
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 13 March 2020
Sec. Cellular Neurophysiology
Volume 14 - 2020 | https://doi.org/10.3389/fncel.2020.00039
This article is part of the Research Topic Epilepsy and Neurodevelopmental Diseases View all 11 articles
 Anna Fassio1,2*†
Anna Fassio1,2*† Antonio Falace3†
Antonio Falace3† Alessandro Esposito1,4
Alessandro Esposito1,4 Davide Aprile1,4Renzo Guerrini3,5
Davide Aprile1,4Renzo Guerrini3,5 Fabio Benfenati2,4
Fabio Benfenati2,4Autophagy is a highly conserved degradative process that conveys dysfunctional proteins, lipids, and organelles to lysosomes for degradation. The post-mitotic nature, complex and highly polarized morphology, and high degree of specialization of neurons make an efficient autophagy essential for their homeostasis and survival. Dysfunctional autophagy occurs in aging and neurodegenerative diseases, and autophagy at synaptic sites seems to play a crucial role in neurodegeneration. Moreover, a role of autophagy is emerging for neural development, synaptogenesis, and the establishment of a correct connectivity. Thus, it is not surprising that defective autophagy has been demonstrated in a spectrum of neurodevelopmental disorders, often associated with early-onset epilepsy. Here, we discuss the multiple roles of autophagy in neurons and the recent experimental evidence linking neurodevelopmental disorders with epilepsy to genes coding for autophagic/lysosomal system-related proteins and envisage possible pathophysiological mechanisms ranging from synaptic dysfunction to neuronal death.
Macroautophagy (henceforth autophagy) is a highly conserved cellular process that tackles dysfunctional proteins, lipids, and organelles to lysosomes for degradation. Substrates are initially isolated by a double membrane, the phagophore, which subsequently elongates and surrounds the substrates with a membranous structure, the autophagosome (AP; Dikic and Elazar, 2018). APs are transient organelles destined to fuse with the lysosome for the degradation of their contents. Autophagy is virtually active in all cell types to ensure homeostasis and has been implicated in protein and organelle quality control, development and differentiation, aging, and immunity. Autophagy is modulated by nutrients and growth factors and levels of AMP/ATP sensed by mammalian target of rapamycin complex1 (mTORC1) and AMP-dependent protein kinase (AMPK), respectively (Menzies et al., 2017). A scheme of the autophagy pathway is reported in Figure 1.
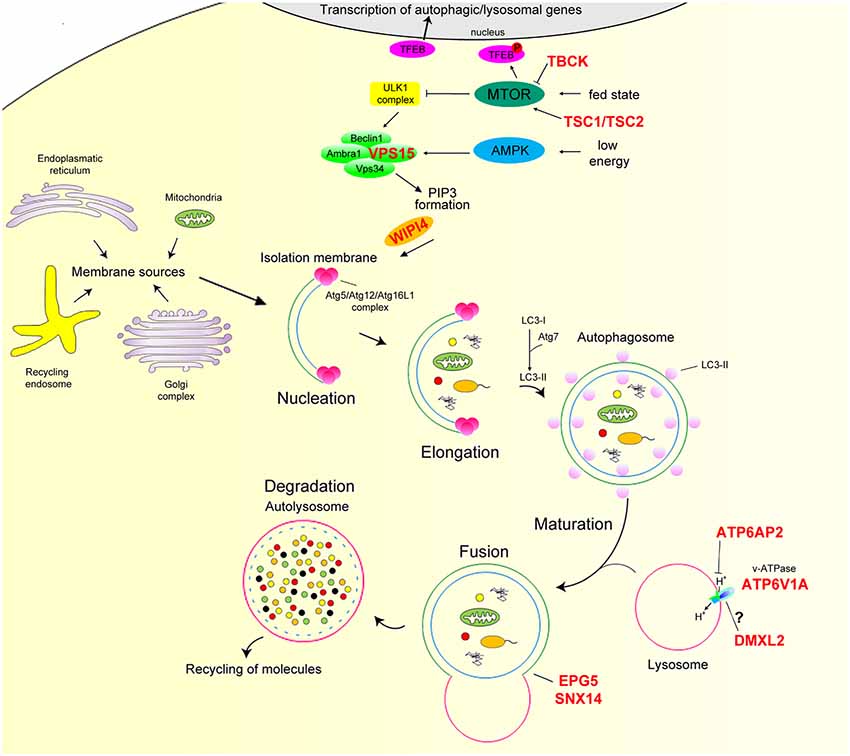
Figure 1. Overview of the mammalian autophagy pathway. In nutrient-rich conditions (fed state) mTOR phosphorylates TFEB preventing its nuclear translocation. In nutrient depletion or low-energy state, mTOR is inactive, and TFEB translocates to the nucleus leading to the transcription of many autophagy and lysosomal genes. mTOR inhibition together with AMPK activation positively regulate the ULK1 complex. Induction of this complex regulates the recruitment of the Beclin1/Ambta1/VSP34/VPS15 complex to the phagophore and, hence, the production of PI3P and downstream autophagy effectors Atg5/Atg12/Atg16L1 via the binding of WIPI proteins. This step is essential for the conversion of LC3-I to LC3-II through Atg7 and its conjugation to phagophore membrane. The membranes of these structures appear to have multiple sources, such as the endoplasmic reticulum, Golgi apparatus and trans-Golgi network, endosomal compartment, and mitochondria. LC3-II attracts components of the autophagy machinery and is required for elongation and closure of the phagophore membrane. Mature autophagosome finally fuses with the lysosome, forming the autolysosome, where autophagic cargo is degraded and then released back to the cytoplasm to be re-used by the cell. The proton gradient imposed by the lysosomal v-ATPase is essential for proteolysis as hydrolase activity strictly relies on acidic pH. Autophagy genes mutated in neurodevelopmental disorders with epilepsy are marked in red. AMPK, AMP-dependent protein kinase; mTORC1, mammalian target of rapamycin complex 1; TFEB, transcription factor EB; ULK, mammalian homologs of the Caenorhabditis elegans uncoordinated-51 kinase.
Dysfunctional autophagy has been associated with several pathologies and most neurodegenerative diseases. Neurons appear to be particularly dependent on autophagy since their post-mitotic nature makes them highly sensitive to the accumulation of toxic proteins and damaged organelles. The complex and polarized neuronal architecture poses specific challenges for an efficient cargo recycling. In neurons, APs are preferentially formed at synaptic terminals, and are transported to the cell soma, where they fuse to lysosomes for degradation. Here, we review the physiological role of autophagy in neurons and discuss recent experimental evidence linking neurodevelopmental disorders with epilepsy to genes of the autophagy/lysosomal systems (Table 1).
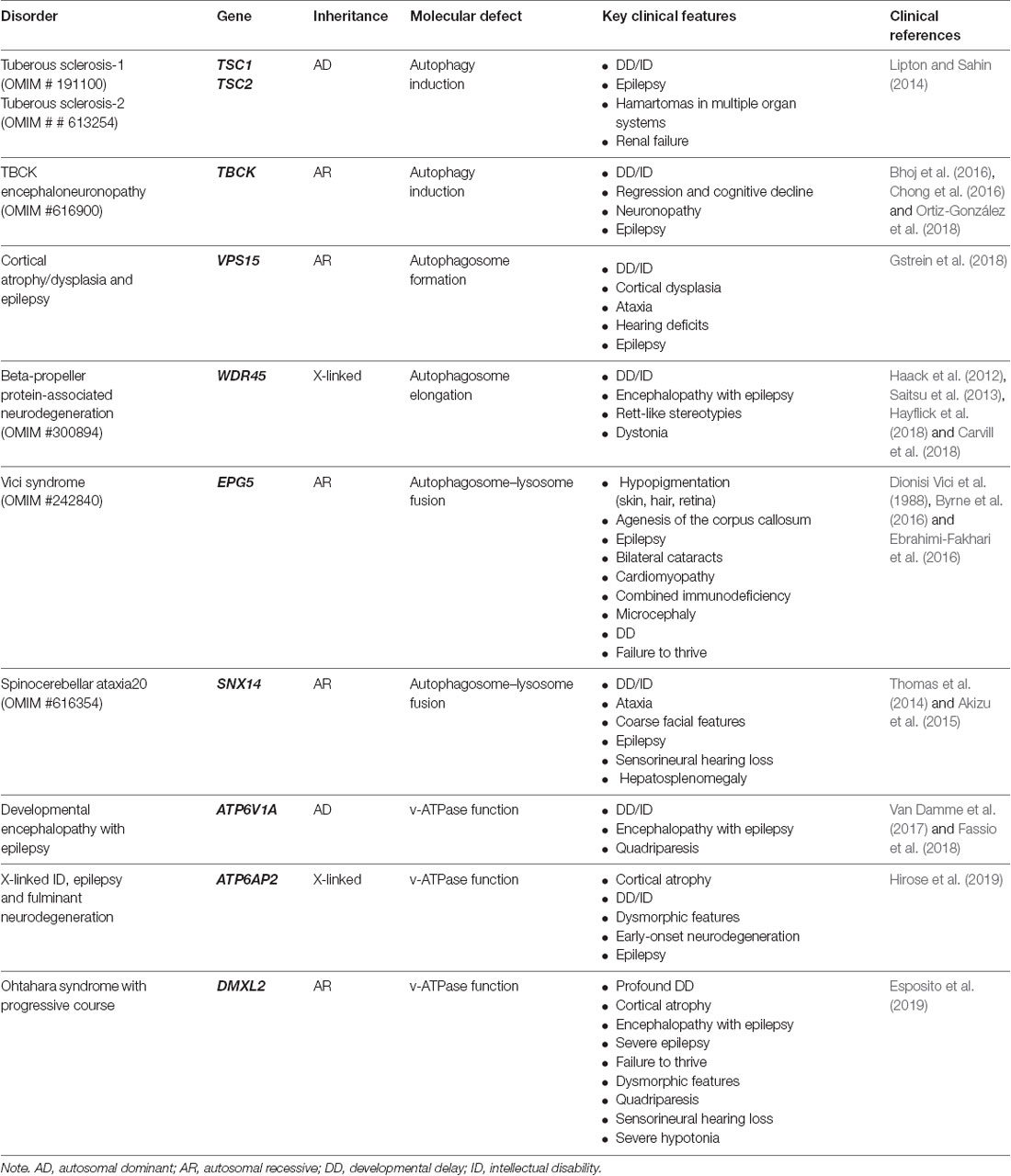
Table 1. List autophagy/lysosomal genes involved in neurodevelopmental disorders with epilepsy.
The central nervous system (CNS) requires autophagy to maintain its homeostasis. Since the ubiquitous deletion of core autophagy genes is lethal at the embryonic or perinatal stages, several nervous system-specific knockout mouse models have been developed to explore the roles of autophagy in CNS. Removal of Atg5 and Atg7 in neuronal precursor cells (NPCs) leads to the accumulation of cytoplasmic inclusion bodies, with neurodegeneration and progressive motor deficits, further pointing to autophagy as a key quality control system in neurons over their lifespan (Hara et al., 2006; Komatsu et al., 2006). In subsequent models, generated by targeting distinct genes of the core autophagic pathway (Fimia et al., 2007; Liang et al., 2010; Joo et al., 2016), similarly reduced survival and early-onset, progressive neurodegeneration occurred, although the underlying pathophysiological basis varied according to the specific gene targeted. Altogether these models emphasize autophagy as a major cellular process protecting against neurodegeneration, in line with the evidence that in human defective autophagy underlies the accumulation of protein aggregates in several neurodegenerative disorders (Rubinsztein et al., 2005; Kiriyama and Nochi, 2015).
Balanced differentiation, proliferation, and cell death rates in the developing brain are essential for neurogenesis. The autophagy machinery interacts with developmental signals involved in cell fate decisions, including Wnt, Sonic hedgehog, TGFB, and FGF (Kiyono et al., 2009; Gao et al., 2010; Jimenez-Sanchez et al., 2012; Zhang et al., 2012). Studies in animal models have disclosed the pivotal role of autophagy in neuronal proliferation and in sustaining the post-natal pool of NPCs. Loss of Ambra1, a Beclin1 activator, resulted in severe defects of the neural tube development, ubiquitinated protein accumulation, unbalanced cell proliferation, and excessive apoptosis, as a consequence of autophagy impairment (Fimia et al., 2007). In this model, impairment of basal autophagy induced hyperproliferation as indirect consequence of misregulated recycling of transcriptional factors (Cecconi et al., 2007; Fimia et al., 2007). Notch signaling is a master regulator of neurogenesis and neuronal development (Bray, 2006; Ables et al., 2011). Autophagy regulates Notch degradation and defects in the autophagy machinery impact on NPC fate (Wu et al., 2016). Impairing AP formation by in utero knockdown of Atg5 harms β-Catenin stability, thus, leading to inhibited differentiation and increased proliferation of NPCs in the developing cortex (Lv et al., 2015). These in vivo findings suggest that a multi-level interaction between autophagy, cell proliferation, and cell death occurs during mammalian neural development. As reviewed elsewhere, an important role for autophagy in adult neurogenesis has also emerged (Dhaliwal et al., 2017; Menzies et al., 2017).
Neurons have a uniquely polarized morphology characterized by extended and highly elaborated axonal and dendritic arborizations, and neuronal homeostasis is critical for establishment and maintenance of their polarized structures (Lee et al., 2013; Maday, 2016). From the earlier phases of neurodevelopment, a highly efficient autophagy is required to allow membrane trafficking events, axonal guidance, and establishment of brain connectivity (Dragich et al., 2016). Axonal APs undergo robust retrograde motility toward the soma, driven by the active motor dynein. Moreover, mutations in autophagy genes cause pathological processes associated with long-range white matter defects.
While autophagy in axonal development and homeostasis has been extensively studied, recent findings have also pointed out the key role of autophagy in ciliogenesis. In neurons, cilia are involved in cortical patterning, neurogenesis, neuronal maturation, and cerebellar development (Lee and Gleeson, 2010). The autophagy impairment due to somatic-activating mutations in MTOR leads to abnormal accumulation of the OFD1 protein at centriolar satellites and disruption of neuronal ciliogenesis. Impaired ciliogenesis abrogates Wnt signaling, which is required for neuronal polarization, and underlies cortical dyslamination reported in patients (Park et al., 2018).
Autophagy not only regulates early neuronal development but also plays specific, multiple, and largely unexplored roles at synapses. As recently reviewed, dysfunctional autophagy at both pre- and post-synaptic sites leads to aging and neurodegeneration (Nikoletopoulou et al., 2015; Vijayan and Verstreken, 2017; Azarnia Tehran et al., 2018; Liang and Sigrist, 2018). Degradation of postsynaptic neurotransmitter receptors involves trafficking in autophagosomal structures (Rowland, 2006; Shehata et al., 2012, 2018; Hui et al., 2019). At dopaminergic presynaptic sites, autophagy shapes synapse ultrastructure and modulates neurotransmitter release, while at glutamatergic synapses mTOR-regulated autophagy promotes spine pruning during development (Hernandez et al., 2012; Tang et al., 2014). The presynaptic proteins endophilin and its partner synaptojanin, known to regulate synaptic vesicle (SV) endocytosis and recycling, turned out to positively regulate synaptic autophagy, suggesting a functional link between SV cycling and autophagy. Endophilin induces the membrane curvature that recruits Atg3 and Atg8 to initiate synaptic AP generation (Murdoch et al., 2016; Soukup et al., 2016). Synaptojanin promotes synaptic autophagy by removing Atg18 from preautophagosomal structures necessary for AP maturation (George et al., 2014; Vanhauwaert et al., 2017). Conversely, the active zone protein Bassoon has been proposed to inhibit autophagy (Okerlund et al., 2017). At presynaptic sites, a role for autophagy in the degradation of SV proteins has been suggested, and the small GTPase Rab26 was reported to cluster SVs and direct them to preautophagosomal structures for degradation (Binotti et al., 2015; Lüningschrör et al., 2017). In addition, endosomal microautophagy, a chaperone-mediated form of autophagy, which directly targets proteins to the endo-lysosomal system, has been described to degrade misfolded synaptic proteins and regulate neurotransmission at the Drosophila neuromuscular junction (Uytterhoeven et al., 2015). The biogenesis of APs occurs in nerve terminals (Katsumata et al., 2010; Shehata et al., 2012), and synaptic APs retrogradely transport endocytosed elements to the neuronal soma for signaling (Wang et al., 2015). Whether SV cycling and synaptic autophagy are reciprocally regulated and how they crosstalk with somatic autophagy is a matter of investigation. In a recent article, selective induction of autophagy at the presynaptic site has been shown to specifically target damaged proteins, thus maintaining synapse integrity and function (Hoffmann et al., 2019). The discovery that neuronal autophagy and formation of APs at synapses are activity dependent (Shehata et al., 2018) suggests that synaptic autophagy may be regulated by long-term synaptic plasticity underlying learning and memory formation. However, whether autophagy stimulates or suppresses memory processes and its relationship with nutrient signaling pathways is still controversial. Fasting has been shown to induce autophagy in the hypothalamus, but to inhibit it in the hippocampus and cerebral cortex, where memory formation and consolidation occur. BDNF-mediated suppression of autophagy is required for the growth factor effects on synaptic plasticity and memory enhancement both in vitro and in vivo (Nikoletopoulou et al., 2017). Glatigny and coworkers recently showed that, in hippocampal neurons, autophagy is induced by synaptic plasticity paradigms and necessary for novel memory formation (Glatigny et al., 2019). These recent data suggest that autophagy is involved in the regulation of synaptic strength and that its dysregulation may impact on the plasticity of the network and on the excitation/inhibition balance.
In the last decade, several single-gene disorders of the autophagy pathway—defined as “congenital disorders of autophagy”—have been identified through next-generation sequencing. Genetic defects affect a range of functional steps from early phases of autophagy induction to autolysosome formation. The associated disorders, which are clinically heterogeneous, mainly affect the central and peripheral nervous systems, but often cause multi-systemic involvement (Ebrahimi-Fakhari et al., 2016). Structural brain abnormalities, developmental delay, intellectual disability, severe epilepsy, and progressive impairment in relation to neurodegeneration are common features of this class of disorders. Members of the autophagy process involved in neurodevelopmental disorders with epilepsy are highlighted in Figure 1 and discussed below.
A direct link between autophagy and epileptogenesis was first supported by studies showing that rapamycin, an inhibitor of the mTOR pathway and a powerful autophagy inducer, strongly modulates seizures in several models (Giorgi et al., 2015). Germline and somatic mutations in genes of the mTOR pathway have been identified in patients with various epileptic disorders (Parrini et al., 2016), and a direct contribution of defective autophagy has been confirmed (Yasin et al., 2013; Park et al., 2018). Hypofunctional mutations in TSC1 or TSC2 in tuberous sclerosis result in the uncontrolled activation of the mTORC1 pathway (Lipton and Sahin, 2014) and subsequent inhibition of autophagy directly linked to epileptogenesis in a forebrain-specific conditional TSC1 mouse model (McMahon et al., 2012).
Mutations in TBCK cause a neurodevelopmental syndrome with intellectual disability, coarse face, congenital hypotonia, leukoencephalopathy, progressive motor neuronopathy, and seizures (Bhoj et al., 2016; Chong et al., 2016; Ortiz-González et al., 2018). As suggested by bioinformatic analysis, TBCK encodes a putative Rab GTPase-activating protein, although its function remains elusive. Loss-of-function mutations in TBCK lead to inhibition of mTORC1 and, thus, to uncontrolled autophagy induction in patient-derived fibroblasts (Bhoj et al., 2016; Ortiz-González et al., 2018). In this model, loss of TBCK results in increased number of APs accompanied by an augmented autophagic flux insensitive to pro-autophagic stimuli (Ortiz-González et al., 2018). Glycosylated proteins were not properly degraded in TBCK patients’ fibroblasts, and storage of lipofuscin was observed in patient’s neurons (Beck-Wödl et al., 2018; Ortiz-González et al., 2018), suggesting that dysregulated autophagy leads to a storage disease phenotype (Teinert et al., 2020).
The signaling pathway adapting the autophagic response to nutrients and energy levels focuses on the phosphorylation of the ULK1 complex, a process which controls the recruitment of the VPS34/VPS15/Ambra1/Beclin1 complex to the phagophore and the formation of PI3P and downstream autophagy effectors through the binding of WD-repeat phosphoinositide-interacting (WIPI) proteins (Menzies et al., 2017; Figure 1). Gstrein et al. (2018) identified a recessive homozygous mutation in VSP15 (L1224R) in a single patient with severe cortical atrophy and dysplasia, optic nerve atrophy, intellectual disability, spasticity, ataxia, muscle wasting, and seizures. The L1224R mutation leads to an accumulation of autophagy substrates in patient’s fibroblasts. In the same study, a forebrain-specific conditional Vps15 mouse model was developed revealing that loss of Vps15 resulted in severe cortical atrophy accompanied by autophagic impairment and progressive degeneration of the hippocampus and cortex (Gstrein et al., 2018).
Mutations in X-linked gene WDR45, encoding WIPI4, cause beta-propeller protein-associated neurodegeneration (BPAN; Haack et al., 2012; Saitsu et al., 2013). BPAN is a variant of neurodegeneration with brain iron accumulation spectrum (Hayflick et al., 2018) and is characterized by a bi-phasic development. After an initial epileptic encephalopathy in the childhood, progressive neurodegeneration and Parkinsonism develop in adulthood (Carvill et al., 2018). Studies in WDR45 patients’ fibroblasts and neurons derived from their reprogramming showed that loss of WDR45 leads to higher levels of cell iron and oxidative stress, accompanied by mitochondrial abnormalities, autophagic impairment, and dysfunctional lysosomes (Seibler et al., 2018). In mice, deletion of Wdr45 in the brain results in axonal pathology and accumulation of autophagy substrates in neurons (Zhao et al., 2015).
Biallelic EPG5 mutations in the Vici syndrome, together with recessive SNX14 variants in cerebellar ataxia and intellectual disability syndrome, affect the late stages of autophagy. Vici syndrome is a severe progressive neurodevelopmental multisystem disorder featuring agenesis of the corpus callosum, bilateral cataracts, hypertrophic and/or dilated cardiomyopathy, combined immunodeficiency, and hypopigmentation (Dionisi Vici et al., 1988; Byrne et al., 2016; Ebrahimi-Fakhari et al., 2016). Profound developmental delay, progressive microcephaly, and failure to thrive are common features and suggest a neurodegenerative component following the prominent neurodevelopmental defect. Two-thirds of patients develop seizures, often evolving as epileptic encephalopathy (Byrne et al., 2016). The EPG5 protein acts as a tethering factor that determines the fusion specificity of APs with late endosomes/lysosomes (Wang et al., 2016), and in vivo loss of EPG5 results in block of the autophagic pathway, progressive motor deficit, and neurodegeneration (Zhao et al., 2013). Bi-allelic mutations in SNX14 are the cause of autosomal-recessive childhood-onset spinocerebellar ataxia 20 (Thomas et al., 2014; Akizu et al., 2015). Patients showed progressive cerebellar neurodegeneration, developmental delay, intellectual disability, and seizures (Akizu et al., 2015). SNX14 encodes a protein, of the sorting nexin family and binds lysosomal membrane phosphatidylinositol residues, that is enriched in AP-containing cell fraction where it mediates lysosome–AP fusion (Mas et al., 2014). In SNX14 patient-derived neurons, lysosomal enlargement and autophagic dysfunction were reported. This phenotype was also observed in the Snx14-zebrafish model, where it leads to progressive Purkinje cell degeneration, suggesting that impaired autophagy finally results in neuronal cell death (Akizu et al., 2015). In cultured mouse neurons, loss of Snx14 decreases intrinsic excitability and impairs both excitatory and inhibitory synaptic transmission (Huang et al., 2014).
The vacuole H+-adenosine triphosphatases (v-ATPase) is a proton pump responsible for acidification of intracellular organelles and secretory granules that regulates several cellular processes such as protein trafficking, maturation, and degradation (Forgac, 2007). Acidification of lysosomes by v-ATPase is essential for autophagy progression, and inhibiting v-ATPase activity is a widely used treatment to mimic a block of autophagy. In neurons, v-ATPase is expressed by SVs and allows neurotransmitter loading and SV trafficking (Bodzęta et al., 2017). V-ATPase is a multimeric complex composed by a cytosolic domain (v1), responsible for ATP hydrolysis, and a transmembrane domain (v0), responsible for H+ transport. Recessive mutations in ATP6V1A, coding for the “A” subunit of the v1 sub-complex, have been first described in patients with cutis laxa, dysmorphic features, and seizures in the context of a severe condition with premature lethality (Van Damme et al., 2017). Subsequently, we described de novo heterozygous mutations in ATP6V1A in four patients with developmental delay and epilepsy with variable of severity, ranging from mild intellectual disability and epilepsy to early-onset epileptic encephalopathies accompanied by myelination defects and brain atrophy. Pathogenic mutations affect lysosomal homeostasis in patients’ cells and impair neurite development and synaptic contacts when expressed in murine neurons. The mutations associated with the severe phenotype result in loss of function and autophagy impairment (Fassio et al., 2018). On the contrary, covalent targeting of ATP6V1A has been recently shown to activate autophagy by increasing v-ATPase catalytic activity and inhibiting mTORC1 activation (Chung et al., 2019).
A de novo deletion variant of the v-ATPase accessory protein ATP6AP2 has been found in a patient with neurodevelopmental disorder characterized by fulminant degeneration (Hirose et al., 2019). This patient exhibited mild facial dysmorphisms, early-onset intractable seizures, and spasticity. Sequential MRI scans documented progressive brain shrinkage with thin corpus callosum and hypomyelination. The authors, by employing both patient’s iPSC-derived neural cells and murine knockdown models, demonstrated that ATP6AP2 is a key regulator of v-ATPase function in the CNS, and that its loss results in lysosomal and autophagic defects. In these models, the loss of ATP6AP2 impairs stem cell self-renewal and neuronal survival with a strong dependence on the dosage of the transcripts.
In addition to ATP6AP2, v-ATPase assembly and activity relies on several parameters, including kinase activity, nutrient and stress levels, extra- and intra-cellular pH, and accessory proteins that interact with v0 and v1 components (McGuire et al., 2017). We recently demonstrated that DMXL2, a member of WD40 protein family known to regulate v-ATPase trafficking and activity (Yan et al., 2009; Einhorn et al., 2012; Tuttle et al., 2014), is mutated in children with severe developmental and epileptic encephalopathy, associating Ohtahara syndrome, and profound developmental delay with a progressive course leading to premature mortality. MRI scans in these patients showed thin corpus callosum, hypomyelination, and progressive brain shrinkage. Loss of DMXL2 protein in patients’ fibroblasts results in impaired autophagy, and modeling DMXL2 loss in murine neurons recapitulates defective autophagy and affects neurite development and synaptic connectivity (Esposito et al., 2019). While the complete loss of Dmxl2 is embryonically lethal in mice (Tata et al., 2014; Gobé et al., 2019), heterozygous Dmxl2 mice show macrocephaly and corpus callosum dysplasia, confirming the DMXL2 role in brain development (Kannan et al., 2017).
Altogether, these pieces of evidence support a primary role of autophagy dysregulation in epileptogenesis and suggest that severity of the clinical manifestations variably evolving in a neurodegenerative disorder might depend on different timing and specificity of molecular events underlying epilepsy and neurodegeneration. Defects altering early stages of neuronal development and, therefore, synaptic activity could underlie pro-epileptogenic changes in neuronal circuitries followed by progressive accumulation of autophagy substrates and consequent neuronal stress and degeneration. Our hypothesis is that the spectrum of phenotypes and clinical severities of the epileptic syndromes associated with mutations of autophagy genes primarily derive from an initial synaptic dysfunction, with structural and functional synaptic alterations that, depending on gene dosage and/or severity of the pathogenic mutations, may turn into neuronal damage with degeneration and death. Future work on disease murine models and/or patient-derived neurons needs to be performed to unravel the cellular and molecular mechanisms linking autophagy failure to brain hyperexcitability, seizures, and fulminant neurodegeneration and to evaluate the ability of autophagy inducers as novel therapeutic strategies for these intractable disorders.
AFas and AFal wrote the manuscript and prepared the table and the figure. AE, DA, and RG revised the manuscript. FB coordinated the preparation of the review and revised the manuscript for submission.
This work was supported by Fondazione Telethon, GGP19120 2019 and ERA-NET-NEURON SNAREOPATHIES 2017 to FB; IRCCS San Martino 5x100 2016 to AFas.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ables, J. L., Breunig, J. J., Eisch, A. J., and Rakic, P. (2011). Not(ch) just development: notch signalling in the adult brain. Nat. Rev. Neurosci. 12, 269–283. doi: 10.1038/nrn3024
Akizu, N., Cantagrel, V., Zaki, M. S., Al-Gazali, L., Wang, X., Rosti, R. O., et al. (2015). Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat. Genet. 47, 528–534. doi: 10.1038/ng.3256
Azarnia Tehran, D., Kuijpers, M., and Haucke, V. (2018). Presynaptic endocytic factors in autophagy and neurodegeneration. Curr. Opin. Neurobiol. 48, 153–159. doi: 10.1016/j.conb.2017.12.018
Beck-Wödl, S., Harzer, K., Sturm, M., Buchert, R., Rieß, O., Mennel, H.-D., et al. (2018). Homozygous TBC1 domain-containing kinase (TBCK) mutation causes a novel lysosomal storage disease—a new type of neuronal ceroid lipofuscinosis (CLN15)? Acta Neuropathol. Commun. 6:145. doi: 10.1186/s40478-018-0646-6
Bhoj, E. J., Li, D., Harr, M., Edvardson, S., Elpeleg, O., Chisholm, E., et al. (2016). Mutations in TBCK, encoding TBC1-domain-containing kinase, lead to a recognizable syndrome of intellectual disability and hypotonia. Am. J. Hum. Genet. 98, 782–788. doi: 10.1016/j.ajhg.2016.03.016
Binotti, B., Pavlos, N. J., Riedel, D., Wenzel, D., Vorbrüggen, G., Schalk, A. M., et al. (2015). The GTPase Rab26 links synaptic vesicles to the autophagy pathway. Elife 4:e05597. doi: 10.7554/eLife.05597
Bodzęta, A., Kahms, M., and Klingauf, J. (2017). The presynaptic v-ATPase reversibly disassembles and thereby modulates exocytosis but is not part of the fusion machinery. Cell Rep. 20, 1348–1359. doi: 10.1016/j.celrep.2017.07.040
Bray, S. J. (2006). Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689. doi: 10.1038/nrm2009
Byrne, S., Jansen, L., U-King-Im, J.-M., Siddiqui, A., Lidov, H. G. W., Bodi, I., et al. (2016). EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain 139, 765–781. doi: 10.1093/brain/awv393
Carvill, G. L., Liu, A., Mandelstam, S., Schneider, A., Lacroix, A., Zemel, M., et al. (2018). Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR45. Epilepsia 59, e5–e13. doi: 10.1111/epi.13957
Cecconi, F., Di Bartolomeo, S., Nardacci, R., Fuoco, C., Corazzari, M., Giunta, L., et al. (2007). A novel role for autophagy in neurodevelopment. Autophagy 3, 505–507. doi: 10.4161/auto.4616
Chong, J. X., Caputo, V., Phelps, I. G., Stella, L., Worgan, L., Dempsey, J. C., et al. (2016). Recessive inactivating mutations in TBCK, encoding a Rab GTPase-activating protein, cause severe infantile syndromic encephalopathy. Am. J. Hum. Genet. 98, 772–781. doi: 10.1016/j.ajhg.2016.01.016
Chung, C. Y.-S., Shin, H. R., Berdan, C. A., Ford, B., Ward, C. C., Olzmann, J. A., et al. (2019). Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 15, 776–785. doi: 10.1038/s41589-019-0308-4
Dhaliwal, J., Trinkle-Mulcahy, L., and Lagace, D. C. (2017). Autophagy and adult neurogenesis: discoveries made half a century ago yet in their infancy of being connected. Brain Plast. 3, 99–110. doi: 10.3233/bpl-170047
Dikic, I., and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364. doi: 10.1038/s41580-018-0003-4
Dionisi Vici, C., Sabetta, G., Gambarara, M., Vigevano, F., Bertini, E., Boldrini, R., et al. (1988). Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am. J. Med. Genet. 29, 1–8. doi: 10.1002/ajmg.1320290102
Dragich, J. M., Kuwajima, T., Hirose-Ikeda, M., Yoon, M. S., Eenjes, E., Bosco, J. R., et al. (2016). Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. Elife 5:e14810. doi: 10.7554/eLife.14810
Ebrahimi-Fakhari, D., Saffari, A., Wahlster, L., Lu, J., Byrne, S., Hoffmann, G. F., et al. (2016). Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain 139, 317–337. doi: 10.1093/brain/awv371
Einhorn, Z., Trapani, J. G., Liu, Q., and Nicolson, T. (2012). Rabconnectin3 promotes stable activity of the H+ pump on synaptic vesicles in hair cells. J. Neurosci. 32, 11144–11156. doi: 10.1523/JNEUROSCI.1705-12.2012
Esposito, A., Falace, A., Wagner, M., Gal, M., Mei, D., Conti, V., et al. (2019). Biallelic DMXL2 mutations impair autophagy and cause Ohtahara syndrome with progressive course. Brain 142, 3876–3891. doi: 10.1093/brain/awz326
Fassio, A., Esposito, A., Kato, M., Saitsu, H., Mei, D., Marini, C., et al. (2018). De novo mutations of the ATP6V1A gene cause developmental encephalopathy with epilepsy. Brain 141, 1703–1718. doi: 10.1093/brain/awy092
Fimia, G. M., Stoykova, A., Romagnoli, A., Giunta, L., Di Bartolomeo, S., Nardacci, R., et al. (2007). Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125. doi: 10.1038/nature05925
Forgac, M. (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929. doi: 10.1038/nrm2272
Gao, C., Cao, W., Bao, L., Zuo, W., Xie, G., Cai, T., et al. (2010). Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol. 12, 781–790. doi: 10.1038/ncb2082
George, A. A., Hayden, S., Holzhausen, L. C., Ma, E. Y., Suzuki, S. C., and Brockerhoff, S. E. (2014). Synaptojanin 1 is required for endolysosomal trafficking of synaptic proteins in cone photoreceptor inner segments. PLoS One 9:e84394. doi: 10.1371/journal.pone.0084394
Giorgi, F. S., Biagioni, F., Lenzi, P., Frati, A., and Fornai, F. (2015). The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J. Neural Transm. 122, 849–862. doi: 10.1007/s00702-014-1312-1
Glatigny, M., Moriceau, S., Rivagorda, M., Ramos-Brossier, M., Nascimbeni, A. C., Lante, F., et al. (2019). Autophagy is required for memory formation and reverses age-related memory decline. Curr. Biol. 29, 435.e8–448.e8. doi: 10.1016/j.cub.2018.12.021
Gobé, C., Elzaiat, M., Meunier, N., André, M., Sellem, E., Congar, P., et al. (2019). Dual role of DMXL2 in olfactory information transmission and the first wave of spermatogenesis. PLoS Genet. 15:e1007909. doi: 10.1371/journal.pgen.1007909
Gstrein, T., Edwards, A., Přistoupilová, A., Leca, I., Breuss, M., Pilat-Carotta, S., et al. (2018). Mutations in Vps15 perturb neuronal migration in mice and are associated with neurodevelopmental disease in humans. Nat. Neurosci. 21, 207–217. doi: 10.1038/s41593-017-0053-5
Haack, T. B., Hogarth, P., Kruer, M. C., Gregory, A., Wieland, T., Schwarzmayr, T., et al. (2012). Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 91, 1144–1149. doi: 10.1016/j.ajhg.2012.10.019
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Hayflick, S. J., Kurian, M. A., and Hogarth, P. (2018). “Neurodegeneration with brain iron accumulation,” in Handbook of Clinical Neurology, eds D. H. Geschwind, H. L. Paulson and C. Klein (New York, NY: Elsevier), 293–305.
Hernandez, D., Torres, C. A., Setlik, W., Cebrián, C., Mosharov, E. V., Tang, G., et al. (2012). Regulation of presynaptic neurotransmission by macroautophagy. Neuron 74, 277–284. doi: 10.1016/j.neuron.2012.02.020
Hirose, T., Cabrera-Socorro, A., Chitayat, D., Lemonnier, T., Féraud, O., Cifuentes-Diaz, C., et al. (2019). ATP6AP2 variant impairs CNS development and neuronal survival to cause fulminant neurodegeneration. J. Clin. Invest. 129, 2145–2162. doi: 10.1172/jci79990
Hoffmann, S., Orlando, M., Andrzejak, E., Bruns, C., Trimbuch, T., Rosenmund, C., et al. (2019). Light-activated ROS production induces synaptic autophagy. J. Neurosci. 39, 2163–2183. doi: 10.1523/JNEUROSCI.1317-18.2019
Huang, H.-S., Yoon, B.-J., Brooks, S., Bakal, R., Berrios, J., Larsen, R. S., et al. (2014). Snx14 regulates neuronal excitability, promotes synaptic transmission, and is imprinted in the brain of mice. PLoS One 9:e98383. doi: 10.1371/journal.pone.0098383
Hui, K. K., Takashima, N., Watanabe, A., Chater, T. E., Matsukawa, H., Nekooki-Machida, Y., et al. (2019). GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABAA receptor trafficking and social behavior. Sci. Adv. 5:eaau8237. doi: 10.1126/sciadv.aau8237
Jimenez-Sanchez, M., Menzies, F. M., Chang, Y.-Y., Simecek, N., Neufeld, T. P., and Rubinsztein, D. C. (2012). The Hedgehog signalling pathway regulates autophagy. Nat. Commun. 3:1200. doi: 10.1038/ncomms2212
Joo, J. H., Wang, B., Frankel, E., Ge, L., Xu, L., Iyengar, R., et al. (2016). The noncanonical role of ULK/ATG1 in ER-to-golgi trafficking is essential for cellular homeostasis. Mol. Cell 62, 491–506. doi: 10.1016/j.molcel.2016.04.020
Kannan, M., Bayam, E., Wagner, C., Rinaldi, B., Kretz, P. F., Tilly, P., et al. (2017). WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc. Natl. Acad. Sci. U S A 114, E9308–E9317. doi: 10.1073/pnas.1713625114
Katsumata, K., Nishiyama, J., Inoue, T., Mizushima, N., Takeda, J., and Yuzaki, M. (2010). Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy 6, 378–385. doi: 10.4161/auto.6.3.11262
Kiriyama, Y., and Nochi, H. (2015). The function of autophagy in neurodegenerative diseases. Int. J. Mol. Sci. 16, 26797–26812. doi: 10.3390/ijms161125990
Kiyono, K., Suzuki, H. I., Matsuyama, H., Morishita, Y., Komuro, A., Kano, M. R., et al. (2009). Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 69, 8844–8852. doi: 10.1158/0008-5472.can-08-4401
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Lee, J. H., and Gleeson, J. G. (2010). The role of primary cilia in neuronal function. Neurobiol. Dis. 38, 167–172. doi: 10.1016/j.nbd.2009.12.022
Lee, K.-M., Hwang, S.-K., and Lee, J.-A. (2013). Neuronal autophagy and neurodevelopmental disorders. Exp. Neurobiol. 22, 133–142. doi: 10.5607/en.2013.22.3.133
Liang, C.-C., Wang, C., Peng, X., Gan, B., and Guan, J.-L. (2010). Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 285, 3499–3509. doi: 10.1074/jbc.m109.072389
Liang, Y., and Sigrist, S. (2018). Autophagy and proteostasis in the control of synapse aging and disease. Curr. Opin. Neurobiol. 48, 113–121. doi: 10.1016/j.conb.2017.12.006
Lipton, J. O., and Sahin, M. (2014). The neurology of mTOR. Neuron 84, 275–291. doi: 10.1016/j.neuron.2014.09.034
Lüningschrör, P., Binotti, B., Dombert, B., Heimann, P., Perez-Lara, A., Slotta, C., et al. (2017). Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Nat. Commun. 8:678. doi: 10.1038/s41467-017-00689-z
Lv, X., Jiang, H., Li, B., Liang, Q., Wang, S., Zhao, Q., et al. (2015). The crucial role of Atg5 in cortical neurogenesis during early brain development. Sci. Rep. 4:6010. doi: 10.1038/srep06010
Maday, S. (2016). Mechanisms of neuronal homeostasis: autophagy in the axon. Brain Res. 1649, 143–150. doi: 10.1016/j.brainres.2016.03.047
Mas, C., Norwood, S. J., Bugarcic, A., Kinna, G., Leneva, N., Kovtun, O., et al. (2014). Structural basis for different phosphoinositide specificities of the PX domains of sorting nexins regulating G-protein signaling. J. Biol. Chem. 289, 28554–28568. doi: 10.1074/jbc.m114.595959
McGuire, C., Stransky, L., Cotter, K., and Forgac, M. (2017). Regulation of V-ATPase activity. Front. Biosci. 22, 609–622. doi: 10.2741/4506
McMahon, J., Huang, X., Yang, J., Komatsu, M., Yue, Z., Qian, J., et al. (2012). Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J. Neurosci. 32, 15704–15714. doi: 10.1523/JNEUROSCI.2392-12.2012
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi: 10.1016/j.neuron.2017.01.022
Murdoch, J. D., Rostosky, C. M., Gowrisankaran, S., Arora, A. S., Soukup, S.-F., Vidal, R., et al. (2016). Endophilin-A deficiency induces the Foxo3a-Fbxo32 network in the brain and causes dysregulation of autophagy and the ubiquitin-proteasome system. Cell Rep. 17, 1071–1086. doi: 10.1016/j.celrep.2016.09.058
Nikoletopoulou, V., Papandreou, M.-E., and Tavernarakis, N. (2015). Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 22, 398–407. doi: 10.1038/cdd.2014.204
Nikoletopoulou, V., Sidiropoulou, K., Kallergi, E., Dalezios, Y., and Tavernarakis, N. (2017). Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metab. 26, 230.e5–242.e5. doi: 10.1016/j.cmet.2017.06.005
Okerlund, N. D., Schneider, K., Leal-Ortiz, S., Montenegro-Venegas, C., Kim, S. A., Garner, L. C., et al. (2017). Bassoon controls presynaptic autophagy through Atg5. Neuron 93, 897.e7–913.e7. doi: 10.1016/j.neuron.2017.01.026
Ortiz-González, X. R., Tintos-Hernández, J. A., Keller, K., Li, X., Foley, A. R., Bharucha-Goebel, D. X., et al. (2018). Homozygous boricua TBCK mutation causes neurodegeneration and aberrant autophagy. Ann. Neurol. 83, 153–165. doi: 10.1002/ana.25130
Park, S. M., Lim, J. S., Ramakrishina, S., Kim, S. H., Kim, W. K., Lee, J., et al. (2018). Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron 99, 83.e7–97.e7. doi: 10.1016/j.neuron.2018.05.039
Parrini, E., Conti, V., Dobyns, W. B., and Guerrini, R. (2016). Genetic basis of brain malformations. Mol. Syndromol. 7, 220–233. doi: 10.1159/000448639
Rowland, A. M. (2006). Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J. Neurosci. 26, 1711–1720. doi: 10.1523/JNEUROSCI.2279-05.2006
Rubinsztein, D. C., DiFiglia, M., Heintz, N., Nixon, R. A., Qin, Z.-H., Ravikumar, B., et al. (2005). Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1, 11–22. doi: 10.4161/auto.1.1.1513
Saitsu, H., Nishimura, T., Muramatsu, K., Kodera, H., Kumada, S., Sugai, K., et al. (2013). De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449. doi: 10.1038/ng.2562
Seibler, P., Burbulla, L. F., Dulovic, M., Zittel, S., Heine, J., Schmidt, T., et al. (2018). Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain 141, 3052–3064. doi: 10.1093/brain/awy230
Shehata, M., Abdou, K., Choko, K., Matsuo, M., Nishizono, H., and Inokuchi, K. (2018). Autophagy enhances memory erasure through synaptic destabilization. J. Neurosci. 38, 3809–3822. doi: 10.1523/JNEUROSCI.3505-17.2018
Shehata, M., Matsumura, H., Okubo-Suzuki, R., Ohkawa, N., and Inokuchi, K. (2012). Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J. Neurosci. 32, 10413–10422. doi: 10.1523/JNEUROSCI.4533-11.2012
Soukup, S.-F., Kuenen, S., Vanhauwaert, R., Manetsberger, J., Hernández-Díaz, S., Swerts, J., et al. (2016). A LRRK2-dependent endophilina phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 92, 829–844. doi: 10.1016/j.neuron.2016.09.037
Tang, G., Gudsnuk, K., Kuo, S.-H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Tata, B., Huijbregts, L., Jacquier, S., Csaba, Z., Genin, E., Meyer, V., et al. (2014). Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol. 12:e1001952. doi: 10.1371/journal.pbio.1001952
Teinert, J., Behne, R., Wimmer, M., and Ebrahimi-Fakhari, D. (2020). Novel insights into the clinical and molecular spectrum of congenital disorders of autophagy. J. Inherit. Metab. Dis. 43, 51–62. doi: 10.1002/jimd.12084
Thomas, A. C., Williams, H., Setó-Salvia, N., Bacchelli, C., Jenkins, D., O’Sullivan, M., et al. (2014). Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am. J. Hum. Genet. 95, 611–621. doi: 10.1016/j.ajhg.2014.10.007
Tuttle, A. M., Hoffman, T. L., and Schilling, T. F. (2014). Rabconnectin-3a regulates vesicle endocytosis and canonical Wnt signaling in zebrafish neural crest migration. PLoS Biol. 12:e1001852. doi: 10.1371/journal.pbio.1001852
Uytterhoeven, V., Lauwers, E., Maes, I., Miskiewicz, K., Melo, M. N., Swerts, J., et al. (2015). Hsc70–4 deforms membranes to promote synaptic protein turnover by endosomal microautophagy. Neuron 88, 735–748. doi: 10.1016/j.neuron.2015.10.012
Van Damme, T., Gardeitchik, T., Mohamed, M., Guerrero-Castillo, S., Freisinger, P., Guillemyn, B., et al. (2017). Mutations in ATP6V1E1 or ATP6V1A cause autosomal-recessive cutis laxa. Am. J. Hum. Genet. 100, 216–227. doi: 10.1016/j.ajhg.2016.12.010
Vanhauwaert, R., Kuenen, S., Masius, R., Bademosi, A., Manetsberger, J., Schoovaerts, N., et al. (2017). The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 36, 1392–1411. doi: 10.15252/embj.201695773
Vijayan, V., and Verstreken, P. (2017). Autophagy in the presynaptic compartment in health and disease. J. Cell Biol. 216, 1895–1906. doi: 10.1083/jcb.201611113
Wang, T., Martin, S., Papadopulos, A., Harper, C. B., Mavlyutov, T. A., Niranjan, D., et al. (2015). Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type A. J. Neurosci. 35, 6179–6194. doi: 10.1523/JNEUROSCI.3757-14.2015
Wang, Z., Miao, G., Xue, X., Guo, X., Yuan, C., Wang, Z., et al. (2016). The vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol. Cell 63, 781–795. doi: 10.1016/j.molcel.2016.08.021
Wu, X., Fleming, A., Ricketts, T., Pavel, M., Virgin, H., Menzies, F. M., et al. (2016). Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 7:10533. doi: 10.1038/ncomms10533
Yan, Y., Denef, N., and Schüpbach, T. (2009). The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev. Cell 17, 387–402. doi: 10.1016/j.devcel.2009.07.001
Yasin, S. A., Ali, A. M., Tata, M., Picker, S. R., Anderson, G. W., Latimer-Bowman, E., et al. (2013). mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol. 126, 207–218. doi: 10.1007/s00401-013-1135-4
Zhang, J., Liu, J., Liu, L., McKeehan, W. L., and Wang, F. (2012). The fibroblast growth factor signaling axis controls cardiac stem cell differentiation through regulating autophagy. Autophagy 8, 690–691. doi: 10.4161/auto.19290
Zhao, Y. G., Sun, L., Miao, G., Ji, C., Zhao, H., Sun, H., et al. (2015). The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 11, 881–890. doi: 10.1080/15548627.2015.1047127
Keywords: epilepsy, autophagy, lysosome, neuron development, synapse
Citation: Fassio A, Falace A, Esposito A, Aprile D, Guerrini R and Benfenati F (2020) Emerging Role of the Autophagy/Lysosomal Degradative Pathway in Neurodevelopmental Disorders With Epilepsy. Front. Cell. Neurosci. 14:39. doi: 10.3389/fncel.2020.00039
Received: 09 December 2019; Accepted: 10 February 2020;
Published: 13 March 2020.
Edited by:
Eleonora Palma, Sapienza University of Rome, ItalyReviewed by:
Alexander Dityatev, German Center for Neurodegenerative Diseases (DZNE), GermanyCopyright © 2020 Fassio, Falace, Esposito, Aprile, Guerrini and Benfenati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Fassio, YWZhc3Npb0B1bmlnZS5pdA==
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.