 Joana R. Guedes
Joana R. Guedes Taotao Lao4
Taotao Lao4 Joseph El Khoury
Joseph El Khoury- 1Doctoral Programme in Experimental Biology and Biomedicine, CNC - Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
- 2Institute for Interdisciplinary Research, University of Coimbra, Coimbra, Portugal
- 3CNC - Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
- 4Center for Immunology and Inflammatory Diseases, Massachusetts General Hospital, Harvard Medical School, Charlestown, MA, United States
- 5Division of Infectious Diseases, Massachusetts General Hospital, Harvard Medical School, Charlestown, MA, United States
Chemokines and their receptors have been shown to affect amyloid-β (Aβ) and tau pathologies in mouse models of Alzheimer's disease (AD) by regulating microglia and monocyte-associated neuroinflammation, microglial movement and monocyte recruitment into the brain. These cells in turn can promote and mediate Aβ phagocytosis and degradation and tau phosphorylation. In this review we discuss published work in this field in mouse models of AD and review what is known about the contributions of microglial and monocyte chemokines and their receptors to amyloid and tau pathologies. We focus on the roles of the chemokine/chemokine receptor pairs CCL2/CCR2, CX3CL1/CX3CR1, CCL5/CCR5, CXCL10/CXCR3 and CXCL1/CXCR2, highlighting important knowledge gaps in this field. A full understanding of the functions of chemokines and their receptors in AD may guide the development of novel immunotherapies for this devastating disease.
Introduction
Neuroinflammation is an important contributor to Alzheimer's disease (AD) pathogenesis and progression (1, 2). Indeed, several inflammatory mediators such as tumor necrosis factor (TNF) and interleukin-1 (IL-1) are elevated in the brains of AD patients and mouse models of AD (3, 4). In addition, several variants in immune genes such as TREM2, CD33, and CR1 that regulate the inflammatory response have been identified by Genome-Wide Association Studies (GWAS) as genetic risk factors for AD (5–9). Microglia and recruited peripheral blood monocytes are the principal innate immune cells involved in the pathogenesis of AD and extensive evidence indicate that their functions are in part regulated by chemokines and their receptors (1).
Beta Amyloid Deposition and Tau Hyperphosphorylation are Hallmarks of AD
Clinically, AD is associated with dementia and progressive cognitive decline. Pathologically, AD is characterized by the presence of extracellular senile plaques and intraneuronal neurofibrillary tangles (NFTs). These protein deposits contain aggregates of the amyloid-β (Aβ) peptide and hyperphosphorylated microtubule-associated protein tau (p-τ), respectively. The processes of Aβ production and tau hyperphosphorylation involve different pathways. Aβ accumulation is the result of tandem cleavage of the amyloid precursor protein (APP) by secretases, whereas tau hyperphosphorylation results from the activity of several kinases. However, increasing evidence indicate that these abnormal protein deposits influence each other and have an additive effect on disease progression and that Aβ deposition appears to regulate tau pathology (10, 11). Indeed, in experimental animals, Aβ injection in the brain of P301L mice, a model for tau pathology, increases formation of NFTs (12). Furthermore, breeding mice expressing 5 early onset familial AD mutations (5XFAD) with mice expressing the tau P301S mutation (PS19) results in a ~10-fold aggravated tauopathy (13). In contrast, APP-KO mice subjected to subtoxic doses of soluble oligomeric forms of tau protein do not exhibit tau-induced defects in spatial/associative memory and its electrophysiological surrogate long-term potentiation (LTP), suggesting that tau pathology is dependent on APP expression (14). Aβ accumulation appears to promote tau hyperphosphorylation via activation of glycogen synthase kinase-3β (GSK-3β) (15).
The effects of Aβ deposition on tau pathology and neurodegeneration may not be necessarily due only to fibrillar Aβ deposits in senile plaques. In fact, cognitive deficits start before these plaques are visible and it has been proposed that the spectrum of Aβ species, including intraneuronal Aβ, soluble Aβ and Aβ oligomers also contribute to synaptic disruption and tau hyperphosphorylation. Regardless of the exact species of Aβ involved in this process, the co-localization of Aβ and p-τ in synaptic sites (16) suggest that the deposition of these protein aggregates and their contribution to neuronal loss is affected by their interactions. In this regard, strategies for treatment of AD should consider both Aβ and p-τ aggregates and how they influence each other (17).
The Mononuclear Phagocyte System in Alzheimer's Disease
There is strong supporting evidence that the inflammatory response in AD is in part driven by the interaction of Aβ with mononuclear phagocytes, including microglia and recruited peripheral blood monocytes (1, 18, 19). Multiple forms of Aβ accumulate in the AD brain before the development of visible senile plaques and formation of NFT. Since these forms of Aβ can interact with microglia and/or monocytes and lead to an inflammatory response, several groups have suggested that neuroinflammation is an early event in Aβ and tau pathologies (20–22), preceding the accumulation of larger visible protein deposits. The mononuclear phagocyte system has been extensively studied in the context of neuroinflammation in AD. However, the contribution of different cells types of this system, such as microglia and monocytes to AD pathogenesis is only beginning to be understood.
Microglia are the principal resident sentinels in the brain and can rapidly sense changes in their environment such as Aβ deposition, using a set of genes termed the sensome (23). Subsequent to sensing Aβ, the interaction of microglia with Aβ is a double-edged-sword. On one hand, microglia can phagocyte and clear Aβ deposits thereby limiting the progression of AD pathology (24, 25). In support of this pathway, reduced expression of Aβ-binding receptors in microglia from aged mice is associated with reduced Aβ phagocytosis and clearance and increased Aβ accumulation and disease progression (4). In contrast, the continuous interaction of microglia with Aβ induces the activation of the inflammasome pathway and produce several inflammatory mediators and neurotoxins, thereby contributing to the progression of AD in several ways (2, 19, 26–30). It is possible that persistent Aβ-induced microglial activation could also contribute to Aβ deposition, since continuous production of neurotoxic factors by microglia can induce neuronal apoptosis and consequent release of intraneuronal Aβ into the extracellular space. In addition, inflammatory cytokines as IL-1β, INF-γ, and TNF-α have been shown to upregulate β-secretase expression thereby increasing Aβ production (31). Such inflammatory cytokines also affect LTP in the hippocampus (32), suggesting that they can influence synaptic plasticity, which is essential for memory formation. In support of this, during microglia activation, their branches disappear, giving place to an amoeboid morphology which may limit their ability to refine and sustain synapses.
In contrast to what we know about microglia-Aβ interactions, less is known about microglia and NFTs. Microglia appear to promote tau propagation and contribute to the spreading of tau pathology in the brain (33, 34). Microglial activation also induces tau phosphorylation and aggregation (35). Some anti-inflammatory drugs have been shown to reduce p-τ in P301S and 3xTg AD mice (22, 36), but whether the mechanism is microglia-dependent remains to be determined. Published reports also suggest that injured neurons exhibiting tau hyperphosphorylation modulate microglia-mediated neuroinflammation (37). While these studies are compelling, they do not provide evidence of direct activation of microglia by p-τ. However, they suggest that neuroinflammation is closely linked to tau pathology.
In addition to studies on microglia function in AD, monocytes can infiltrate the AD brain and, although their role in AD progression is unclear at this time, it has been proposed that they have both detrimental and beneficial effects depending on the stage of disease development (38–40). Monocytes infiltrate the AD brain and clear perivascular Aβ (39, 41, 42). In support of this, stimulating monocyte infiltration by peripheral challenging with macrophage colony-stimulating factor (M-CSF) and lipopolysaccharide (LPS) or blocking immunosuppressive transforming growth factor beta (TFG-β) can attenuate AD pathology (43). While these studies suggest a protective role for monocytes in AD, monocytes, similar to microglia, can become activated and promote neuroinflammation and neurotoxicity (44).
In contrast to the studies supporting a role of monocytes in AD, a recent report using parabiosis experiments and staining for CD11b and CD45 suggests that monocytes do not infiltrate the brain in APP/PS1 and 5XFAD mouse models of amyloidosis (45). Interestingly, the same group also showed the presence of monocytes in the brain of 5XFAD mice when single cell RNAseq was performed on myeloid cells and more cell specific markers were analyzed (46). Additional mouse and human studies using single cell RNASeq, flow cytometry with multiple microglia and monocyte markers and in situ hybridization will help clarify this apparent discrepancy.
Chemokines are Essential for the Accumulation of Mononuclear Phagocytes in the Alzheimer's Disease Brain
Chemokines are chemotactic cytokines which mediate immune cells migration to sites of inflammation. Initially designated with specific protein names, chemokines are now classified based on the number of amino acids between two cysteine residues: α-chemokines, with the first cysteine residues separated by one amino acid (CXC); β-chemokines, with adjacent cysteine residues (CC); lymphotactin, with only two cysteines, and fractalkine (CX3CL1) in which the first two cysteine residues are separated by three amino acids (47, 48). These small chemoattractant proteins bind to chemokine receptors classified in the same manner. Different chemokine receptors are expressed on different immune cells. Monocytes, for example, express CCR1, CCR2, CCR5, CCR8, CXCR4, and some CX3CR1 (49). In contrast, microglia express very high levels of CX3CR1 and CCR5 and, to a lesser extent CXCR4, CXCR3, and CXCR2 (23).
Since chemokines mediate the infiltration of peripheral monocytes into the inflamed central nervous system (50), we proposed that the same occurs in AD (47, 48). The association between chemokines and AD is supported by studies showing that CCL2 and CCL5 expression are increased in the AD brain (51, 52). In vitro, microglia express CCL2 when incubated with Aβ (19) and neurons from AD brains have been shown to upregulate CCL5 expression (53). Moreover, CCL5 is essential to Aβ induced-microglia chemotaxis (54, 55). Similarly, the CX3CR1 ligand CX3CL1 is highly expressed on neurons. These studies suggest that chemokines are important in AD and possibly mediate the infiltration of peripheral monocytes into the AD brain and/or the accumulation of microglia at sites of Aβ deposition. Additional insights into the roles of chemokines, their receptors and mononuclear phagocytes in AD progression come from animal models where the function of chemokine receptors have been challenged.
CCL2/CCR2 Axis
When combined with Aβ and p-τ levels, CCL2 expression in the brain and cerebrospinal fluid (CSF) is a reliable predictor of AD severity (51, 56). In support of these observations, the CCL2 receptor, CCR2, was the first chemokine receptor shown to be associated with AD. We found that, in Tg2576 AD mice, CCR2 deficiency accelerates early disease progression by impairing the accumulation of mononuclear phagocytes (39). APP-CCR2−/− mice exhibited higher Aβ levels and reduced CD11b+ cell recruitment into the brain. Importantly, these mice showed higher mortality in a CCR2 gene dosage-dependent manner. Subsequent studies showed that the decrease in perivascular Aβ observed in our study was due to the lack of blood monocytes accumulation at these sites and possibly their infiltration in the brain parenchyma (39). These findings were corroborated by the finding that CCR2 deficiency worsened memory deficits and increased soluble Aβ in APP/PS1 mice (57). This study also showed that lack of CCR2 stimulated the expression of TGF-β receptors and CX3CR1 in plaque-associated microglia, implicating another chemokine receptor in AD pathology. The progressive cognitive decline in these mice was associated with a decrease in the numbers of CX3CR1lowLy6-ChighCCR2+Gr1+ circulating inflammatory monocytes (58) and restoring CCR2 expression in bone marrow cells reestablished memory capacities and decreased soluble Aβ accumulation (58). Altogether, these reports show that CCR2+ monocytes can be protective in AD.
We propose that CCR2 promotes the recruitment of monocytes initially from the bone marrow into the blood, then from the blood across the blood-brain barrier (BBB), to the perivascular space, then from the perivascular space to sites of Aβ deposition in the parenchyma where these cells can potentially clear Aβ by phagocytosis (Figure 1). In support of these findings, we recently showed that the CCL2/CCR2 axis was impaired in blood-derived monocytes from AD patients, causing a deficit in cell migration (59).
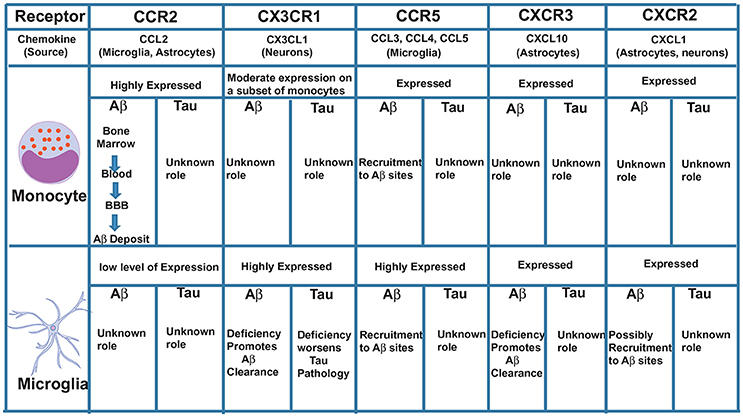
Figure 1. Major chemokine receptors expressed on monocytes and microglia and their possible roles in Aβ and Tau pathologies.
CX3CL1/CX3CR1 Axis
Another chemokine receptor thoroughly studied in AD is CX3CR1. This receptor is highly expressed in microglia and CX3CR1-GFP knock-in mice (where GFP replaced one CX3CR1 allele) have been used to specifically study, in vivo, the role of microglia in AD and other brain diseases. In physiological conditions, disruption of CX3CR1 function affects cognitive functions in a IL-1β-dependent manner (60) and exacerbates LPS-induced inflammation (61), suggesting that CX3CR1 maintains microglial homeostasis, being essential for their function in synaptic support and limiting their activation. In contrast to its clear role in maintaining microglial homeostatic functions under physiological conditions, dysregulation of the CX3CL1/CX3CR1 axis in AD mouse models can have both neuroprotective and neurotoxic effects depending on the mouse model used. CX3CR1 deficiency in three different AD mouse models—APP/PS1, R1.40 and CRND8—reduced amyloid deposits and enhanced Aβ phagocytic ability by microglia (62, 63). These effects were associated with decreased microglia activation and TNF levels and increased IL-1β levels. Similarly, following injection of Aβ1−40 fibrils in the hippocampus, downregulation of CX3CR1 with siRNAs also suppressed microglial activation, increasing synaptic strength and cognitive functions (64). On the other hand, CX3CR1 deficiency in hAPP mice worsened behavioral deficits associated with cytokine production independent of plaques deposition (65). Although these mice do not express tau mutations, the effect of CX3CR1 disruption was associated with enhanced tau pathology. Moreover, hTau mice (which exhibit p-τ) lacking CX3CR1 exhibited enhanced tau phosphorylation and aggregation associated with microglial activation, as well as behavioral impairments (35, 66). Analyzing these studies, it is interesting to observe that the neuroprotective role of CX3CR1 is always associated with tau and not Aβ. In support of these findings, overexpression of soluble CX3CL1 in mice with Tau but not Aβ pathology led to substantial improvements (67). Interestingly, CX3CR1 deletion in 3xTg mice, which have both Aβ and tau pathologies, prevented neuronal loss, suggesting that the effect of CX3CR1 deficiency on Aβ pathology may be more dominant than its effect on tau pathology (68). It is possible, however, that CX3CR1 is involved in the killing of neurons with intracellular tau deposits and the subsequent release of tau (68) (Figure 1). Additional studies with AD models that exhibit both Aβ and tau pathologies are needed to definitively clarify the role of CX3CR1 in AD.
CCL5/CCR5 Axis
CCR5+ reactive microglia are found associated with Aβ deposits in AD patients (69). CCR5−/− mice have higher levels of Aβ, C99 [a product that results from APP cleavage by β-secretase 1 (BACE1)] and BACE1 itself compared to normal mice (70). These levels were associated with astrocyte activation and CCR2 overexpression leading to cognitive impairments. Also, following injection of Aβ1−40 into the lateral ventricle of CCR5−/− mice, a reduced activation of microglia and astrocytes was observed compared to wild-type mice (71). Interestingly, CCR5 appears to be essential for the transendothelial migration of T cells across the BBB in the hippocampus of rats injected with Aβ (72), suggesting that CCR5 may be essential for the infiltration of non-phagocytic peripheral immune cells to the AD brain. Since CCR5 is also expressed on a subset of monocytes and on microglia, it is possible that it plays an additive role to CCR2 in mediating monocyte movement across the BBB, as well as in the movement of microglia toward sites of Aβ deposition in the parenchyma (Figure 1).
In addition to CCL5, we have shown that microglia and macrophages stimulated with Aβ, in vitro, show increased mRNA levels for the chemokines CCL3 and CCL4 also known as MIP1α and MIP1β respectively (19). These two chemokines are also upregulated in adult human microglia isolated from post-mortem brains and stimulated with Aβ (73) and in plaque-associated microglia in AD patients (74). The exact roles of these chemokines in AD is not clear. However, both chemokines are ligands for CCR5, suggesting possible complementary roles to CCL5 in the accumulation of T cells, monocytes or microglia in the AD brain. To date, there are no published reports describing the effects of CCL3, CCL4, or CCL5 deficiency on Aβ deposition or tau pathology in a transgenic Aβ or tau model. Such studies will help clarify the role of CCR5 ligands in AD development and progression, an important current knowledge gap in the field.
CXCL10/CXCR3 Axis
CXCL10 levels in CSF are significantly increased in patients with amnestic mild cognitive impairment (MCI) and patients with mild AD, but not in patients with severe AD (75). Unlike its receptor CXCR3, which is constitutively expressed on microglia and neurons, CXCL10 is expressed in a subpopulation of astrocytes in the normal brain and is markedly elevated in astrocytes in AD (76, 77). Peripheral injection of LPS strongly induces CXCL10 the brain of rats, as well as in cultured astrocytes and microglia (78), indicating its involvement in the response to inflammatory stimuli. Although CXCL10 was found to co-localize with Aβ plaques in APP mice (79), its function in regulating Aβ pathology remains unclear. A recent study using CXCR3-deficient AD mice showed that deletion of CXCR3 significantly reduced plaque burden and Aβ levels in APP/PS1 mice with morphological evidence for microglial activation, but reduced plaque association. This study suggests a possible role in the recruitment of microglia to Aβ deposits (80). CXCR3 deficiency also increased microglial uptake of Aβ, reduced concentrations of proinflammatory cytokines and attenuated behavioral deficits in APP/PS1 mice (80), suggesting an important role for CXCL10/CXCR3 signaling in mediating Aβ-induced pathology in mouse models. The role of CX3CR1 in tau pathology is not clear and needs to be investigated (Figure 1).
CXCL1/CXCR2 Axis
CXCR2 is expressed at low levels on microglia (23). CXCR2 deficiency results in reduction of Aβ with concurrent increases of γ-secretase substrates in the APP/PS1 mice (81). Whether microglia play a direct role in this process is not clear and requires a more detailed analysis of CXCR2- deficient APP/PS1 mice. Insights into a possible microglial role on the effect of CXCR2 depletion came from studies performed in rats injected with Aβ. Administration of a competitive CXCR2 antagonist to Aβ-injected rats significantly reduced expression of CXCR2 and microgliosis (82). Similar studies in mouse models of AD need to be done to further validate these findings.
Chemokines Receptors as Targets for AD Therapy
Based on the above discussion, it may be possible, in the future, to consider chemokine receptors as potential targets for the treatment of AD. One could envision upregulating CCR2 expression to increase influx of monocytes into the AD brain, therefore leading to increased removal of Aβ deposits and reduced Aβ burden. However, it will be difficult to devise such a strategy for CX3CR1 and CCR5, until the effects of deleting these receptors on both Aβ- and tau-associated pathologies are defined in animal models that exhibit both features. Importantly, one must consider that these receptors can display different functions in different stages of AD progression.
Conclusion
The work summarized in this review suggests that chemokines and their receptors are important for AD pathogenesis and in the development of the two main pathological hallmarks of AD—Aβ deposition and tau hyperphosphorylation. These receptors are associated with innate and adaptive responses regulating microglia and peripheral immune cells activation, and are essential for the infiltration of immune cells to the AD brain and movement of recruited monocytes and microglia toward sites of Aβ deposition. These studies clearly point to the importance of the neuroinflammatory component of AD as an active process which contributes to AD development and suggest that the presence of immune cells in the AD brain is tightly regulated during different stages of AD progression, constituting a double-edged sword that may lead to neuroprotective or neurotoxic outcomes. However, in spite of the significant amount of knowledge gained so far, there are important knowledge gaps that limit our understanding of the roles of chemokines and their receptors in AD. Additional studies to explore how these receptors and their ligands influence Aβ deposition, tau hyperphosphorylation, microglia and monocyte accumulation, the overall inflammatory response and neuronal degeneration are needed. A full understanding of the roles of chemokines and their receptors in AD may guide the development of multiple novel immunotherapies targeted to various stages of the disease.
Author Contributions
JRG and JE wrote the initial version of the manuscript, then all authors contributed to writing and editing the submitted manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JE is funded by NIH grants 1RF1 AG051506 and R01 AI119065. JRG was a recipient of a PhD fellowship (SFRH/BD/51677/2011) and is now a recipient of a Post-doctoral fellowship (SFRH/BPD/120611/2016) both funded by the Portuguese Foundation for Science and Technology (FCT). ALC is the recipient of a Post-doctoral fellowship from FCT with reference SFRH/BPD/108312/2015.
References
1. El Khoury J. Neurodegeneration and the neuroimmune system. Nat Med. (2010) 16:1369–70. doi: 10.1038/nm1210-1369
2. Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
3. Lopez-Gonzalez I, Schluter A, Aso E, Garcia-Esparcia P, Ansoleaga B, LLorens F, et al. Neuroinflammatory signals in alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol. (2015) 74:319–44. doi: 10.1097/NEN.0000000000000176
4. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. (2008) 28: 8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
5. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. Alzheimer genetic analysis, TREM2 variants in Alzheimer's disease. N Engl J Med. (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
6. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. (2009) 41:1088–93. doi: 10.1038/ng.440
7. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. (2011) 43:429–35. doi: 10.1038/ng.803
8. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. (2009) 41:1094–9. doi: 10.1038/ng.439
9. Hickman SE, El Khoury J. TREM2 and the neuroimmunology of Alzheimer's disease. Biochem Pharmacol. (2014) 88:495–8. doi: 10.1016/j.bcp.2013.11.021
10. Amar F, Sherman MA, Rush T, Larson M, Boyle G, Chang L, et al. The amyloid-beta oligomer Abeta*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci Signal. (2017) 10:eaal2021. doi: 10.1126/scisignal.aal2021
11. Fagan AM, Vos SJ. Preclinical Alzheimer's disease criteria. Lancet Neurol. (2013) 12:1134. doi: 10.1016/S1474-4422(13)70194-7
12. Gotz J, Chen F, van Dorpe J, Nitsch RM, Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science (2001) 293:1491–5. doi: 10.1126/science.1062097
13. Stancu IC, Ris L, Vasconcelos B, Marinangeli C, Goeminne L, Laporte V, et al. Tauopathy contributes to synaptic and cognitive deficits in a murine model for Alzheimer's disease. FASEB J. (2014) 28:2620–31. doi: 10.1096/fj.13-246702
14. Puzzo D, Piacentini R, Fa M, Gulisano W, Li Puma DD, Staniszewski A, et al. LTP and memory impairment caused by extracellular Abeta and Tau oligomers is APP-dependent. Elife (2017) 6:e26991. doi: 10.7554/eLife.26991
15. Tokutake T, Kasuga K, Yajima R, Sekine Y, Tezuka T, Nishizawa M, et al. Hyperphosphorylation of Tau induced by naturally secreted amyloid-beta at nanomolar concentrations is modulated by insulin-dependent Akt-GSK3beta signaling pathway. J Biol Chem. (2012) 287:35222–33. doi: 10.1074/jbc.M112.348300
16. Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK, Co-occurrence of Alzheimer's disease ss-amyloid and tau pathologies at synapses. Neurobiol Aging (2010) 31:1145–52. doi: 10.1016/j.neurobiolaging.2008.07.021
17. Lanzillotta A, Sarnico I, Benarese M, Branca C, Baiguera C, Hutter-Paier B, et al. The gamma-secretase modulator CHF5074 reduces the accumulation of native hyperphosphorylated tau in a transgenic mouse model of Alzheimer's disease. J Mol Neurosci. (2011) 45:22–31. doi: 10.1007/s12031-010-9482-2
18. Hickman SE, El Khoury J, The neuroimmune system in Alzheimer's disease: the glass is half full. J Alzheimers Dis. (2013) 33(Suppl. 1):S295–302. doi: 10.3233/JAD-2012-129027
19. El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med. (2003) 197:1657–66. doi: 10.1084/jem.20021546
20. Guedes JR, Custodia CM, Silva RJ, de Almeida LP, Pedroso de Lima MC, Cardoso AL. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer's disease triple transgenic mouse model. Hum Mol Genet. (2014) 23:6286–301. doi: 10.1093/hmg/ddu348
21. Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, et al. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer's disease. PLoS ONE (2013) 8:e59586. doi: 10.1371/journal.pone.0059586
22. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron (2007) 53:337–51. doi: 10.1016/j.neuron.2007.01.010
23. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. (2013) 16:1896–905. doi: 10.1038/nn.3554
24. Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, et al. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat Commun. (2013) 4:2030. doi: 10.1038/ncomms3030
25. Cornejo F, Vruwink M, Metz C, Munoz P, Salgado N, Poblete J, et al. Scavenger Receptor-A deficiency impairs immune response of microglia and astrocytes potentiating Alzheimer's disease pathophysiology. Brain Behav Immun. (2018) 69:336–50. doi: 10.1016/j.bbi.2017.12.007
26. El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD, Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature (1996) 382:716–9. doi: 10.1038/382716a0
27. Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, et al. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. (2002) 160:101–12. doi: 10.1016/S0002-9440(10)64354-4
28. Gold M, El Khoury J. Beta-amyloid, microglia, and the inflammasome in Alzheimer's disease. Semin Immunopathol. (2015) 37:607–11. doi: 10.1007/s00281-015-0518-0
29. Heneka MT, Golenbock DT, Latz E, Innate immunity in Alzheimer's disease. Nat Immunol. (2015) 16:229–36. doi: 10.1038/ni.3102
30. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer's disease. Nature (2017) 552:355–61. doi: 10.1038/nature25158
31. Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, et al. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. (2007) 170:680–92. doi: 10.2353/ajpath.2007.060378
32. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. (2011) 25:181–213. doi: 10.1016/j.bbi.2010.10.015
33. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. (2015) 18:1584–93. doi: 10.1038/nn.4132
34. Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain (2015) 138:1738–55. doi: 10.1093/brain/awv081
35. Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron (2010) 68:19–31. doi: 10.1016/j.neuron.2010.08.023
36. Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol. (2009) 183:1375–83. doi: 10.4049/jimmunol.0901005
37. Lastres-Becker I, Innamorato NG, Jaworski T, Rabano A, Kugler S, Van Leuven F, et al. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain (2014) 137:78–91. doi: 10.1093/brain/awt323
38. Hohsfield LA, Humpel C. Migration of blood cells to beta-amyloid plaques in Alzheimer's disease. Exp Gerontol. (2015) 65:8–15. doi: 10.1016/j.exger.2015.03.002
39. El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. (2007) 13:432–8. doi: 10.1038/nm1555
40. Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Li S, Ljubimov V, et al. Therapeutic effects of glatiramer acetate and grafted CD115(+) monocytes in a mouse model of Alzheimer's disease. Brain (2015) 138:2399–422. doi: 10.1093/brain/awv150
41. Lebson L, Nash K, Kamath S, Herber D, Carty N, Lee DC, et al. Trafficking CD11b-positive blood cells deliver therapeutic genes to the brain of amyloid-depositing transgenic mice. J Neurosci. (2010) 30:9651–8. doi: 10.1523/JNEUROSCI.0329-10.2010
42. Michaud JP, Bellavance MA, Prefontaine P, Rivest S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. (2013) 5:646–53. doi: 10.1016/j.celrep.2013.10.010
43. Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci USA. (2009) 106:1261–6. doi: 10.1073/pnas.0805453106
44. Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F. Beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem. (1999) 274:15493–9. doi: 10.1074/jbc.274.22.15493
45. Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. (2016) 213:667–75. doi: 10.1084/jem.20151948
46. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell (2017) 169:1276–1290e17. doi: 10.1016/j.cell.2017.05.018
47. Hickman SE, El Khoury J. Mechanisms of mononuclear phagocyte recruitment in Alzheimer's disease. CNS Neurol Disord Drug Targets (2010) 9:168–73. doi: 10.2174/187152710791011982
48. El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer's disease: therapeutic implications. Trends Pharmacol Sci. (2008) 29:626–32. doi: 10.1016/j.tips.2008.08.004
49. Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med. (1998) 338:436–45. doi: 10.1056/NEJM199802123380706
50. Djukic M, Mildner A, Schmidt H, Czesnik D, Bruck W, Priller J, et al. Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain (2006) 129:2394–403. doi: 10.1093/brain/awl206
51. Sokolova A, Hill MD, Rahimi F, Warden LA, Halliday GM, Shepherd CE. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer's disease. Brain Pathol. (2009) 19:392–8. doi: 10.1111/j.1750-3639.2008.00188.x
52. Tripathy D, Thirumangalakudi L, Grammas P. RANTES upregulation in the Alzheimer's disease brain: a possible neuroprotective role. Neurobiol Aging (2010) 31:8–16. doi: 10.1016/j.neurobiolaging.2008.03.009
53. Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, et al. Ineffective phagocytosis of amyloid-ß by macrophages of Alzheimer's disease patients. J Alzheimer's Dis. (2005) 7:221–32. doi: 10.3233/JAD-2005-7304
54. Huang WC, Yen FC, Shie FS, Pan CM, Shiao YJ, Yang CN, et al. TGF-beta1 blockade of microglial chemotaxis toward Abeta aggregates involves SMAD signaling and down-regulation of CCL5. J Neuroinflamm. (2010) 7:28. doi: 10.1186/1742-2094-7-28
55. Lee JK, Schuchman EH, Jin HK, Bae JS. Soluble CCL5 derived from bone marrow-derived mesenchymal stem cells and activated by amyloid beta ameliorates Alzheimer's disease in mice by recruiting bone marrow-induced microglia immune responses. Stem Cells (2012) 30:1544–55. doi: 10.1002/stem.1125
56. Westin K, Buchhave P, Nielsen H, Minthon L, Janciauskiene S, Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer's disease. PLoS ONE (2012) 7:e30525. doi: 10.1371/journal.pone.0030525
57. Naert G, Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer's disease. J Neurosci. (2011) 31:6208–20. doi: 10.1523/JNEUROSCI.0299-11.2011
58. Naert G, Rivest S. Hematopoietic CC-chemokine receptor 2 (CCR2) competent cells are protective for the cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer's disease. Mol Med. (2012) 18:297–313. doi: 10.2119/molmed.2011.00306
59. Guedes JR, Santana I, Cunha C, Duro D, Almeida MR, Cardoso AM, et al. MicroRNA deregulation and chemotaxis and phagocytosis impairment in Alzheimer's disease. Alzheimers Dement. (2016) 3:7–17. doi: 10.1016/j.dadm.2015.11.004
60. Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. (2011) 31:16241–50. doi: 10.1523/JNEUROSCI.3667-11.2011
61. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. (2006) 9:917–24. doi: 10.1038/nn1715
62. Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. (2010) 177:2549–62. doi: 10.2353/ajpath.2010.100265
63. Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. (2010) 30:17091–101. doi: 10.1523/JNEUROSCI.4403-10.2010
64. Wu J, Bie B, Yang H, Xu JJ, Brown DL, Naguib M. Suppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiency. Neurobiol Aging (2013) 34:2843–52. doi: 10.1016/j.neurobiolaging.2013.06.003
65. Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, et al. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. (2011) 286:32713–22. doi: 10.1074/jbc.M111.254268
66. Bolos M, Llorens-Martin M, Perea JR, Jurado-Arjona J, Rabano A, Hernandez F, et al. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol Neurodegener. (2017) 12:59. doi: 10.1186/s13024-017-0200-1
67. Lee S, Xu G, Jay TR, Bhatta S, Kim KW, Jung S, et al. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J Neurosci. (2014) 34:12538–46. doi: 10.1523/JNEUROSCI.0853-14.2014
68. Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. (2010) 13:411–3. doi: 10.1038/nn.2511
69. Xia M, Qin S, Wu L, Mackay CR, Hyman BT. Immunohistochemical study of the β-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer's disease brains. Am J Pathol. (1998) 153:31–7. doi: 10.1016/S0002-9440(10)65542-3
70. Lee YK, Kwak DH, Oh KW, Nam SY, Lee BJ, Yun YW, et al. CCR5 deficiency induces astrocyte activation, Abeta deposit and impaired memory function. Neurobiology Learn Mem. (2009) 92:356–63. doi: 10.1016/j.nlm.2009.04.003
71. Passos GF, Figueiredo CP, Prediger RD, Pandolfo P, Duarte FS, Medeiros R, et al. Role of the macrophage inflammatory protein-1alpha/CC chemokine receptor 5 signaling pathway in the neuroinflammatory response and cognitive deficits induced by beta-amyloid peptide. Am J Pathol. (2009) 175:1586–97. doi: 10.2353/ajpath.2009.081113
72. Man SM, Ma YR, Shang DS, Zhao WD, Li B, Guo DW, et al. Peripheral T cells overexpress MIP-1alpha to enhance its transendothelial migration in Alzheimer's disease. Neurobiol Aging (2007) 28:485–96. doi: 10.1016/j.neurobiolaging.2006.02.013
73. Walker DG, Lue LF, Beach TG. Gene expression profiling of amyloid beta peptide-stimulated human post-mortem brain microglia. Neurobiol Aging (2001) 22:957–66. doi: 10.1016/S0197-4580(01)00306-2
74. Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, Eggen BJL, et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer's disease. Neurobiol Aging (2017) 55:115–22. doi: 10.1016/j.neurobiolaging.2017.03.021
75. Galimberti D, Schoonenboom N, Scheltens P, Fenoglio C, Bouwman F, Venturelli E, et al. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch Neurol. (2006) 63:538–43. doi: 10.1001/archneur.63.4.538
76. Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer's disease. J Neuroimmunol. (2000) 108:227–35. doi: 10.1016/S0165-5728(00)00285-X
77. Zaheer S, Thangavel R, Wu Y, Khan MM, Kempuraj D, Zaheer A. Enhanced expression of glia maturation factor correlates with glial activation in the brain of triple transgenic Alzheimer's disease mice. Neurochem Res. (2013) 38:218–25. doi: 10.1007/s11064-012-0913-z
78. Ren LQ, Gourmala N, Boddeke HW, Gebicke-Haerter PJ. Lipopolysaccharide-induced expression of IP-10 mRNA in rat brain and in cultured rat astrocytes and microglia. Brain Res Mol Brain Res. (1998) 59:256–63. doi: 10.1016/S0169-328X(98)00170-3
79. Duan RS, Yang X, Chen ZG, Lu MO, Morris C, Winblad B, et al. Decreased fractalkine and increased IP-10 expression in aged brain of APP(swe) transgenic mice. Neurochem Res. (2008) 33:1085–9. doi: 10.1007/s11064-007-9554-z
80. Krauthausen M, Kummer MP, Zimmermann J, Reyes-Irisarri E, Terwel D, Bulic B, et al. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer's disease model. J Clin Invest. (2015) 125:365–78. doi: 10.1172/JCI66771
81. Bakshi P, Margenthaler E, Reed J, Crawford F, Mullan M. Depletion of CXCR2 inhibits gamma-secretase activity and amyloid-beta production in a murine model of Alzheimer's disease. Cytokine (2011) 53:163–9. doi: 10.1016/j.cyto.2010.10.008
Keywords: Alzheimer's disease, amyloid-β peptide, protein tau, microglia, monocytes, chemokine receptors, chemokines, neuroinflammation
Citation: Guedes JR, Lao T, Cardoso AL and El Khoury J (2018) Roles of Microglial and Monocyte Chemokines and Their Receptors in Regulating Alzheimer's Disease-Associated Amyloid-β and Tau Pathologies. Front. Neurol. 9:549. doi: 10.3389/fneur.2018.00549
Received: 31 March 2018; Accepted: 19 June 2018;
Published: 14 August 2018.
Edited by:
Beatriz Gomez Perez-Nievas, King's College London, United KingdomReviewed by:
Tyler Kent Ulland, Washington University in St. Louis, United StatesKiran Bhaskar, University of New Mexico, United States
Copyright © 2018 Guedes, Lao, Cardoso and El Khoury. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph El Khoury, jelkhoury@mgh.harvard.edu