 Nadja Borisow
Nadja Borisow Masahiro Mori2
Masahiro Mori2 Friedemann Paul
Friedemann Paul- 1NeuroCure Clinical Research Center, Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin, Germany
- 2Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan
- 3Department of Neuroradiology, Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin, Germany
- 4Experimental and Clinical Research Center, Max Delbrueck Center for Molecular Medicine and Charité – Universitätsmedizin Berlin, Berlin, Germany
Neuromyelitis optica spectrum disorders (NMOSD) are autoantibody mediated chronic inflammatory diseases. Serum antibodies (Abs) against the aquaporin-4 water channel lead to recurrent attacks of optic neuritis, myelitis and/or brainstem syndromes. In some patients with symptoms of NMOSD, no AQP4-Abs but Abs against myelin-oligodendrocyte-glycoprotein (MOG) are detectable. These clinical syndromes are now frequently referred to as “MOG-encephalomyelitis” (MOG-EM). Here we give an overview on current recommendations concerning diagnosis of NMOSD and MOG-EM. These include antibody and further laboratory testing, MR imaging and optical coherence tomography. We discuss therapeutic options of acute attacks as well as longterm immunosuppressive treatment, including azathioprine, rituximab, and immunoglobulins.
Introduction
Neuromyelitis optica spectrum disorders (NMOSD) are rare chronic inflammatory central nervous system diseases distinct from multiple sclerosis (MS). The French term “neuro-myélite optique aiguë,” which may be translated as “neuromyelitis optica acuta” was first used by Devic in 1894 (1, 2). In the majority of patients with NMOSD, autoantibodies (Abs) against the astrocyte aquaporin-4 (AQP4) water channel are detectable and patients typically suffer from recurrent attacks of severe optic neuritis or/and myelitis (3–7). In rarer cases, brainstem and brain involvement e.g., area postrema syndrome or diencephalic syndrome can occur (8, 9). Patients also frequently suffer from burdensome symptoms like pain, headache, depression, fatigue, and sleep disorders (10–14). Despite treatment, recovery from attacks is often incomplete and disease remission rarely occurs (15, 16). Thus, in relapsing NMOSD, which account for approximately 80–85% of cases, neurologic deficits frequently accumulate during the disease course. Patients without long-term immunosuppressive therapy have a worse prognosis with a higher mortality rate (17). Disease onset ranges between 4 and 88 years with a mean age at onset of 39 years (18–21). Women are disproportionately more often affected and, particularly in AQP4-seropositive patients, female to male-ratio can reach up to 10:1 (19, 22, 23). In 20–30% of patients, depending on the assay used, AQP4-Abs are not detectable (24, 25). Whether AQP4-Ab positive and AQP4-Ab negative diseases are varieties of the same disorder or rather reflect different disease entities is a topic of ongoing research (26–28).
Recently, various publications described the detection of serum-Abs against myelin-oligodendrocyte-glycoprotein (MOG) in AQP4-Ab negative NMOSD patients including pediatric cohorts and few patients with MS (29–41). In the past, MOG-Abs were particularly described in acute disseminated encephalomyelitis (ADEM), an inflammatory CNS disorder that, if it has an pediatric onset, is mostly monophasic and has a favorable outcome in the majority of cases (42, 43). MOG is a glycoprotein localized on the surface of the myelin sheath as well as of the cell body and processes of oligodendrocytes (44, 45). According to the revised 2015 NMOSD diagnostic criteria (46), diseases with or without evidence of AQP4-Abs as well as disorders with MOG-Abs can be assigned to the NMO spectrum. Although there are numerous overlaps in clinical presentation and imaging findings with NMOSD with and without AQP4-Ab, MOG-Ab-associated disease is more and more considered a disease entity in its own (47). Previous studies on NMOSD might have included patients with MOG-Abs and therefore overlapping features could have been reported in these studies. Various terms are used to describe the disease such as “MOG-antibody related disorder,” “MOG-associated disease,” “MOG antibody disease,” “MONEM” or “MOG-encephalomyelitis (MOG-EM)”(40, 47–50). Hereafter, we use the term “MOG-EM,” as it reflects the relevant symptoms of the disease and is used in several recent publications, e.g., (49). Although ADEM can also be accompanied by MOG-Abs (51), in this manuscript we do not regard MOG-Ab positive patients with ADEM-phenotype as part of the “MOG-EM” due to their distinct clinical characteristics. To date, the relevance of MOG-Abs and their nosologic categorization is a topic of current discussion and under further investigation (47, 52, 53).
To give an overview on diagnosis and treatment recommendations in NMOSD and MOG-EM, we here describe our own clinical experiences and give a review on the current literature using the Pubmed online database. We used the search terms “neuromyelitis optica,” “neuromyelitis optica spectrum disorder,” “MOG,” aquaporin-4 antibodies,” “MRI,” “diagnostic criteria,” “therapy,” and combinations of these. To find all relevant publications, we did not restrict the year of publication; however, most reports originate from the last 5 years.
Diagnosis
In NMOSD and MOG-EM, most common symptoms are optic neuritis and longitudinally extensive transverse myelitis (LETM). Signs of brainstem affection like persistent hiccup, nausea or vomiting should explicitly be asked for as they are often attributed to other reasons and are therefore not reported spontaneously by the patient. Rarer clinical manifestations of NMOSD comprise narcolepsy, acute diencephalic syndrome or muscle affection (54, 55), while in MOG-EM extraneural involvement such as reversible paraspinal muscle hyperintensity have been described, as well as MOG-Abs in combined central and peripheral demyelination syndromes (56, 57).
Like NMOSD, MOG-EM can affect optic nerve, spinal cord, and brainstem. However, some studies showed histopathological differences between NMOSD and MOG-EM (58, 59). AQP4-Abs bind to water channels located on astrocytes, whereas MOG-Abs target myelin-forming oligodendrocytes (53). Both types of antibodies may lead to disturbances of the integrity of blood brain barrier and to CNS inflammation (53, 60). However, while inflammation in MOG-EM primarily results in demyelination, demyelination in NMOSD seems to be a secondary phenomenon following astrocytic damage (61, 62).
In patients with AQP4-Abs, the most frequent symptoms at onset are optic neuritis in 37-54% of the patients, and LETM in 30–47% of the patients (26, 63, 64). In patients with MOG-Abs, optic neuritis was the first clinical manifestation in 33–64% whereas myelitis occurred in 18–33% of the patients as initial symptom (33, 48, 65). Also during the further course of the disease, optic neuritis seems to be more frequent in MOG-EM than in NMOSD with myelitis being less common (29, 66). However, in population-based ON studies and unselected cohorts of patients with ON, both the prevalence of AQP4-Abs and MOG-Abs is low (67–69). In MOG-EM, cases of encephalitis and seizures were described whereas these symptoms are rare in NMOSD (70–72). MOG-EM differs from NMOSD in further clinical characteristics e.g., in gender ratio and age at onset. In (relapsing) NMOSD, up to 90% of the patients are female, whereas the proportion of male patients in MOG-EM ranges from 43 to 63% (22, 26, 29–31, 73). The published mean age at onset ranges from 27 and 37 years in patients with MOG-EM (29–31, 73) and between 30 and 46 years for patients with NMOSD (19, 26, 29–31, 73). At onset, patients with MOG-Abs are more likely to suffer from simultaneous or rapidly sequential optic neuritis and LETM compared to patients with AQP4-Abs (31). In AQP4-Ab positive NMOSD, most patients (80–90%) have a relapsing disease course (26, 73, 74). In MOG-EM, monophasic disease course is considered to be more frequent, however, the duration of follow-up and a referral bias might have influenced these results (33, 73–76). Some studies showed lower disability outcomes, measured by the Expanded Disability Status Scale (EDSS), in MOG-EM than in NMOSD, suggesting a presumably more favorable prognosis (29–31, 73). However, long-term data from MOG-EM are scant. Whereas spinal cord lesions frequently affect cervicothoracic segments in NMOSD, they tend to be localized in thoracolumbar parts of the spinal cord including the conus in MOG-EM (29, 31). Table 1 summarizes the epidemiological and clinical features in NMOSD and MOG-EM.
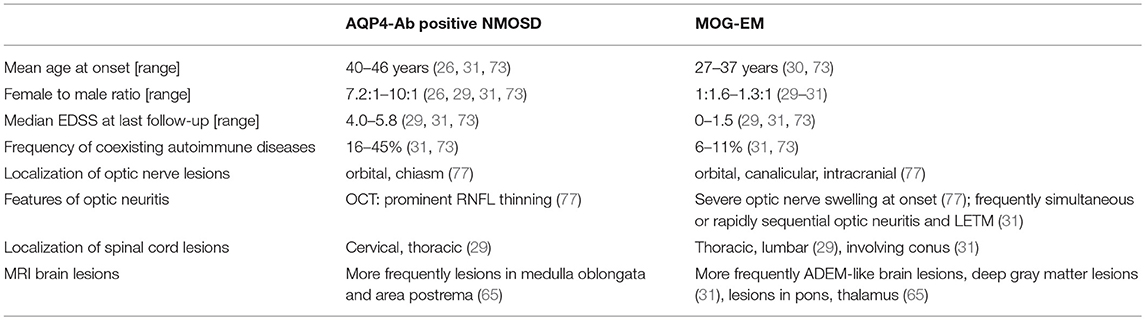
Table 1. Epidemiological and clinical features in NMOSD and MOG-EM.
Antibody Diagnosis
A central component of diagnostics in NMOSD and MOG-EM is the detection of Abs in serum. AQP4-Abs were firstly described in 2004 and made it possible to differentiate NMOSD from MS (78). The best detection rates are provided by cell-based assays (CBA) (24, 32, 79, 80). In NMOSD, the sensitivity of these assays ranges between 80 and 100%, whereas specificity varies between 86 and 100% (24). Contrarily, enzyme-linked immunosorbent assays (ELISA) may lead to false-positive results and should not be used as sole method (81–83).
Specific antibodies against MOG are detectable in pediatric patients with acute disseminated encephalomyelitis (ADEM) (84–86). In MS patients, MOG-Abs were described for the first time at the beginning of the 1990s (87). Further studies confirmed these findings (88–90). Later, MOG-Abs were found in AQP4-Ab negative patients with clinical symptoms of NMOSD (91, 92). Like in NMOSD, CBA are the current gold standard to detect MOG-Abs (39, 49). Formerly used assays had a low MOG specificity, which led to high rates of false positive results (39, 49). Therefore to date, cell-based assays targeting at full-length human MOG and the use of IgG1-specific secondary antibodies is highly recommended to avoid cross-reactivity with IgM and IgA antibodies (39, 49).
AQP4- and MOG-serostatus and Ab-level may change during the disease course. In patients with suspected NMOSD or MOG-EM without initial evidence for seropositivity, further Ab-testing may be required during the course of the disease, especially during acute attacks and intervals without treatment. AQP4-Abs usually stay detectable during remission, although the titer may be lower with immunosuppression (some patients even seroconvert to negative over time) and during acute attacks (93). In MOG-EM, approximately 80% of patients with evidence of MOG-Abs during acute attack remained seropositive during remission (33). However, the rate was lower with only 50% of patients remaining seropositive in a study from Korea (76). As in AQP4-Ab positive NMOSD, MOG-Ab serum titers are significantly higher during acute attack than during remission (33). In some studies, Ab-titers were associated with relapses and treatment status (32, 93, 94). However, the level of AQP4-titer does not seem to be predictive for long term disease course (95), and AQP4-Ab serostatus is not predictive of response to immunotherapy (96). Testing of patients with progressive MS for MOG antibodies is not warranted under most circumstances (97).
Testing CSF for AQP4- or MOG Abs is not routinely recommended as it does not seem to provide an additional benefit for diagnosing NMOSD or MOG-EM (98, 99). AQP4-Abs in CSF can be detected in only 70% of Aqp4-Ab seropositive patients and in none of the AQP4 seronegative patients (100). Like AQP4-Abs, MOG-Abs are produced mainly extrathecally and are therefore less frequent in CSF than in serum (32).
Comorbidity with other autoimmune disorders is frequent in NMOSD patients (101–105). Therefore, further tests for autoantibodies should comprise Abs associated with rheumatologic diseases e.g., ANA, ANCA, Anti-ds-DNA-Abs, and lupus anticoagulant. If there are clinical signs in anamnesis or examination, Ab testing for myasthenia gravis, coeliac disease, or paraneoplastic disorders should be performed (101, 106–110). In MOG-EM, the frequency of coexistent autoimmune diseases seems to be lower than reported for AQP4-Ab positive patients (33, 66).
Further Laboratory Diagnosis
Other laboratory tests are recommended to diagnose coexisting autoimmune disorders and to exclude other differential diagnoses. Next to routine laboratory tests, this includes differential blood count, blood sedimentation, folic acid, and vitamin B12 (111). To exclude sarcoidosis which is a relevant differential diagnosis as it can also manifest with optic neuropathy or myelopathy (112, 113), tests on hypercalcemia and hypercalciuria, interleukin-2-rezeptor (sIL-2 R), and angiotensin-converting enzyme (ACE) should be performed (112, 114).
Analysis of cerebrospinal fluid (CSF) might be helpful to exclude other diagnoses, especially to differentiate between NMOSD/MOG-EM and MS. In NMOSD, white cell counts were elevated in up to 50% of the patients, especially during acute attack, and in approximately 10% of the patients CSF-restricted oligoclonal IgG bands (OCB) can be detected (73, 100). Increased CSF/serum albumin ratio as a marker of dysfunction of blood brain barrier was found in 51% of NMOSD patients (100).
In MOG-EM, elevated white cell counts were found in 25–70% of the patients, whereas there was no differentiation between tests during acute attack and remission. (33, 66, 73). Like in NMOSD, OCB were detected in 10% of the MOG-EM patients and CSF/serum albumin ratio was elevated in 32% (33, 66, 73).
Magnetic Resonance Imaging
Next to the AQP4-Abs, MRI is an essential element to diagnose NMOSD. It helps to differentiate NMOSD from MS and other CNS disorders (115).
Spinal cord imaging was already included in the 2006 NMO diagnostic criteria (116). These criteria require MRI spinal cord lesion extending over ≥3 vertebral segments (116). However, in 15 percent of myelitis attacks, spinal cord lesions do not extend over ≥3 vertebral segments which may lead to misdiagnosis or delayed diagnosis of NMOSD (117). Typical NMOSD lesions are located centrally in the spinal cord and involve more than the half of spinal cord cross-section area (118). It was suggested by Yonezu et al.(119) that “bright spotty lesions” are characteristic for NMOSD and might reflect microcystic defects of the spinal cord (113). The specificity of this sign however still needs to be confirmed in further studies. The interval between clinical symptoms and the MRI is influencing the MRI presentation of LETM lesions. They may not be present from relapse onset and may change into multiple short lesions or into spinal cord atrophy during the disease course (120, 121). Hence, there is the risk miss a typical MRI presentation of the LETM when the MRI is performed too early or too late (72). Other causes for longitudinally extensive spinal cord LETM lesions include sarcoidosis or spondylotic myelopathy or rarely MS and need to be considered (112, 122, 123). In addition longitudinally extensive myelitis lesions were recently described in patients with symptoms of meningitis, encephalitis and/or myelitis that were tested positive for glial fibrillary acidic protein (GFAP)-IgG (124–126).
Brain MRI at first presentation often shows no lesions which has been the reason to define normal brain MRI as one NMO diagnostic criterion in 2006 (116). However, more recent studies showed that the presence of cerebral lesions is not uncommon in the clinical course of NMOSD (127–129). Hence, the NMOSD 2015 diagnostic criteria have incorporated findings of cerebral MRI and define NMOSD-typical brain lesions (46). These lesions can be located at the periependymal surfaces of the third and fourth ventricle, in the area postrema, corpus callosum, hypothalamus or thalamus (130–132). In addition, subcortical or deep white matter lesions are possible. Meningeal enhancement has been reported in some cases, although this does not appear to be a very frequent imaging finding in NMOSD (133). Orbital MRI may show increased T2 signal and gadolinium enhancement of the optic nerve as signs of an optic neuritis. This can be helpful to diagnose MOG-EM or NMOSD in patients without AQP4-Abs (46, 77, 131, 134, 135). Chiasmal involvement is more common in AQP4-NMOSD than in MOG-EM (134).
A study by Ramanathan et al. showed no MRI brain lesions in a large proportion of MOG-EM patients (66). Conversely, other authors found supra- and infratentorial MRI abnormalities in 40–50% of the patients (33, 65). Brain imaging allows to distinguish MOG-EM from MS, but shows many overlaps with AQP4-Ab NMOSD (136–138). Moreover, a relevant number of patients show pathologic findings in MRI of optic nerve and spinal cord, comparable to NMOSD patients (33, 74). However, one study revealed a more frequent occurrence of optic nerve head swelling and retrobulbar affection of the optic nerve in MOG-EM compared to NMOSD (134).
Figure 1 shows MRI features of NMOSD and MOG-EM. Studies investigating non-conventional MR imaging in NMOSD will not be reviewed further as they currently lack implications for clinical management (139–141).
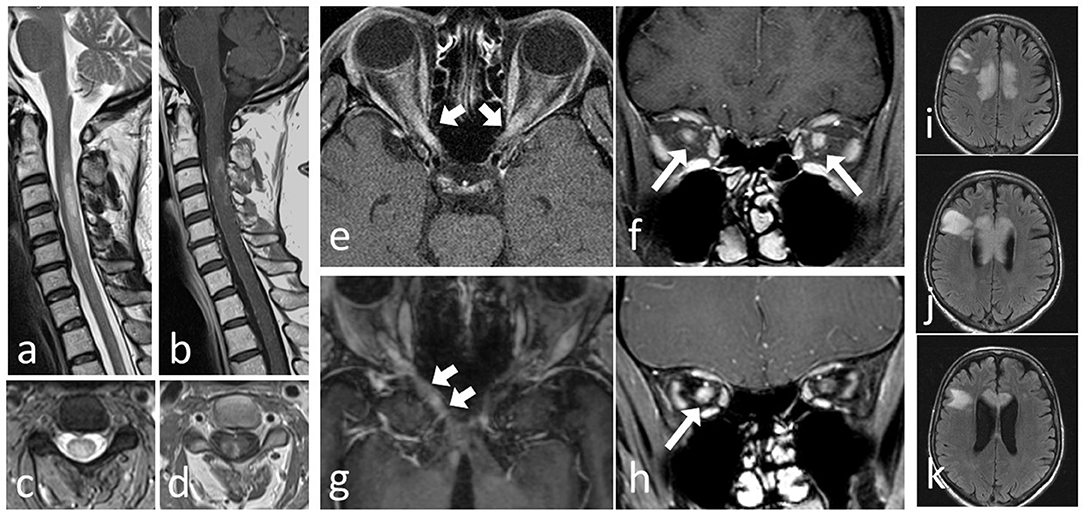
Figure 1. MRI of patients with AQP4-Ab positive NMOSD and patients with MOG-EM. Severe cervical LETM in a NMOSD AQP+ patient: (a) T2 sagittal and (b) T2 axial of a cervical myelon lesion with ring Gd-Enhancement and T1 hypointense center in (c) T1+Gd sagittal and (d) T1+Gd axial. Bilateral opticusneuritis in a MOG-EM patient (e) T1+Gd axial and (f) T1+Gd coronar. Unilateral optic neuritis with chiasmal involvement in a NMOSD AQP4+ patient: (g) T1+Gd axial and (h) T1+Gd coronar. Tumefactive lesion involving the corpus callosum in a NMOSD AQP4+ patient (i–k) T2 axial.
Optical Coherence Tomography
Optical coherence tomography is an interferometric technique using near infra-red backscattered light to generate high resolution images of the retina and its various layers, that is increasingly applied in various neuroimmunological disorders (142–148). OCT displays severe damage to the retinal nerve fiber layer and the ganglion cell layer following attacks of optic neuritis in both AQP4 NMOSD and MOG-EM that correlates with visual function and quality of life (149–158). It is currently a matter of debate if retinal damage following optic neuritis is equally severe in AQP4-NMOSD and MOG-EM (75, 77, 152, 159–162) and to which extent structural retinal alterations occur in NMOSD independently of optic neuritis attacks (143, 157, 163–167). Although the utility of OCT in patient management requires further investigation, it may help quantify the extent of structural retinal damage following optic neuritis attacks and thus hopefully inform treatment decisions (168–170), and support differential diagnosis in the near future.
Diagnostic Criteria
Current NMOSD diagnostic criteria were published by the International Panel for NMO Diagnosis in 2015 (46) and were aimed at taking recent advances in the field following the 2006 Wingerchuk criteria into consideration (116). They differentiate between NMOSD with AQP4-Abs and NMOSD without AQP4-Abs or unknown AQP4-Ab status.
In the case of positive AQP4-Ab status, one of the following clinical core symptoms is required:
1. Optic neuritis
2. Acute myelitis
3. Area postrema syndrome: episode of otherwise unexplained hiccups or nausea and vomiting
4. Acute brainstem syndrome
5. Symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions
6. Symptomatic cerebral syndrome with NMOSD-typical brain lesions
NMOSD-typical brain lesions may involve the dorsal medulla, especially the area postrema, the periependymal surfaces of the third or fourth ventricle, the hypothalamus, thalamus, the corpus callosum, cerebral peduncles, and the internal capsule. Moreover, subcortical or deep white matter lesions and corticospinal tract lesions are possible. Alternative diagnoses e.g., MS, sarcoidosis, infectious or neoplastic diseases have to be excluded.
In patients without evidence of AQP4-Ab two of the above mentioned core clinical characteristics are necessary for NMOSD diagnosis. At least one of these core clinical characteristics has to be ON, LETM or area postrema syndrome. Moreover, supportive characteristics in cerebral, spinal cord or optic nerve MRI are required. These are
- normal brain MRI or long optic nerve lesions with increased T2 signal or gadolinium enhancement of the optic nerve or the chiasm in patients with ON,
- spinal cord MRI lesion or focal spinal cord atrophy extending over ≥3 segments in patients with myelitis and
- lesions involving dorsal medulla oblongata/area postrema in patients with area postrema syndrome
- periependymal brainstem lesions in patients with acute brainstem syndrome.
Using the 2015 instead of the 2006 criteria led to a significant increase in the number of patients diagnosed with NMOSD (138, 171, 172)
For MOG-EM, to date, no evidence based diagnostic criteria exist. However, NMOSD 2015 diagnostic criteria allow to include cases of NMOSD associated with other specific autoantibodies (46).
Treatment of Acute Attacks
In NMOSD as well as in MOG-EM, acute attacks are usually treated with 1,000 mg intravenous methylprednisolone (IVMP) for 3–5 days. Jarius et al. showed complete or almost complete recovery in 50% of IVMP treated MOG-EM attacks (33). In NMOSD, IVMP led to complete recovery in 17–35% of the attacks (15, 173). In case of poor response, treatment escalation with 2,000 mg IVMP may improve outcome, for further therapy escalation plasma exchange (PLEX) or immunoadsorption are possible (15, 173–175). PLEX and immunoadsorption did not show a difference in their efficacy in the therapy of NMOSD attacks (176). They can also be used as first-line therapy (in particular in myelitis attacks) if response to methylprednisolone during previous attacks was poor. An early initiation of PLEX seems to improve the clinical outcome (176, 177).
Preventative Immunosuppressive Therapy
Attacks in NMOSD as well as in MOG-EM are often characterized by severe neurologic deficits with poor recovery. Frequently, a relevant disability persists after an attack. However, there are indications that MOG-EM has a less severe course than NMOSD and relapse risk depends on Ab status (30, 65, 66). In some patients with evidence of MOG-Abs, seroconversion to an Ab- negative status may occur during the disease course (30, 32, 76).
There is increasing evidence that immunosuppressive therapy is essential to reduce disease activity and to avoid further attacks. However, to date no placebo controlled trial has been published and only one open randomized clinical trial has been performed (178). Thus, the current treatment paradigm is based on case series, (retrospective) observational studies as well as expert opinion. Hereafter, we describe the to-date used treatments in NMOSD and MOG-EM (179).
Low Dose Prednisone/Prednisolone
Low dose oral corticosteroids are used in many neurologic diseases. Oral prednisone/prednisolone can be given subsequent to attack therapy with IVMP in decreasing dose levels and as comedication during the first months of azathioprine (AZA) or mycophenolate mofetil (MMF) treatment until these drugs exert their full efficacy. Possible side effects are weight gain, hypertension, thrombosis, osteoporosis, fungal and viral infections, hyperglycemia, gastritis and peptic ulcer, psychiatric disturbances and a Cushing syndrome (180).
Data on long-term treatment with oral prednisone/prednisolone in NMOSD are limited. A few studies could show a decrease in ARR by low dose steroid therapy (181, 182). Moreover, it is known from treatment experiences with AZA that additional oral prednisone is effective to reduce disease activity during the first 3–6 months until AZA reaches its full efficacy.
In MOG-EM, low treatment failure rates were achieved with oral prednisone (66). The occurrence of relapses during tapering or after cessation of subsequent oral prednisone after IVMP attack treatment supports the beneficial effects of corticosteroid therapy in MOG-EM (33), at least in patients with persistence of MOG Abs (66). However, due to the known side effects and the existence of other treatment alternatives, a long-term therapy with low dose prednisone should be critically weighed.
Azathioprine
AZA is a purine analog, acts as antimetabolite and inhibits the differentiation of lymphocytes. Thereby it has antiproliferative and immunosuppressive effects. It is administered in a dose of 2–3 mg/kg body weight per day and reaches its full effectiveness after 3–6 months. During the initial period, additional oral prednisone [1 mg/kg/day] is necessary and can be slowly tapered when AZA becomes fully effective.
The most important side effect is a bone marrow depression with anemia, leuko- and/or thrombopenia. The risk of bacterial, viral or fungal infection is increased. Moreover, elevation of liver enzymes, nausea or emesis can appear. Rare side effects especially after long treatment duration include malignomas e.g., of the skin, and a progressive multifocal leukoencephalopathy (PML). Furthermore, add-on therapy with prednisone enhances the risk of side effects, like a diabetogenic metabolic state, thrombosis or psychiatric symptoms.
Patients with a congenital deficiency of thiopurinmethyltransferase (TPMT), an enzyme responsible for metabolisation of AZA, have a high risk of bone marrow depression. Therefore, it is recommended to test for TPMT-deficiency in patients with pronounced deterioration of blood count after initiation of AZA-therapy.
A recently published prospective randomized controlled trial compared the efficacy of AZA and rituximab (RTX) in NMOSD. It showed a significant decrease in mean ARR from 1 to 0.51 and a decrease in mean EDSS from 2.40 to 1.95 by AZA (178). 54% of the patient treated with AZA became relapse free after 1 year (178). A prospective study including 77 NMOSD patients (183) and other retrospective studies (181, 184–186) showed comparable results.
In a study by Jarius et al. 14 out of 17 MOG-patients (82%) suffered from at least one attack while treated with AZA (33). Attacks occurred mainly in patients that were not co-treated with oral prednisone and during the first 6 months. This highlights the need for co-treatment with oral prednisone until AZA reaches its full efficacy.
Rituximab
RTX is a monoclonal Ab directed against the surface molecule CD20 on B-lymphocytes. RTX leads to a depletion of CD20+B-lymphocytes, which act as precursor cells of antibody producing plasma cells (187). A thereby triggered reduction of antibody formation is presumably the RTX mechanism of action.
The most frequently used dose regimen is the intravenous administration of each 1,000mg with an interval of 2 weeks followed by 6-monthly dosages of 1,000mg (179, 188). Alternatively, initially 375 mg/m2 body surface every week over a period of 4 weeks can be administered. As an alternative to a fixed dosage regimen every 6 months, monitoring of CD19+/CD20+ B-lymphocytes and administration of RTX in the case of reconstitution of these cells is possible (189, 190). Another option is the administration of RTX depending on monitoring of CD27+ memory B-cells which might in some cases allow to lower the cumulative RTX dose (191). An evidence that one of these regimens has therapeutic superiority over the other does not exist to date.
Before first administration, active infections like tuberculosis or hepatitis B have to be excluded (192). An update of vaccination status and anti-pneumococcal vaccination is recommended (192).
Side effects include infusion-related symptoms like pruritus, headache, rash or fever. To reduce the risk of these symptoms, a premedication with an analgesic/antipyretic and an antihistamine is recommended. The risk of infections and severe skin reactions like the Lyell-syndrome or the Stevens-Johnson-Syndrome is elevated. Cardiac symptoms e.g., arrhythmia or cardiac insufficiency were reported (193). Moreover, neurologists must be aware of hypogammaglobulinemia that may occur with long-term RTX treatment (194).
In 2005, an open label study described for the first time a significant reduction in disease activity in eight NMO patients treated with RTX (195). Since then, an increasing number of patients was treated with RTX. However, to date only a few prospective studies investigating the effect of RTX on NMOSD exist. The above mentioned study by Nikoo et al. showed a reduction of the ARR by 83 percent as mean ARR decreased from 1.30 to 0.21 (178). Mean EDSS decreased from 3.55 to 2.56. Other prospective and retrospective trials found significant reductions of ARR to values between 0.1 and 0.46 in adult and pediatric patients treated with RTX (181, 196–202). A further overview on efficacy and safety profile of RTX in NMOSD is given in the topical literature (203–205).
AZA and RTX are the most frequently used immunosuppressants in NMOSD. With regards to ARR and EDSS, comparison studies between both drugs seem to suggest a superiority of RTX compared to AZA (178, 184). Thus, currently RTX seems to be the most effective treatment in NMOSD, although some studies describe a rebound in disease activity shortly after RTX induction (199, 206). Treatment effect does not seem to depend on AQP4-serostatus (96).
In MOG-EM, treatment with RTX led to a decline in relapse rate in only 3 out of 9 patients (33). Most of the attacks occurred shortly after RTX infusion. Some authors recommend RTX as second-line-therapy if preventative treatment with low-dose prednisone or monthly intravenous immunoglobulins (IVIG) is not effective (66). In patients with myelitis, RTX is recommended from an earlier stage as a myelitis often leads to severe residual deficits (66). Whether RTX is indeed less effective in MOG-EM than in NMOSD has to be analyzed in further studies (207).
Mycophenolate Mofetil
MMF is an immunosuppressant that inhibits the inosine monophosphate dehydrogenase. Thereby, the synthesis of guanosin nucleotide and subsequently, the proliferation of B- and T-lymphocytes is inhibited. The administered daily dose ranges between 750 and 3,000 mg/d (179, 208, 209).
The most common side effects are leucopenia, diarrhea, vomiting and sepsis. The risk of malignomas can be increased especially if MMF is combined with other immunosuppressants.
A retrospective observational study investigated the effect of MMF in NMOSD and MOG-EM. 33/67 (49%) of the patients were relapse-free, in 44/53 (83%) the EDSS improved or stabilized (208). Other observational studies showed similar results with proportions of relapse-free patients between 56 and 60% in NMOSD (210, 211) In comparison to AZA, MMF showed fewer side effects with equal efficacy (211, 212). As in treatment with RTX, response to MMF does not differ in dependence on AQP4-serostatus (96).
In MOG-EM patients, a combined therapy with MMF and steroids appeared to have a positive effect; however, this effect diminished after steroid tapering (66). As MMF may take several months to reach its full efficacy, add-on prednisone should be tapered only very slowly.
Intravenous Immunoglobulins
Even less data is available for treatment with IVIG in NMOSD. A small retrospective study including six patients with NMO/NMOSD treated with IVIG 2–3- monthly showed a decrease in ARR from 0.75 to 0.15 (213). One study investigated IVIG treatment of acute NMOSD relapses (214), however, further data on preventive IVIG therapy is lacking.
In a study by Ramanathan et al, 4 out of 7 MOG-EM patients treated with IVIG were relapse-free (66). The authors recommend prophylaxis with low-dose prednisone or monthly IVIG with MMF or RTX as a next step for treating MOG-EM. Jarius et al. reported data of one MOG-EM patient who was relapse-free during 11 months of IVIG treatment and 12 months after IVIG discontinuation (33).
Methotrexate
Methotrexate (MTX) is an analog of folate, acts as folate antagonist and inhibits the dihydrofolate reductase. Hereby it inhibits DNA and RNA synthesis and has an immunosuppressive and anti-inflammatory effect. Side effects include gastrointestinal symptoms like nausea or diarrhea, bone marrow depression and an increase of liver enzymes.
Retrospective studies in NMOSD showed a decrease of ARR between 64 and 87% (215–217). In MOG-EM, MTX led to disease stabilization in 5/6 patients (33). Therefore, MTX seems to be a treatment option in patients that do not respond to first-line-therapy or suffer from side effects of other treatments (216).
MS Immunomodulatory Medication and Rarer Treatment Options
Treatment with MS medications like interferon-beta, glatiramer acetate, fingolimod, alemtuzumab, natalizumab, and presumably also dimethyl fumarate is known to have no or even harmful effects in NMOSD (181, 218–230). Similar results were found in patients with MOG-EM which were treated with one of these drugs for suspected MS (33); however, studies on treatment effects of these drugs are even rarer than in NMOSD.
Mitoxantrone is able to significantly reduce ARR in NMOSD patients (231, 232), nevertheless, due to its cardio- and myelotoxic side effects and the availability of alternatives with fewer adverse events its use should be considered very critically (233–235). Cyclophosphamide does not seem to be effective in NMOSD (236). Data about the effects of mitoxantrone or cyclophosphamide in MOG-EM are missing.
Ongoing Studies
To date, various clinical trials are ongoing to investigate the effect of new drugs in NMOSD. A placebo-controlled clinical trial is testing the effect of inebelizumab (MEDI-551), a humanized monoclonal antibody against CD19+ B-cells on NMOSD relapse rate (237–239). The efficacy of B-cell-depleting therapy in NMOSD is well known from treatment with RTX. AQP4-Ab positive as well as AQP4-Ab negative patients with at least one relapse during the last year or with at least two relapses during the last 2 years before screening can be included in this study (237).
Another agent under investigation is eculizumab, a monoclonal antibody inhibiting the complement protein C5. There were encouraging findings from an open label study where 12 out of 14 highly active patients became relapse-free by eculizumab treatment (240, 241). A subsequent double-blind placebo-controlled phase 3 trial aiming to enroll approximately 130 patients is now in the open-label extension stage (242).
Tocilizumab, an inhibitor of the IL-6 signaling pathway, showed significant reduction of disease activity in two pilot studies including in total 15 patients with high-active NMOSD (243, 244). Moreover, it might be an option in NMOSD patients with concomitant cancer or paraneoplastic syndrome (245). To date, an open label randomized controlled trial comparing tocilizumab and AZA is recruiting patients (246). Satralizumab (SA237), a follow-on monoclonal antibody of tocilizumab, is under investigation in a placebo-controlled double-blind phase 3 study (247). Efforts to restore immune tolerance as novel therapeutic endeavor are in preparation, however, various technical and conceptual issues hamper prompt implementation in clinical trials and practice (248, 249).
Further information on ongoing or completed (pilot) studies as well as non-conventional treatment approaches, e.g., with cetirizine, regulatory dentritic cells or autologous bone marrow derived stem cells in NMOSD may be found at the website https://clinicaltrials.gov and in current literature (188, 250, 251).
Summary
The diagnosis and treatment in NMOSD and MOG-EM require special clinical expertise. The 2015 NMOSD diagnostic criteria and the availability of antibody testing and MRI are the basis to diagnose and differentiate NMOSD or MOG-EM. Early diagnosis and initiation of adequate therapy are essential—at least in seropositive patients–to avoid disease attacks and persistent deficits. Long term immunosuppressive treatment, e.g., with RTX or AZA, has emerged to be the most effective therapies to reduce disease activity. Further therapeutic options, in particular various monoclonal antibodies are currently under clinical investigation in NMOSD.
Author Contributions
NB performed literature research and drafted the manuscript. FP, MM, MS, and SK critically reviewed the manuscript.
Conflict of Interest Statement
FP serves on the scientific advisory board for Novartis; received speaker honoraria and travel funding from Bayer, Novartis, Biogen Idec, Teva, Sanofi-Aventis/Genzyme, Merck Serono, Alexion, Chugai, MedImmune, and Shire; is an academic editor for PLoS One, is an associate editor for Neurology® Neuroimmunology & Neuroinflammation; consulted for Sanofi-Genzyme, Biogen Idec, MedImmune, Shire, and Alexion; received research support from Bayer, Novartis, Biogen Idec, Teva, Sanofi-Aventis/Genzyme, Alexion, Merck Serono, German Research Council, Werth Stiftung of the City of Cologne, German Ministry of Education and Research, Arthur Arnstein Stiftung Berlin, EU FP7 Framework Program, Arthur Arnstein Foundation Berlin, Guthy Jackson Charitable Foundation, and National Multiple Sclerosis of the USA. SK serves as a Deputy Editor of Journal of Neurology, Neurosurgery, and Psychiatry and an Editorial Board member of Journal of the Neurological Sciences.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Jarius S, Wildemann B. The history of neuromyelitis optica. J Neuroinflammation (2013) 10:8. doi: 10.1186/1742-2094-10-8
3. Pache F, Wildemann B, Paul F, Jarius S. [Neuromyelitis optica]. Fortschr Neurol Psychiatr. (2017) 85:e1. doi: 10.1055/s-0035-1567186
4. Weinshenker BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc. (2017) 92:663–79. doi: 10.1016/j.mayocp.2016.12.014
5. Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. (2014) 176:149–64. doi: 10.1111/cei.12271
6. Metz I, Beißbarth T, Ellenberger D, Pache F, Stork L, Ringelstein M, et al. Serum peptide reactivities may distinguish neuromyelitis optica subgroups and multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e204. doi: 10.1212/NXI.0000000000000204
7. Mori M, Kuwabara S, Paul F. Worldwide prevalence of neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry (2018) 89:555–6. doi: 10.1136/jnnp-2017-317566
8. Kremer L, Mealy M, Jacob A, Nakashima I, Cabre P, Bigi S, et al. Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler. (2014) 20:843–7. doi: 10.1177/1352458513507822
9. Wang KC, Lee CL, Chen SY, Lin KH, Tsai CP. Prominent brainstem symptoms/signs in patients with neuromyelitis optica in a Taiwanese population. J Clin Neurosci. (2011) 18:1197–200. doi: 10.1016/j.jocn.2010.12.052
10. Chavarro VS, Mealy MA, Simpson A, Lacheta A, Pache F, Ruprecht K, et al. Insufficient treatment of severe depression in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e286. doi: 10.1212/NXI.0000000000000286
11. Penner IK, Paul F. Fatigue as a symptom or comorbidity of neurological diseases. Nat Rev Neurol. (2017) 13:662–75. doi: 10.1038/nrneurol.2017.117
12. Song Y, Pan L, Fu Y, Sun N, Li YJ, Cai H, et al. Sleep abnormality in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflammation (2015) 2:e94. doi: 10.1212/NXI.0000000000000094
13. Asseyer S, Schmidt F, Chien C, Scheel M, Ruprecht K, Bellmann-Strobl J, et al. Pain in AQP4-IgG-positive and MOG-IgG-positive neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin. (2018) 4:2055217318796684. doi: 10.1177/2055217318796684
14. Mizuno Y, Shinoda K, Watanabe M, Matsushita T, Yamasaki R, Kira JI. Short-lasting unilateral neuralgiform headache attacks with cranial autonomic symptoms in NMOSD. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e447. doi: 10.1212/NXI.0000000000000447
15. Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Wegner B, et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. (2016) 79:206–16. doi: 10.1002/ana.24554
16. Pandit L, Mustafa S. Spontaneous remission lasting more than a decade in untreated AQP4 antibody-positive NMOSD. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e351. doi: 10.1212/NXI.0000000000000351
17. Mealy MA, Kessler RA, Rimler Z, Reid A, Totonis L, Cutter G, et al. Mortality in neuromyelitis optica is strongly associated with African ancestry. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e468. doi: 10.1212/NXI.0000000000000468
18. Collongues N, Marignier R, Zéphir H, Papeix C, Blanc F, Ritleng C, et al. Neuromyelitis optica in France: a multicenter study of 125 patients. Neurology (2010) 74:736–42. doi: 10.1212/WNL.0b013e3181d31e35
19. Pandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, et al. Demographic and clinical features of neuromyelitis optica: A review. Mult Scler. (2015) 21:845–53. doi: 10.1177/1352458515572406
20. Krumbholz M, Hofstadt-van Oy U, Angstwurm K, Kleiter I, Jarius S, Paul F, et al. Very late-onset neuromyelitis optica spectrum disorder beyond the age of 75. J Neurol. (2015) 262:1379–84. doi: 10.1007/s00415-015-7766-8
21. Bove R, Elsone L, Alvarez E, Borisow N, Cortez MM, Mateen FJ, et al. Female hormonal exposures and neuromyelitis optica symptom onset in a multicenter study. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e339. doi: 10.1212/NXI.0000000000000339
22. Borisow N, Kleiter I, Gahlen A, Fischer K, Wernecke KD, Pache F, et al. Influence of female sex and fertile age on neuromyelitis optica spectrum disorders. Mult Scler. (2017) 23:1092–103. doi: 10.1177/1352458516671203
23. Wingerchuk DM. Neuromyelitis optica: effect of gender. J Neurol Sci. (2009) 286:18–23. doi: 10.1016/j.jns.2009.08.045
24. Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry (2016) 87:1005–15. doi: 10.1136/jnnp-2015-312601
25. Melamed E, Levy M, Waters PJ, Sato DK, Bennett JL, John GR, et al. Update on biomarkers in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e134. doi: 10.1212/NXI.0000000000000134
26. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J Neuroinflammation (2012) 9:14. doi: 10.1186/1742-2094-9-14
27. Jiao Y, Fryer JP, Lennon VA, Jenkins SM, Quek AML, Smith CY, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology (2013) 81:1197–204. doi: 10.1212/WNL.0b013e3182a6cb5c
28. Kiyat-Atamer A, Ekizoglu E, Tüzün E, Kürtüncü M, Shugaiv E, Akman-Demir G, et al. Long-term MRI findings in neuromyelitis optica: seropositive versus seronegative patients. Eur J Neurol. (2013) 20:781–7. doi: 10.1111/ene.12058
29. Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology (2014) 82:474–81. doi: 10.1212/WNL.0000000000000101
30. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain J Neurol. (2017) 140:3128–38. doi: 10.1093/brain/awx276
31. Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. (2014) 71:276–83. doi: 10.1001/jamaneurol.2013.5857
32. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm. (2016) 13:279. doi: 10.1186/s12974-016-0717-1
33. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm. (2016) 13:280. doi: 10.1186/s12974-016-0718-0
34. Körtvélyessy P, Breu M, Pawlitzki M, Metz I, Heinze HJ, Matzke M, et al. ADEM-like presentation, anti-MOG antibodies, and MS pathology: TWO case reports. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e335. doi: 10.1212/NXI.0000000000000335
35. Spadaro M, Gerdes LA, Krumbholz M, Ertl-Wagner B, Thaler FS, Schuh E, et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e257. doi: 10.1212/NXI.0000000000000257
36. Hamid SHM, Whittam D, Mutch K, Linaker S, Solomon T, Das K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. (2017) 264:2088–94. doi: 10.1007/s00415-017-8596-7
37. Sepúlveda M, Armangué T, Sola-Valls N, Arrambide G, Meca-Lallana JE, Oreja-Guevara C, et al. Neuromyelitis optica spectrum disorders: Comparison according to the phenotype and serostatus. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e225. doi: 10.1212/NXI.0000000000000225
38. Pandit L, Sato DK, Mustafa S, Takahashi T, D'Cunha A, Malli C, et al. Relapsing optic neuritis and isolated transverse myelitis are the predominant clinical phenotypes for patients with antibodies to myelin oligodendrocyte glycoprotein in India. Mult Scler J Exp Transl Clin. (2016) 2:2055217316675634. doi: 10.1177/2055217316675634
39. Waters P, Woodhall M, O'Connor KC, Reindl M, Lang B, Sato DK, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e89. doi: 10.1212/NXI.0000000000000089
40. Hacohen Y, Wong YY, Lechner C, Jurynczyk M, Wright S, Konuskan B, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol. (2018) 75:478–87. doi: 10.1001/jamaneurol.2017.4601
41. Hacohen Y, Absoud M, Deiva K, Hemingway C, Nytrova P, Woodhall M, et al. Myelin oligodendrocyte glycoprotein antibodies are associated with a non-MS course in children. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e81. doi: 10.1212/NXI.0000000000000081
42. Pohl D, Alper G, Van Haren K, Kornberg AJ, Lucchinetti CF, Tenembaum S, et al. Acute disseminated encephalomyelitis: Updates on an inflammatory CNS syndrome. Neurology (2016) 87:S38–45. doi: 10.1212/WNL.0000000000002825
43. Berzero G, Cortese A, Ravaglia S, Marchioni E. Diagnosis and therapy of acute disseminated encephalomyelitis and its variants. Expert Rev Neurother. (2016) 16:83–101. doi: 10.1586/14737175.2015.1126510
44. Hjelmström P, Penzotti JE, Henne RM, Lybrand TP. A molecular model of myelin oligodendrocyte glycoprotein. J Neurochem. (1998) 71:1742–9.
45. Vourc'h P, Andres C. Oligodendrocyte myelin glycoprotein (OMgp): evolution, structure and function. Brain Res Brain Res Rev (2004) 45:115–24. doi: 10.1016/j.brainresrev.2004.01.003
46. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
47. Dos Passos GR, Oliveira LM, da Costa BK, Apostolos-Pereira SL, Callegaro D, Fujihara K, et al. MOG-IgG-associated optic neuritis, encephalitis, and myelitis: lessons learned from neuromyelitis optica spectrum disorder. Front Neurol. (2018) 9:217. doi: 10.3389/fneur.2018.00217
48. Cobo-Calvo Á, Ruiz A, D'Indy H, Poulat AL, Carneiro M, Philippe N, et al. MOG antibody-related disorders: common features and uncommon presentations. J Neurol. (2017) 264:1945–55. doi: 10.1007/s00415-017-8583-z
49. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflamm. (2018) 15:134. doi: 10.1186/s12974-018-1144-2
50. Narayan R, Simpson A, Fritsche K, Salama S, Pardo S, Mealy M, et al. MOG antibody disease: a review of MOG antibody seropositive neuromyelitis optica spectrum disorder. Mult Scler Relat Disord. (2018) 25:66–72. doi: 10.1016/j.msard.2018.07.025
51. Dale RC, Tantsis EM, Merheb V, Kumaran RYA, Sinmaz N, Pathmanandavel K, et al. Antibodies to MOG have a demyelination phenotype and affect oligodendrocyte cytoskeleton. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e12. doi: 10.1212/NXI.0000000000000012
52. Reindl M, Rostasy K. MOG antibody-associated diseases. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e60. doi: 10.1212/NXI.0000000000000060
53. Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm. (2015) 2:e62. doi: 10.1212/NXI.0000000000000062
54. Chen HX, Zhang Q, Lian ZY, Liu J, Shi ZY, Miao XH, et al. Muscle damage in patients with neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e400. doi: 10.1212/NXI.0000000000000400
55. Baba T, Nakashima I, Kanbayashi T, Konno M, Takahashi T, Fujihara K, et al. Narcolepsy as an initial manifestation of neuromyelitis optica with anti-aquaporin-4 antibody. J Neurol. (2009) 256:287–8. doi: 10.1007/s00415-009-0139-4
56. Pandit L, Mustafa S, Uppoor R, Nakashima I, Takahashi T, Kaneko K. Reversible paraspinal muscle hyperintensity in anti-MOG antibody-associated transverse myelitis. Neurol Neuroimmunol Neuroinflamm. (2018)5:e412. doi: 10.1212/NXI.0000000000000412
57. Vazquez Do Campo R, Stephens A, Marin Collazo IV, Rubin DI. MOG antibodies in combined central and peripheral demyelination syndromes. Neurol Neuroimmunol Neuroinflamm. (2018) doi: 10.1212/NXI.0000000000000503
58. Jarius S, Metz I, König FB, Ruprecht K, Reindl M, Paul F, et al. Screening for MOG-IgG and 27 other anti-glial and anti-neuronal autoantibodies in “pattern II multiple sclerosis” and brain biopsy findings in a MOG-IgG-positive case. Mult Scler. (2016) 22:1541–49. doi: 10.1177/1352458515622986
59. Spadaro M, Gerdes LA, Mayer MC, Ertl-Wagner B, Laurent S, Krumbholz M, et al. Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol. (2015) 2:295–301. doi: 10.1002/acn3.164
60. Takeshita Y, Obermeier B, Cotleur AC, Spampinato SF, Shimizu F, Yamamoto E, et al. Effects of neuromyelitis optica-IgG at the blood-brain barrier in vitro. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e311. doi: 10.1212/NXI.0000000000000311
61. Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol. (2012) 123:861–72. doi: 10.1007/s00401-012-0986-4
62. Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain J Neurol. (2007) 130:1224–34. doi: 10.1093/brain/awm047
63. Aboul-Enein F, Seifert-Held T, Mader S, Kuenz B, Lutterotti A, Rauschka H, et al. Neuromyelitis optica in Austria in 2011: to bridge the gap between neuroepidemiological research and practice in a study population of 8.4 million people. PloS ONE (2013) 8:e79649. doi: 10.1371/journal.pone.0079649
64. ZhangBao J, Zhou L, Li X, Cai T, Lu J, Lu C, et al. The clinical characteristics of AQP4 antibody positive NMO/SD in a large cohort of Chinese Han patients. J Neuroimmunol. (2017) 302:49–55. doi: 10.1016/j.jneuroim.2016.11.010
65. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology (2018) 90:e1858–69. doi: 10.1212/WNL.0000000000005560
66. Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
67. Soelberg K, Jarius S, Skejoe H, Engberg H, Mehlsen JJ, Nilsson AC, et al. A population-based prospective study of optic neuritis. Mult Scler. (2017) 23:1893–901. doi: 10.1177/1352458517734070
68. Soelberg K, Specovius S, Zimmermann HG, Grauslund J, Mehlsen JJ, Olesen C, et al. Optical coherence tomography in acute optic neuritis: A population-based study. Acta Neurol Scand. (2018) doi: 10.1111/ane.13004 [Epub ahead of print].
69. Jarius S, Frederikson J, Waters P, Paul F, Akman-Demir G, Marignier R, et al. Frequency and prognostic impact of antibodies to aquaporin-4 in patients with optic neuritis. J Neurol Sci. (2010) 298:158–62. doi: 10.1016/j.jns.2010.07.011
70. Fujimori J, Takai Y, Nakashima I, Sato DK, Takahashi T, Kaneko K, et al. Bilateral frontal cortex encephalitis and paraparesis in a patient with anti-MOG antibodies. J Neurol Neurosurg Psychiatry (2017) 88:534–6. doi: 10.1136/jnnp-2016-315094
71. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
72. Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs aquaporin 4 IgG disease. JAMA Neurol. (2018) 75:65–71. doi: 10.1001/jamaneurol.2017.3196
73. Höftberger R, Sepulveda M, Armangue T, Blanco Y, Rostásy K, Calvo AC, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler. (2015) 21:866–74. doi: 10.1177/1352458514555785
74. van Pelt ED, Wong YYM, Ketelslegers IA, Hamann D, Hintzen RQ. Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands. Eur J Neurol. (2016) 23:580–7. doi: 10.1111/ene.12898
75. Pandit L, Mustafa S, Nakashima I, Takahashi T, Kaneko K. MOG-IgG-associated disease has a stereotypical clinical course, asymptomatic visual impairment and good treatment response. Mult Scler J Exp Transl Clin (2018) 4:2055217318787829. doi: 10.1177/2055217318787829
76. Hyun JW, Woodhall MR, Kim SH, Jeong IH, Kong B, Kim G, et al. Longitudinal analysis of myelin oligodendrocyte glycoprotein antibodies in CNS inflammatory diseases. J Neurol Neurosurg Psychiatry (2017) 88:811–7. doi: 10.1136/jnnp-2017-315998
77. Akaishi T, Sato DK, Nakashima I, Takeshita T, Takahashi T, Doi H, et al. MRI and retinal abnormalities in isolated optic neuritis with myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies: a comparative study. J Neurol Neurosurg Psychiatry (2016) 87:446–8. doi: 10.1136/jnnp-2014-310206
78. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet (2004) 364:2106–12. doi: 10.1016/S0140-6736(04)17551-X
79. Waters PJ, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, Wingerchuk DM. Evaluation of aquaporin-4 antibody assays. Clin Exp Neuroimmunol. (2014) 5:290–303. doi: 10.1111/cen3.12107
80. Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, et al. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. (2008) 4:202–14. doi: 10.1038/ncpneuro0764
81. Lang K, Prüss H. Frequencies of neuronal autoantibodies in healthy controls: Estimation of disease specificity. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e386. doi: 10.1212/NXI.0000000000000386
82. Pittock SJ, Lennon VA, Bakshi N, Shen L, McKeon A, Quach H, et al. Seroprevalence of aquaporin-4-IgG in a northern California population representative cohort of multiple sclerosis. JAMA Neurol. (2014) 71:1433–6. doi: 10.1001/jamaneurol.2014.1581
83. Kister I, Paul F. Pushing the boundaries of neuromyelitis optica: does antibody make the disease? Neurology (2015) 85:118–9. doi: 10.1212/WNL.0000000000001749
84. Duignan S, Wright S, Rossor T, Cazabon J, Gilmour K, Ciccarelli O, et al. Myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies are highly specific in children with acquired demyelinating syndromes. Dev Med Child Neurol. (2018) 60:958–62. doi: 10.1111/dmcn.13703
85. Brilot F, Dale RC, Selter RC, Grummel V, Kalluri SR, Aslam M, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol. (2009) 66:833–42. doi: 10.1002/ana.21916
86. Lalive PH, Häusler MG, Maurey H, Mikaeloff Y, Tardieu M, Wiendl H, et al. Highly reactive anti-myelin oligodendrocyte glycoprotein antibodies differentiate demyelinating diseases from viral encephalitis in children. Mult Scler. (2011) 17:297–302. doi: 10.1177/1352458510389220
87. Xiao BG, Linington C, Link H. Antibodies to myelin-oligodendrocyte glycoprotein in cerebrospinal fluid from patients with multiple sclerosis and controls. J Neuroimmunol. (1991) 31:91–6.
88. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. (1999) 5:170–5. doi: 10.1038/5532
89. Reindl M, Linington C, Brehm U, Egg R, Dilitz E, Deisenhammer F, et al. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: a comparative study. Brain J Neurol. (1999) 122 (Pt 11):2047–56.
90. Klawiter EC, Piccio L, Lyons JA, Mikesell R, O'Connor KC, Cross AH. Elevated intrathecal myelin oligodendrocyte glycoprotein antibodies in multiple sclerosis. Arch Neurol. (2010) 67:1102–8. doi: 10.1001/archneurol.2010.197
91. Haase CG, Schmidt S. Detection of brain-specific autoantibodies to myelin oligodendrocyte glycoprotein, S100beta and myelin basic protein in patients with Devic's neuromyelitis optica. Neurosci Lett. (2001) 307:131–3. doi: 10.1016/S0304-3940(01)01949-8
92. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology (2012) 79:1273–7. doi: 10.1212/WNL.0b013e31826aac4e
93. Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain (2008) 131:3072–80. doi: 10.1093/brain/awn240
94. Valentino P, Marnetto F, Granieri L, Capobianco M, Bertolotto A. Aquaporin-4 antibody titration in NMO patients treated with rituximab: a retrospective study. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e317. doi: 10.1212/NXI.0000000000000317
95. Kessler RA, Mealy MA, Jimenez-Arango JA, Quan C, Paul F, López R, et al. Anti-aquaporin-4 titer is not predictive of disease course in neuromyelitis optica spectrum disorder: a multicenter cohort study. Mult Scler Relat Disord. (2017) 17:198–201. doi: 10.1016/j.msard.2017.08.005
96. Mealy MA, Kim SH, Schmidt F, López R, Jimenez Arango JA, Paul F, et al. Aquaporin-4 serostatus does not predict response to immunotherapy in neuromyelitis optica spectrum disorders. Mult Scler. (2017). doi: 10.1177/1352458517730131 [Epub ahead of print].
97. Jarius S, Ruprecht K, Stellmann JP, Huss A, Ayzenberg I, Willing A, et al. MOG-IgG in primary and secondary chronic progressive multiple sclerosis: a multicenter study of 200 patients and review of the literature. J Neuroinflamm. (2018) 15:88. doi: 10.1186/s12974-018-1108-6
98. Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflamm. (2010) 7:52. doi: 10.1186/1742-2094-7-52
99. Majed M, Fryer JP, McKeon A, Lennon VA, Pittock SJ. Clinical utility of testing AQP4-IgG in CSF: guidance for physicians. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e231. doi: 10.1212/NXI.0000000000000231
100. Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci. (2011) 306:82–90. doi: 10.1016/j.jns.2011.03.038
101. Iyer A, Elsone L, Appleton R, Jacob A. A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity (2014) 47:154–61. doi: 10.3109/08916934.2014.883501
102. Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, et al. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol. (2008) 65:78–83. doi: 10.1001/archneurol.2007.17
103. Sergio P, Mariana B, Alberto O, Claudia U, Oscar R, Pablo M, et al. Association of neuromyelitis optic (NMO) with autoimmune disorders: report of two cases and review of the literature. Clin Rheumatol. (2010) 29:1335–8. doi: 10.1007/s10067-010-1502-7
104. Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e110. doi: 10.1212/NXI.0000000000000110
105. Jarius S, Jacobi C, de Seze J, Zephir H, Paul F, Franciotta D, et al. Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler. (2011) 17:1067–73. doi: 10.1177/1352458511403958
106. Jarius S, Paul F, Franciotta D, de Seze J, Münch C, Salvetti M, Ruprecht K, Liebetrau M, Wandinger KP, Akman-Demir G, et al. Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler. (2012) 18:1135–43. doi: 10.1177/1352458511431728
107. Uzawa A, Mori M, Iwai Y, Kobayashi M, Hayakawa S, Kawaguchi N, et al. Association of anti-aquaporin-4 antibody-positive neuromyelitis optica with myasthenia gravis. J Neurol Sci. (2009) 287:105–7. doi: 10.1016/j.jns.2009.08.040
108. Beauchemin P, Iorio R, Traboulsee AL, Field T, Tinker AV, Carruthers RL. Paraneoplastic neuromyelitis optica spectrum disorder: a single center cohort description with two cases of histological validation. Mult Scler Relat Disord. (2018) 20:37–42. doi: 10.1016/j.msard.2017.12.012
109. Soelberg K, Larsen SR, Moerch MT, Thomassen M, Brusgaard K, Paul F, et al. Aquaporin-4 IgG autoimmune syndrome and immunoreactivity associated with thyroid cancer. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e252. doi: 10.1212/NXI.0000000000000252
110. Sudo A, Chihara N, Takenaka Y, Nakamura T, Ueda T, Sekiguchi K, et al. Paraneoplastic NMOSD associated with EG junction adenocarcinoma expressing unprotected AQP4. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e482. doi: 10.1212/NXI.0000000000000482
111. Jarius S, Paul F, Ruprecht K, Wildemann B. Low vitamin B12 levels and gastric parietal cell antibodies in patients with aquaporin-4 antibody-positive neuromyelitis optica spectrum disorders. J Neurol. (2012) 259:2743–5. doi: 10.1007/s00415-012-6677-1
112. Flanagan EP, Kaufmann TJ, Krecke KN, Aksamit AJ, Pittock SJ, Keegan BM, et al. Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann Neurol. (2016) 79:437–47. doi: 10.1002/ana.24582
113. Kidd DP, Burton BJ, Graham EM, Plant GT. Optic neuropathy associated with systemic sarcoidosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e270. doi: 10.1212/NXI.0000000000000270
114. Sawaya R, Radwan W. Sarcoidosis associated with neuromyelitis optica. J Clin Neurosci. (2013) 20:1156–8. doi: 10.1016/j.jocn.2012.09.030
115. Geraldes R, Ciccarelli O, Barkhof F, De Stefano N, Enzinger C, Filippi M, et al. The current role of MRI in differentiating multiple sclerosis from its imaging mimics. Nat Rev Neurol. (2018) 14:199–213. doi: 10.1038/nrneurol.2018.14
116. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology (2006) 66:1485–9. doi: 10.1212/01.wnl.0000216139.44259.74
117. Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol. (2015) 72:81–7. doi: 10.1001/jamaneurol.2014.2137
118. Pekcevik Y, Mitchell CH, Mealy MA, Orman G, Lee IH, Newsome SD, et al. Differentiating neuromyelitis optica from other causes of longitudinally extensive transverse myelitis on spinal magnetic resonance imaging. Mult Scler. (2016) 22:302–11. doi: 10.1177/1352458515591069
119. Yonezu T, Ito S, Mori M, Ogawa Y, Makino T, Uzawa A, et al. “Bright spotty lesions” on spinal magnetic resonance imaging differentiate neuromyelitis optica from multiple sclerosis. Mult Scler. (2014) 20:331–7. doi: 10.1177/1352458513495581
120. Asgari N, Skejoe HPB, Lillevang ST, Steenstrup T, Stenager E, Kyvik KO. Modifications of longitudinally extensive transverse myelitis and brainstem lesions in the course of neuromyelitis optica (NMO): a population-based, descriptive study. BMC Neurol. (2013) 13:33. doi: 10.1186/1471-2377-13-33
121. Asgari N, Skejoe HPB, Lennon VA. Evolution of longitudinally extensive transverse myelitis in an aquaporin-4 IgG-positive patient. Neurology (2013) 81:95–6. doi: 10.1212/WNL.0b013e318297ef07
122. Flanagan EP, Krecke KN, Marsh RW, Giannini C, Keegan BM, Weinshenker BG. Specific pattern of gadolinium enhancement in spondylotic myelopathy. Ann Neurol. (2014) 76:54–65. doi: 10.1002/ana.24184
123. Whittam D, Bhojak M, Das K, Jacob A. Longitudinally extensive myelitis in MS mimicking neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e333. doi: 10.1212/NXI.0000000000000333
124. Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol. (2017) 81:298–309. doi: 10.1002/ana.24881
125. Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, Lennon VA. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. (2016) 73:1297–307. doi: 10.1001/jamaneurol.2016.2549
126. Luessi F, Engel S, Spreer A, Bittner S, Zipp F. GFAPα IgG-associated encephalitis upon daclizumab treatment of MS. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e481. doi: 10.1212/NXI.0000000000000481
127. Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol. (2006) 63:390–6. doi: 10.1001/archneur.63.3.390
128. Cabrera-Gómez JA, Quevedo-Sotolongo L, González-Quevedo A, Lima S, Real-González Y, Cristófol-Corominas M, et al. Brain magnetic resonance imaging findings in relapsing neuromyelitis optica. Mult Scler. (2007) 13:186–92. doi: 10.1177/1352458506070725
129. Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, Klawiter EC, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology (2015) 84:1165–73. doi: 10.1212/WNL.0000000000001367
130. Wang KY, Chetta J, Bains P, Balzer A, Lincoln J, Uribe T, et al. Spectrum of MRI brain lesion patterns in neuromyelitis optica spectrum disorder: a pictorial review. Br J Radiol. (2018) 91:20170690. doi: 10.1259/bjr.20170690
131. Carnero Contentti E, Daccach Marques V, Soto de Castillo I, Tkachuk V, Antunes Barreira A, Armas E, et al. Frequency of brain MRI abnormalities in neuromyelitis optica spectrum disorder at presentation: A cohort of Latin American patients. Mult Scler Relat Disord. (2018) 19:73–8. doi: 10.1016/j.msard.2017.11.004
132. Fan M, Fu Y, Su L, Shen Y, Wood K, Yang L, et al. Comparison of brain and spinal cord magnetic resonance imaging features in neuromyelitis optica spectrum disorders patients with or without aquaporin-4 antibody. Mult Scler Relat Disord. (2017) 13:58–66. doi: 10.1016/j.msard.2017.02.003
133. Asgari N, Flanagan EP, Fujihara K, Kim HJ, Skejoe HP, Wuerfel J, et al. Disruption of the leptomeningeal blood barrier in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e343. doi: 10.1212/NXI.0000000000000343
134. Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson APD, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler. (2016) 22:470–82. doi: 10.1177/1352458515593406
135. Akaishi T, Nakashima I, Takeshita T, Mugikura S, Sato DK, Takahashi T, et al. Lesion length of optic neuritis impacts visual prognosis in neuromyelitis optica. J Neuroimmunol. (2016) 293:28–33. doi: 10.1016/j.jneuroim.2016.02.004
136. Jurynczyk M, Geraldes R, Probert F, Woodhall MR, Waters P, Tackley G, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain J Neurol. (2017) 140:617–27. doi: 10.1093/brain/aww350
137. Jurynczyk M, Tackley G, Kong Y, Geraldes R, Matthews L, Woodhall M, et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry (2017) 88:132–6. doi: 10.1136/jnnp-2016-314005
138. Hyun JW, Jeong IH, Joung A, Kim SH, Kim HJ. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology (2016) 86:1772–9. doi: 10.1212/WNL.0000000000002655
139. Kremer S, Renard F, Achard S, Lana-Peixoto MA, Palace J, Asgari N, et al. Use of advanced magnetic resonance imaging techniques in neuromyelitis optica spectrum disorder. JAMA Neurol. (2015) 72:815–22. doi: 10.1001/jamaneurol.2015.0248
140. Finke C, Heine J, Pache F, Lacheta A, Borisow N, Kuchling J, et al. Normal volumes and microstructural integrity of deep gray matter structures in AQP4+ NMOSD. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e229. doi: 10.1212/NXI.0000000000000229
141. Pache F, Zimmermann H, Finke C, Lacheta A, Papazoglou S, Kuchling J, et al. Brain parenchymal damage in neuromyelitis optica spectrum disorder - A multimodal MRI study. Eur Radiol. (2016) 26:4413–22. doi: 10.1007/s00330-016-4282-x
142. Oberwahrenbrock T, Traber GL, Lukas S, Gabilondo I, Nolan R, Songster C, et al. Multicenter reliability of semiautomatic retinal layer segmentation using OCT. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e449. doi: 10.1212/NXI.0000000000000449
143. Oertel FC, Zimmermann H, Paul F, Brandt AU. Optical coherence tomography in neuromyelitis optica spectrum disorders: potential advantages for individualized monitoring of progression and therapy. EPMA J. (2018) 9:21–33. doi: 10.1007/s13167-017-0123-5
144. Zimmermann H, Oberwahrenbrock T, Brandt AU, Paul F, Dörr J. Optical coherence tomography for retinal imaging in multiple sclerosis. Degener Neurol Neuromuscul Dis. (2014) 2014:4:153-62. doi: 10.2147/DNND.S73506
145. Oertel FC, Zimmermann H, Brandt AU, Paul F. [Optical coherence tomography in neuromyelitis optica spectrum disorders]. Nervenarzt (2017) 88:1411–20. doi: 10.1007/s00115-017-0444-6
146. Ayadi N, Dörr J, Motamedi S, Gawlik K, Bellmann-Strobl J, Mikolajczak J, et al. Temporal visual resolution and disease severity in MS. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e492. doi: 10.1212/NXI.0000000000000492
147. You Y, Graham EC, Shen T, Yiannikas C, Parratt J, Gupta V, et al. Progressive inner nuclear layer dysfunction in non-optic neuritis eyes in MS. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e427. doi: 10.1212/NXI.0000000000000427
148. Waldman AT, Liu GT, Lavery AM, Liu G, Gaetz W, Aleman TS, et al. Optical coherence tomography and visual evoked potentials in pediatric MS. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e356. doi: 10.1212/NXI.0000000000000356
149. Bennett JL, de Seze J, Lana-Peixoto M, Palace J, Waldman A, Schippling S, et al. Neuromyelitis optica and multiple sclerosis: seeing differences through optical coherence tomography. Mult Scler. (2015) 21:678–88. doi: 10.1177/1352458514567216
150. Petzold A, Wattjes MP, Costello F, Flores-Rivera J, Fraser CL, Fujihara K, et al. The investigation of acute optic neuritis: a review and proposed protocol. Nat Rev Neurol. (2014) 10:447–58. doi: 10.1038/nrneurol.2014.108
151. Finke C, Zimmermann H, Pache F, Oertel FC, Chavarro VS, Kramarenko Y, et al. Association of visual impairment in neuromyelitis optica spectrum disorder with visual network reorganization. JAMA Neurol. (2018) 75:296–303. doi: 10.1001/jamaneurol.2017.3890
152. Pache F, Zimmermann H, Mikolajczak J, Schumacher S, Lacheta A, Oertel FC, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 4: afferent visual system damage after optic neuritis in MOG-IgG-seropositive versus AQP4-IgG-seropositive patients. J Neuroinflammation (2016) 13:282. doi: 10.1186/s12974-016-0720-6
153. Schneider E, Zimmermann H, Oberwahrenbrock T, Kaufhold F, Kadas EM, Petzold A, et al. Optical coherence tomography reveals distinct patterns of retinal damage in neuromyelitis optica and multiple sclerosis. PloS ONE (2013) 8:e66151. doi: 10.1371/journal.pone.0066151
154. Havla J, Kümpfel T, Schinner R, Spadaro M, Schuh E, Meinl E, et al. Myelin-oligodendrocyte-glycoprotein (MOG) autoantibodies as potential markers of severe optic neuritis and subclinical retinal axonal degeneration. J Neurol. (2017) 264:139–51. doi: 10.1007/s00415-016-8333-7
155. Schmidt F, Zimmermann H, Mikolajczak J, Oertel FC, Pache F, Weinhold M, et al. Severe structural and functional visual system damage leads to profound loss of vision-related quality of life in patients with neuromyelitis optica spectrum disorders. Mult Scler Relat Disord. (2017) 11:45–50. doi: 10.1016/j.msard.2016.11.008
156. Bouyon M, Collongues N, Zéphir H, Ballonzoli L, Jeanjean L, Lebrun C, et al. Longitudinal follow-up of vision in a neuromyelitis optica cohort. Mult Scler. (2013) 19:1320–2. doi: 10.1177/1352458513476562
157. Oertel FC, Zimmermann H, Mikolajczak J, Weinhold M, Kadas EM, Oberwahrenbrock T, et al. Contribution of blood vessels to retinal nerve fiber layer thickness in NMOSD. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e338. doi: 10.1212/NXI.0000000000000338
158. Gelfand JM, Cree BA, Nolan R, Arnow S, Green AJ. Microcystic inner nuclear layer abnormalities and neuromyelitis optica. JAMA Neurol. (2013) 70:629–33. doi: 10.1001/jamaneurol.2013.1832
159. Jelcic I, Hanson JVM, Lukas S, Weber KP, Landau K, Pless M, et al. Unfavorable structural and functional outcomes in myelin oligodendrocyte glycoprotein antibody-associated optic neuritis. J Neuro-Ophthalmol. (2018) doi: 10.1097/WNO.0000000000000669
160. Akaishi T, Kaneko K, Himori N, Takeshita T, Takahashi T, Nakazawa T, et al. Subclinical retinal atrophy in the unaffected fellow eyes of multiple sclerosis and neuromyelitis optica. J Neuroimmunol. (2017) 313:10–5. doi: 10.1016/j.jneuroim.2017.10.001
161. Stiebel-Kalish H, Lotan I, Brody J, Chodick G, Bialer O, Marignier R, et al. Retinal nerve fiber layer may be better preserved in MOG-IgG versus AQP4-IgG optic neuritis: a cohort study. PloS ONE (2017) 12:e0170847. doi: 10.1371/journal.pone.0170847
162. Martinez-Lapiscina EH, Sepulveda M, Torres-Torres R, Alba-Arbalat S, Llufriu S, Blanco Y, et al. Usefulness of optical coherence tomography to distinguish optic neuritis associated with AQP4 or MOG in neuromyelitis optica spectrum disorders. Ther Adv Neurol Disord. (2016) 9:436–40. doi: 10.1177/1756285616655264
163. Yamamura T, Nakashima I. Foveal thinning in neuromyelitis optica: A sign of retinal astrocytopathy? Neurol Neuroimmunol Neuroinflamm. (2017) 4:e347. doi: 10.1212/NXI.0000000000000347
164. Dalmau J. Precision in neuroimmunology. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e345. doi: 10.1212/NXI.0000000000000345
165. Jeong IH, Kim HJ, Kim N-H, Jeong KS, Park CY. Subclinical primary retinal pathology in neuromyelitis optica spectrum disorder. J Neurol. (2016) 263:1343–8. doi: 10.1007/s00415-016-8138-8
166. Oertel FC, Kuchling J, Zimmermann H, Chien C, Schmidt F, Knier B, et al. Microstructural visual system changes in AQP4-antibody-seropositive NMOSD. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e334. doi: 10.1212/NXI.0000000000000334
167. Oertel FC, Havla J, Roca-Fernández AI, Lizak N1, Zimmermann H, Motamedi S, et al. Retinal ganglion cell loss in neuromyelitis optica: a longitudinal study. J Neurol Neurosurg Psychiatry (2018) doi: 10.1136/jnnp-2018-318382 [Epub ahead of print].
168. Nakamura M, Nakazawa T, Doi H, Hariya T, Omodaka K, Misu T, et al. Early high-dose intravenous methylprednisolone is effective in preserving retinal nerve fiber layer thickness in patients with neuromyelitis optica. Graefes Arch Clin Exp Ophthalmol. (2010) 248:1777–85. doi: 10.1007/s00417-010-1344-7
169. Merle H, Olindo S, Jeannin S, Valentino R, Mehdaoui H, Cabot F, Donnio A, et al. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol. (2012) 130:858–62. doi: 10.1001/archophthalmol.2012.1126
170. Brandt AU, Specovius S, Oberwahrenbrock T, Zimmermann HG, Paul F, Costello F. Frequent retinal ganglion cell damage after acute optic neuritis. Mult Scler Relat Disord. (2018) 22:141–7. doi: 10.1016/j.msard.2018.04.006
171. Hamid SH, Elsone L, Mutch K, Solomon T, Jacob A. The impact of 2015 neuromyelitis optica spectrum disorders criteria on diagnostic rates. Mult Scler. (2017) 23:228–33. doi: 10.1177/1352458516663853
172. Sepúlveda M, Aldea M, Escudero D, Llufriu S, Arrambide G, Otero-Romero S, et al. Epidemiology of NMOSD in Catalonia: Influence of the new 2015 criteria in incidence and prevalence estimates. Mult Scler. (2017) doi: 10.1177/1352458517735191 [Epub ahead of print].
173. Abboud H, Petrak A, Mealy M, Sasidharan S, Siddique L, Levy M. Treatment of acute relapses in neuromyelitis optica: Steroids alone versus steroids plus plasma exchange. Mult Scler. (2016) 22:185–92. doi: 10.1177/1352458515581438
174. Aungsumart S, Apiwattanakul M. Clinical outcomes and predictive factors related to good outcomes in plasma exchange in severe attack of NMOSD and long extensive transverse myelitis: Case series and review of the literature. Mult Scler Relat Disord. (2017) 13:93–7. doi: 10.1016/j.msard.2017.02.015
175. Faissner S, Nikolayczik J, Chan A, Gold R, Yoon M-S, Haghikia A. Immunoadsorption in patients with neuromyelitis optica spectrum disorder. Ther Adv Neurol Disord. (2016) 9:281–6. doi: 10.1177/1756285616646332
176. Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Hellwig K, et al. Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e504. doi: 10.1212/NXI.0000000000000504
177. Bonnan M, Valentino R, Debeugny S, Merle H, Fergé JL, Mehdaoui H, Cabre P. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry (2018) 89:346–51. doi: 10.1136/jnnp-2017-316286
178. Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. (2017) 264:2003–9. doi: 10.1007/s00415-017-8590-0
179. Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol. (2014) 261:1–16. doi: 10.1007/s00415-013-7169-7
180. Patt H, Bandgar T, Lila A, Shah N. Management issues with exogenous steroid therapy. Indian J Endocrinol Metab. (2013) 17:S612–7. doi: 10.4103/2230-8210.123548
181. Stellmann JP, Krumbholz M, Friede T, Gahlen A, Borisow N, Fischer K, et al. Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatry (2017) 88:639–47. doi: 10.1136/jnnp-2017-315603
182. Watanabe S, Misu T, Miyazawa I, Nakashima I, Shiga Y, Fujihara K, et al. Low-dose corticosteroids reduce relapses in neuromyelitis optica: a retrospective analysis. Mult Scler. (2007) 13:968–74. doi: 10.1177/1352458507077189
183. Qiu W, Kermode AG, Li R, Dai Y, Wang Y, Wang J, et al. Azathioprine plus corticosteroid treatment in Chinese patients with neuromyelitis optica. J Clin Neurosci. (2015) 22:1178–82. doi: 10.1016/j.jocn.2015.01.028
184. Zhang M, Zhang C, Bai P, Xue H, Wang G. Effectiveness of low dose of rituximab compared with azathioprine in Chinese patients with neuromyelitis optica: an over 2-year follow-up study. Acta Neurol Belg. (2017) 117:695–702. doi: 10.1007/s13760-017-0795-6
185. Elsone L, Kitley J, Luppe S, Lythgoe D, Mutch K, Jacob S, et al. Long-term efficacy, tolerability and retention rate of azathioprine in 103 aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder patients: a multicentre retrospective observational study from the UK. Mult Scler. (2014) 20:1533–40. doi: 10.1177/1352458514525870
186. Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, et al. Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology (2011) 77:659–66. doi: 10.1212/WNL.0b013e31822a2780
187. Bennett JL, O'Connor KC, Bar-Or A, Zamvil SS, Hemmer B, Tedder TF, et al. B lymphocytes in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e104. doi: 10.1212/NXI.0000000000000104
188. Kleiter I, Gold R. Present and future therapies in neuromyelitis optica spectrum disorders. Neurother J Am Soc Exp Neurother. (2016) 13:70–83. doi: 10.1007/s13311-015-0400-8
189. Ellwardt E, Ellwardt L, Bittner S, Zipp F. Monitoring B-cell repopulation after depletion therapy in neurologic patients. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e463. doi: 10.1212/NXI.0000000000000463
190. Evangelopoulos ME, Andreadou E, Koutsis G, Koutoulidis V, Anagnostouli M, Katsika P, et al. Treatment of neuromyelitis optica and neuromyelitis optica spectrum disorders with rituximab using a maintenance treatment regimen and close CD19 B cell monitoring. A six-year follow-up. J Neurol Sci. (2017) 372:92–6. doi: 10.1016/j.jns.2016.11.016
191. Cohen M, Romero G, Bas J, Ticchioni M, Rosenthal M, Lacroix R, Brunet C, et al. Monitoring CD27+ memory B-cells in neuromyelitis optica spectrum disorders patients treated with rituximab: results from a bicentric study. J Neurol Sci. (2017) 373:335–8. doi: 10.1016/j.jns.2017.01.025
192. Ciron J, Audoin B, Bourre B, Brassat D, Durand-Dubief F, Laplaud D, et al. Recommendations for the use of Rituximab in neuromyelitis optica spectrum disorders. Rev Neurol. (2018) 174:255–64. doi: 10.1016/j.neurol.2017.11.005
193. Rommer PS, Dörner T, Freivogel K, Haas J, Kieseier BC, Kümpfel T, et al. Safety and clinical outcomes of rituximab treatment in patients with multiple sclerosis and neuromyelitis optica: experience from a national online registry (GRAID). J Neuroimmune Pharmacol. (2016) 11:1–8. doi: 10.1007/s11481-015-9646-5
194. Marcinnò A, Marnetto F, Valentino P, Martire S, Balbo A, Drago A. Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e498. doi: 10.1212/NXI.0000000000000498
195. Cree BAC, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology (2005) 64:1270–2. doi: 10.1212/01.WNL.0000159399.81861.D5
196. Zéphir H, Bernard-Valnet R, Lebrun C, Outteryck O, Audoin B, Bourre B, et al. Rituximab as first-line therapy in neuromyelitis optica: efficiency and tolerability. J Neurol. (2015) 262:2329–35. doi: 10.1007/s00415-015-7852-y
197. Radaelli M, Moiola L, Sangalli F, Esposito F, Barcella V, Ferrè L, et al. Neuromyelitis optica spectrum disorders: long-term safety and efficacy of rituximab in Caucasian patients. Mult Scler. (2016) 22:511–9. doi: 10.1177/1352458515594042
198. Torres J, Pruitt A, Balcer L, Galetta S, Markowitz C, Dahodwala N. Analysis of the treatment of neuromyelitis optica. J Neurol Sci. (2015) 351:31–5. doi: 10.1016/j.jns.2015.02.012
199. Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. (2013) 70:1110–7. doi: 10.1001/jamaneurol.2013.3071
200. Cabre P, Mejdoubi M, Jeannin S, Merle H, Plumelle Y, Cavillon G, et al. Treatment of neuromyelitis optica with rituximab: a 2-year prospective multicenter study. J Neurol. (2018) 265:917–25. doi: 10.1007/s00415-018-8771-5
201. Kim SH, Jeong IH, Hyun JW, Joung A, Jo HJ, Hwang SH, et al. Treatment outcomes with rituximab in 100 patients with neuromyelitis optica: influence of FCGR3A polymorphisms on the therapeutic response to rituximab. JAMA Neurol. (2015) 72:989–95. doi: 10.1001/jamaneurol.2015.1276
202. Nosadini M, Alper G, Riney CJ, Benson LA, Mohammad SS, Ramanathan S, et al. Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e188. doi: 10.1212/NXI.0000000000000188
203. Damato V, Evoli A, Iorio R. Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. JAMA Neurol. (2016) 73:1342–8. doi: 10.1001/jamaneurol.2016.1637
204. Kim SH, Kim HJ. Rituximab in neuromyelitis optica spectrum disorders: why not as first-line therapy. JAMA Neurol. (2017) 74:482. doi: 10.1001/jamaneurol.2016.5933
205. Das G, Damotte V, Gelfand JM, Bevan C, Cree BAC, Do L, et al. Rituximab before and during pregnancy: A systematic review, and a case series in MS and NMOSD. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e453. doi: 10.1212/NXI.0000000000000453
206. Perumal JS, Kister I, Howard J, Herbert J. Disease exacerbation after rituximab induction in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e61. doi: 10.1212/NXI.0000000000000061
207. Whittam D, Cobo-Calvo A, Lopez-Chiriboga AS, Pardo S, Dodd J, Brandt A, et al. Treatment of MOG-IgG-associated demyelination with Rituximab: a multinational study of 98 patients (S13.003). Neurology (2018) 90.
208. Montcuquet A, Collongues N, Papeix C, Zephir H, Audoin B, Laplaud D, et al. Effectiveness of mycophenolate mofetil as first-line therapy in AQP4-IgG, MOG-IgG, and seronegative neuromyelitis optica spectrum disorders. Mult Scler. (2017) 23:1377–84. doi: 10.1177/1352458516678474
209. Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol. (2009) 66:1128–33. doi: 10.1001/archneurol.2009.175
210. Chen H, Zhang Y, Shi Z, Feng H, Yao S, Xie J, Zhou H. The Efficacy and tolerability of mycophenolate mofetil in treating neuromyelitis optica and neuromyelitis optica spectrum disorder in Western China. Clin Neuropharmacol. (2016) 39:81–7. doi: 10.1097/WNF.0000000000000131
211. Yang Y, Wang CJ, Wang BJ, Zeng ZL, Guo SG. Comparison of efficacy and tolerability of azathioprine, mycophenolate mofetil, and lower dosages of rituximab among patients with neuromyelitis optica spectrum disorder. J Neurol Sci. (2018) 385:192–7. doi: 10.1016/j.jns.2017.12.034
212. Chen H, Qiu W, Zhang Q, Wang J, Shi Z, Liu J, et al. Comparisons of the efficacy and tolerability of mycophenolate mofetil and azathioprine as treatments for neuromyelitis optica and neuromyelitis optica spectrum disorder. Eur J Neurol. (2017) 24:219–26. doi: 10.1111/ene.13186
213. Viswanathan S, Wong AHY, Quek AML, Yuki N. Intravenous immunoglobulin may reduce relapse frequency in neuromyelitis optica. J Neuroimmunol. (2015) 282:92–6. doi: 10.1016/j.jneuroim.2015.03.021
214. Elsone L, Panicker J, Mutch K, Boggild M, Appleton R, Jacob A. Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler. (2014) 20:501–4. doi: 10.1177/1352458513495938
215. Tackley G, O'Brien F, Rocha J, Woodhall M, Waters P, Chandratre S, et al. Neuromyelitis optica relapses: Race and rate, immunosuppression and impairment. Mult Scler Relat Disord. (2016) 7:21–5. doi: 10.1016/j.msard.2016.02.014
216. Kitley J, Elsone L, George J, Waters P, Woodhall M, Vincent A, et al. Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J Neurol Neurosurg Psychiatry (2013) 84:918–21. doi: 10.1136/jnnp-2012-304774
217. Ramanathan RS, Malhotra K, Scott T. Treatment of neuromyelitis optica/neuromyelitis optica spectrum disorders with methotrexate. BMC Neurol. (2014) 14:51. doi: 10.1186/1471-2377-14-51
218. Papeix C, Vidal JS, de Seze J, Pierrot-Deseilligny C, Tourbah A, Stankoff B, et al. Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler. (2007) 13:256–9. doi: 10.1177/1352458506070732
219. Palace J, Leite MI, Nairne A, Vincent A. Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. (2010) 67:1016–7. doi: 10.1001/archneurol.2010.188
220. Ayzenberg I, Schöllhammer J, Hoepner R, Hellwig K, Ringelstein M, Aktas O, et al. Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: a multicenter retrospective study. J Neurol. (2016) 263:575–82. doi: 10.1007/s00415-015-7991-1
221. Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung HP, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. (2012) 69:239–45. doi: 10.1001/archneurol.2011.216
222. Kowarik MC, Hoshi M, Hemmer B, Berthele A. Failure of alemtuzumab as a rescue in a NMOSD patient treated with rituximab. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e208. doi: 10.1212/NXI.0000000000000208
223. Gelfand JM, Cotter J, Klingman J, Huang EJ, Cree BAC. Massive CNS monocytic infiltration at autopsy in an alemtuzumab-treated patient with NMO. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e34. doi: 10.1212/NXI.0000000000000034
224. Fujihara K, Nakashima I. Secondary progression and innate immunity in NMO: A possible link to alemtuzumab therapy? Neurol Neuroimmunol Neuroinflamm. (2014) 1:e38. doi: 10.1212/NXI.0000000000000038
225. Azzopardi L, Cox AL, McCarthy CL, Jones JL, Coles AJ. Alemtuzumab use in neuromyelitis optica spectrum disorders: a brief case series. J Neurol. (2016) 263:25–9. doi: 10.1007/s00415-015-7925-y
226. Kira JI. Unexpected exacerbations following initiation of disease-modifying drugs in neuromyelitis optica spectrum disorder: Which factor is responsible, anti-aquaporin 4 antibodies, B cells, Th1 cells, Th2 cells, Th17 cells, or others? Mult Scler. (2017) 23:1300–2. doi: 10.1177/1352458517703803
227. Yoshii F, Moriya Y, Ohnuki T, Ryo M, Takahashi W. Fingolimod-induced leukoencephalopathy in a patient with neuromyelitis optica spectrum disorder. Mult Scler Relat Disord. (2016) 7:53–7. doi: 10.1016/j.msard.2016.03.004
228. Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. (2012) 18:113–5. doi: 10.1177/1352458511431973
229. Yamout BI, Beaini S, Zeineddine MM, Akkawi N. Catastrophic relapses following initiation of dimethyl fumarate in two patients with neuromyelitis optica spectrum disorder. Mult Scler. (2017) 23:1297–300. doi: 10.1177/1352458517694086
230. Gahlen A, Trampe AK, Haupeltshofer S, Ringelstein M, Aktas O, Berthele A, et al. Aquaporin-4 antibodies in patients treated with natalizumab for suspected MS. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e363. doi: 10.1212/NXI.0000000000000363
231. Cabre P, Olindo S, Marignier R, Jeannin S, Merle H, Smadja D, et al. Efficacy of mitoxantrone in neuromyelitis optica spectrum: clinical and neuroradiological study. J Neurol Neurosurg Psychiatry (2013) 84:511–6. doi: 10.1136/jnnp-2012-303121
232. Kim SH, Kim W, Park MS, Sohn EH, Li XF, Kim HJ. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol. (2011) 68:473–9. doi: 10.1001/archneurol.2010.322
233. Stroet A, Hemmelmann C, Starck M, Zettl U, Dörr J, Friedemann P, et al. Incidence of therapy-related acute leukaemia in mitoxantrone-treated multiple sclerosis patients in Germany. Ther Adv Neurol Disord. (2012) 5:75–9. doi: 10.1177/1756285611433318
234. Dörr J, Bitsch A, Schmailzl KJG, Chan A, von Ahsen N, Hummel M, et al. Severe cardiac failure in a patient with multiple sclerosis following low-dose mitoxantrone treatment. Neurology (2009) 73:991–3. doi: 10.1212/WNL.0b013e3181b878f6
235. Paul F, Dörr J, Würfel J, Vogel HP, Zipp F. Early mitoxantrone-induced cardiotoxicity in secondary progressive multiple sclerosis. J Neurol Neurosurg Psychiatry (2007) 78:198–200. doi: 10.1136/jnnp.2006.091033
236. Bichuetti DB, Oliveira EML, Boulos Fde C, Gabbai AA. Lack of response to pulse cyclophosphamide in neuromyelitis optica: evaluation of 7 patients. Arch Neurol. (2012) 69:938–9. doi: 10.1001/archneurol.2012.545
237. A Double-masked, Placebo-controlled Study With Open Label Period to Evaluate MEDI-551 in Neuromyelitis Optica and Neuromyelitis Optica Spectrum Disorders - Full Text View - ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT02200770 (Accessed April 18, 2018).
238. Chen D, Gallagher S, Monson NL, Herbst R, Wang Y. Inebilizumab, a B Cell-Depleting anti-CD19 antibody for the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med. (2016) 5:E107. doi: 10.3390/jcm5120107
239. Cree BA, Bennett JL, Sheehan M, Cohen J, Hartung HP, Aktas O, et al. Placebo-controlled study in neuromyelitis optica-Ethical and design considerations. Mult Scler. (2016) 22:862–72. doi: 10.1177/1352458515620934
240. Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. (2013) 12:554–62. doi: 10.1016/S1474-4422(13)70076-0
241. Paul F. Hope for a rare disease: eculizumab in neuromyelitis optica. Lancet Neurol. (2013) 12:529–31. doi: 10.1016/S1474-4422(13)70089-9
242. An Open Label Extension Trial of Eculizumab in Relapsing NMO Patients - Full Text View - ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT02003144 (Accessed April 17, 2018).
243. Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology (2014) 82:1302–6. doi: 10.1212/WNL.0000000000000317
244. Ringelstein M, Ayzenberg I, Harmel J, Lauenstein AS, Lensch E, Stögbauer F, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. (2015) 72:756–63. doi: 10.1001/jamaneurol.2015.0533
245. Tanaka M, Yanagida H, Suzumura A. Treatment for paraneoplastic neuromyelitis optica spectrum disorder (NMOsd): Probable effects of tocilizumab for both cancer and NMOsd. Mult Scler Relat Disord. (2018) 20:82–3. doi: 10.1016/j.msard.2017.12.002
246. Tocilizumab vs Azathioprine in Neuromyelitis Optica Spectrum Disorders - Full Text View - ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT03350633 (Accessed August 14, 2018)
247. Efficacy and Safety Study as Add-on Therapy of SA237 to Treat NMO and NMOSD - Full Text View - ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT02028884 (Accessed April 18, 2018).
248. Steinman L, Bar-Or A, Behne JM, Benitez-Ribas D, Chin PS, Clare-Salzler M, et al. Restoring immune tolerance in neuromyelitis optica: part I. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e276. doi: 10.1212/NXI.0000000000000276
249. Bar-Or A, Steinman L, Behne JM, Benitez-Ribas D, Chin PS, Clare-Salzler M, et al. Restoring immune tolerance in neuromyelitis optica: Part II. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e277. doi: 10.1212/NXI.0000000000000277
250. Paul F, Murphy O, Pardo S, Levy M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin Investig Drugs (2018) 27:265–71. doi: 10.1080/13543784.2018.1443077
Keywords: neuromyelitis optica, aquaporin-4 antibodies, MOG-encephalomyelitis, diagnostic criteria, immunosuppressive treatment
Citation: Borisow N, Mori M, Kuwabara S, Scheel M and Paul F (2018) Diagnosis and Treatment of NMO Spectrum Disorder and MOG-Encephalomyelitis. Front. Neurol. 9:888. doi: 10.3389/fneur.2018.00888
Received: 27 June 2018; Accepted: 01 October 2018;
Published: 23 October 2018.
Edited by:
Johann Sellner, Universitätsklinikum Salzburg, AustriaReviewed by:
Barbara M. P. Willekens, Antwerp University Hospital, BelgiumNicolas Collongues, Hôpitaux Universitaires de Strasbourg, France
Copyright © 2018 Borisow, Mori, Kuwabara, Scheel and Paul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadja Borisow, bmFkamEuYm9yaXNvd0BjaGFyaXRlLmRl