 Rosangela Ferese1
Rosangela Ferese1 Simona Scala1Francesca Biagioni1Emiliano Giardina2,3Stefania Zampatti1,2Nicola Modugno1Claudio Colonnese1,4Marianna Storto1
Simona Scala1Francesca Biagioni1Emiliano Giardina2,3Stefania Zampatti1,2Nicola Modugno1Claudio Colonnese1,4Marianna Storto1 Francesco Fornai1,5Giuseppe Novelli1,3Stefano Ruggieri1
Francesco Fornai1,5Giuseppe Novelli1,3Stefano Ruggieri1 Stefano Gambardella1*
Stefano Gambardella1*- 1IRCCS Neuromed, Pozzilli, Italy
- 2Molecular Genetics Laboratory UILDM, Santa Lucia Foundation, Rome, Italy
- 3Department of Biomedicine and Prevention, University of Rome Tor Vergata, Rome, Italy
- 4DAI Neurology and Psichiatry, Department of Neuroradiology, Policlinico Umberto I, Sapienza University of Rome, Rome, Italy
- 5Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Pisa, Italy
Mutations of PLA2G6 gene are responsible for PARK14, an autosomal recessive L-DOPA responsive dystonia/parkinsonism with early/adult onset. This phenotype possesses an high clinical variability, which consists in the occurrence of cerebral and cerebellar atrophy, iron accumulation in the basal ganglia, and cognitive decline. This report describes a PD patient carrying an heterozygous PLA2G6 mutation, which was identified also in his PD affected sister. This patient is characterized by a L-DOPA responsive typical parkinsonian syndrome without the occurrence of dystonia, a slight cognitive decline, presence of iron accumulation both in neo and paleostriatum while cerebellar atrophy was absent. Clinical and imaging features are compatible with the PARK14 phenotype. Although PARK14 has been previously reported to be inherited as a recessive disorder, clinical and genetic analysis of this proband and his family rise the hypothesis that even heterozygous PLA2G6 mutations may cause PARK14. It remains to be analyzed whether these heterozygous variants may act as dominant mutations, or they merely increase the risk to develop PD by acting within a context of synergistic genetic and/or environmental backgrounds.
Background
Phospholipase A2-associated neurodegeneration (PLAN), a syndrome of Neurodegeneration with Brain Iron Accumulation (NBIA), is an autosomal recessive neurological disorder caused by mutations in the PLA2G6 gene (1).
PLAN syndrome encompasses a group of phenotypes like classic infantile neuroaxonal dystrophy (INAD), and atypical neuroaxonal dystrophy of childhood-onset (atypical NAD), characterized by high levels of iron, especially in the globus pallidus (2, 3).
PLA2G6 gene encodes a calcium-independent group VI phospholipase A2 (iPLA-VI), which is critical in regulating cell membrane homeostasis, mitochondrial function and membrane remodeling (4, 5). Moreover, elevated expression of α-Synuclein (α-Syn) in neuronal mitochondria occurs constantly in PLA2G6-deficiency (6).
Because of its role in the generation of reactive oxygen species and its association with neurodegeneration with brain iron accumulation, an involvement of PLA2G6 in the pathogenesis of Parkinson's disease (PD) was proposed. In fact, oxidative stress and increased brain iron deposits play a pivotal role in the genesis of PD (7).
In line with this, in 2008 PLA2G6 gene was shown to be the causative gene underlying PARK14, an autosomal recessive L-DOPA responsive dystonia/parkinsonism with early or adult-onset (8–12). These patients suffer from dystonia, bradykinesia, rigidity, and marked cognitive decline. Subsequently, a range of phenotypes has been reported, from pure Parkinsonism to severe generalized dystonia. While brain MRI shows cerebellar atrophy in almost all well-established PLAN cases, in adult-onset PARK14 little cerebellar atrophy is observed (13–19).
To date, there are about 80 different PLA2G6 gene abnormalities, and no mutation hot spots have been reported so far. PLA2G6 genotype-phenotype correlation is described in INAD patients with homozygous null mutations, and in some PARK14 patients, where truncated mutations produce a complex phenotype with cortical and cerebellar atrophy (8, 20–25).
Autosomal recessive inheritance of PARK14 suggests that PLA2G6 mutations cause the loss of PLA2G6 function and impair the ability of PLA2G6 to protect mitochondria against apoptotic stimuli and exert a neuroprotective effect on dopaminergic cells (4).
In this manuscript, we describe a patient carrying a PLA2G6 mutation which is affected by familial PD, with some clinical and imaging features which are compatible with the PARK14 phenotype. Unexpectedly, this patient carries an heterozygous mutation in the PLA2G6 gene, despite the evidence provided so far indicates the occurrence of PD only in homozygosis. Strengthening this unique genotype-phenotype correlation, PD was present also in the sister of the proband, who is also a carrier of the heterozygous mutation. Remarkably, in non-carrier sibling PD was not present.
Clinical Presentation
The proband, a 69 years old caucasian man (II:7), was diagnosed with familial PD at the age of 55. Both parents (I:1 and I:2, died, respectively at 85 and 91) and two siblings (II:3, 72 years old and II:6, 71 years old) did not show any neurological sign so far. In contrast, his 74 years old sister (II:2) was recently diagnosed with PD at the age 67 (Figure 1A).
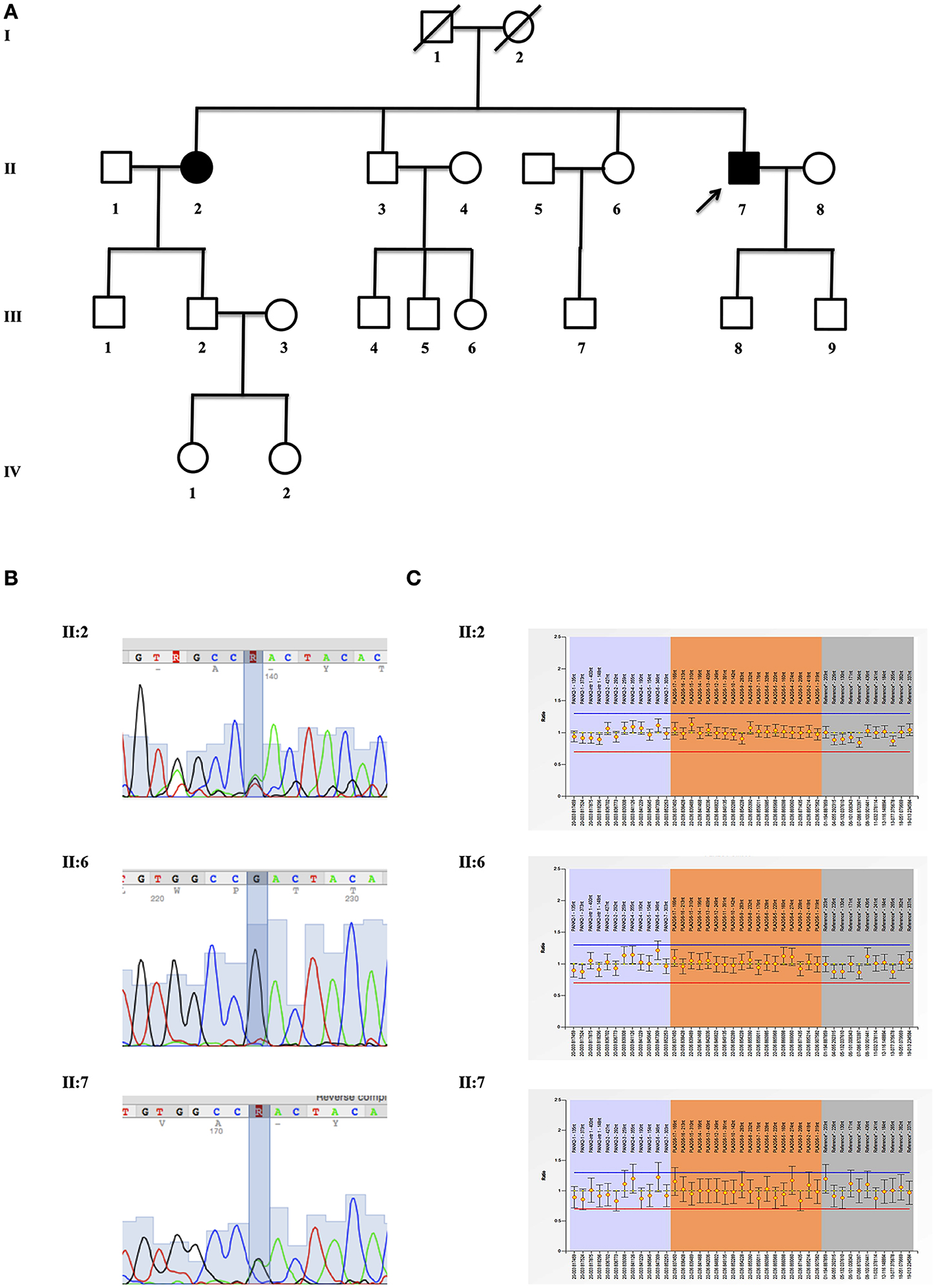
Figure 1. Genetic evaluation. (A) Pedigrees of the investigated kindred. Proband (II:7), his affected sister (II:2) and healthy sibling (II:6) are indicated with an arrow; (B) mutation analysis of p.Asp31Asn (NP_003551.2), c.91G > A (NM_003560.2:) (rs150024227) in exon 2 of PLA2G6 gene (OMIM #603604). Sequence analysis is shown for proband (II:7), his affected sister (II:2), and healthy sibling (II:6); (C) MLPA analysis of PLA2G6 gene shows the results for proband (II:7), his affected sister (II:2) and healthy sibling (II:6).
In the medical history of the proband (II:7), no noticeable acute or chronic disorders potentially related to parkinsonism has been reported. In 2004, he presented rigidity of upper and lower left limbs, freezing of gait, and non-motor symptoms (urinary incontinence and reduced olfaction). He was responsive to L-DOPA+benserazide 250+25, which led the UPDRS III score from 17 to 9 during the on- and off-phase respectively. Brain MRI (1,5 Tesla, GE Healthcare), spinal MRI, ECG and evoked potentials were normal at the time of PD diagnosis. The electromyogram (EMG) showed a slight denervation at perineal level.
Three years later a DAT-Scan was carried out, suggesting a marked asymmetric reduction of dopamine (DA) uptake sites within the right basal ganglia (Figure 2A). Five years later, in 2012, DAT-Scan consistently indicated nigro-striatal denervation. Neuropsychological assessment (MMSE) was reported within the normal range.
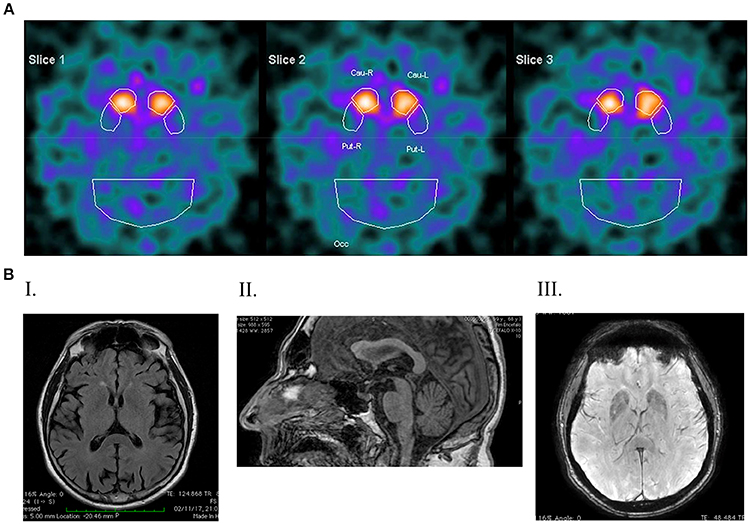
Figure 2. DaTSCAN and Magnetic Resonance Imaging (1,5 Tesla). (A) DATSCAN image showing a reduced tracer uptake in both striatal regions; (B) magnetic resonance imaging. (I) T2 FLAIR shows post-central atrophy and bilateral temporo-insular atrophy; (II) SWAN shows a reduced thickness of the mesencephalic tagmen; (III) T1 FSPGR shows signs of iron deposition in the globus pallidum and in both putamen.
In 2014 brain MRI showed enlargement of sub-aracnoid spaces bilaterally, surrounding frontal lobes. ECG reported abnormal heart rate recovery. The UPDRS-III registered a deterioration (off = 42, on = 12), and electroneurography was consistent with mild axonal sensory polyneuropathy. There was still no cognitive deterioration at MMSE, while neither mood nor obsessive disorders were reported.
In 2017, the UPDRS-III in the on phase was 20 and a gait disturbance appeared along with involuntary oro-facial dyskinesia. Brain MRI revealed post-central atrophy and bilateral temporo-insular atrophy (Figure 2B-I), along with a reduced thickness of the mesencephalic tegmentum (Figure 2B-II). There was no cerebellar atrophy, while in the basal ganglia brain iron accumulation was remarkable involving the head of the caudate nucleus, putamen and globus pallidus (Figure 2B-III). Spinal MRI, electrocardiogram, electromyogram and electroneuronography were unchanged compared with 2014. At this time a slight cognitive deterioration appeared (MMSE 23/30).
A written informed consent for genetic analysis was obtained, and clinical exome sequencing considered 4800 human genes including 17 genes related to Parkinson disease (PARK1:SNCA; PARK2:PRKN; PARK3:SPR; PARK5:UCHL1; PARK6:PINK1; PARK7:DJ1; PARK8: LRRK2; PARK9:ATP13A2; PARK10:ELAVL4; PARK11:GIGYF2; PARK12:TAF1; PARK13:HTRA2; PARK14:PLA2G6; PARK15:FBXO7; PARK16:ADORA1; PARK17:VPS35; PARK18:EIF4GI) (TruSight One Sequencing Panels, Illumina) was performed. Sequence analysis identified the mutation p.Asp31Asn (NP_003551.2), c.91G>A (NM_003560.2:) (rs150024227) in PLA2G6 gene (OMIM #603604), confirmed by Sanger sequencing (Figure 1B). No mutations in other PD-related genes were identified. Multiple-ligation probe amplification (MLPA) (SALSA MLPA kit P120-B2, MRC-Holland) ruled out the presence of duplication or deletion as second mutation (Figure 1C). After a genetic counseling, the presence of p.Asp31Asn was evaluated in II:2 (PD affected sister just diagnosed) and II:6 (healthy sister). The same p.Asp31Asn variant was confirmed in II:2, while it was not present in asymptomatic II:6. Unfortunately, it was impossible to evaluate this variant in the parents (I:1 and I:2) and in one healthy brother (II:3).
Discussion
This manuscript describes a patient with familial PD, who carries a p.Asp31Asn variant in PLA2G6, the causative gene of PARK14. This PD phenotype is characterized by parkinsonism with dystonia. Clinical variability consists in the occurrence of cerebral and cerebellar atrophy, iron accumulation in the basal ganglia, age at onset and cognitive decline.
This patient is characterized by a L-DOPA responsive typical parkinsonian syndrome without the occurrence of dystonia even at 15 years after PD diagnosis. At longer time interval, a slight cognitive decline appeared. MRI showed the presence of iron accumulation both in neo and paleostriatum while cerebellar atrophy was absent. No data about disease progression are available so far for the newly diagnosed sister (II:2) who had a delayed disease onset compared with the patient reported (67 and 55, respectively).
PARK14 was been described as a recessive disease, where two mutations (homozygous or compound heterozygous) in PLA2G6 gene were always present. The patient described here is uniques since he carries a single mutation affecting the PLA2G6. This variant segregated with PD in II:2 (PD affected sister just diagnosed), while it was not present II:6 (healthy sister).
These findings pose the question about the role of heterozygosity in genes related with PARK 14 and, in general, autosomal recessive parkinsonism. In fact, specific allelic variants in the PARK14 locus may lead to PD with an autosomal dominant inheritance (being still unknown the penetrance). In keeping with this, a recent report describes an heterozygous mutation in PARK14 causing PD in a 69 years old patient (23). This patient was reported to carry all typical features of L-DOPA responsive PD associated with dystonia, but no evidence of iron deposition. It could be speculated that only a few mutations in PLP2G6 could act as dominant, producing a specific kind of parkinsonism as witnessed by the phenotype described here compared with other PARK14 patients with two PLA2G6 variants. Further analysis are required to firmly establish whether specific heterozygous mutations of PARK14 may lead to PD.
Another potential explanation is that heterozygote mutations in genes related with autosomal recessive parkinsonism lead to increased risk of PD. In these scenario, the present data contribute to the working hypothesis that PLA2G6 heterozygote mutations may represent a risk factor for PD, as reported for some PLA2G6 mutations in idiopathic PD patients (25). This is supported by the frequency of this variant in different populations (ExAC database: minor allele frequency = 0.0004), and by the presence of single variants reported in sporadic PD (14). It is fascinating that recent studies report PLA2G6 immunostaining in the core of brainstem-type Lewy bodies from PARK14 PD patients and, most remarkably, in idiopathic PD (26, 27). This witnesses for a role of PLA2G6 in the physiopathology of PD thus remarking the potential role of environmental interactions as well as genetic background.
However, it is still debated whether heterozygous carriers of mutations of genes related with autosomal recessive parkinsonism own an increased risk to develop PD (28). Although more than 50% of patients with mutations in parkin or PINK1 have only a single heterozygous mutation, the few large studies that assessed the frequency of heterozygotes remain controversial (29, 30). In fact, while a retrospective analysis of the occurrence of PINK1 heterozygous rare variants in PD could not detect significant differences compared with controls, another study reported that heterozygous CNVs (Copy Number Variations) in PARK2 gene could confer an higher risk to develop PD (28, 30).
Therefore, although the pathogenic significance of these variants remains uncertain, they may represent a risk factor to develop PD, supporting the hypothesis that haplo-insufficiency is a more likely explanation than compound heterozygosity through unidentified recessive mutations.
Conclusions
In this manuscript, we describe a patient carrying a PLA2G6 mutation which is affected by familial PD, with some clinical and imaging features which are compatible with the PARK14 phenotype. This patient is characterized by a L-DOPA responsive typical parkinsonian syndrome without the occurrence of dystonia, a slight cognitive decline, presence of iron accumulation both in neo and paleostriatum while cerebellar atrophy was absent.
Although PARK14 has been reported so far as a recessive disease, clinical and genetic analysis of the proband and his family rise the hypothesis of a potential role of some heterozygous PLA2G6 mutations in causing PARK14. It remains to be analyzed in detail whether these variants act really as dominant PD-inducing mutations, or they play a role as risk factors acting within synergistic genetic and/or environment backgrounds. Deciphering the role of heterozygosity in parkinsonism is important for the development of guidelines, for genetic testing and counseling, and for the understanding of late-onset Parkinson's disease.
Ethics Statement
Approved by the IRCCS Neuromed INM Ethics Committee. Written informed consent was obtained from the participant for the publication of this case report. Protocol ID:CGM-01 Clinical Trials ID:NCT03084224
Author Contributions
SG, RF, and FB planned and designed the study. RF and SS test execution, acquired and analyzed the data. RF and SG drafting of manuscript. SZ, NM, CC, EG, and MS pathology and clinical data. FF, GN, and SR critical revisions. All authors approved submission of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with several of the authors EG and GN.
Acknowledgments
The authors wish to thank the patients enrolled in this research.
References
1. Sadeh M. Neurodegeneration associated with genetic defects in phospholipase A2. Neurology (2009) 73:819. doi: 10.1212/WNL.0b013e3181b2851b
2. Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet. (2006) 79:942–8. doi: 10.1086/508572
3. Illingworth MA, Meyer E, Chong WK, Manzur AY, Carr LJ, Younis R, et al. PLA2G6-associated neurodegeneration (PLAN): further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab. (2014) 112:183–9. doi: 10.1016/j.ymgme.2014.03.008
4. Chiu CC, Yeh TH, Lu CS, Huang YC, Cheng YC, Huang YZ, et al. PARK14 PLA2G6 mutants are defective in preventing rotenone-induced mitochondrial dysfunction, ROS generation and activation of mitochondrial apoptotic pathway. Oncotarget (2017) 8:79046–60. doi: 10.18632/oncotarget.20893
5. Kinghorn KJ, Castillo-Quan JI. Mitochondrial dysfunction and defects in lipid homeostasis as therapeutic targets in neurodegeneration with brain iron accumulation. Rare Dis. (2016) 4:e1128616. doi: 10.1080/21675511.2015.1128616
6. Sumi-Akamaru H, Beck G, Shinzawa K, Kato S, Riku Y, Yoshida M, et al. High expression of α-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol Commun. (2016) 4:27. doi: 10.1186/s40478-016-0298-3
7. Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons (2013) 3:461–91. doi: 10.3233/JPD-130230
8. Kauther KM, Höft C, Rissling I, Oertel WH, Möller JC. The PLA2G6 gene in early-onset Parkinson's disease. Mov Disord. (2011) 26:2415–7. doi: 10.1002/mds.23851
9. Paisán-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. (2009) 65:19–23. doi: 10.1002/ana.21415
10. Paisán-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, et al. Early-onset L-doparesponsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord. (2010) 25:1791–800. doi: 10.1002/mds.23221
11. Sina F, Shojaee S, Elahi E, Paisan-Ruiz C. R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol. (2009) 16:101–4. doi: 10.1111/j.1468-1331.2008.02356.x
12. Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology (2010) 75:1356–61. doi: 10.1212/WNL.0b013e3181f73649
13. Blake RB, Gilbert DL, Schapiro MB. Child Neurology: two sisters with dystonia and regression PLA2G6-associated neurodegeneration. Neurology (2016) 87:e1-3. doi: 10.1212/WNL.0000000000002804
14. Lu CS, Lai SC, Wu RM, Weng YH, Huang CL, Chen RS, et al. PLA2G6 mutations in PARK14-linked young-onset Parkinsonism and sporadic Parkinson's Disease. Am J Med Genet B Neuropsychiatr Genet. (2012) 159B:183–91. doi: 10.1002/ajmg.b.32012
15. Paisán-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging (2012) 33:814–23. doi: 10.1016/j.neurobiolaging.2010.05.009
16. Karkheiran S, Shahidi GA, Walker RH, Paisán-Ruiz C. PLA2G6-associated Dystonia-Parkinsonism: Case Report and Literature Review. Tremor Other Hyperkinet Mov. (2015). 5:317. doi: 10.7916/D84Q7T4W
17. Malaguti MC, Melzi V, Di Giacopo R, Monfrini E, Di Biase E, Franco G, et al. A novel homozygous PLA2G6 mutation causes dystonia-parkinsonism. Parkinsonism Relat Disord. (2015) 21:337–9. doi: 10.1016/j.parkreldis.2015.01.001
18. Wirth T, Weibel S, Montaut S, Bigaut K, Rudolf G, Chelly J, et al. Severe early-onset impulsive compulsive behavior and psychosis in PLA2G6-related juvenile Parkinson's disease. Parkinsonism Relat Disord. (2017) 41:127–9. doi: 10.1016/j.parkreldis.2017.05.014
19. Michelis JP, Hattingen E, Gaertner FC, Minnerop M, Träber F, Biskup S, et al. Expanded phenotype and hippocampal involvement in a novel compound heterozygosity of adult PLA2G6 associated neurodegeneration (PARK14). Parkinsonism Relat Disord. (2017) 37:111–3. doi: 10.1016/j.parkreldis.2017.01.005
20. Kurian MA, Morgan NV, MacPherson L, Foster K, Peake D, Gupta R, et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology (2008) 70:1623–9. doi: 10.1212/01.wnl.0000310986.48286.8e
21. Wu Y, Jiang Y, Gao Z, Wang J, Yuan Y, Xiong H, et al. Clinical study and PLA2G6 mutation screening analysis in Chinese patients with infantile neuroaxonal dystrophy. Eur J Neurol. (2009) 16:240–5. doi: 10.1111/j.1468-1331.2008.02397.x
22. Crompton D, Rehal PK, MacPherson L, Foster K, Lunt P, Hughes I, et al. Multiplex ligation-dependent probe amplification (MLPA) analysis is an effective tool for the detection of novel intragenic PLA2G6 mutations: implications for molecular diagnosis. Mol Genet Metab. (2010) 100:207–12. doi: 10.1016/j.ymgme.2010.02.009
23. Tan EK, Ho P, Tan L, Prakash KM, Zhao Y. PLA2G6 mutations and Parkinson's disease. Ann Neurol. (2010) 67:148. doi: 10.1002/ana.21663
24. Tonelli A, Romaniello R, Grasso R, Cavallini A, Righini A, Bresolin N, et al. Novel splice-site mutations and a large intragenic deletion in PLA2G6 associated with a severe and rapidly progressive form of infantile neuroaxonal dystrophy. Clin Genet. (2010) 78:432–40. doi: 10.1111/j.1399-0004.2010.01417.x
25. Bower MA, Bushara K, Dempsey MA, Das S, Tuite PJ. Novel mutations in siblings with later-onset PLA2G6-associated neurodegeneration (PLAN). Mov Disord. (2011) 26:1768–9. doi: 10.1002/mds.23617
26. Miki Y, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. PLA2G6 accumulates in Lewy bodies in PARK14 and idiopathic Parkinson's disease. Neurosci Lett. (2017) 645:40–5. doi: 10.1016/j.neulet.2017.02.027
27. Miki Y, Yoshizawa T, Morohashi S, Seino Y, Kijima H, Shoji M, et al. Neuropathology of PARK14 is identical to idiopathic Parkinson's disease. Mov Disord. (2017) 32:799–800. doi: 10.1002/mds.26952
28. Huttenlocher J, Stefansson H, Steinberg S, Helgadottir HT, Sveinbjörnsdóttir S, Riess O, et al. Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson's disease. Hum Mol Genet. (2015) 24:5637–43. doi: 10.1093/hmg/ddv277
29. Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. (2007) 6:652–62. doi: 10.1016/S1474-4422(07)70174-6
Keywords: genetic of Parkinson's disease, incomplete penetrance, PARK14, Parkinson and iron accumulation, L-DOPA responsive
Citation: Ferese R, Scala S, Biagioni F, Giardina E, Zampatti S, Modugno N, Colonnese C, Storto M, Fornai F, Novelli G, Ruggieri S and Gambardella S (2018) Heterozygous PLA2G6 Mutation Leads to Iron Accumulation Within Basal Ganglia and Parkinson's Disease. Front. Neurol. 9:536. doi: 10.3389/fneur.2018.00536
Received: 06 April 2018; Accepted: 18 June 2018;
Published: 10 July 2018.
Edited by:
Antonio Pisani, Università degli Studi di Roma Tor Vergata, ItalyReviewed by:
Graziella Madeo, National Institutes of Health (NIH), United StatesRachel Paes Guimarães, Universidade Estadual de Campinas, Brazil
Copyright © 2018 Ferese, Scala, Biagioni, Giardina, Zampatti, Modugno, Colonnese, Storto, Fornai, Novelli, Ruggieri and Gambardella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Gambardella, c3RlZmFuby5nYW1iYXJkZWxsYUBuZXVyb21lZC5pdA==