 Sandra O. Braz
Sandra O. Braz Julien Acquaire1,2
Julien Acquaire1,2 Geneviève Gourdon
Geneviève Gourdon Mário Gomes-Pereira
Mário Gomes-Pereira
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 10 July 2018
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00519
This article is part of the Research Topic Beyond Borders: Myotonic Dystrophies – A European Perception View all 11 articles
Intensive effort has been directed toward the modeling of myotonic dystrophy (DM) in mice, in order to reproduce human disease and to provide useful tools to investigate molecular and cellular pathogenesis and test efficient therapies. Mouse models have contributed to dissect the multifaceted impact of the DM mutation in various tissues, cell types and in a pleiotropy of pathways, through the expression of toxic RNA transcripts. Changes in alternative splicing, transcription, translation, intracellular RNA localization, polyadenylation, miRNA metabolism and phosphorylation of disease intermediates have been described in different tissues. Some of these events have been directly associated with specific disease symptoms in the skeletal muscle and heart of mice, offering the molecular explanation for individual disease phenotypes. In the central nervous system (CNS), however, the situation is more complex. We still do not know how the molecular abnormalities described translate into CNS dysfunction, nor do we know if the correction of individual molecular events will provide significant therapeutic benefits. The variability in model design and phenotypes described so far requires a thorough and critical analysis. In this review we discuss the recent contributions of mouse models to the understanding of neuromuscular aspects of disease, therapy development, and we provide a reflective assessment of our current limitations and pressing questions that remain unanswered.
Animal models offer experimental tools to investigate the causes and mechanisms of disease, when the access to human samples is limited. The remarkable progresses in genetic engineering allowed the introduction of human mutations in the mouse genome, to reproduce molecular, cellular and physiological disease manifestations. The resulting phenotypes provide insight to confirm starting hypotheses, reveal novel pathogenic mechanisms and evaluate new therapies. Myotonic dystrophy (DM) illustrates the cardinal contribution of mouse models to the systematic dissection of a complex disease mechanism, from genetic mutation to the design of clinical trials.
DM is the most common form of adult muscular dystrophy, characterized by pleiotropic symptoms, which are highly variable in their nature and severity (1). Major muscular features include myotonia, muscle weakness, atrophy and smooth muscle dysfunction. Cardiac conduction defects and arrhythmias are associated with cardiomyopathy and may lead to sudden death (1). Brain involvement is illustrated by predominant structural abnormalities of the white matter, cognitive impairment (such as executive dysfunction, visuospatial deficits and abnormal social cognition), behavioral changes (such as apathy and social avoidance) and excessive daytime sleepiness (2). Other peripheral disease manifestations include insulin resistance, iridescent posterior subcapsular cataracts, and gastrointestinal complications (such as constipation/diarrhea) (1).
Two different autosomal dominant mutations in two unrelated genes cause DM and define two genetically distinct forms of the condition. DM type 1 (DM1) is caused by the expansion of a CTG trinucleotide repeat in the 3′-untranslated region (UTR) of the DM protein kinase (DMPK) gene (3). The DM type 2 (DM2) mutation consists in the expansion of an intronic CCTG tetranucleotide in the CCHC-type zinc finger nucleic acid binding protein (CNBP) gene (4). Although genetically distinct, DM1 and DM2 share a toxic RNA gain of function mechanism. In both conditions, expanded CUG/CCUG transcripts accumulate in the cell nucleus to form RNA aggregates or RNA foci (4–6), which perturb the function of RNA-binding proteins and a number of downstream events (7). Although clinically similar, disease symptoms are usually milder in DM2 than in DM1 (1, 8).
In a scenario where the expansion of simple non-coding DNA repeats has a broad deleterious impact on multiple tissues and physiological processes the generation of mouse models that faithfully reproduce the disease presents unique challenges. In order to be clinically relevant mouse models must have construct, face and predictive value (9). In other words, relevant mouse models must recapitulate the genetics and molecular pathogenesis (construct value); they must mimic clinical human features, both molecularly and physiologically (face value); and they must provide a platform to determine the effectiveness of new therapeutic interventions on a clinical population (predictive value). However, mouse models rarely, if ever, completely recapitulate all aspects of human disease. This is particularly applicable to DM, given the clinical variability of the disease, the involvement of multiple tissues and the complexity of the underlying molecular pathways. Even with this caveat, mouse models, alone or in combination, have been instrumental to understand fundamental molecular pathomechanisms (10). Importantly, they have allowed molecular and cellular analyses at various developmental stages, as well as in cell types and tissues that are not easily accessible in humans. We have previously reviewed the contribution of mouse models to decipher the grounds of RNA toxicity and to evaluate promising preclinical assays (10), but there is little doubt that mouse models have continued to provide in-depth understanding of DM disease mechanisms over the last years.
Here we discuss how recent mouse data refined our understanding of RNA toxicity and unfolded numerous roles and pathogenic implications of the RNA-binding proteins dysregulated in DM. We review other emerging disease intermediates and dysregulated signaling pathways recently uncovered. Pre-clinical therapeutic developments are discussed in light of their contribution to reinforce fundamental aspects of disease pathogenesis. We focus primarily on the neuromuscular aspects of the disease to establish correlations between mouse data and human pathology. We point out some contradictory findings between mouse models to illustrate the challenges, complexity and variability of DM disease pathogenesis.
The toxicity of RNA repeats was unequivocally demonstrated in HSALR transgenic mice, through the insertion of an expanded CTG sequence in the 3′UTR of an unrelated gene: the human actin, alpha 1 (ACTA1) gene. The expression of CUG-containing ACTA1 transcripts in mouse skeletal muscle generated genuine myotonia and histological signs of myopathy (11). The elimination of the expanded transcripts by antisense oligonucleotides reduced myotonia in these mice (12), confirming the toxicity of CUG RNA repeats.
The absence of muscle weakness in the HSALR mouse line that expressed the highest transgene levels and showed pronounced muscle histopathology was intriguing and suggested the dissociation between the toxicity of RNA foci and the etiology of muscle weakness (11), an hypothesis that persisted for some years. However, the later analysis of a second HSALR line, which also expressed high levels of the transgene and showed myotonia, revealed reduced grip strength (13). Contrary to the initial reports, these findings corroborate the view that the expression of toxic RNA repeats is sufficient to trigger muscle weakness. CUG RNA toxicity was further demonstrated and confirmed in other mouse lines, listed in Table 1.
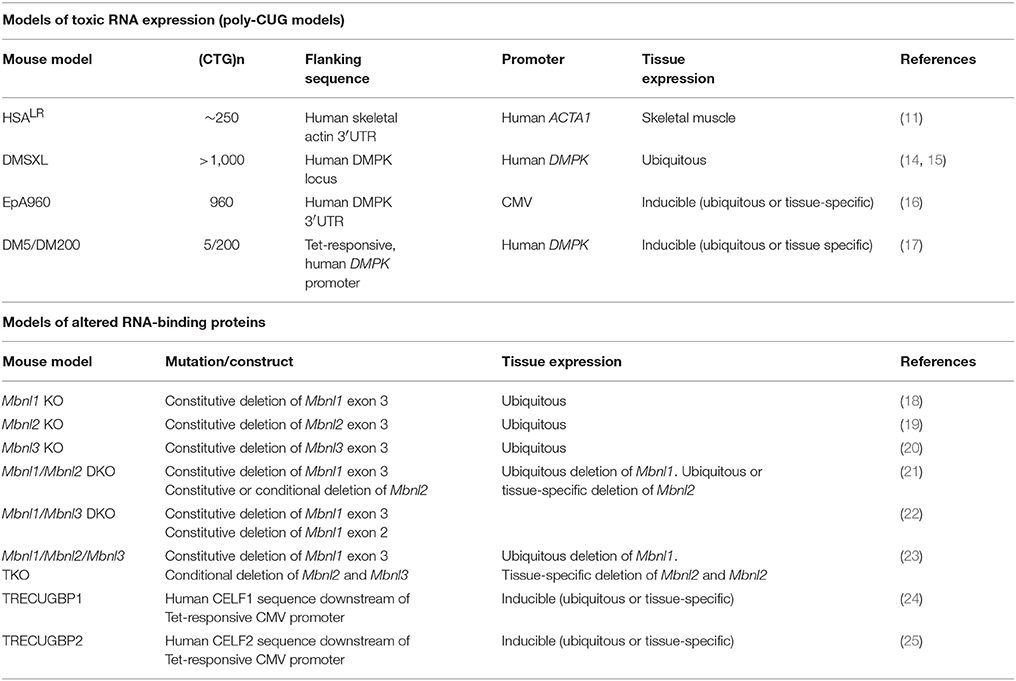
Table 1. Summary of transgene design and expression in the DM mouse models most extensively studied.
The ubiquitous expression of expanded DMPK transcripts from the human DM1 locus resulted in multisystemic phenotypes in DMSXL mice carrying more than 1000 CTG repeats. These phenotypes include reduced muscle strength, lower motor performances, peripheral neuropathy, respiratory impairment, abnormal cognition and behavior, and cardiac conduction defects (26–29). Similarly, the inducible expression of a large, interrupted CTG repeat flanked by the 3′UTR of the DMPK gene produced cardiac, muscular and neurological phenotypes in EpA960 mice (16, 30, 31). Surprisingly, high expression of short (CTG)5 repeats within the DMPK 3′UTR was pathogenic in DM5 mice, causing DM1-like myotonia and cardiac conduction defects (17). Hence, the expression of many copies of a short CUG repeat may have functional outcomes that are comparable to the expression of a few copies of large CUG RNA repeats. In other words, the toxicity of repetitive RNA is two-fold: it is determined not only by the sequence length but also by the abundance of the repeat transcripts in the cell. While HSALR, DMSXL and EpA960 animals accumulate foci, nuclear RNA aggregates were not detected in DM5 mice, raising the possibility that submicroscopic RNA foci can cause disease, or that soluble CUG RNA is also pathogenic (32). The DM1 molecular hallmarks reported in the main poly-CUG mouse models are summarized in Table 2. No poly-CCUG DM2 mouse model has been fully characterized yet.
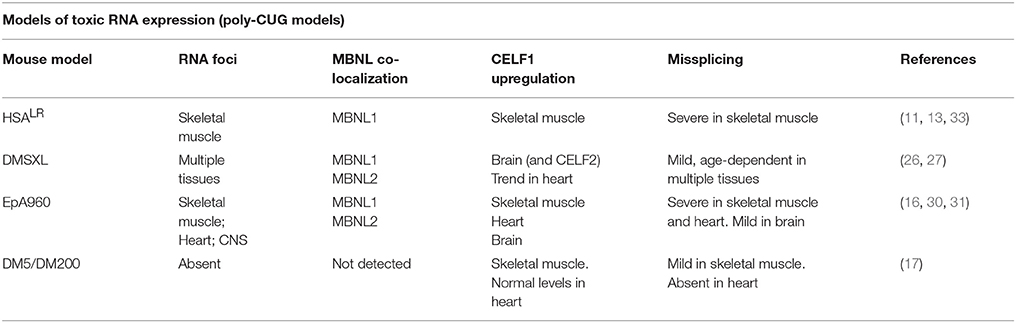
Table 2. Molecular hallmarks of RNA toxicity in the mouse models expressing CUG RNA repeats.
RNA foci are dynamic ribonucleoproteic structures that disrupt important RNA-binding proteins (Figure 1). Members of the MBNL (muscleblind-like) family of splicing factors are sequestered and partially inactivated by the RNA foci in DM1 and DM2 (34, 35), while CELF (CUGBP Elav-like family) proteins are abnormally upregulated, at least in DM1 (16, 36, 37). MBNL sequestration, CELF upregulation and missplicing have been detected to different extents in mouse models expressing poly-CUG RNA transcripts (Table 2). MBNL and CELF proteins bind independently to RNA targets and functionally compete to regulate their downstream processing (25). The two protein families comprise key regulators of developmental splicing transitions. The combined MBNL sequestration and CELF upregulation results in the pathogenic expression of fetal isoforms in adult DM tissues (7). In other words, DM spliceopathy does not produce “unusual” splicing isoforms; instead, it is associated with the expression of normal splicing products that are not well-suited to adult tissue function, leading to the onset of typical disease manifestations. In this context myotonia is the consequence of the abnormal splicing of the CLCN1 chloride channel (38, 39), while insulin resistance is most likely associated with the missplicing of the insulin receptor (38, 40). It is important to note the significant overlap between the splicing abnormalities in DM and other muscular dystrophies (41). The similarities depict a scenario in which splicing dysregulation in DM is not only a primary disease process, but also a secondary event caused by general tissue degeneration.
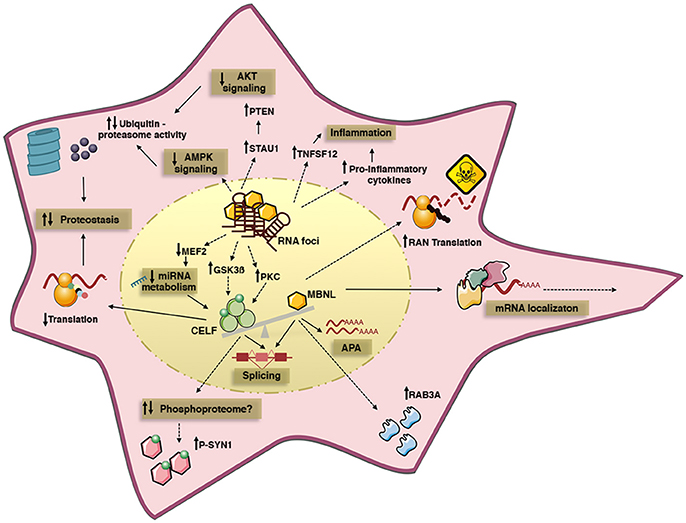
Figure 1. Summary of some of the cell pathways and signaling cascades dysregulated by toxic RNA repeats in DM cells. The expression of toxic RNA transcripts sequesters MBNL proteins into nuclear RNA foci, and upregulates CELF proteins. Different mechanisms may account for CELF upregulation, such as altered PKC and GSK3ß kinase activity, or changes in miRNA levels due to altered MEF2 transcription program. MBNL inactivation and CELF gain-of-function cause pathogenic missplicing. Functional MBNL inactivation alone disrupts alternative polyadenylation and intracellular localization of mRNA targets; it is also believed to dysregulate protein expression, independently of splicing, and to promote RAN translation of toxic peptides. In turn, CELF1 upregulation affects translation efficacy and it may affect the phosphorylation of a subset of proteins through unidentified mechanisms. Protein homeostasis is also perturbed by the downregulation of AKT and AMPK signaling pathways, which likely promotes protein catabolism by increased ubiquitin-proteasome activity, hence contributing to muscle atrophy and weakness. Finally, the increased expression of pro-inflammatory cytokines suggests ongoing inflammation in DM. Solid lines represent well defined disease mechanisms, while dashed lines represent circumstantial data with poorly defined mechanistic links.
In addition to the canonical sense transcripts, both DM1 and DM2 loci produce antisense transcripts, a feature shared with many microsatellite repeat loci and that has been suggested to regulate local gene expression (42). The CTG expansion interferes with the relative levels of sense and antisense RNA in DM1 patients (43) and in transgenic mice carrying the human DM1 locus (44). The pathogenic impact of these changes on local gene expression and disease mechanisms requires further studies in experimental models.
Humans and mice (as well as most vertebrates) express three MBNL genes (MBNL1, MBNL2, and MBNL3) (45). Endogenous MBNL1 and MBNL2 co-localize with CUG and CCUG RNA foci in DM1 and DM2 cells, respectively (4, 46–48), whereas MBNL3 protein was not detected in adult tissues (48).
The three MBNL paralogs show differences in spatial distribution in adult mouse tissues. Mbnl1 and Mbnl2 transcripts are ubiquitously expressed, but Mbnl1 RNA levels are higher in heart, whereas Mbnl2 is more homogenously distributed (49). The steady-state levels of MBNL2 protein, however, are low in adult skeletal muscle (19). Mbnl3 transcript levels are very low in adult mice (49). Differences in protein distribution extend to cell types: the analysis of primary mouse cultures revealed higher relative levels of MBNL1 in astrocytes, while MBNL2 was more abundant in primary neurons (50).
The involvement of MBNL proteins in DM was tested in knockout lines generated either through the deletion of Mbnl genes alone, or the combined inactivation of multiple Mbnl genes (Table 1). These mice revealed some degree of functional specialization between individual members of the MBNL family and clarified their roles in disease molecular pathogenesis.
Direct evidence of detrimental MBNL sequestration was provided by the generation of Mbnl1 KO mice. Mbnl1 inactivation impacted primarily the skeletal muscle and caused pronounced myotonia, but it also resulted in DM1-like subcapsular cataracts, lack of motivation and apathy in knockout mice (18, 51). The impact on cardiac function was less obvious and dependent on the genetic background of Mbnl1 KO mice: cardiac conduction defects were more pronounced on a homogenous 129/Sv background (52), relative to a mixed 129/Sv x C57BL6 background (21). The reasons behind strain-specific cardiac differences between the homogenous and the mixed background have not yet been resolved, but the comparison between these two lines may provide unique insight into the modifiers of disease severity. It is important to note that DM is a highly variable condition, and that variability in disease manifestations may be explained by a complex interplay between genetic modifiers and environmental factors. The backcrossing of different mouse models onto different genetic backgrounds may facilitate the identification of relevant genetic modifiers of disease.
Although reproducing critical muscular and cardiac phenotypes, Mbnl1 KO mice did not develop prominent muscle weakness/wasting or marked cognitive deficits, aside from decreased motivation (51). Additional MBNL members may therefore serve as key disease intermediates. Indeed, the inactivation of Mbnl2 yielded mild muscle pathology, but marked CNS phenotypes, suggesting a tissue-specific impact of Mbnl gene inactivation. Neurological phenotypes of Mbnl2 KO include sleep disturbance, defective spatial memory, abnormal synaptic plasticity and seizure susceptibility (19).
The deleterious effect of Mbnl1 inactivation on muscle physiology was accompanied by splicing defects that are more severe in skeletal muscle and in heart than in the CNS (18, 52, 53), and it related to the role of MBNL1 in the control of fetal-to-adult splicing transitions in muscle (18, 33). Similarly, MBNL2 appears to serve a similar function in the CNS (19). As a result of this regional specialization, Mbnl1 KO mice express embryonic splicing isoforms predominantly in the muscle (18, 33), while Mbnl2 KO mice exhibit embryonic splicing profiles mainly in the CNS (19). Still, we cannot exclude other significant roles of MBNL1 in the CNS independent of splicing: MBNL1 controls the steady-state levels of RAB3A, and possibly other synaptic proteins (27, 54); and it also determines the length of neuronal dendrites and axons (31) (Figure 2).
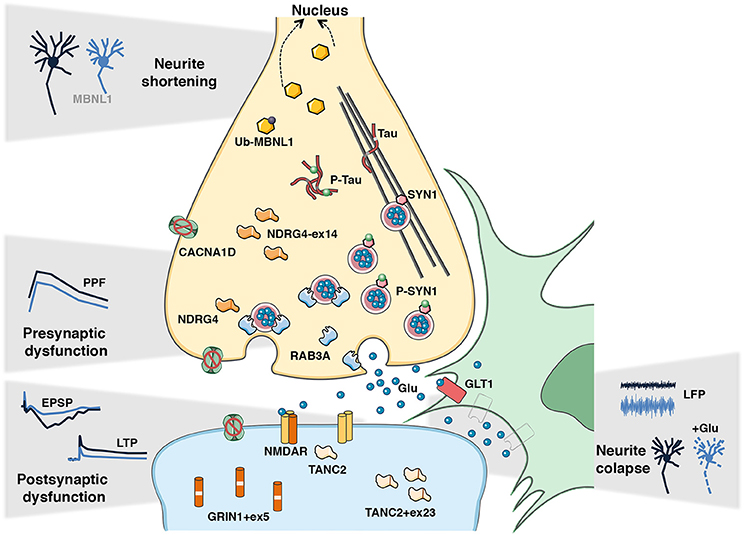
Figure 2. Candidate disease intermediates of DM synaptic dysfunction and learning deficits. The dysregulated disease mechanisms in the CNS of DM1 mouse models appear to involve both pre- and postsynaptic events, which lead to global synaptic dysfunction and consequent cognitive and memory deficits. In the pre-synaptic compartment the hyperphosphorylation of SYN1 and upregulation of RAB3A, together with the missplicing of Mapt/Tau, Ndrg4, and Cacna1d may contribute to impaired short-term synaptic plasticity, notably through decreased paired-pulse facilitation (PPF) detected in DMSXL mice. In the postsynaptic counterpart, the missplicing of Grin1, Tanc2, and Cacna1d may disrupt the functioning of the voltage-gated NMDA receptor, and consequently NMDAR-mediated mechanisms of long-term potentiation (LTP) detected in Mbnl2 KO and EpA960 mice. Reduced GLT1 levels in neighboring astrocytes likely result in neuronal hyperexcitability, demonstrated by increased local field potentiation (LFP) in DMSXL mice, and it can ultimately lead to neuronal damage and neurite collapse in the presence of excessive glutamate. The mislocalization of MBNL1 into the nucleus following abnormal de-ubiquitination decreases neuritogenesis and affects neuronal morphology in EpA960 mice. Together these events likely mediate defective synaptic transmission and abnormal brain connectivity behind DM cognitive and behavioral changes.
The inactivation of Mbnl3 yielded intriguing results: despite low Mbnl3 expression in adult muscle, Mbnl3 KO mice exhibited reduced grip strength and age-dependent decline in skeletal muscle regeneration (20). Other age-associated phenotypes were described in an independent Mbnl3 KO line, such as glucose intolerance, cardiac deficits and subcapsular cataracts (55). MBNL3 loss of function may therefore contribute to the accelerated aging suggested in DM (56). Interestingly, the phenotypes of Mbnl3 KO mice are not accompanied by significant changes in alternative splicing (20, 55). Together with the primary localization of MBNL3 in the cytoplasm (23), these findings predict roles of MBNL proteins other than splicing regulation.
Despite the significant phenotypes of Mbnl1 and Mbnl2 single gene knockout lines, they do not model the full disease spectrum, possibly due to compensatory mechanisms of the remaining Mbnl genes (21). In Mbnl1 KO mice, Mbnl2 expression is upregulated and MBNL2 protein binds to target transcripts that are normally regulated by MBNL1 (21). In order to recreate a situation that resembles more closely the human disease, in which the three MBNL paralogs are sequestered by toxic RNA foci (35), compound knockout mice were generated (Table 1). While Mbnl1/Mbnl2 double knockout (DKO) mice were embryonic lethal, the inactivation of one Mbnl2 copy in a Mbnl1 KO background was sufficient to exacerbate myotonia and trigger muscle weakness, loss of mature neuromuscular junctions and cardiac conduction defects, which were absent in single Mbnl1 KO mice (21, 57). The aggravated phenotypes were accompanied by an increasing severity in spliceopathy and significant changes in alternative polyadenylation (APA) (58). The molecular analysis of Mbnl1/Mbnl2 DKO mice was instrumental to reveal the role of MBNL proteins in the regulation of APA: MBNL proteins and the APA machinery compete to bind to APA sites of a subset of transcripts, in a mechanism that regulates the processing and length of the 3′ end of target transcripts, with subsequent implications for their stability and localization (Figure 1). Similar to DM splicing abnormalities, the sequestration and functional inactivation of MBNL proteins by toxic RNA results in the persistence of fetal APA profiles in adult muscle and brain of DM1 and DM2 patients (58, 59). The direct contribution of individual APA defects to specific symptoms is unclear (60), but the mouse models available offer unique tools to address this question, not only in DM but also in other conditions in which MBNL proteins are sequestered by toxic RNA repeats.
Dual depletion of Mbnl1 and Mbnl3 also enhanced myotonia, muscle weakness and myopathy in skeletal muscle (22). However, in contrast to Mbnl1/Mbnl2 DKO, increased myotonia was not associated with a greater extent of splicing dysregulation in Mbnl1/Mbnl3 DKO mice. Instead, enhanced myotonia was the result of the synergy between Clcn1 missplicing caused by Mbnl1 deletion alone, and defective CLCN1 translation, caused by combined inactivation of MBNL1 and MBNL3 proteins (22).
More recently, conditional triple knockout (TKO) mice were generated by muscle-specific deletion Mbnl2 and Mbnl3 on an Mbnl1 knockout background (Table 1). Mbnl1/Mbnl2/Mbnl3 TKOs present high neonatal mortality, growth defects, respiratory distress, muscle weakness and wasting in association with pronounced splicing and gene expression defects (23). Interestingly, the total spliceopathy in muscle was only modestly increased in Mbnl1/Mbnl2/Mbnl3 TKO mice, relative to Mbnl1/Mbnl2 DKOs, supporting the view that congenital spliceopathy is primarily due to compound loss of MBNL1 and MBNL2, and further pointing to MBNL3 functions, other than splicing regulation. Hence, the congenital form of the disease seems to require combined inactivation of the three Mbnl paralogs from an early developmental stage. DMSXL mice, which also show growth retardation from birth, and DM1 individuals express both sense and anti-sense DMPK transcripts from embryonic and fetal stages (44), confirming that the toxic RNA mechanisms behind congenital cases could operate early on during development.
Overall the generation and characterization of single and compound Mbnl KO mouse models, demonstrated that the simultaneous sequestration of various MBNL proteins is instrumental for the development of clinical manifestations of DM. Given the sparse availability and technical difficulties of working with human DM tissue, constitutive and conditional Mbnl KO mice grant the possibility to investigate abnormal RNA processing in different tissues, cell types and developmental stages.
MBNL1 loss of function accounts more than 80% of missplicing events and nearly 70% of expression defects in the skeletal muscle of HSALR mice (61, 62), strongly anticipating the benefits of therapeutic gene replacement. Overexpression of MBNL1 through viral infection or genetic manipulation ameliorated myotonia and splicing abnormalities in the tibialis anterior of HSALR mice, however it was insufficient to fully correct muscle histopathology (63, 64). While further confirming the role of Mbnl1 loss of function in the onset of myotonia, these findings also hint at the involvement of additional disease intermediates in muscle pathology. It is conceivable that other MBNL proteins might be necessary to fully reverse muscle phenotypes. Prior to the further development of MBNL replacement strategies, it is important to evaluate to which extent MBNL proteins are interchangeable and capable to functionally replace each other in muscle and in other tissues of DM1 mouse models expressing expanded CUG transcripts. Alternatively, the incomplete rescuing of muscle physiology in HSALR mice by MBNL1 overexpression points to the involvement of other families of disease intermediates alongside MBNL proteins.
MBNL proteins are also present in the cytoplasm (23, 48), where they likely regulate mRNA stability (61, 62, 65), as well as the intracellular localization of mRNA transcripts through binding to the 3′UTR of their targets (66, 67). The role of MBNL proteins in mRNA trafficking might be particularly relevant in highly polarized brain cells, such as neurons. Altered MBNL activity or intracellular localization in DM1 could be detrimental for correct transport of mRNAs toward specialized cell compartments (such as axons, dendrites and synapses), which would subsequently affect local translation and ultimately cell function.
In further support of a cytoplasmic function of MBNL proteins, the expression of CUG RNA in the forebrain of EpA960 mice (Table 1) affects MBNL1 ubiquitination and distribution between the nucleus and the cytoplasm (Figure 2), prior to the shortening of neuronal dendrites and axons (31, 68). Morphological impairments occurred in the absence of missplicing, suggesting a contribution of cytoplasmic MBNL1 to disease process.
MBNL proteins have been recently proposed to act as guardians against proteotoxicity. CUG and CCUG RNA generate toxic peptides through non-conventional repeat-associated non-ATG (RAN) translation (69, 70). The combination of bidirectional transcription with RAN translation of expanded repeats produces multiple toxic species, which co-localize with markers of apoptosis, supporting a role in disease pathology (71). Interestingly, RNA accumulation seems to co-exist and exacerbate RAN translation in the same cell, in a mechanism mediated by MBNL proteins: MBNL sequestration and inactivation by nuclear RNA foci promotes RAN translation in DM1 and DM2 cell models (70, 72). RAN products have been reported in DMSXL mice (69). Future mouse studies are required to elucidate the relationship between RNA foci, MBNL protein and RAN translation, and the pathogenicity of RAN peptides in multiple tissues and cell types.
CELF1 upregulation correlates with muscle histopathology in DM1 patients and DM5 transgenic mice (Table 1) (73), pointing to a direct role of CELF1 in disease pathogenesis (Figure 1). The upregulation of CELF1 in DM2 skeletal muscle is contentious, with conflicting reports of normal and increased protein levels (33, 74–76).
To directly address the role of CELF1 gain of function, overexpressing mice were generated. Ubiquitous CELF1 upregulation resulted in severe developmental phenotypes and muscle histopathology (77), which correlated with transgene expression levels (77, 78). The high mortality of these mice limited their face and predictive value. Conditional mouse lines were more informative, since they offered the opportunity to focus on individual tissues and assess the pathogenic contribution of CELF protein overexpression alone (Table 1). Induction of CELF1 transgene expression in mouse skeletal muscle was sufficient to reproduce muscle wasting, defective motor performance and myopathy (79); while CELF1 upregulation in heart caused cardiac conduction defects, cardiomyopathy with hypertrophy and early mortality (24). As expected, muscular and cardiac phenotypes were accompanied by missplicing events in muscle and heart, respectively (24, 79). A splicing-mediated effect was further supported by the expression of a dominant-negative Celf1 variant in HSALR mice, which partially corrected missplicing in skeletal muscle (80).
CELF proteins can also regulate the alternative splicing of transcripts involved in neuronal function (81). Therefore, the upregulation of CELF1 and CELF2 reported in human DM1 brains (27, 82) may have a substantial contribution to the etiology of neurological dysfunction. Conditional overexpressing models could help investigate the cognitive and behavior consequences of CELF1 or CELF2 upregulation, and identify subsets of transcripts that specifically respond to these two RNA-binding proteins. In support of target discrimination between CELF proteins, it was shown that the splicing of MAPT exon 10 responds specifically to CELF2, but not to CELF1 upregulation (82).
Although already generated, CELF2-overexpressing mice have only been used in molecular approaches to study the antagonistic role of MBNL and CELF proteins in splicing regulation (25). The phenotypic consequences of CELF2 overexpression have not been reported yet.
In addition to regulating alternative splicing in the nucleus, CELF proteins have cytoplasmic roles in the regulation of mRNA stability, translation and deadenylation (83). The distribution of CELF1 between the nucleus and the cytoplasm is regulated by AKT phosphorylation (84). As a result, CELF gain of function in DM1 has intricate consequences that affect multiple cellular pathways in different cell compartments.
CELF1 activity is controlled by multiple phosphorylation events. The role of CELF1 in translation depends on the phosphorylation of Serine-302: phosphorylated CELF1 acts as an activator, while unphosphorylated CELF1 represses translation (84, 85). In DM1, the increase in the total levels of CELF1 is accompanied by the elevation of both phosphorylated and unphosphorylated forms of CELF1 at Serine-302 (40, 86, 87). This results in the reprogramming of protein translation, altered proteostasis and global cell stress, which ultimately affects cell function (75, 78, 84).
DM5 mice have corroborated the cytoplasmic functions of CELF1 in DM1 pathogenesis. The genetic inactivation of Celf1 in DM5 mice did not mitigate missplicing, but instead corrected the expression of CELF1 translational targets in skeletal muscle (73); a tissue that shows CELF1 upregulation in DM5 mice (17). The molecular changes were sufficient to improve motor performance, grip strength and histopathology, but left myotonia unchanged (73). These results demonstrate the pathogenic relevance of CELF1-regulated translation, and point to CELF1-independent myotonia mechanisms in DM. Interestingly, in line with the absence of CELF1 upregulation in DM5 hearts (17), Celf1 deletion did not ameliorate the cardiac function (73).
Finally, it is worth noting that CELF1 overexpression alone is associated with the hyperphosphorylation of Synapsin-1 in cell culture (27) (Figure 2), suggesting a contributing role of this RNA-binding protein in the regulation of the phosphoproteome.
CELF1 upregulation in DM1 operates at protein level, since transcript load remains unchanged in skeletal muscle (73). In the heart of DM1 patients and induced EpA960 mice, upregulation correlates with CELF1 protein hyperphosphorylation, higher protein stability and increased PKC activity (87) (Figure 1). Consistent with a direct role of PKC in CELF1 metabolism, treatment of EpA960 mice with PKC inhibitors immediately after transgene induction avoided CELF1 upregulation, reduced mouse mortality and improved cardiac function (88). These findings provided pharmacological evidence of the involvement of PKC in CELF1 function and in DM1 cardiac phenotypes. Surprisingly, the genetic inactivation of Pkc did not lower CELF1 expression or correct histopathology in the skeletal muscle of DM5 mice (89).
Different reasons may account for the differing outcomes of CELF1 results obtained with EpA960 and DM5 mice. First, the inherent differences between the two mouse models: while in EpA960 interrupted large repeats are expressed under the control of a non-DMPK promoter (16), DM5 mice express short (CUG)5 RNA repeat in multiple tissues and cell types under the control of the human DMPK promoter (17). Second, it is conceivable that the molecular mechanisms of CELF1 upregulation differ between heart and skeletal muscle: CELF1 upregulation in the skeletal muscle might be independent of PKC. Third, off-target effects of kinase inhibition might have introduced confounding factors in the analysis. Indeed, the PKC inhibitor Ro 31-8220 used in EpA960 mice has since then been found to reduce RNA foci, release MBNL1 and correct MBNL1-dependent splicing events in a cell model of DM1 (90). Furthermore, Ro 31-8220 can also inhibit other kinases, including GSK3ß (91).
Experimental evidence of the role of GSK3ß in DM1 muscle pathology was obtained in HSALR mice: GSK3ß inhibition restored CELF1 protein levels and translational activity, improved muscle strength and corrected histopathological changes (13, 92). The possibility remains that different aspects of CELF1 metabolism are controlled by different phosphorylation events: phosphorylation by PKC increases protein stability (87, 92), GSK3ß controls the translational activity of CELF1 (13) and AKT regulates the nucleus-cytoplasm distribution of CELF1 (84) (Figure 1). We currently do not know the molecular link between the repeat expansion and altered kinase activity.
Kinase-independent mechanisms of CELF1 upregulation have been proposed. Under physiological conditions, CELF1 protein decreases in adult mouse tissues in response to a developmental increase in a subset of microRNA (miRNA) species. Transgene induction in EpA960 revealed that CUG RNA toxicity disrupts the MEF2 transcription network, lowers miRNA expression reversing the physiological miRNA developmental program and causing CELF1 upregulation (93). Therefore altered levels of miRNA in DM1 tissues could explain CELF1 upregulation.
To discard the possibility of a direct or indirect regulation of CELF1 by MBNL proteins, CELF1 expression was measured in Mbnl1 KO mice, and revealed no changes (18). In contrast, induction of CELF1 over-expression in transgenic mice yielded MBNL1 upregulation, possibly mediated by tissue regeneration (79).
Despite progress in the understanding of the multifaceted metabolism of CELF1 in DM1, the jury is still out on the molecular mechanisms of upregulation in DM1 and the extent of the therapeutic benefits of CELF targeting in tissues, other than the heart. The mechanisms behind CELF2 upregulation in the CNS of DMSXL mice are less clear (27). Useful mouse models are available to address these questions (Table 1 and Table 2), through pharmacological or genetic manipulation of CELF1 and CELF2 levels, as well as the activity of candidate kinases and miRNA metabolism.
Additional layers of DM1 molecular pathogenesis, beyond the canonical involvement MBNL and CELF RNA-binding proteins, have emerged from recent mouse studies of muscle and heart phenotypes. Hereditary myotonia is usually caused by the malfunction of ion channels (94). In line with this view, compelling evidence has demonstrated the direct role of CLCN1 chloride channel missplicing in the onset of DM1 and DM2 myotonia (39, 63). The mechanisms behind muscle weakness/wasting and cardiac dysfunction can be more diverse, and mediated by a combination of interacting intermediates. In this section we first discuss some critical splicing events, whose contribution to muscle and heart pathology has been corroborated by mouse studies. Then we review the emerging role of additional pathways, whose mechanistic link with MBNL and CELF canonical disease intermediates has not yet been elucidated and deserves further attention.
Progressive muscle weakness and wasting are among the most prominent clinical features of DM1, in association with centralized nuclei and myofiber atrophy, without overt regeneration, fibrosis or necrosis (1). Previous studies have shown associations between muscle weakness and MBNL1-dependent splicing of BIN1 (95), CACNA1S (96) and DMD (97). The recreation of the DM1 missplicing of Bin1, Cacna1s or Dmd in wild-type mice, through RNA antisense technology, corroborated the contribution of these events to muscle weakness and myopathy (95–98). However, it is still unclear if the combined inactivation of multiple MBNL proteins is the sole responsible for muscle weakness. Elevation of CELF1 protein may certainly play a determinant role too, as suggested by the muscle phenotype of CELF1-overexpressing mice (30) and by the improved muscle strength following CELF1 downregulation in HSALR mice (13). Some CELF1-responsive splicing events may provide connecting dots in the mechanisms of muscle pathology: while RYR1 missplicing alters excitation-contraction coupling in skeletal muscle (99), the shift of PKM splicing to an embryonic isoform results in less efficient energy production, likely associated with muscle weakness and wasting (98).
An expected role for splicing dysregulation has also been suggested in DM heart disease. In spite of the confirmed contribution of MBNL1/MBNL2 loss of function (21) and CELF1 upregulation toward cardiac conduction defects (24), the downstream disease intermediates remained elusive. MBNL1-dependent missplicing of SCN5A was found in the heart of DM1 patients and Mbnl1/Mbnl2 DKO mice. When the DM1 splicing isoform is expressed in wild-type mice, it causes DM1-like cardiac conduction defects and arrhythmias (100). The influential role of SCN5A does not rule out the contribution of other yet unidentified splicing events that may reinforce heart spliceopathy and aggravate cardiac disease in DM.
The RNA binding protein Staufen1 is significantly upregulated in DM1 muscle biopsies, in the absence of missplicing of the corresponding transcript, and it correlates with disease severity (101). Sustained expression of Staufen1 in the skeletal muscle of overexpressing transgenic mice causes muscle weakness and myopathy, characterized by an increase in the frequency of small fibers and central nuclei. Staufen1 impairs muscle differentiation through enhanced translation of c-myc (102), which in turn upregulates the transcription of the PTEN tumor suppressor gene and ultimately inhibits downstream AKT signaling (103). The AKT pathway promotes cell survival, proliferation and growth and mediates cell metabolism, transcription and translation in response to extracellular stimuli and changes in energy balance (104). The increased expression of atrogenes in Staufen1-overexpressing mice was linked to AKT signaling inactivation and PTEN upregulation, which interfere with the activity of the ubiquitin-proteasome system to promote catabolic protein degradation, which likely contributes to the muscular phenotypes (103). In further support of elevated protein degradation in DM1 muscle weakness and myopathy, DMSXL mice show enhanced proteasome activity in association with muscle weakness and myopathy (26, 105).
The dysregulation of the adaptive switch between catabolic and anabolic states in DM may extend beyond AKT missignaling, and encompass other intermediates. Maintaining an adequate supply of energy is an essential requirement for cell function, notably in muscle and CNS, which depends on the cross talk between AKT and AMPK signaling pathways (104). Interestingly, the activation of AMPK signaling is also impaired in the skeletal muscle of HSALR mice following fasting (106), corroborating the idea that DM perturbs cell master sensors of energy balance. Importantly, pharmacological treatments to normalize this pathway improved muscle strength and corrected myotonia in these mice (106). Although these data suggest a role of the AMPK cascade in DM1 muscle pathology, it was also noted that the pharmacological activation of AMPK reduced RNA foci in HSALR mice. Hence, it is possible that rather than a direct role on the etiology of muscle pathology, AMPK dysregulation perturbs the dynamics of CUG RNA, stabilizes foci and accentuates spliceopathy, thereby aggravating muscle manifestations. Conversely, the AMPK activator alone may simply destabilize RNA foci and lead to an amelioration of mouse phenotypes through a restoration of splicing.
Given the role of Staufen1 in neuronal dendrite arborization and synaptic development (107), it will be of interest to study the implication of Staufen1 in the neurological deficits of DM1. Both AKT and AMPK signaling pathways are implicated in multiple aspects of brain development and function, and their dysregulation has been associated with neurological disease (104, 108). Their role in DM may, however, be restricted to muscle, since no altered AKT/AMPK signaling activity was detected in DM1 neural stem cells (109). Nonetheless, these results must be confirmed in relevant DM mouse models of brain dysfunction.
Tumor necrosis factor superfamily member 12 (TNFSF12) was found upregulated in the skeletal muscle of DM5 and DM200 mice (Table 1), shortly after transgene induction and prior to the onset of muscle pathology (110). Genetic deletion of Tnfsf12 or the inhibition of the downstream signaling cascade by anti-TWEAK antibodies improved the muscle strength of DM5 mice, demonstrating the physiological relevance of TWEAK signaling in DM1. The binding of TWEAK to its receptor, TNFSF12, regulates cell proliferation, differentiation, inflammation and apoptosis (111). In muscle, the TWEAK-TNFSF12 complex becomes particularly engaged in response to disease, triggering the activation of pro-inflammatory responses that can contribute to DM1 myopathy (110). Further support of ongoing inflammation in muscle was provided by global analysis of gene expression in congenital DM1, which revealed significant upregulation of pro-inflammatory genes (112).
It is conceivable that muscle weakness and atrophy in DM1 is multifactorial process, resulting not only from simultaneous dysregulation of splicing, unbalanced protein synthesis/degradation, but also inflammation.
miRNA profiling revealed significant changes in the heart (113), skeletal muscle (114–118) and serum (119) of DM1 and/or DM2 patients. Despite the divergence of some of the results reported, miRNA dysregulation emerged as a disease feature, which could either be a direct consequence of RNA toxicity, or a lateral event secondary to altered cell physiology. The investigation of miR-1 dysregulation favored the former. Mature miR-1 appears to be downregulated in DM1 and DM2 hearts, in association with an expected increase in miR-1 downstream targets: the upregulation of GJA1 (connexin 43) gap junction protein and CACNA1C calcium channel might subsequently contribute to heart phenotypes (113). In an effort to shed light onto the mechanisms of miR-1 misregulation, MBNL1 knocking down in cell culture blocked the maturation of pre-miR-1, which suggested a role of MBNL1 in miRNA processing and biogenesis, in agreement with the normal or elevated levels of pre-miR-1 found in DM1 and DM2 patients, respectively (113). However, this hypothesis is at odds with subsequent findings. First, miR-1 remained unaltered in Mbnl1 KO mice (93). It is possible that MBNL2 upregulation in these mice (21), which compensates for the lack of MBNL1, could avoid miR-1 downregulation. To answer this question it would be important to study miR-1 levels in Mbnl1/Mbnl2 DKO. Second, global analysis of miRNA species revealed that CUG-associated changes occurred already at the precursor stage in the induced EpA960 mouse model, arguing against a primary defect in subsequent miRNA processing and maturation. Instead, these results were consistent with defects in miRNA transcription and were attributed to the dysregulation of the MEF2 transcriptional program (93). It is possible that the high expression levels of the expanded (and interrupted) transgene in EpA960 mice trigger severe molecular defects and more pronounced dysregulation of miR-1 transcription, upstream from processing and maturation, relative to DM1 and DM2 patients. Finally, recent findings on CELF1-overexpressing mice did not fully match previous results in human tissue either. In contrast with the upregulation of miR-1 targets reported in DM1 and DM2 hearts (113), GJA1 protein levels decrease in the heart of CELF1-overexpressing mice (120). The discrepancy between patients and these mice might be explained by a combined effect of the heterogeneous regional distribution of GJA1 in disease hearts, and the study of different disease stages: GJA1 levels may show an initial compensatory increase during the early adaptation disease stages studied in human samples (113), followed by a late decrease during maladaptation disease stages, like in CELF1-overexpressing mice (120). Further studies are required to clarify these questions and to extend the implications of miRNA metabolism to other affected tissues, notably the CNS.
The sequestration of expanded DMPK RNA in the nucleus of DM1 cells causes a 50% reduction in protein levels (121). Initial reports suggested a role of DMPK haploinsufficiency in disease etiology, a hypothesis corroborated by a dose-dependent effect in mouse heart: the deletion of one copy of the murine Dmpk gene was sufficient to disrupt cardiac conduction (122, 123). In contrast, late and mild myopathy in skeletal muscle required full deletion of both Dmpk copies in knockout mice (124). These early findings suggest that therapeutic hopes aiming to eliminate DMPK transcripts may aggravate some aspects of the disease pathology, particularly in heart. In this context, it is worth reviewing our actual knowledge on the contribution of DMPK protein to disease.
The recent re-evaluation of the impact of Dmpk deletion in knockout mice, bred onto homogeneous genetic backgrounds, showed no functional impact on cardiac or skeletal muscle, thereby excluding a role of DMPK loss of function in muscle phenotypes (125). The reasons behind the diverging results relative to early findings may relate to the strain background and the role of unidentified modifiers. Alternatively, the differences may relate to the replacement strategy used to inactivate the Dmpk gene, which might have interfered with the expression of flanking genes in the knockout lines previously generated (125). In summary, these data provide evidence of the limited functional impact of DMPK inactivation on heart and skeletal muscle, and validate the anti-sense therapies being developed, which are discussed below. Nonetheless, the role of DMPK protein in the CNS, as well as in other tissues, needs to be further explored.
The burden of CNS dysfunction has shifted DM research from an initial focus on muscle pathology, to the investigation of brain disease mechanisms. Sophisticated imaging techniques have characterized structural and metabolic abnormalities in human brains (2, 126). Molecular studies have also been performed in the nervous system, but they rely on samples collected at the end-stage of the disease. Animal models overcome this critical limitation, as they provide tissue samples throughout disease progression, offering the possibility to characterize molecular, cellular and electrophysiological changes in the nervous system prior to the onset of disease symptoms. In this section we critically review relevant neurological phenotypes of various DM mouse models, and the insight they provide to the understanding of disease mechanisms in the central and peripheral nervous system.
Two DM1 mouse models express large CUG RNA transcripts in the CNS: the ubiquitous DMSXL line and the inducible EpA960 mice (Table 1). Both DMSXL and forebrain-induced EpA960 mice show impaired spatial learning and memory in the Morris Water Maze, resembling the visuoconstructive defects in DM1 patients (27, 31). DMSXL mice have also shown signs of anhedonia and novelty inhibition of exploratory activity (27). The electrophysiological profiling of the hippocampus revealed synaptic dysfunction behind these phenotypes: while DMSXL mice show impaired short-term paired-pulse facilitation (27), suggestive of pre-synaptic dysfunction; EpA960 exhibit reduced long-term potentiation (LTP) (31), which is more often associated with post-synaptic abnormalities (Figure 2). The diverging effects on pre- and post-synaptic neuronal plasticity between the mouse lines may be accounted for, at least partly, by their intrinsic differences: DMSXL mice express pure CUG repeats in multiple brain cell types from an early embryonic stage; while induced EpA960 mice express higher levels of interrupted CUG repeats post-natally, in the neurons of the forebrain (Table 1).
Typical RNA foci accumulation and co-localization with MBNL1 and MBNL2 were detected in various cell types of DMSXL brains (27) and in EpA960 neurons (31). Still, both lines showed only limited spliceopathy (27, 31). In contrast, Mbnl2 KO and Mbnl DKO displayed more pronounced splicing dysregulation, which may contribute to impaired LTP and spatial learning of Mbnl2 knockout mice (19, 59): the missplicing of Grin1 may reduce dendritic localization of the glutamate receptor, which may be further aggravated by Tanc2 abnormalities (127, 128); while Cacna1d and Ndrg4 misregulation might impair neuronal activity and learning (129, 130) (Figure 2). Other MBNL-dependent pathways may, however, contribute to brain disease, such as defects in APA (59) and changes in the expression and phosphorylation of synaptic proteins (27, 50, 54).
MAPT/Tau protein has long been associated with DM1 brain disease. Abnormal MAPT isoform distribution was first described at the protein level (131), in association with the intranuclear accumulation of hyperphosphorylated protein fibers, or tangles in patients (Figure 2). Abnormal missplicing was later described in patients (46, 132) and in the brain of DMSXL mice (27). The pronounced Mapt RNA missplicing in Mbnl1/Mbnl2 DKO indicates the critical role of the spliceopathy resulting from the dual loss of these two RNA-binding proteins (59). The DM1 tauopathy has been suggested to interfere with axonal transport and neurosecretion (133), but further animal studies are required to decipher the mechanisms.
Imaging and neuropsychological assessment have uncovered candidate brain regions primarily affected by DM. The identification of critical brain areas will be important to direct future therapies toward the most relevant brain territories, and it will likely depend on an intricate interplay of factors, such as somatic repeat length, levels of toxic RNA, foci abundance and the activity of RNA-binding proteins. A small number of studies has investigated repeat instability (132) and DMPK gene expression in different brain areas in a limited number of human patients (134). DM1 mice offer the possibility to surmount the limited availability of human tissue and perform more detailed analyses. Transgenic DM1 mice expressing ~500 CTG repeats under the control of the human DMPK promoter and the regulatory regions of the DM1 locus (14) showed age-dependent accumulation of larger repeat sizes in most brain regions (Figure 3). The semi-quantitative results did not reveal brain regions with exceptionally high somatic mosaicism, in which we could anticipate the accumulation of very long CUG repeats. The cerebellum, however, exhibited lower levels of somatic instability, as reported in humans (135) and in another model of CTG repeat instability (136). The average repeat size in the cerebellum was nonetheless within the disease-associated range. It is possible that future analyses of somatic repeat instability in smaller brain areas or individual cell types of these mice will reveal susceptible cell populations that accumulate significantly longer repeat expansions.
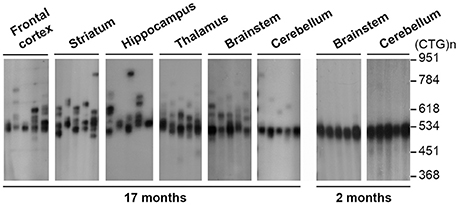
Figure 3. Analysis of CTG somatic mosaicism in the CNS of transgenic mice carrying the DM1 locus. The autoradiographs show representative SP-PCR analyses of 10–20 transgene molecules per reaction in dissected brain regions of old and young DMSXL hemizygotes, aged 17 and 2 months, respectively. The size markers, converted into repeat number are displayed on the right.
Similarly, the expression levels of the DMPK transgene showed modest variation between CNS regions (54). In contrast, RNA foci were not homogenously distributed and accumulated preferentially in the frontal cortex and certain areas of the brainstem of DMSXL mice (26, 54), and they appeared to be more abundant in cortical astrocytes relative to neurons (27). The analysis of well-defined histological layers of the mouse cerebellum has also shown greater foci accumulation and more severe spliceopathy in Bergmann astrocytes, relative to the neighboring Purkinje cells (50). Together these findings demonstrate more pronounced pathologic events in defined brain cell populations and cell types, a view further supported by the preferential accumulation of anti-sense RAN-translated products in the oligodendrocytes of DM2 brains (70).
The factors governing the distribution of DM pathology in the brain remain elusive and must be addressed in future mouse studies, but variations in the expression of MBNL and other RNA-binding proteins between brain regions (54) and cell types should be considered (50).
While waiting for efficient gene therapy to correct the causing genetic defect (the DNA repeat expansion) or neutralize the pathogenic molecule (the toxic RNA), one can imagine pharmacological means to ameliorate or prevent progression of neurological symptoms. Such strategies require comprehensive characterization of neuronal activity and integrative brain dysfunction.
Perturbed balance between excitatory and inhibitory neurons disrupts cognition in neurological diseases. Several mouse studies favor a scenario of neuronal excitability in DM1. Both DMSXL and Mbnl2 KO mice present elevated susceptibility to PTZ-induced seizures, suggesting GABA-mediated hyperexcitability (19). In line with elevated neuronal excitability, Mbnl2 KO mice show augmented responsiveness to intracortical train stimulation, in a mechanism partially mediated by abnormal glutamate neurotransmission (137). Reduced expression of the glial GLT1 glutamate transporter in DMSXL brains is associated with elevated neuronal firing in vivo (50) (Figure 2). This finding supports a role of defective glutamatergic transmission and neuronal excitability in DM1, mediated by abnormal neuroglial interactions. Neuronal hyperexcitability is a frequent cause of epilepsy. Although epileptic episodes are rare in DM, patients present high sensitivity to GABA agonists (138) as well as abnormalities in glutamatergic transmission in the frontal lobe (139).
In addition to GABA and glutamate, circumstantial evidence points to the involvement of other signaling molecules. HPLC quantification revealed region-specific defects in dopamine and serotonin neurochemicals in DMSXL brains, in association with high foci content in dopaminergic and serotonergic brain centers (27). Importantly, DM1 brains have shown loss of neurons signaling through these two types of neurotransmitters (140, 141).
Today optogenetics allows the neuronal manipulation of neuronal circuits in vivo. In combination with electrophysiology, imaging and behavior assays, these techniques can provide insight into the contribution of neuronal activity to the cognitive performance of DM mice, and elucidate the neuronal circuits most profoundly affected by the disease.
The brain structural changes found in DM1 and DM2 are mainly characterized by white matter hyperintensities, some general atrophy and dispersed gray matter reduction across the four cortical lobes, the basal ganglia, and cerebellum. Importantly, white matter abnormalities correlate with disease duration and cognitive deficits (2, 126). Functional imaging revealed low glucose uptake and cerebral hypoperfusion, as well as abnormal connectivity patterns that correlate with atypical personality traits and executive dysfunction (142, 143). The correlation between imaging data and neuropsychological profiles hints to the involvement of complex neuronal networks, through defective neurodevelopment, neurodegeneration or neurodysfuntion. Today we still do not know the contributing weight of each of these components to DM brain disease. The molecular and histological mouse studies have shed some light on this question.
Higher expression of embryonic splicing isoforms in the brains of DMSXL (27), Mbnl2 KO and Mbnl1/Mbnl2 DKO mice (19, 59) points to a disrupted developmental program. In contrast, the dysregulation of synaptic proteins does not recreate embryonic events (54), supporting functional deficits in DM brains, rather than a developmental delay.
Inducible EpA960 mice have recently given further insight. Transgene induction in adult forebrain (after the completion of CNS development) yielded progressive loss of axonal and dendritic integrity, together with brain atrophy (31)—a sign of ongoing neurodegeneration in adults, possibly in line with the reported premature and accelerated cognitive decline in DM1 patients (144). However, the EpA960 mouse data do not exclude developmental disruption, should toxic RNA be expressed during early embryonic stages.
Understanding the contribution of defective development, neurodysfunction and neurodegeneration is critical to design therapeutic schemes: we must intervene prior to the establishment of irreversible developmental defects, irreparable cell damage or permanent network dysfunction. Mouse models, and in particular the inducible lines (Table 1), will help assess the reversibility of neurological phenotypes and whether neurological disease progression can be halted and even reversed.
The involvement of the peripheral nervous system (PNS) and the presence of peripheral neuropathy in DM1 has been open to debate (145). The scarce availability of human samples has slowed down research on this topic, but mice expressing toxic CUG repeats in the PNS and in the neuromuscular junction (NMJ) have surpassed this limitation.
Axonopathy was detected in the DMSXL sciatic nerves, characterized by smaller nerve sections, loss and reduced size of myelinated fibers, in association with thinner myelin sheaths, which may highlight ongoing pathogenicity in myelinating cells. The neuronopathy extends to the spinal cord of DMSXL, where a reduction in the number of motor neurons was reported (146).
The analysis of the neuromuscular junction (NMJ) in DM1 muscle biopsies revealed abundant accumulation of RNA foci both in pre-synaptic motoneurons and in post-synaptic nuclei, with pronounced MBNL1 sequestration (147). As a result the NMJ is at risk of developing DM1-associated spliceopathy, but we currently do not know which MBNL1-dependent targets and pathways are dysregulated. In addition, the expression of two members of the SLITRK family of membrane proteins is dysregulated in DM1, in a MBNL1-independent manner, affecting neuromuscular connections (148). Together these findings suggest that both MBNL-dependent and MBNL-independent mechanisms may disturb the organization, stability and function of the NMJ, thereby contributing to PNS pathology and, importantly, to muscle pathology. In support of this view, the expression of expanded CUG RNA in the diaphragmatic NMJ of DMSXL mice is associated with disorganized endplates, lower density of postsynaptic acetylcholine receptors and reduced number of myelinated neurons, possibly mediating the respiratory impairment of these mice (28). In contrast, HSALR transgenic mice exhibit poor foci accumulation in subsynaptic nuclei (147), indicating that the muscle phenotypes of this line (such as myotonia, central nuclei and ring fibers) do not require the expression of toxic RNA in the NMJ. Subsynaptic RNA toxicity in the NMJ would preferably contribute to DM1 muscle features that are not detected in HSALR mice, such as angular fiber atrophy and pyknotic nuclear clumps (147). In conclusion, defective communication between nerve endings and skeletal muscle might be a common feature in DM1, likely contributing to muscle pathology.
Following the identification of CUG repeats as the pathogenic element in DM1, expanded RNA transcripts became an attractive therapeutic target, endorsed by the reversion of disease phenotypes in an inducible mouse model of DM1 (17). Hence, the neutralization of CUG repeats has been tested in relevant DM1 mouse models, taking advantage of antisense oligonucleotides (ASO) or small molecules (Table 3).
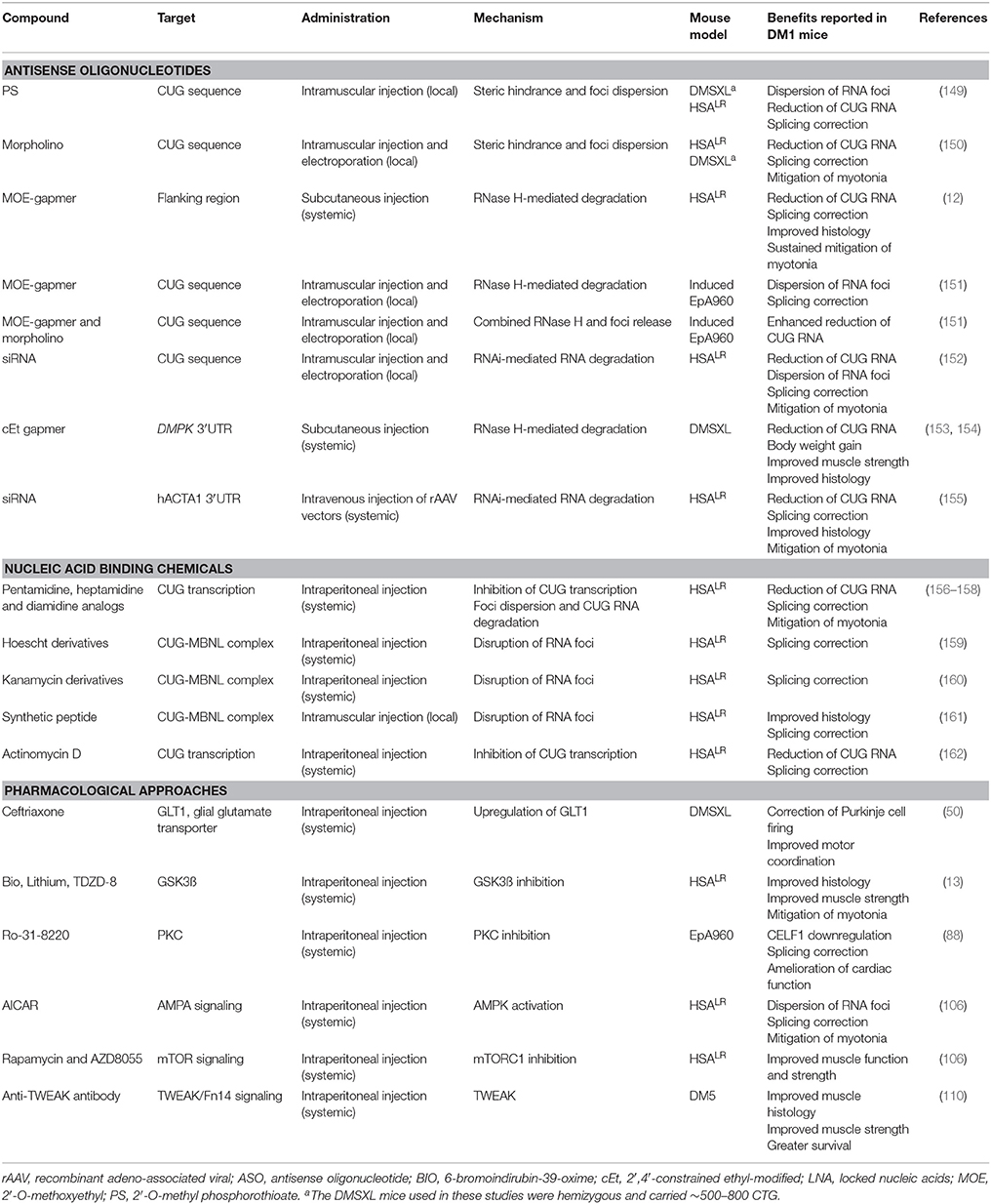
Table 3. Therapeutic strategies tested in DM1 mouse models.
ASO have been designed to disperse nuclear RNA foci and redistribute MBNL proteins, or to induce the degradation of expanded transcripts. Early approaches aimed to destabilize CUG RNA foci by direct injection of morpholino-type ASO into the skeletal muscle of HSALR mice. The reduction in nuclear foci, redistribution of MBNL1 protein and splicing correction was sufficient to improve muscle histology and myotonia (150). Similarly, 2′-O-methyl phosphorothioate (PS) modified ASO reduced foci number and corrected missplicing in two independent mouse models (149); unfortunately the molecular benefits were insufficient to improve muscle phenotypes (Table 3). Both strategies reduced the levels of toxic transcripts without RNase H activation, likely through the degradation of expanded transcripts released from nuclear foci. Alternative approaches used RNase H-active ASO to enhance nuclear RNA degradation of CUG repeats. Intramuscular injection and electroporation of 2′-O-methoxyethyl (MOE) gapmers knocked-down expanded CUG transcripts in EpA960 mice and reduced RNA foci (151). Further reduction in toxic RNA was achieved by the combination of RNase H-active MOE gampers and morpholinos (151). However, local injection caused some degree of muscle damage, which aggravated histopathology and splicing dysregulation in these mice. The systemic delivery of ASO overcomes this problem and is particularity attractive given the vast number of tissues and organs affected in DM1: systemic administration of MOE gapmers reduced expanded CUG RNA, corrected global transcriptome, ameliorated histopathology and resulted in long-term suppression of myotonia in HSALR mice (12). Similarly, 2′-4′-constrained-ethyl (cEt) ASO administrated systemically yielded robust reduction of expanded DMPK transcripts, improved body weight, muscle strength and histology of DMSXL mice (154). The demonstration that expanded CUG RNA is a potential target for the RNA interference (RNAi) pathway (163) suggested the therapeutic use of siRNA. Both intramuscular injection and viral delivery of siRNA molecules activated toxic CUG degradation, reduced molecular signs of RNA toxicity and improved the phenotypes of HSALR mice (152, 155).
ASO offer today a promising pipeline for therapeutic development, but their efficient delivery and biodistribution are still critical hurdles to overcome.
Small soluble chemicals with high biodistribution and low toxicity may provide an alternative to ASO. Some of these compounds were tested in DM1 mouse models (Table 3). Derivatives of pentamidine (and other diamidines), hoescht and aminoglycoside, as well as synthetic peptides yielded limited correction of missplicing in HSALR mice (156, 159, 160). While diamidines inhibit the transcription of toxic CUG RNA, the others likely disrupt RNA-protein complexes, releasing MBNL proteins from nuclear CUG foci. Although the benefits of some of these molecules were modest in mice, the results established a scaffold for chemical redesign to optimize biodistribution, reduce toxicity and increase efficacy.
Approaches limited to restoring MBNL function are unlikely to fully address the consequences of RNA toxicity and additional intermediates should also be targeted. The dissection of the molecular pathways implicated in DM1 pathogenesis revealed some of these targets and hinted at novel routes of pharmacological intervention (Table 3). In the future, therapeutic combination of multiple approaches to eliminate the primary offending RNA with approaches to correct downstream pathogenic events might be required.
Strategies targeting the DNA repeat expansion mutation were previously tested in DM1 mouse cell culture systems (164) or directly in HSALR skeletal muscle (165), and proved capable of stabilizing the trinucleotide CTG repeat tract. Although substantial effort has concentrated on the deleterious accumulation of toxic RNA, recent gene editing tools provide new means to target the upstream DNA mutation that causes DM1. CRISPR/Cas9 systems were tested in DMSXL mouse cells to induce repeat contractions (43), while modified Cas9 was used in HSALR mice to block the transcription of toxic RNA (166).
By definition, an animal model provides a simplification of the complex human system, or at least, part of it. Mouse models offer a good compromise between easy manipulation, affordable research cost and similarity to the complex physiology of humans. However, mice have limitations too, and today there is no perfect DM1 mouse model that fully recreates all disease aspects. Conversely, reduced body mass has been repeatedly reported in mice (21, 23, 26, 77, 78) but no direct parallel has been established with human clinical symptoms, nor is it known to what extend this phenotype reflects a DM1-associated developmental delay.
Given the nature of the constructs used, transgene expression varies between models and introduces some drawbacks that should not be overlooked. Some constitutive models (such as the DMSXL mice) express low transgene levels, and require breeding to homozygosity to develop disease phenotypes. In contrast, the high expression levels in tissue-specific models (such as the EpA960 and DM5 mice) may trigger some non-specific disease features. Finally, tissue and cell type-specific expression in HSALR, EpA960 and DM5/DM200 mice can mask non-cell-autonomous mechanisms, critical for some features of disease pathogenesis. In summary, the collection of mice available today covers different aspects of DM1 pathology to a certain extent, partially fulfilling the absence of a perfect mouse model, and providing means for data validation by independent laboratories.
Simple organisms can also provide complementary models for basic, translational and pre-clinical research. Although phylogenetically distant from humans, Drosophila melanogaster, Caenorhabditis elegans or zebrafish (Danio rerio), have multiple advantages over mice, including their easy manipulation, low maintenance cost and fast generation of large offspring. The expression of toxic RNA in D. melanogaster recreated molecular features of DM1, such as RNA foci accumulation, muscleblind protein sequestration and missplicing (167–169). Some lines showed eye degeneration (167, 168), a general readout of neurotoxicity, but which does not necessarily relate to human pathology. The development of muscle phenotypes, such as muscle wasting (167) and hypercontraction (169) seems more relevant. The expression of expanded CUG repeats also resulted in RNA foci and muscle phenotypes in zebrafish (170, 171) and C. elegans (172, 173). Together, these data suggest the conservation of the core mechanisms of RNA toxicity across species, and corroborate the use of simple organisms in large screenings for disease modifiers. Such studies have already resulted in the identification if genetic modifiers (167, 174, 175), chemicals that correct DM1 splicing abnormalities (176) and miRNA sponges that regulate MBNL protein levels and rescue fly phenotypes (177). The physiology of small organisms and humans are nonetheless substantially different, and therefore parallels must be established with care.
Transgenic mouse models, alone or in combination, have been key to understanding fundamental molecular pathomechanisms of DM. Over the last decade, the progress in mouse studies and the advances in high throughput approaches (e.g., transcriptomics and proteomics) have led to the identification of hundreds of misregulated genes and proteins, through changes in alternative splicing, polyadenylation, protein translation and phosphorylation. Understanding the contribution of these molecular events to the etiology of DM will help depict the course between repeat expansion and the onset of disease manifestations. Future studies should continue to address “which” disease intermediates and cell populations, “where” in the tissue and “when” during disease course experience the most pronounced abnormalities. Linking these variables will identify critical events and developmental windows during which specific cell pathways are particularly sensitive to pathological insults and targetable by corrective therapies. A better understanding of pathophysiological trajectories will guide the development of efficient therapeutic approaches.
Some models have deliberately focused on specific disease features and recapitulated a small number of disease phenotypes (e.g. muscle pathology in HSALR models, cardiac function in inducible CELF1-overexpressing mice). Although oversimplifying the situation, this reductionist approach has offered the opportunity to break the complexity of disease down to tractable “building blocks” and to unravel the mechanisms behind individual aspects of the disease. The future combination of these different models, by intercrossing different transgenic lines, might be considered to “rebuild” the convoluted human disease and to explore the interdependence of individual factors. The complexity of DM pathobiology and variation in mouse models design require, however, a critical approach in the interpretation and comparison of the results obtained with different lines.
There is little doubt that mouse models will continue to provide in-depth understanding of disease. One of their major advantages is the opportunity to monitor early pathological changes, prior to the onset of disease symptoms, which is difficult to achieve in humans with the current diagnostic standards. We anticipate that future studies will uncover additional cellular pathways impacted during the disease course, while revealing targetable events to reverse disease.
JA: experimental work and data acquisition; GG and MG-P: study design, interpretation, data analysis; SB and MG-P: preparation of figures; SB, GG, and MG-P: manuscript preparation.
We would like to acknowledge the AFM-Téléthon (France; Project Grant 19920), ANR (France), Imagine Foundation (France), INSERM (France) and Paris Descartes—Sorbonne Paris Cité University (France) for financial support. Our research has received a state subsidy managed by the National Research Agency, under the Investments or the Future program bearing the reference ANR-10-IAHU-01 and under the program ANR-10 BLAN-1121-01.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to apologize in advance to many authors and colleagues whose work was not directly discussed in this review owing to space limitations. We are grateful to the ethics committees that reviewed our protocols and procedures. All animal experiments were conducted according to the ARRIVE guidelines (Animal Research: Reporting In Vivo Experiments). This project has been conducted with the authorization for animal experimentation No. 75 003 in the animal facility with the approval No. B 91 228 107, both delivered by Prefecture de Police and the French Veterinary Department.
1. Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. (2012) 11:891–905. doi: 10.1016/S1474-4422(12)70204-1
2. Gourdon G, Meola G. Myotonic dystrophies: state of the art of new therapeutic developments for the CNS. Front Cell Neurosci. (2017) 11:101. doi: 10.3389/fncel.2017.00101
3. Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell (1992) 68:799–808. doi: 10.1016/0092-8674(92)90154-5
4. Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science (2001) 293:864–7. doi: 10.1126/science.1062125
5. Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol. (1995) 128:995–1002. doi: 10.1083/jcb.128.6.995
6. Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3' untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci USA (1997) 94:7388–93. doi: 10.1073/pnas.94.14.7388
7. Sicot G, Gourdon G, Gomes-Pereira M. Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: new findings and future challenges. Hum Mol Genet. (2011) 20:R116–123. doi: 10.1093/hmg/ddr343
8. Meola G, Cardani R. Myotonic dystrophy type 2: an update on clinical aspects, genetic and pathomolecular mechanism. J Neuromuscul Dis. (2015) 2:S59–71. doi: 10.3233/JND-150088
9. Willner P. Validation criteria for animal models of human mental disorders: learned helplessness as a paradigm case. Prog Neuropsychopharmacol Biol Psychiatry (1986) 10:677–90. doi: 10.1016/0278-5846(86)90051-5
10. Gomes-Pereira M, Cooper TA, Gourdon G. Myotonic dystrophy mouse models: towards rational therapy development. Trends Mol Med. (2011) 17:506–17. doi: 10.1016/j.molmed.2011.05.004
11. Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science (2000) 289:1769–73. doi: 10.1126/science.289.5485.1769
12. Wheeler TM, Leger AJ, Pandey SK, Macleod AR, Nakamori M, Cheng SH, et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature (2012) 488:111–5. doi: 10.1038/nature11362
13. Jones K, Wei C, Iakova P, Bugiardini E, Schneider-Gold C, Meola G, et al. GSK3beta mediates muscle pathology in myotonic dystrophy. J Clin Invest. (2012) 122:4461–72. doi: 10.1172/JCI64081
14. Seznec H, Lia-Baldini AS, Duros C, Fouquet C, Lacroix C, Hofmann-Radvanyi H, et al. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum Mol Genet. (2000) 9:1185–94. doi: 10.1093/hmg/9.8.1185
15. Gomes-Pereira M, Foiry L, Nicole A, Huguet A, Junien C, Munnich A, et al. CTG trinucleotide repeat “big jumps”: large expansions, small mice. PLoS Genet. (2007) 3:e52. doi: 10.1371/journal.pgen.0030052
16. Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. (2007) 117:2802–11. doi: 10.1172/JCI32308
17. Mahadevan MS, Yadava RS, Yu Q, Balijepalli S, Frenzel-McCardell CD, Bourne TD, et al. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet. (2006) 38:1066–70. doi: 10.1038/ng1857
18. Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, et al. A muscleblind knockout model for myotonic dystrophy. Science (2003a) 302:1978–80. doi: 10.1126/science.1088583
19. Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron (2012) 75:437–50. doi: 10.1016/j.neuron.2012.05.029
20. Poulos MG, Batra R, Li M, Yuan Y, Zhang C, Darnell RB, et al. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum Mol Genet. (2013) 22:3547–58. doi: 10.1093/hmg/ddt209
21. Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med. (2013) 5:1887–900. doi: 10.1002/emmm.201303275
22. Choi J, Personius KE, Difranco M, Dansithong W, Yu C, Srivastava S, et al. Muscleblind-like 1 and muscleblind-like 3 depletion synergistically enhances myotonia by altering Clc-1 RNA translation. EBioMed. (2015) 2:1034–47. doi: 10.1016/j.ebiom.2015.07.028
23. Thomas JD, Sznajder LJ, Bardhi O, Aslam FN, Anastasiadis ZP, Scotti MM, et al. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. (2017) 31:1122–33. doi: 10.1101/gad.300590.117
24. Koshelev M, Sarma S, Price RE, Wehrens XH, and Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet. (2010) 19:1066–75. doi: 10.1093/hmg/ddp570
25. Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res (2015) 25:858–871. doi: 10.1101/gr.184390.114
26. Huguet A, Medja F, Nicole A, Vignaud A, Guiraud-Dogan C, Ferry A, et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. (2012) 8:e1003043. doi: 10.1371/journal.pgen.1003043
27. Hernandez-Hernandez O, Guiraud-Dogan C, Sicot G, Huguet A, Luilier S, Steidl E, et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain (2013a) 136:957–70. doi: 10.1093/brain/aws367
28. Panaite PA, Kuntzer T, Gourdon G, Lobrinus JA, and Barakat-Walter I. Functional and histopathological identification of the respiratory failure in a DMSXL transgenic mouse model of myotonic dystrophy. Dis Model Mech. (2013) 6:622–31. doi: 10.1242/dmm.010512
29. Algalarrondo V, Wahbi K, Sebag F, Gourdon G, Beldjord C, Azibi K, et al. Abnormal sodium current properties contribute to cardiac electrical and contractile dysfunction in a mouse model of myotonic dystrophy type 1. Neuromuscul Disord. (2014) 25:308–20. doi: 10.1016/j.nmd.2014.11.018
30. Orengo JP, Chambon P, Metzger D, Mosier DR, Snipes GJ, Cooper TA. Expanded CTG repeats within the DMPK 3' UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci USA (2008) 105:2646–51. doi: 10.1073/pnas.0708519105
31. Wang PY, Lin YM, Wang LH, Kuo TY, Cheng SJ, Wang GS. Reduced cytoplasmic MBNL1 is an early event in a brain-specific mouse model of myotonic dystrophy. Hum Mol Genet. (2017) 26:2247–57. doi: 10.1093/hmg/ddx115
32. Mahadevan MS. Myotonic dystrophy: is a narrow focus obscuring the rest of the field? Curr Opin Neurol. (2012) 25:609–13. doi: 10.1097/WCO.0b013e328357b0d9
33. Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. (2006) 15:2087–97. doi: 10.1093/hmg/ddl132
34. Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J. (2000) 19:4439–48. doi: 10.1093/emboj/19.17.4439
35. Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. (2002) 11:805–14. doi: 10.1093/hmg/11.7.805
36. Timchenko LT, Miller JW, Timchenko NA, Devore DR, Datar KV, Lin L, et al. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucl Acids Res. (1996) 24:4407–14. doi: 10.1093/nar/24.22.4407
37. Roberts R, Timchenko NA, Miller JW, Reddy S, Caskey CT, Swanson MS, et al. Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice. Proc Natl Acad Sci USA (1997) 94:13221–6. doi: 10.1073/pnas.94.24.13221
38. Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell (2002) 10:45–53. doi: 10.1016/S1097-2765(02)00572-5
39. Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. (2007b) 117:3952–57. doi: 10.1172/JCI33355
40. Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. (2001) 29:40–7. doi: 10.1038/ng704
41. Orengo JP, Ward AJ, Cooper TA. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann Neurol. (2011) 69:681–90. doi: 10.1002/ana.22278
42. Batra R, Charizanis K, Swanson MS. Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum Mol Genet. (2010) 19:R77–82. doi: 10.1093/hmg/ddq132
43. Gudde A, Van Heeringen SJ, De Oude AI, Van Kessel IDG, Estabrook J, Wang ET, et al. Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol. (2017) 14:1374–88. doi: 10.1080/15476286.2017.1279787
44. Michel L, Huguet-Lachon A, Gourdon G. Sense and antisense DMPK RNA foci accumulate in DM1 tissues during development. PLoS ONE (2015) 10:e0137620. doi: 10.1371/journal.pone.0137620
45. Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation (2006) 74:65–80. doi: 10.1111/j.1432-0436.2006.00060.x
46. Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. (2004) 13:3079–88. doi: 10.1093/hmg/ddh327
47. Mankodi A, Lin X, Blaxall BC, Swanson MS, Thornton CA. Nuclear RNA foci in the heart in myotonic dystrophy. Circ Res (2005) 97:1152–5. doi: 10.1161/01.RES.0000193598.89753.e3
48. Holt I, Jacquemin V, Fardaei M, Sewry CA, Butler-Browne GS, Furling D, et al. Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle. Am J Pathol. (2009) 174:216–27. doi: 10.2353/ajpath.2009.080520
49. Kanadia RN, Urbinati CR, Crusselle VJ, Luo D, Lee YJ, Harrison JK, et al. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr Patterns (2003b) 3:459–62. doi: 10.1016/S1567-133X(03)00064-4
50. Sicot G, Servais L, Dinca DM, Leroy A, Prigogine C, Medja F, et al. (2017). Downregulation of the glial GLT1 glutamate transporter and purkinje cell dysfunction in a mouse model of myotonic dystrophy. Cell Rep. 19:2718–29. doi: 10.1016/j.celrep.2017.06.006
51. Matynia A, Ng CH, Dansithong W, Chiang A, Silva AJ, Reddy S. Muscleblind1, but not Dmpk or Six5, contributes to a complex phenotype of muscular and motivational deficits in mouse models of myotonic dystrophy. PLoS ONE (2010) 5:e9857. doi: 10.1371/journal.pone.0009857
52. Dixon DM, Choi J, El-Ghazali A, Park SY, Roos KP, Jordan MC, et al. Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci Rep. (2015) 5:9042. doi: 10.1038/srep09042
53. Suenaga K, Lee KY, Nakamori M, Tatsumi Y, Takahashi MP, Fujimura H, et al. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS ONE (2012) 7:e33218. doi: 10.1371/journal.pone.0033218
54. Hernandez-Hernandez O, Sicot G, Dinca DM, Huguet A, Nicole A, Buee L, et al. Synaptic protein dysregulation in myotonic dystrophy type 1: disease neuropathogenesis beyond missplicing. Rare Dis. (2013b) 1:e25553. doi: 10.4161/rdis.25553
55. Choi J, Dixon DM, Dansithong W, Abdallah WF, Roos KP, Jordan MC, et al. Muscleblind-like 3 deficit results in a spectrum of age-associated pathologies observed in myotonic dystrophy. Sci Rep. (2016) 6:30999. doi: 10.1038/srep30999
56. Mateos-Aierdi AJ, Goicoechea M, Aiastui A, Fernandez-Torron R, Garcia-Puga M, Matheu A, et al. Muscle wasting in myotonic dystrophies: a model of premature aging. Front Aging Neurosci. (2015) 7:125. doi: 10.3389/fnagi.2015.00125
57. Chou CC, Chang PC, Wei YC, and Lee KY. Optical mapping approaches on muscleblind-like compound knockout mice for understanding mechanistic insights into ventricular arrhythmias in myotonic dystrophy. J Am Heart Assoc. (2017) 6:e005191. doi: 10.1161/JAHA.116.005191
58. Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell (2014) 56:311–22. doi: 10.1016/j.molcel.2014.08.027
59. Goodwin M, Mohan A, Batra R, Lee KY, Charizanis K, Gomez FJ, et al. MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep. (2015) 12:1159–68. doi: 10.1016/j.celrep.2015.07.029
60. Curinha A, Oliveira Braz S, Pereira-Castro I, Cruz A, Moreira A. Implications of polyadenylation in health and disease. Nucleus (2014) 5:508–19. doi: 10.4161/nucl.36360
61. Osborne RJ, Lin X, Welle S, Sobczak K, O'rourke JR, Swanson MS, et al. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet. (2009) 18:1471–81. doi: 10.1093/hmg/ddp058
62. Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. (2010) 17:187–93. doi: 10.1038/nsmb.1720
63. Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci USA (2006) 103:11748–53. doi: 10.1073/pnas.0604970103
64. Chamberlain CM, Ranum LP. Mouse model of muscleblind-like 1 overexpression: skeletal muscle effects and therapeutic promise. Hum Mol Genet. (2012) 21:4645–54. doi: 10.1093/hmg/dds306
65. Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, et al. CUGBP1 and MBNL1 preferentially bind to 3' UTRs and facilitate mRNA decay. Sci Rep. (2012) 2:209. doi: 10.1038/srep00209
66. Adereth Y, Dammai V, Kose N, Li R, Hsu T. RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat Cell Biol. (2005) 7:1240–7. doi: 10.1038/ncb1335
67. Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell (2012) 150:710–24. doi: 10.1016/j.cell.2012.06.041
68. Wang PY, Chang KT, Lin YM, Kuo TY, Wang GS. Ubiquitination of MBNL1 is required for its cytoplasmic localization and function in promoting neurite outgrowth. Cell Rep. (2018) 22:2294–306. doi: 10.1016/j.celrep.2018.02.025
69. Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA (2011) 108:260–5. doi: 10.1073/pnas.1013343108
70. Zu T, Cleary JD, Liu Y, Banez-Coronel M, Bubenik JL, Ayhan F, et al. RAN translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron (2017) 95:1292–305 e1295. doi: 10.1016/j.neuron.2017.08.039
71. Cleary JD, Ranum LP. New developments in RAN translation: insights from multiple diseases. Curr Opin Genet Dev. (2017) 44:125–34. doi: 10.1016/j.gde.2017.03.006
72. Kino Y, Washizu C, Kurosawa M, Oma Y, Hattori N, Ishiura S, et al. Nuclear localization of MBNL1: splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum Mol Genet. (2015) 24:740–56. doi: 10.1093/hmg/ddu492
73. Kim YK, Mandal M, Yadava RS, Paillard L, Mahadevan MS. Evaluating the effects of CELF1 deficiency in a mouse model of RNA toxicity. Hum Mol Genet. (2014) 23:293–302. doi: 10.1093/hmg/ddt419
74. Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, et al. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis. (2009) 36:181–90. doi: 10.1016/j.nbd.2009.07.009
75. Salisbury E, Schoser B, Schneider-Gold C, Wang GL, Huichalaf C, Jin B, et al. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol. (2009) 175:748–62. doi: 10.2353/ajpath.2009.090047
76. Cardani R, Bugiardini E, Renna LV, Rossi G, Colombo G, Valaperta R, et al. Overexpression of CUGBP1 in skeletal muscle from adult classic myotonic dystrophy type 1 but not from myotonic dystrophy type 2. PLoS ONE (2013) 8:e83777. doi: 10.1371/journal.pone.0083777
77. Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet. (2005) 14:1539–547. doi: 10.1093/hmg/ddi162
78. Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem. (2004) 279:13129–39. doi: 10.1074/jbc.M312923200
79. Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet. (2010) 19:3614–22. doi: 10.1093/hmg/ddq277
80. Berger DS, Ladd AN. Repression of nuclear CELF activity can rescue CELF-regulated alternative splicing defects in skeletal muscle models of myotonic dystrophy. PLoS Curr. (2012) 4:RRN1305. doi: 10.1371/currents.RRN1305
81. Ladd AN. CUG-BP, Elav-like family (CELF)-mediated alternative splicing regulation in the brain during health and disease. Mol Cell Neurosci. (2012) 56:456–64. doi: 10.1016/j.mcn.2012.12.003
82. Dhaenens CM, Tran H, Frandemiche ML, Carpentier C, Schraen-Maschke S, Sistiaga A, et al. Mis-splicing of Tau exon 10 in myotonic dystrophy type 1 is reproduced by overexpression of CELF2 but not by MBNL1 silencing. Biochim Biophys Acta (2011) 1812:732–42. doi: 10.1016/j.bbadis.2011.03.010
83. Meola G, Jones K, Wei C, Timchenko LT. Dysfunction of protein homeostasis in myotonic dystrophies. Histol Histopathol. (2013) 28:1089–98. doi: 10.14670/HH-28.1089
84. Huichalaf C, Sakai K, Jin B, Jones K, Wang GL, Schoser B, et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J. (2010) 24:3706–19. doi: 10.1096/fj.09-151159
85. Timchenko NA, Wang GL, Timchenko LT. RNA CUG-binding protein 1 increases translation of 20-kDa isoform of CCAAT/enhancer-binding protein beta by interacting with the alpha and beta subunits of eukaryotic initiation translation factor 2. J Biol Chem. (2005) 280:20549–57. doi: 10.1074/jbc.M409563200
86. Timchenko NA, Iakova P, Cai ZJ, Smith JR, and Timchenko LT. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol Cell Biol. (2001) 21:6927–38. doi: 10.1128/MCB.21.20.6927-6938.2001
87. Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. (2007) 28:68–78. doi: 10.1016/j.molcel.2007.07.027
88. Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, Cooper TA. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest. (2009) 119:3797–806. doi: 10.1172/JCI37976
89. Kim YK, Yadava RS, Mandal M, Mahadevan K, Yu Q, Leitges M, et al. Disease phenotypes in a mouse model of RNA toxicity are independent of protein kinase calpha and protein kinase cbeta. PLoS ONE (2016) 11:e0163325. doi: 10.1371/journal.pone.0163325
90. Ketley A, Chen CZ, Li X, Arya S, Robinson TE, Granados-Riveron J, et al. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum Mol Genet. (2014) 23:1551–62. doi: 10.1093/hmg/ddt542
91. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. (2000) 351:95–105. doi: 10.1042/bj3510095
92. Wei C, Stock L, Valanejad L, Zalewski ZA, Karns R, Puymirat J, et al. Correction of GSK3beta at young age prevents muscle pathology in mice with myotonic dystrophy type 1. FASEB J. (2018) 32:2073–85. doi: 10.1096/fj.201700700R
93. Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, Cooper TA. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep. (2014) 6:336–45. doi: 10.1016/j.celrep.2013.12.025
94. Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum Mutat. (2002) 19:423–34. doi: 10.1002/humu.10063
95. Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. (2011) 17:720–5. doi: 10.1038/nm.2374
96. Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, et al. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum Mol Genet. (2012) 21:1312–24. doi: 10.1093/hmg/ddr568
97. Rau F, Laine J, Ramanoudjame L, Ferry A, Arandel L, Delalande O, et al. Abnormal splicing switch of DMD's penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat Commun. (2015) 6:7205. doi: 10.1038/ncomms8205
98. Gao Z, Cooper TA. Reexpression of pyruvate kinase M2 in type 1 myofibers correlates with altered glucose metabolism in myotonic dystrophy. Proc Natl Acad Sci USA (2013) 110:13570–5. doi: 10.1073/pnas.1308806110
99. Kimura T, Lueck JD, Harvey PJ, Pace SM, Ikemoto N, Casarotto MG, et al. Alternative splicing of RyR1 alters the efficacy of skeletal EC coupling. Cell Calcium (2009) 45:264–74. doi: 10.1016/j.ceca.2008.11.005
100. Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat Commun. (2016) 7:11067. doi: 10.1038/ncomms11067
101. Ravel-Chapuis A, Belanger G, Yadava RS, Mahadevan MS, Desgroseillers L, Cote J, et al. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol. (2012) 196:699–712. doi: 10.1083/jcb.201108113
102. Ravel-Chapuis A, Crawford TE, Blais-Crepeau ML, Belanger G, Richer CT, Jasmin BJ. The RNA-binding protein Staufen1 impairs myogenic differentiation via a c-myc-dependent mechanism. Mol Biol Cell. (2014) 25:3765–78. doi: 10.1091/mbc.e14-04-0895
103. Crawford Parks TE, Ravel-Chapuis A, Bondy-Chorney E, Renaud JM, Cote J, Jasmin BJ. Muscle-specific expression of the RNA-binding protein Staufen1 induces progressive skeletal muscle atrophy via regulation of phosphatase tensin homolog. Hum Mol Genet. (2017) 26:1821–38. doi: 10.1093/hmg/ddx085
104. Manning BD, and Toker A. (2017). AKT/PKB signaling: navigating the network. Cell 169:381–405. doi: 10.1016/j.cell.2017.04.001
105. Vignaud A, Ferry A, Huguet A, Baraibar M, Trollet C, Hyzewicz J, et al. Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin-proteasome pathway. Neuromuscul Disord. (2010) 20:319–25. doi: 10.1016/j.nmd.2010.03.006
106. Brockhoff M, Rion N, Chojnowska K, Wiktorowicz T, Eickhorst C, Erne B, et al. Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. J Clin Invest. (2017) 127:549–63. doi: 10.1172/JCI89616
107. Vessey JP, Macchi P, Stein JM, Mikl M, Hawker KN, Vogelsang P, et al. A loss of function allele for murine Staufen1 leads to impairment of dendritic Staufen1-RNP delivery and dendritic spine morphogenesis. Proc Natl Acad Sci USA (2008) 105:16374–9. doi: 10.1073/pnas.0804583105
108. Amato S, and Man HY. (2011). Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle 10:3452–60. doi: 10.4161/cc.10.20.17953
109. Denis JA, Gauthier M, Rachdi L, Aubert S, Giraud-Triboult K, Poydenot P, et al. mTOR-dependent proliferation defect in human ES-derived neural stem cells affected by myotonic dystrophy type 1. J Cell Sci. (2013) 126:1763–72. doi: 10.1242/jcs.116285
110. Yadava RS, Foff EP, Yu Q, Gladman JT, Kim YK, Bhatt KS, et al. TWEAK/Fn14, a pathway and novel therapeutic target in myotonic dystrophy. Hum Mol Genet. (2015) 24:2035–48. doi: 10.1093/hmg/ddu617
111. Sato S, Ogura Y, Kumar A. TWEAK/Fn14 signaling axis mediates skeletal muscle atrophy and metabolic dysfunction. Front Immunol. (2014) 5:18. doi: 10.3389/fimmu.2014.00018
112. Nakamori M, Hamanaka K, Thomas JD, Wang ET, Hayashi YK, Takahashi MP, et al. Aberrant myokine signaling in congenital myotonic dystrophy. Cell Rep. (2017) 21:1240–52. doi: 10.1016/j.celrep.2017.10.018
113. Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol. (2011) 18:840–5. doi: 10.1038/nsmb.2067
114. Gambardella S, Rinaldi F, Lepore SM, Viola A, Loro E, Angelini C, et al. Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J Transl Med. (2010) 8:48. doi: 10.1186/1479-5876-8-48
115. Perbellini R, Greco S, Sarra-Ferraris G, Cardani R, Capogrossi MC, Meola G, et al. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord. (2011) 21:81–8. doi: 10.1016/j.nmd.2010.11.012
116. Greco S, Perfetti A, Fasanaro P, Cardani R, Capogrossi MC, Meola G, et al. Deregulated microRNAs in myotonic dystrophy type 2. PLoS ONE (2012) 7:e39732. doi: 10.1371/journal.pone.0039732
117. Fernandez-Costa JM, Garcia-Lopez A, Zuniga S, Fernandez-Pedrosa V, Felipo-Benavent A, Mata M, et al. Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum Mol Genet. (2013) 22:704–716. doi: 10.1093/hmg/dds478
118. Fritegotto C, Ferrati C, Pegoraro V, Angelini C. Micro-RNA expression in muscle and fiber morphometry in myotonic dystrophy type 1. Neurol Sci. (2017) 38:619–25. doi: 10.1007/s10072-017-2811-2
119. Koutsoulidou A, Kyriakides TC, Papadimas GK, Christou Y, Kararizou E, Papanicolaou EZ, et al. Elevated muscle-specific miRNAs in serum of myotonic dystrophy patients relate to muscle disease progress. PLoS ONE (2015) 10:e0125341. doi: 10.1371/journal.pone.0125341
120. Chang KT, Cheng CF, King PC, Liu SY, Wang GS. CELF1 mediates connexin 43 mRNA degradation in dilated cardiomyopathy. Circ Res. (2017) 121:1140–52. doi: 10.1161/CIRCRESAHA.117.311281
121. Fu YH, Friedman DL, Richards S, Pearlman JA, Gibbs RA, Pizzuti A, et al. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science (1993) 260:235–38. doi: 10.1126/science.8469976
122. Berul CI, Maguire CT, Aronovitz MJ, Greenwood J, Miller C, Gehrmann J, et al. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J Clin Invest. (1999) 103:R1–7. doi: 10.1172/JCI5346
123. Berul CI, Maguire CT, Gehrmann J, Reddy S. Progressive atrioventricular conduction block in a mouse myotonic dystrophy model. J Interv Card Electrophysiol. (2000) 4:351–8. doi: 10.1023/A:1009842114968
124. Reddy S, Smith DB, Rich MM, Leferovich JM, Reilly P, Davis BM, et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet. (1996) 13:325–35. doi: 10.1038/ng0796-325
125. Carrell ST, Carrell EM, Auerbach D, Pandey SK, Bennett CF, Dirksen RT, et al. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum Mol Genet. (2016) 25:4328–38. doi: 10.1093/hmg/ddw266
126. Okkersen K, Monckton DG, Le N, Tuladhar AM, Raaphorst J, Van Engelen BGM. Brain imaging in myotonic dystrophy type 1: a systematic review. Neurology (2017) 89:960–9. doi: 10.1212/WNL.0000000000004300
127. Pal R, Agbas A, Bao X, Hui D, Leary C, Hunt J, et al. Selective dendrite-targeting of mRNAs of NR1 splice variants without exon 5: identification of a cis-acting sequence and isolation of sequence-binding proteins. Brain Res. (2003) 994:1–18. doi: 10.1016/j.brainres.2003.08.046
128. Han S, Nam J, Li Y, Kim S, Cho SH, Cho YS, et al. Regulation of dendritic spines, spatial memory, and embryonic development by the TANC family of PSD-95-interacting proteins. J Neurosci. (2010) 30:15102–12. doi: 10.1523/JNEUROSCI.3128-10.2010
129. McKinney BC, Sze W, Lee B, Murphy GG. Impaired long-term potentiation and enhanced neuronal excitability in the amygdala of Ca(V)1.3 knockout mice. Neurobiol Learn Mem. (2009) 92:519–28. doi: 10.1016/j.nlm.2009.06.012
130. Yamamoto H, Kokame K, Okuda T, Nakajo Y, Yanamoto H, Miyata T. NDRG4 protein-deficient mice exhibit spatial learning deficits and vulnerabilities to cerebral ischemia. J Biol Chem. (2011) 286:26158–65. doi: 10.1074/jbc.M111.256446
131. Vermersch P, Sergeant N, Ruchoux MM, Hofmann-Radvanyi H, Wattez A, Petit H, et al. Specific tau variants in the brains of patients with myotonic dystrophy. Neurology (1996) 47:711–7. doi: 10.1212/WNL.47.3.711
132. Sergeant N, Sablonniere B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet. (2001) 10:2143–55. doi: 10.1093/hmg/10.19.2143
133. Caillet-Boudin ML, Fernandez-Gomez FJ, Tran H, Dhaenens CM, Buee L, and Sergeant N. Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci. (2014) 6:57. doi: 10.3389/fnmol.2013.00057
134. Furuta M, Kimura T, Nakamori M, Matsumura T, Fujimura H, Jinnai K, et al. Macroscopic and microscopic diversity of missplicing in the central nervous system of patients with myotonic dystrophy type 1. Neuroreport (2018) 29:235–40. doi: 10.1097/WNR.0000000000000968
135. Ishii S, Nishio T, Sunohara N, Yoshihara T, Takemura K, Hikiji K, et al. Small increase in triplet repeat length of cerebellum from patients with myotonic dystrophy. Hum Genet. (1996) 98:138–40. doi: 10.1007/s004390050176
136. Fortune MT, Vassilopoulos C, Coolbaugh MI, Siciliano MJ, Monckton DG. Dramatic, expansion-biased, age-dependent, tissue-specific somatic mosaicism in a transgenic mouse model of triplet repeat instability. Hum Mol Genet. (2000) 9:439–45. doi: 10.1093/hmg/9.3.439
137. Chen G, Carter RE, Cleary JD, Reid TS, Ranum LP, Swanson MS, et al. Altered levels of the splicing factor muscleblind modifies cerebral cortical function in mouse models of myotonic dystrophy. Neurobiol Dis. (2018) 112:35–48. doi: 10.1016/j.nbd.2018.01.003
138. Meola G, Sansone V. Cerebral involvement in myotonic dystrophies. Muscle Nerve (2007) 36:294–306. doi: 10.1002/mus.20800
139. Takado Y, Terajima K, Ohkubo M, Okamoto K, Shimohata T, Nishizawa M, et al. Diffuse brain abnormalities in myotonic dystrophy type 1 detected by 3.0 T proton magnetic resonance spectroscopy. Eur Neurol. (2015) 73:247–56. doi: 10.1159/000371575
140. Ono S, Takahashi K, Jinnai K, Kanda F, Fukuoka Y, Kurisaki H, et al. Loss of serotonin-containing neurons in the raphe of patients with myotonic dystrophy: a quantitative immunohistochemical study and relation to hypersomnia. Neurology (1998a) 50:535–8. doi: 10.1212/WNL.50.2.535
141. Ono S, Takahashi K, Jinnai K, Kanda F, Fukuoka Y, Kurisaki H, et al. Loss of catecholaminergic neurons in the medullary reticular formation in myotonic dystrophy. Neurology (1998b) 51:1121–4. doi: 10.1212/WNL.51.4.1121
142. Serra L, Cercignani M, Bruschini M, Cipolotti L, Mancini M, Silvestri G, et al. “I know that you know that i know”: neural substrates associated with social cognition deficits in DM1 patients. PLoS ONE (2016) 11:e0156901. doi: 10.1371/journal.pone.0156901
143. Peric S, Brajkovic L, Belanovic B, Ilic V, Salak-Djokic B, Basta I, et al. Brain positron emission tomography in patients with myotonic dystrophy type 1 and type 2. J Neurol Sci. (2017) 378:187–92. doi: 10.1016/j.jns.2017.05.013
144. Gallais B, Gagnon C, Mathieu J, Richer L. Cognitive decline over time in adults with myotonic dystrophy type 1: A 9-year longitudinal study. Neuromuscul Disord. (2017) 27:61–72. doi: 10.1016/j.nmd.2016.10.003
145. Ashizawa T, Sarkar PS. Myotonic dystrophy types 1 and 2. Handb Clin Neurol. (2011) 101:193–237. doi: 10.1016/B978-0-08-045031-5.00015-3
146. Panaite PA, Kielar M, Kraftsik R, Gourdon G, Kuntzer T, Barakat-Walter I. Peripheral neuropathy is linked to a severe form of myotonic dystrophy in transgenic mice. J Neuropathol Exp Neurol. (2011) 70:678–85. doi: 10.1097/NEN.0b013e3182260939
147. Wheeler TM, Krym MC, Thornton CA. Ribonuclear foci at the neuromuscular junction in myotonic dystrophy type 1. Neuromuscul Disord. (2007a) 17:242–7. doi: 10.1016/j.nmd.2006.12.015
148. Marteyn A, Maury Y, Gauthier MM, Lecuyer C, Vernet R, Denis JA, et al. Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell (2011) 8:434–44. doi: 10.1016/j.stem.2011.02.004
149. Mulders SA, Van Den Broek WJ, Wheeler TM, Croes HJ, Van Kuik-Romeijn P, De Kimpe SJ, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci USA (2009) 106:13915–20. doi: 10.1073/pnas.0905780106
150. Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science (2009) 325:336–9. doi: 10.1126/science.1173110
151. Lee JE, Bennett CF, Cooper TA. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc Natl Acad Sci USA (2012) 109:4221–6. doi: 10.1073/pnas.1117019109
152. Sobczak K, Wheeler TM, Wang W, Thornton CA. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol Ther. (2012) 21:380–7. doi: 10.1038/mt.2012.222
153. Pandey SK, Wheeler TM, Justice SL, Kim A, Younis HS, Gattis D, et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J Pharmacol Exp Ther. (2015) 355:329–40. doi: 10.1124/jpet.115.226969
154. Jauvin D, Chretien J, Pandey SK, Martineau L, Revillod L, Bassez G, et al. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol Ther Nucleic Acids (2017) 7:465–74. doi: 10.1016/j.omtn.2017.05.007
155. Bisset DR, Stepniak-Konieczna EA, Zavaljevski M, Wei J, Carter GT, Weiss MD, et al. Therapeutic impact of systemic AAV-mediated RNA interference in a mouse model of myotonic dystrophy. Hum Mol Genet. (2015) 24:4971–83. doi: 10.1093/hmg/ddv219
156. Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci USA (2009) 106:18551–6. doi: 10.1073/pnas.0903234106
157. Coonrod LA, Nakamori M, Wang W, Carrell S, Hilton CL, Bodner MJ, et al. Reducing levels of toxic RNA with small molecules. ACS Chem Biol. (2013) 8:2528–37. doi: 10.1021/cb400431f
158. Siboni RB, Bodner MJ, Khalifa MM, Docter AG, Choi JY, Nakamori M, et al. Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J Med Chem. (2015a) 58:5770–80. doi: 10.1021/acs.jmedchem.5b00356
159. Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, et al. Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database and chemical similarity searching. J Am Chem Soc. (2012) 134:4731–42. doi: 10.1021/ja210088v
160. Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem Biol. (2012) 7:856–62. doi: 10.1021/cb200408a
161. Garcia-Lopez A, Llamusi B, Orzaez M, Perez-Paya E, Artero RD. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc Natl Acad Sci USA (2011) 108:11866–71. doi: 10.1073/pnas.1018213108
162. Siboni RB, Nakamori M, Wagner SD, Struck AJ, Coonrod LA, Harriott SA, et al. Actinomycin D specifically reduces expanded CUG repeat rna in myotonic dystrophy models. Cell Rep. (2015b) 13:2386–94. doi: 10.1016/j.celrep.2015.11.028
163. Krol J, Fiszer A, Mykowska A, Sobczak K, De Mezer M, and Krzyzosiak WJ. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell (2007) 25:575–86. doi: 10.1016/j.molcel.2007.01.031
164. Gomes-Pereira M, Monckton DG. Chemically induced increases and decreases in the rate of expansion of a CAG∙CTG triplet repeat. Nucleic Acids Res. (2004) 32:2865–72. doi: 10.1093/nar/gkh612
165. Nakamori M, Pearson CE, and Thornton CA. Bidirectional transcription stimulates expansion and contraction of expanded (CTG).(CAG) repeats. Hum Mol Genet. (2011) 20:580–8. doi: 10.1093/hmg/ddq501
166. Pinto BS, Saxena T, Oliveira R, Mendez-Gomez HR, Cleary JD, Denes LT, et al. Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol Cell (2017) 68:479–90 e475. doi: 10.1016/j.molcel.2017.09.033
167. Garcia-Lopez A, Monferrer L, Garcia-Alcover I, Vicente-Crespo M, Alvarez-Abril MC, Artero RD. Genetic and chemical modifiers of a CUG toxicity model in drosophila. PLoS ONE (2008) 3:e1595. doi: 10.1371/journal.pone.0001595
168. Yu Z, Teng X, Bonini NM. Triplet repeat-derived siRNAs enhance RNA-mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet. (2011) 7:e1001340. doi: 10.1371/journal.pgen.1001340
169. Picchio L, Plantie E, Renaud Y, Poovthumkadavil P, Jagla K. Novel Drosophila model of myotonic dystrophy type 1: phenotypic characterization and genome-wide view of altered gene expression. Hum Mol Genet. (2013) 22:2795–810. doi: 10.1093/hmg/ddt127
170. Delorimier E, Coonrod LA, Copperman J, Taber A, Reister EE, Sharma K, et al. Modifications to toxic CUG RNAs induce structural stability, rescue mis-splicing in a myotonic dystrophy cell model and reduce toxicity in a myotonic dystrophy zebrafish model. Nucleic Acids Res. (2014) 42:12768–78. doi: 10.1093/nar/gku941
171. Todd PK, Ackall FY, Hur J, Sharma K, Paulson HL, Dowling JJ. Transcriptional changes and developmental abnormalities in a zebrafish model of myotonic dystrophy type 1. Dis Model Mech. (2014) 7:143–55. doi: 10.1242/dmm.012427
172. Chen KY, Pan H, Lin MJ, Li YY, Wang LC, Wu YC, et al. Length-dependent toxicity of untranslated CUG repeats on Caenorhabditis elegans. Biochem Biophys Res Commun. (2007) 352:774–779. doi: 10.1016/j.bbrc.2006.11.102
173. Wang LC, Chen KY, Pan H, Wu CC, Chen PH, Liao YT, et al. Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell Mol Life Sci. (2011) 68:1255–67. doi: 10.1007/s00018-010-0522-4
174. Garcia SM, Tabach Y, Lourenco GF, Armakola M, Ruvkun G. Identification of genes in toxicity pathways of trinucleotide-repeat RNA in C. elegans. Nat Struct Mol Biol. (2014) 21:712–20. doi: 10.1038/nsmb.2858
175. Bargiela A, Cerro-Herreros E, Fernandez-Costa JM, Vilchez JJ, Llamusi B, Artero R. Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis Model Mech. (2015) 8:679–90. doi: 10.1242/dmm.018127
176. Garcia-Alcover I, Colonques-Bellmunt J, Garijo R, Tormo JR, Artero R, Alvarez-Abril MC, et al. Development of a Drosophila melanogaster spliceosensor system for in vivo high-throughput screening in myotonic dystrophy type 1. Dis Model Mech. (2014) 7:1297–306. doi: 10.1242/dmm.016592
Keywords: myotonic dystrophy, mouse, trinucleotide DNA repeat, skeletal muscle, cardiac muscle, central nervous system, brain, RNA toxicity
Citation: Braz SO, Acquaire J, Gourdon G and Gomes-Pereira M (2018) Of Mice and Men: Advances in the Understanding of Neuromuscular Aspects of Myotonic Dystrophy. Front. Neurol. 9:519. doi: 10.3389/fneur.2018.00519
Received: 17 March 2018; Accepted: 12 June 2018;
Published: 10 July 2018.
Edited by:
Giovanni Meola, Università degli Studi di Milano, ItalyReviewed by:
Raghav Govindarajan, University of Missouri, United StatesCopyright © 2018 Braz, Acquaire, Gourdon and Gomes-Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mário Gomes-Pereira, bWFyaW8ucGVyZWlyYUBpbnNlcm0uZnI=.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.