullock RM and Gajavelli S (2019) Enduring Neuroprotective Effect of Subacute Neural Stem Cell Transplantation After Penetrating TBI. Front. Neurol. 9:1097. doi: 10.3389/fneur.2018.01097
Received: 11 June 2018; Accepted: 03 December 2018;
Published: 17 January 2019.
Edited by:
Stefania Mondello, Università degli Studi di Messina, ItalyReviewed by:
Yumin Zhang, Uniformed Services University of the Health Sciences, United StatesIbrahim Jalloh, University of Cambridge, United Kingdom
Copyright © 2019 Kassi, Mahavadi, Clavijo, Caliz, Lee, Ahmed, Yokobori, Hu, Spurlock, Wasserman, Rivera, Nodal, Powell, Di, Torres, Leung, Rubiano, Bullock and Gajavelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shyam Gajavelli, c2dhamF2ZWxAbWVkLm1pYW1pLmVkdQ==