 Matthew D. Howe1
Matthew D. Howe1 Liang Zhu1
Liang Zhu1 Lauren H. Sansing2Nicole R. Gonzales1Louise D. McCullough1
Lauren H. Sansing2Nicole R. Gonzales1Louise D. McCullough1 Nancy J. Edwards3*
Nancy J. Edwards3*- 1Department of Neurology, McGovern Medical School, University of Texas Health Science Center, Houston, TX, United States
- 2Department of Neurology, Yale University School of Medicine, New Haven, CT, United States
- 3Neuroscience Department, Kaiser Permanente, Redwood City, CA, United States
Background: Intracerebral hemorrhage (ICH) is a stroke subtype associated with high disability and mortality. There is a clinical need for blood-based biomarkers that can aid in diagnosis, risk stratification, and prognostication. Given their role in the pathophysiology of ICH, we hypothesized markers of blood-brain barrier disruption and fibrosis would associate with neurologic deterioration and/or long-term functional outcomes. We also hypothesized these markers may be unique in patients with ICH due to cerebral amyloid angiopathy (CAA) vs. other etiologies.
Methods: Seventy-nine patients enrolled in prospective ICH registries at two separate hospitals (the University of Texas Health Science Center at Houston and Hartford Hospital) were included in this study. We assessed initial injury severity and admission variables along with measures of inpatient deterioration (hematoma expansion, perihematomal edema (PHE), and early and delayed neurologic deterioration) and functional outcome [modified Rankin Scale (mRS) score at discharge and 90 days]. Serial biospecimens were obtained at 5 pre-specified timepoints (within 24 h, 1–2, 3–5, 6–8, and 10 days); serum samples were analyzed for fibronectin, all three TGF-β isoforms, and 7 matrix metalloproteinases (MMPs).
Results: In our initial correlation analysis, MMP 10 and 3 were associated with hematoma expansion and early neurologic deterioration, whereas MMP 8 and MMP 1 were associated with PHE and delayed neurologic deterioration (respectively). Subacute levels of MMP 8 (sampled from day 6–10) positively correlated with PHE even after adjusting for multiple comparisons (p = 0.02). Acute levels of MMP 1, TGF-β1, and TGF-β3 were predictive of functional outcome, with TGF-β1 and TGF-β3 associating with 90 day mRS independent of age, hematoma volume, hemorrhage location, GCS, and IVH [p = 0.02; OR 1.03 (95% CI 1.0–1.05); p = 0.03; OR 3.1 (95% CI 1.1–8.8)]. When evaluated together as a panel, the cytokines distinguished patients with ICH due to CAA vs. ICH due to hypertension (AUC 0.81).
Conclusions: Serum levels of fibronectin, TGF-β, and MMPs may be useful in refining ICH etiology and prognosis. Further large-scale studies are needed to confirm these findings, particularly regarding patients with CAA.
Introduction
Intracerebral hemorrhage (ICH) accounts for approximately 15% of strokes and is a major cause of morbidity and mortality worldwide, affecting approximately 2 million individuals each year (1). Nearly 40% of ICH patients will die within 30 days, and of survivors, functional independence within 6 months is achieved by only 20% (2, 3). For spontaneous ICH, hypertension remains the most frequent etiology, though certain subsets of patients (such as elderly patients) may have other ICH causes such as antithrombotic use or cerebral amyloid angiopathy [CAA; (3, 4)]. In ICH, the exposure of brain tissue to blood components results in various neuroinflammatory cascades that lead to secondary injury after ICH. For instance, thrombin induces the activation of matrix metalloproteases (MMPs) that can then degrade basement membrane components of the blood-brain barrier (BBB), such as the protein fibronectin (5–9). BBB disruption can result in an influx of peripheral immune cells into the brain along with the formation of cerebral edema, referred to as perihematomal edema (PHE). Other mediators, such as transforming growth factor-β (TGF-β), may instead regulate neuroinflammation and promote repair processes such as the formation of glial scars (10, 11).
While these ICH injury and repair processes have been studied to a far greater extent in experimental models of ICH, there is less literature on the role of these mediators in patients with ICH. Several studies have suggested MMPs, fibronectin, and/or TGF-β are elaborated in both the brains and peripheral blood of ICH patients, but it is unclear if serum levels of these mediators could serve as diagnostic or prognostic markers. For instance, TGF-β1 can induce amyloid deposition in cerebral blood vessels but as of yet, peripheral levels of TGF-β have not reliably distinguished CAA hemorrhage patients from other ICH patients (12, 13). In addition, the identification of prognostic serum markers could guide individualized patient care and rehabilitation strategies and hopefully aid in the improvement of ICH outcomes.
In this study, we performed an exploratory analysis of serum levels of MMPs, fibronectin, and TGF-β across time in patients with ICH in the hope of ascertaining if and when these cytokines were predictive of clinically relevant short- and long-term outcomes. We also examined whether these cytokines could be useful (either individually or as a grouped panel) as diagnostic of ICH etiology, specifically ICH due to CAA.
Materials and Methods
Study Population
For this study, we used ICH patient data and serum samples from the University of Texas Health Science Center at Houston (UTH) and Hartford Hospital (HH). The study was approved by the Institutional Review Boards at both institutions, and all patients (or their surrogates) were consented. We abstracted data from prospective ICH databases detailing admission data, radiologic findings, hospital course, and functional outcomes. Seventy-nine patients with spontaneous, supratentorial cerebral hemorrhages of moderate size were included in the study. All patients were enrolled within 24 h of ICH ictus. Patient exclusions were the following: underlying vascular lesion (aneurysm, arteriovenous malformation), traumatic brain injury, venous sinus thrombosis, infratentorial hemorrhage location, and/or inadequate clinical follow-up.
Clinical and Radiologic Assessments
ICH severity was assessed via the National Institutes of Health Stroke Scale (NIHSS), Glasgow Coma Scale (GCS), and ICH score (14–16). Functional outcomes were assessed at discharge and 90 days post-injury by the modified Rankin Scale [mRS, (17)]. Our primary radiologic outcomes were hematoma volume (HV) and perihematomal edema (PHE), as quantified via computer-based analysis with MIPAV (Medical Image Processing, Analysis, and Visualization) software. Serial volumes were obtained by review of every head CT obtained during the patient's admission as a part of routine clinical care. ICH etiology was determined by the clinical team with independent verification by an experienced neurologist (NJE); patients were subdivided into deep hypertensive ICH, lobar ICH due to probable CAA (as defined via the modified Boston criteria), and lobar ICH due to “other” causes, such as coagulopathy (18). In the UTH cohort, the majority of CAA ICH patients had probable CAA with supporting pathological evidence.
Serum Sample Collection
Serum samples were obtained from our study cohort serially and at pre-specified timepoints: timepoint 1 (within 24 h), timepoint 2 (1–2 days post-injury), timepoint 3 (3–5 days), timepoint 4 (6–8 days), and timepoint 5 (10 days). All biospecimens were sampled by a team blinded to the clinical status of the patient. Samples were processed within 1 h of collection and stored at−80 degrees Celsius.
Measurement of Serum Analytes
Samples were thawed on ice for 1 h and thoroughly vortexed prior to beginning any assays. Serum fibronectin was measured using the Human Fibronectin Quantikine ELISA Kit (R&D Systems). Latent and active TGF-β1, TGF-β2, and TGF-β3 were measured using the Bio-Plex Pro TGF-β 3-PLEX assay (Bio-Rad). Similarly, MMP 1, MMP 2, MMP 3, MMP 7, MMP 8, MMP 9, and MMP 10 were measured using the Bio-Plex Pro Human MMP Panel (Bio-Rad). All assays were performed and read according to the manufacturer's instructions.
Statistical Analysis
Descriptive statistics were provided for demographic and baseline variables in our ICH patients. Nonparametric analysis (Spearman correlation, Wilcoxon rank sum test) was used to define serum cytokine associations with our outcomes of interest (hematoma expansion, early and delayed neurologic deterioration, PHE, discharge and 90 day mRS). For patients with serial samples (n = 36), we also grouped cytokine levels across acute (0–5 days) and subacute (6–10 days) timepoints, adjusting for multiple comparisons using the Benjamini and Hochberg procedure to control the false discovery rate (FDR) at 0.05 (19). Further analysis of cytokine association with functional outcome was performed via a multivariable logistic regression analysis adjusting for age, hematoma volume, hemorrhage location, admission GCS, and IVH. Logistic regression analysis was also used to generate a receiver operating characteristic (ROC) curve comparing cytokine signatures in CAA ICH patients vs. deep hypertensive ICH patients, with adjustment for hematoma volume. All data analyses were performed in SAS software 9.4 (Cary, NC).
Results
Baseline Cohort Findings
Baseline clinical variables are outlined in Table 1 (n = 79). The mean age of our cohort was 66.8 years old (± 14.3 years) and 39.7% of patients were female. Mean peak hematoma volume was 40.8 cm3 (± 26 cm3) and mean peak PHE volume was 26.5 cm3 (± 17.9 cm3). Intraventricular extension of hemorrhage was present in 65% of patients. 42 patients had deep hypertensive hemorrhages, and 37 had lobar hemorrhages.
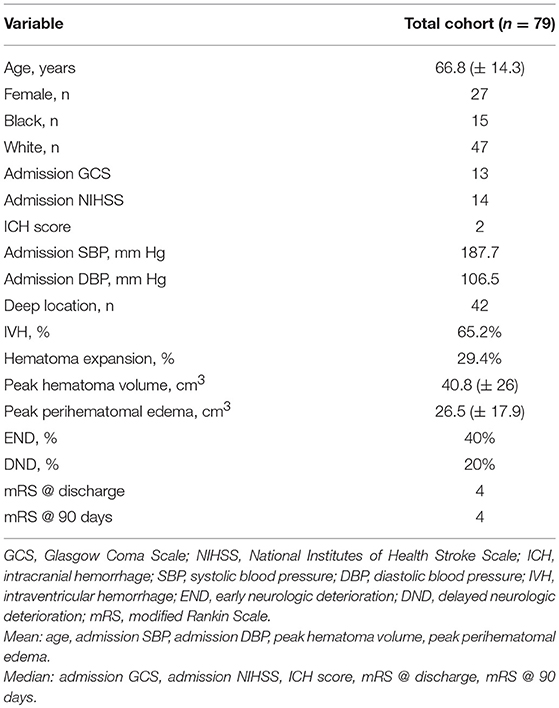
Table 1. Baseline patient demographics and ICH variables.
Temporal Association of Serum Markers With Clinical Deterioration and Functional Outcome
We characterized serum cytokine levels in our ICH patients across our 5 prespecified timepoints. 36 of our 79 patients had serial samples throughout. Univariate analysis was performed to ascertain cytokine associations with clinically relevant outcomes—hematoma expansion, early neurologic deterioration (END), delayed neurologic deterioration (DND), perihematomal edema volume, discharge mRS, and 90 day mRS. In Figure 1, we have displayed boxplots of serum cytokine levels across time in patients with or without our outcomes of interest; statistically significant associations are highlighted in Table 2. MMP 10 at an early timepoint (within 2 days of ICH ictus) was the only variable significantly associated with hematoma expansion (p = 0.03) and MMP 3 at timepoint 1 was the only variable significantly associated with END (p = 0.05). Interestingly, the “delayed” clinical deterioration variables—PHE and DND—were only associated with markers at later timepoints (MMP 8 and fibronectin at day 6–8 for PHE, p = 0.005 and 0.006; MMP 1 at day 3–5 for DND, p = 0.02). Serum markers associated with discharge disposition and mRS at discharge included MMP 1, MMP 8, and MMP 10. MMP 1 (p = 0.01), TGF-β1 (p = 0.05), and TGF-β2 (0.02) were associated with mRS at 90 days.
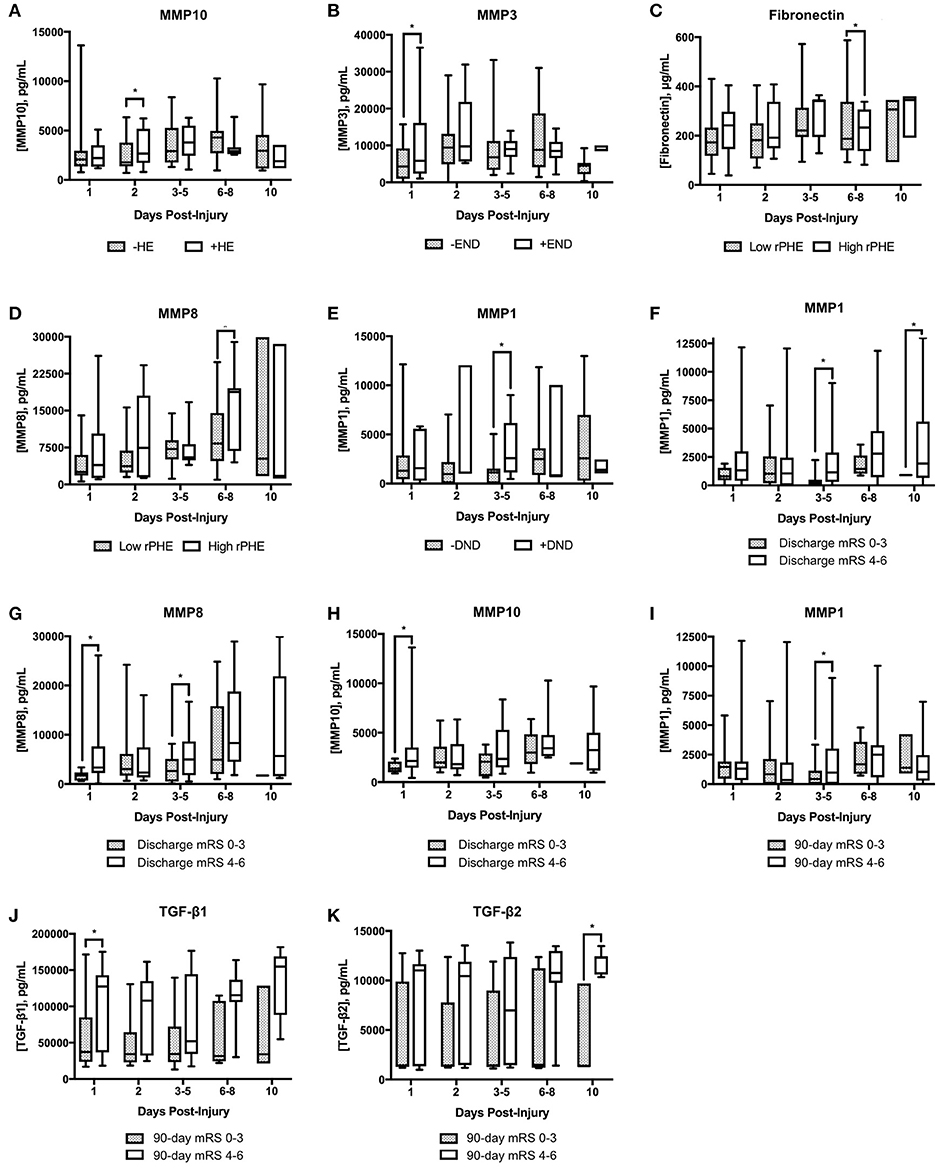
Figure 1. Box plots of serum cytokine levels across timepoints as separated by ICH outcomes. (A) MMP10 levels in patients without or with hematoma expansion (HE); (B) MMP3 in patients without or with early neurologic deterioration (END); (C) fibronectin in patients with low or high relative perihematomal edema (rPHE); (D) MMP8 levels in patients with low or high rPHE; (E) MMP1 in patients without or with delayed neurologic deterioration (DND); (F–H) cytokine levels in patients with favorable (0–3) vs. unfavorable (4–6) modified Rankin Scale (mRS) score at discharge; (I–K) cytokine levels in patients with favorable versus unfavorable mRS at 90 days. *p ≤ 0.05. MMP, matrix metalloproteinase; TGF, transforming growth factor.
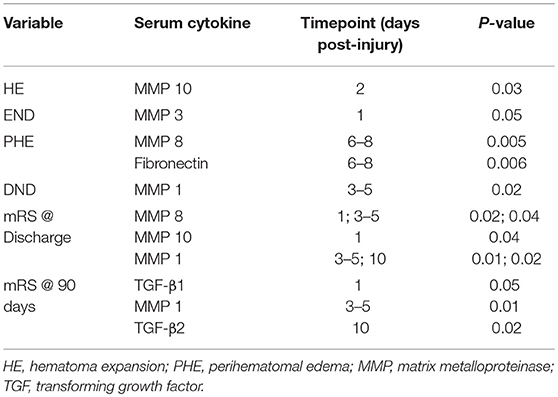
Table 2. Univariate analysis of serum marker associations with clinical and/or radiologic outcome measures.
Acute vs. Subacute Groupings
To determine if any of these markers would be useful if obtained within a temporal window rather than at one specific timepoint, we grouped acute (day 0–5) and subacute (day 6–10) timepoints together and reassessed the above associations. In this analysis, we corrected for multiple comparisons as described in the Methods section. Regarding our measures of short-term deterioration (hematoma expansion, END, DND, and PHE), the only association that remained significant upon adjustment was the association of subacute MMP 8 with PHE (p = 0.02). Functional outcomes at both discharge and at 90 days were significantly associated with acute levels of MMP 1 (p = 0.04, p = 0.007), TGF-β1 (p = 0.04, p = 0.003), and TGF-β3 (p = 0.04, p = 0.004). And, as displayed in Table 3, higher levels of TGF-β1 and TGF-β3 were predictive of poorer long-term functional outcome independent of patient age, hematoma volume, hemorrhage location, admission GCS, and IVH [p = 0.02, OR 1.03 (95% CI 1–1.05) and p = 0.03, OR 3.1 (95% CI 1.1–8.8), respectively].
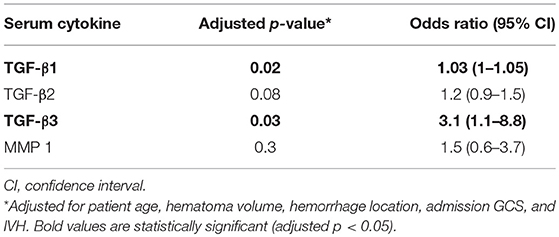
Table 3. Multivariate logistic regression analysis of marker association with poorer long-term functional outcome.
Molecular Signature Associated With CAA Hemorrhages
We next sought to determine if ICH etiology (ICH due to CAA, in particular) was associated with any particular serum marker or network of markers. When each marker was analyzed independently (adjusting for multiple comparisons), we did not find any statistically significant associations with ICH etiology. However, when we examined MMPs, fibronectin, and TGF-β together, the cytokine panel as a whole was able to distinguish CAA ICH patients from hypertensive ICH patients. Figure 2 demonstrates the receiver operating characteristic (ROC) curve, adjusted for hematoma volume, with an area under the curve (AUC) of 0.81.
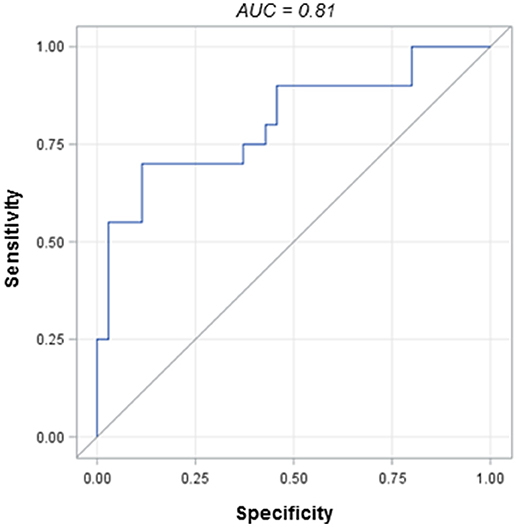
Figure 2. Receiver operating characteristic (ROC) curve for the cytokine panel in CAA vs. hypertensive ICH patients.
Discussion
The main findings of this study include the association of several of our candidate serum markers with various measures of ICH severity, deterioration, and functional outcome along with the identification of a serum molecular signature for patients with ICH due to CAA as compared to patients with ICH due to hypertensive hemorrhage. Several of the statistically significant associations presented here are novel, such as the association of MMP 8 with PHE. In addition, our study demonstrates certain associations being distributed throughout time, suggesting the time course of biosampling is relevant (the first several days vs. the first week or beyond). And though several of these markers have been previously demonstrated in other studies to be prognostic, this is the first study (to our knowledge) where these markers, when examined as a network, were useful as diagnostic markers for CAA-related ICH.
Numerous studies of ICH in both preclinical models and in patients have suggested a role for MMPs in the pathogenesis of ICH. For instance, in patients with ICH, MMP 3 and MMP 9 have been associated with deterioration due to cerebral edema and poorer functional outcomes (7, 8, 20, 21). In our study, MMP 3 and 9 were primarily associated with initial injury severity, hematoma volume, and early neurologic deterioration. We did not find a statistically significant association of MMP 3 or MMP 9 with PHE or long-term functional outcome. This may have been due to (1) the larger panel of markers being tested, and (2) our search was for outcome associations independent of previously demonstrated clinical prognosticators. For instance, in the study by Li et al, though initial analyses suggested MMP 3 was associated with absolute PHE volume, when a multivariable model including hematoma volume was used, MMP 3 was no longer an independent predictor of PHE—only HV was (20).
In our study, TGF-β1 and TGF-β3 were independently predictive of 90 day mRS, suggesting serum evaluations of these markers could be prognostically additive to scoring systems such as the ICH score. Taylor et al also reported an independent association of TGF-β1 with functional outcome in patients with ICH (10). In this study, though, rebounds in TGF-β1 levels by 72 h correlated with favorable functional outcome whereas in our study, higher absolute levels of TGF-β1/TGF-β3 throughout our sampling period correlated with poorer outcome. There are several potential explanations for the divergent findings: in the previous study, TGF-β1 was measured in plasma whereas we measured our cytokines in the serum; we did not compare cytokine levels to those of non-neurologic controls (whereas the previous study did); our patients were “sicker” overall—with higher hematoma volumes and ICH scores; we sampled additional timepoints through day 10. Interestingly, in our univariate analysis, higher TGF-β2 at day 10 in particular was associated with unfavorable 90 day mRS. Numerous studies of neuroinflammation in ICH have revealed a bitemporal time course where initially damaging cytokines may become helpful during subacute/chronic repair processes, and vice versa (22–25). Prolonged elevations of TGF-β peripherally may signal prolonged injury in the central nervous system; as a result, prolonged elevations of serum TGF-β may correlate with poorer recovery. Also, in preclinical models of stroke, absolute levels of TGF-β signaling was significantly higher in aged animals and associated with larger infarct size (11). All this highlights the need for large, multicenter biosampling studies in ICH, with standardized protocols and serial sampling including sampling weeks to months from ICH ictus (26).
Our study produced several additional novel findings. First was the association of the collagenases—MMP 1 and MMP 8–with clinicoradiologic deterioration. In particular, subacute levels of MMP 8 were positively correlated with absolute PHE in our cohort. These findings warrant further study in preclinical models of ICH and in ICH patients. In studies of bacterial meningitis, MMP 8 degrades occludin, an integral plasma-membrane protein present in the tight junctions that form the BBB (27, 28). And other studies have demonstrated a role for MMP 8 in modulating Th1/Th2 polarization and the production of TNFα by activated microglia (29–31).
The other novel finding of this study regards the potential use of these markers in diagnosing ICH due to CAA. Previous work has identified brain tissue changes in TGF-β, fibronectin, and MMP activation in CAA models (12, 32–36). For instance, in one study of aged transgenic mice, TGF-β1 was central to the deposition of beta amyloid in cerebral blood vessels and the meninges in response to tissue injury (12). There is far less literature examining the expression of these cytokines peripherally (e.g., in the serum) of CAA patients. Similar to the study by Greenberg et al. TGF-β levels alone were not diagnostic for CAA-related ICH (13). When evaluated in conjunction with fibronectin and our MMP panel, though, there was a unique marker signature in CAA ICH patients compared to hypertensive hemorrhage patients, in 2 independent study site cohorts. There is a need for a serum marker of CAA to complement clinical diagnostic criteria such as the MRI-based modified Boston criteria [especially as their sensitivity/specificity is decreased in patients who have yet to present with a symptomatic ICH, (37, 38)].
There are several limitations to this study. These are preliminary findings, and the size of our patient cohort is small. That being said, our patients were sampled in a serial fashion (essentially increasing our n) and the results demonstrated here could be considered hypothesis-generating. We did not find as many statistically significant associations of markers obtained during later timepoints, likely in part due to the fact that our sample numbers decreased if patients were discharged or died prior to day 10. We included patients with primarily moderate-sized, supratentorial hemorrhages only, thereby limiting the generalizability of our findings. Finally, regarding our ROC analysis, we excluded patients with lobar hemorrhages clinically determined to be due to causes other than CAA; that being said, there may be a subset of these patients who, upon obtaining tissue from hematoma evacuation or at autopsy, indeed have evidence of CAA.
Ethics Statement
This study was carried out in accordance with the recommendations of the Institutional Review Boards at the University of Texas Health Science Center at Houston and Hartford Hospital. The protocol was approved by the Institutional Review Boards at both institutions. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
MH: performing assays, organizing data, constructing initial manuscript drafts, editing drafts. LZ: all statistical analyses, editing drafts. LS: data acquisition/interpretation, editing drafts. NG: editing drafts. LM: study design, funding/sourcing assays, editing drafts. NE: study design, data acquisition/interpretation, writing the manuscript, editing drafts.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mukherjee D, Patil CG. Epidemiology and the global burden of stroke. World Neurosurg. (2011) 76(Suppl. 6):S85–90. doi: 10.1016/j.wneu.2011.07.023
2. Rost NS, Smith EE, Chang Y, Snider RW, Chanderraj R, Schwab K, et al. Prediction of functional outcome in patients with primary intracerebral hemorrhage: the FUNC score. Stroke (2008) 39:2304–9. doi: 10.1161/STROKEAHA.107.512202
3. Grysiewicz RA, Thomas K, Pandey DK. Epidemiology of ischemic and hemorrhagic stroke: incidence, prevalence, mortality, and risk factors. Neurol Clin. (2008) 26:871–95. doi: 10.1016/j.ncl.2008.07.003
4. Lovelock CE, Cordonnier C, Naka H, Al-Shahi Salman R, Sudlow CL, Edinburgh Stroke Study Group, et al. Antithrombotic drug use, cerebral microbleeds, and intracerebral hemorrhage: a systematic review of published and unpublished studies. Stroke (2010) 41:1222–8. doi: 10.1161/STROKEAHA.109.572594
5. Machida T, Takata F, Matsumoto J, Takenoshita H, Kimura I, Yamauchi A, et al. Brain pericytes are the most thrombin-sensitive matrix metalloproteinase-9-releasing cell type constituting the blood–brain barrier in vitro. Neurosci Lett. (2015) 599:109–14. doi: 10.1016/j.neulet.2015.05.028
6. Carmichael ST, Vespa PM, Saver JL, Coppola G, Geschwind DH, Starkman S, et al. Genomic profiles of damage and protection in human intracerebral hemorrhage. J Cereb Blood Flow Metab. (2008) 28:1860–75. doi: 10.1038/jcbfm.2008.77
7. Florczak-Rzepka M, Grond-Ginsbach C, Montaner J, Steiner T. Matrix metalloproteinases in human spontaneous intracerebral hemorrhage: an update. Cerebrovasc Dis. (2012) 34:249–62. doi: 10.1159/000341686
8. Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Davalos A. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke (2005) 36:86–91. doi: 10.1161/01.STR.0000149615.51204.0b
9. Sonni S, Lioutas VA, Selim MH. New avenues for treatment of intracranial hemorrhage. Curr Treat Options Cardiovasc Med. (2014) 16:277. doi: 10.1007/s11936-013-0277-y
10. Taylor RA, Chang CF, Goods BA, Hammond MD, Mac Grory B, Ai Y, et al. TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Invest. (2017) 127:280–92. doi: 10.1172/JCI88647
11. Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation. (2010) 7:62. doi: 10.1186/1742-2094-7-62
12. Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, et al. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease. Nature (1997) 389:603–6.
13. Greenberg SM, Cho HS, O'Donnell HC, Rosand J, Segal AZ, Younkin LH, et al. Plasma beta-amyloid peptide, transforming growth factor-beta 1, and risk for cerebral amyloid angiopathy. Ann N Y Acad Sci. (2000) 903:144–9. doi: 10.1111/j.1749-6632.2000.tb06361
14. Spilker J, Kongable G, Barch C, Braimah J, Brattina P, Daley S, et al. Using the NIH Stroke Scale to assess stroke patients. The NINDS rt-PA Stroke Study Group. J Neurosci Nurs. (1997) 29:384–92. doi: 10.1097/01376517-199712000-00008
15. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet (1974) 2:81–4. doi: 10.1016/S0140-6736(74)91639-0
16. Hemphill JC, Bonovich DC, Besmertis L, Manley GT, Johnston SC. The ICH score: a simple, reliable grading scale for intracerebral hemorrhage. Stroke (2001) 32:891–7. doi: 10.1161/01.STR.32.4.891
17. Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: implications for stroke clinical trials: a literature review and synthesis. Stroke (2007) 38:1091–6. doi: 10.1161/01.STR.0000258355.23810.c6
18. Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston Criteria. Neurology (2001) 56:537–9. doi: 10.1212/WNL.56.4.537
19. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Source J R Stat Soc Ser B. (1995) 57:289–300.
20. Li N, Liu YF, Ma L, Worthmann H, Wong YL, Wang YJ, et al. Association of molecular markers with perihematomal edema and clinical outcome in intracerebral hemorrhage. Stroke (2013) 44:658–63. doi: 10.1161/STROKEAHA.112.673590
21. Chang JJ, Emanuel BA, Mack WJ, Tsivgoulis G, Alexandrov AV. Matrix metalloproteinase-9: dual role and temporal profile in intracerebral hemorrhage. J Stroke Cerebrovasc Dis. (2014) 23:2498–505. doi: 10.1016/j.jstrokecerebrovasdis.2014.07.005
22. Doyle KP, Buckwalter MS. The double-edged sword of inflammation after stroke: what sharpens each edge? Ann Neurol. (2012) 71:729–31. doi: 10.1002/ana.23579
23. Mracsko E, Veltkamp R. Neuroinflammation after intracerebral hemorrhage. Front Cell Neurosci. (2014) 8:388. doi: 10.3389/fncel.2014.00388
24. Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke (2011) 42:1781–6. doi: 10.1161/STROKEAHA.110.596718
25. Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease – a double-edged sword. Neuron (2002) 35:419–32. doi: 10.1016/S0896-6273(02)00794-8
26. Senn R, Elkind MS V, Montaner J, Christ-Crain M, Katan M. Potential role of blood biomarkers in the management of nontraumatic intracerebral hemorrhage. Cerebrovasc Dis. (2014) 38:395–409. doi: 10.1159/000366470
27. Vandenbroucke RE, Dejonckheere E, Van Lint P, Demeestere D, Van Wonterghem E, Vanlaere I, et al. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-CSF barrier contributes to lethality during systemic inflammatory diseases. J Neurosci. (2012) 32:9805–16. doi: 10.1523/JNEUROSCI.0967-12.2012
28. Schubert-Unkmeir A, Konrad C, Slanina H, Czapek F, Hebling S, Frosch M. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog. (2010) 6:e10000874. doi: 10.1371/journal.ppat.1000874
29. Lee EJ, Han JE, Woo MS, Shin JA, Park EM, Kang JL, et al. Matrix metalloproteinase-8 plays a pivotal role in neuroinflammation by modulating TNF-α activation. J Immunol. (2014) 193:2384–93. doi: 10.4049/jimmunol.1303240
30. Wen G, Zhang C, Chen Q, Luong Ie A, Mustafa A, Ye S, et al. A novel role of matrix metalloproteinase-8 in macrophage differentiation and polarization. J Biol Chem. (2015) 290:19158–72. doi: 10.1074/jbc.M114.634022
31. Rella JM, Jilma B, Fabry A, Kaynar AM, Mayr FB. MMP-8 genotypes influence the inflammatory response in human endotoxemia. Inflammation (2014) 37:451–6 doi: 10.1007/s10753-013-9758-0
32. Zhao L, Arbel-Ornath M, Wang X, Betensky RA, Greenberg SM, Frosch MP, et al. Matrix metalloproteinase 9-mediated intracerebral hemorrhage induced by cerebral amyloid angiopathy. Neurobiol Aging. (2015) 36:2963–71. doi: 10.1016/j.neurobiolaging.2015.07.016
33. Hernandez-Guillamon M, Martinez-Saez E, Delgado P, Domingues-Montanari S, Boada C, Penalba A, et al. MMP-2/MMP-9 plasma level and brain expression in cerebral amyloid angiopathy-associated hemorrhagic stroke. Brain Pathol. (2012) 22:133-141. doi: 10.1111/j.1750-3639.2011.00512.x
34. Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C, et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta(2016) 1862:1037–46. doi: 10.1016/j.bbadis.2015.08.024
35. Hamaguchi T, Okino S, Sodeyama N, Itoh Y, Takahashi A, Otomo E, et al. Association of a polymorphism of the transforming growth factor-beta1 gene with cerebral amyloid angiopathy. J Neurol Neurosurg Psychiatry (2005) 76:696–9. doi: 10.1136/jnnp.2003.034454
36. Weiss R, Lifshitz V, Frenkel D. TGF-β1 affects endothelial cell interaction with macrophages and T cells leading to the development of cerebrovascular amyloidosis. Brain Behav Immun. (2011) 25:1017–24. doi: 10.1016/j.bbi.2010.11.012
37. van Rooden S, van der Grond J, van den Boom R, Haan J, Linn J, Greenberg SM, et al. Descriptive analysis of the boston criteria applied to a dutch-type cerebral amyloid angiopathy population. Stroke (2009) 40:3022–7. doi: 10.1161/STROKEAHA.109.554378
Keywords: intracerebral hemorrhage, biomarker, matrix metalloproteinase, fibronectin, TGF beta, cerebral amyloid angiopathy
Citation: Howe MD, Zhu L, Sansing LH, Gonzales NR, McCullough LD and Edwards NJ (2018) Serum Markers of Blood-Brain Barrier Remodeling and Fibrosis as Predictors of Etiology and Clinicoradiologic Outcome in Intracerebral Hemorrhage. Front. Neurol. 9:746. doi: 10.3389/fneur.2018.00746
Received: 10 May 2018; Accepted: 17 August 2018;
Published: 07 September 2018.
Edited by:
Anna Rosell, Vall d'Hebron Research Institute (VHIR), SpainReviewed by:
Christian Foerch, Universitätsklinikum Frankfurt, GermanyCraig S. Anderson, University of New South Wales, Australia
Copyright © 2018 Howe, Zhu, Sansing, Gonzales, McCullough and Edwards. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nancy J. Edwards, bmFuY3kuai5lZHdhcmRzQGtwLm9yZw==