 Deqiang Han1,2,3
Deqiang Han1,2,3 Wei Zheng
Wei Zheng Zhiguo Chen
Zhiguo Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 10 March 2020
Sec. Cellular Neuropathology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00045
This article is part of the Research Topic Neurodegeneration: From Genetics to Molecules, Volume II View all 18 articles
Aggregation of α-Synuclein, possibly caused by disturbance of proteostasis, has been identified as a common pathological feature of Parkinson’s disease (PD). However, the initiating events of aggregation have not been fully illustrated, and this knowledge may be critical to understanding the disease mechanisms of PD. Proteostasis is essential in maintaining normal cellular metabolic functions, which regulate the synthesis, folding, trafficking, and degradation of proteins. The toxicity of the aggregating proteins is dramatically influenced by its physical and physiological status. Genetic mutations may also affect the metastable phase transition of proteins. In addition, neuroinflammation, as well as lipid metabolism and its interaction with α-Synuclein, are likely to contribute to the pathogenesis of PD. In this review article, we will highlight recent progress regarding α-Synuclein proteostasis in the context of PD. We will also discuss how the phase transition status of α-Synuclein could correlate with different functional consequences in PD.
Parkinson’s disease (PD) is a common progressive neurodegenerative disorder that affects 1–3% of the aging population (Pringsheim et al., 2014; Kalia and Lang, 2015). The clinical symptoms of PD include resting tremor, rigidity, bradykinesia and postural instability (Jankovic, 2008). One of the neuropathological hallmarks of PD is reduced dopaminergic neurons in the substantia nigra (SN) pars compacta, which causes striatal dopamine deficiency (Obeso et al., 2010; Poewe et al., 2017; Hyung Ho Yoon and Jeon, 2018). Another pathological feature is intraneuronal inclusions, such as Lewy bodies and Lewy neurites in residual dopaminergic neurons. The major constituent of Lewy body is aggregated α-Synuclein (Spillantini et al., 1997; Wakabayashi et al., 2013). Abnormally folded proteins may present with different forms, such as small oligomers, aggregates, and complex inclusions; accumulation of misfolded proteins contributes to the progression of neurodegenerative diseases including PD (Hetz and Mollereau, 2014). Proteostasis is essential in maintaining normal cellular metabolic functions that regulate the synthesis, folding, trafficking, and degradation of proteins. The toxicity of the aggregating proteins is dramatically influenced by the physical and physiological status. In addition, genetic mutations may affect the metastable phase transition of proteins. In addition to playing a central role in the neurodegenerative process of PD, α-Synuclein contributes to the initiation and persistence of inflammatory responses—another important feature of PD. Also detected in Lewy body aggregates in post-mortem brain tissues are lipid vesicles and membrane fragments (Shahmoradian et al., 2019), suggesting that lipid metabolism may be implicated in PD. In this review, we will highlight recent progress in the research area regarding α-Synuclein proteostasis and its role in the pathogenesis of PD. We will also discuss how phase transition and posttranslational modification (PTM) of α-Synuclein may correlate with different functional consequences. Additionally, we will take into account neuroinflammation and lipid metabolism and discuss its interaction with α-Synuclein in the context of PD (Figure 1).
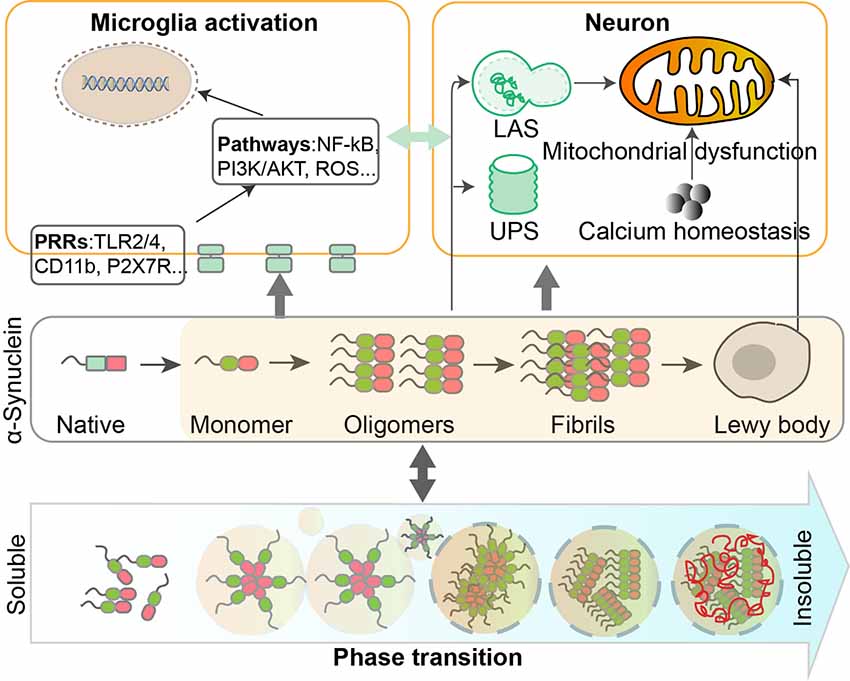
Figure 1. Dynamic regulation and phase transition of α-Synuclein are involved in the pathogenesis of Parkinson’s disease (PD). α-Synuclein consists of three domains and can undergo post-translational modifications (PTMs) such as phosphorylation, SUMOylation, nitration, and O-GlcNAcylation. The transition of α-Synuclein from a monomeric to oligomeric state and further to fibrils is related to the pathological gain of toxicity in PD. Intracellular homeostasis of α-Synuclein is maintained under the surveillance of the ubiquitin-proteasome system (UPS) and lysosomal autophagy system (LAS). Accumulation of α-Synuclein will take place when these degradation systems are damaged. α-Synuclein aggregates could impair mitochondrial functions in human dopaminergic neurons, by altering calcium homeostasis. Different forms of α-Synuclein could activate microglia through different receptors and downstream pathways, leading to inflammatory responses that contribute to neurodegeneration.
α-Synuclein is the major component of intraneuronal protein aggregates in patients with PD and plays a key function in the progression of this disease (Poewe et al., 2017). Intracellular homeostasis of α-Synuclein is maintained under intrinsic surveillance mechanisms including the ubiquitin-proteasome system (UPS) and lysosomal autophagy system (LAS). LAS seems to be more important than UPS in clearing the oligomeric assemblies. A recent study showed that inclusion formation is dependent on the concentration of α-Synuclein, whereas clearance of inclusion is mediated by the autophagy pathway, as revealed by quantitative measurement of the formation and clearance of α-Synuclein inclusions in a yeast model of PD (Perrino et al., 2019). Accumulation of α-Synuclein is largely associated with the impairment of these degradation systems (Xilouri et al., 2013). In turn, abnormal proteins can directly or indirectly interfere with the UPS mechanisms and further affect the function of related pathways, leading to irreversible changes in neuronal protein homeostasis and degeneration (Cookson, 2009; Sato et al., 2018).
α-Synuclein is normally expressed at high levels in neurons and possibly in oligodendrocytes in the CNS (Asi et al., 2014; Mehra et al., 2019). It is an intrinsically disordered protein that exists in both a soluble and a membrane-bound form in neurons (Mehra et al., 2019). The structure of α-Synuclein includes three domains. N-terminal region (1–60 Aa) that enables the protein to bind to membranes, contains seven conserved repeat sequences which form an amphipathic α-helix (Davidson et al., 1998). Non-amyloid component (NAC; 61–95) containing a highly hydrophobic motif that regulates the oligomerization and fibrillogenesis process is necessary for aggregation of α-Synuclein. The C-terminal tail (96–140) is involved in nuclear localization as well as interactions with other proteins, small molecules, and metals (Eliezer et al., 2001; Ulmer et al., 2005). The pathological gain of neurotoxicity related to α-Synuclein involves multiple biological processes. Initially, soluble α-Synuclein monomers form oligomers, then gradually accumulate into insoluble mature fibrils; eventually, α-Synuclein aggregates into large insoluble fibrils which is more toxic to neurons and can cause cell death and progressive motor impairment (Melki, 2015; Peelaerts et al., 2015; Mor et al., 2016; Karpowicz et al., 2019; Ma et al., 2019). The fibrillar form of α-Synuclein assemblies is capable of promoting aggregation of monomeric α-Synuclein in vitro and this phenomenon can spread across cells in a prion-like fashion in cell cultures and animal models (Karpowicz et al., 2019; Ma et al., 2019).
Phase separation occurs when single-phase complexes divide into a concentrated phase and a diluted phase. Eukaryotic cells use phase transition strategies to facilitate the formation of membraneless intracellular territories (Verdile et al., 2019). Through weak intermolecular interactions, multivalent proteins reach a solubility limit to form liquid condensates (Banani et al., 2017; Shin and Brangwynne, 2017). Several proteins undergoing phase transition include intrinsically disordered regions (IDRs). The IDRs often contain prion-like domains (PLDs) and low complexity domains (LCDs; Hughes et al., 2018; Maharana et al., 2018; Wang et al., 2018). Further, the N-terminal domain of α-Synuclein that mediates the aggregation process has two LCDs, indicating that α-Synuclein may, under appropriate conditions, undergo phase separation (Guerrero-Ferreira et al., 2018; Li et al., 2018).
α-Synuclein in monomeric ensembles can exist as distinct conformational phases (Jónsson et al., 2012). Aggregation of abnormally folded proteins is generally considered as a sequential oligomerization process, adding monomers to the already formed nuclei (Serio et al., 2000). The process of aggregation displays an initial lag phase in which precursor clusters assemble spontaneously. To examine such early steps in aggregation, Narayanan et al. (2019) developed a quantitative methodology that employs super-resolution imaging of fixed cells and light-sheet imaging of living cells. They found that mammalian cells have precursor clusters even under normal growth conditions, suggesting that early aggregates behave like condensates.
To further shed light on the early events of aggregation formation of α-Synuclein, recent studies showed that liquid-liquid phase separation of α-Synuclein precedes aggregation by using in vitro reconstitution and cellular models. Liquid-like droplets of α-Synuclein generated in vitro eventually transition from a liquid to a solid form that contains oligomers and fibrils. Consistently, some aggravation-related factors like low pH, phosphomimetics, and familial PD mutations also promote α-Synuclein liquid-liquid phase separation and its subsequent maturation. Furthermore, in vivo evidence demonstrated that liquid droplets of α-Synuclein transform into perinuclear aggresomes under oxidative stress. The phase transition of natural unstructured α-Synuclein and its transformation to an aggregated disease-associated state is closely correlated with the pathogenesis of PD (Ray et al., 2019).
α-Synuclein also undergoes substantial PTMs (Zhang et al., 2019), which include phosphorylation, SUMOylation, Nitration, and O-GlcNAcylation, et al. Toxicity and aggregation of α-Synuclein are largely related to these PTMs.
α-Synuclein can be phosphorylated at serine 129; phosphorylation at this position is toxic and can enhance the formation of α-Synuclein oligomers and accelerate neuronal loss. α-Synuclein can also be modified by PIAS2 that adds a small ubiquitin-like modifier (SUMO) at lysine residues. However, how SUMOylation may influence the property of α-Synuclein remains inconclusive and seemingly contradictory results have been reported. Rott et al. (2017) showed that SUMOylation can promote α-Synuclein aggregation, and meantime inhibit α-Synuclein ubiquitination and reduce its degradation. In contrast, Krumova et al. (2011) reported that SUMOylation can inhibit α-Synuclein aggregation and promote protein solubility. Further studies are needed to address this issue.
Oxidative stress may be another contributor to the pathogenesis of PD. Widespread accumulation of nitrated α-Synuclein in inclusions have been detected by using antibodies specific to nitrated tyrosine residues of α-Synuclein (Giasson et al., 2000). Further studies showed that four locations of tyrosine are susceptible to nitration (Y39, Y125, Y133, and Y136; Giasson et al., 2000; Schapira and Jenner, 2011; Bose and Beal, 2016). Abundant evidence suggests that nitration of α-Synuclein is implicated in the toxicity of aggregates. Nitration of Y-39 speeds up the oligomerization of α-Synuclein. Interestingly, a mutation of this site to cysteine residue leads to high levels of fibrillation (Zhou and Freed, 2004; Danielson et al., 2009). Nitrated α-Synuclein in the form of a monomer or dimer accelerates fibril formation and can seed the fibrillation of unmodified α-Synuclein. Further study showed that nitration-induced α-Synuclein oligomerization involves interactions between the N- and C-terminal regions of different α-Synuclein molecules. Nitration on the N- or C-terminal regions impact the order of oligomerization; for example, only dimers are formed when Y39 is not available for nitration (Burai et al., 2015). Another study showed that nitration of α-Synuclein can stabilize the formation of lower molecular weight oligomers, resulting in decreased fibril formation (Hodara et al., 2004). In all, nitration may be part of the complicated regulatory mechanisms that control the proteostasis of α-Synuclein (Burai et al., 2015).
O-GlcNAcylation is a dynamic process, in which GlcNAc is transferred by O-GlcNAc transferase (OGT) from UDP-GlcNAc to the serine and threonine residues, and subsequently removed by O-GlcNAcase (OGA; Hart et al., 2007). α-Synuclein can be O-GlcNAcylated at nine different positions (Marotta et al., 2015). Proper O-GlcNAcylation of certain proteins prevents their aggregation, and loss of this modification is a contributing factor in the development of neurodegenerative diseases. It has been verified that the O-GlcNAcylation of α-Synuclein stabilizes the monomeric state of proteins and alters the structure of α-Synuclein aggregates (Marotta et al., 2015; Zhang et al., 2019). In general, O-GlcNAcylation inhibits the aggregation of α-Synuclein. Furthermore, α-Synuclein with three O-GlcNAc modifications can prevent the aggregation of unmodified proteins. O-GlcNAcylation of α-Synuclein peptide also inhibits the toxicity of extracellular α-Synuclein fibrils (Levine et al., 2019).
The transition of α-Synuclein from a monomeric to oligomeric state leads to a pathological toxic function in PD. α-Synuclein as monomers interacts and regulates ATP synthase to augment the efficiency of ATP production under physiological conditions (Ludtmann et al., 2016). In contrast, α-Synuclein in the state of beta sheet-rich oligomers interacts with mitochondrial proteins such as ATP synthase and disturbs complex I-dependent functions. Interaction with oligomers induces oxidation of the beta subunit of ATP synthase and peroxidation of mitochondrial lipid. These events further augment the likelihood to form permeability transition pore (PTP) which is toxic to cells (Ludtmann et al., 2018). α-Synuclein fibrils also cause neurotoxicity and cell death by activating nitric oxide synthase (NOS), leading to DNA damage and polymerase-1 (PARP-1) activation. Reciprocally, PAR accelerates the fibrillation of α-Synuclein (Kam et al., 2018). It seems that different states of α-Synuclein exert various, even opposing effects in cells. The soluble monomers are harmless, whereas oligomers and fibrils are toxic, although the extent of toxicity may be different. Exactly how the order of oligomerization and change of the configuration may impact on the toxicity of α-Synuclein requires further studies.
Microglia are the major abundant CNS-specific immune cells that participate in the maintenance of brain homeostasis through mediating inflammation and/or phagocytosis. Chronic microgliosis is considered a pathological feature of PD, and the levels of inflammatory factors secreted by microglia correlate with the progression of PD (Labzin et al., 2018). Different forms of α-Synuclein exhibit distinct effects in triggering microglial phagocytosis and inflammation. Rather than oligomers of α-Synuclein which only induce upregulation of IL-1β (Krashia et al., 2019), fibrillar α-Synuclein is able to elicit strong pro-inflammatory responses in a microglial cell line (Gustot et al., 2015; Hoffmann et al., 2016; Zhou et al., 2016). Fibrillar α-Synuclein can activate NLRP3 inflammasome which leads to the cleaving of pro-inflammatory cytokines, such as IL-1β and IL-18 (Zhou et al., 2016; Chatterjee et al., 2020; Haque et al., 2020). Also, using a rat model that overexpresses human α-Synuclein, Krashia et al. found that α-Synuclein-induced inflammation precedes nigral degeneration, and administration of resolving D1, a potent lipid mediator that can resolve inflammation to promote restoration of tissue homeostasis, prevents neuronal dysfunction and motor deficits (Krashia et al., 2019). Multiple receptors and pathways are responsible for inducing pro-inflammatory responses in microglia in PD (Table 1). Both TLR2 and TLR4, whose expression levels are increased in PD patients and MPTP-administrated models, can be activated by α-Synuclein to induce sterile inflammation in PD (Kaur et al., 2017; Ferreira and Romero-Ramos, 2018). The oligomer of α-Synuclein can bind to the P2X7 receptor to activate the PI3K/AKT pathway in BV2 cells, a microglial cell line. α-Synuclein aggregates can also be internalized in autophagosomes via FcγR on microglia, which leads to activation of NF-kB pathway (Cao et al., 2012). CD36 is possibly involved in α-Synuclein-induced microglial activation but the mechanism remains elusive (Ferreira and Romero-Ramos, 2018). Prostaglandin E2 receptor subtype 2 (EP2) on microglia seems to play a critical part in neurotoxicity caused by α-Synuclein aggregation, based on in vivo and in vitro evidence (Jin et al., 2007). Aggregated nitrated α-Synuclein can induce ROS production from microglia, which is inhibited by blockade of potassium channels (Thomas et al., 2007). In turn, activated microglia and inflammation might promote α-Synuclein misfolding and aggregation. Wang et al. (2016) reported that inflammation-induced caspase 1 activation directly cleaves wild-type α-Synuclein, and the truncated α-Synuclein is more prone to aggregation and leads to toxicity in a neuronal PD cellular model. The effect of immune cells and inflammation on the spread of prion-like α-Synuclein remains largely unexplored. On the one hand, spread of α-Synuclein is determined by the dynamic net sum of seeding, propagation, and phagocytosis of fibrillar α-Synuclein, and microglia are beneficial in a sense that they can facilitate clearance of α-Synuclein aggregates and damaged neurons. On the other hand, inflammation-induced reactive species might have a negative influence on the conformation changes of α-Synuclein; but the detailed mechanisms require further investigation.
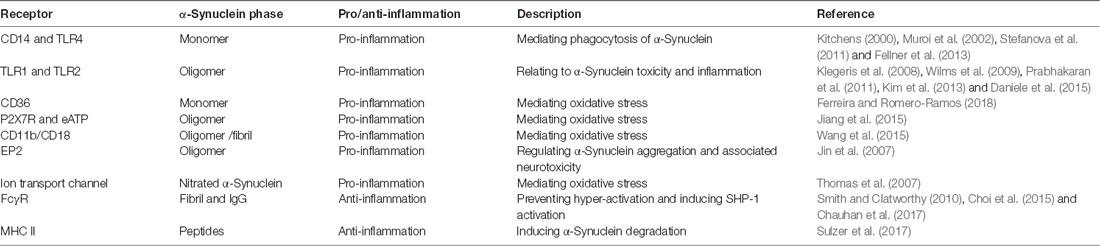
Table 1. Receptors on microglia that are involved in α-Synuclein-induced responses.
Accumulating evidence has suggested that lipid metabolism and its interaction with α-Synuclein are implicated in many aspects of PD pathogenesis. Upon binding of α-Synuclein to synthetic lipid membranes in an in vitro test, α-Synuclein undergoes a structural transition from random coil to alpha-helical secondary conformation (Davidson et al., 1998). α-Synuclein binds to small vesicles containing acidic phospholipids preferentially instead of to those with a net neutral charge. The membrane-bound form may have a higher aggregation propensity than the cytosolic form, and membrane-bound α-Synuclein can generate nuclei which are able to seed the more abundant cytosolic form (Lee et al., 2002). Interaction with the membrane is in agreement with the proposed biological functions of α-Synuclein, such as regulation of synaptic plasticity. The binding affinity of α-Synuclein to model membranes is much higher when the membrane is in a fluid phase vs. in a gel phase (Galvagnion et al., 2016). The solubility, but not the fluidity, determine the magnitude by which membrane prompts fibril formation of α-Synuclein (Galvagnion et al., 2016). This evidences demonstrated that the chemical properties of lipids determine the balance between functional and deleterious interactions of α-Synuclein with lipid membranes, allowing for a deeper understanding of how this interaction may contribute to neurodegeneration. These data implicate a possible way of involvement of inflammation in aggregate formation since inflammation-induced reactive species can directly alter the property of membranes. Another factor that may influence membrane property is aging. Hallett et al. reported, in the aging brain, an aberrant association between α-Synuclein and dopamine vesicular membrane, which was concurrent with synaptic destabilization (Hallett et al., 2019). However, overexpression of α-Synuclein without lipid deregulation does not result in the otherwise observed abnormality, suggesting that the aberrant association is lipid-dependent (Brekk et al., 2018). Lipid trafficking also seems to be involved in PD pathogenesis. Lipids are transported by Rab proteins. Chung et al. (2009) found that Rab3b is more highly expressed in A10 vs. A9 dopaminergic neurons, which could be one reason accountable for the relatively greater vulnerability of A9 neurons compared with A10 neurons; overexpression of Rab3b in A9 neurons in rats confers a protective effect and leads to improved motor functions in a PD model. In another study, overexpression of a different Rab protein, Rab1a, normalizes expression of α-Synuclein in patient neurons on a genetic background of SNCA triplication, possibly through enhanced trafficking to lysosomes (Mazzulli et al., 2016). The study points out an interesting approach to cope with the accumulation of α-Synuclein in PD.
Multiple studies have demonstrated that α-Synuclein aggregation and mitochondrial dysfunction are both important in the pathogenesis of PD. Accumulating data showed that α-Synuclein aggregation and mitochondrial defects may have a bidirectional interaction. α-Synuclein interacts with mitochondria by specifically binding to the TOM20 receptor, inhibiting mitochondrial protein import machinery, and impairing mitochondrial functions (Di Maio et al., 2016). α-Synuclein aggregation in mitochondria impairs complex I in human dopaminergic neuronal cells, consequently interfering with mitochondrial functions (Devi et al., 2008). Additionally, it was suggested that soluble, prefibrillar α-Synuclein, but not α-Synuclein monomers, impairs the retention of Ca2+ in mitochondria, and induces mitochondrial depolarization and swelling, leading to Ca2+ dependent mitochondrial dysfunction (Luth et al., 2014). On the other hand, mitochondrial defects result in oligomerization and accumulation of α-Synuclein, which in turn aggravates the dysfunction of mitochondria. Growing evidence suggests the involvement of α-Synuclein in the dynamics of mitochondria, such as mitochondrial fission, fusion, and mitophagy. Overexpression of α-Synuclein in Caenorhabditis elegans and cultured cells reduces mitochondrial fusion, resulting in fragmentation of mitochondria (Kamp et al., 2010). And these fusion deficits and mitochondrial fragmentation are rescued by overexpression of PINK1, PARKIN or DJ-1 (Kamp et al., 2010). However, the detailed picture illustrating the interaction between α-Synuclein accumulation and mitochondrial dysfunction remains obscure and requires further investigation.
The majority of PD are sporadic, and around 5–10% of PD are familial, presenting with monogenic forms of the disease (Rocha et al., 2018). More than 20 PD-related genes have been identified, including SNCA, leucine-rich repeat kinase 2 (LRRK2), glucocerebrosidase (GBA), PINK1, DJ-1 and Parkin (Li et al., 2014; Kalinderi et al., 2016; Balestrino and Schapira, 2018).
Studies showed that accumulation of insoluble α-Synuclein plays a significant role not only in the neurodegenerative process of sporadic PD but also in familial PD; the interaction between α-Synuclein and mutant genes contributes to neuronal death, dysfunction, and loss of connectivity (Stojkovska et al., 2018). The genetic mutations are mostly involved in α-Synuclein aggregation or clearance pathways, often leading to early-onset PD.
Mutations in the GBA is the single largest risk factor associated with PD (Balestrino and Schapira, 2018). A lot of studies have indicated a key role for α-Synuclein in the pathogenesis of GBA-PD. GBA encodes lysosomal enzyme GCase that catalyzes the hydrolysis of glucosylceramide (GlcCer), and mutation of this gene normally results in dysfunction of the autophagy-lysosomal pathway (Robak et al., 2017). Since degradation of α-Synuclein partially depends on autophagy, lysosomal dysfunction caused by GBA mutation could lead to α-Synuclein accumulation and aggregation (Vogiatzi et al., 2008). Induced pluripotent stem cell (iPSC)-derived neurons from GBA-deficient PD patients show a reduced GCase activity and higher levels of GlcCer as well as α-Synuclein aggregation due to lysosomal defects (Schöndorf et al., 2014). On the other hand, α-Synuclein aggregation, in turn, inhibits GCase activity, and the toxic oligomeric form of α-Synuclein is stabilized by an increased level of GlcCer, the substrate of GCase (Mazzulli et al., 2011). These data demonstrated that α-Synuclein and GCase may form a bidirectional pathogenic loop, participating in a self-propagating feedback process that eventually leads to neurodegeneration (Mazzulli et al., 2011). In addition, GBA mutations can elevate the level of α-Synuclein via altered interaction with lipid membranes in addition to lysosomal defects. It was proposed that GlcCer and another glycosphingolipid could induce toxic conversion of α-Synuclein (Suzuki et al., 2018). Recently, a study revealed that GBA deficiency may impact on the formation of α-Synuclein tetramers and related multimers (Kim et al., 2018). The major normal structure of α-Synuclein is a folded tetramer that is resistant to aggregation (Bartels et al., 2011). In comparison with tetramers, the monomer of α-Synuclein tends to aggregate and transition to insoluble deposits such as Lewy bodies (Bartels et al., 2011). Thus, the authors concluded that the accumulation of GlcCer due to GBA mutation destabilizes α-Synuclein tetramers and related multimers and increases the level of α-Synuclein monomers, eventually contributing to α-Synuclein aggregation and neurodegeneration in PD (Kim et al., 2018). Taken together, these studies highlight the mechanistic connection between GBA deficiency and α-Synuclein properties, providing unique therapeutic opportunities for reducing neurotoxicity of α-Synuclein for treatment of PD.
In this review article, we summarized how phase transition, posttranslational modifications, physical and physiological status of α-Synuclein, inflammation, lipid metabolism, and genetic mutations may correlate with different functional consequences in PD (Figure 1). Proteostasis of α-Synuclein, as involved in maintaining normal cellular metabolic functions, plays a key role in the neurodegenerative process. The evolution and transformation of different species of α-Synuclein significantly affect the pathogenesis of PD.
Further work is required to elucidate the detailed mechanisms that regulate the proteostasis of α-Synuclein, particularly the initiating events of aggregation. Genetic mutations involved in early-onset familial PD may contribute to the phase transition of α-Synuclein, especially in the early stage of PD development; and mechanistic insight into this field may help develop novel therapeutic strategies to target α-Synucleinopathy. iPSC-derived dopaminergic neurons with LRRK2 G2019S mutation present with the accelerated accumulation of α-Synuclein. Treatment with terazosin, which can activate phosphoglycerate kinase 1 (PGK1) and subsequently increase cellular ATP level, reverses the elevation of α-Synuclein (Cai et al., 2019). Importantly, a lower frequency and slower progression of PD, and reduced disease-related complications are found in individuals taking the prescribed drug terazosin (Cai et al., 2019). The molecular mechanism remains elusive but one possibility is that ATP has property of a hydrotrope and can inhibit the formation and facilitate the dissolving of aggregates (Patel et al., 2017; Hayes et al., 2018). The study exemplifies that a molecule targeting the phase transition of α-Synuclein may slow down the progression of PD clinically. With the discovery of more molecules/drugs that target different aspects of α-Synuclein phase transition and with a better understanding of the genetic regulation of α-Synucleinopathy, new interventional opportunities are promised to emerge for treatment of PD.
DH, WZ, XW, and ZC wrote the manuscript together.
Grant Sponsors: Stem Cell and Translation National Key Project (2016YFA0101403), National Natural Science Foundation of China (81973351, 81661130160, 81422014, 81561138004), Beijing Municipal Natural Science Foundation (5142005), Beijing Talents Foundation (2017000021223TD03), Support Project of High-level Teachers in Beijing Municipal Universities in the Period of 13th Five-year Plan (CIT and TCD20180333), Beijing Medical System High-Level Talent Award (2015-3-063), Beijing Municipal Health Commission Fund (PXM 2018_026283_000002), Beijing One Hundred, Thousand, and Ten Thousand Talents Fund (2018A03), Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201706), and the Royal Society-Newton Advanced Fellowship (NA150482).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Asi, Y. T., Simpson, J. E., Heath, P. R., Wharton, S. B., Lees, A. J., Revesz, T., et al. (2014). α-synuclein mRNA expression in oligodendrocytes in MSA. Glia 62, 964–970. doi: 10.1002/glia.22653
Balestrino, R., and Schapira, A. H. V. (2018). Glucocerebrosidase and Parkinson disease: molecular, clinical, and therapeutic implications. Neuroscientist 24, 540–559. doi: 10.1177/1073858417748875
Banani, S. F., Lee, H. O., Hyman, A. A., and Rosen, M. K. (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298. doi: 10.1038/nrm.2017.7
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). α-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Bose, A., and Beal, M. F. (2016). Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 139, 216–231. doi: 10.1111/jnc.13731
Brekk, O. R., Moskites, A., Isacson, O., and Hallett, P. J. (2018). Lipid-dependent deposition of α-synuclein and Tau on neuronal Secretogranin II-positive vesicular membranes with age. Sci. Rep. 8:15207. doi: 10.1038/s41598-018-33474-z
Burai, R., Ait-Bouziad, N., Chiki, A., and Lashuel, H. A. (2015). Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J. Am. Chem. Soc. 137, 5041–5052. doi: 10.1021/ja5131726
Cai, R., Zhang, Y., Simmering, J., Schultz, J., Li, Y., Fernandez-Carasa, I., et al. (2019). Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Invest. 129, 4539–4549. doi: 10.1172/jci129987
Cao, S., Standaert, D. G., and Harms, A. S. (2012). The γ chain subunit of Fc receptors is required for α-synuclein-induced pro-inflammatory signaling in microglia. J. Neuroinflammation 9:259. doi: 10.1186/1742-2094-9-259
Chatterjee, K., Roy, A., Banerjee, R., Choudhury, S., Mondal, B., Halder, S., et al. (2020). Inflammasome and α-synuclein in Parkinson’s disease: a cross-sectional study. J. Neuroimmunol. 338:577089. doi: 10.1016/j.jneuroim.2019.577089
Chauhan, P., Hu, S., Sheng, W. S., Prasad, S., and Lokensgard, J. R. (2017). Modulation of microglial cell Fcγ receptor expression following viral brain infection. Sci. Rep. 7:41889. doi: 10.1038/srep41889
Choi, Y. R., Kang, S. J., Kim, J. M., Lee, S. J., Jou, I., Joe, E. H., et al. (2015). FcγRIIB mediates the inhibitory effect of aggregated α-synuclein on microglial phagocytosis. Neurobiol. Dis. 83, 90–99. doi: 10.1016/j.nbd.2015.08.025
Chung, C. Y., Koprich, J. B., Hallett, P. J., and Isacson, O. (2009). Functional enhancement and protection of dopaminergic terminals by RAB3B overexpression. Proc. Natl. Acad. Sci. U S A 106, 22474–22479. doi: 10.1073/pnas.0912193106
Cookson, M. R. (2009). α-synuclein and neuronal cell death. Mol. Neurodegener. 4:9. doi: 10.1186/1750-1326-4-9
Daniele, S. G., Béraud, D., Davenport, C., Cheng, K., Yin, H., and Maguire-Zeiss, K. A. (2015). Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 8:ra45. doi: 10.1126/scisignal.2005965
Danielson, S. R., Held, J. M., Schilling, B., Oo, M., Gibson, B. W., and Andersen, J. K. (2009). Preferentially increased nitration of α-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal. Chem. 81, 7823–7828. doi: 10.1021/ac901176t
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. doi: 10.1074/jbc.273.16.9443
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.m710012200
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). α-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8:342ra78. doi: 10.1126/scitranslmed.aaf3634
Eliezer, D., Kutluay, E., Bussell, R. Jr., and Browne, G. (2001). Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073. doi: 10.1006/jmbi.2001.4538
Fellner, L., Irschick, R., Schanda, K., Reindl, M., Klimaschewski, L., Poewe, W., et al. (2013). Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 61, 349–360. doi: 10.1002/glia.22437
Ferreira, S. A., and Romero-Ramos, M. (2018). Microglia response during Parkinson’s disease: α-synuclein intervention. Front. Cell. Neurosci. 12:247. doi: 10.3389/fncel.2018.00247
Galvagnion, C., Brown, J. W., Ouberai, M. M., Flagmeier, P., Vendruscolo, M., Buell, A. K., et al. (2016). Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. U S A 113, 7065–7070. doi: 10.1073/pnas.1601899113
Giasson, B. I., Duda, J. E., Murray, I. V. J., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Guerrero-Ferreira, R., Taylor, N. M., Mona, D., Ringler, P., Lauer, M. E., Riek, R., et al. (2018). Cryo-EM structure of α-synuclein fibrils. Elife 7:e36402. doi: 10.7554/eLife.36402
Gustot, A., Gallea, J. I., Sarroukh, R., Celej, M. S., Ruysschaert, J.-M., and Raussens, V. (2015). Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem. J. 471, 323–333. doi: 10.1042/bj20150617
Haque, M. E., Akther, M., Jakaria, M., Kim, I. S., Azam, S., and Choi, D. K. (2020). Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov. Disord. 35, 20–33. doi: 10.1002/mds.27874
Hart, G. W., Housley, M. P., and Slawson, C. (2007). Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022. doi: 10.1038/nature05815
Hallett, P. J., Engelender, S., and Isacson, O. (2019). Lipid and immune abnormalities causing age-dependent neurodegeneration and Parkinson’s disease. J. Neuroinflammation 16, 153. doi: 10.1186/s12974-019-1532-2
Hayes, M. H., Peuchen, E. H., Dovichi, N. J., and Weeks, D. L. (2018). Dual roles for ATP in the regulation of phase separated protein aggregates in Xenopus oocyte nucleoli. Elife 7:e35224. doi: 10.7554/elife.35224
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hodara, R., Norris, E. H., Giasson, B. I., Mishizen-Eberz, A. J., Lynch, D. R., Lee, V. M.-Y., et al. (2004). Functional consequences of α-synuclein tyrosine nitration diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 279, 47746–47753. doi: 10.1074/jbc.M408906200
Hoffmann, A., Ettle, B., Bruno, A., Kulinich, A., Hoffmann, A.-C., Von Wittgenstein, J., et al. (2016). α-synuclein activates BV2 microglia dependent on its aggregation state. Biochem. Biophys. Res. Commun. 479, 881–886. doi: 10.1016/j.bbrc.2016.09.109
Hughes, M. P., Sawaya, M. R., Boyer, D. R., Goldschmidt, L., Rodriguez, J. A., Cascio, D., et al. (2018). Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 359, 698–701. doi: 10.1126/science.aan6398
Hyung Ho Yoon, J. M., and Jeon, S. R. (2018). Optogenetics to restore neural circuit function in Parkinson’s disease. J. Neurorestoratol. 6, 88–92. doi: 10.26599/JNR.2018.9040007
Jankovic, J. (2008). Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. doi: 10.1136/jnnp.2007.131045
Jiang, T., Hoekstra, J., Heng, X., Kang, W., Ding, J., Liu, J., et al. (2015). P2X7 receptor is critical in α-synuclein—mediated microglial NADPH oxidase activation. Neurobiol. Aging 36, 2304–2318. doi: 10.1016/j.neurobiolaging.2015.03.015
Jin, J., Shie, F.-S., Liu, J., Wang, Y., Davis, J., Schantz, A. M., et al. (2007). Prostaglandin E 2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated α-synuclein. J. Neuroinflammation 4:2. doi: 10.1186/1742-2094-4-2
Jónsson, S. A., Mohanty, S., and Irbäck, A. (2012). Distinct phases of free α-synuclein—a Monte Carlo study. Proteins 80, 2169–2177. doi: 10.1002/prot.24107
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kalinderi, K., Bostantjopoulou, S., and Fidani, L. (2016). The genetic background of Parkinson’s disease: current progress and future prospects. Acta Neurol. Scand. 134, 314–326. doi: 10.1111/ane.12563
Kam, T. I., Mao, X., Park, H., Chou, S. C., Karuppagounder, S. S., Umanah, G. E., et al. (2018). Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 362:eaat8407. doi: 10.1126/science.aat8407
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Karpowicz, R. J. Jr., Trojanowski, J. Q., and Lee, V. M. (2019). Transmission of α-synuclein seeds in neurodegenerative disease: recent developments. Lab. Invest. 99, 971–981. doi: 10.1038/s41374-019-0195-z
Kaur, K., Gill, J. S., Bansal, P. K., and Deshmukh, R. (2017). Neuroinflammation-A major cause for striatal dopaminergic degeneration in Parkinson’s disease. J. Neurol. Sci. 381, 308–314. doi: 10.1016/j.jns.2017.08.3251
Kim, C., Ho, D. H., Suk, J. E., You, S., Michael, S., Kang, J., et al. (2013). Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4:1562. doi: 10.1038/ncomms2534
Kim, S., Yun, S. P., Lee, S., Umanah, G. E., Bandaru, V. V. R., Yin, X., et al. (2018). GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. U S A 115, 798–803. doi: 10.1073/pnas.1700465115
Kitchens, R. L. (2000). Role of CD14 in cellular recognition of bacterial lipopolysaccharides. Chem. Immunol. 74, 61–82. doi: 10.1159/000058750
Klegeris, A., Pelech, S., Giasson, B. I., Maguire, J., Zhang, H., Mcgeer, E. G., et al. (2008). α-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol. Aging 29, 739–752. doi: 10.1016/j.neurobiolaging.2006.11.013
Krashia, P., Cordella, A., Nobili, A., La Barbera, L., Federici, M., Leuti, A., et al. (2019). Blunting neuroinflammation with resolvin D1 prevents early pathology in a rat model of Parkinson’s disease. Nat. Commun. 10:3945. doi: 10.1038/s41467-019-11928-w
Krumova, P., Meulmeester, E., Garrido, M., Tirard, M., Hsiao, H.-H., Bossis, G., et al. (2011). Sumoylation inhibits α-synuclein aggregation and toxicity. J. Cell Biol. 194, 49–60. doi: 10.1083/jcb.201010117
Labzin, L. I., Heneka, M. T., and Latz, E. (2018). Innate immunity and neurodegeneration. Annu. Rev. Med. 69, 437–449. doi: 10.1146/annurev-med-050715-104343
Lee, H. J., Choi, C., and Lee, S. J. (2002). Membrane-bound α-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 277, 671–678. doi: 10.1074/jbc.m107045200
Levine, P. M., Galesic, A., Balana, A. T., Mahul-Mellier, A. L., Navarro, M. X., De Leon, C. A., et al. (2019). α-synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 116, 1511–1519. doi: 10.1073/pnas.1808845116
Li, B., Ge, P., Murray, K. A., Sheth, P., Zhang, M., Nair, G., et al. (2018). Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 9:3609. doi: 10.1038/s41467-018-05971-2
Li, J. Q., Tan, L., and Yu, J. T. (2014). The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 9:47. doi: 10.1186/1750-1326-9-47
Ludtmann, M. H., Angelova, P. R., Horrocks, M. H., Choi, M. L., Rodrigues, M., Baev, A. Y., et al. (2018). α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 9:2293. doi: 10.1038/s41467-018-04422-2
Ludtmann, M. H., Angelova, P. R., Ninkina, N. N., Gandhi, S., Buchman, V. L., and Abramov, A. Y. (2016). Monomeric α-synuclein exerts a physiological role on brain ATP synthase. J. Neurosci. 36, 10510–10521. doi: 10.1523/jneurosci.1659-16.2016
Luth, E. S., Stavrovskaya, I. G., Bartels, T., Kristal, B. S., and Selkoe, D. J. (2014). Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 289, 21490–21507. doi: 10.1074/jbc.m113.545749
Ma, J., Gao, J., Wang, J., and Xie, A. (2019). Prion-like mechanisms in Parkinson’s disease. Front. Neurosci. 13:552. doi: 10.3389/fnins.2019.00552
Maharana, S., Wang, J., Papadopoulos, D. K., Richter, D., Pozniakovsky, A., Poser, I., et al. (2018). RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921. doi: 10.1126/science.aar7366
Marotta, N. P., Lin, Y. H., Lewis, Y. E., Ambroso, M. R., Zaro, B. W., Roth, M. T., et al. (2015). O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson’s disease. Nat. Chem. 7, 913–920. doi: 10.1038/nchem.2361
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., Mclean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and β-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Mazzulli, J. R., Zunke, F., Isacson, O., Studer, L., and Krainc, D. (2016). α-synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. U S A 113, 1931–1936. doi: 10.1073/pnas.1520335113
Mehra, S., Sahay, S., and Maji, S. K. (2019). α-synuclein misfolding and aggregation: implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 1867, 890–908. doi: 10.1016/j.bbapap.2019.03.001
Melki, R. (2015). Role of different α-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J. Parkinsons Dis. 5, 217–227. doi: 10.3233/jpd-150543
Mor, D. E., Ugras, S. E., Daniels, M. J., and Ischiropoulos, H. (2016). Dynamic structural flexibility of α-synuclein. Neurobiol. Dis. 88, 66–74. doi: 10.1016/j.nbd.2015.12.018
Muroi, M., Ohnishi, T., and Tanamoto, K. (2002). Regions of the mouse CD14 molecule required for toll-like receptor 2- and 4-mediated activation of NF-κ B. J. Biol. Chem. 277, 42372–42379. doi: 10.1074/jbc.m205966200
Narayanan, A., Meriin, A., Andrews, J. O., Spille, J.-H., Sherman, M. Y., and Cisse, I. I. (2019). A first order phase transition mechanism underlies protein aggregation in mammalian cells. Elife 8:e39695. doi: 10.7554/eLife.39695
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., et al. (2010). Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 16, 653–661. doi: 10.1038/nm.2165
Patel, A., Malinovska, L., Saha, S., Wang, J., Alberti, S., Krishnan, Y., et al. (2017). ATP as a biological hydrotrope. Science 356, 753–756. doi: 10.1126/science.aaf6846
Peelaerts, W., Bousset, L., Van der Perren, A., Moskalyuk, A., Pulizzi, R., Giugliano, M., et al. (2015). α-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. doi: 10.1038/nature14547
Perrino, G., Wilson, C., Santorelli, M., and Di Bernardo, D. (2019). Quantitative characterization of α-synuclein aggregation in living cells through automated microfluidics feedback control. Cell Rep. 27, 916.e5–927.e5. doi: 10.1016/j.celrep.2019.03.081
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Prabhakaran, K., Chapman, G. D., and Gunasekar, P. G. (2011). α-Synuclein overexpression enhances manganese-induced neurotoxicity through the NF-κB-mediated pathway. Toxicol. Mech. Methods 21, 435–443. doi: 10.3109/15376516.2011.560210
Pringsheim, T., Jette, N., Frolkis, A., and Steeves, T. D. (2014). The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov. Disord. 29, 1583–1590. doi: 10.1002/mds.25945
Ray, S., Singh, N., Pandey, S., Kumar, R., Gadhe, L., Datta, D., et al. (2019). Liquid-liquid phase separation and liquid-to-solid transition mediate α-synuclein amyloid fibril containing hydrogel formation. bioRxiv [Preprint]. doi: 10.1101/619858
Robak, L. A., Jansen, I. E., van Rooij, J., Uitterlinden, A. G., Kraaij, R., Jankovic, J., et al. (2017). Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203. doi: 10.1093/brain/awx285
Rocha, E. M., De Miranda, B., and Sanders, L. H. (2018). α-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109, 249–257. doi: 10.1016/j.nbd.2017.04.004
Rott, R., Szargel, R., Shani, V., Hamza, H., Savyon, M., Abd Elghani, F., et al. (2017). SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. U S A 114, 13176–13181. doi: 10.1073/pnas.1704351114
Sato, S., Uchihara, T., Fukuda, T., Noda, S., Kondo, H., Saiki, S., et al. (2018). Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 8:2813. doi: 10.1038/s41598-018-21325-w
Schapira, A. H., and Jenner, P. (2011). Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 26, 1049–1055. doi: 10.1002/mds.23732
Schöndorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5:4028. doi: 10.1038/ncomms5028
Serio, T. R., Cashikar, A. G., Kowal, A. S., Sawicki, G. J., Moslehi, J. J., Serpell, L., et al. (2000). Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289, 1317–1321. doi: 10.1126/science.289.5483.1317
Shahmoradian, S. H., Lewis, A. J., Genoud, C., Hench, J., Moors, T. E., Navarro, P. P., et al. (2019). Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22, 1099–1109. doi: 10.1038/s41593-019-0423-2
Shin, Y., and Brangwynne, C. P. (2017). Liquid phase condensation in cell physiology and disease. Science 357:eaaf4382. doi: 10.1126/science.aaf4382
Smith, K. G., and Clatworthy, M. R. (2010). FcγRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat. Rev. Immunol. 10, 328–343. doi: 10.1038/nri2762
Spillantini, M. G., Schmidt, M. L., Lee, V. M.-Y., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-synuclein in lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stefanova, N., Fellner, L., Reindl, M., Masliah, E., Poewe, W., and Wenning, G. K. (2011). Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 179, 954–963. doi: 10.1016/j.ajpath.2011.04.013
Stojkovska, I., Krainc, D., and Mazzulli, J. R. (2018). Molecular mechanisms of α-synuclein and GBA1 in Parkinson’s disease. Cell Tissue Res. 373, 51–60. doi: 10.1007/s00441-017-2704-y
Sulzer, D., Alcalay, R. N., Garretti, F., Cote, L., Kanter, E., Agin-Liebes, J., et al. (2017). T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546, 656–661. doi: 10.1038/nature22815
Suzuki, M., Sango, K., Wada, K., and Nagai, Y. (2018). Pathological role of lipid interaction with α-synuclein in Parkinson’s disease. Neurochem. Int. 119, 97–106. doi: 10.1016/j.neuint.2017.12.014
Thomas, M. P., Chartrand, K., Reynolds, A., Vitvitsky, V., Banerjee, R., and Gendelman, H. E. (2007). Ion channel blockade attenuates aggregated α synuclein induction of microglial reactive oxygen species: relevance for the pathogenesis of Parkinson’s disease. J. Neurochem. 100, 503–519. doi: 10.1111/j.1471-4159.2006.04315.x
Ulmer, T. S., Bax, A., Cole, N. B., and Nussbaum, R. L. (2005). Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 280, 9595–9603. doi: 10.1074/jbc.M411805200
Verdile, V., De Paola, E., and Paronetto, M. P. (2019). Aberrant phase transitions: side effects and novel therapeutic strategies in human disease. Front. Genet. 10:173. doi: 10.3389/fgene.2019.00173
Vogiatzi, T., Xilouri, M., Vekrellis, K., and Stefanis, L. (2008). Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283, 23542–23556. doi: 10.1074/jbc.M801992200
Wakabayashi, K., Tanji, K., Odagiri, S., Miki, Y., Mori, F., and Takahashi, H. (2013). The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 47, 495–508. doi: 10.1007/s12035-012-8280-y
Wang, W., Nguyen, L. T., Burlak, C., Chegini, F., Guo, F., Chataway, T., et al. (2016). Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc. Natl. Acad. Sci. U S A 113, 9587–9592. doi: 10.1073/pnas.1610099113
Wang, M., Tao, X., Jacob, M. D., Bennett, C. A., Ho, J. D., Gonzalgo, M. L., et al. (2018). Stress-induced low complexity RNA activates physiological amyloidogenesis. Cell Rep. 24, 1713.e4–1721.e4. doi: 10.1016/j.celrep.2018.07.040
Wang, Q., Qian, L., Chen, S. H., Chu, C. H., Wilson, B., Oyarzabal, E., et al. (2015). Post-treatment with an ultra-low dose of NADPH oxidase inhibitor diphenyleneiodonium attenuates disease progression in multiple Parkinson’s disease models. Brain 138, 1247–1262. doi: 10.1093/brain/awv034
Wilms, H., Rosenstiel, P., Romero-Ramos, M., Arlt, A., Schäfer, H., Seegert, D., et al. (2009). Suppression of MAP kinases inhibits microglial activation and attenuates neuronal cell death induced by α-synuclein protofibrils. Int. J. Immunopathol. Pharmacol. 22, 897–909. doi: 10.1177/039463200902200405
Xilouri, M., Brekk, O. R., and Stefanis, L. (2013). α-synuclein and protein degradation systems: a reciprocal relationship. Mol. Neurobiol. 47, 537–551. doi: 10.1007/s12035-012-8341-2
Zhang, J., Li, X., and Li, J. D. (2019). The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s diseases. Front. Neurosci. 13:381. doi: 10.3389/fnins.2019.00381
Zhou, W., and Freed, C. R. (2004). Tyrosine-to-cysteine modification of human α-synuclein enhances protein aggregation and cellular toxicity. J. Biol. Chem. 279, 10128–10135. doi: 10.1074/jbc.m307563200
Keywords: α-synuclein, phase transition, Parkinson’s disease, proteostasis, genetic mutations, inflammation
Citation: Han D, Zheng W, Wang X and Chen Z (2020) Proteostasis of α-Synuclein and Its Role in the Pathogenesis of Parkinson’s Disease. Front. Cell. Neurosci. 14:45. doi: 10.3389/fncel.2020.00045
Received: 24 September 2019; Accepted: 18 February 2020;
Published: 10 March 2020.
Edited by:
Karla Guadalupe Carvajal, National Institute of Pediatrics (Mexico), MexicoReviewed by:
Tatsuro Mutoh, Fujita Health University, JapanCopyright © 2020 Han, Zheng, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiguo Chen, Y2hlbnpoaWd1b0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.