
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Neurol., 26 June 2018
Sec. Neurocritical and Neurohospitalist Care
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00497
 William A. Kofke1*
William A. Kofke1* Yue Ren2John G. Augoustides1Hongzhe Li2Katherine Nathanson3
Yue Ren2John G. Augoustides1Hongzhe Li2Katherine Nathanson3 Robert Siman4Qing Cheng Meng1Weiming Bu1
Robert Siman4Qing Cheng Meng1Weiming Bu1 Sukanya Yandrawatthana1
Sukanya Yandrawatthana1 Guy Kositratna1Cecilia Kim5Joseph E. Bavaria6
Guy Kositratna1Cecilia Kim5Joseph E. Bavaria6Background: Neuroprotection studies are generally unable to demonstrate efficacy in humans. Our specific hypothesis is that multiple pathophysiologic pathways, of variable importance, contribute to ischemic brain damage. As a corollary to this, we discuss the broad hypothesis that a multifaceted approach will improve the probability of efficacious neuroprotection. But to properly test this hypothesis the nature and importance of the multiple contributing pathways needs elucidation. Our aim is to demonstrate, using functional genomics, in human cardiac surgery procedures associated with cerebral ischemia, that the pathogenesis of perioperative human ischemic brain damage involves the function of multiple variably weighted proteins involving several pathways. We then use these data and literature to develop a proposal for rational design of human neuroprotection protocols.
Methods: Ninety-four patients undergoing deep hypothermic circulatory arrest (DHCA) and/or aortic valve replacement surgery had brain damage biomarkers, S100β and neurofilament H (NFH), assessed at baseline, 1 and 24 h post-cardiopulmonary bypass (CPB) with analysis for association with 92 single nucleotide polymorphisms (SNPs) (selected by co-author WAK) related to important proteins involved in pathogenesis of cerebral ischemia.
Results: At the nominal significance level of 0.05, changes in S100β and in NFH at 1 and 24 h post-CPB were associated with multiple SNPs involving several prospectively determined pathophysiologic pathways, but were not individually significant after multiple comparison adjustments. Variable weights for the several evaluated SNPs are apparent on regression analysis and, notably, are dissimilar related to the two biomarkers and over time post CPB. Based on our step-wise regression model, at 1 h post-CPB, SOD2, SUMO4, and GP6 are related to relative change of NFH while TNF, CAPN10, NPPB, and SERPINE1 are related to the relative change of S100B. At 24 h post-CPB, ADRA2A, SELE, and BAX are related to the relative change of NFH while SLC4A7, HSPA1B, and FGA are related to S100B.
Conclusions: In support of the proposed hypothesis, association SNP data suggest function of specific disparate proteins, as reflected by genetic variation, may be more important than others with variation at different post-insult times after human brain ischemia. Such information may support rational design of post-insult time-sensitive multifaceted neuroprotective therapies.
Donnan (1) in the 2007 Feinberg lecture made this remarkable statement about neuroprotection research:
“We have reached a stage at which research in this area should stop altogether or radical new approaches adopted.”
Donnan's challenge was to develop a radical, transformative, new approach to studying neuroprotection. However, little has changed since 2007 in the approach to clinical neuroprotection studies other than efforts to do a better job with the process of monotherapy preclinical research (2, 3). Over 23,000 publications can be found dealing with stroke and its treatment in various preclinical models. Over 4,700 clinical trials with over 6,300 interventions are listed on the Internet Stroke Trials Registry (4) based on this extensive volume of preclinical work. However, unfortunately there are few apparent reproducible and fully implemented results of any demonstrable efficacy in the acute context in humans. This constitutes a massive failure to translate preclinical findings to humans. Various causes of this futility in neuroprotection research have been suggested (5–14). These include the use of animal models lacking the concomitant anatomy and diseases seen in humans, overly homogeneous animal model insults, inbred animal strains without co-morbid diseases, timing and dosing of therapy in relation to ischemia (before, during, after), the problematic use of monotherapy for a pathophysiologically multifaceted disease (5, 8, 11, 13, 15–17), and variations in the health care system with varying approaches to overall care (16, 18–25).
Several high level commissions were tasked to solve this problem. This has resulted in the STAIR (3) and ARRIVE (26) recommendations for proper standards in the conduct and transparent reporting of preclinical neuroprotective drug development (Notably the STAIR guidelines do not discount so-called “cocktail” combination therapies). The International Mission on Prognosis and Clinical Trial Design (IMPACT) (27–29) was also assembled to evaluate comparable issues in traumatic brain injury neuroprotection trials. In addition, the NINDS sponsored a workshop on Improving the Quality of NINDS-Supported Preclinical and Clinical Research through Rigorous Study Design and Transparent Reporting was held in 2012 (30) with promulgation of the RIGOR guidelines (31). In addition the Multicentre Preclinical Animal Research Team (Multi-Part) (32) provides a detailed prescription for the conduct of multi-institutional randomized pre-clinical trials. Others provide support for this notion (33, 34) and have also written reviews and editorials urging better quality preclinical research in support of the concepts espoused in the STAIR and ARRIVE guidelines (35–37). It is notable that all of these efforts to improve the translation of preclinical research generally entail simply improving existing approaches without truly innovative approaches to the problem, which we believe we are herein suggesting.
Neuroprotection, since Donnan's comments in 2007, notwithstanding the aforementioned several commissions (STAIR, ARRIVE, IMPACT, Multi-Part) addressing the problem, continues as a major challenge, and opportunity, in clinical medicine. The massive failure to translate preclinical findings to humans is the essence of the challenge facing us. Presently, it is apparent that the new transformative approach suggested as needed by Donnan has not yet been developed. Of the numerous reasons for the many failed human neuroprotection studies that have been suggested, one important contributor is certainly the notion that many time-dependent pathophysiologic processes are undoubtedly involved in the final outcome of a cerebral insult (16). Notably, these pathways likely interact (38), sometimes merging into so called “hub” pathways (39), and their import likely varies in relation to each other and over time after the ischemic insult (40). Thus, it follows that using a therapy oriented to a single or a few molecules or pathways of unknown relative importance and with no consideration of temporal changes in importance after an ischemic insult is problematic (16). This report explores an alternate way to think about neuroprotection…a new frame… taking into consideration multiple pathways and their relative varying time-dependent importance, which may support development of a transformative strategy to more reliably achieve human neuroprotection.
Following Donnan's charge, a potentially radical new approach would be to implement multifaceted therapy to address the multiple pathways involved in the pathogenesis of ischemic brain injury (16). Notably, the efficacious use of combination therapy is already employed in management of hypertension (41), cancer (42, 43), coronary artery disease (44), AIDS (45), and even postoperative nausea (46). However, this approach has not penetrated in a rational manner into neuroprotection. Given the time-related complexity of acute cerebral ischemia, in order to do that rationally, one needs to have information in humans of the post-ischemia time-related various pathways and their relative importance which leads to damage. The purpose of this Hypothesis and Theory report is two-fold:
(1) To test the hypothesis, in humans after cardiopulmonary bypass, using functional genomics, that multiple biochemical pathways already known to be involved in ischemic brain damage have disparate importance which varies with time post-insult; and
(2) Use our observations as the basis for a proposal to reframe the manner in which neuroprotection research is developed, designed, and translated.
The data presented and associated hypothesis and theory delineated is an answer to Donnan's challenge to develop a radical, transformative, new approach to studying and delivering neuroprotection in humans.
The protocol was approved by the institutional biomedical review board of the University of Pennsylvania. After obtaining written informed consent, 98 patients were enrolled. Four non-Caucasian patients were not included in the analysis leaving 94 patients for downstream data analysis. Patients undergoing aortic surgery with deep hypothermic circulatory arrest (DHCA) or aortic valve replacement (AVR) surgery were enrolled. Each had blood drawn for biomarkers of brain damage, S100β (47) and neurofilament H (NFH) (48, 49), immediately after induction of anesthesia (baseline-BL), just prior to CPB (aCPB), 1 h after the end of CPB (pCPB), and 24 h (24H) postoperatively. The group of patients we studied, undergoing cardiac surgery with cardiopulmonary bypass, constitute relatively homogeneous and reproducible clinical situations associated with subtle focal and global ischemic neurologic injury (50).
Each patient underwent analysis for status of 92 single nucleotide polymorphisms (SNPs) thought to be related to proteins and pathways described in the pathogenesis of ischemic brain damage. This approach was employed to avoid the need for an extremely large sample size that would be required with a more standard Genome Wide Association Study. The SNPs chosen were based on a literature review of pathogenesis of ischemic brain damage followed by determination of presence of SNPs related to relevant proteins identified from this literature search. SNPs had to have been demonstrated to have biological impact (not necessarily neurologic) on a human phenotype, and have a distribution of base-pair incidences which support valid statistical inference.
Candidate SNP's proteins include those with roles in: lipoprotein metabolism; nucleotide metabolism; vascular regulation; inflammation; protein chaperone/repair; peroxidation; calcium regulation; 2nd messenger/cell signaling; energy metabolism; platelets/coagulation; apoptotic factors; neurotransmitters; acid-base/cell volume regulation; and estrogen. The candidate proteins and the specific associated SNPs (and bibliographic citations) are fully detailed in the Supplementary Material File (Table I) and summarized in Table 1.
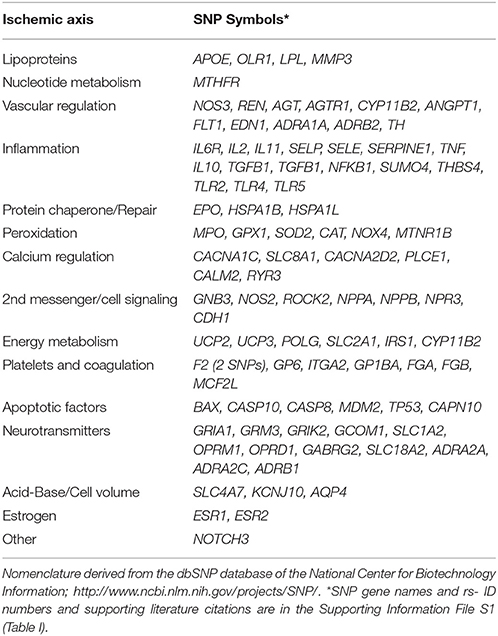
Table 1. SNPs evaluated; according to ischemic axes.
The DHCA protocol used at the University of Pennsylvania has been described (51). Briefly, patients undergo balanced general endotracheal anesthesia with direct intra-arterial blood pressure monitoring and cardiac output monitoring via an oximetric pulmonary arterial catheter (Baxter/Edwards, Deerfield, IL). Temperature is continuously measured in the nasopharynx and bladder. For DHCA patients retrograde cerebral perfusion is initiated via superior vena cava cannula with its tip cephalad to the azygos vein and continued for the duration of DHCA at 10°C with perfusion pressure 25 mmHg, flow 200–300 mL/min and 10° Trendelenburg position. DHCA is < 1 h, after which withdrawal of cardiopulmonary bypass is effected. Cardiotomy suction is routinely returned to the cardiopulmonary bypass circuit and hence to the systemic circulation. AVR patients were similarly managed with hypothermic cardiopulmonary bypass to 30–34°C but without retrograde cerebral perfusion.
Samples were assayed for S100β by radioimmunoassay using human S100β ELISA kits obtained from EMD Millipore Corp. (Billerica, MA, USA). The detection limit of S100β is 2.7 pg/mL using a 50 μL sample size. The sensitivity was determined by plotting the standard curve and then measuring the point of the curve at a distance of three SD from the standard. The isoform S100β is the 21,000 Dalton homodimer ββ. It is present in high concentration in glial cells and Schwann cells (52). The S100 test kit is specific for the β-subunit of the S100 protein, and it measures the β-subunit concentration in both the ββ and αβ isoforms of the protein.
NFH was quantified from human sera using fluorescence sandwich ELISAs developed, described, and validated by coauthor Siman (48). For the sensitive and specific measurement of our target protein analytes in protein-rich and highly complex serum samples, we employ fluorescence-based sandwich immunoassays (ELIFAs), in which alkaline phosphatase is the reporter enzyme and 4-methylumbelliferyl-phosphate is the fluorogenic substrate. This approach increases sensitivity over standard colorimetric ELISA by at least an order of magnitude (53). For the described studies, we measured serum levels of pNFH by ELIFA. We used a modified version of a well-established immunoassay to measure a hypophosphorylated form of the high molecular weight neurofilament subunit. For sensitive and specific detection of pNFH, separate mouse and rabbit antibodies were used for the capture and detection of a hypophosphorylated form of the protein.
The ELIFAs are standardized across experiments so that treatment effects on the relative amounts of each biomarker may be compared directly. For pNFH we employed as standard a mouse spinal cord extract. Fluorescence signals from serially diluted standard samples were fit by nonlinear regression using Graph Pad Prism to create standard curves. In all cases, the fluorescence signal for the serum protein is represented as the relative fluorescence units per unit volume of standard, which in turn is normalized to the volume of input serum. Negative controls were tested to ensure the fidelity of each ELIFA by systematic deletion of either the capture reagent, antigen, or detecting antibody.
Custom targeted SNP assays, designed in collaboration with Illumina technical service, based on the Illumina GoldenGate system were used. The GoldenGate custom multiplex platform genotypes between 96 and 1536 SNPs (in increments of 96) using primer extension and ligation reactions (54, 55). The design process, resulted in the customized list of analyzed SNPs, which were incorporated into the custom GoldenGate reagent kit. Call rates are typically over 99%, with very high reproducibility and accuracy.
Due to the small number of African-American and Asian patients, our analysis focused only on Caucasians.
Linear regression was used to detect SNPs that are differentially associated with changes in NFH or S100β values. The model takes the form:
where YaCPB stands for the value of NFH or S100B at aCPB, YBL stands for the value of NFH or S100B at baseline. Similar forms of the equation were used for times pCPB and 24 H.
Bonferroni p-values for Studentized residuals were calculated based on t-distribution for each observation. If the p < 0.05, this observation was considered as an outlier and was deleted. Benjamini-Hochberg procedure was used for the multiple comparison problem.
GEE model was used to analyze the effect between different treatment groups and the time effect for S100β and NFH. In this study, observations of the sample patients were correlated with each other while those from different patients were assumed to be independent. GEE model can take into account the dependency of within group observations by specifying a working correlation structure.
Akaike information criterion (AIC) based backward stepwise regression was used to select a group of differentially associated SNPs. The model starts with a list of candidate variables which includes significant SNPs (raw p < 0.05) from previous linear regression result and other covariates. At each step, AIC, which measures the information lost for the model, is calculated. If deleting a variable results in a lower AIC, then the variable is dropped. Otherwise, it is kept in the model. Similar outlier detection methods are applied and the outliers are removed in the final model.
Ninety-four enrolled patients underwent analysis. Eleven underwent DHCA only, 65 underwent AVR only with hypothermic cardiopulmonary bypass, and 18 underwent both procedures. Patient characteristics are summarized in Table 2.
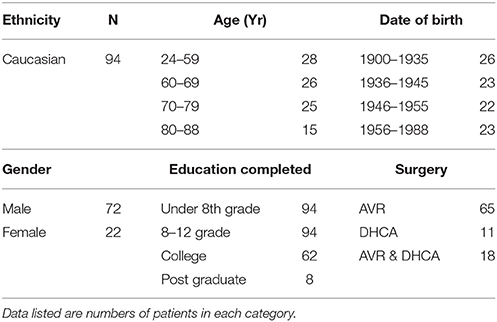
Table 2. Patient characteristics.
The result of the GEE model shows that the surgical group difference of S100β concentration between DHCA patients and AVR-only patients is significant (P = 0.0014) while no group differences are apparent for NFH concentration (P = 0.78; Figure 1) NFH concentration has a large variability in both surgery groups and the error bars have overlap. As for the S100β concentrations, patients undergoing DHCA procedure have higher S100β concentrations than those undergoing only AVR surgery. Moreover, S100β concentration changes significantly (P < 2 × 10−16) over time.
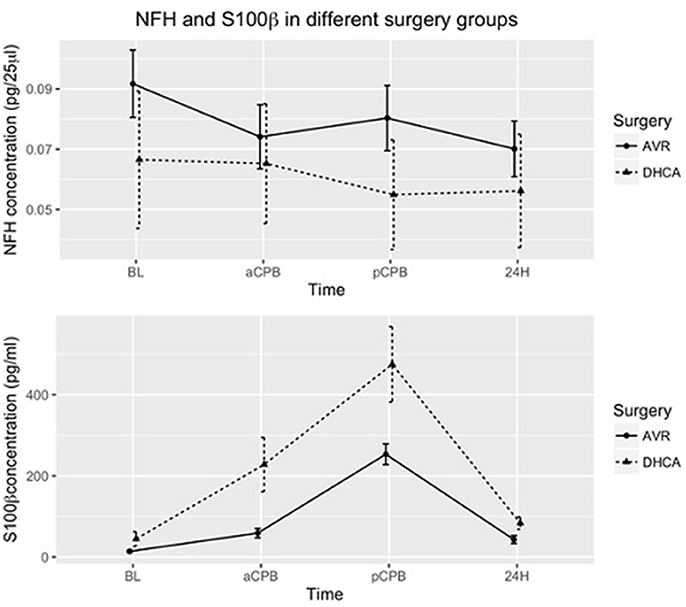
Figure 1. Mean of NFH and S100β concentrations in patients undergoing DHCA (with or without AVR) and patients undergoing AVR only. The error bars show the mean ± standard error. Group differences for S100 β are significant (P = 0.0014). In addition, S100β concentrations change significantly over time (P < 2 × 10−16). Group assignments were: immediately after induction of anesthesia (baseline-BL), just prior to CPB (aCPB), 1 h after the end of CPB (pCPB), and 24 h (24 H) postoperatively.
In the combined group analysis, S100β values compared to baseline were elevated before (P = 2.99 × 10−6) and after (P = 3.33 × 10−13) CPB but not at 24 h (P = 0.136). NFH values overall were minimally changed from baseline (Figure 2).
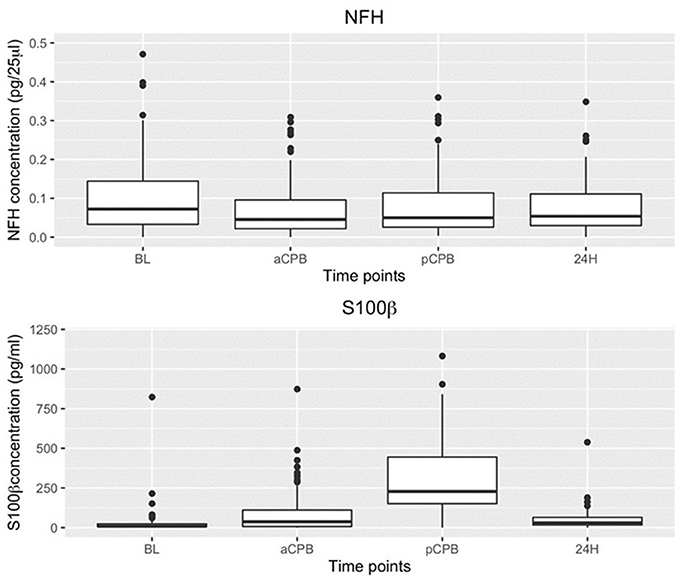
Figure 2. Boxplot of NFH and S100β concentrations at baseline (BL) before (aCPB) and after (pCPB)CPB and 24 h post CPB(24 h) for AVR and DHCA patients combined. Compared to BL NFH values were not increased before or after CPB. S100β increased from baseline before (aCPB) (P = 2.99 × 10−6) and after (pCPB) (P = 3.33 × 10−13) CPB but not at 24 h.
Genotyping call scores were acceptable in 85 subjects. Among these patients the median call score was 1.0 with IQR 0.989–1.0 and range 0.213–1.0; and mean call score was 0.926 ± 0.194 (SD).
At the nominal significance level of P = 0.05, absolute levels of S100β and NFH and relative changes in S100β and NFH compared to baseline were associated with SNPs in multiple genes involving several pathways with different patterns over time. However, the associations were not significant after multiple comparison adjustments. These nominally significant associations are listed in Table 3 (presented without multiple comparison considerations in order to demonstrate the multiplicity of involved ischemic axes and support future specific SNP and pathway hypothesis testing). Among these SNPs for which a nominal association with biomarkers was present, specific base pair combinations related to elevations in biomarker levels were observed. Figures depicting these can be found in the Supplementary Material File (Figure I).
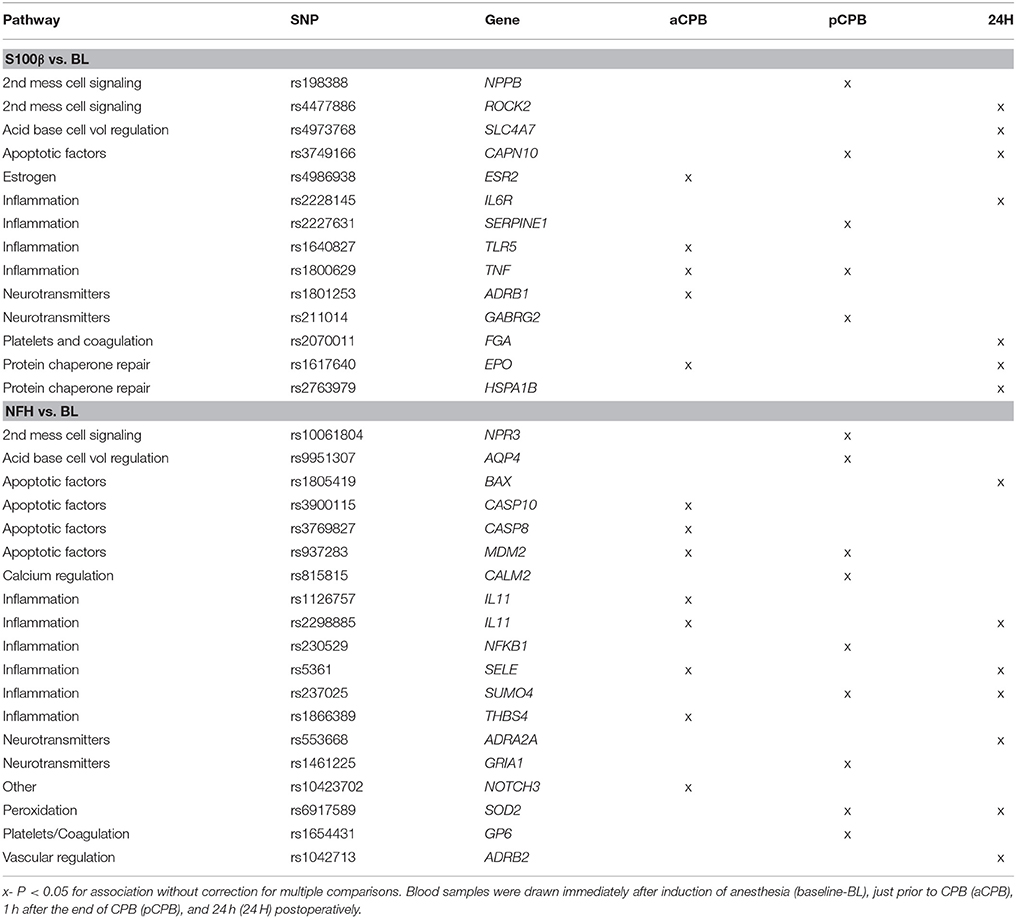
Table 3. Nominally significant SNP associations for relative changes in biomarkers.
In order to make a preliminary assessment of relative weights of specific genes in the contribution to biomarker elevations at different times, multiple regression equations were calculated for changes in NFH and S100β at pCPB and 24H times related to SNPs. The results of the multiple regression analyses depicting the most highly weighted SNPs are shown in Table 4, indicating disparate genes (and presumably downstream proteins) with varying weights associating with changes in biomarkers and with a different pattern of associations at the two times post-CPB.
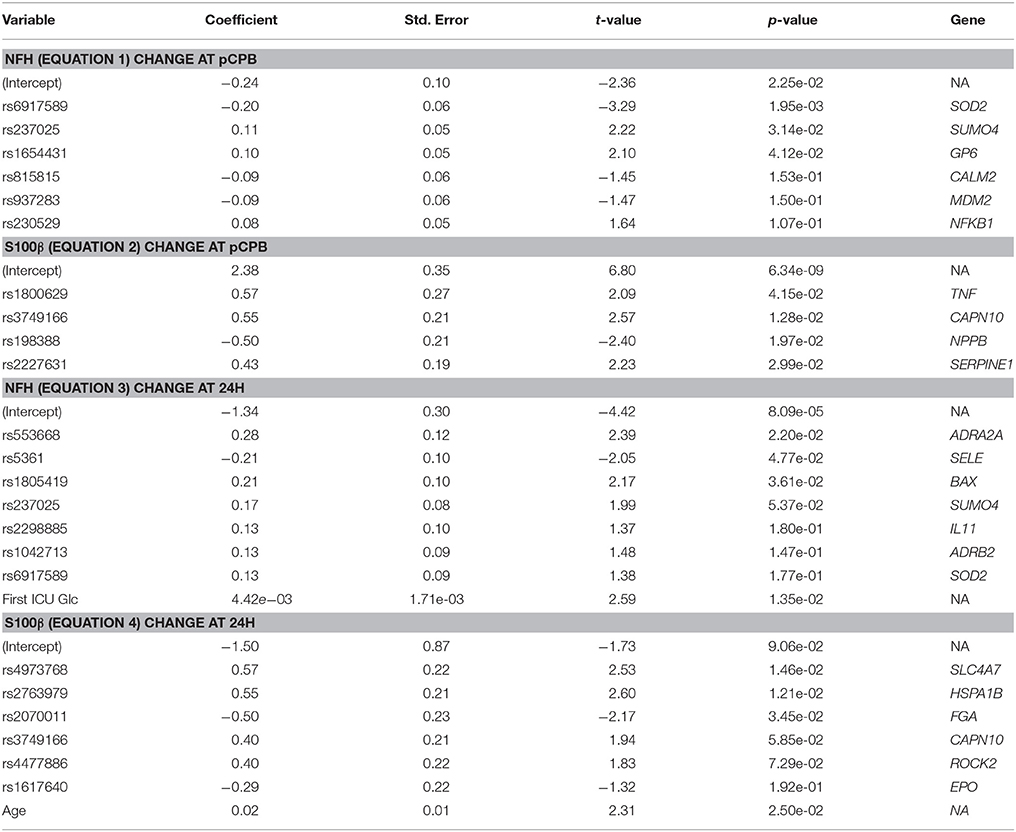
Table 4. Regression data: change in biomarkers from BL: pCPB and 24H.
Our biomarker data in humans after cardiopulmonary bypass, a procedure associated with subtle ischemic brain damage (50), support, but do not prove, the notion that multiple specific pathophysiologic processes, with variable contributing importance, are likely involved in the pathogenesis of ischemic brain damage in humans. Nonetheless, our data suggest that the importance of different pathways differ according to biomarker and may change with time post insult. From these data we hypothesize that neuroprotection studies, in order to be successful, must be multifaceted, focused on demonstrably important pathways, and, moreover, the nature of the multifaceted therapy may change with time after the onset of the ischemic insult.
The human model we chose was based on the relative homogeneity and reproducibility of the ischemic insult with previously reported increases in neural biomarkers which have been associated with neurologic outcomes. Cardiopulmonary bypass such as we studied, is associated with cerebral ischemia which can have relatively silent impact but produces abnormalities in postoperative MRI (50), biomarkers of brain injury which vary with specific SNPs (56), and neuropsychiatric changes (57).
S100β and hypophosphorylated neurofilament H (NFH) were assessed as biomarkers of brain injury for this study. Both biomarkers were chosen based on neural element origins and prior reports indicating elevations after ischemic insults and associations of such elevations with neurologic outcome. The S100β protein is thought to derive from astrocytes (52, 58). Increases in S100β have been associated with neurologic outcomes after cardiac surgery (47, 59) cardiac arrest (60) and stroke (61). The major weakness with S100β in our study is observations of very high levels reported in cardiotomy and mediastinal blood with suggestions that injured pericardial or myocardial tissues may be contributing to early post-CPB elevations (62), likely an important element in the S100β increases we observed at pCPB. Notwithstanding these reasonable concerns there are ample reports in other contexts of S100β clearly being related to volume of brain injury on imaging and with clinical outcomes (47, 59, 60, 63), such that the increase in mean S100β we observed at pCPB likely represents both neural and non-neural release of the protein. Notably NFH, based on structural neural proteins released with neuronal degeneration (48), would not be expected to be associated with cardiotomy suction admixture, although this has not been formally evaluated. The lack of a comparable abrupt rise in mean NFH concentration at pCPB in our data supports the notion that NFH is not contaminated by cardiotomy suctioned blood.
NFH was developed as a biomarker based on a review of abundant structural neural proteins by coauthor Siman with preclinical studies indicating NFH release from degenerating cultured neurons (48). NFH elevations in cerebrospinal fluid have been observed in previous studies involving cardiac surgery (49). Moreover other studies in humans report increases in NFH in CSF of patients sustaining traumatic brain injury (TBI) (64), DHCA (49), and subarachnoid hemorrhage (SAH) with vasospasm (65), and with outcome in SAH linked to CSF NFH levels (65). Some increase in NFH has been observed in serum of patients after TBI relative to controls but link to neurological outcome was not evaluated (64).
In an effort to avoid an extremely large sample size that would be required with a more standard genome wide association study, biologically active SNPs representing protein function and pathways already reported to be involved in the pathogenesis of ischemic brain damage were studied. This of course provides an inherent bias of the investigator choosing the SNPs (WAK) but nonetheless allows us to better ascertain the primary goal of demonstrating involvement of multiple pathways and proteins contributing to post-ischemic brain damage. Clearly, previously unknown biological contributors could be missed with this approach and is a consideration for future studies. Nonetheless, this study is underpowered to confidently offer any valid conclusions about specific SNPs studied but does suggest important hypotheses regarding the notions of multiple involved genes and function of their downstream proteins, varying relative weights, and temporal changes in disparate proteins involved in pathogenesis of ischemic brain damage. Moreover, the nominal SNP associations before multiple comparison adjustment, although possibly a random finding, may suggest a smaller more focused SNP sample for future studies.
Our protocol was designed to explore the feasibility of a novel approach, using functional genomics to evaluate the role of genetic variation in specific multifactorial vulnerability to ischemia. This sort of approach should contribute to the theoretical underpinnings for rational design of multifaceted clinical neuroprotection trials. Notably, the main obstacle in devising a multifaceted approach to neuroprotection is determining the physiological and biochemical processes to target…. logically expected to be those which are most heavily weighted in terms of impact on ischemic outcome and the correct timing of such therapy. Our regression analyses were done in an endeavor to address this issue, suggesting that multiple disparate genes (and likely disparate function of downstream proteins) involved in pathogenesis of ischemic brain damage have varying contributing weights which vary temporally post insult. This forms the basis for the theoretical rationale subsequently described in more detail.
In a biological system undergoing a complex injury characterized by severity, S, an equation can be derived comprised of numerous pathophysiologic factors, Fi, and weighting factors, Wi that vary with post-insult time (66, 67), t:
This overall concept, originally presented by Kofke (16) was the basis for performing the multiple regression analysis of our data, where the weighting factors are our regression coefficients.
DNA based systems have inherent variation in genes and in translation of important proteins thereby leading to a large number of disparate and interacting factors (14, 68–72) contributing to S with also an unknown number of as yet unknown other factors with correspondingly unknown variability and weighting. Such uncertainty supports the need to do proper foundational studies before clinical efficacy trials.
This functional genomics approach lays the groundwork for larger studies; perhaps targeting conventional cerebral ischemia syndromes such as stroke, vasospasm, and cardiac arrest; to determine more conclusively the most important pathways and proteins contributing to susceptibility to neural damage after such ischemic insults. This can be expected to lead to a systems biology (67, 73) type detailed genome-linked description of the pathophysiology of human cerebral ischemia. This approach will likely include vertical gene-linked phenotypes assessments (74) of the pathophysiology of cerebral ischemia, including the potentially pivotal role of so called “hub” pathways (39). Such validation in larger studies will involve mechanistically different biomarkers demonstrated to be predictive of outcome, e.g., microdialysis data, magnetic resonance imaging and spectroscopy, various other blood/CSF biomarkers, and so on. Moreover, if and when methods become available that indicate changes in the cerebral transcriptome (75, 76) post-insult then potentially even more relevant information may become available. Neuroprotection based on these concepts will necessarily be multifaceted with the specific combinations of therapies based on weighting factors which also vary with time post insult, as suggested by the pathophysiologic multiple regression equations parameters in Table 4 and further illustrated in Table 5.
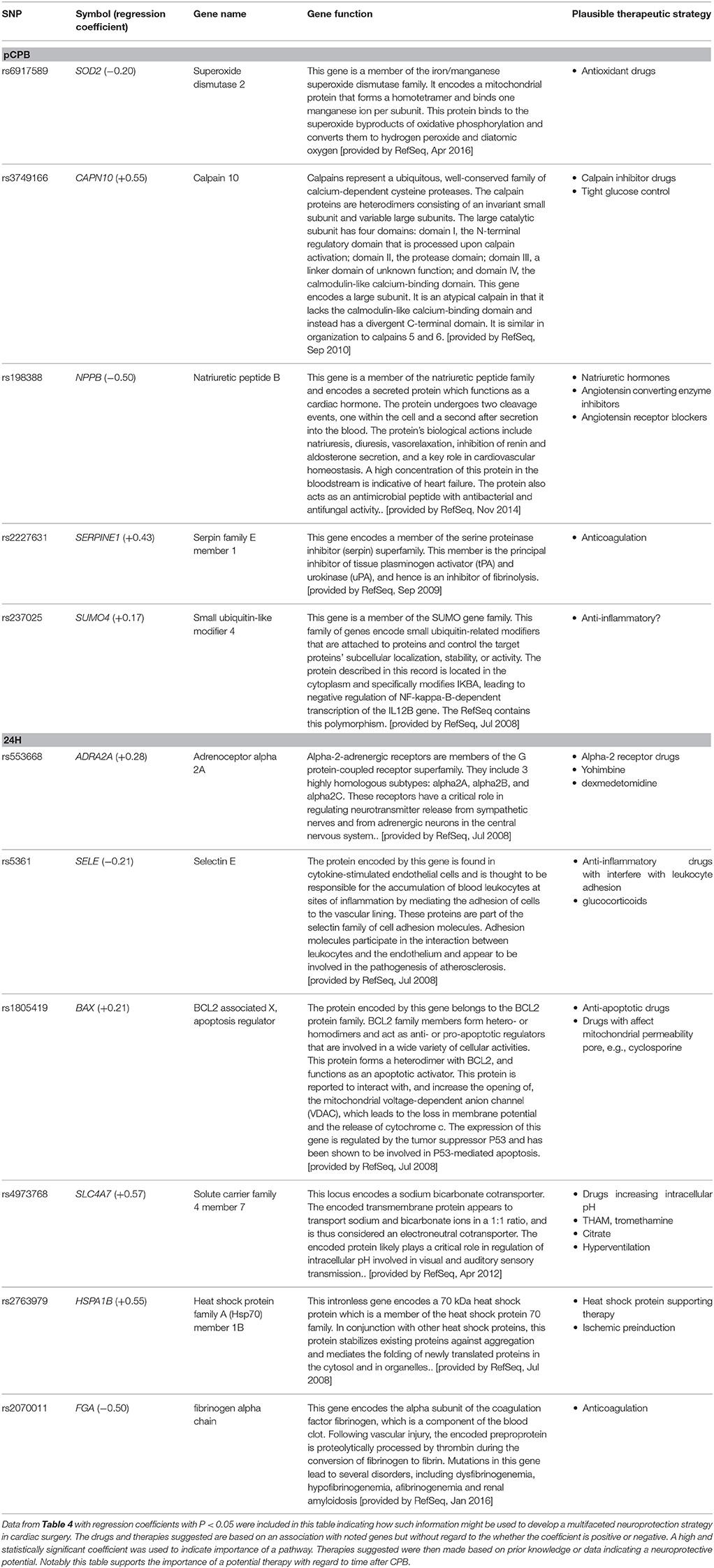
Table 5. Example of possible multifaceted therapy derived from post CPB-based functional genomic analysis.
A further complicating factor includes the notion that there are endogenous biological accommodation factors which may attenuate or worsen an insult. Such factors will need to be considered in future studies exploring pathophysiologic pathways, understanding that the impact of a pathway may be exacerbated or attenuated over time post insult based on such endogenous responses. For example cerebral ischemia/acidosis begets spontaneous hyperventilation (77–80) and systemic hypertension is a typical concomitant of brainstem ischemia (81). Other examples include ischemia-mediated adjustments in protein transcription and ischemic preconditioning (82–90) and issues in resilience or plasticity of pathophysiological networks, e.g., apoptosis networks (91, 92). In addition, other peri-insult time related epigenetic processes that may affect the transcription and translation of a gene (93, 94) may also be included in these issues.
Although not specifically addressed in our analyses many authors have suggested that biological complexity is further embellished by social factors e.g., nursing numbers relative to patient numbers (and complexity), health care worker experience, availability of pharmaceuticals and innovative equipment, quality of emergency teams, local cultures and ethics related to safety, cost containment, and morale, practice variation, and so on (19–21, 95, 96), which are also important to the outcome severity of injury. Notably, inter-center variance has been reported as a very important element in the quality, complexity, and reproducibility of clinical research (19, 20, 36, 97). This is one of the factors underlying the Multi-PART initiative (32) which one may surmise will simulate such inter-center variation in creating a realistic real-word environment for preclinical trials (33, 98).
Such multiple interactions and complexity are summarized in Figure 3 and have been suggested in pathophysiology reviews by Lipton (68) and others (14, 20, 40, 70–72, 91, 92, 99). Given the numerous highly variable, possibly nonlinear, biological and health system/social factors that contribute to post insult brain damage, it is unsurprising that clinical studies directed at improving only one of numerous complex interacting factors tend to show no or little effect. This is particularly true in multi-institutional trials (increasing social variation), unless it is truly a breakthrough phenomenon [large W for brain blood flow as seen in early endovascular thrombectomy in ischemic stroke (100)] or if the therapy itself exerts a multifaceted effect [e.g., hypothermia (101)]. This then leads to the notion that the current widely practiced methods of advancing clinical therapy for complex problems is generally a fruitless waste of public resources and that an alternate method is needed which is based on a multifactorial approach. Rogalewski et al. (8), Candelario et al. (15), Ginsberg (11), and O'Collins et al. (13) have reviewed and endorsed this concept; however, they and others who endorse the notion of combination therapy (5, 6, 10) do not suggest a rational method for establishing foundational information regarding time-dependent weights of various facets, and then building a multimodal approach rather than trying everything at once…another prescription for failure. A rational method for introducing neuroprotective therapies in research protocols is needed…indeed, the entire field of neuroprotection research needs to be reframed in a manner which accounts for these important pathophysiological principles.
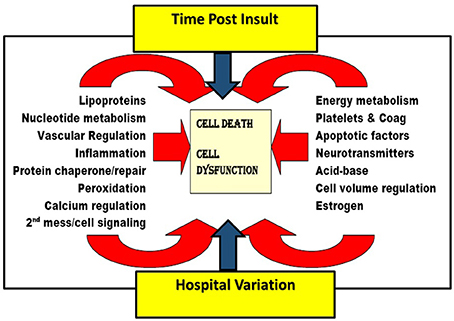
Figure 3. Schematic depicting many of the pathways involved in the pathogenesis of ischemic brain injury, illustrating the impact of time post ischemia and health system variation on multiple involved pathways. Figure adapted and altered from Kofke (16).
We propose fairly abstract concepts relating to biological complexity which may be used to guide development of neuroprotection strategies. We therefore, suggest that foundational human research is needed to first define the importance of potential target pathways and proteins at various times after insult. And only after such information is garnered should any further neuroprotection trials be attempted. Such trials optimally will be multifaceted based on the recommended foundational research. An example of how this might happen using our data follows.
The most important genes from the regression tables with P < 0.05 are delineated in Table 5 with a description of their function taken from the www.ncbi.nlm.nih.gov web pages describing various aspects of individual genes. From these descriptions one can make inferences, and generate concrete ideas regarding the most important proteins and pathways involved in the development of post-CPB brain damage as indicated by the biomarker surrogates used in these studies. Overall, if further validated, this suggests that a multifaceted approach to neuroprotection in the context of cardiac surgery would be different early post CPB vs. the next day and that specific elements of a multifaceted approach could be as suggested in Table 5. Based on this table one might imagine a neuroprotective cocktail, grounded also in preclinical studies, being developed comprised at CPB of tirilazad (102) (antioxidant), to-be-developed anti-apoptotic drug, tight glucose control, nasiritide (103), enalopril (104), and tranexamic acid (105) and 24 h later provide a neuroprotective cocktail comprised of dexmedetomidine (106), cyclosporine (107), glucocorticoids, tromethamine (THAM) (108, 109), and to-be-developed heat shock protein supporting drugs or gene therapy (110). Note that this is an entirely theoretical construct based on our data which we know is not compelling and is in need of future studies corroborating this approach to building a foundation for logically designed neuroprotection. We are simply describing a concrete application example of the notions presented.
This hypothesized strategy to developing neuroprotective therapies is necessarily population-based, i.e., definition of important factors and their time-dependent weighting factors are derived from and applied to populations of patients. The possibility also exists, if genomics analyses could be done quickly, or a patient's genome is available in the medical record (111), to design individualized multifaceted neuroprotection strategies on hospital admission or preoperatively. Given current work to include patients' genomes in their medical records (111, 112), and link such data to phenotypes (74) this may be the time-based multifaceted personalized neuroprotection strategy of the future.
We describe putative associations of biomarkers of ischemic brain damage with variation in polymorphisms of multiple genes, suggesting expected variation in downstream protein function, relevant to the pathogenesis of ischemic brain damage. Phenotype-linked genetic and epigenetic variation may be useful to indicate the most important time-dependent biochemical proteins and pathways involved in the pathogenesis of ischemic damage. This approach, if amplified with larger population-based or personalized innovations, may provide a theoretical basis for the transformation and reframing in the approach to neuroprotection as advocated by Donnan a decade ago.
WK: study design, oversight, data interpretation, primary author of manuscript, sole author of most of the concepts in introduction and discussion; YR: statistical analysis; JA and JB: study design and implementation; HL: study design, statistical genomics strategy, data interpretation; KN: genomics oversight and interpretation; RS: NFH analysis; QM: S100 analysis; WB: S100 analysis; SY and GK: data analysis; CK: genomic analysis. All authors contributed to manuscript revision, read and approved the submitted version.
The research described was funded by the US Public Health Service: NIH grant 1R21NS061074-01A2.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The University of Pennsylvania Department of Anesthesiology and Critical Care Clinical Research Trials group provided valuable assistance in conduct of the studies. Sherman Stein, University of Pennsylvania, contributed to the complexity and severity equation concepts.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2018.00497/full#supplementary-material
1. Donnan GA. The 2007 Feinberg lecture: a new road map for neuroprotection. Stroke (2008) 39:242. doi: 10.1161/STROKEAHA.107.493296
2. Lapchak PA, Zhang JH. Data standardization and quality management. Transl Stroke Res. (2018) 9:4–8. doi: 10.1007/s12975-017-0531-9
3. Fisher M. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke (1999) 30:2752–8. doi: 10.1161/01.STR.30.12.2752
4. Anonymous. Stroke Trials Registry [Online]. St Louis (2017). Available online at: http://www.strokecenter.org/trials/index.aspx (Accessed November 30, 2017).
5. Gisvold SE, Sterz F, Abramson NS, Bar-Joseph G, Ebmeyer U, Gervais H, et al. Cerebral resuscitation from cardiac arrest: treatment potentials. Crit Care Med. (1996) 24:S69–80. doi: 10.1097/00003246-199602000-00049
6. Cheng YD, Al-Khoury L, Zivin JA. Neuroprotection for ischemic stroke: two decades of success and failure. NeuroRx (2004) 1:36–45. doi: 10.1602/neurorx.1.1.36
7. Danton GH, Dietrich WD. The search for neuroprotective strategies in stroke. AJNR Am J Neuroradiol. (2004) 25:181–94.
8. Rogalewski A, Schneider A, Ringelstein EB, Schabitz WR. Toward a multimodal neuroprotective treatment of stroke. Stroke (2006) 37:1129–36. doi: 10.1161/01.STR.0000209330.73175.34
9. Adams HP, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Circulation (2007) 115:478–534. doi: 10.1161/CIRCULATIONAHA.107.181486
10. Fisher M, Hanley DF, Howard G, Jauch EC, Warach S. Recommendations from the STAIR V meeting on acute stroke trials, technology and outcomes. Stroke (2007) 38:245–48. doi: 10.1161/01.STR.0000255951.37434.aa
11. Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology (2008) 55:363–89. doi: 10.1016/j.neuropharm.2007.12.007
12. Ducruet AF, Grobelny BT, Zacharia BE, Hickman ZL, Yeh ML, Connolly ES. Pharmacotherapy of cerebral ischemia. Expert Opin Pharmacother. (2009) 10:1895–906. doi: 10.1517/14656560903055095
13. O'Collins VE, MacLeod MR, Donnan GA, Howells DW. Evaluation of combination therapy in animal models of cerebral ischemia. J Cereb Blood Flow Metab. (2012) 32:585–97. doi: 10.1038/jcbfm.2011.203
14. Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascades in ischemic stroke. Int J Stroke (2012) 7:378–85. doi: 10.1111/j.1747-4949.2012.00839.x
15. Candelario-Jalil E. Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Invest Drugs (2009) 10:644–654.
16. Kofke WA. Incrementally applied multifaceted therapeutic bundles in neuroprotection clinical trials time for change. Neurocrit Care (2010) 12:438–44. doi: 10.1007/s12028-010-9332-7
17. Baker WB, Parthasarathy AB, Gannon KP, Kavuri VC, Busch DR, Abramson K, et al. Noninvasive optical monitoring of critical closing pressure and arteriole compliance in human subjects. J Cereb Blood Flow Metab. (2017) 37:2691–705. doi: 10.1177/0271678X17709166
18. Cebul RD, Snow RJ, Pine R, Hertzer NR, Norris DG. Indications, outcomes, and provider volumes for carotid endarterectomy. J Am Med Assoc. (1998) 279:1282–7. doi: 10.1001/jama.279.16.1282
19. Clifton G, Choi S, Miller E, Levin H, Smith KJ, Muizelaar J, et al. Intercenter variance in clinical trials of head trauma–experience of the National Acute Brain Injury Study: hypothermia. J Neurosurg. (2001) 95:751–5. doi: 10.3171/jns.2001.95.5.0751
20. Dong Y, Chbat NW, Gupta A, Hadzikadic M, Gajic O. Systems modeling and simulation applications for critical care medicine. Ann Intens Care (2012) 2:1–14. doi: 10.1186/2110-5820-2-18
21. Kosty JA, LeRoux PD, Levine J, Park S, Kumar MA, Frangos S, et al. Comparison of clinical and research practices in measuring cerebral perfusion pressure (CPP): a literature review and practitioner survey. Anes Analg. (2013) 117:694–8. doi: 10.1213/ANE.0b013e31829cc765
22. Romero Sevilla R, Portilla Cuenca JC, López Espuela F, Redondo Peñas I, Bragado Trigo I, Yerga Lorenzana B, et al. A stroke care management system prevents outcome differences related to time of stroke unit admission. Neurologia (2016) 31:149–56. doi: 10.1016/j.nrl.2015.07.011
23. Meltzer AJ, Agrusa C, Connolly PH, Schneider DB, Sedrakyan A. Impact of provider characteristics on outcomes of carotid endarterectomy for asymptomatic carotid stenosis in New York state. Ann Vasc Surg. (2017) 45:56–61. doi: 10.1016/j.avsg.2017.05.015
24. Perri JL, Nolan BW, Goodney PP, DeMartino RR, Brooke BS, Arya S, et al. Factors affecting operative time and outcome of carotid endarterectomy in the Vascular Quality Initiative. J Vasc Surg. (2017) 66:1100–8. doi: 10.1016/j.jvs.2017.03.426
25. Pross C, Busse R, Geissler A. Hospital quality variation matters – A time-trend and cross-section analysis of outcomes in German hospitals from 2006 to 2014. Health Policy (2017) 121:842–52. doi: 10.1016/j.healthpol.2017.06.009
26. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the arrive guidelines for reporting animal research. PLoS Biol. (2010) 8:e1000412. doi: 10.1371/journal.pbio.1000412
27. Kochanek PM, Bell MJ. Making an IMPACT in traumatic brain injury research. Lancet Neurol. (2013) 12:1132–3. doi: 10.1016/S1474-4422(13)70245-X
28. Maas AIR, Murray GD, Roozenbeek B, Lingsma HF, Butcher I, McHugh GS, et al. Advancing care for traumatic brain injury: findings from the IMPACT studies and perspectives on future research. Lancet Neurol. (2013) 12:1200–10. doi: 10.1016/S1474-4422(13)70234-5
29. Maas AIR. International Mission on Prognosis and ClinicalTrialDesign [Online]. Antwerp: TBI-IMPACT.org (2018). Available online at: http://www.tbi-impact.org/ (Accessed March 16, 2018).
30. NINDS. Improving the Quality of NINDS-Supported Preclinical and Clinical Research through Rigorous Study Design and Transparent Reporting [Online]. Bethesda, MD: PHS (2012). Available online at: https://www.ninds.nih.gov/sites/default/files/transparency_in_reporting_guidance_1.pdf (Accessed March 16, 2018).
31. Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res. (2013) 4:279–85. doi: 10.1007/s12975-012-0209-2
32. Howells DW, Allan S, Dirnagl U, MacLeod MR, Sena E, Planas A, et al. Final Report Summary - MULTI-PART (Multicentre Preclinical Animal Research Team) [Online]. University of Edinburgh, UK: EU Publications Office (2016). Available: https://cordis.europa.eu/result/rcn/177100_en.html (Accessed March 16, 2018).
33. Kleikers PW, Hooijmans C, Göb E, Langhauser F, Rewell SS, Radermacher K, et al. A combined pre-clinical meta-analysis and randomized confirmatory trial approach to improve data validity for therapeutic target validation. Sci Rep. (2015) 5:13428. doi: 10.1038/srep13428
34. Fraser JF. Standardisation of research strategies in acute ischaemic stroke. Lancet Neurol. (2016) 15:784–5. doi: 10.1016/S1474-4422(16)30080-1
35. Grupke S, Hall J, Dobbs M, Bix GJ, Fraser JF. Understanding history, and not repeating it. Neuroprotection for acute ischemic stroke: from review to preview. Clin. Neurol. Neurosurg. (2015) 129:1–9. doi: 10.1016/j.clineuro.2014.11.013
36. Hawryluk GWJ, Bullock MR. Design of acute neuroprotection studies. Handbook of Clin Neurol. (2015). 128:761–78. doi: 10.1016/B978-0-444-63521-1.00047-9
37. Hermann DM, Doeppner TR. From bedside to bench: how clinical reality should instruct stroke modeling. Neuromethods (2016), 120:1–6. doi: 10.1007/978-1-4939-5620-3_1
38. Sams-Dodd F. Is poor research the cause of the declining productivity of the pharmaceutical industry? An industry in need of a paradigm shift. Drug Discovery Today (2013) 18:211–7. doi: 10.1016/j.drudis.2012.10.010
39. Langfelder P, Mischel PS, Horvath S. When is hub gene selection better than standard meta-analysis? PLoS ONE (2013) 8:e61505. doi: 10.1371/journal.pone.0061505
40. Dirnagl U, Endres M. Found in translation: preclinical stroke research predicts human pathophysiology, clinical phenotypes, and therapeutic outcomes. Stroke (2014) 45:1510–8. doi: 10.1161/STROKEAHA.113.004075
41. Brown MJ, Cruickshank JK, Dominiczak AF, MacGregor GA, Poulter NR, Russell GI, et al. Better blood pressure control: how to combine drugs. J Hum Hypertens. (2003) 17:81–6. doi: 10.1038/sj.jhh.1001511
42. Corn BW. Advances in the combination of radiation therapy and chemotherapy against cancer. Drug News Perspect. (2004) 17:705–12.
43. Chura JC, Marushin R, Boyd A, Ghebre R, Geller MA, Argenta PA. Multimodal therapy improves survival in patients with CNS metastasis from uterine cancer: a retrospective analysis and literature review. Gynecol Oncol. (2007) 107:79–85. doi: 10.1016/j.ygyno.2007.05.027
44. Ramanath VS, Eagle KA. Evidence-based medical therapy of patients with acute coronary syndromes. Am J Cardiovasc Drugs (2007) 7:95–9116. doi: 10.2165/00129784-200707020-00002
45. Bartlett JA, Fath MJ, Demasi R, Hermes A, Quinn J, Mondou E, et al. An updated systematic overview of triple combination therapy in antiretroviral-naive HIV-infected adults.[see comment]. AIDS (2006) 20:2051–64. doi: 10.1097/01.aids.0000247578.08449.ff
46. Apfel CC, Korttila K, Abdalla M, Kerger H, Turan A, Vedder I, et al. A factorial trial of six interventions for the prevention of postoperative nausea and vomiting. N Engl J Med. (2004) 350:2441–51. doi: 10.1056/NEJMoa032196
47. Shaaban Ali M, Harmer M, Vaughan R. Serum S100 protein as a marker of cerebral damage during cardiac surgery. Br J Anaesth. (2000) 85:287–98. doi: 10.1093/bja/85.2.287
48. Siman R, McIntosh TK, Soltesz KM, Chen Z, Neumar RW, Roberts VL. Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol Dis. (2004) 16:311–20. doi: 10.1016/j.nbd.2004.03.016
49. Siman R, Roberts VL, McNeil E, Dang A, Bavaria JE, Ramchandren S, et al. Biomarker evidence for mild central nervous system injury after surgically-induced circulation arrest. Brain Res. (2008) 1213:1–11. doi: 10.1016/j.brainres.2008.03.034
50. Floyd TF, Shah PN, Price CC, Harris F, Ratcliffe SJ, Acker MA, et al. Clinically silent cerebral ischemic events after cardiac surgery: their incidence, regional vascular occurrence, and procedural dependence. Ann Thorac Surg. (2006) 81:2160–6. doi: 10.1016/j.athoracsur.2006.01.080
51. Appoo JJ, Augoustides JG, Pochettino A, Savino JS, McGarvey ML, Cowie DC, et al. Perioperative outcome in adults undergoing elective deep hypothermic circulatory arrest with retrograde cerebral perfusion in proximal aortic arch repair: evaluation of protocol-based care. J Cardiothorac Vasc Anesth. (2006) 20:3–7. doi: 10.1053/j.jvca.2005.08.005
52. Maas MB, Furie KL. Molecular biomarkers in stroke diagnosis and prognosis. Biomark Med. (2009) 3:363–83. doi: 10.2217/bmm.09.30
53. Porstmann T, Kiessig ST. Enzyme immunoassay techniques an overview. J Immunol Methods (1992) 150:5–21. doi: 10.1016/0022-1759(92)90061-W
54. Corder E, Saunders A, Strittmatter W, Schmechel D, Gaskell P, Small G, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science (1993) 261:921–3. doi: 10.1126/science.8346443
55. Skoog I, Hesse C, Aevarsson O, Landahl S, Wahlstrom J, Fredman P, et al. A population study of apoE genotype at the age of 85: relation to dementia, cerebrovascular disease, and mortality. J Neuro Neurosurg Psychiatr. (1998) 64:37–43. doi: 10.1136/jnnp.64.1.37
56. Kofke W, Konitzer P, Meng Q, Guo J, Cheung A. Effect of Apolipoprotein E genotype on NSE and S-100 levels after cardiac and vascular Surgery. Anesth Analg. (2004) 99:1323–5. doi: 10.1213/01.ANE.0000135345.03635.6A
57. Newman MF, Mathew JP, Grocott HP, Mackensen GB, Monk T, Welsh-Bohmer KA, et al. Central nervous system injury associated with cardiac surgery. Lancet (2006) 368:694–703. doi: 10.1016/S0140-6736(06)69254-4
58. Rickmann M, Wolff JR. S100 protein expression in subpopulations of neurons of rat brain. Neuroscience (1995) 67:977–91. doi: 10.1016/0306-4522(94)00615-C
59. Westaby S, Johnsson P, Parry A, Blomqvist S, Solem J, Alling C, et al. Serum S100 protein: a potential marker for cerebral events during cardiopulmonary bypass. Ann Thorac Surg. (1996) 61:88–92. doi: 10.1016/0003-4975(95)00904-3
60. Einav S, Kaufman N, Algur N, Kark JD. Modeling serum biomarkers S100 beta and neuron-specific enolase as predictors of outcome after out-of-hospital cardiac arrest: an aid to clinical decision making. J Am Coll Cardiol. (2012) 60:304–11. doi: 10.1016/j.jacc.2012.04.020
61. Nash DL, Bellolio MF, Stead LG. S100 as a marker of acute brain ischemia: a systematic review. Neurocrit Care (2008) 8:301–7. doi: 10.1007/s12028-007-9019-x
62. Yuan SM. S100 and s100ß: Biomarkers of cerebral damage in cardiac surgery with or without the use of cardiopulmonary bypass. Braz J Cardiovasc Surg. (2014) 29:630–41. doi: 10.5935/1678-9741.20140084
63. Brea D, Sobrino T, Blanco M, Cristobo I, Rodríguez-González R, Rodríguez-Yañez M, et al. Temporal profile and clinical significance of serum neuron-specific enolase and S100 in ischemic and hemorrhagic stroke. Clin Chem Lab Med. (2009) 47:1513–8. doi: 10.1515/CCLM.2009.337
64. Siman R, Toraskar N, Dang A, McNeil E, McGarvey M, Plaum J, et al. A panel of neuron-enriched proteins as markers for traumatic brain injury in humans. J Neurotrauma (2009) 26:1867–77. doi: 10.1089/neu.2009.0882
65. Siman R, Giovannone N, Toraskar N, Frangos S, Stein SC, Levine JM, et al. Evidence that a panel of neurodegeneration biomarkers predicts vasospasm, infarction, and outcome in aneurysmal subarachnoid hemorrhage. PLoS ONE (2011) 6:e28938. doi: 10.1371/journal.pone.0028938
66. Wang LY, Liu J, Li Y, Li B, Zhang YY, Jing ZW, et al. Time-dependent variation of pathways and networks in a 24-hour window after cerebral ischemia-reperfusion injury. BMC Syst Biol. (2015) 9:4. doi: 10.1186/s12918-015-0152-4
67. Nagy Z, Nardai S. Cerebral ischemia/repefusion injury: From bench space to bedside. Brain Res Bull. (2017) 134:30–7. doi: 10.1016/j.brainresbull.2017.06.011
68. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. (1999) 79:1431–568. doi: 10.1152/physrev.1999.79.4.1431
69. Deb P, Sharma S, Hassan KM. Pathophysiologic mechanisms of acute ischemic stroke: an overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology (2010) 17:197–218. doi: 10.1016/j.pathophys.2009.12.001
70. Quan Z, Quan Y, Wei B, Fang D, Yu W, Jia H, et al. Protein-protein interaction network and mechanism analysis in ischemic stroke. Mol Med Rep. (2015) 11:29–36. doi: 10.3892/mmr.2014.2696
71. Dang H, Li K, Yu Y, Zhang Y, Liu J, Wang P, et al. Variation of pathways and network profiles reveals the differential pharmacological mechanisms of each effective component to treat middle cerebral artery ischemia-reperfusion mice. Exp Biol Med. (2016) 241:79–89. doi: 10.1177/1535370215594584
72. Yu Y, Zhang X, Li B, Zhang Y, Liu J, Li H, et al. Entropy-based divergent and convergent modular pattern reveals additive and synergistic anticerebral ischemia mechanisms. Exp Biol Med. (2016) 241:2063–74. doi: 10.1177/1535370216662361
73. Monk J, Nogales J, Palsson BO. Optimizing genome-scale network reconstructions. Nat Biotechnol. (2014) 32:447–52. doi: 10.1038/nbt.2870
74. Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet. (2015) 16:85–97. doi: 10.1038/nrg3868
75. Tang Y, Xu H, Du X, Lit L, Walker W, Lu A, et al. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: a microarray study. J Cereb Blood Flow Metab. (2006) 26:1089–102. doi: 10.1038/sj.jcbfm.9600264
76. Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature (2011) 478:483–9. doi: 10.1038/nature10523
77. Christensen S, Brodersen P, Olesen J, Paulson O.B. Cerebral apoplexy (stroke) treated with or without prolonged artificial hyperventilation: cerebrospinal fluid acid-base balance and intracranial pressure. Stroke (1973) 4:620–31. doi: 10.1161/01.STR.4.4.620
78. Fujishima M, Nakatomi Y, Tamaki K, Ogata J. Cerebral ischaemia induced by bilateral carotid occlusion in spontaneously hypertensive rats. Supra- and infratentorial metabolism and arterial acid-base balance. J Neurol Sci. (1977) 33:1–11. doi: 10.1016/0022-510X(77)90176-9
79. Straver JS, Keunen RWM, Stam CJ, Tavy DLJ, De Ruiter GR, Smith SJ, et al. Transcranial Doppler and systemic hemodynamic studies in septic shock. Neurol Res. (1996) 18:313–8. doi: 10.1080/01616412.1996.11740427
80. Williamson CA, Sheehan KM, Tipirneni R, Roark CD, Pandey AS, Thompson BG, et al. The association between spontaneous hyperventilation, delayed cerebral ischemia, and poor neurological outcome in patients with subarachnoid hemorrhage. Neurocrit Care (2015) 23:330–8. doi: 10.1007/s12028-015-0138-5
81. Cushing H. Concerning a definite regulatory mechanism of the vaso-motor entre which controls blood pressure during cerebral compression. Johns Hopkins Hospital Bulletin No. (1901) 126:290–2.
82. Gidday J, Fitzgibbons J, Shah A, Park T. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Lett. (1994) 168:221–4. doi: 10.1016/0304-3940(94)90455-3
83. Abas F, Alkan T, Goren B, Taskapilioglu O, Sarandol E, Tolunay S, et al. Neuroprotective effects of postconditioning on lipid peroxidation and apoptosis after focal cerebral ischemia/reperfusion injury in rats. Turkish Neurosurg. (2010) 20:1–8.
84. Zhan X, Ander BP, Jickling G, Turner R, Stamova B, Xu H, et al. Brief focal cerebral ischemia that simulates transient ischemic attacks in humans regulates gene expression in rat peripheral blood. J Cereb Blood Flow Metab. (2010) 30:110–8. doi: 10.1038/jcbfm.2009.189
85. Scornavacca G, Gesuete R, Orsini F, Pastorelli R, Fanelli R, De Simoni MG, et al. Proteomic analysis of mouse brain cortex identifies metabolic down-regulation as a general feature of ischemic pre-conditioning. J Neurochem. (2012) 122:1219–29. doi: 10.1111/j.1471-4159.2012.07874.x
86. Gong J, Gong S, Zhang M, Zhang L, Hu Y, Liu Y, et al. Cerebral ischemic preconditioning reduces glutamate excitotoxicity by up-regulating the uptake activity of GLT-1 in rats. Amino Acids (2014) 46:1537–45. doi: 10.1007/s00726-014-1723-1
87. Jiang T, Yu JT, Zhu XC, Zhang QQ, Tan MS, Cao L, et al. Ischemic preconditioning provides neuroprotection by induction of AMP-activated protein kinase-dependent autophagy in a rat model of ischemic stroke. Mol Neurobiol. (2014) 51:220–9. doi: 10.1007/s12035-014-8725-6
88. Li H, Jin M, Lv T, Guan J. Mechanism of focal cerebral ischemic tolerance in rats with ischemic preconditioning involves MyD88- and TRIF-dependent pathways. Exp Ther Med (2014) 6:1375–9. doi: 10.3892/etm.2013.1318
89. Shah NH, Aizenman E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl Stroke Res. (2014) 5:38–58. doi: 10.1007/s12975-013-0297-7
90. Stetler RA, Leak RK, Gan Y, Li P, Zhang F, Hu X, et al. Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Progr Neurobiol. (2014) 114:58–83. doi: 10.1016/j.pneurobio.2013.11.005
91. Mai Z, Liu H. Boolean network-based analysis of the apoptosis network: irreversible apoptosis and stable surviving. J Theor Biol. (2009) 259:760–9. doi: 10.1016/j.jtbi.2009.04.024
92. Mai Z, Liu H. Corrigendum to “Boolean network-based analysis of the apoptosis network: Irreversible apoptosis and stable surviving” [J. Theor. Biol. 259 (2009) 760-769]. J Theor Biol. (2012) 313:217. doi: 10.1016/j.jtbi.2012.08.012
94. Gräff J, Kim D, Dobbin MM. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. (2011) 91:603–49. doi: 10.1152/physrev.00012.2010
95. Kofke WA, Rie MA. Research ethics and law of healthcare system quality improvement: the conflict of cost containment and quality. Crit Care Med. (2003) 31:S143–52. doi: 10.1097/01.CCM.0000054902.85329.50
96. Rie MA, Kofke WA. Nontherapeutic quality improvement: the conflict of organizational ethics and societal rule of law. Crit Care Med. (2007) 35:S66–84. doi: 10.1097/01.CCM.0000253307.18478.54
97. Gelpke GJ, Braakman R, Habbema JDF. Comparison of outcome in two series of patients with severe head injuries. J Neurosurg. (1983) 59:745–50. doi: 10.3171/jns.1983.59.5.0745
98. Howells DW, Sena ES, O'Collins V, MacLeod MR. Improving the efficiency of the development of drugs for stroke. Int J Stroke (2012) 7:371–7. doi: 10.1111/j.1747-4949.2012.00805.x
99. Patel RAG, McMullen PW. Neuroprotection in the treatment of acute ischemic stroke. Progr Cardiovasc Dis. (2017) 59:542–8. doi: 10.1016/j.pcad.2017.04.005
100. Berkhemer OA, Fransen PSS, Beumer D, Van Den Berg LA, Lingsma HF, Yoo AJ, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. (2015) 372:11–20. doi: 10.1056/NEJMoa1411587
101. Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, et al. Targeted temperature management at 33°C versus 36°C after cardiac arrest. N Engl J Med. (2013) 369:2197–206. doi: 10.1056/NEJMoa1310519
102. Hall E, Pazara K, Braughler J. Effects of tirilazad mesylate on postischemic brain lipid peroxidation and recovery of extracellular calcium in gerbils. Stroke (1991) 22:361. doi: 10.1161/01.STR.22.3.361
103. James ML, Wang H, Venkatraman T, Song P, Lascola CD, Laskowitz DT. Brain natriuretic peptide improves long-term functional recovery after acute CNS injury in mice. J Neurotrauma (2010) 27:217–28. doi: 10.1089/neu.2009.1022
104. Werner C, Hoffman W, Kochs E, Rabito S, Miletich D. Captopril improves neurologic outcome from incomplete cerebral ischemia in rats. Stroke (1991) 22:910. doi: 10.1161/01.STR.22.7.910
105. Medcalf RL. Fibrinolysis: from blood to the brain. J Thromb Haemost (2017) 15:2089–98. doi: 10.1111/jth.13849
106. Chen Y, Zhang X, Zhang B, He G, Zhou L, Xie Y. Dexmedetomidine reduces the neuronal apoptosis related to cardiopulmonary bypass by inhibiting activation of the JAK2–STAT3 pathway. Drug Design Dev Ther. (2017) 11:2787–99. doi: 10.2147/DDDT.S140644
107. Akdemir G, Ergungor MF, Sezer M, Albayrak L, Daglioglu E, Kilinc K, et al. Therapeutic efficacy of intraventricular cyclosporine A and methylprednisolone on a global cerebral ischemia model in rats. Neurol Res. (2005) 27:827–34. doi: 10.1179/016164105X63610
108. Yoshida K, Marmarou A. Effects of tromethamine and hyperventilation on brain injury in the cat. J Neurosurg (1991) 74:87–96. doi: 10.3171/jns.1991.74.1.0087
109. Kiening KL, Schneider GH, Unterberg AW, Lanksch WR. Effect of Tromethamine (THAM) on infarct volume following permanent middle cerebral artery occlusion in rats. Acta Neurochirurg Suppl. (1997) 1997:188–90. doi: 10.1007/978-3-7091-6837-0_58
110. Hecker JG, Hall LL, Irion VR, Hecker JG, Hall LL, Irion VR. Nonviral gene delivery to the lateral ventricles in rat brain: initial evidence for widespread distribution and expression in the central nervous system. Mol Ther. (2001) 3:375–84. doi: 10.1006/mthe.2001.0272
111. Rasmussen-Torvik LJ, Stallings SC, Gordon AS, Almoguera B, Basford MA, Bielinski SJ, et al. Design and anticipated outcomes of the eMERGE-PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin Pharmacol Ther. (2014) 96:482–9. doi: 10.1038/clpt.2014.137
Keywords: neuroprotection, functional genomics, cerebral ischemia, cardiac surgery, cardiopulmonary bypass, biomarkers, systems biology, clinical trials
Citation: Kofke WA, Ren Y, Augoustides JG, Li H, Nathanson K, Siman R, Meng QC, Bu W, Yandrawatthana S, Kositratna G, Kim C and Bavaria JE (2018) Reframing the Biological Basis of Neuroprotection Using Functional Genomics: Differentially Weighted, Time-Dependent Multifactor Pathogenesis of Human Ischemic Brain Damage. Front. Neurol. 9:497. doi: 10.3389/fneur.2018.00497
Received: 07 December 2017; Accepted: 07 June 2018;
Published: 26 June 2018.
Edited by:
Barak Bar, Loyola University Medical Center, United StatesReviewed by:
Jason D. Hinman, University of California, Los Angeles, United StatesCopyright © 2018 Kofke, Ren, Augoustides, Li, Nathanson, Siman, Meng, Bu, Yandrawatthana, Kositratna, Kim and Bavaria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William A. Kofke, a29ma2VhQHVwaHMudXBlbm4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.